Abstract
Cytochromes P450 (CYP450s) promote the biosynthesis of steroid hormones with major impact on the onset of diseases such as breast and prostate cancers. By merging distinct functions into the same catalytic scaffold, steroidogenic CYP450s enhance complex chemical transformations with extreme efficiency and selectivity. Mammalian CYP450s and their redox partners are membrane-anchored proteins, dynamically associating to form functional machineries. Mounting evidence signifies that environmental factors are strictly intertwined with CYP450s catalysis. Atomic-level simulations have the potential to provide insights into the catalytic mechanism of steroidogenic CYP450s and on its regulation by environmental factors, furnishing information often inaccessible to experimental means. In this review, after an introduction of computational methods commonly employed to tackle these systems, we report the current knowledge on three steroidogenic CYP450s—CYP11A1, CYP17A1, and CYP19A1—endowed with multiple catalytic functions and critically involved in cancer onset. In particular, besides discussing their catalytic mechanisms, we highlight how the membrane environment contributes to (i) regulate ligand channeling through these enzymes, (ii) modulate their interactions with specific protein partners, (iii) mediate post-transcriptional regulation induced by phosphorylation. The results presented set the basis for developing novel therapeutic strategies aimed at fighting diseases originating from steroid metabolism dysfunction.
1. Introduction
Cytochromes P450 (CYP450s) catalyze a variety of reactions over a broad range of substrates, being among the most versatile enzymes in Nature. These promote the biotransformation of a wide spectrum of xenobiotic and endogenous compounds [], in addition to the biosynthesis of steroid hormones and the conversion of polyunsaturated fatty acids. Hence, CYP450s play a regulatory role as the basis of homeostasis and development, and promote the metabolism of carcinogenic species and of many marketed drugs []. The nomenclature employed to categorize this complex and growing family of enzymes is based on the name of the genes, which uses the italicized prefix “CYP” for humans, followed by an Arabic number to denote the family, a letter to designate the subfamily, and, finally, another number referring to the specific cytochrome. The encoded proteins are identified by the same name of the gene without the use of italics (e.g., the CYP19A1 genes encodes for CYP19A1 protein) [].
In humans six CYP450s participate in steroidogenesis, a fundamental metabolic pathway that leads to the formation of biologically active steroid hormones, starting from cholesterol (see Figure 1) []. These are divided in type I and II enzymes carrying out their action in the mitochondria and in the endoplasmic reticulum (ER), respectively. An altered regulation of steroidogenesis induces the onset of several pathologies, such as polycystic ovary syndrome, hypertension, and of distinct cancer types (i.e., breast and prostate cancers). Hence, understanding the mechanism of steroidogenesis has innumerable biological and pharmacological implications and is rooted into unraveling their biochemistry as well as the multiple regulatory mechanisms finely tuning steroid production. CYP450s are typically classified as monooxygenases, which catalyze the hydroxylation of organic molecules, requiring molecular oxygen, protons, and electrons supplied from water molecules and specific protein partners, respectively. It is now commonly accepted that the hydroxylation reaction is mediated by Compound I (Cpd I) via an oxygen rebound mechanism [,]. However, besides the canonical mono-oxygenation, CYP450s can promote a variety of distinct chemical reactions, including deformylation, desaturation, and C–C coupling [,]. While, only two enzymes (CYP11B1 and CYP21A2, involved in the synthesis of both glucocorticoids and mineralocorticoids) are endowed with exclusive hydroxylase activity, the others are characterized by intricate catalytic mechanisms, in which different functions are merged within the same catalytic scaffold, adding fascinating complexity to their catalytic mechanism. These are: (i) CYP11A1, also called cholesterol side-chain cleavage enzyme (P450scc), the first enzyme of steroid biosynthesis; (ii) CYP19A1, commonly called Aromatase (AR), which catalyzes estrogen biosynthesis and (iii) CYP17A1, which remarkably joins a 17α-hydroxylase with 17,20-lyase function [].
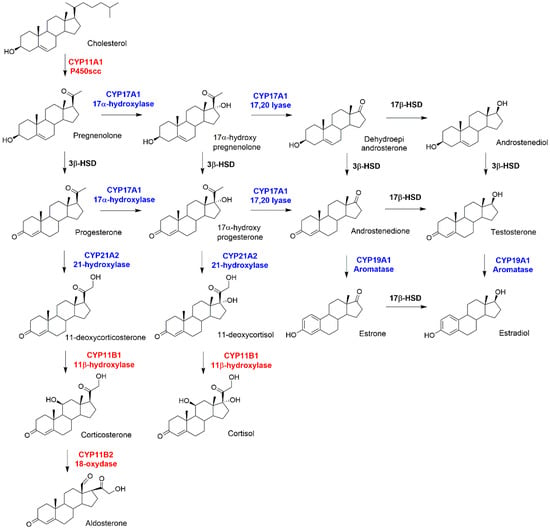
Figure 1.
Schematic picture of steroidogenesis. Mitochondrial and microsomal Cytochromes P450 (CYP450s) are highlighted in red and blue, respectively. Two dehydrogenases, 3β and 17β, are highlighted in black.
All mammalian CYP450s are attached and partially immersed to ER or mitochondrial membranes via an N-terminal helix spanning the lipidic bilayer []. Their membrane-bound nature makes it challenging to obtain crystal structures []. These latter represent the main sources of structural information needed to disclose CYP450s’ function.
The electrons are key ingredients of the chemical reactions catalyzed by CYP450s. For microsomal CYP450s these are provided by the cytochrome P450 reductase (CPR), and Cytochrome b5 (CYb5), membrane-attached proteins located in the ER, whereas in the mitochondrial membrane they are supplied by the ferredoxin system (Adrenodoxin (Adx), a Fe2S2 containing ferredoxin, and Adrenodoxin Reductase (AdR)). Mounting evidence has indicated how the function of CYP450s machinery is intrinsically connected with their membrane bound nature. Indeed, protein-lipid interactions modulate CYP450s flooding and diffusion on the top of membranes, finely regulating their catalysis as well as substrate selectivity, and interaction with essential protein partners [].
This review focuses in particular on CYP19A1, CYP17A1, and CYP11A1 as a proxy of steroidogenic CYP450s. These enzymes have been selected for their intricate catalytic mechanisms merging different functions, their crucial involvement in severe pathologies (cancer, pre-eclampsia etc), and for their relevance as pharmaceutical targets. Due to its captivating complexity, unraveling the mechanism of the steroidogenic machinery requires the complementary use of biochemical and biophysical experimental techniques along with atomic-level simulations. This review is mostly focused on the achievements attained in understanding the different facets of CYP450s’ mechanism by means of all-atom simulations. Hence, after a brief introduction on computational methods employed to tackle these systems, we discuss: (i) the general CYP450 catalytic mechanism, highlighting its specificity in steroidogenesis; (ii) the role of biological membranes in affecting substrate/drug channeling and their substrate selectivity by sculpting distinct access/egress channels to the active site, and, finally, (iii) a common post-transcriptional regulation mechanism of CYP450s induced by phosphorylation. Unraveling the inner-working mechanism of this intricate and utmost important machinery has broad biological and pharmacological implications to tackle disorders linked to steroid metabolism dysfunction, setting the basis for novel inhibitory strategies against cancer (breast and prostate) disease.
2. Computational Methods to Dissect the Mechanism of CYP450s
2.1. Force Field Based Molecular Dynamics (MD)
The intricate chemical and regulatory mechanisms underlining CYP450s’ functions requires the use of several complementary biochemical and biophysical techniques to be disentangled. Among the available tools, computer simulations have the potential to tackle such mechanisms reaching a temporal and spatial resolution inaccessible, yet complementary, to experimental methods. In this respect, molecular dynamics (MD) simulations have often been used to study CYP450s. This method allows the dynamic evolution of a system to be followed at near-physiological conditions, while its thermodynamic properties are calculated from averages over sufficiently long trajectories []. By calculating the forces acting on a system’s particles, these can be propagated via a suitable equation of motions. In classical MD the interactions between atoms are described with empirical force fields (FFs). In these, the potential energy is expressed as the sum of bonded contributions (bonds, angles, dihedrals, improper dihedrals) and nonbonded terms (Coulomb potential to model electrostatics and Lennard–Jones to describe atom–atom attraction/repulsion and dispersion interactions). Besides providing information on the dynamic evolution of the system, classical MD allows the interaction of biomolecules to be characterized with the surrounding environment at the atomic-scale and is routinely employed to study large biomolecular machineries comprising up to millions of atoms [], and reaching timescales of the order of 10–100 of microseconds [].
For CYP450s extensive MD simulations have contributed to unravel the model and the principles driving the interaction between these proteins and their associated membranes [,,,], predicting how CYP450s float on the ER or mitochondrial membranes, assessing their orientation with respect to the membrane plane, and their immersion inside the lipidic bilayer, as well as assessing the influence of the lipid type on these properties, on the slow functional motion of the enzymes, and on ligand-channeling to the active site from the protein surface [,,]. In this latter case, the typical simulation time-scale of classical MD is, however, not sufficient to identify possible ligand routes, and to identify the most viable channels by calculating the relative free energy profiles for ligand passage through the protein. Hence, methods capable of estimating thermodynamic quantities (free energy, entropy, and enthalpy) and kinetic parameters (free energy barriers) rigorously from MD simulations have been developed. These allow predicting equilibrium observables from non-equilibrium “biased” MD simulations.
