The Use of a γ-Al2O3 and MgO Mixture in the Catalytic Conversion of 1,1,1,2-Tetrafluoroethane (HFC-134a)



 and
and
Abstract
:1. Introduction
2. Results and Discussion
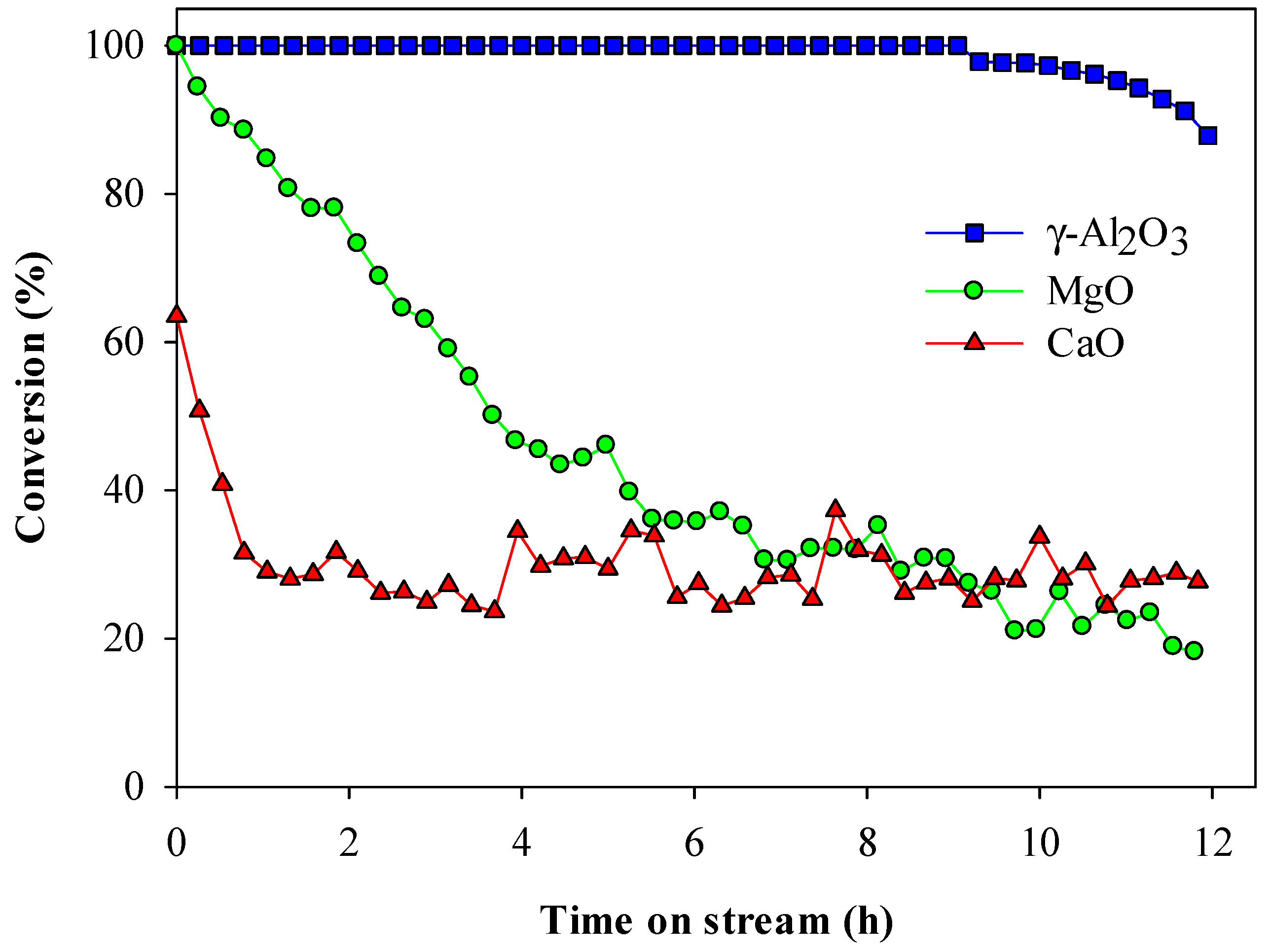
