Before analyzing the isotopic exchange processes that took place for the selected reactions, we will first discuss the methodological basis utilized for the analysis of isotope labelling using mass spectrometry (MS) and infrared spectroscopy (IR).
2.1. Analysis of Plasmas Induced Isotope Exchange Reactions
For the ammonia reaction, we reported a maximum nitrogen conversion of 7% according to Reaction (1) [
9], where the nitrogen and hydrogen acting as reactants give rise to ammonia, as follows:
For the reported conditions of the maximum nitrogen conversion in the literature [
11], for each 100 molecules of nitrogen and 100 of hydrogen in the inlet mixture, seven nitrogen molecules would transform into the ammonia. The outlet gas mixture would consist of 14 molecules of ammonia (product of Reaction (1)), 93 molecules of unreacted nitrogen, and 79 molecules of unreacted hydrogen. We attributed this relatively low reaction yield, in comparison with that attained in conventional catalytic processes [
21] (yet the 7% found is one of the highest reported for the DBD synthesis of ammonia [
7,
8,
9]), to the existence of back reactions leading to the formation of nitrogen and hydrogen from the formed ammonia (i.e., the inverse of Reaction (1)) or other intermediate processes, which result inefficient in rendering ammonia molecules. The isotope reaction experiment carried out in the present work does not pretend to increase the reaction yield, but instead evaluates the occurrence of intermediate processes that are neutral with respect to the formation of new ammonia molecules. For this purpose, we used a ternary gas mixture of nitrogen, hydrogen, and ammonia, approaching the composition of the outlet gas mixture of Reaction (1) reported in the literature [
11], and ensuring that the amount of ammonia in the inlet and outlet mixtures remains invariable (i.e., to meet conditions under which there is no net production of ammonia). This does not mean that there are not intermediate reaction processes in the plasma, but that the formation of one ammonia molecule according to Reaction (1) is compensated with the decomposition of another one (i.e., inverse to Reaction (1)). A negligible formation of ammonia was indeed proved in the experiment, because the final concentration of N
2 detected by MS in the outlet gases remained constant after plasma activation (see below). As D
2 substituted H
2 in the inlet reaction mixture, the only source of H during the plasma activation of the ternary mixture was the NH
3 inlet gas feeding the reactor.
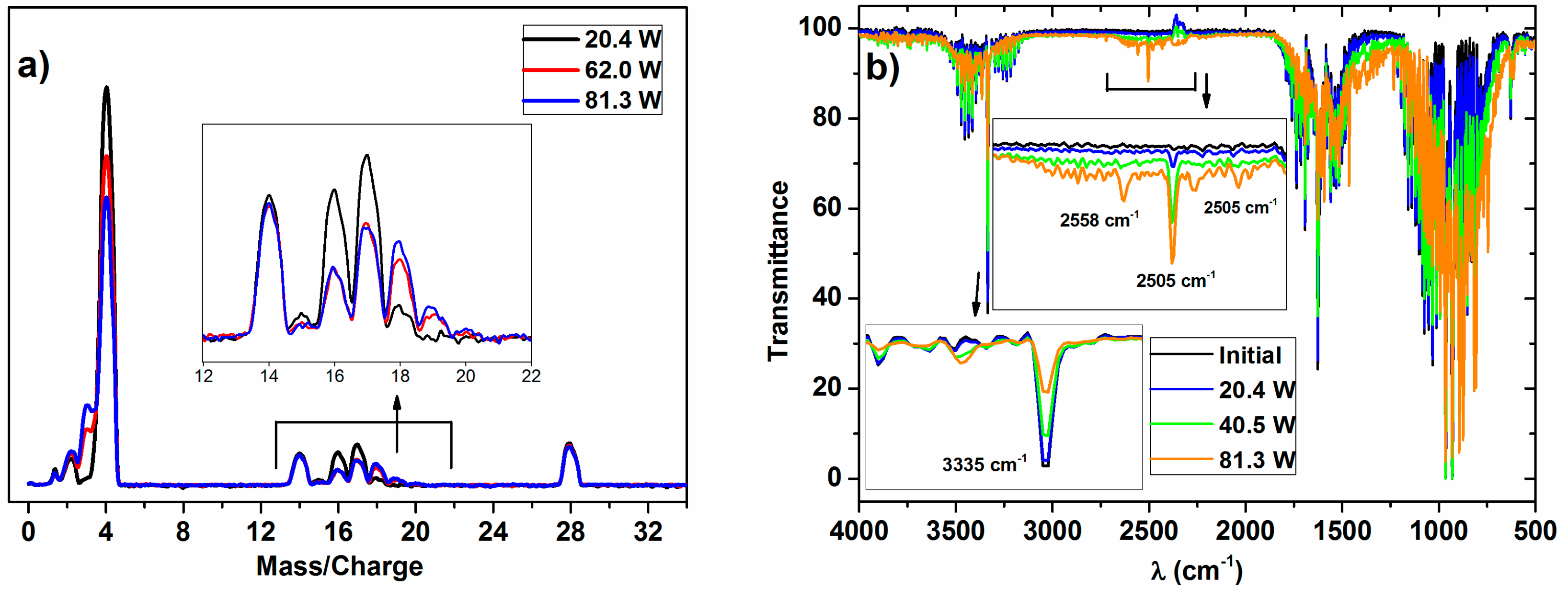
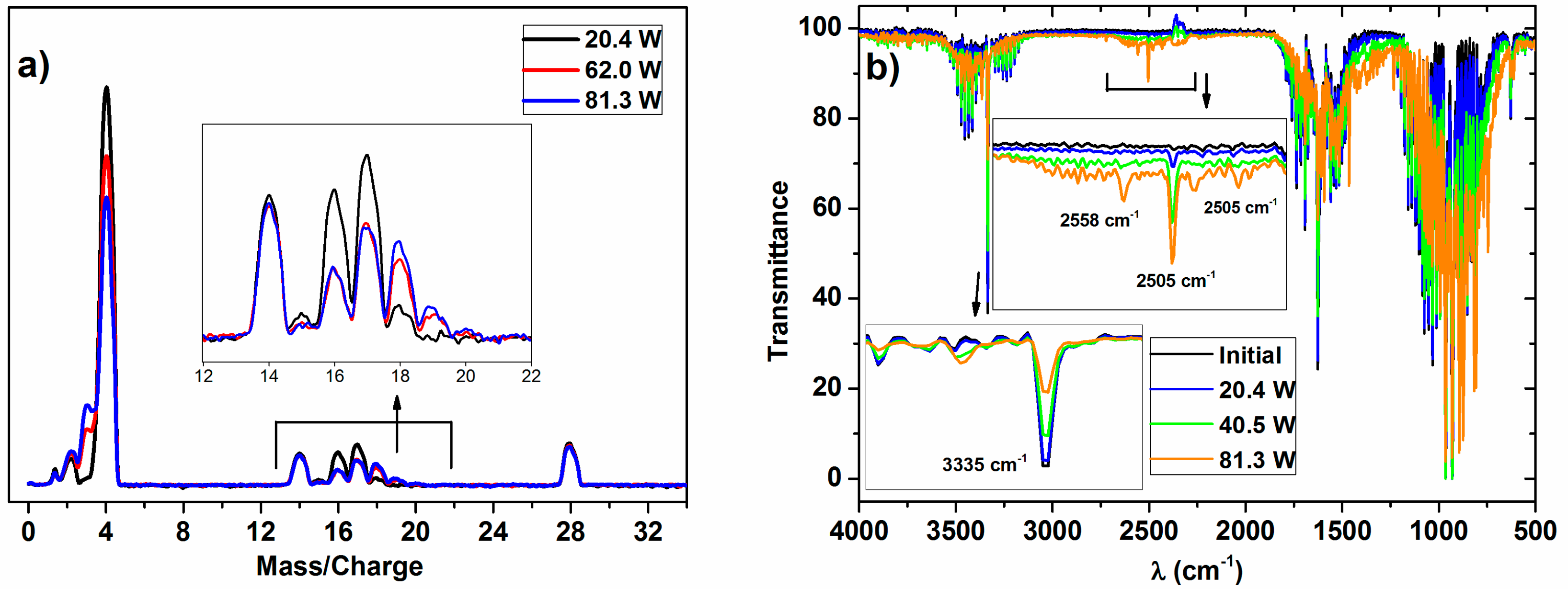
The typical MS and IR spectra of the outlet gases after the plasma activation of the NH
