Altering Conversion and Product Selectivity of Dry Reforming of Methane in a Dielectric Barrier Discharge by Changing the Dielectric Packing Material



Abstract
1. Introduction
2. Results
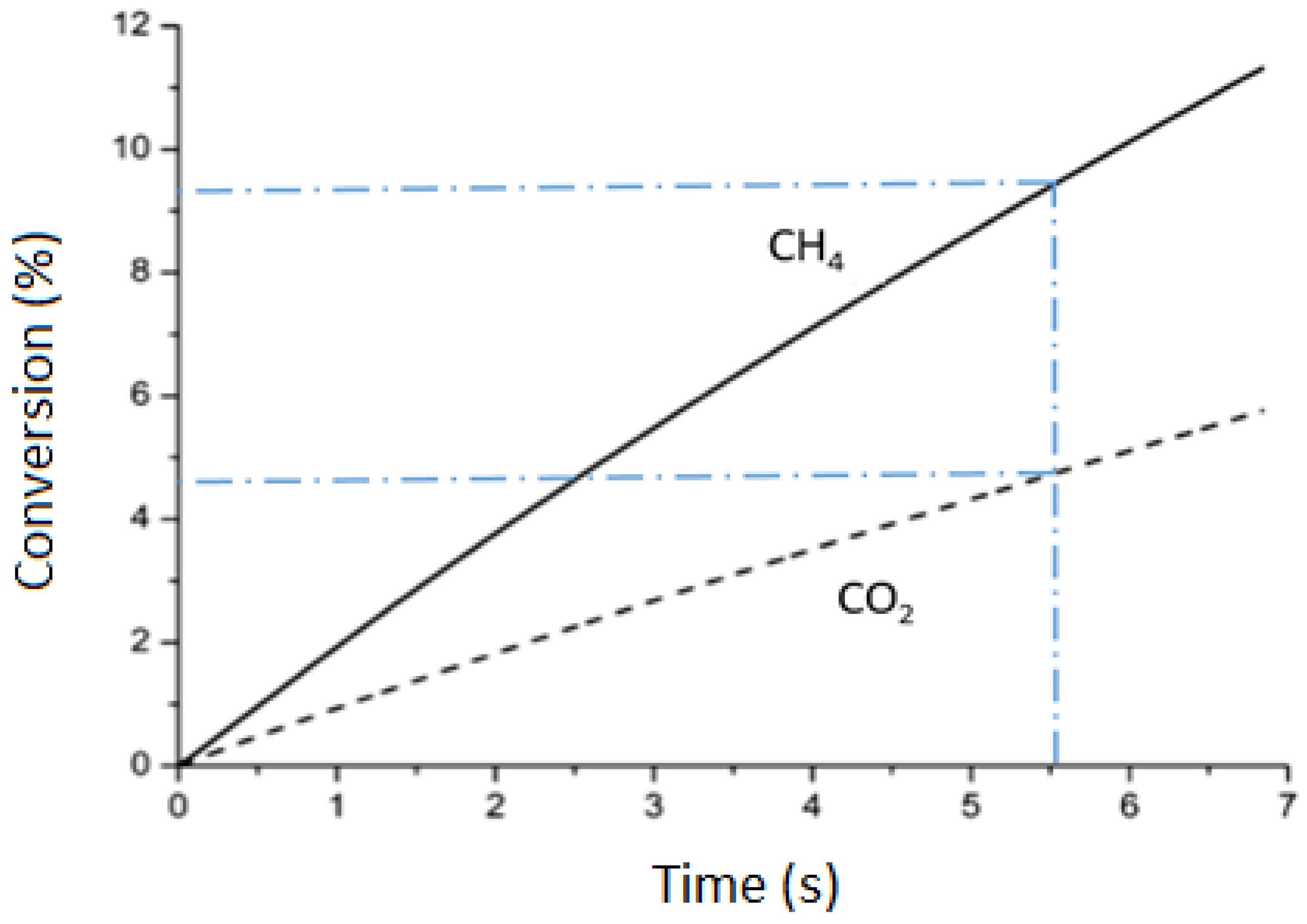
2.1. CO2 Conversion in DRM and Comparison with CO2 Splitting
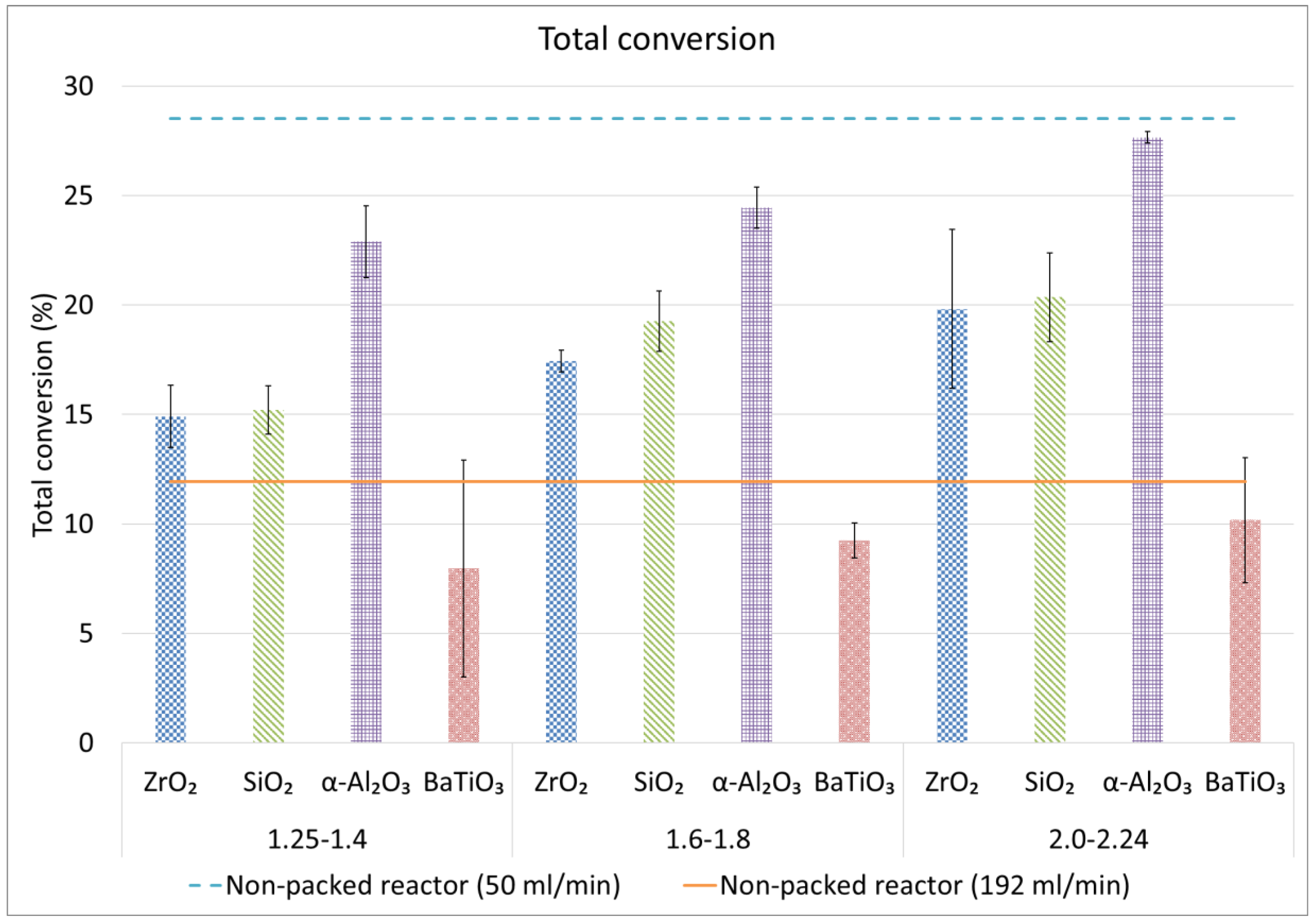
2.2. CH4 and Total Conversion
2.3. Comparison Studies α/γ-Al2O3
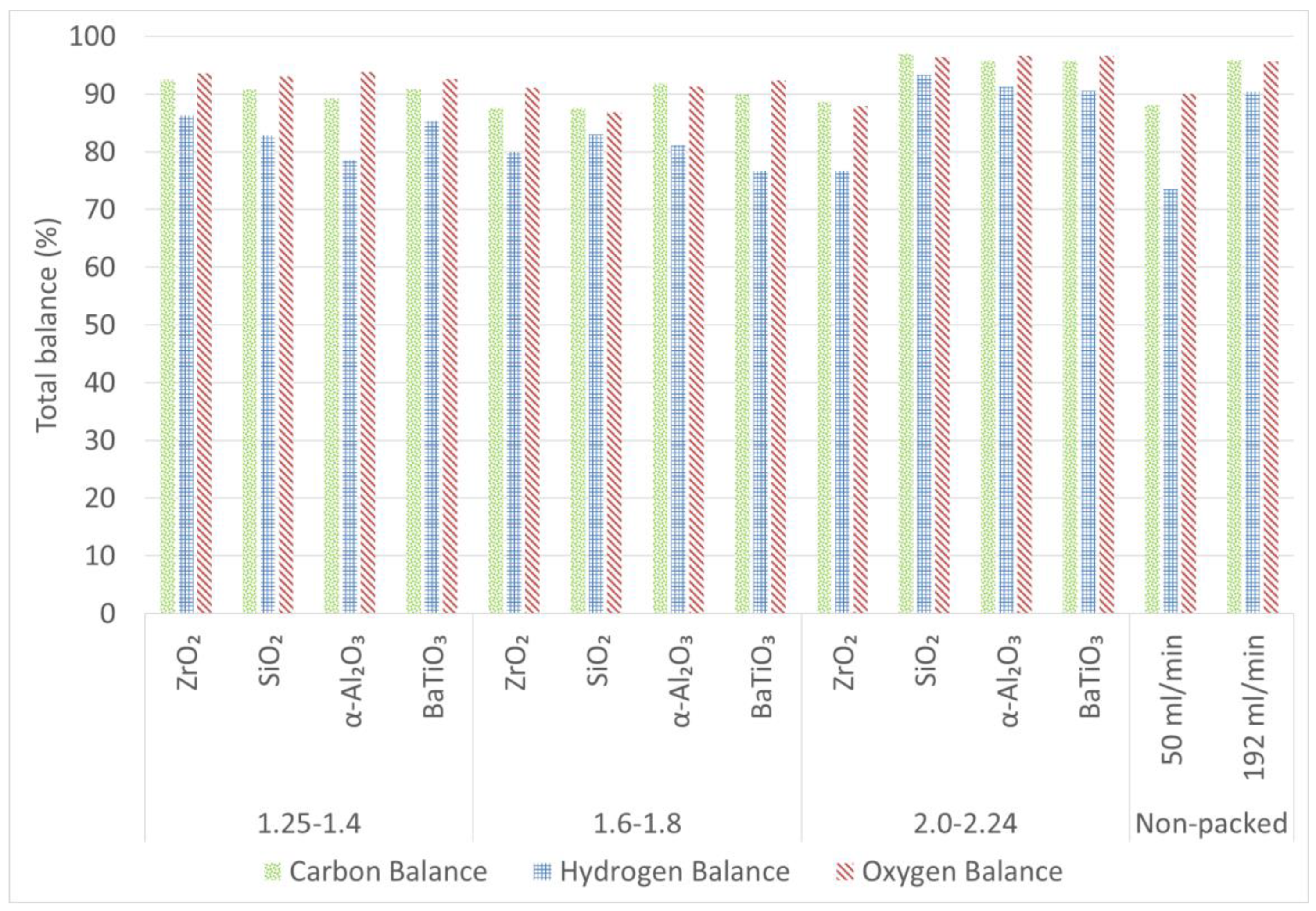
2.4. Carbon, Hydrogen and Oxygen Balances
2.5. Product Fractions
- For the non-packed reactor at 50 mL/min and all α-Al2O3 spheres, the order is: ethane > H2 > propane > ethyne.
- For the non-packed reactor at 192 mL/min, the order is: ethane > H2 > ethyne > propane > ethene.
- For the smallest ZrO2 and BaTiO3 spheres and all SiO2 spheres, the order is: ethyne ≊ ethane > H2 > propane (formaldehyde in case of BaTiO3).
- For the two largest BaTiO3 spheres and the intermediate ZrO2 spheres, the order is: ethane > ethyne ≊ H2 > propane > ethene.