Random expulsion molecular dynamics (RAMD) simulations have been often applied to sample ligands travelling along CYP450 channels []. By applying randomly oriented forces to the ligand atoms, one can quickly identify possible escape routes which from the active site reach the protein surface. By repeating these ligand dissociation events several times, it is possible to identify the most likely dissociation paths at statistical level. Alternatively, Steered MD (SMD) can be employed to induce the dissociation of a ligand by applying an external force. This is usually done along predefined routes (such as those identified by RAMD) [,]. Nevertheless, obtaining a convergent free energy profile by using the SMD simulations and the Jarzinski’s equality is challenging [,]. Hence, information on free energy profiles can be obtained by complementary methods, able to force the exploration along a predefined reaction coordinate. In any MD simulation, the exploration of the phase space is limited by Boltzmann statistics, so that at equilibrium the system will sample only a limited portion of the conformational space. In Umbrella Sampling (US) this probability is modified by adding a bias to the potential in order to force the exploration of important regions, which would be rarely accessible by Boltzmann statistics. This enhanced non-Boltzmann sampling is forced around specific values of the selected reaction coordinate. Thus, in order to get a reliable estimate of the potential of mean force, one needs to run several simulations centered on distinct points of the reaction coordinate, and then reconstruct the unbiased potential of mean force by the Weighted Histogram Analysis Method (WHAM) [,].
Conversely, metadynamics represents an efficient approach to enhance the sampling of conformational space, without a priory guess of the reaction coordinate. By means of a history-dependent biasing potential, the system is encouraged to visit new (Boltzmann disfavored) states, which allow it to evolve toward different stable/metastable states, and the (negative of the) biasing potential provides an estimate of the underlying free energy surface. In the original implementation, the method relies on the identification of two reaction coordinates (named collective variables). Several variants of this original scheme [] have been introduced to allow faster exploration and convergence of the free energy [,].
2.2. Mixed Quantum-Classical (QM/MM) Static or Dynamics Simulations
Relying on a predefined potential energy surface, FFs cannot be used to investigate processes in which a change of the electronic structure occurs (i.e., chemical reactions [,,], polarization, and charge transfer effects [], as well as rearrangements in the sphere of the transition metals []). In these cases, a quantum mechanical (QM)-based approach is mandatory. Despite the significant improvement of computational software, algorithms, and computer architectures, QM calculations are still too expensive for tackling biomolecules in their physiological environment. Therefore, QM studies of enzymatic reactions are typically performed with a mixed Quantum Mechanics–Molecular Mechanics approach (QM/MM) []. In this multiscale method, the reaction site is treated by using the QM level of theory, while the rest of the system is treated by using Molecular Mechanics (i.e., Force Field). As a result, the QM/MM method allows a limited portion of the system to be treated with QM accuracy, while still accounting for the rest of the protein and its environment (water solvent, biological membrane, ions) at the atomic-level of detail. When the partition between the QM and the MM region cuts chemical bonds, the QM subsystem is often saturated by using a hydrogen link atom [,].
Typically for CYP450s the QM region is treated at fist principle level by using Density Functional Theory (DFT), since this accounts for correlation effects at a reasonable level of accuracy. These are, in fact, of remarkable importance for the correct description of the transition metal moiety. The accuracy of DFT is, however, hampered by the approximated form of the exchange correlation (XC) functional. Some of the well-known deficiencies of the commonly employed XC functionals include the neglection of dispersion interactions, which critically affects the interactions between, e.g., aliphatic or aromatic fragments, and the so-called self-interaction error, which may determine an unphysical electron delocalization in open shell systems [,]. Modern XC functionals include empirical correction terms to improve the description of van der Waals complexes, while a more accurate description of radical systems can be achieved by including a fraction of exact (Hartree–Fock) exchange in the XC functional. However, in problematic cases, specific corrections have to be introduced in order to correct the self-interaction error []. The inaccuracy of the XC functional certainly affects the chemistry of CYP450s in which the iron atom alternates among distinct possible oxidation states, which may correspond to open or closed shell configurations and for which distinct ground spin states may become accessible along the catalytic reaction. The coupling between the QM and the MM layers represents the most challenging part of every mixed quantum classical method. While van der Waals interactions, stretching, bending, and torsional terms between QM and MM regions are accounted for by the MM terms, the electrostatic coupling is difficult, and is the bottleneck of the QM/MM calculation. However, this is probably the most important contribution to the mixed QM/MM Hamiltonian, as long-range electrostatic interactions can markedly affect the electronic properties of the metal center [,].
QM/MM simulations can be performed by using a static or a dynamic approach. In the first case, to study enzymatic reactions one needs to derive the dependency of the energy on the nuclear coordinates by computing the energy for different atomic configurations via efficient algorithms, which allow the stationary points of the potential energy surface to be located, corresponding to ground or transition states. In the dynamical contest the system moves, instead, on a potential energy surface which is calculated both at QM and MM level (for the two regions considered). This allows ab initio calculations (in the QM region) to be performed at finite temperature, significantly increasing the computational cost of the calculation with respect to classical MD. As such, the time-scale accessible by QM/MM MD runs is limited to 100 s of ps. This clearly hampers a direct simulation of chemical processes and enhanced sampling simulations techniques are needed to dissect possible reaction paths and their associated free energy landscape.
3. Catalytic Mechanism of CYP450s
Considering the lack of reactivity of molecular oxygen at physiological temperatures, Nature has supplied living systems with enzymes able to perform oxidation reactions at mild conditions. Among these, CYP450s typically act as monooxygenase, inserting one oxygen atom into endogenous/exogenous substrates (RH), and reducing the second oxygen to a water molecule (see Equation (1)).
NADPH + H+ + O2 + RH → NADP+ + H2O + ROH
Nowadays, it is well established that CYP450s share a general and common catalytic cycle, as displayed in Figure 2 [,].
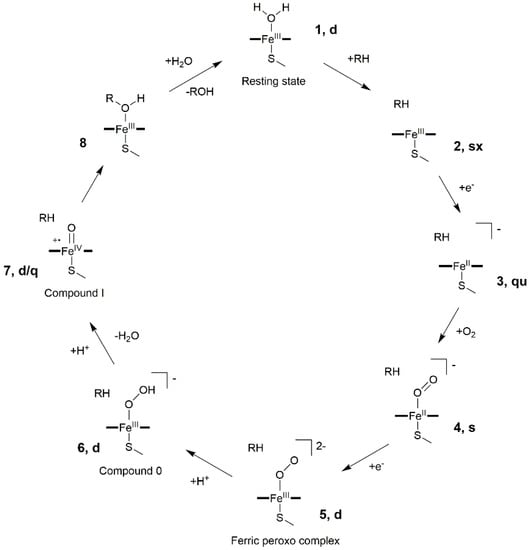
Figure 2.
General CYP450 catalytic cycle. Spin multiplicities for each heme intermediate (s = singlet, d = doublet, q = quartet, qu = quintet and sx = sextet) are summarized according to ref [].
The resting state (1) of the enzyme coordinates is a water molecule in the heme distal site, which is then displaced upon entrance of the substrate (RH) inside the binding cavity. Water departure leaves a pentacoordinated ferric-porphyrin complex (2), which undergoes the first electron transfer (ET) from the redox partner, leading to the formation of a high spin ferrous complex (3). This species can easily bind molecular oxygen, forming an oxy-ferrous complex (4). After a second ET, a ferric peroxo complex (5) is formed, which, being a strong base, can be easily protonated to form compound 0 (Cpd 0, 6). A second proton transfer induces the formation of the highly reactive iron-oxo species compound I (Cpd I), a FeIV-oxo porphyrin radical cation (7). In the prototypical CYP450 monooxygenase cycle, Cpd I performs substrate hydroxylation via an oxygen rebound mechanism (8) to form an alcohol, ROH, restoring ultimately the heme resting state (1). Whereas the protons necessary for the reactions are supplied either from the substrate or from water molecules trapped into the binding site, the two electrons, required at each cycle, are furnished by a specific redox partner, which by floating on biological membrane can associate/dissociate to/from their partner proteins (among which CYP450s) in a highly dynamic and competitive scenario.
3.1. The Oxygen Rebound Mechanism
The prototypical reaction carried out by CYP450s is the C–H hydroxylation, for which the generally accepted catalytic mechanism [] begins with the abstraction by Cpd I of a substrate’s hydrogen atom. This leads to a radical intermediate and a Cpd II-like iron species, bearing a hydroxyl radical. Then, the substrate and the hydroxyl radical join forming the hydroxylated product (Figure 3).
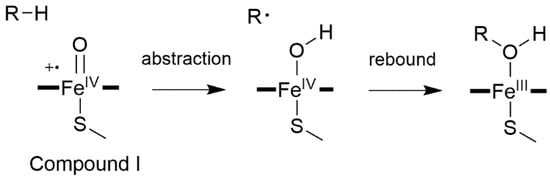
Figure 3.
General CYP450 oxygen rebound mechanism for hydroxylations, with R corresponding to a generic alkane.
Remarkably, this mechanism can also be invoked to explain other reaction types, such as desaturation, epoxidation, heteroatom oxygenation, and dealkylation reactions []. Due to its importance in exogenous and endogenous compounds metabolism, CYP450-catalyzed C–H hydroxylation has been the object of intense experimental and computational interest, as discussed in many comprehensive reviews [,].
3.2. Other Reaction Mechanisms
Although it is well established that the reactive species for most of the CYP450 oxidations is the radical cation complex, Cpd I, an alternative mechanism, involving the ferric peroxo complex, FeO2−, has been at times proposed to account for atypical reactions, such as the breaking of a C–C bond (Figure S1) []. Furthermore, in order to rationalize some of these unusual chemical reactions, a dual hydrogen abstraction (DHA) mechanism, relying on two subsequent hydrogen abstraction steps, has been put forward [,]. In this case, a first C–H abstraction is followed by a second H-abstraction from the hydroxyl group of the radical intermediate, leading to the aldehyde formation. A reversed order of events, called reversed-DHA (Figure S2) may also take place.
In the following, we limit our description to reactions invoked to explain steroid biosynthesis, for more detailed and comprehensive description on other possible reaction mechanisms promoted by all CYP450s the reader is invited to refer to more specialized and comprehensive reviews [,,].