3/D
2/N
2 mixture for three power values is shown in
Figure 1.
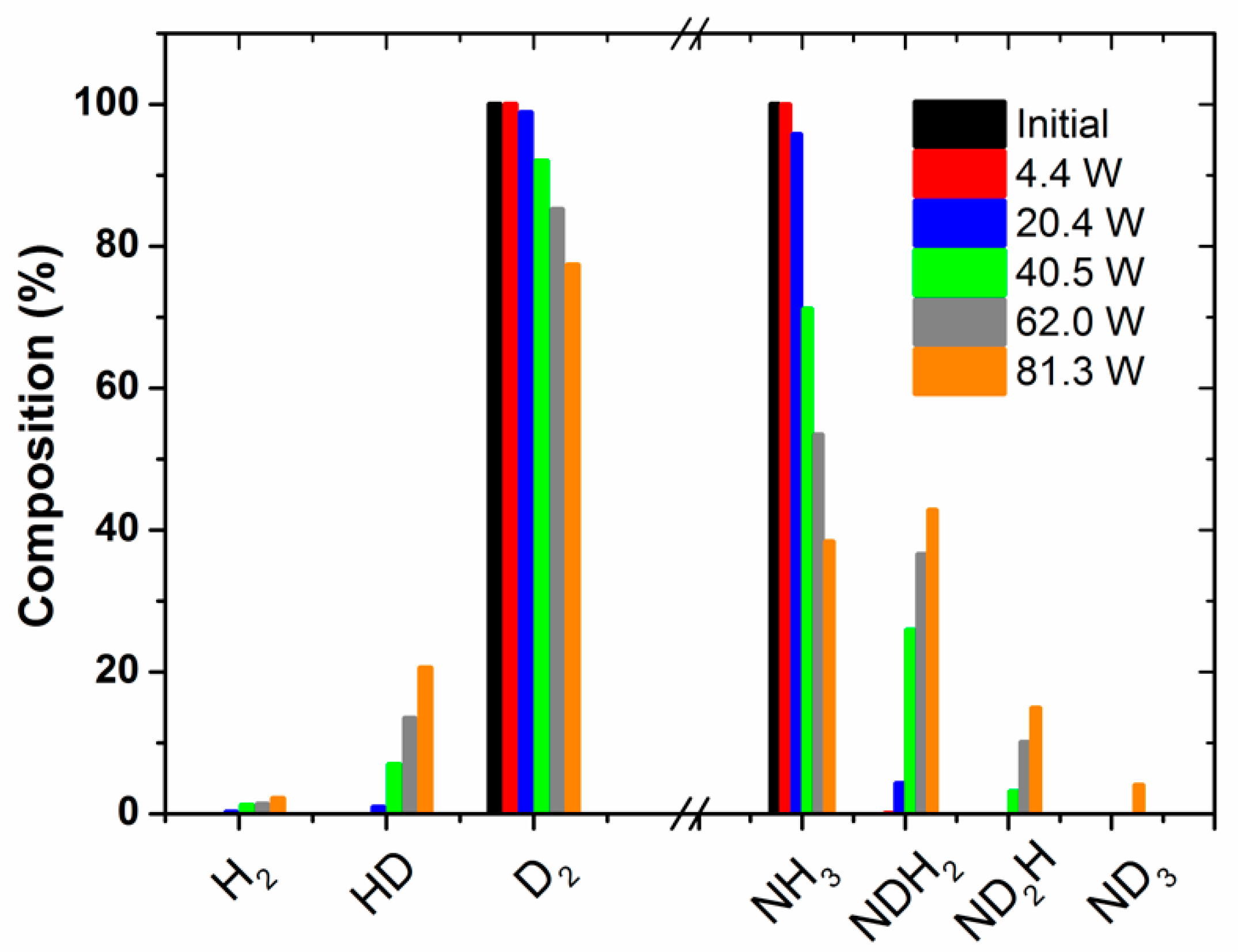
These MS spectra clearly show changes in the relative height of the peaks for the range of
m/
z values 0–4 (i.e., hydrogen–deuterium zone) and 13–20 (water and ammonia zones), which must be attributed to a progressively higher isotope exchange of H by D atoms at increasing powers. In concrete, the mass spectra reveal a clear increase of peaks at
m/
z = 3, attributable to HD, and others at
m/
z = 18 and 19, which must be attributed to NDH
2 and ND
2H (the fragmented ions are reported in
Table 1). A minor contribution at
m/
z = 20, due to ND
3 molecules, could also be found in the spectra of the mixture activated with the maximum power. Meanwhile, a decrease in the intensity of the
m/
z = 17 peak indicates a parallel decrease in the NH
3 concentration in the outlet gases. In this set of experiments, no significant intensity change could be detected for the
m/
z = 28 (and
m/
z = 14) peak because of N
2, a behavior indicating that the concentration of nitrogen remains constant, and that, therefore, practically no new ammonia (i.e., including all of the labelled ND
xH
y molecules) is formed according to Reaction (1) during plasma activation.
A first account of the isotope labelling experiments for the wet reforming reaction was reported by the authors of [
3], for a process that complied with the following stoichiometry:
The outlet gases consisted of unreacted methane and water, carbon monoxide, and hydrogen. A conversion up to 50% of the initial methane flow was achieved under maximum operating power conditions (i.e., for 100 molecules of methane in the inlet mixture, there would be 50 molecules in of CO and 50 of methane in the outlet gas mixture). For the isotope labelling experiments, conventional water (i.e., H
2O) was substituted by deuterated water (D
2O). For this reaction, the MS reported in the literature [
3] showed the appearance of
m/
z peaks, due to H
2 + D
2 + HD (as hydrogen), CH
4 + CH
3D + CH
2D
2 (as methane), and D
2O + DHO + H
2O (as water).
A quantitative evaluation of the percentages of the labelled molecules (i.e., incorporating D in their structure) in ammonia and hydrogen (Reaction (1)), and in hydrogen, methane, and water (Reaction (2)) are possible from the intensity of the MS peaks, taking into account the contributions to a particular peak of the molecular and molecular fragmented ions with this mass to charge (
m/
z) ratio. A summary of the different contributions to each particular
m/
z peak is reported in
