Value-Added Products from Urea Glycerolysis Using a Heterogeneous Biosolids-Based Catalyst




Abstract
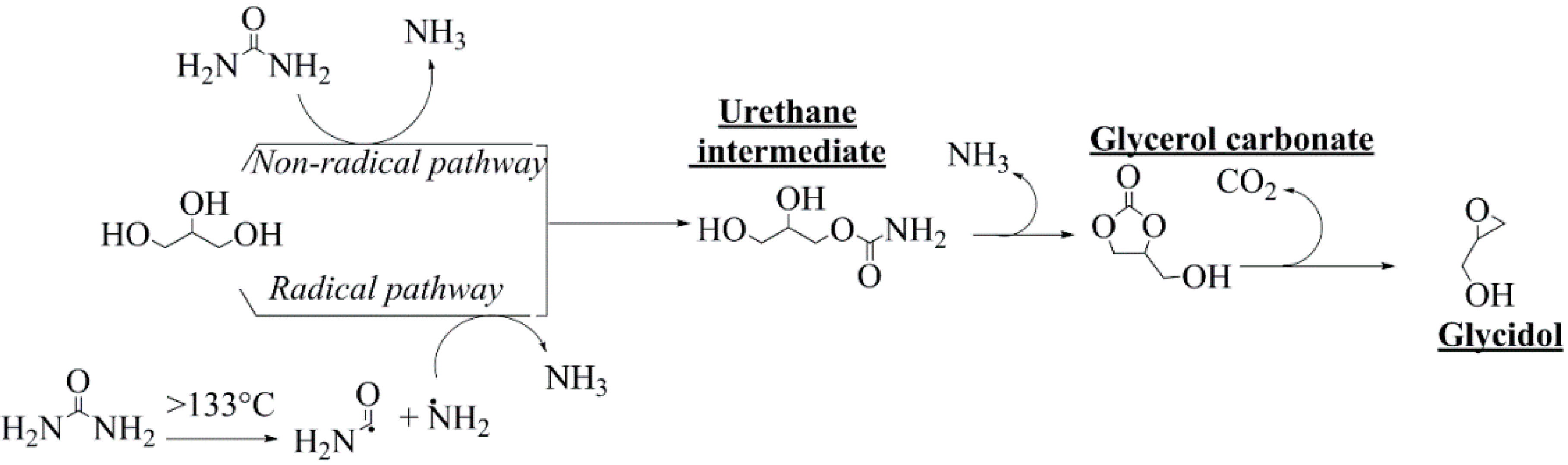
1. Introduction
2. Results and Discussion
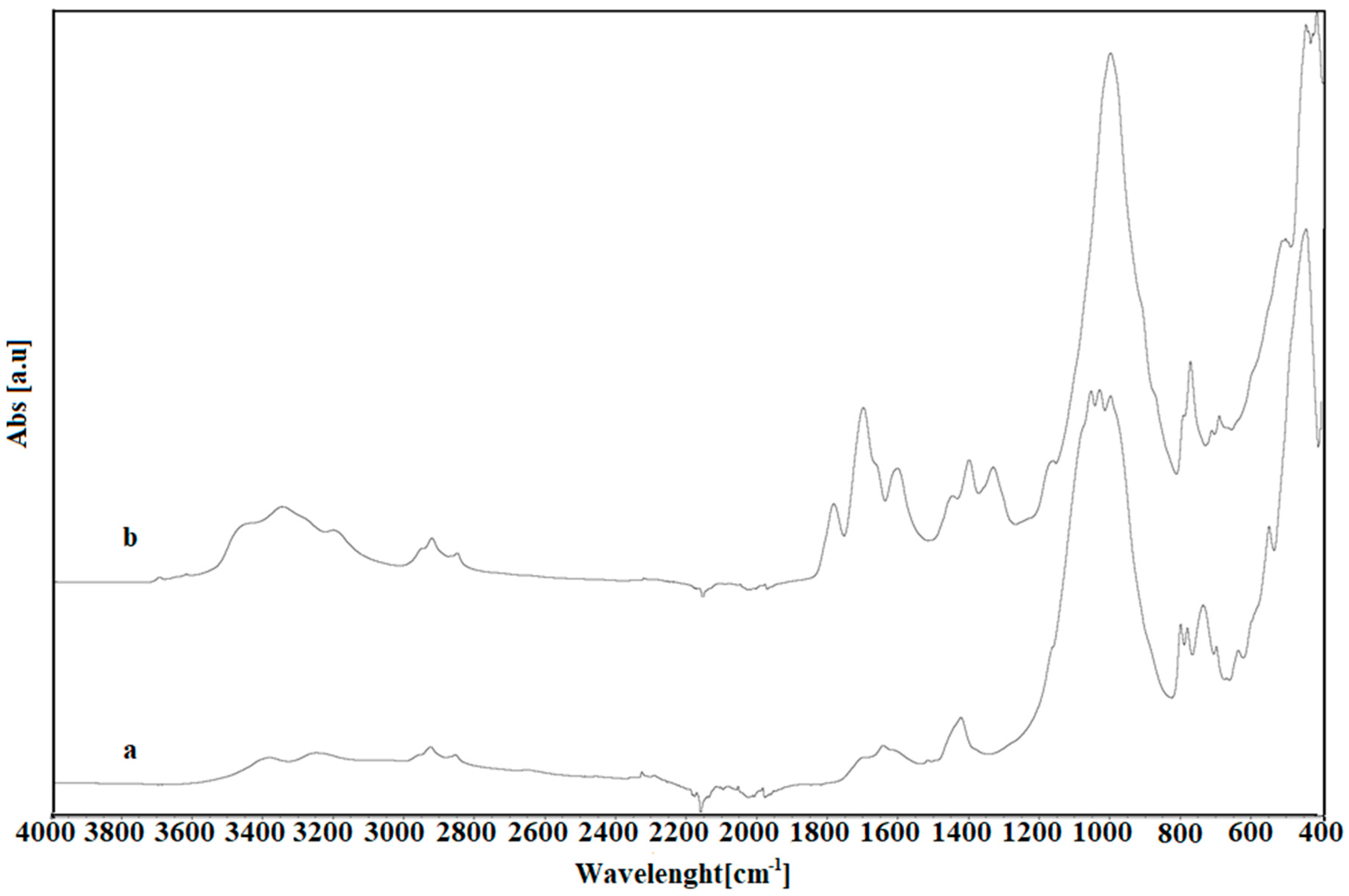
2.1. Assessment of the Biosolids-Based Catalyst
2.2. Influence of Catalyst Loading
2.3. Influence of Temperature
2.4. Influence of the Molar Ratio of Urea/Glycerol
2.5. Influence of Residual Atmosphere
2.6. Influence of Recycling on Urea Glycerolysis on the Conversion Rate
2.7. Comparison between Biosolids Based Catalysts and Heterogenous Catalysts
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Catalytic Acetylation of Glycerol
3.2.2. Gas Chromatography and Mass Spectrometry
3.2.3. Characterization of Inorganic Content in the Biosolids-Based Catalyst
3.2.4. Quantification of Total Surface Acidic Groups
3.2.5. Attenuated Total Reflection Fourier Transform Infrared Spectroscopy
3.2.6. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leiserowitz, A.A.; Kates, R.W.; Parris, T.M. Sustainability values, attitudes, and behaviors: A review of multinational and global trends. Annu. Rev. Environ. Resour. 2006, 31, 413–444. [Google Scholar] [CrossRef]
- United, N. Johannesburg declaration on sustainable development. In Proceedings of the World Summit on Sustainable Development, Johannesburg, South Africa, 26 August–4 September 2002. [Google Scholar]
- Anastas, P.; Eghbali, N. Green chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Manzer, L.E. Recent developments in the conversion of biomass to renewable fuels and chemicals. Top. Catal. 2010, 53, 1193–1196. [Google Scholar] [CrossRef]
- Hill, J.; Nelson, E.; Tilman, D.; Polasky, S.; Tiffany, D. Environmental, economic, and energetic costs and benefits of biodiesel and ethanol biofuels. Proc. Natl. Acad. Sci. USA 2006, 103, 11206–11210. [Google Scholar] [CrossRef] [PubMed]
- Ryan, L.; Convery, F.; Ferreira, S. Stimulating the use of biofuels in the European Union: Implications for climate change policy. Energy Policy 2006, 34, 3184–3194. [Google Scholar] [CrossRef]
- Yang, F.; Hanna, M.A.; Sun, R. Value-added uses for crude glycerol—A byproduct of biodiesel production. Biotechnol. Biofuels 2012, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.H. Reactive applications of cyclic alkylene carbonates. Ind. Eng. Chem. Res. 2003, 42, 663–674. [Google Scholar] [CrossRef]
- Rokicki, G.; Rakoczy, P.; Parzuchowski, P.; Sobiecki, M. Hyperbranched aliphatic polyethers obtained from environmentally benign monomer: Glycerol carbonate. Green Chem. 2005, 7, 529–539. [Google Scholar] [CrossRef]