3.3. Reaction Mechanisms of Steroidogenic CYP450s
Among the CYP450s involved in steroidogenesis only two possess exclusively a monooxygenase function. The first, CYP21A2 (steroid 21-hydroxylase), catalyzes the 21-hydroxylation of progesterone and 17α-hydroxyprogesterone to deoxycorticosterone and 11-deoxycortisol, respectively. The second, CYP11B1 (steroid 11β-hydroxylase), performs the subsequent oxidation to obtain corticosterone and cortisol, respectively. Corticosterone is later converted to aldosterone, the most potent human mineralocorticoid, by CYP11B2 (aldosterone synthase) via a three-step catalysis. The first two, 11β- and 18-hydroxylations are canonical monooxygenase reactions. The last step, leading to 18-oxidation, may be rationalized with an oxygen rebound mechanism. In this chapter, we focus on the remaining steroidogenic CYP450s, which are typified by intricate catalytic mechanisms, in which multiple functions are merged within the same catalytic scaffold. These include: (i) CYP11A1, commonly called cholesterol side-chain cleavage enzyme (P450scc), which catalyzes the very first step of steroid biosynthesis; (ii) CYP17A1, which joins a 17α-hydroxylase with 17,20-lyase function catalyzing androgens production, and (iii) CYP19A1 (commonly called Aromatase, AR), which is the only enzyme in vertebrates able to catalyze the formation of a six-membered aromatic ring such as in estrone or estradiol [].
3.3.1. CYP11A1—Cholesterol Side-Chain Cleavage Enzyme (P450scc)
In humans CYP11A1 catalyzes the synthesis of pregnenolone, the precursor of all steroid hormones in the mitochondria. This is the first, and rate limiting step in the synthesis of all steroid hormones, thus quantitatively regulating steroid production, and qualitatively determining the type of hormones produced by the downstream enzymes. Pregnenolone, is synthesized in the inner membrane of mitochondria by a complex enzymatic machinery comprising CYP11A1 and its redox partners. CYP11A1 deficiency has been associated with lipoid congenital adrenal hyperplasia, affecting the development of sex characteristics, while its overexpression impairs the placentation process and contributes to the pathogenesis of pre-eclampsia []. Pregnenolone biosynthesis occurs in three steps (Figure 4a). The first two are stereospecific hydroxylations leading, respectively, to the formation of 22R-hydroxycholesterol and 20R,22R-dihydroxycholesterol, followed by the oxidative scission of the C20–22 bond to obtain pregnenolone and isocaproic aldehyde [].
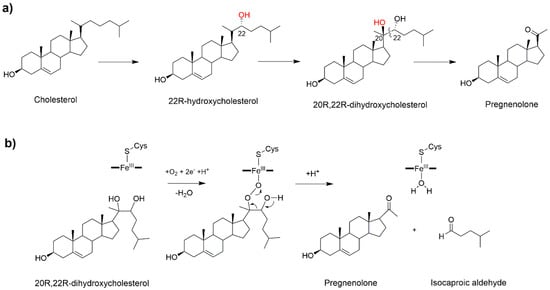
Figure 4.
(a) The three-step biosynthesis of pregnenolone catalyzed by CYP11A1 (P450scc). (b) Proposed mechanism for the last catalytic step [].
In line with other steroidogenic CYP450s, CP11A1 is characterized by a high substrate-specificity limited to cholesterol, 7-dehydrocolesterol and vitamin D []. Cryoreduction in combination with EPR spectroscopy showed that the reactive species responsible for the first catalytic step is Cpd I, pinpointing a canonical hydroxylation reaction mediated by oxygen rebound []. By analogy, it is probable to assume that the same mechanism may also be operative in the second step leading to the formation of 20R,22R-dihydroxycholesterol. Despite the third step having been studied for decades, a clear mechanistic picture remains elusive [,,]. A C20-peroxy intermediate was invoked for the C–C bond cleavage step (Figure 4b) on the basis of experiments showing the retention of the C22 hydrogen and C20 oxygen in the products [,]. Considering the crystal structure [] and a recent EPR study, Cpd I was declared as the active species of the second and third catalytic steps []. Yet, in spite of its pivotal importance as critical regulator of all human steroids production, a theoretical study of this important reaction mechanism has never been attempted so far.
3.3.2. CYP17A1—Steroid 17α-Hydroxylase/17,20 Lyase (CYP450c17)
CYP17A1 is a microsomal cytochrome promoting the biosynthesis of androgens or precursor of glucocorticoids, hence representing a critical cross point of steroid metabolism []. This enzyme is a dual-function monooxygenase, exhibiting both 17-hydroxylase and 17,20 lyase functions, playing a vital role in both adrenal and gonadal steroidogenesis. CYP17A1 converts pregnenolone and progesterone to their 17α-hydroxylated products, and, subsequently, to dehydroepiandrosterone and androstenedione (Figure 5a). Since prostate cancer cells proliferate in response to androgens, CYP17A1 is an important target to fight against this type of hormone-dependent cancer [].
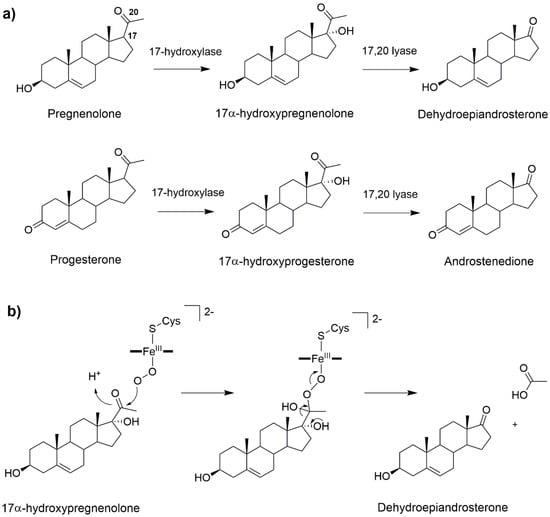
Figure 5.
(a) 17α-hydroxylation and 17α-20-lyase reactions catalyzed by CYP17A1. (b) Possible reaction mechanism of CYP17A1 lyase reaction involving the FeO2− intermediate [].
The 17-hydroxylation of pregnenolone and progesterone occurs via a canonical oxygen rebound mechanism. Nevertheless, the lyase step remains object of controversy. This reaction may be either mediated by Cpd I [] or by the nucleophilic attack of FeO2− on the steroid C20 atom [,,]. Recent solvent isotope effect experiments confirmed that the ferric peroxo intermediate is most likely operative [] (Figure 5b). Consistently, a DFT-based study, complemented by classical MD simulations, has characterized the reaction mechanism of both the hydroxylase and lyase reactions, using Cpd I and peroxo anion as reactive species of the two steps, respectively. For the hydroxylase step the reaction was studied starting from different binding poses of the substrate, as obtained from classical MD simulations. For all these poses, similar reaction free energy barriers of 13–14 kcal/mol were obtained. In contrast, for the lyase step two distinct mechanisms (stepwise or concerted) were discovered both having an activation free energy of 20 kcal/mol []. Future studies accounting explicitly for the influence of the biological environment and finite temperature effects may further elucidate this catalytic mechanism, aiding the development of inhibitors which should exclusively hamper its lyase activity to fight prostate cancer.
3.3.3. CYP19A1—Aromatase
CYP19A1, also called Aromatase (AR), is another key player in the steroid hormone biosynthetic pathway. This performs the conversion of androgens (androstenedione or testosterone), into estrogens (estrone or estradiol) in a three-step reaction (Figure 6a) occurring at the inner side of the ER membrane [].
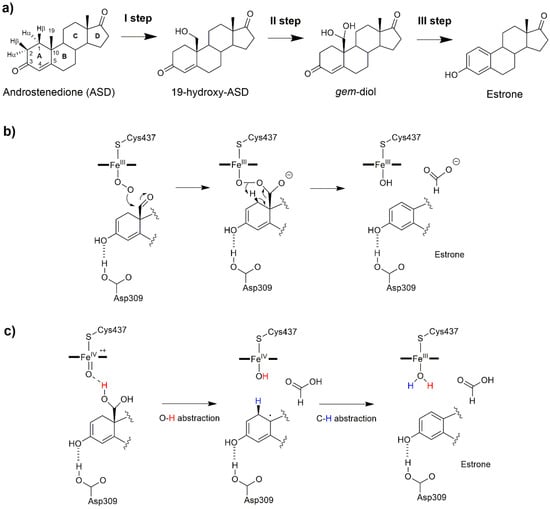
Figure 6.
(a) The sequential oxidation steps catalyzed by the aromatase (AR) enzyme. Aromatization mechanism involving (b) the ferric peroxo complex intermediate, FeO2−, as the reactive species, and (c) a dual hydrogen abstraction mechanism [].
Considering that abnormal estrogen production stimulates the proliferation of estrogen dependent breast cancer, this enzyme represents a key pharmacological target against this disease. For this reason, AR has been the object of several studies aimed at tackling distinct aspects of its reaction mechanism [,] and searching for novel competitive inhibitors [,,]. As for the other steroidogenic CYP450s, AR catalysis has been the object of debate and initially believed to be different from the canonical CYP450s due to the highly hydrophobic nature and substrate specificity of the active site. Nowadays, it is well established that the first two hydroxylation steps are mediated by Cpd I, involving a canonical hydrogen abstraction and hydroxyl radical rebound mechanism (Figure 3). After the two hydroxylation steps a 19,19-gem-diol (gem-diol, Figure 6a) is formed. Several reaction mechanisms have been proposed for the last catalytic step in which the aromatization of the substrate A ring takes place: among these, the Baeyer–Villiger oxidation of C19 [], 4,5 epoxidation [], and 2β hydroxylation [,]. On the basis of 18O labelling experiments [,,], the commonly accepted mechanism relied on the nucleophilic attack of the ferric peroxo complex, FeO2−, to the aldehyde group of a dehydrated gem-diol, with estrone being formed by the decomposition of a peroxo hemiacetal (Figure 6b). Pure QM and QM/MM studies, based on DFT, identified distinct mechanisms in which the peroxo ferric intermediate was operative overcoming free energy barriers in line with experiments [,]. Yet none of these studies provided a clear comprehensive picture of the whole catalytic cycle. Conversely, recent experimental studies, based on resonance Raman spectroscopy [], Kinetic Solvent Isotope Effect [], and new 18O labelling studies [] definitively pinpointed Cpd I as the reactive species of the last step. In a recent work, by performing a sophisticated computational protocol relying on extensive FF-based and QM/MM metadynamics simulations, a comprehensive atomic-level picture of AR catalysis was provided (Figure 7), disclosing that, after the first two hydroxylation steps, in which also a 2,3-enol formation takes place, a non-canonical DHA mechanism completes substrate aromatization (Figure 6c) [,].