Table 1. The contributions of the doubly ionized species are disregarded in our analysis because of their very low probability. A quantitative evaluation of the percentage of labelled molecules can be made discounting the intensity of the residual masses always present in the MS analysis chamber (e.g., due to residual hydrogen, water, and hydrocarbons, an example of this can be seen in the
Supporting Information, Figure S1) and also that of ionized molecular fragments with contributions that can be taken from fragmentation pattern libraries [
22]. For example, to estimate the relative amount of NDH
2 in the outlet mixture, we proceed by assuming that I (
m/
z = 18) = I (H
2O
+) + I (ND
2+) + I (NDH
2+), where I (H
2O
+) is the intensity due to the residual water and is determined when measuring the spectrum of the initial mixture before switching on the plasma (see
Figure S1), and I (ND
2+) is determined from the intensity of the ND
2H
+ (
m/
z = 19) and ND
3+ (
m/
z = 20) peaks and their reported fragmentation patterns [
21]. The results using this quantification procedure for the two investigated reactions will be presented in the next section.
For the investigated ammonia reaction, the evaluation of the isotope exchange was confirmed by the data retrieved from the IR spectra. A rough evaluation of the series of spectra in
Figure 1b indicates that the intensity profile of the vibrational/rotational band systems in the regions 3600–3100 cm
−1, 1800–1300 cm
−1, and 1250–700 cm
−1 is characteristic of NH
3 (a typical spectrum of NH
3 gas is reported in the
supported information, in Figure S2), and varies and becomes progressively shifted to lower wave numbers as the plasma power increases. In addition, a series of new little bands appear in the region of 2740–2400 cm
−1. This progressive shift and the appearance of new bands agree with the progressive formation of ND
2H and NDH
2 (and traces of ND
3 at the highest power), substituting the NH
3 molecules (note that the overall amount of ammonia remained invariable). This qualitative assessment of the spectral evolution and attribution of the bands coincides with the reported analysis of the IR spectra of NH
3, NH
2D, NHD
2, and ND
3 in the gas phase [
23,
24,
25]. After a careful evaluation of these series of spectra, we could identify some specific bands that can be associated with NH
3 (3335 cm
−1), NH
2D (2505 cm
−1), NHD
2 (2558 cm
−1), and ND
3 (2420 cm
−1), which have been used to confirm the isotopic exchange deduced by the mass spectrometry analysis of the plasma activated mixture. However, as the extinction coefficient for each particular band is not easily accessible, the results will be semiquantitative and will be used just to confirm the tendencies deduced by MS.