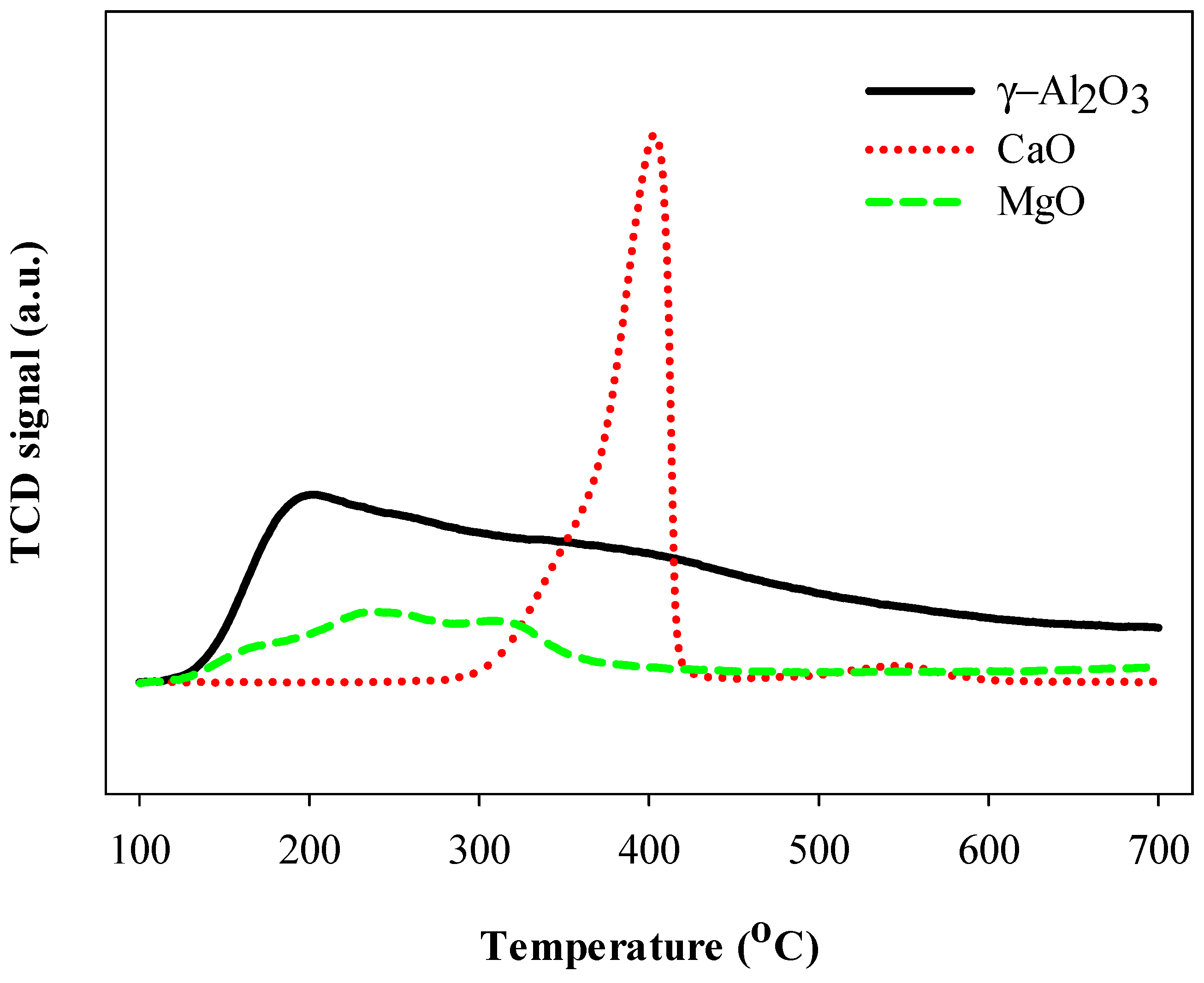
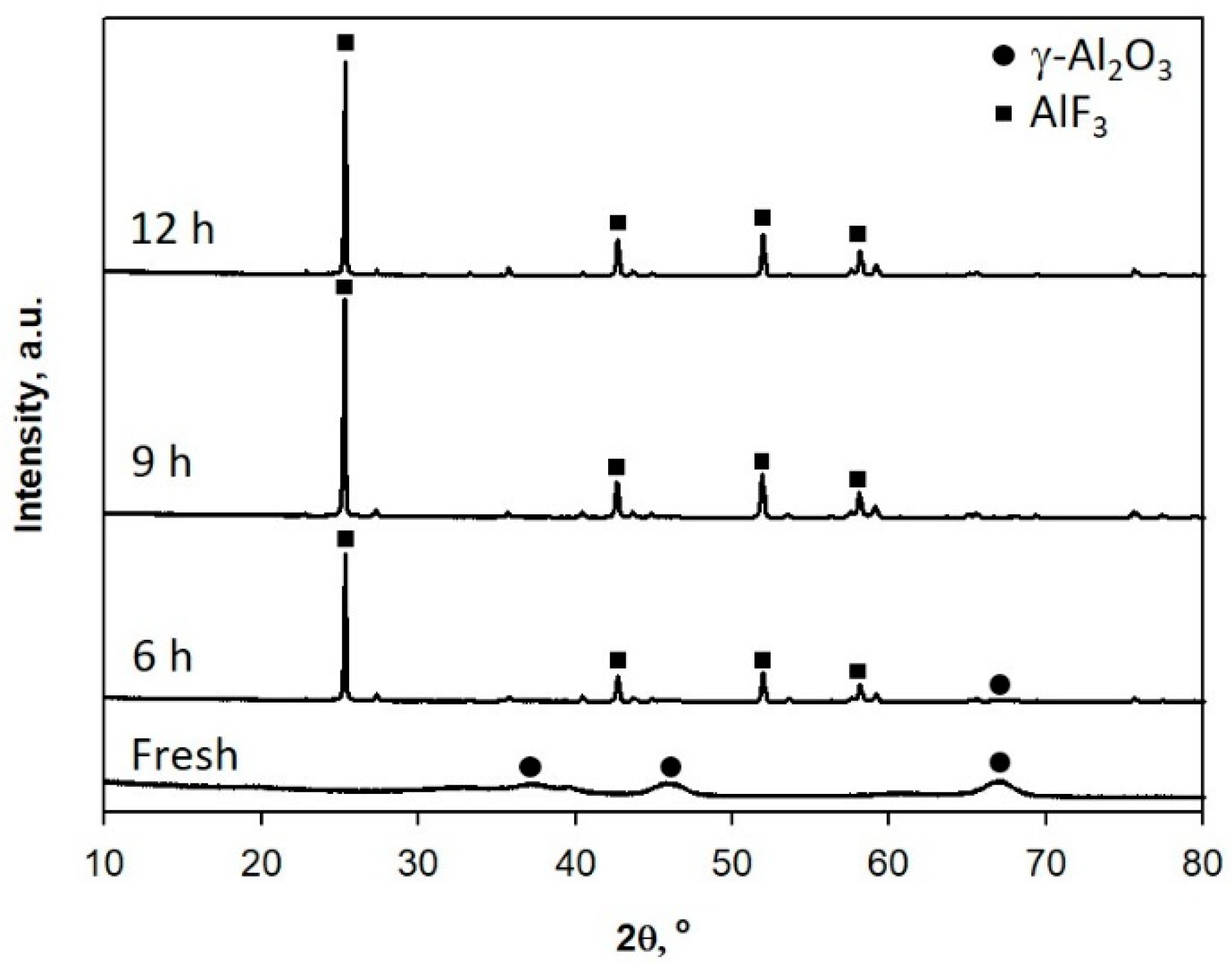
2.1. Thermal and Catalytic Decomposition of HFC-134a over Single Bed Catalyst
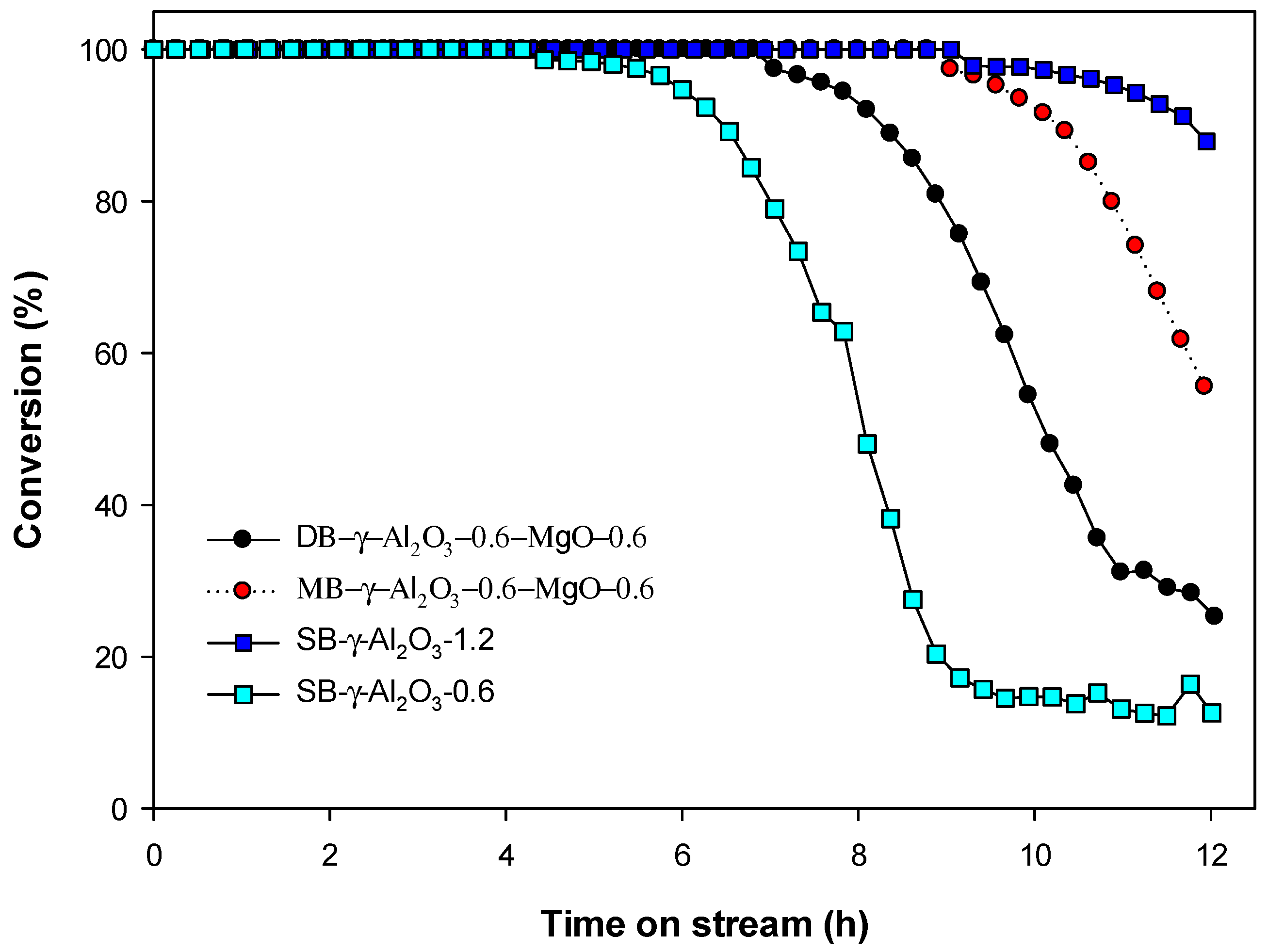
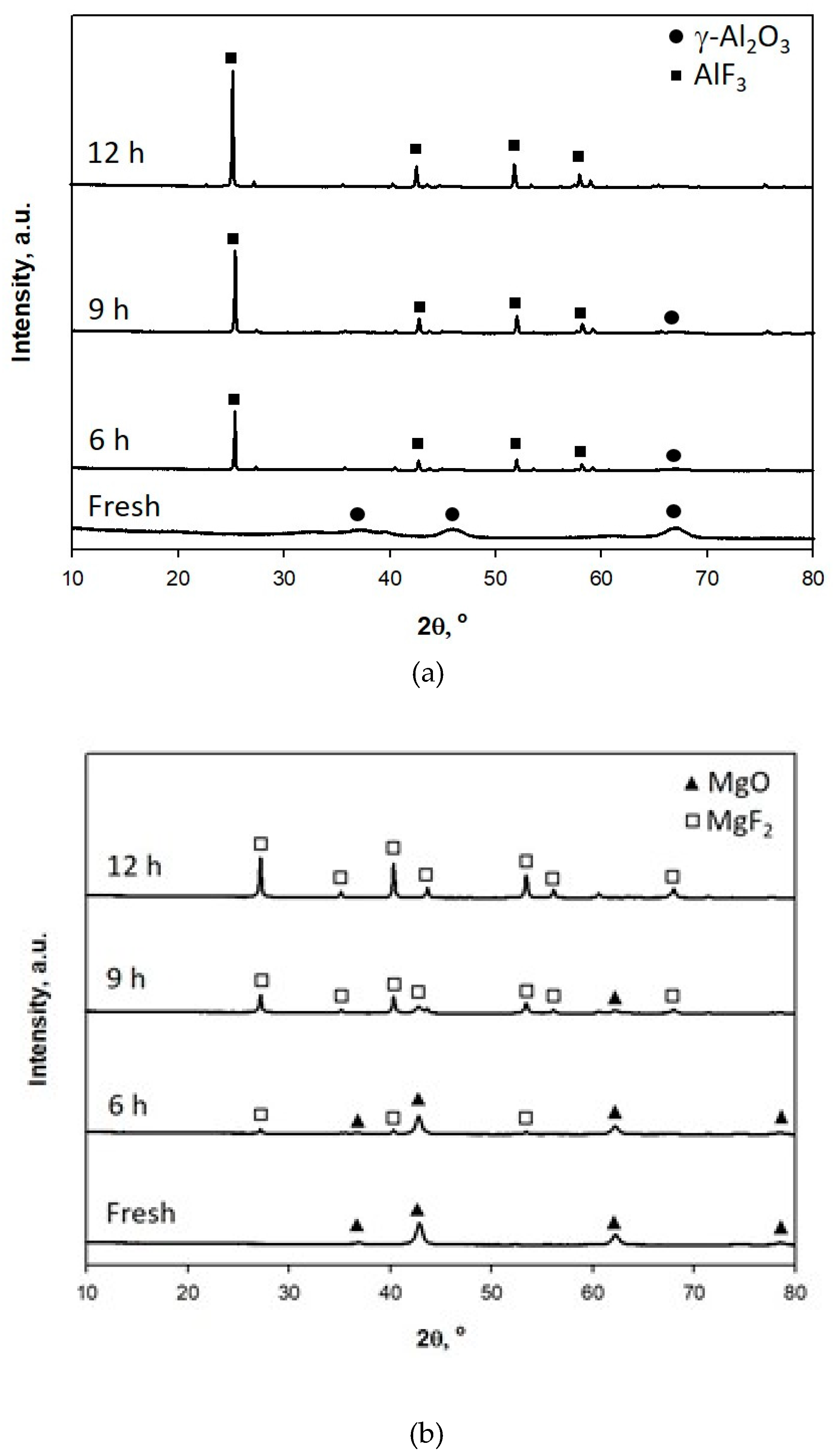