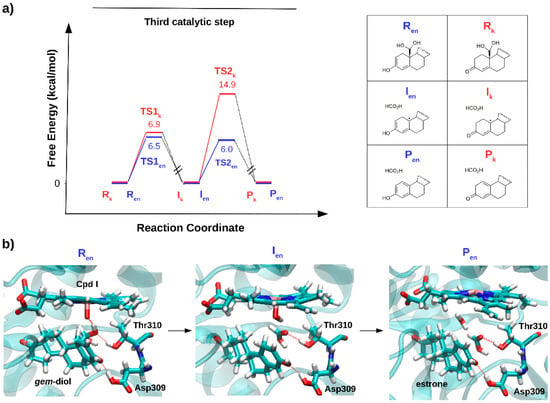
Figure 7.
(a) The relative free energy profile (kcal/mol) for the aromatization (third catalytic) step, considering the keto (Rk) and enol (Ren) form of androstenedione as a reagent. (b) Representative snapshots, as obtained in ref [], for the reagent (Ren), intermediate (Ien) and product (Pen) states formed along the lowest energy path, are reported.
Thus, the mysterious third AR catalytic step may be classified within a class of non-canonical reactions promoted by CYP450s, contributing to enlarge the range of AR functions beyond that of canonical hydroxylation reactions. In summary, from the studies performed so far on steroidogenic CYP450s it is tempting to suggest that Nature may have engineered selected steroidogenic CYP450s to merge multiple functions to selectively and efficiently promote complex and distinct chemical transformations within the same catalytic scaffold.
4. CYP450s’ Choreography on the Biological Membranes’ Surface
Human CYP450s are attached via an N-terminal anchor to the ER and mitochondrial membranes. These play an active role in regulating the function of CYP450s. As an example, membranes (i) affect partitioning and accumulation of hydrophobic substrates [], which should travel into the buried CYP450s active site facing the membrane (Figure 8), (ii) influence the structural and dynamical traits, such as the penetration and orientation of CYP450 with respect to the lipidic bilayer plane, inducing a fine tuning of the proteins slow functional motions underlining the opening and closing of substrate egress/access channels, (iii) condition the CYP450s’ ability to dynamically diffuse on the top of the membrane in search of its redox or other protein partners involved in catalysis as well as in its post-transcriptional regulation [,]. Despite their critical impact on CYP450 function, membranes have only recently been considered in computational studies [,].
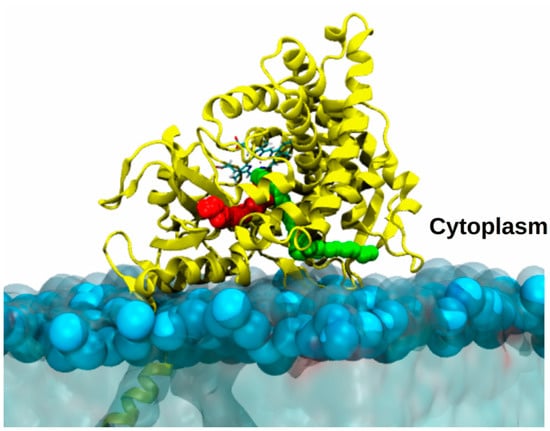
Figure 8.
Model of aromatase embedded in a membrane mimic. Most likely access/egress channels to enter/exit aromatase catalytic site, travelled by androstenedione or drugs, are highlighted as red or green balls. AR is shown as yellow ribbons, with the heme shown in licorice and colored by the atom name. The membrane is displayed as light-blue transparent surface with the phosphate atoms highlighted by light blue van der Waals spheres.
MD simulations, in agreement with experimental studies, provided an atomic-level structural model of many CYP450s embedded in distinct membrane types, consistently showing that the proteins extend above the membrane surface, while remaining anchored via its N-terminal helix. Conversely the proximal side, above the heme moiety, remains exposed towards the solvent in order to establish interactions with the CYP450 specific redox partner. CYP450s are partly immersed in the hydrophobic framework of the membrane (with the F/G-loop being deeply buried in the bilayer, while the B/C-loop and β1, β2, β4–β5 sheets interact with it) and assume a characteristic heme tilt angle (the tilt angle accounts for the orientation of the heme moiety with respect to the membrane plane) oscillating between 30–89 degrees []. These interactions and, in turn, CYP450s’ orientation are also remarkably affected by the membrane composition [,].
Access Channels
CYP450s are characterized by buried active sites connected to the protein surface via multiple arduous channels []. Identifying and characterizing the size and shape of those channels, as well as the structural and dynamic traits of ligands (substrate and inhibitors) channeling through them, to reach the catalytic site, is of pivotal importance from either a fundamental biochemical understanding or a pharmaceutical point of view []. These properties may indeed affect CYP450s ligand selectivity (kcat/km) and drug efficacy (residence time of a drug at the target site). In spite of its importance, unraveling ligand channeling energetics remains a daunting challenge to be addressed by experimental techniques, since the free energy landscape is most likely characterized by multiple metastable states. In this respect, computational methods can shed light on this biological process and several computational tools have been developed to this end (i.e., CAVER [], MOLE [] and MOLEonline []). Moreover, recently even a database of all known CYP450 channels, called ChannelDB, has been released [].
CYP19A1 and CYP17A1 on the ER Membrane
Among steroidogenic CYP450s, this issue has been faced for AR attached to a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane []. A first study of this enzyme, immersed in water solution and in the membrane mimic, revealed an increased flexibility of the protein residues in contact with the membrane in comparison to the simulation performed in water. Moreover, RAMD simulations revealed that the membrane markedly affected the type and the number of viable channels []. A different work investigated also the influence of distinct membrane compositions on CYP19A1, obtaining analogous results []. Similarly, an MD simulation study supplied a structural model of the binding of CYP17A1, and, consistently with other CYP450s, disclosed a significant impact of the membrane interface on the structure and dynamics and on the opening/closing of different tunnels []. Nevertheless, to understand the viability of those channels by substrates/inhibitors their ranking according to cost of ligands passage is required []. This can be achieved only by computing the relative free energy profile of ligand channeling by enhanced sampling simulations. In a recent study [], AR channels were mapped using multiple SMD simulations, which allowed only the relative probability of travelling along them to be assessed, while the profiles were computed by US simulations. Strikingly, as a result, ligands differing in terms of size, hydrophobicity, shape (the substrate androstenedione and a potent clinically used inhibitor letrozole), preferentially travel along the same channels (Figure 8) to get into or dissociate from the active site. A bioinformatics analysis revealed that these preferential paths may be even conserved among distinct CYP450s.
5. Interaction with Specific Redox Partners
The biotransformation processes catalyzed by all mammalian CYP450s depend on their redox partners, which supply the electrons necessary for catalysis (two electrons are needed for each mono-oxygenation reaction) []. In the microsomal electron transport chain, CPR and CYb5 are the main electron donors for those CYP450s anchored to the ER membrane []. Conversely, at the mitochondrial membrane the electrons are donated by the ferredoxin redox system (adrenodoxin reductase (AdR) or adrenodoxin (Adx)) [].
5.1. NADPH-Cythochrome P450 Reductase (CPR)
CPR is a membrane-bound protein composed of three domains separated by a linker domain and a flexible hinge, each binding a cofactor crucial for its function. These cofactors are the nicotinamide adenine dinucleotide phosphate (NADPH), the flavin adenine dinucleotide (FAD) and the flavin mononucleotide (FMN) [,]. CPR promotes the electron transfer (ET) from NADPH to CYP450s with CYP:CPR ratio of 3–15:1. However, CPR also supplies electrons to many other proteins besides CYP450s, in a highly competitive scenario [].
Experimental studies revealed that CPR exists in equilibrium between two conformational (closed and open) states [] and that the balance between the two conformations depends on the redox state of the NADPH cofactor. In this scenario, the open conformation serves to facilitate ET within the CPR in two steps: (i) two electrons, are consecutively transferred from NADPH to FAD and, (ii) then they move from FAD to the FMN group. In the open conformation, the FMN cofactor is exposed to the solvent, thus, upon diffusing on the ER membrane, CPR can recruit its target protein, inducing the ET from FMN domain to the heme iron. During the transition from a closed to open conformation NADPH and FAD domains change their position, moving significantly away from the membrane surface, while the FMN remains in its proximity, with the FMN cofactor being solvent exposed []. This is a necessary prerequisite for the successful interaction with CYP450s.
The importance of the interaction between CYP450s and their CPR redox partner for catalysis may even go beyond the mere supplement of electrons. Recent MD simulations complemented by QM/MM of the CYP450 BM3 in complex with FMN domain, with and without the substrate bound in the CYP450 active site, elucidated that the FMN domain must undergo significant conformational changes to approach the heme moiety, accelerating in this manner the ET []. This study revealed that the interaction between the α1-helix of CPR and the C-helix of the heme of CYP450 BM3 was enhanced by a reorientation of this helix to a thermochemically stable form gated and choreographed by substrate binding []. It is, however, unclear if this ligand-induced allosteric enhancement of ET may be operative even in steroidogenic enzymes, whose high substrate specificity is often intertwined with active site rigidity.