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Snoeckx, R.; Bogaerts, A. Plasma technology—A novel solution for CO2 conversion? Chem. Soc. Rev. 2017, 46, 5805–5863. [Google Scholar] [CrossRef] [PubMed]
- Song, C. Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing. Catal. Today 2006, 115, 2–32. [Google Scholar] [CrossRef]
- Chung, W.C.; Chang, M.B. Review of catalysis and plasma performance on dry reforming of CH4 and possible synergistic effects. Renew. Sustain. Energy Rev. 2016, 62, 13–31. [Google Scholar] [CrossRef]
- Usman, M.; Wan Daud, W.M.A.; Abbas, H.F. Dry reforming of methane: Influence of process parameters—A review. Renew. Sustain. Energy Rev. 2015, 45, 710–744. [Google Scholar] [CrossRef]
- Jarvis, S.M.; Samsatli, S. Technologies and infrastructures underpinning future CO2 value chains: A comprehensive review and comparative analysis. Renew. Sustain. Energy Rev. 2018, 85, 46–68. [Google Scholar] [CrossRef]
- Chung, W.; Pan, K.; Lee, H.; Chang, M. Dry Reforming of Methane with Dielectric Barrier Discharge and Ferroelectric Packed-Bed Reactors. Energy Fuels 2014, 28, 7621–7631. [Google Scholar] [CrossRef]
- Arkatova, L.A. The deposition of coke during carbon dioxide reforming of methane over intermetallides. Catal. Today 2010, 157, 170–176. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef] [PubMed]
- Samukawa, S.; Hori, M.; Rauf, S.; Tachibana, K.; Bruggeman, P.; Kroesen, G.; Whitehead, J.C.; Murphy, A.B.; Gutsol, A.F.; Starikovskaia, S.; et al. The 2012 Plasma Roadmap. J. Phys. D Appl. Phys. 2012, 45, 253001. [Google Scholar] [CrossRef]
- Lavoie, J.-M. Review on dry reforming of methane, a potentially more environmentally-friendly approach to the increasing natural gas exploitation. Front. Chem. 2014, 2, 81. [Google Scholar] [CrossRef] [PubMed]
- Kogelschatz, U. Dielectric-barrier discharges: Their history, discharge physics, and industrial applications. Plasma Chem. Plasma Process. 2003, 23, 1–46. [Google Scholar] [CrossRef]
- Wang, L.; Yi, Y.; Wu, C.; Guo, H.; Tu, X. One-Step Reforming of CO2 and CH4 into High-Value Liquid Chemicals and Fuels at Room Temperature by Plasma-Driven Catalysis. Angew. Chem. Int. Ed. 2017, 56, 13679–13683. [Google Scholar] [CrossRef] [PubMed]
- Neyts, E.C.; Bogaerts, A. Understanding plasma catalysis through modelling and simulation—A review. J. Phys. D Appl. Phys. 2014, 47, 224010. [Google Scholar] [CrossRef]
- Aerts, R.; Somers, W.; Bogaerts, A. Carbon Dioxide Splitting in a Dielectric Barrier Discharge Plasma: A Combined Experimental and Computational Study. ChemSusChem 2015, 8, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Paulussen, S.; Verheyde, B.; Tu, X.; De Bie, C.; Martens, T.; Petrovic, D.; Bogaerts, A.; Sels, B. Conversion of carbon dioxide to value-added chemicals in atmospheric pressure dielectric barrier discharges. Plasma Sources Sci. Technol. 2010, 19, 034015. [Google Scholar] [CrossRef]
- Ozkan, A.; Bogaerts, A.; Reniers, F. Routes to increase the conversion and the energy efficiency in the splitting of CO2 by a dielectric barrier discharge. J. Phys. D Appl. Phys. 2017, 50, 084004. [Google Scholar] [CrossRef]
- Zhang, A.-J.; Zhu, A.-M.; Guo, J.; Xu, Y.; Shi, C. Conversion of greenhouse gases into syngas via combined effects of discharge activation and catalysis. Chem. Eng. J. 2010, 156, 601–606. [Google Scholar] [CrossRef]
- Zheng, X.; Tan, S.; Dong, L.; Li, S.; Chen, H. Plasma-assisted catalytic dry reforming of methane: Highly catalytic performance of nickel ferrite nanoparticles embedded in silica. J. Power Sources 2015, 274, 286–294. [Google Scholar] [CrossRef]
- Zheng, X.; Tan, S.; Dong, L.; Li, S.; Chen, H. LaNiO3@SiO2 core–shell nano-particles for the dry reforming of CH4 in the dielectric barrier discharge plasma. Int. J. Hydrog. Energy 2014, 39, 11360–11367. [Google Scholar] [CrossRef]
- Karuppiah, J.; Manoj Kumar Reddy, P.; Linga Reddy, E.; Subrahmanyam, C. Catalytic non-thermal plasma reactor for decomposition of dilute chlorobenzene. Plasma Process. Polym. 2013, 10, 1074–1080. [Google Scholar] [CrossRef]
- Krawczyk, K.; Młotek, M.; Ulejczyk, B.; Schmidt-Szałowski, K. Methane conversion with carbon dioxide in plasma-catalytic system. Fuel 2014, 117, 608–617. [Google Scholar] [CrossRef]
- Wang, Q.; Cheng, Y.; Jin, Y. Dry reforming of methane in an atmospheric pressure plasma fluidized bed with Ni/γ-Al2O3 catalyst. Catal. Today 2009, 148, 275–282. [Google Scholar] [CrossRef]
- Zeng, Y.; Zhu, X.; Mei, D.; Ashford, B.; Tu, X. Plasma-catalytic dry reforming of methane over-Al2O3 supported metal catalysts. Catal. Today 2015, 256, 80–87. [Google Scholar] [CrossRef]
- Zhang, K.; Mukhriza, T.; Liu, X.; Greco, P.P.; Chiremba, E. A study on CO2 and CH4 conversion to synthesis gas and higher hydrocarbons by the combination of catalysts and dielectric-barrier discharges. Appl. Catal. A Gen. 2015, 502, 138–149. [Google Scholar] [CrossRef]
- Tu, X.; Gallon, H.J.; Twigg, M.V.; Gorry, P.A.; Whitehead, J.C. Dry reforming of methane over a Ni/Al2O3 catalyst in a coaxial dielectric barrier discharge reactor. J. Phys. D Appl. Phys. 2011, 44, 274007. [Google Scholar] [CrossRef]