2.2. The Use of Al2O3 -MgO Catalysts on the Catalytic Decomposition of HFC-134a
3. Experimental
3.1. Catalysts
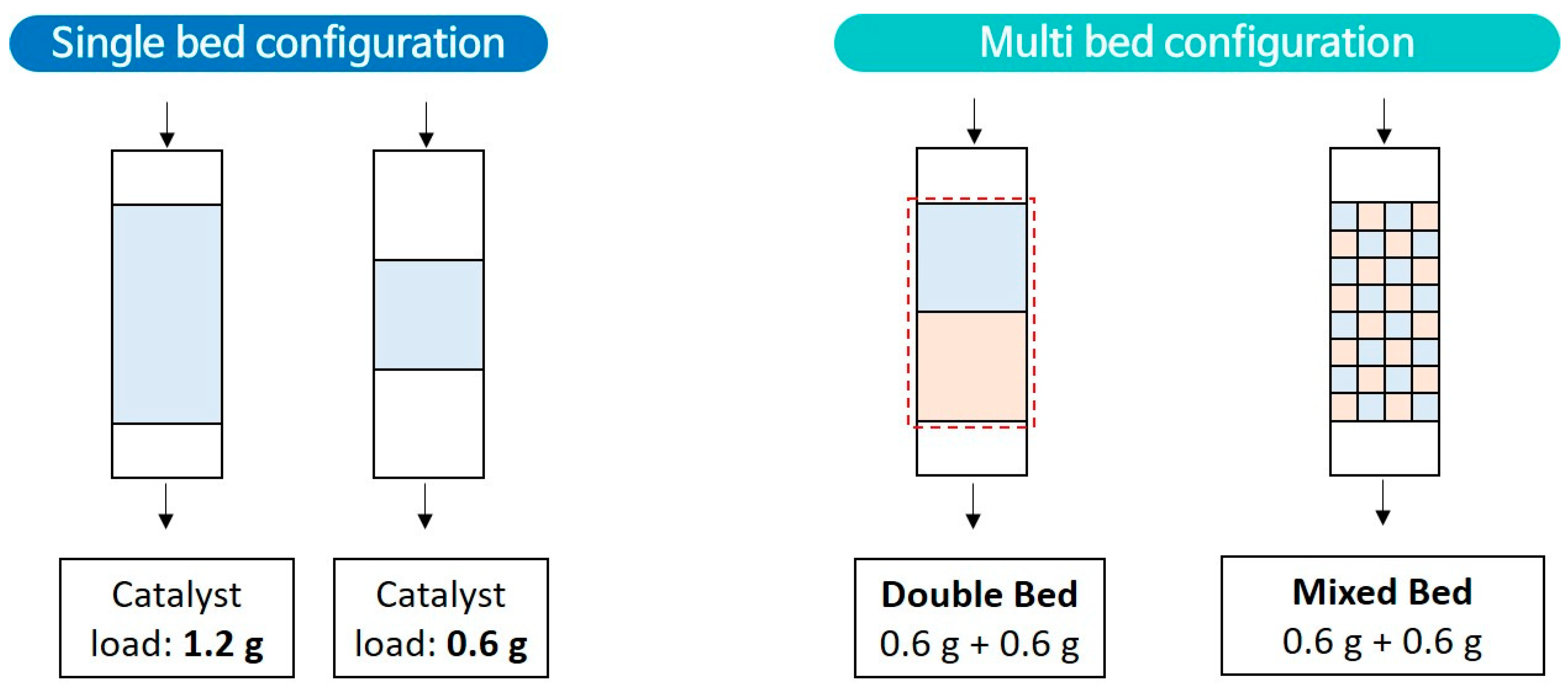
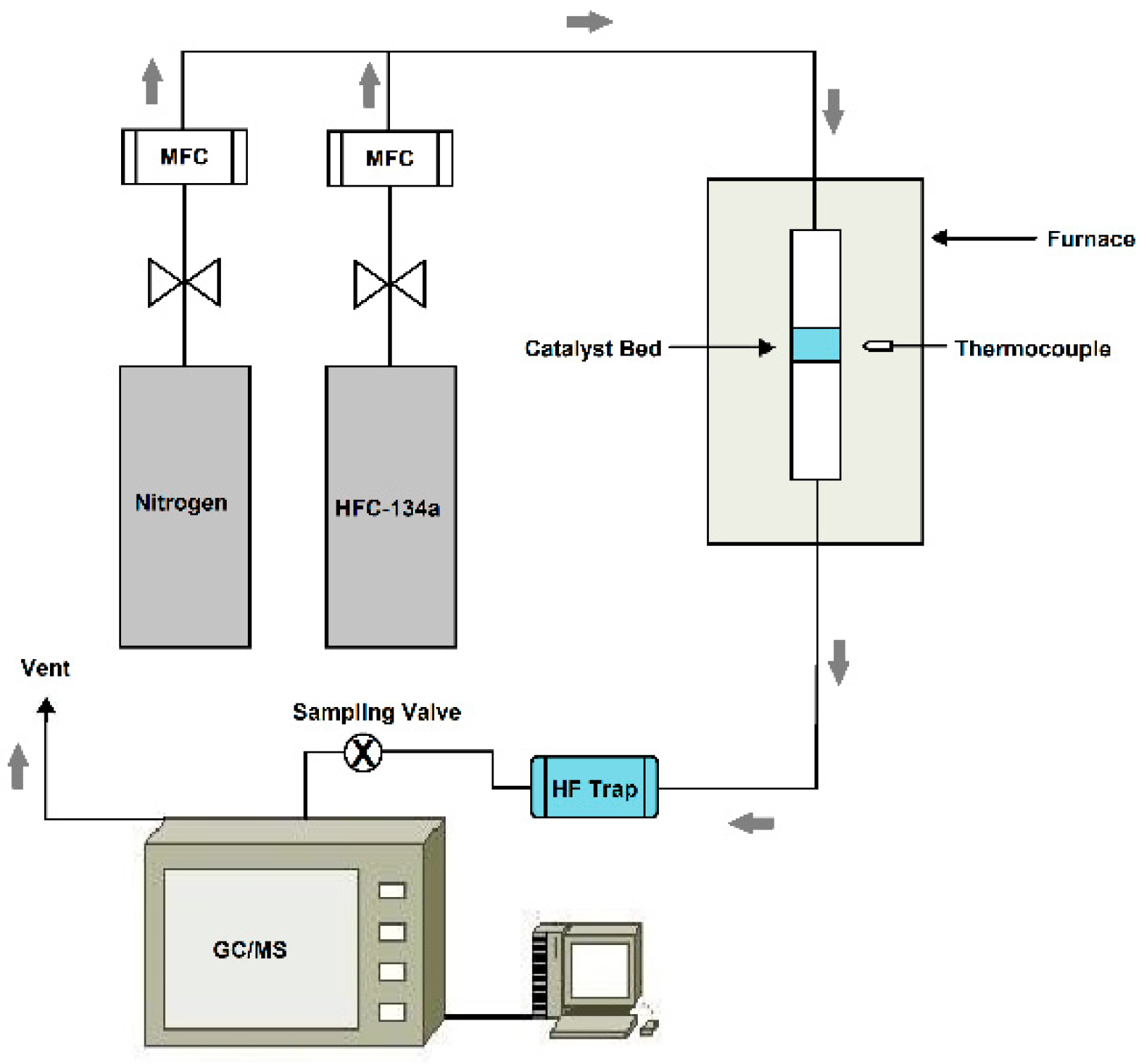
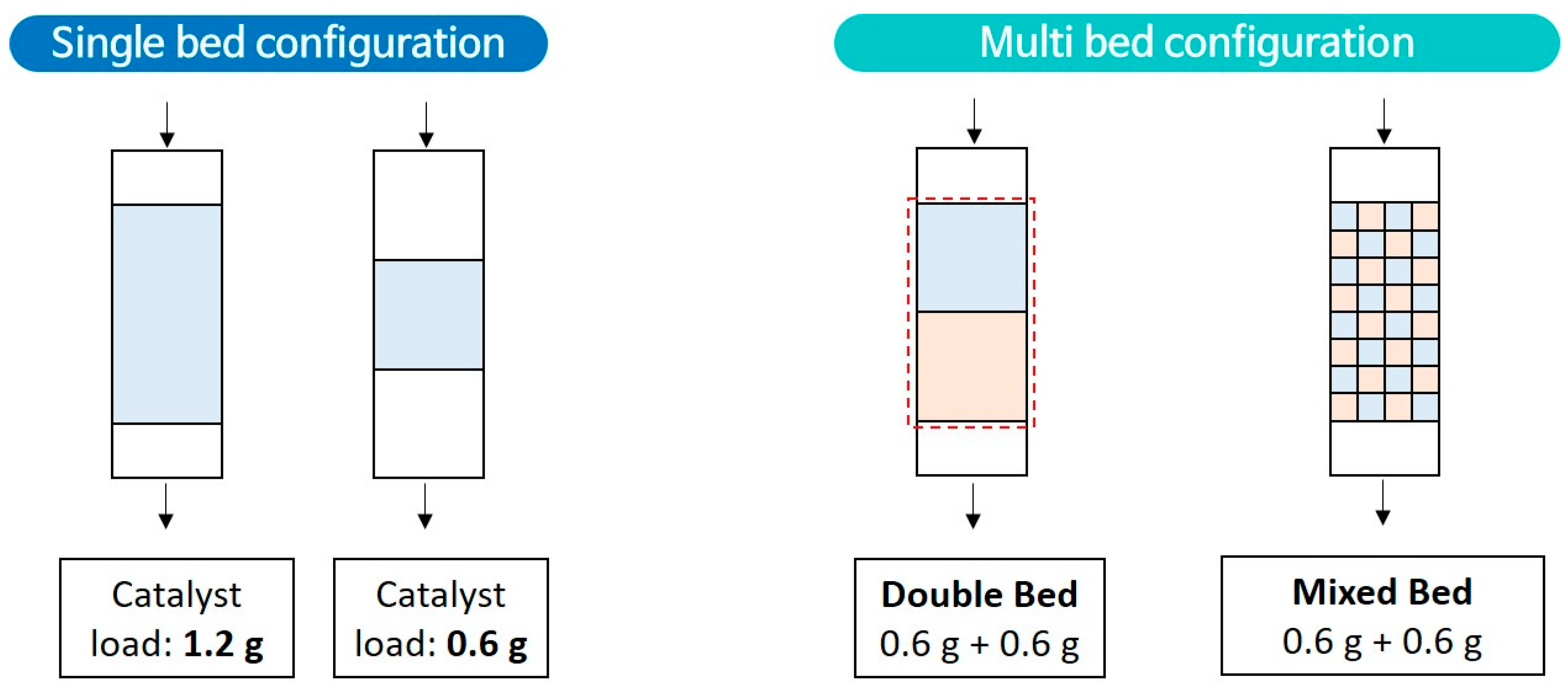
3.2. Catalytic Decomposition of HFC-134a Using a Lab Scale Fixed-Bed Reactor
3.3. Characterizations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Intergovernmental Panels on Climate Change (IPCC). Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change; Solomon, S., Qin, D., Manning, M., Chen, Z., Marquis, M., Averyt, K.B., Tignor, M., Miller, H.L., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- European Environment Agency (EEA). Emissions and Supply of Fluorinated Greenhouse Gases. 2018. Available online: https://www.eea.europa.eu/data-and-maps/indicators/emissions-and-consumption-of-fluorinated-2/assessment (accessed on 27 October 2019).