CPR/CYP19A1 Adduct
Because of its intricacy and the lack of structural information on the CPR/CYP450s adduct, the mechanism of ET and its modulation by the membrane environment remains elusive. The only structure of a CPR/CYP450 adduct was solved for the heme and FMN-binding domain of bacterial P450 (PDB: IBVY) reported by Sevrioukova et al. []. The structure was used as a starting point to build and relax, via a 7 ns-long MD simulation, the adduct between CPR and CYP19A1. This, along with other studies, revealed that the interaction between CPR and CYP450 is essentially based on electrostatic interactions [] and suggested that ET may be here mediated by water molecules lying at the interface of the two proteins []. Distinct studies showed that the FMN domain of the CPR is rich in acidic amino acids, while that of AR is positively charged by lysine and arginine residues at the proximal site where CPR binds []. This was confirmed by mutagenesis experiments showing that upon mutations of key Lys residues, such as Lys108, AR loses its catalytic activity. Similarly, a study done by us on either the FMN domain/AR adduct or the complete CPR/AR model (Figure 9), both embedded in a membrane mimic, carried out by consensus protein docking studies (i.e., use of distinct docking programs based on different scoring functions and search methods) resulted in only one stable binding pose stabilized by electrostatic interaction [].
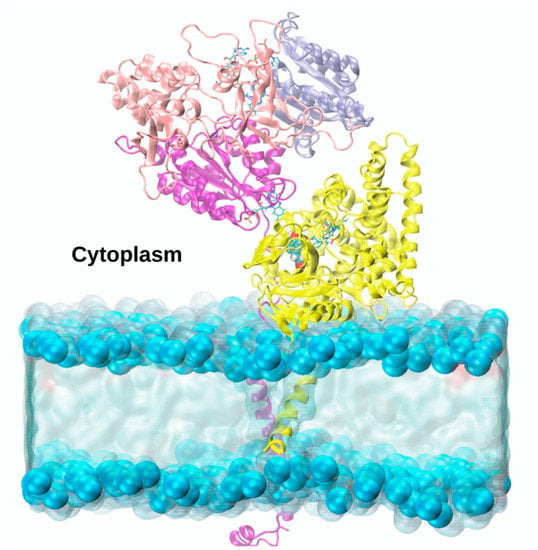
Figure 9.
Atomic-level model of the adduct between Aromatase (yellow ribbons) and CYP450s Reductase, as obtained from consensus docking studies, whose nicotinamide adenine dinucleotide phosphate (NADPH), flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN) domains (shown as blue, pink and purple ribbons, respectively) are embedded in a lipid bilayer [].
5.2. Cythochrome b5 (CYb5)
CYb5 is a heme protein bound to the ER, which actively participates in many reactions mediated by CYP450s, providing the electrons necessary for their catalysis []. According to experimental data, CYb5 can be reduced by accepting electrons from the NADH-cytochrome b5 reductase (CYbR), or from CPR (Figure 10a), acting therefore as an electron shuttle. After the reduction, CYb5 is able to modulate CYP450 catalysis [].
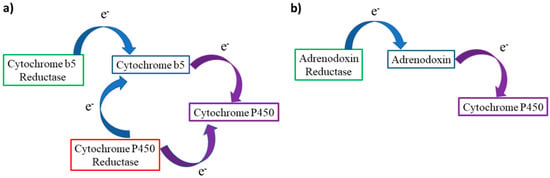
Figure 10.
(a) Electron transport path from CYb5 or CPR to CYP450s at the endoplasmic reticulum membrane. (b) Simplified transport chain in mitochondrial CYP450s, Adx can act as a shuttle transferring one electron at a time from AdR to the target cytochrome.
CYPb5 can reduce many CYP450s, among which the oxy-ferrous form of CYP17A1, is 10-fold faster than the CPR. As detailed above, CYP17A1 catalyzes the synthesis of androgens from the steroid precursors, pregnenolone and progesterone, in two steps: allylic hydroxylation and lyase reaction. CYb5 has been suggested to promote the latter reaction, transferring only one electron due to its low redox potential (+20 mV) []. Therefore, considering the CYP450 catalytic cycle (Figure 2), CYb5 intervenes after the formation of the oxy-ferrous complex (4), providing the second electron to complete the reduction cycle. At this point, the lyase reaction of 17-hydroxy-pregnenolone, as well as 17-hydroxy-progesterone, proceeds through a nucleophilic attack on the carbonyl group of the substrate, forming a peroxo-hemiketal intermediate. This can undergo homolytic or heterolytic cleavage of the O–O bond and rearrange to release the dehydroepiandrosterone product and a molecule of acetic acid [,].
5.3. Adrenodoxin (Adx)
Adx is [2Fe-2S] ferredoxin, which, on receiving electrons from a FAD-containing Adx reductase (AdR), transfers them to mitochondrial CYP450s (Figure 10b). The electrons are supplied to AdR by NADPH. The molecular mechanism of complex formation and electron transport within this system has remained unclear: AdR, Adx, and CYP450 have been proposed to form 1:1:1 or 1:2:1 complexes, but Adx has also been suggested to act as a shuttle, sequentially transporting one electron at a time from AdR to CYP450. In both the complex and shuttle models the interactions between the protein pairs are mainly electrostatically driven. Brownian dynamics simulations attempted to characterize the Adx/CYP11A1 adduct, confirming this hypothesis [].
6. Post-Transcriptional Regulation of CYP450s Activity
Kinase enzymes promote wide spread regulatory mechanisms by phosphorylating their target proteins, and inducing, as a result, their activation/inactivation and/or functional changes. Among their possible targets, also CYP450s can be post-transcriptional regulated by kinases [].
6.1. CYP19A1 Phosphorylation
Phosphorylation of AR acts as functional switch mechanism for estrogen modulation. It has been shown that phosphorylation by insulin-like growth factor-1 increases AR activity in breast cancer cells []. Additionally, it was reported that exposure of estrogen-dependent MCF-7 and ZR75 BC cells to estrogens (i.e., 17β-estradiol) increases the phosphorylation status, and, in turn, the activity of AR. Phosphorylation occurs at Tyr361 and it is induced by c-Src kinase [,]. Additionally, exposure of estrogen receptor positive breast cancer cells to estrogens even inhibits the phosphatase PTP1B ability to de-phosphorylate AR. Hence, estrogens perform a dual function convergently leading their enhanced biosynthesis via AR activation by phosphorylation, and, simultaneous inhibition of PTP1B []. As a result, estrogens, synthesized by AR, bind the estrogen receptor α, and send signals to c-Src. These signals promote the phosphorylation of Tyr361, which increases AR activity leading to an augmented local estrogen production with possible implications in breast cancer onset and progression. Tyr361 lies in the heme proximal cavity of AR, and, since its mutation to Phe dramatically decreases AR activity, it is likely involved in the ET from CPR to AR. Despite mounting evidences on phosphorylation of AR in multiple tissues, the mechanism underlying this post-transcriptional regulation of estrogen biosynthesis remains poorly understood. Recent computational studies highlighted how the phosphorylation of Tyr361 enhances the stabilization of the AR/CPR adduct, facilitating AR in the competition with other CYP450s for the CPR [].
6.2. CYP17A1 Phosphorylation
Human CYP17A1 is phosphorylated on serine and threonine residues by Protein Kinase A (PKA). Phosphorylation increases enzyme activity, affecting exclusively the lyase reaction, while having no effect on allylic hydroxylation []. As for AR, dephosphorylation has an opposite regulatory effect []. Mutagenesis studies pinpointed Ser427 and Thr341 as possible phosphorylation sites []. Despite its importance, how phosphorylation affects the mechanism of this enzyme remains elusive and has never been addressed at computational level.
6.3. CYP11A1 Phosphorylation
Remarkably, even the synthesis of pregnenolone at inner mitochondrial membrane by CYP11A1 is regulated by phosphorylation. Experimental data indicate that PKA can phosphorylate CYP11A1 serine and threonine amino acids, enhancing as a result their enzymatic activity []. In addition, CYP11A1 is actively and selectively phosphorylated by the phospholipid and calcium dependent protein kinase C (PKC) in vitro []. In mitochondria, the electrons necessary for cholesterol metabolism biosynthesis are shuttled to CYP11A1 by Adx. A combined experimental and computational study relying on stopped-flow experiments and Brownian dynamics has addressed this steroid hydroxylating system from bovine adrenal glands. Despite this study being focused on the phosphorylation of Adx at Thr71 by casein kinase 2 (CK2), it disclosed that upon phosphorylation the binding affinity of Adx towards CYP11A1 increases, consistently with the findings illustrated above. The Adx/CYP450 complex adopts a geometry able to increase the ET rate with respect to the wild type counterpart. These findings, evidence the existence of a general mechanism of post-transcriptional regulation in CYP450s, which remains to be investigated in more detail.
7. Conclusions and Perspectives
Understanding the mechanism of steroidogenesis at atomic level is of pivotal importance in disclosing the fundamental biological principles leading to sexual differentiation, reproduction, and fertility; to dissect the multiple finely tuned regulatory mechanisms controlling these pathways and to exploit this information for therapeutic intervention against disorders caused by steroid deregulation [].
In recent years, a growing body of experimental and theoretical studies have contributed to unravel controversial steps of the complex catalytic mechanisms of steroidogenic CYP450s [,,,,,]. Nonetheless, a comprehensive theoretical investigation of CYP17A1 and CYP11A1 catalytic mechanism is still missing in order to further confirm the functional classification of these enzymes. Instead, computational studies have remarkably contributed in disclosing the central role of membranes in the CYP450s functions. A consensus general structural model of CYP450s on the membrane surface has now been reached, as well as the establishment of the crucial role of membranes in affecting channel opening. Conversely, how membranes modulate the traveling of distinct substrate/inhibitor through protein channels has only lately started to be tackled. Furthermore, several aspects remain to be investigated, such as the role of the membrane in fine tuning the CYP450s redox potential [].
Being membranes vital for CYP450s, a detailed atomic-level picture of the association/dissociation and competition with the distinct protein partners occurring at the membrane interface is also of utmost importance. Current research efforts are devoted to unravel how protein–protein interactions modulate CYP450s catalysis []. These mainly involve the interactions of CYP450s with their reducing partners as well as even more complex structures formed by CYP450s’ dimers or polymers [].