2.2. Evaluation of Inefficient Reaction Events
The plasma induced reactions are triggered by the interaction of the plasma electrons with the gas molecules, giving rise to a series of activated intermediate species, radicals, and ions, which, through the intervention of a series of intermediate reactions, will eventually give rise to the product molecules of Reactions (1) and (2) detected in the outlet mixture. It is noteworthy that such intermediate reactions may involve not only “reactants”, but also “products” molecules if they are present in the reaction medium. In the course of the DBD plasma processes, energy is wasted whenever these intermediate reactions do not give rise to “products” molecules. The use of labelled reactants tries to monitor the occurrence of the intermediate processes that are ineffective in producing “product” molecules. Some examples illustrating this type of inefficient intermediate reactions are as follows:
Reaction (1)
where one NH
3 molecule is being transformed into one NHD
2 molecule (or another indistinguishable NH
3 molecule when using H
2 instead of D
2) after three intermediate reactions, a set of processes that from the point of view of the reaction yield do not contribute to increasing the ammonia production (although it spends a considerable amount of the energy associated to plasma electrons). We must stress that in DBD process, particularly if they involve the use of catalysts, the intermediate reactions not involving electrons may take place either in the plasma phase or on the surface of the interelectrode pellets used to moderate the discharge (see reference [
11], where we suggest this possibility for the synthesis of ammonia).
Reaction (2)
or alternatively
or
In this case, the energy of the electrons would be spent in dissociating the water or the methane used as reactants and, through a series of intermediate reactions, in creating intermediate species that react to form new CHxDy molecules without giving rise to hydrogen and CO as reaction products (Reaction (2)).
Some of the intermediate species quoted above in the examples of the intermediate reactions for Reactions (1) and (2) (e.g., H*, HO*, or NH*), have been effectively detected by optical emission spectroscopy during these reactions [
9,
11,
26].
One of the purposes of the present study using labelled reactants is to develop a methodology to semi-quantitatively assess the occurrence of intermediate reactions. For this end, we will proceed in the following two steps: (i) determine the percentual distribution of the distinct labelled molecules of a given compound (e.g., the percentage of CH
4, CDH
3, and CD
2H
2) in the outlet gases for each experimental condition, (ii) approach the relative number of exchange processes that take place in each experiment. For step (i), we used the evaluation procedure based on the
m/
z peak intensities described in
Section 2.1. For step (ii), we proceed as follows: we defined a relative number of reaction events (REs) for each compound as the sum of the percentage of a given isotopically marked molecule, multiplied by the number of exchanged isotope atoms, corrected by the flow ratio of this particular molecule in the outlet flow (note that in the case of the wet reforming Reaction (2), the total outlet flow increases with respect to the inlet). For example, the REs for methane (i.e., including all forms of isotopically labelled molecules) will be determined as follows:
where, for example, %CD
3H
3 is the percentage of this labelled molecule, referred to the total number of methane molecules. In this case, it is multiplied by three, because three H atoms in a parent CH
4 reactant molecule have been substituted by D. A similar argument holds for the other isotopically exchanged molecules.
Similarly, specific RE numbers can be defined for a specific isotopic molecular form. For example, for CD
3H we will have the following:
Similar definitions of REs can be applied to the other labelled molecules (e.g., CD2H2) detected for Reaction (2) and for the ammonia Reaction (1), although, in this case, the inlet and outlet flows are the same, as there is practically no net formation or decomposition of ammonia.
Similar relative numbers of the exchange reaction events can be determined for water (REwater) and hydrogen (REhydrogen), and for their specific labelled molecules. For a given reaction, the total number of exchange reaction events (TRE) can be then calculated as the sum of all REs. We will make the assumption that TRE is as an indication of the number inefficient intermediate reactions occurring during the plasma process and, therefore, of the amount of energy wasted in the overall DBD plasma process. This assumption is somehow arbitrary and clearly underestimates the actual number of inefficient intermediate processes taking place to yield a given labelled molecule. This is so, because the RE and TRE numbers defined as in Reactions (3) or (4) do not take into account all of the possible intermediate processes contributing to isotope exchange reactions, see, for example, the intermediate reactions shown as the examples in “Reaction (2)” above. Moreover, these definitions of RE and TRE do not take into account the intermediate reactions involving either H2 or H* atoms that are present in the system, and therefore do not give rise to isotopically exchanged molecules in the final isotopic molecular mixture.
In the following sections, we will determine the REs and TREs for Reactions (1) and (2) as a function of the power consumption, and discuss the evolution found in these parameters as a way to semi-quantitatively estimate the energy ratio wasted in the intermediate reactions, which are inefficient to render product molecules from the reactant molecules. These considerations rely on the assumption that elementary reaction rates are not significantly affected by the type of isotope bonded to the excited molecules. In reality, in conventional low pressure plasma, the rate of elementary reactions can be little affected by the type of isotope [
27]. However, in atmospheric pressure, non-equilibrium plasmas excited with high AC voltages at relatively low frequency (i.e., conditions utilized in DBD discharges) energy are mainly used to induce very high electron temperatures and high vibrational temperatures [
28], where the effect of the isotope mass will be negligible and therefore the above hypothesis is fully justified.