- Sentek, J.; Krawczyk, K.; Młotek, M.; Kalczewska, M.; Kroker, T.; Kolb, T.; Schenk, A.; Gericke, K.-H.; Schmidt-Szałowski, K. Plasma-catalytic methane conversion with carbon dioxide in dielectric barrier discharges. Appl. Catal. B Environ. 2010, 94, 19–26. [Google Scholar] [CrossRef]
- Pham, M.H.; Goujard, V.; Tatibouët, J.M.; Batiot-Dupeyrat, C. Activation of methane and carbon dioxide in a dielectric-barrier discharge-plasma reactor to produce hydrocarbons-Influence of La2O3/γ-Al2O3 catalyst. Catal. Today 2011, 171, 67–71. [Google Scholar] [CrossRef]
- Gallon, H.J.; Tu, X.; Whitehead, J.C. Effects of Reactor Packing Materials on H2 Production by CO2 Reforming of CH4 in a Dielectric Barrier Discharge. Plasma Process. Polym. 2012, 9, 90–97. [Google Scholar] [CrossRef]
- Song, H.K.; Choi, J.-W.; Yue, S.H.; Lee, H.; Na, B.-K.; Songu, H.K. Synthesis gas production via dielectric barrier discharge over Ni/γ-Al2O3 catalyst. Catal. Today 2004, 89, 27–33. [Google Scholar] [CrossRef]
- Tu, X.; Whitehead, J.C. Plasma-catalytic dry reforming of methane in an atmospheric dielectric barrier discharge: Understanding the synergistic effect at low temperature. Appl. Catal. B Environ. 2012, 125, 439–448. [Google Scholar] [CrossRef]
- Wang, Q.; Yan, B.; Jin, Y.; Cheng, Y. Dry Reforming of Methane in a Dielectric Barrier Discharge Reactor with Ni/Al2O3 Catalyst: Interaction of Catalyst and Plasma. Energy Fuels 2009, 23, 4196–4201. [Google Scholar] [CrossRef]
- Michielsen, I.; Uytdenhouwen, Y.; Pype, J.; Michielsen, B.; Mertens, J.; Reniers, F.; Meynen, V.; Bogaerts, A. CO2 dissociation in a packed bed DBD reactor: First steps towards a better understanding of plasma catalysis. Chem. Eng. J. 2017, 326, 477–488. [Google Scholar] [CrossRef]
- Vandenbroucke, A.M.; Morent, R.; De Geyter, N.; Leys, C. Non-thermal plasmas for non-catalytic and catalytic VOC abatement. J. Hazard. Mater. 2011, 195, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Futamura, S.; Zhang, A.; Einaga, H.; Kabashima, H. Involvement of catalyst materials in nonthermal plasma chemical processing of hazardous air pollutants. Catal. Today 2002, 72, 259–265. [Google Scholar] [CrossRef]
- Kim, H.-H. Nonthermal Plasma Processing for Air-Pollution Control: A Historical Review, Current Issues, and Future Prospects. Plasma Process. Polym. 2004, 1, 91–110. [Google Scholar] [CrossRef]
- Subrahmanyam, C.; Magureanu, M.; Renken, A.; Kiwi-Minsker, L. Catalytic abatement of volatile organic compounds assisted by non-thermal plasma. Appl. Catal. B Environ. 2006, 65, 150–156. [Google Scholar] [CrossRef]
- Kim, H.H.; Ogata, A. Interaction of Nonthermal Plasma with Catalyst for the Air Pollution Control. Int. J. Plasma Environ. Sci. Technol. 2012, 6, 43–48. [Google Scholar]
- Francke, K.-P.; Miessner, H.; Rudolph, R. Plasmacatalytic processes for environmental problems. Catal. Today 2000, 59, 411–416. [Google Scholar] [CrossRef]
- Pasquiers, S. Removal of pollutants by plasma catalytic processes. Eur. Phys. J. Appl. Phys 2004, 28, 319–324. [Google Scholar] [CrossRef]
- Guaitella, O.; Thevenet, F.; Puzenat, E.; Guillard, C.; Rousseau, A. C2H2 oxidation by plasma/TiO2 combination: Influence of the porosity, and photocatalytic mechanisms under plasma exposure. Appl. Catal. B Environ. 2008, 80, 296–305. [Google Scholar] [CrossRef]
- Van Durme, J.; Dewulf, J.; Leys, C.; Van Langenhove, H. Combining non-thermal plasma with heterogeneous catalysis in waste gas treatment: A review. Appl. Catal. B Environ. 2008, 78, 324–333. [Google Scholar] [CrossRef]
- Aerts, R.; Somers, W.; Bogaerts, A. A detailed description of the CO2 splitting by dielectric barrier discharges. ChemSusChem 2015, 8, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Kong, M.; Liu, T.; Fei, J.; Zheng, X. Characteristics of the Decomposition of CO2 in a Dielectric Packed-Bed Plasma Reactor. Plasma Chem. Plasma Process. 2012, 32, 153–163. [Google Scholar] [CrossRef]
- Mei, D.; Zhu, X.; He, Y.-L.Y.Y.; Yan, J.D.; Tu, X. Plasma-assisted conversion of CO2 in a dielectric barrier discharge reactor: Understanding the effect of packing materials. Plasma Sources Sci. Technol. 2015, 24, 15011. [Google Scholar] [CrossRef]
- Ramakers, M.; Michielsen, I.; Aerts, R.; Meynen, V.; Bogaerts, A. Effect of argon or helium on the CO2 conversion in a dielectric barrier discharge. Plasma Process. Polym. 2015, 12, 755–763. [Google Scholar] [CrossRef]
- Snoeckx, R.; Aerts, R.; Tu, X.; Bogaerts, A. Plasma-Based Dry Reforming: A Computational Study Ranging From Nanoseconds to Seconds Timescale. J. Phys. Chem. 2013, 117, 4957–4970. [Google Scholar] [CrossRef]
- Atkins, P.; Jones, L. Chemical Principles: The Quest for Insight, 4th ed.; Craig Bleyer: New York, NY, USA, 2008. [Google Scholar]
- Wang, W.; Berthelot, A.; Zhang, Q.; Bogaerts, A. Modelling of plasma-based dry reforming: How do uncertainties in the input data affect the calculation results? J. Phys. D Appl. Phys. 2018, 51, 204003. [Google Scholar] [CrossRef]