Of relevance is the so far largely unexplored mechanism of post-translational regulation of CYP450 catalysis, by phosphorylation/de-phosphorylation. Distinct studies converge to a scenario in which by altering the electrostatic potential of the CYP450 (or its redox partner) the resulting adduct gains stability by establishing stronger electrostatic interactions. This may increase either the redox potential of the process or simply limit the competition among the phosphorylated CYP450s and the other proteins contending for the same redox partner. Understanding the fascinating regulatory process of steroidogenesis has also potential implications for developing knowledge-based small-molecule regulators able to damp/control CYP450 catalysis enhancement with the aim of counteracting cancer diseases (mainly prostate and breast).
We expect that atomic-level understanding of the intricate molecular framework underlining steroidogenic CYP450s’ function in their native environment [] will be significantly expanded in forthcoming years, allowing further steps to be taken towards the clinical management of diseases associated with steroidogenic disorders [].
Supplementary Materials
The following are available online at http://www.mdpi.com/2073-4344/9/1/81/s1, Figure S1: Reaction between the ferric peroxo complex, FeO2−, and a generic aldehyde, Figure S2: Dual hydrogen abstraction proposed, as an example, to rationalize ethanol oxidation carried out by CYP2E1.
Author Contributions
A.S., I.R. and A.M. wrote the paper, A.S. and A.M. designed research.
Funding
Italian Association for Cancer Research: My First AIRC Grant (17134) and Gianni Bonadonna Fellowship.
Acknowledgments
A.M. and I.R. thank the Italian Association for Cancer Research Association for Financial Support via the My First AIRC grant no. 17134 and the Gianni Bonadonna Fellowship. The authors thank Jacopo Sgrignani for useful suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ortiz de Montellaro, P.R. Cytochrome P450: Structure, Mechanism, and Biochemistry; Springer: San Francisco, CA, USA, 2015. [Google Scholar]
- Srejber, M.; Navratilova, V.; Paloncyova, M.; Bazgier, V.; Berka, K.; Anzenbacher, P.; Otyepka, M. Membrane-attached mammalian cytochromes P450: An overview of the membrane’s effects on structure, drug binding, and interactions with redox partners. J. Inorg. Biochem. 2018, 183, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R.; Koymans, L.; Kamataki, T.; Stegeman, J.J.; Feyereisen, R.; Waxman, D.J.; Waterman, M.R.; Gotoh, O.; Coon, M.J.; Estabrook, R.W.; et al. P450 superfamily: Update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 1996, 6, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.T.; McClusky, G.A. Aliphatic hydroxylation via oxygen rebound. Oxygen transfer catalyzed by iron. J. Am. Chem. Soc. 1976, 98, 859–861. [Google Scholar] [CrossRef]
- Groves, J.T.; McClusky, G.A. Aliphatic hydroxylation by highly purified liver microsomal cytochrome P-450. Evidence for a carbon radical intermediate. Biochem. Biophys. Res. Commun. 1978, 81, 154–160. [Google Scholar] [CrossRef]
- Guengerich, F.P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef]
- Black, S.D. Membrane topology of the mammalian P450 cytochromes. FASEB J. 1992, 6, 680–685. [Google Scholar] [CrossRef]
- Williams, P.A.; Cosme, J.; Sridhar, V.; Johnson, E.F.; McRee, D.E. Mammalian microsomal cytochrome P450 monooxygenase: Structural adaptations for membrane binding and functional diversity. Mol. Cell 2000, 5, 121–131. [Google Scholar] [CrossRef]
- Barnaba, C.; Gentry, K.; Sumangala, N.; Ramamoorthy, A. The catalytic function of cytochrome P450 is entwined with its membrane-bound nature. F1000Research 2017, 6, 662. [Google Scholar] [CrossRef]
- Vargiu, A.V.; Magistrato, A. Atomistic-level portrayal of drug-DNA Interplay: A history of courtships and meetings revealed by molecular simulations. ChemMedChem 2014, 9, 1966–1981. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Palermo, G.; Spinello, A.; Rothlisberger, U.; Magistrato, A. All-atom simulations disentangle the functional dynamics underlying gene maturation in the intron lariat spliceosome. Proc. Natl. Acad. Sci. USA 2018, 115, 6584–6589. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Berka, K.; Hendrychova, T.; Anzenbacher, P.; Otyepka, M. Membrane position of ibuprofen agrees with suggested access path entrance to cytochrome P450 2C9 active site. J. Phys. Chem. A 2011, 115, 11248–11255. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, V.; Balali-Mood, K.; Sansom, M.S.; Wade, R.C. Structure and dynamics of the membrane-bound cytochrome P450 2C9. PLoS Comput. Biol. 2011, 7, e1002152. [Google Scholar] [CrossRef] [PubMed]
- Sgrignani, J.; Magistrato, A. Influence of the membrane lipophilic environment on the structure and on the substrate access/egress routes of the human aromatase enzyme. A computational study. J. Chem. Inf. Model. 2012, 52, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Comer, J.; Herndon, C.; Leung, N.; Pavlova, A.; Swift, R.V.; Tung, C.; Rowley, C.N.; Amaro, R.E.; Chipot, C.; et al. Simulation-Based Approaches for Determining Membrane Permeability of Small Compounds. J. Chem. Inf. Model. 2016, 56, 721–733. [Google Scholar] [CrossRef]
- Jiang, W.; Ghosh, D. Motion and flexibility in human cytochrome p450 aromatase. PLoS ONE 2012, 7, e32565. [Google Scholar] [CrossRef]
- Sgrignani, J.; Bon, M.; Colombo, G.; Magistrato, A. Computational approaches elucidate the allosteric mechanism of human aromatase inhibition: A novel possible route to Small-molecule regulation of CYP450s activities? J. Chem. Inf. Model. 2014, 54, 2856–2868. [Google Scholar] [CrossRef]
- Magistrato, A.; Sgrignani, J.; Krause, R.; Cavalli, A. Single or Multiple Access Channels to the CYP450s Active Site? An Answer from Free Energy Simulations of the Human Aromatase Enzyme. J. Phys. Chem. Lett. 2017, 8, 2036–2042. [Google Scholar] [CrossRef]
- Colizzi, F.; Bussi, G. RNA unwinding from reweighted pulling simulations. J. Am. Chem. Soc. 2012, 134, 5173–5179. [Google Scholar] [CrossRef] [PubMed]
- Vargiu, A.V.; Ruggerone, P.; Magistrato, A.; Carloni, P. Sliding of alkylating anticancer drugs along the minor groove of DNA: New insights on sequence selectivity. Biophys. J. 2008, 94, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Piana, S.; Laio, A. A bias-exchange approach to protein folding. J. Phys. Chem. B 2007, 111, 4553–4559. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Gervasio, F.L.; Laio, A.; Parrinello, M. Free-energy landscape for beta hairpin folding from combined parallel tempering and metadynamics. J. Am. Chem. Soc. 2006, 128, 13435–13441. [Google Scholar] [CrossRef]
- Casalino, L.; Palermo, G.; Rothlisberger, U.; Magistrato, A. Who Activates the Nucleophile in Ribozyme Catalysis? An Answer from the Splicing Mechanism of Group II Introns. J. Am. Chem. Soc. 2016, 138, 10374–10377. [Google Scholar] [CrossRef]
- Sgrignani, J.; Magistrato, A. QM/MM MD Simulations on the Enzymatic Pathway of the Human Flap Endonuclease (hFEN1) Elucidating Common Cleavage Pathways to RNase H Enzymes. ACS Catal. 2015, 139, 3864–3875. [Google Scholar] [CrossRef]
- Alonso-Gil, S.; Males, A.; Fernandes, P.Z.; Williams, S.J.; Davies, G.J.; Rovira, C. Computational Design of Experiment Unveils the Conformational Reaction Coordinate of GH125 alpha-Mannosidases. J. Am. Chem. Soc. 2017, 139, 1085–1088. [Google Scholar] [CrossRef]
- Casalino, L.; Palermo, G.; Abdurakhmonova, N.; Rothlisberger, U.; Magistrato, A. Development of Site-Specific Mg(2+)-RNA Force Field Parameters: A Dream or Reality? Guidelines from Combined Molecular Dynamics and Quantum Mechanics Simulations. J. Chem. Theory Comput. 2017, 13, 340–352. [Google Scholar] [CrossRef]
- Spinello, A.; Magistrato, A. An omics perspective to the molecular mechanisms of anticancer metallo-drugs in the computational microscope era. Expert Opin. Drug Discov. 2017, 12, 813–825. [Google Scholar] [CrossRef]
- Vidossich, P.; Magistrato, A. QM/MM molecular dynamics studies of metal binding proteins. Biomolecules 2014, 4, 616–645. [Google Scholar] [CrossRef] [PubMed]
- Sgrignani, J.; Magistrato, A. First-principles modeling of biological systems and structure-based drug-design. Curr. Comput. Aided Drug. Des. 2013, 9, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Brunk, E.; Rothlisberger, U. Mixed Quantum Mechanical/Molecular Mechanical Molecular Dynamics Simulations of Biological Systems in Ground and Electronically Excited States. Chem. Rev. 2015, 115, 6217–6263. [Google Scholar] [CrossRef] [PubMed]