- Environmental Investigation Agency (EIA). Kigali Amendment to the Montreal Protocol: A Crucial Step in the Fight against Catastrophic Climate Change. 2016. Available online: https://eia-international.org/wp-content/uploads/EIA-Kigali-Amendment-to-the-Montreal-Protocol-FINAL.pdf (accessed on 27 October 2019).
- Han, E.; Li, Y.; Tang, H.; Liu, H. Treatment of the potent greenhouse gas, CHF3-an overview. J. Fluorine Chem. 2012, 140, 7–16. [Google Scholar] [CrossRef]
- Jasinski, M.; Dors, M.; Mizeraczyk, J. Destruction of Freon HFC-134a Using Nozzless Microwave Plasma Source. Plasma Chem. Plasma P 2009, 29, 363–372. [Google Scholar] [CrossRef]
- Ohno, M.; Ozawa, Y.; Ono, T. Decomposition of HFC134a Using Arc Plasma. Int. J. Plasma Environ. Sci. Technol. 2007, 1, 159–165. [Google Scholar]
- Han, T.U.; Yoo, B.-S.; Kim, Y.-M.; Hwang, B.; Subidya, G.L.; Park, Y.-K.; Kim, S. Catalytic conversion of 1,1,1,2-tetrafluoroethane (HFC-134a). Korean J. Chem. Eng. 2018, 35, 1611–1619. [Google Scholar] [CrossRef]
- Ryu, J.; No, K.; Kim, Y.; Park, E.; Hong, S. Synthesis and application of ferroelectric poly (vinylidene fluoride-co-trifluoroethylene) films using electrophoretic deposition. Sci. Rep. 2016, 6, 36176. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-Y.; Kwak, K.; Seo, J.-W.; Park, M.; Paik, H.; Lee, J.-Y.; Hong, J.; No, K. Ferroelectric nanodot formation from spin-coated poly(vinylidene fluoride-co-trifluoroethylene) films and their application to organic solar cells. J. Appl. Polym. Sci. 2015, 132, 41230. [Google Scholar] [CrossRef]
- Brown, L.F.; Carlson, R.L.; Sempsrott, J.M. Spin-cast P (VDF-TrFE) films for high performance medical ultrasound transducers. In Proceedings of the 1997 IEEE Ultrasonics Symposium Proceedings. An International Symposium (Cat. No.97CH36118), Toronto, ON, Canada, 5–8 October 1997; pp. 1725–1727. [Google Scholar]
- Higashi, Y.; Sakoda, N.; Islam, M.A.; Takada, Y.; Koyama, S.; Akasaka, R. Measurements of Saturation Pressures for Trifluoroethene (R1123) and 3,3,3-Trifluoropropene (R1243zf). J. Chem. Eng. Data 2018, 63, 417–421. [Google Scholar] [CrossRef]
- Jia, W.; Liu, M.; Lang, X.; Hu, C.; Li, J.; Zhu, Z. Catalytic dehydrofluorination of 1,1,1,2-tetrafluoroethane to synthesize trifluoroethylene over a modified NiO/Al2O3 catalyst. Catal. Sci. Technol. 2015, 5, 3103–3107. [Google Scholar] [CrossRef]
- Song, J.-Y.; Chung, S.-H.; Kim, M.-S.; Seo, M.-G.; Lee, Y.-H.; Lee, K.-Y.; Kim, J.-S. The catalytic decomposition of CF4 over Ce/Al2O3 modified by a cerium sulfate precursor. J. Mol. Catal. A Chem. 2013, 370, 50–55. [Google Scholar] [CrossRef]
- Xu, X.; Sun, L.; Wang, Y. NF3 decomposition over Al2O3 reagents without water. J. Nat. Gas Chem. 2015, 20, 418–422. [Google Scholar] [CrossRef]
- Gandhi, M.S.; Mok, Y.S. Effect of packing materials on the decomposition of tetrafluoroethane in a packed-bed dielectric barrier discharge plasma reactor. Int. J. Environ. Sci. Technol. 2015, 12, 499–506. [Google Scholar] [CrossRef]