- Berthelot, A.; Bogaerts, A. Modeling of CO2 plasma: Effect of uncertainties in the plasma chemistry. Plasma Sources Sci. Technol. 2017, 26, 115002. [Google Scholar] [CrossRef]
- De Bie, C.; Van Dijk, J.; Bogaerts, A. The Dominant Pathways for the Conversion of Methane into Oxygenates and Syngas in an Atmospheric Pressure Dielectric Barrier Discharge. J. Phys. Chem. C 2015, 119, 22331–22350. [Google Scholar] [CrossRef]
- Wang, W.; Kim, H.H.; Van Laer, K.; Bogaerts, A. Streamer propagation in a packed bed plasma reactor for plasma catalysis applications. Chem. Eng. J. 2018, 334, 2467–2479. [Google Scholar] [CrossRef]
- Van Laer, K.; Bogaerts, A. Influence of Gap Size and Dielectric Constant of the Packing Material on the Plasma Behaviour in a Packed Bed DBD Reactor: A Fluid Modelling Study. Plasma Process. Polym. 2017, 14, e1600129. [Google Scholar] [CrossRef]
- Van Laer, K.; Bogaerts, A. How bead size and dielectric constant affect the plasma behaviour in a packed bed plasma reactor: A modelling study. Plasma Sources Sci. Technol. 2017, 26, 085007. [Google Scholar] [CrossRef]
- Butterworth, T.D. The Effects of Particle Size on CO2 reduction in Packed Bed Dielectric Barrier Discharge Plasma Reactors. Ph.D. Thesis, University of Sheffield, Sheffield, UK, 2015. [Google Scholar]
- Aerts, R.; Snoeckx, R.; Bogaerts, A. In-Situ Chemical Trapping of Oxygen in the Splitting of Carbon Dioxide by Plasma. Plasma Process. Polym. 2014, 11, 985–992. [Google Scholar] [CrossRef]
- Snoeckx, R.; Ozkan, A.; Reniers, F.; Bogaerts, A. The Quest for Value-Added Products from Carbon Dioxide and Water in a Dielectric Barrier Discharge: A Chemical Kinetics Study. ChemSusChem 2017, 10, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Uytdenhouwen, Y.; Van Alphen, S.; Michielsen, I.; Meynen, V.; Cool, P.; Bogaerts, A. A packed-bed DBD micro plasma reactor for CO2 dissociation: Does size matter? Chem. Eng. J. 2018, 348, 557–568. [Google Scholar] [CrossRef]
- Whitehead, J.C. Plasma–catalysis: The known knowns, the known unknowns and the unknown unknowns. J. Phys. D Appl. Phys. 2016, 49, 243001. [Google Scholar] [CrossRef]
- Pinhão, N.; Moura, A.; Branco, J.B.; Neves, J. Influence of gas expansion on process parameters in non-thermal plasma plug-flow reactors: A study applied to dry reforming of methane. Int. J. Hydrog. Energy 2016, 41, 9245–9255. [Google Scholar] [CrossRef]
- Snoeckx, R.; Heijkers, S.; Van Wesenbeeck, K.; Lenaerts, S.; Bogaerts, A. CO2 conversion in a dielectric barrier discharge plasma: N2 in the mix as a helping hand or problematic impurity? Energy Environ. Sci. 2016, 9, 30–39. [Google Scholar] [CrossRef]
- Dullien, F.A. Porous Media-Fluid Transport and Pore Structure; Academic Press: Cambridge, MA, USA, 1991; ISBN 9780122236518. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Reactor | Operating Conditions | Implementing Packing and/or Catalysts | Conclusion | Highest Conversion | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gap (mm) | Power (Watt) | Flow (mL/min) | Reactor Volume (cm3) | Frequency | SEI (kJ/L) | Packing/Catalyst | Shape | Packing Size | ||||
| CO2 splitting/non-packed | 1.5 | 70 | 150 | 21.9 | 5–65 kHz | 28 | No influence of frequency | 10% | [42] | |||
| 2 | 100–200 | 50–500 | 13.56 | 10–90 kHz | 12–240 | Conversion  when flow rate when flow rate  , Tgas , P . Best frequency depends on power , Tgas , P . Best frequency depends on power | 30% | [15] | ||||
| 2 | 10–97 | 50–2000 | 15.1 | 16.2–28.6 kHz | 0.3–116 | Conversion when flow rate , barrier thickness , frequency and power | 35% | [16] | ||||
| 4 | 21.6–35.3 | 40 | 30.17 | 13 kHz | 32.4–53 | Conversion when power and discharge length Cokes: small on inner electrode | 13% | [43] | ||||
| 4.5, 3.5, 2.5 or 2 | 60 | 50 | 17.67 | 26.5 kHz | 72 | Conversion when flow rate | 12% | [32] | ||||
| CO2 splitting/packed | 4 | 21.6–35.3 | 40 | 30.17 | 13 kHz | 32.4–53 | Silica gel | Beads | 20–40 mesh | silica gel < α-Al2O3 < quartz ≈ γ-Al2O3 < CaTiO3 Cokes: limited on inner electrode | 14% | [43] |
| Quartz | Pellets with rigid edges | 20–40 mesh | silica gel < α-Al2O3 < quartz ≈ γ-Al2O3 < CaTiO3 Cokes: limited on inner electrode | 16% | ||||||||
| γ-Al2O3 | Beads | 20–40 mesh | silica gel < α-Al2O3 < quartz ≈ γ-Al2O3 < CaTiO3 Cokes: limited on inner electrode | 16% | ||||||||
| α-Al2O3 | Beads | 20–40 mesh | silica gel < α-Al2O3 < quartz ≈ γ-Al2O3 < CaTiO3 Cokes: limited on inner electrode | 15% | ||||||||
| CaTiO3 | Beads | 20–40 mesh | silica gel < α-Al2O3 < quartz ≈ γ-Al2O3 < CaTiO3 Cokes: limited on inner electrode | 20.5% | ||||||||
| 3 | 20–50 | 50 | 10.1 | 9 kHz | 24–60 | Glass | Beads | 1 mm | Glass < BaTiO3 | 22% (16% without packing) | [44] | |
| BaTiO3 | Beads | 1 mm | Glass < BaTiO3 | 28% (16% without packing) | ||||||||
| 4.5, 3.5, 2.5 or 2 | 60 | 50 | 17.67 | 26.5 kHz | 72 | Glass wool | Beads | 1.25–2.24 mm | Conversion when # contact points , void space volumes and bead/gap size ratio . Impact of the packing material (chemistry and physical), also influenced by setup | 10% | [32] | |