- Cascella, M.; Magistrato, A.; Tavernelli, I.; Carloni, P.; Rothlisberger, U. Role of protein frame and solvent for the redox properties of azurin from Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2006, 103, 19641–19646. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 enzymes: Their structure, reactivity, and selectivity-modeled by QM/MM calculations. Chem. Rev. 2010, 110, 949–1017. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Mechanisms of cytochrome P450 substrate oxidation: MiniReview. J. Biochem. Mol. Toxicol. 2007, 21, 163–168. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Munro, A.W. Unusual cytochrome p450 enzymes and reactions. J. Biol. Chem. 2013, 288, 17065–17073. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, C.; Wang, H.; Han, K.; Shaik, S. A new mechanism for ethanol oxidation mediated by cytochrome P450 2E1: Bulk polarity of the active site makes a difference. ChemBioChem 2007, 8, 277–281. [Google Scholar] [CrossRef]
- Spinello, A.; Pavlin, M.; Casalino, L.; Magistrato, A. A Dehydrogenase Dual Hydrogen Abstraction Mechanism Promotes Estrogen Biosynthesis: Can We Expand the Functional Annotation of the Aromatase Enzyme? Chem. Eur. J. 2018, 24, 10840–10849. [Google Scholar] [CrossRef]
- Li, J.; Papadopoulos, V.; Vihma, V. Steroid biosynthesis in adipose tissue. Steroids 2015, 103, 89–104. [Google Scholar] [CrossRef]
- Pan, T.; He, G.; Chen, M.; Bao, C.; Chen, Y.; Liu, G.; Zhou, M.; Li, S.; Xu, W.; Liu, X. Abnormal CYP11A1 gene expression induces excessive autophagy, contributing to the pathogenesis of preeclampsia. Oncotarget 2017, 8, 89824–89836. [Google Scholar] [CrossRef]
- Shikita, M.; Hall, P.F. Cytochrome P-450 from bovine adrenocortical mitochondria: An enzyme for the side chain cleavage of cholesterol. II. Subunit structure. J. Biol. Chem. 1973, 248, 5605–5609. [Google Scholar] [PubMed]
- Strushkevich, N.; MacKenzie, F.; Cherkesova, T.; Grabovec, I.; Usanov, S.; Park, H.W. Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc. Natl. Acad. Sci. USA 2011, 108, 10139–10143. [Google Scholar] [CrossRef] [PubMed]
- Tuckey, R.C.; Li, W.; Zjawiony, J.K.; Zmijewski, M.A.; Nguyen, M.N.; Sweatman, T.; Miller, D.; Slominski, A. Pathways and products for the metabolism of vitamin D3 by cytochrome P450scc. FEBS J. 2008, 275, 2585–2596. [Google Scholar] [CrossRef]
- Davydov, R.; Gilep, A.A.; Strushkevich, N.V.; Usanov, S.A.; Hoffman, B.M. Compound I is the reactive intermediate in the first monooxygenation step during conversion of cholesterol to pregnenolone by cytochrome P450scc: EPR/ENDOR/cryoreduction/annealing studies. J. Am. Chem. Soc. 2012, 134, 17149–17156. [Google Scholar] [CrossRef] [PubMed]
- Duque, C.; Morisaki, M.; Ikekawa, N.; Shikita, M.; Tamaoki, B. The final step of side-chain cleavage of cholesterol by adrenocortical cytochrome P-450(scc) studied with [22(-18)O]20,22-dihydroxycholesterols, [18O]isocaproaldehyde, [18O]water and atmospheric [18O]oxygen. Biochem. Biophys. Res. Commun. 1978, 85, 317–325. [Google Scholar] [CrossRef]
- Lieberman, S.; Warne, P.A. 17-Hydroxylase: An evaluation of the present view of its catalytic role in steroidogenesis. J. Steroid Biochem. Mol. Biol. 2001, 78, 299–312. [Google Scholar] [CrossRef]
- Byon, C.Y.; Gut, M. Steric considerations regarding the biodegradation of cholesterol to pregnenolone.-exclusion of (22S)-22-hydroxycholesterol and 22-ketocholesterol as intermediates. Biochem. Biophys. Res. Commun. 1980, 94, 549–552. [Google Scholar] [CrossRef]
- Davydov, R.; Strushkevich, N.; Smil, D.; Yantsevich, A.; Gilep, A.; Usanov, S.; Hoffman, B.M. Evidence That Compound I Is the Active Species in Both the Hydroxylase and Lyase Steps by Which P450scc Converts Cholesterol to Pregnenolone: EPR/ENDOR/Cryoreduction/Annealing Studies. Biochemistry 2015, 54, 7089–7097. [Google Scholar] [CrossRef]
- Attard, G.; Reid, A.H.; Olmos, D.; de Bono, J.S. Antitumor activity with CYP17 blockade indicates that castration-resistant prostate cancer frequently remains hormone driven. Cancer Res. 2009, 69, 4937–4940. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, F.K.; Gonzalez, E.; Auchus, R.J.; Guengerich, F.P. Mechanism of 17alpha,20-Lyase and New Hydroxylation Reactions of Human Cytochrome P450 17A1: 18O Labeling and Oxygen Surrogate Evidence for a Role of a Perferryl Oxygen. J. Biol. Chem. 2016, 291, 17143–17164. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.C.; Denisov, I.G.; Grinkova, Y.V.; Khatri, Y.; Sligar, S.G. Kinetic solvent isotope effect in human P450 CYP17A1-mediated androgen formation: Evidence for a reactive peroxoanion intermediate. J. Am. Chem. Soc. 2013, 135, 16245–16247. [Google Scholar] [CrossRef] [PubMed]
- Khatri, Y.; Gregory, M.C.; Grinkova, Y.V.; Denisov, I.G.; Sligar, S.G. Active site proton delivery and the lyase activity of human CYP17A1. Biochem. Biophys. Res. Commun. 2014, 443, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Mak, P.J.; Gregory, M.C.; Denisov, I.G.; Sligar, S.G.; Kincaid, J.R. Unveiling the crucial intermediates in androgen production. Proc. Natl. Acad. Sci. USA 2015, 112, 15856–15861. [Google Scholar] [CrossRef] [PubMed]
- Bonomo, S.; Jorgensen, F.S.; Olsen, L. Mechanism of Cytochrome P450 17A1-Catalyzed Hydroxylase and Lyase Reactions. J. Chem. Inf. Model. 2017, 57, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Sgrignani, J.; Cavalli, A.; Colombo, G.; Magistrato, A. Enzymatic and Inhibition Mechanism of Human Aromatase (CYP19A1) Enzyme. A Computational Perspective from QM/MM and Classical Molecular Dynamics Simulations. Mini Rev. Med. Chem. 2016, 16, 1112–1124. [Google Scholar] [CrossRef]
- Hackett, J.C.; Brueggemeier, R.W.; Hadad, C.M. The final catalytic step of cytochrome p450 aromatase: A density functional theory study. J. Am. Chem. Soc. 2005, 127, 5224–5237. [Google Scholar] [CrossRef]
- Sgrignani, J.; Iannuzzi, M.; Magistrato, A. Role of Water in the Puzzling Mechanism of the Final Aromatization Step Promoted by the Human Aromatase Enzyme. Insights from QM/MM MD Simulations. J. Chem. Inf. Model. 2015, 55, 2218–2226. [Google Scholar] [CrossRef]
- Caporuscio, F.; Rastelli, G.; Imbriano, C.; Del Rio, A. Structure-based design of potent aromatase inhibitors by high-throughput docking. J. Med. Chem. 2011, 54, 4006–4017. [Google Scholar] [CrossRef]
- Ghosh, D.; Lo, J.; Morton, D.; Valette, D.; Xi, J.; Griswold, J.; Hubbell, S.; Egbuta, C.; Jiang, W.; An, J.; et al. Novel aromatase inhibitors by structure-guided design. J. Med. Chem. 2012, 55, 8464–8476. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.; Calder, M.R.; Corina, D.L.; Wright, J.N. Mechanistic studies on C-19 demethylation in oestrogen biosynthesis. Biochem. J. 1982, 201, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Morand, P.; Williamson, D.G.; Layne, D.S.; Lompa-Krzymien, L.; Salvador, J. Conversion of an androgen epoxide into 17beta-estradiol by human placental microsomes. Biochemistry 1975, 14, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, H.; Fishman, J. Unusually facile aromatization of 2 beta-hydroxy-19-oxo-4-androstene-3, 17-dione to estrone. Implications in estrogen biosynthesis. J. Am. Chem. Soc. 1974, 96, 7325–7329. [Google Scholar] [CrossRef] [PubMed]
- Goto, J.; Fishman, J. Participation of a nonenzymatic transformation in the biosynthesis of estrogens from androgens. Science 1977, 195, 80–81. [Google Scholar] [CrossRef] [PubMed]
- Caspi, E.; Wicha, J.; Arunachalam, T.; Nelson, P.; Spiteller, G. Estrogen biosynthesis: Concerning the obligatory intermediacy of 2.beta.-hydroxy-10.beta.-formyl androst-4-ene-3,17-dione. J. Am. Chem. Soc. 1984, 106, 7282–7283. [Google Scholar] [CrossRef]
- Akhtar, M.; Corina, D.; Pratt, J.; Smith, T. Studies on the removal of C-19 in oesterogen biosynthesis using 18O2. J. Chem. Soc. Chem. Commun. 1976, 854–856. [Google Scholar] [CrossRef]
- Mak, P.J.; Luthra, A.; Sligar, S.G.; Kincaid, J.R. Resonance Raman spectroscopy of the oxygenated intermediates of human CYP19A1 implicates a compound i intermediate in the final lyase step. J. Am. Chem. Soc. 2014, 136, 4825–4828. [Google Scholar] [CrossRef]
- Khatri, Y.; Luthra, A.; Duggal, R.; Sligar, S.G. Kinetic solvent isotope effect in steady-state turnover by CYP19A1 suggests involvement of Compound 1 for both hydroxylation and aromatization steps. FEBS Lett. 2014, 588, 3117–3122. [Google Scholar] [CrossRef]
- Yoshimoto, F.K.; Guengerich, F.P. Mechanism of the third oxidative step in the conversion of androgens to estrogens by cytochrome P450 19A1 steroid aromatase. J. Am. Chem. Soc. 2014, 136, 15016–15025. [Google Scholar] [CrossRef]
- Xu, K.; Wang, Y.; Hirao, H. Estrogen Formation via H-Abstraction from the O–H Bond of gem-Diol by Compound I in the Reaction of CYP19A1: Mechanistic Scenario Derived from Multiscale QM/MM Calculations. ACS Catal. 2015, 5, 4175–4179. [Google Scholar] [CrossRef]