- Iengo, P.; Serio, M.D.; Sorrentino, A.; Solinas, V.; Santacesaria, E. Preparation and properties of new acid catalysts obtained by grafting alkoxides and derivatives on the most common supports. Appl. Catal. A Gen. 1998, 167, 85–101. [Google Scholar] [CrossRef]
- Jia, W.; Wu, Q.; Lang, X.; Hu, C.; Zhao, G.; Li, J.; Zhu, Z. Influence of Lewis Acidity on Catalytic Activity of the Porous Alumina for Dehydrofluorination of 1,1,1,2-Tetrafluoroethane to Trifluoroethylene. Catal. Lett. 2014, 145, 654–661. [Google Scholar] [CrossRef]
- Krahl, T.; Keminitz, E. Aluminium fluoride—The strongest solid Lewis acid: Structure and reactivity. Catal. Sci. Technol. 2017, 7, 773–796. [Google Scholar] [CrossRef]
- Kemnitz, E.; Menz, D.-H. Fluorinated metal oxides and metal fluorides as heterogeneous catalysts. Prog. Solid State Chem. 1998, 26, 97–153. [Google Scholar] [CrossRef]
- Han, J.-Y.; Kim, C.-H.; Lee, B.; Nam, S.-C.; Jung, H.-Y.; Lim, H.; Lee, K.-Y.; Ryi, S.-K. Sorption enhanced catalytic CF4 hydrolysis with a three-stage catalyst-adsorbent reactor. Front. Chem. Sci. Eng. 2017, 11, 537–544. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Amount of Acid Sites (mmol-NH3/g-Catalyst) | |||
|---|---|---|---|---|
| Weak | Intermediate | Strong | Total | |
| γ-Al2O3 | 1.776 | 2.345 | 2.623 | 6.744 |
| MgO | 0.614 | 0.753 | 0.397 | 1.764 |
| CaO | 0.004 | 1.696 | 0.797 | 2.497 |
| Sample Conditions | γ-Al2O3 (Element, wt%) | |||
|---|---|---|---|---|
| Al | O | F | C | |
| Fresh | 43.5 | 52.0 | 0 | 4.5 |
| After 6 h | 21.1 | 26.7 | 36.1 | 16.1 |
| After 9 h | 18.8 | 2.4 | 61.1 | 17.7 |
| After 12 h | 10.3 | 1.5 | 40.8 | 47.4 |
| Sample Conditions | γ-Al2O3 (Element, wt%) | MgO (Element, wt%) | ||||||
|---|---|---|---|---|---|---|---|---|
| Al | O | F | C | Mg | O | F | C | |
| Fresh | 43.5 | 52.0 | 0 | 4.5 | 46.3 | 48.6 | 0 | 5.1 |
| After 6 h | 40.5 | 30.9 | 20.1 | 8.5 | 44.5 | 25.5 | 30.0 | 0 |
| After 9 h | 36.8 | 30.8 | 14.7 | 17.7 | 32.7 | 8.8 | 55.3 | 3.2 |
| After 12 h | 12.5 | 4.8 | 43.6 | 39.1 | 31.5 | 1.6 | 59.3 | 7.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, S.; Sudibya, G.L.; Jeon, J.-K.; Kim, Y.-M.; Swamidoss, C.M.A.; Kim, S. The Use of a γ-Al2O3 and MgO Mixture in the Catalytic Conversion of 1,1,1,2-Tetrafluoroethane (HFC-134a). Catalysts 2019, 9, 901. https://doi.org/10.3390/catal9110901
Jeong S, Sudibya GL, Jeon J-K, Kim Y-M, Swamidoss CMA, Kim S. The Use of a γ-Al2O3 and MgO Mixture in the Catalytic Conversion of 1,1,1,2-Tetrafluoroethane (HFC-134a). Catalysts. 2019; 9(11):901. https://doi.org/10.3390/catal9110901
Chicago/Turabian StyleJeong, Sangjae, Gamal Luckman Sudibya, Jong-Ki Jeon, Young-Min Kim, Caroline Mercy Andrew Swamidoss, and Seungdo Kim. 2019. "The Use of a γ-Al2O3 and MgO Mixture in the Catalytic Conversion of 1,1,1,2-Tetrafluoroethane (HFC-134a)" Catalysts 9, no. 11: 901. https://doi.org/10.3390/catal9110901
APA StyleJeong, S., Sudibya, G. L., Jeon, J.-K., Kim, Y.-M., Swamidoss, C. M. A., & Kim, S. (2019). The Use of a γ-Al2O3 and MgO Mixture in the Catalytic Conversion of 1,1,1,2-Tetrafluoroethane (HFC-134a). Catalysts, 9(11), 901. https://doi.org/10.3390/catal9110901