2.3. Ammonia Synthesis: Intermediate Exchange Reactions
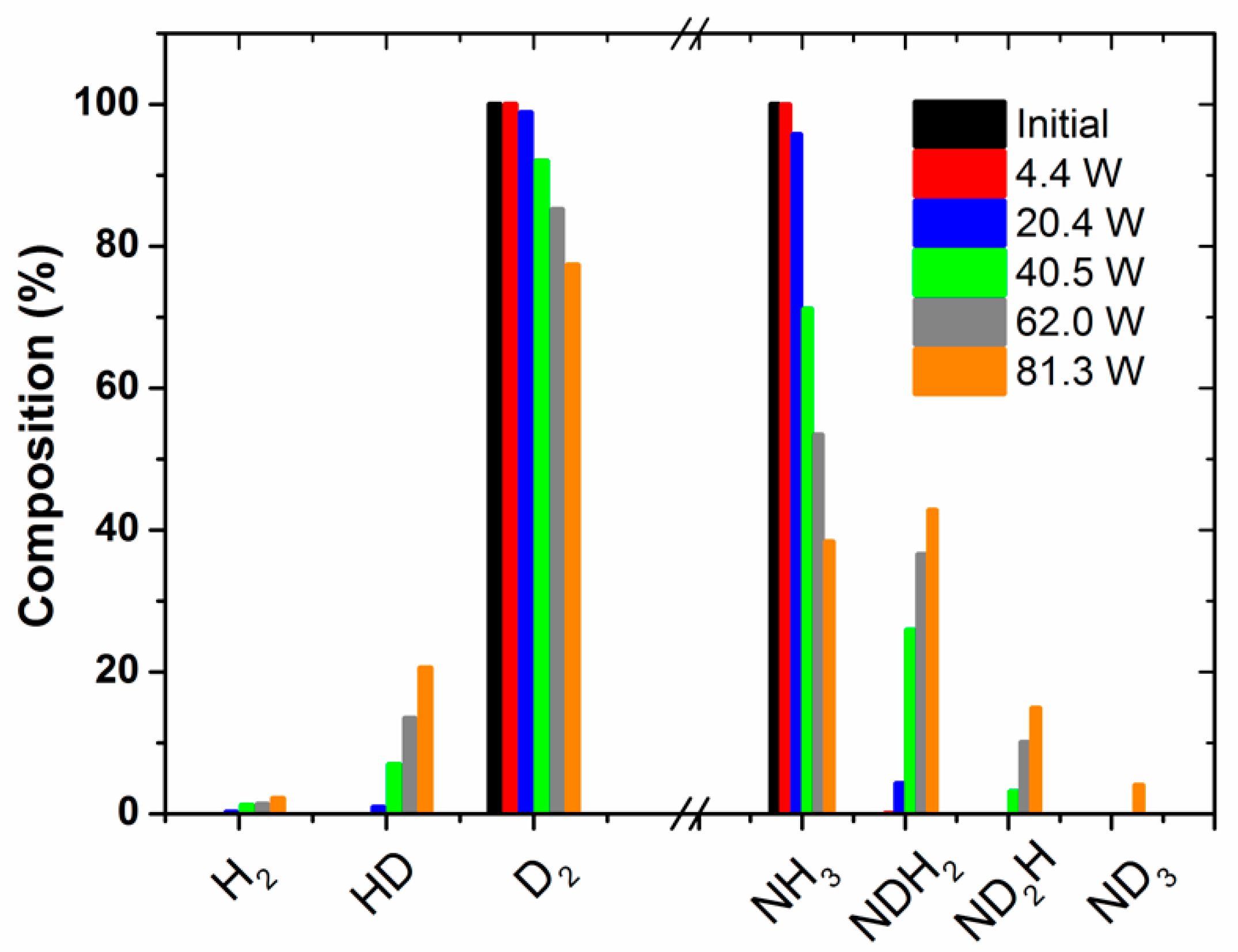
Figure 2 shows the percentages of the isotopic labelled species produced as a function of the applied power. These percentages have been estimated by the analysis of the MS spectra in
Figure 1a, recorded at increasingly higher powers. This plot clearly shows that the percentage of NDH
2 and ND
2H molecules relatively increases with the power. Simultaneously, the percentages of HD and, to a lesser extent, H
2 also increase with this parameter. A similar tendency was obtained by evaluating the evolution of the absorbance intensity of the specific IR bands attributed to the different ammonia species (see
Figure S3 in the Supporting Information). This evaluation has only a semi-quantitative character, because the absorption coefficient of each band should be taken into account for quantification. As practically no change in the amount of ammonia (i.e., including all forms of labelled ammonia molecules) occurs during this series of experiments (as already mentioned, for the selected nitrogen/hydrogen/ammonia mixture, ammonia molecules are formed and decomposed at equivalent rates), we must assume that all of the energy applied to the reactor is used in inducing intermediate REs that are inefficient from the point of view of the ammonia synthesis.