- Paloncyova, M.; DeVane, R.; Murch, B.; Berka, K.; Otyepka, M. Amphiphilic drug-like molecules accumulate in a membrane below the head group region. J. Phys. Chem. B 2014, 118, 1030–1039. [Google Scholar] [CrossRef]
- Denisov, I.G.; Shih, A.Y.; Sligar, S.G. Structural differences between soluble and membrane bound cytochrome P450s. J. Inorg. Biochem. 2012, 108, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Navratilova, V.; Paloncyova, M.; Kajsova, M.; Berka, K.; Otyepka, M. Effect of cholesterol on the structure of membrane-attached cytochrome P450 3A4. J. Chem. Inf. Model. 2015, 55, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, V.; Winn, P.J.; Wade, R.C. The ins and outs of cytochrome P450s. Biochim. Biophys. Acta 2007, 1770, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Petrek, M.; Otyepka, M.; Banas, P.; Kosinova, P.; Koca, J.; Damborsky, J. CAVER: A new tool to explore routes from protein clefts, pockets and cavities. BMC Bioinform. 2006, 7, 316. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Svobodova Varekova, R.; Berka, K.; Pravda, L.; Navratilova, V.; Banas, P.; Ionescu, C.M.; Otyepka, M.; Koca, J. MOLE 2.0: Advanced approach for analysis of biomacromolecular channels. J. Cheminform. 2013, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Berka, K.; Hanak, O.; Sehnal, D.; Banas, P.; Navratilova, V.; Jaiswal, D.; Ionescu, C.M.; Svobodova Varekova, R.; Koca, J.; Otyepka, M. MOLEonline 2.0: Interactive web-based analysis of biomacromolecular channels. Nucleic Acids Res. 2012, 40, W222–W227. [Google Scholar] [CrossRef] [PubMed]
- Pravda, L.; Sehnal, D.; Svobodova Varekova, R.; Navratilova, V.; Tousek, D.; Berka, K.; Otyepka, M.; Koca, J. ChannelsDB: Database of biomacromolecular tunnels and pores. Nucleic Acids Res. 2018, 46, D399–D405. [Google Scholar] [CrossRef] [PubMed]
- Berka, K.; Paloncyova, M.; Anzenbacher, P.; Otyepka, M. Behavior of human cytochromes P450 on lipid membranes. J. Phys. Chem. B 2013, 117, 11556–11564. [Google Scholar] [CrossRef]
- Park, J.; Czapla, L.; Amaro, R.E. Molecular simulations of aromatase reveal new insights into the mechanism of ligand binding. J. Chem. Inf. Model. 2013, 53, 2047–2056. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.L.; Xue, Q.; Zheng, Q.C.; Zhang, J.L.; Kong, C.P.; Fan, J.R.; Zhang, H.X. Structural features and dynamic investigations of the membrane-bound cytochrome P450 17A1. Biochim. Biophys. Acta 2015, 1848, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Paloncyova, M.; Navratilova, V.; Berka, K.; Laio, A.; Otyepka, M. Role of Enzyme Flexibility in Ligand Access and Egress to Active Site: Bias-Exchange Metadynamics Study of 1,3,7-Trimethyluric Acid in Cytochrome P450 3A4. J. Chem. Theory Comput. 2016, 12, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, P.P.; Schenkman, J.B. Differences in the mechanism of functional interaction between NADPH-cytochrome P-450 reductase and its redox partners. Mol. Pharmacol. 1986, 30, 178–185. [Google Scholar] [PubMed]
- Porter, T.D. The roles of cytochrome b5 in cytochrome P450 reactions. J. Biochem. Mol. Toxicol. 2002, 16, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Iyanagi, T. Structure and function of NADPH-cytochrome P450 reductase and nitric oxide synthase reductase domain. Biochem. Biophys. Res. Commun. 2005, 338, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.Y.; Junk, K.W.; Coon, M.J. Resolution of the cytochrome P-450-containing omega-hydroxylation system of liver microsomes into three components. J. Biol. Chem. 1969, 244, 3714–3721. [Google Scholar] [PubMed]
- Sugishima, M.; Sato, H.; Higashimoto, Y.; Harada, J.; Wada, K.; Fukuyama, K.; Noguchi, M. Structural basis for the electron transfer from an open form of NADPH-cytochrome P450 oxidoreductase to heme oxygenase. Proc. Natl. Acad. Sci. USA 2014, 111, 2524–2529. [Google Scholar] [CrossRef]
- Ellis, J.; Gutierrez, A.; Barsukov, I.L.; Huang, W.C.; Grossmann, J.G.; Roberts, G.C. Domain motion in cytochrome P450 reductase: Conformational equilibria revealed by NMR and small-angle x-ray scattering. J. Biol. Chem. 2009, 284, 36628–36637. [Google Scholar] [CrossRef]
- Laursen, T.; Jensen, K.; Moller, B.L. Conformational changes of the NADPH-dependent cytochrome P450 reductase in the course of electron transfer to cytochromes P450. Biochim. Biophys. Acta 2011, 1814, 132–138. [Google Scholar] [CrossRef]
- Dubey, K.D.; Wang, B.; Shaik, S. Molecular Dynamics and QM/MM Calculations Predict the Substrate-Induced Gating of Cytochrome P450 BM3 and the Regio- and Stereoselectivity of Fatty Acid Hydroxylation. J. Am. Chem. Soc. 2016, 138, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.F.; Li, H.; Zhang, H.; Peterson, J.A.; Poulos, T.L. Structure of a cytochrome P450-redox partner electron-transfer complex. Proc. Natl. Acad. Sci. USA 1999, 96, 1863–1868. [Google Scholar] [CrossRef] [PubMed]
- Zollner, A.; Pasquinelli, M.A.; Bernhardt, R.; Beratan, D.N. Protein phosphorylation and intermolecular electron transfer: A joint experimental and computational study of a hormone biosynthesis pathway. J. Am. Chem. Soc. 2007, 129, 4206–4216. [Google Scholar] [CrossRef]
- Dai, Y.; Zhen, J.; Zhang, X.; Zhong, Y.; Liu, S.; Sun, Z.; Guo, Y.; Wu, Q. Analysis of the complex formation, interaction and electron transfer pathway between the “open” conformation of NADPH-cytochrome P450 reductase and aromatase. Steroids 2015, 101, 116–124. [Google Scholar] [CrossRef]
- Ritacco, I.; Spinello, A.; Magistrato, A. The post-translational regulation of steroidogenic CYP450s metabolism as revealed by all-atoms simulations of the aromatase enzyme. 2019; to be published. [Google Scholar]
- Yablokov, E.; Florinskaya, A.; Medvedev, A.; Sergeev, G.; Strushkevich, N.; Luschik, A.; Shkel, T.; Haidukevich, I.; Gilep, A.; Usanov, S.; et al. Thermodynamics of interactions between mammalian cytochromes P450 and b5. Arch. Biochem. Biophys. 2017, 619, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Duggal, R.; Denisov, I.G.; Sligar, S.G. Cytochrome b5 enhances androgen synthesis by rapidly reducing the CYP17A1 oxy-complex in the lyase step. FEBS Lett. 2018, 592, 2282–2288. [Google Scholar] [CrossRef]
- Oesch-Bartlomowicz, B.; Oesch, F. Cytochrome-P450 phosphorylation as a functional switch. Arch. Biochem. Biophys. 2003, 409, 228–234. [Google Scholar] [CrossRef]
- Su, B.; Wong, C.; Hong, Y.; Chen, S. Growth factor signaling enhances aromatase activity of breast cancer cells via post-transcriptional mechanisms. J. Steroid Biochem. Mol. Biol. 2011, 123, 101–108. [Google Scholar] [CrossRef]
- Catalano, S.; Barone, I.; Giordano, C.; Rizza, P.; Qi, H.; Gu, G.; Malivindi, R.; Bonofiglio, D.; Ando, S. Rapid estradiol/ERalpha signaling enhances aromatase enzymatic activity in breast cancer cells. Mol. Endocrinol. 2009, 23, 1634–1645. [Google Scholar] [CrossRef]
- Barone, I.; Giordano, C.; Malivindi, R.; Lanzino, M.; Rizza, P.; Casaburi, I.; Bonofiglio, D.; Catalano, S.; Ando, S. Estrogens and PTP1B function in a novel pathway to regulate aromatase enzymatic activity in breast cancer cells. Endocrinology 2012, 153, 5157–5166. [Google Scholar] [CrossRef]
- Zhang, L.H.; Rodriguez, H.; Ohno, S.; Miller, W.L. Serine phosphorylation of human P450c17 increases 17,20-lyase activity: Implications for adrenarche and the polycystic ovary syndrome. Proc. Natl. Acad. Sci. USA 1995, 92, 10619–10623. [Google Scholar] [CrossRef] [PubMed]
- Tee, M.K.; Dong, Q.; Miller, W.L. Pathways leading to phosphorylation of p450c17 and to the posttranslational regulation of androgen biosynthesis. Endocrinology 2008, 149, 2667–2677. [Google Scholar] [CrossRef]
- Tee, M.K.; Miller, W.L. Phosphorylation of human cytochrome P450c17 by p38alpha selectively increases 17,20 lyase activity and androgen biosynthesis. J. Biol. Chem. 2013, 288, 23903–23913. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.G.; Goldstein, S.; Savard, K.; Marsh, J.M. Protein kinase stimulation of a reconstituted cholesterol side chain cleavage enzyme system in the bovine corpus luteum. J. Biol. Chem. 1975, 250, 5137–5143. [Google Scholar] [PubMed]
- Vilgrain, I.; Defaye, G.; Chambaz, E.M. Adrenocortical cytochrome P-450 responsible for cholesterol side chain cleavage (P-450scc) is phosphorylated by the calcium-activated, phospholipid-sensitive protein kinase (protein kinase C). Biochem. Biophys. Res. Commun. 1984, 125, 554–561. [Google Scholar] [CrossRef]
- Pikuleva, I.A.; Waterman, M.R. Cytochromes p450: Roles in diseases. J. Biol. Chem. 2013, 288, 17091–17098. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).