- Algoufi, Y.T.; Hameed, B.H. Synthesis of glycerol carbonate by transesterification of glycerol with dimethyl carbonate over K-zeolite derived from coal fly ash. Fuel Process. Technol. 2014, 126, 5–11. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Nocito, F.; Pastore, C. A study on the carboxylation of glycerol to glycerol carbonate with carbon dioxide: The role of the catalyst, solvent and reaction conditions. J. Mol. Catal. A Chem. 2006, 257, 149–153. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; De Frutos, P.; Iborra, S.; Noy, M.; Velty, A.; Concepción, P. Chemicals from biomass: Synthesis of glycerol carbonate by transesterification and carbonylation with urea with hydrotalcite catalysts. The role of acid-base pairs. J. Catal. 2010, 269, 140–149. [Google Scholar] [CrossRef]
- Teng, W.K.; Ngoh, G.C.; Yusoff, R.; Aroua, M.K. Microwave-assisted transesterification of industrial grade crude glycerol for the production of glycerol carbonate. Chem. Eng. J. 2016, 284, 469–477. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A. Carbon dioxide fixation into organic compounds. In Carbon Dioxide Recovery and Utilization; Springer: Dordrecht, The Netherlands, 2003; pp. 211–260. [Google Scholar]
- Nguyen, N.; Demirel, Y. Economic analysis of biodiesel and glycerol carbonate production plant by glycerolysis. J. Sustain. Bioenergy Syst. 2013, 3, 209. [Google Scholar] [CrossRef]
- Sienel, G.; Rieth, R.; Rowbottom, K.T. Epoxides. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000; Volume A13, pp. 141–154. [Google Scholar]
- Sonnati, M.O.; Amigoni, S.; de Givenchy, E.P.T.; Darmanin, T.; Choulet, O.; Guittard, F. Glycerol carbonate as a versatile building block for tomorrow: Synthesis, reactivity, properties and applications. Green Chem. 2013, 15, 283–306. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, X.; Liu, C.; Cheng, T. Synthesis of glycerol carbonate from glycerol and urea over lanthanum compounds. React. Kinet. Mech. Catal. 2015, 115, 597–609. [Google Scholar] [CrossRef]
- Clegg, W.; Harrington, R.W.; North, M.; Pasquale, R. Cyclic carbonate synthesis catalysed by bimetallic aluminium–salen complexes. Chem.-A Eur. J. 2010, 16, 6828–6843. [Google Scholar] [CrossRef] [PubMed]
- Gade, S.M.; Munshi, M.K.; Chherawalla, B.M.; Rane, V.H.; Kelkar, A.A. Synthesis of glycidol from glycerol and dimethyl carbonate using ionic liquid as a catalyst. Catal. Commun. 2012, 27, 184–188. [Google Scholar] [CrossRef]