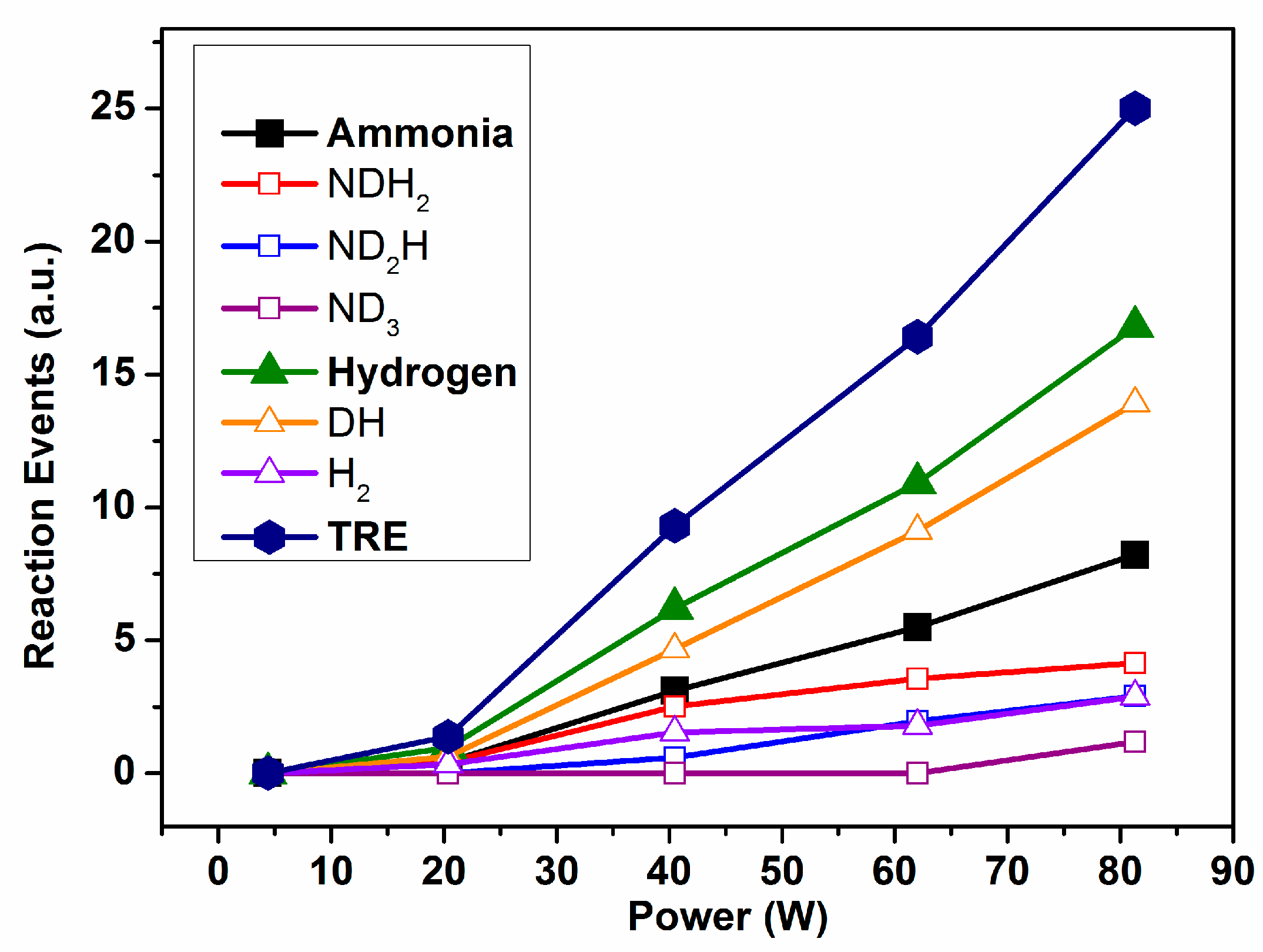
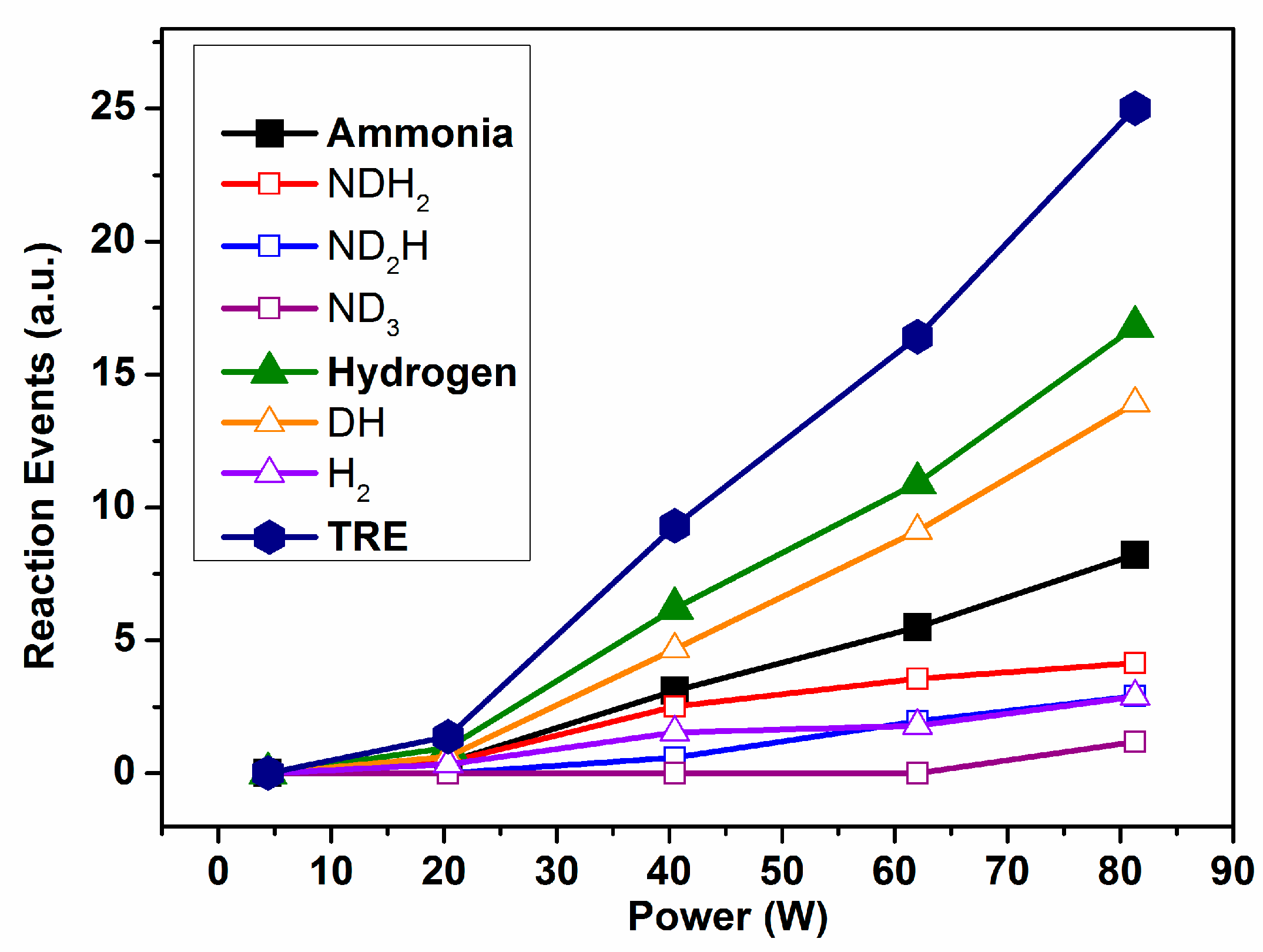
Figure 3 shows the evolution of the individual REs and TRE calculated for Reaction (1). It is apparent in these plots that, for a power higher than 20 W, the RE values for the ammonia and hydrogen species increase with power in a rather lineal way. The final result is that the TRE values present a continuously increasing tendency when the applied power increases. A translation of this evidence for a real process aiming at the synthesis of ammonia from a binary mixture N
2/H
2 (Reaction (1)) is that although increasing the power could contribute to an increase in the ammonia yield by promoting the direct reaction between nitrogen and hydrogen, this increase would likely be damped by the progressive waste of energy involved in promoting an increasingly higher number of intermediate reaction events, as evidenced in