| Quartz wool | Beads | 1.25–2.24 mm | Conversion when # contact points , void space volumes and bead/gap size ratio . Impact of the packing material (chemistry and physical), also influenced by setup | 10% | ||||||||
| SiO2 | Beads | 1.25–2.24 mm | Conversion when # contact points , void space volumes and bead/gap size ratio . Impact of the packing material (chemistry and physical), also influenced by setup | 16% | ||||||||
| ZrO2 | Beads | 1.25–2.24 mm | Conversion when # contact points , void space volumes and bead/gap size ratio . Impact of the packing material (chemistry and physical), also influenced by setup | 19% | ||||||||
| α-Al2O3 | Beads | 1.25–2.24 mm | Conversion when # contact points , void space volumes and bead/gap size ratio . Impact of the packing material (chemistry and physical), also influenced by setup | 17% | ||||||||
| BaTiO3 | Beads | 1.25–2.24 mm | Conversion when # contact points , void space volumes and bead/gap size ratio . Impact of the packing material (chemistry and physical), also influenced by setup | 26% | ||||||||
| Study | Reactor | Operating Conditions | Implementing Packing and/or Catalysts | Selectivity | Conclusion | Highest Conversion | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gap (mm) | Power (Watt) | Flow (mL/min) | Reactor volume (cm3) | Frequency | Ratio CO2/CH4 | T | SEI (kJ/L) | Packing/Catalyst | Shape | Packing size | Highest achieved selectivity per component | ||||
| DRM/ non-packed | 3 | 30–60 | 25–100 | 11.4 | 30–40 kHz | 1 | / | 18–144 | 45% CO, 29% H2, 5% C2H2/C2H4, 22% C2H6, 2% C3H6, 12% C3H8 (estimation) | Conversion when flow rate and P | 50.4% CH4, 30.5% CO2 | [30] | |||
| 1 | 25–75 | 30–75 | 4.4 | 30 kHz | 0.66–3 | / | 20–150 | 76% CO, 57% H2 | Conversion when flow rate , P . CH4 conversion , CO2 conversion ≈ when ratio | 59.7% CH4, 36.9% CO2 | [17] | ||||
| 1 | 80–130 | 10–40 | 4.7 | 20 kHz | 0.25–1 | / | 120–780 | 73.8% CO, 65.9% H2, 18.0% C2, 10.2% C3, 6.2% C4 | Conversion when flow rate , P . CH4 conversion , CO2 conversion ≈ when ratio | 64% CH4, 34% CO2 | [29] | ||||
| 3 | 10 | 40 | 2.12 | 12 kHz | 4 | / | 15 | 20% CO, 34% H2, <1% C2H2, <1% C2H4, 12% C2H6, 1% C3H8, <1% C4H10, 11.9% methanol, 11.9% ethanol, 33.7% acetic acid, 1.6% acetone, 0% HCHO | Impact depends on catalyst, both and conversion and differs from pure packing | 18% CH4, 15% CO2 | [12] | ||||
| DRM/ packed | 3 | 30–60 | 25–100 | 11.4 | 30–40 kHz | 1 | / | 18–144 | 10 wt% Ni@γ-Al2O3 | Pellets | 0.5–1.7 mm | 55% CO, 33% H2, 10% C2H2/C2H4, 47% C2H6, 2% C3H6, 25% C3H8 (estimation) | Conversion when pellet size , quartz wool is best, impact packing on selectivity | 40.2% CH4, 30.5% CO2 | [30] |
| 1 | 25–75 | 30–75 | 4.4 | 30 kHz | 1 | 450 °C | 20–150 | 12% Ni/γ-Al2O3 | ? | ? | 43% CO, 53% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 30% CH4, 24% CO2 | [17] | |
| 12% Cu/γ-Al2O3 | ? | ? | 50% CO, 31% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 7% CH4, 5% CO2 | ||||||||||
| 1%Cu-12% Ni/γ-Al2O3 | ? | ? | 45% CO, 51% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 33% CH4, 25% CO2 | ||||||||||
| DRM/ packed | 5%Cu-12% Ni/γ-Al2O3 | ? | ? | 47% CO, 54% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 37% CH4, 24% CO2 | |||||||||
| 12%Cu-12% Ni/γ-Al2O3 | ? | ? | 75% CO, 56% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 69% CH4, 75% CO2 | ||||||||||
| 16%Cu-12% Ni/γ-Al2O3 | ? | ? | 64% CO, 57% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 43% CH4, 47% CO2 | ||||||||||
| 5% Ni-12%Cu/γ-Al2O3 | ? | ? | 75% CO, 56% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 43% CH4, 45% CO2 | ||||||||||
| 16% Ni-12%Cu/γ-Al2O3 | ? | ? | 71% CO, 58% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 57% CH4, 57% CO2 | ||||||||||
| 20% Ni-12%Cu/γ-Al2O3 | ? | ? | 62% CO, 58% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 35% CH4, 32% CO2 | ||||||||||
| 1 | 130 | 30 | 4.7 | 20 kHz | 1 | / | 260 | γ-Al2O3 | Crushed flakes | 10–20 mesh | 49.2% CO, 51% H2, 9.7% C2, 5.5% C3, 3% C4 | Packing: CO2 conversion , CH4 conversion . After activation: conversion , selectivity for H2 and C2 | 57.6% CH4, 30.9% CO2 | [29] | |
| 2 wt% Ni @ γ-Al2O3 | Crushed flakes | 10–20 mesh | 60.6% CO, 52.3% H2, 9.8% C2, 5.9% C3, 3.2% C4 | Packing: CO2 conversion , CH4 conversion . After activation: conversion , selectivity for H2 and C2 | 55.4% CH4, 32.7% CO2 | ||||||||||
| 5 wt% Ni @ γ-Al2O3 | Crushed flakes | 10–20 mesh | 60.9% CO, 51.9% H2, 10.1% C2, 5.9% C3, 3.2% C4 | Packing: CO2 conversion , CH4 conversion . After activation: conversion , selectivity for H2 and C2 | 55.7% CH4, 33.5% CO2 | ||||||||||
| 7 wt% Ni @ γ-Al2O3 | Crushed flakes | 10–20 mesh | 63.9%CO, 53.5% H2, 10.6% C2, 6.1% C3, 3.6% C4 | Packing: CO2 conversion , CH4 conversion . After activation: conversion , selectivity for H2 and C2 | 55.5% CH4, 32.6% CO2 | ||||||||||
| 10 wt% Ni @ γ-Al2O3 | Crushed flakes | 10–20 mesh | 61.4% CO, 53% H2, 10.6% C2, 6.2% C3, 3.4% C4 | Packing: CO2 conversion , CH4 conversion . After activation: conversion , selectivity for H2 and C2 | 55.2% CH4, 32.7% CO2 | ||||||||||