- Meessen, J.H.; Petersen, H. Urea. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000; Volume A27, pp. 333–350. [Google Scholar]
- Undri, A.; Frediani, M.; Rosi, L.; Frediani, P. Reverse polymerization of waste polystyrene through microwave assisted pyrolysis. J. Anal. Appl. Pyrolysis 2014, 105, 35–42. [Google Scholar] [CrossRef]
- Shukla, K.; Srivastava, V.C. Synthesis of organic carbonates from alcoholysis of urea: A review. Catal. Rev. 2017, 59, 1–43. [Google Scholar] [CrossRef]
- Fernandes, G.P.; Yadav, G.D. Selective glycerolysis of urea to glycerol carbonate using combustion synthesized magnesium oxide as catalyst. Catal. Today 2017, 309, 153–160. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, W.; Zhao, N.; Wei, W.; Sun, Y. Synthesis of cyclic carbonates from urea and diols over metal oxides. Catal. Today 2006, 115, 111–116. [Google Scholar] [CrossRef]
- Mohedano, A.; Monsalvo, V.; Bedia, J.; Lopez, J.; Rodriguez, J. Highly stable iron catalysts from sewage sludge for cwpo. J. Environ. Chem. Eng. 2014, 2, 2359–2364. [Google Scholar] [CrossRef]
- Bedia, J.; Monsalvo, V.; Rodriguez, J.; Mohedano, A. Iron catalysts by chemical activation of sewage sludge with FeCl3 for CWPO. Chem. Eng. J. 2017, 318, 224–230. [Google Scholar] [CrossRef]
- Arnold, R.A.; Habibi, R.; Kopyscinski, J.; Hill, J.M. Interaction of potassium and calcium in the catalytic gasification of biosolids and switchgrass. Energy Fuels 2017, 31, 6240–6247. [Google Scholar] [CrossRef]
- Liu, Z.; McNamara, P.; Zitomer, D. Biochar production and bio-oil upgrading by synergistic catalytic pyrolysis of wastewater biosolids and industrial wastes. Proc. Water Environ. Fed. 2016, 2016, 3182–3187. [Google Scholar] [CrossRef]
- Tu, Y.; Xiong, Y.; Tian, S.; Kong, L.; Descorme, C. Catalytic wet air oxidation of 2-chlorophenol over sewage sludge-derived carbon-based catalysts. J. Hazard. Mater. 2014, 276, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Chae, M.; Xia, L.; Zhu, C.; Bressler, D.C. Accelerating settling rates of biosolids lagoons through thermal hydrolysis. J. Environ. Manag. 2018, 220, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A.; Nocito, F.; Ferragina, C. Valorization of bio-glycerol: New catalytic materials for the synthesis of glycerol carbonate via glycerolysis of urea. J. Catal. 2009, 268, 106–114. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Quaranta, E. Reaction mechanisms in the direct carboxylation of alcohols, polyols, cyclic ethers, and cyclic amines to afford monomeric compounds and polymeric materials. In Reaction Mechanisms in Carbon Dioxide Conversion; Springer: Berlin/Heidelberg, Germany, 2016; pp. 183–235. [Google Scholar]