Figure 3. An obvious conclusion of this evidence is that in order to increase the ammonia production and decrease the energy consumption, it would be required to change other reaction parameters and/or to modify the design and operational mode of the reactor.
2.4. Wet Reforming of Methane: Intermediate Exchange Reactions and Reaction Efficiency
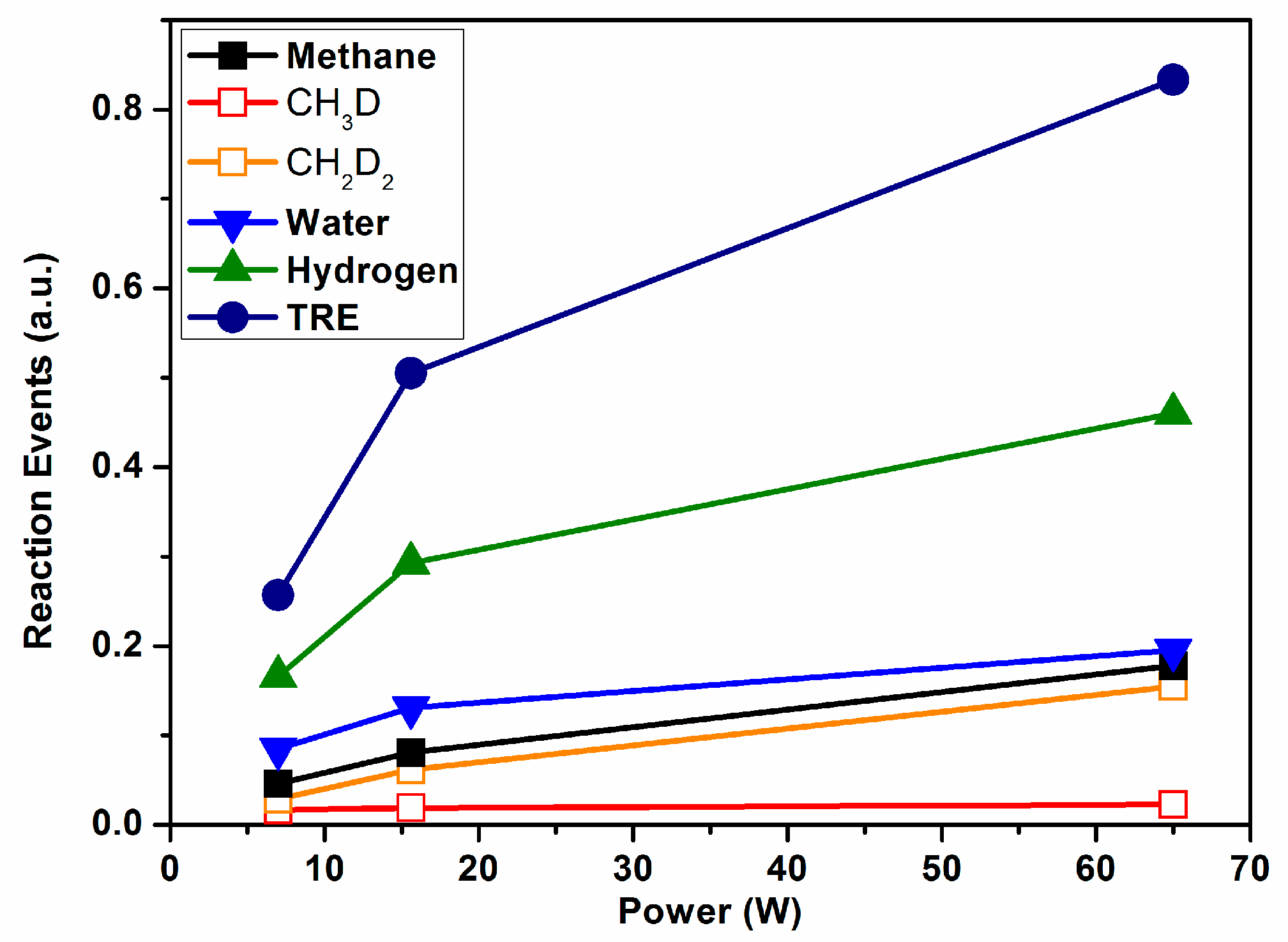
The percentages of the different molecules detected by MS were reported in the literature [
3], together with the percentage of methane transformed into carbon monoxide and hydrogen in each case (percentages of converted methane through Reaction (2) were 11%, 21%, and 50% for the three applied plasma powers).