| 3 | 10 | 40 | 2.12 | 9 kHz | 1 | / | 15 | γ-Al2O3 | ? | ? | 23% CO, 55% H2, <1% C2H2, <1% C2H4, 20% C2H6, 2% C3H8, <1% C4H10, 13% methanol, 9% ethanol, 20% acetic acid, 2% acetone, 0% HCHO | Impact depends on catalyst, both and conversion and differs from pure packing | 15% CH4, 12.5% CO2 | [12] | |
| DRM/packed | Cu/γ-Al2O4 | ? | ? | 14% CO, 35% H2, <1% C2H2, <1% C2H4, 15% C2H6, 2% C3H8, <1% C4H10, 11% methanol, 11% ethanol, 42% acetic acid, 2% acetone, 0% HCHO | Impact depends on catalyst, both and conversion and differs from pure packing | 16% CH4, 7.5% CO2 | |||||||||
| Au/γ-Al2O5 | ? | ? | 20% CO, 42% H2, <1% C2H2, <1% C2H4, 16% C2H6, 2% C3H8, <1% C4H10, 10% methanol, 10% ethanol, 30% acetic acid, 2% acetone, 5% HCHO | Impact depends on catalyst, both and conversion and differs from pure packing | 16% CH4, 15% CO2 | ||||||||||
| Pt/γ-Al2O6 | ? | ? | 20% CO, 40% H2, <1% C2H2, <1% C2H4, 17% C2H6, 2% C3H8, <1% C4H10, 10% methanol, 9% ethanol, 25% acetic acid, 2% acetone, 11% HCHO | Impact depends on catalyst, both and conversion and differs from pure packing | 17.5% CH4, 13% CO2 | ||||||||||
| 5.9 | 40 | 80 | ? | 300 Hz | 0.07–1 | RT-600 °C | 30 | Glass | Beads | 2 mm | 70% CO, 19.5% H2, 42.9% C2, 15% C3, 8.7% C4 | CH4 concentration = C2 . Influence catalyst only > 200 ◦C, for CO2. Effect glass = Al2O3 | 25% CH4, 56.1% CO2 | [27] | |
| γ-Al2O3 | Beads | 2 mm | 70% CO, 19.5% H2, 42.9% C2, 15% C3, 8.7% C4 | CH4 concentration = C2 . Influence catalyst only > 200 ◦C, for CO2. Effect glass = Al2O3 | 25% CH4, 56.1% CO2 | ||||||||||
| La2O3/γ-Al2O3 | Beads | 2 mm | 70% CO, 19.5% H2, 42.9% C2, 15% C3, 8.7% C4 | CH4 concentration = C2 . Influence catalyst only > 200 ◦C, for CO2. | 25% CH4, 56.1% CO2 | ||||||||||
| 2 | 40–240 | 40 | ? | 5–20 kHz | 1 | / | 60–360 | Ni/γ-Al2O3 | Nano-particles | 100 nm | 86% CO, 73% H2 | NiFe2O4#SiO2 conversion and selectivity , carbon deposit | 64.6% CH4, 58% CO2 | [18] | |
| Ni-Fe/γ-Al2O3 | Nano-particles | 100 nm | 87% CO, 74% H2 | NiFe2O4#SiO2 conversion and selectivity , carbon deposit | 68.7% CH4, 60.5% CO2 | ||||||||||
| Ni-Fe/SiO2 | Nano-particles | 100 nm | 88% CO, 75% H2 | NiFe2O4#SiO2 conversion and selectivity , carbon deposit | 73.5% CH4, 62.7% CO2 | ||||||||||
| NiFe2O4 | Nano-particles | 100 nm | 89% CO, 77% H2 | NiFe2O4#SiO2 conversion and selectivity , carbon deposit | 77.4% CH4, 67.1% CO2 | ||||||||||
| NiFe2O4#SiO2 | Nano-particles | 100 nm | 90% CO, 81% H2 | NiFe2O4#SiO2 conversion and selectivity , carbon deposit | 80% CH4, 70.3% CO2 | ||||||||||
| 2 | 150 | 40 | ? | 5–100 kHz | 1 | / | 225 | Ni/SiO2 | ? | ? | 87% CO, 73% H2 | Packing: conversion , selectivity | 65% CH4, 52% CO2 | [19] | |
| LaNiO3/SiO2 | ? | ? | 89% CO, 79% H2 | Packing: conversion , selectivity | 82% CH4, 69% CO2 | ||||||||||
| DRM/packed | LaNiO3 | ? | ? | 90% CO, 81% H2 | Packing: conversion , selectivity | 84% CH4, 72% CO2 | |||||||||
| LaNiO3@SiO2 | ? | ? | 92% CO, 84% H2 | Packing: conversion , selectivity | 88% CH4, 78% CO2 | ||||||||||
| 4.5 | 50 | 50 | ? | 30–40 kHz | 1 | / | 60 | Ni/Al2O3 | Pellets | 0.85–5 mm | 25% CO, 45% H2, 10% C2, 5% C3 | non-packed: filamentary discharge, packed: combination of surface discharges microdischarges, breakdown voltage and conversion | 18% CH4, 13% CO2 | [25] | |
| 3.5 | 1.4–4.8 | 40 | 27.2 | 50 Hz | 0.5–2 | / | 2–7.2 | Ni/Al2O3 | Pellets | 1 mm | 35% CO, 56% H2 | Conversion with packing. Conversion when ratio | 52% CH4, 43% CO2 (38% CH4, 23% CO2 non-packed) | [20] | |
| 3 | 19 | 16.7–33.3 | ? | 6 kHz | 1 | 130–340 °C | 34–68 | Al2O3 | ? | 1–2 mm | 19% CO, 24% H2, 0.6% C2H2/C2H4, 10% C2H6, 0.3% C3H6, 6% C3H8, 1.3% CH3OH | Conversion with packing | 52% CH4, 31% CO2 | [21] | |
| Fe/Al2O3 | ? | 1–2 mm | 14% CO, 21% H2, 1.3% C2H2/C2H4, 9% C2H6, 0.3% C3H6, 5% C3H8, 1% CH3OH | No effect of T or flow rate, Conversion with packing | 46% CH4, 20% CO2 | ||||||||||
| zeolite NaY | ? | ? | 10% CO, 21% H2, 1% C2H2/C2H4, 6% C2H6, 0.2% C3H6, 3% C3H8, 0% CH3OH | No effect of T or flow rate, Conversion with packing | 49% CH4, 19% CO2 | ||||||||||
| zeolite Na ZSM-5 | ? | ? | 5% CO, 21% H2, 0.1% C2H2/C2H4, 9% C2H6, 0% C3H6, 5% C3H8, 0% CH3OH | Conversion with packing | 65% CH4, 40% CO2 | ||||||||||
| 4 | 15–60 | 5–50 | ? | 1–100 kHz | 1 | 325–525 °C | 18–720 | Ni/γ-Al2O3 | Grains | 70–100 mesh | ? | Conversion with packing (fluidized bed) | 48% CH4, 40% CO2 | [22] | |
| 3 | 19 | 16.7–33.3 | ? | 5.7–6 kHz | 1–2 | 120–290 °C | 34–68 | Al2O3 | ? | 1–2 mm | 38% CO, 28% H2, 11% C2, 6% C3, 4% C4, 2% CH3OH | Conversion with packing | 55% CH4, 31% CO2 | [26] | |
| Pd/Al2O3 | ? | 1–2 mm | 40% CO, 29% H2, 15% C2, 5% C3, 3% C4, 1% CH3OH | Conversion with packing | 51% CH4, 28% CO2 | ||||||||||
| Ag/Al2O3 | ? | 1–2 mm | 38% CO, 29% H2, 10% C2, 5% C3, 4% C4, 2% CH3OH | Conversion with packing | 52% CH4, 30% CO2 | ||||||||||
| 4.5 | 10–40 | 50 | 16.5 | 30–40 kHz | 1 | / | 12–48 | Quartz wool | ? | ? | 28% CO, 22% H2, 1% C2H2/C2H4, 7% C2H6, 0.5% C3H6, 4% C3H8 (estimation) | CH4 conversion: quartz wool> no packing > Al2O3 > zeolite 3A | 30% CH4, 12% CO2 | [28] | |
| γ-Al2O3 | pellets | 500–850 μm | 32% CO, 18% H2, 2% C2H2/C2H4, 8% C2H6, 0.5% C3H6, 4% C3H8 (estimation) | CH4 conversion: quartz wool> no packing > Al2O3 > zeolite 3A | 23% CH4, 8% CO2 | ||||||||||
| DRM/ packed | zeolite 3A | beads | 2 mm | 22% CO, 30% H2, 19% C2H2/C2H4, 8% C2H6, 1% C3H6, 6% C3H8 (estimation) | CH4 conversion: quartz wool> no packing > Al2O3 > zeolite 3A | 7% CH4, 3% CO2 | |||||||||
| 2.5 | 7.5–15 | 25–200 | 11.6 | 50 Hz | 0.11–9 | / | 2–36 | Ni/γ-Al2O3 | ? | ? | 37% CO, 33% H2, 22% C2H6 | Ni/γ-Al2O3 and Mn/γ-Al2O3: CH4 conversion , yields CO and H2 . | 19% CH4, 9% CO2 | [23] | |
| Co/γ-Al2O4 | ? | ? | 42% CO, 43% H2, 30% C2H6 | Ni/γ-Al2O3 and Mn/γ-Al2O3: CH4 conversion , yields CO and H2 | 15% CH4, 8% CO2 | ||||||||||
| Cu/γ-Al2O5 | ? | ? | 43% CO, 44% H2, 30% C2H6 | Ni/γ-Al2O3 and Mn/γ-Al2O3: CH4 conversion , yields CO and H2 | 14% CH4, 8% CO2 | ||||||||||
| Mn/γ-Al2O6 | ? | ? | 35% CO, 34% H2, 24% C2H6 | Ni/γ-Al2O3 and Mn/γ-Al2O3: CH4 conversion , yields CO and H2 | 18% CH4, 10% CO2 | ||||||||||
| 7.5 | 46–106 | 25–100 | 100 | 25 kHz | 1 | 110 °C | 28–254 | BaTiO3 | Beads | 3 mm | 50% CO, 56% H2 | BaTiO3 size = conversions | 33% CH4, 20% CO2 | [24] | |
| Ni/SiO2 | Pellets | 2–3 mm | 56% CO, 54% H2 | Packing = conversions | 20% CH4, 12% CO2 | ||||||||||
| NiFe/SiO2 | Pellets | 2–3 mm | 54% CO, 56% H2 | Packing = conversions | 28% CH4, 15% CO2 | ||||||||||
| CH4 Conversion/CO2 Conversion | CO/H2 | ||
|---|---|---|---|
| 1.25–1.4 mm | ZrO2 | 1.7 | 5.5 |
| SiO2 | 1.7 | 4.8 | |
| α-Al2O3 | 1.5 | 9.5 | |
| BaTiO3 | 1.6 | 6.0 | |
| 1.6–1.8 mm | ZrO2 | 1.8 | 5.9 |
| SiO2 | 1.9 | 4.7 | |
| α-Al2O3 | 1.8 | 8.8 | |
| BaTiO3 | 1.9 | 6.3 | |
| 2.0–2.24 mm | ZrO2 | 2.2 | 6.4 |
| SiO2 | 1.2 | 5.3 | |
| α-Al2O3 | 1.5 | 9.0 | |
| BaTiO3 | 2.0 | 6.9 | |
| γ-Al2O3 | 2.3 | 8.3 | |
| Non-packed reactor | 50 mL/min | 1.9 | 7.9 |
| 192 mL/min | 2.0 | 7.2 |
| CO | H2 | C2H6 | C2H4 | C2H2 | C3H8 | C2H6O | C2H5OH | CH2O | CH3OH | CO/H2 Ratio | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ethane | Ethene | Ethyne | Propane | DME | Ethanol | Formaldehyde | Methanol | |||||
| Non-packed (50 mL/min) | 64.2 | 8.1 | 15.9 | 2.7 | 4.1 | 4.3 | 0.2 | 0.22 | 0.14 | 0.15 | 7.9 | |
| Non-packed (192 mL/min) | 53.5 | 7.4 | 23.8 | 2.8 | 5.6 | 4.0 | 0.7 | 0.27 | 1.63 | 0.28 | 7.2 | |
| ZrO2 | 1.25–1.4 | 58.8 | 10.8 | 11.6 | 1.6 | 11.9 | 2.9 | 0.9 | 0.33 | 0.87 | 0.36 | 5.5 |
| 1.6–1.8 | 60.4 | 10.2 | 12.2 | 1.5 | 10.9 | 3.1 | 0.7 | 0.33 | 0.42 | 0.31 | 5.9 | |
| 2.0–2.24 | 63.4 | 9.8 | 12.0 | 1.2 | 8.6 | 3.3 | 0.5 | 0.34 | 0.32 | 0.32 | 6.4 | |
| SiO2 | 1.25–1.4 | 49.3 | 10.3 | 11.7 | 2.7 | 20.3 | 2.9 | 0.9 | 0.24 | 1.44 | 0.26 | 4.8 |
| 1.6–1.8 | 51.9 | 11.0 | 11.8 | 2.0 | 18.0 | 3.3 | 0.8 | 0.28 | 0.51 | 0.26 | 4.7 | |
| 2.0–2.24 | 56.3 | 10.6 | 13.0 | 13.1 | 3.7 | 0.6 | 0.31 | 0.41 | 0.33 | 5.3 | ||
| α-Al2O3 | 1.25–1.4 | 71.3 | 7.5 | 14.0 | 0.5 | 2.2 | 2.9 | 0.5 | 0.28 | 0.29 | 0.43 | 9.5 |
| 1.6–1.8 | 70.9 | 8.0 | 13.6 | 0.6 | 2.7 | 2.7 | 0.4 | 0.25 | 0.36 | 0.41 | 8.8 | |
| 2.0–2.24 | 72.2 | 8.0 | 12.9 | 0.5 | 2.2 | 2.9 | 0.4 | 0.29 | 0.29 | 0.46 | 9 | |
| BaTiO3 | 1.25–1.4 | 53.1 | 8.9 | 11.1 | 2.5 | 13.8 | 2.4 | 1.6 | 0.40 | 5.28 | 0.86 | 6 |
| 1.6–1.8 | 59.0 | 9.3 | 12.4 | 2.1 | 11.4 | 2.5 | 1.3 | 0.37 | 0.96 | 0.69 | 6.3 | |
| 2.0–2.24 | 61.4 | 8.9 | 13.7 | 1.8 | 9.0 | 2.8 | 0.8 | 0.33 | 0.59 | 0.57 | 6.9 | |
| γ-Al2O3 | 2.0–2.24 | 70.1 | 8.5 | 15.4 | 0.6 | 2.0 | 0.4 | 0.0 | 3.0 | 0.0 | 0.0 | 8.3 |
| Parameter | Specification |
|---|---|
| Gap (mm) | 4.5 |
| Frequency (kHz) | 23.5 |
| Power (Watt) | 100 |
| Gas flow rate (mL/min) | 50 (or 192, for non-packed, to have the same residence time as in the packed reactor) |
| Type of material | Non-packed reactor versus SiO2, α-Al2O3, γ-Al2O3, ZrO2 and BaTiO3 |
| Diameter spheres (mm) a | 1.25–1.4; 1.6–1.8; 2.0–2.24 |
| CO2/CH4 ratio | 1/1 |
| Temperature | Ambient (no external heating) |
| Pressure | Atmospheric (±1.2 atm) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michielsen, I.; Uytdenhouwen, Y.; Bogaerts, A.; Meynen, V. Altering Conversion and Product Selectivity of Dry Reforming of Methane in a Dielectric Barrier Discharge by Changing the Dielectric Packing Material. Catalysts 2019, 9, 51. https://doi.org/10.3390/catal9010051
Michielsen I, Uytdenhouwen Y, Bogaerts A, Meynen V. Altering Conversion and Product Selectivity of Dry Reforming of Methane in a Dielectric Barrier Discharge by Changing the Dielectric Packing Material. Catalysts. 2019; 9(1):51. https://doi.org/10.3390/catal9010051
Chicago/Turabian StyleMichielsen, Inne, Yannick Uytdenhouwen, Annemie Bogaerts, and Vera Meynen. 2019. "Altering Conversion and Product Selectivity of Dry Reforming of Methane in a Dielectric Barrier Discharge by Changing the Dielectric Packing Material" Catalysts 9, no. 1: 51. https://doi.org/10.3390/catal9010051
APA StyleMichielsen, I., Uytdenhouwen, Y., Bogaerts, A., & Meynen, V. (2019). Altering Conversion and Product Selectivity of Dry Reforming of Methane in a Dielectric Barrier Discharge by Changing the Dielectric Packing Material. Catalysts, 9(1), 51. https://doi.org/10.3390/catal9010051