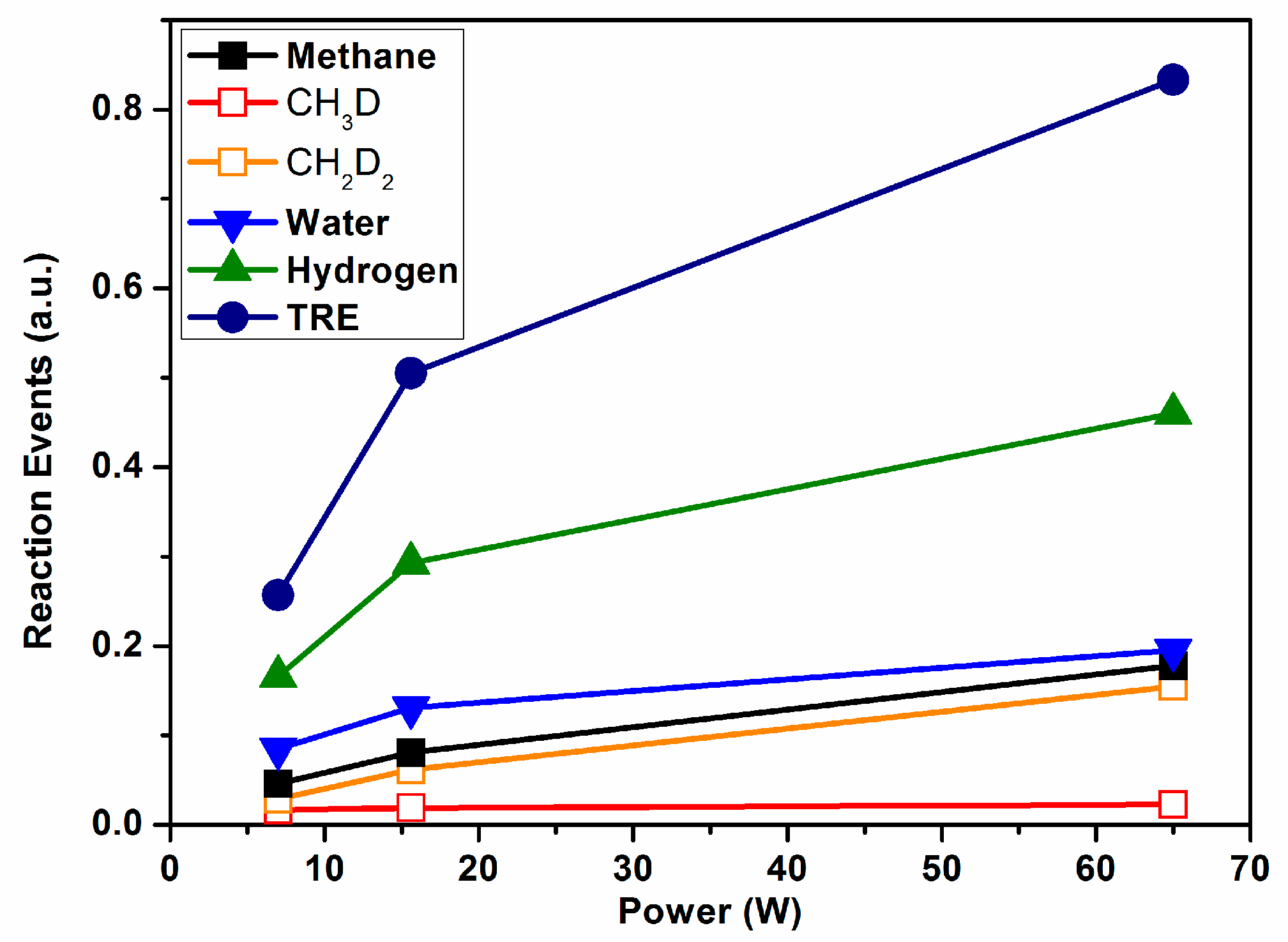
Figure 4 shows the REs for methane (summing their two labelled variants, also presented in this plot), water (sum of HDO and H
2O), and hydrogen (sum of HD and D
2), and the total value of TRE as a function of power consumption. The TRE depicts an increasing evolution with power that reveals a progressive increase in the number of inefficient events with this parameter. However, unlike the rather lineal evolution of the TRE reported for ammonia in
Figure 3, the evolution of TRE in
Figure 4 is characterized by a decreasing slope with power, indicating a certain saturation of the isotopic exchange probability. This tendency suggests that increasing the power may increase the reaction yield, as the number of REs does not progress lineally with it. In agreement with this, a reaction yield of 50% was found for the highest power applied in this experiment [
3]. Another interesting feature deduced from
Figure 4 is that, independently of the applied power, the number of events involved in the formation of CH
2D
2 is much higher than that rendering CH
3D. Tentatively, we associated this difference to the known relatively higher stability of CH
3* radicals [
29] and its smaller tendency to saturate the carbon bonds more than other reactive species such as CH
2* (in other words, the intermediate reaction CH
3* + D* → CH
3D would have a much lower cross section probability than CH
2* + D* → CH
2D*). This isotope labelling result agrees with the current mechanistic models of the wet reforming reaction [
30].

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}