- Suyatmo, R.I.D.; Sulistyo, H.; Sediawan, W.B. The synthesis of glycerol carbonate from biodiesel by product glycerol and urea over amberlyst 15. Jurnal Bahan Alam Terbarukan 2017, 6, 1–5. [Google Scholar]
- Lertlukkanasuk, N.; Phiyanalinmat, S.; Kiatkittipong, W.; Arpornwichanop, A.; Aiouache, F.; Assabumrungrat, S. Reactive distillation for synthesis of glycerol carbonate via glycerolysis of urea. Chem. Eng. Process. Process Intensif. 2013, 70, 103–109. [Google Scholar] [CrossRef]
- Park, J.-H.; Choi, J.S.; Woo, S.K.; Lee, S.D.; Cheong, M.; Kim, H.S.; Lee, H. Isolation and characterization of intermediate catalytic species in the Zn-catalyzed glycerolysis of urea. Appl. Catal. A Gen. 2012, 433, 35–40. [Google Scholar] [CrossRef]
- Rubio-Marcos, F.; Calvino-Casilda, V.; Bañares, M.A.; Fernandez, J.F. Control of the interphases formation degree in Co3O4/ZnO catalysts. ChemCatChem 2013, 5, 1431–1440. [Google Scholar] [CrossRef]
- Manjunathan, P.; Ravishankar, R.; Shanbhag, G.V. Novel bifunctional Zn–Sn composite oxide catalyst for the selective synthesis of glycerol carbonate by carbonylation of glycerol with urea. ChemCatChem 2016, 8, 631–639. [Google Scholar] [CrossRef]
- Ab Rahim, M.H.; He, Q.; Lopez-Sanchez, J.A.; Hammond, C.; Dimitratos, N.; Sankar, M.; Carley, A.F.; Kiely, C.J.; Knight, D.W.; Hutchings, G.J. Gold, palladium and gold–palladium supported nanoparticles for the synthesis of glycerol carbonate from glycerol and urea. Catal. Sci. Technol. 2012, 2, 1914–1924. [Google Scholar] [CrossRef]
- Kim, D.-W.; Park, M.-S.; Selvaraj, M.; Park, G.-A.; Lee, S.-D.; Park, D.-W. Catalytic performance of polymer-supported ionic liquids in the synthesis of glycerol carbonate from glycerol and urea. Res. Chem. Intermed. 2011, 37, 1305. [Google Scholar] [CrossRef]
- Wang, L.; Ma, Y.; Wang, Y.; Liu, S.; Deng, Y. Efficient synthesis of glycerol carbonate from glycerol and urea with lanthanum oxide as a solid base catalyst. Catal. Commun. 2011, 12, 1458–1462. [Google Scholar] [CrossRef]
- Omidghane, M.; Jenab, E.; Chae, M.; Bressler, D.C. Production of renewable hydrocarbons by thermal cracking of oleic acid in the presence of water. Energy Fuels 2017, 31, 9446–9454. [Google Scholar] [CrossRef]
- Benitez, E.; Romero, E.; Gomez, M.; Gallardo-Lara, F.; Nogales, R. Biosolids and biosolids-ash as sources of heavy metals in a plant-soil system. Water Air Soil Pollut. 2001, 132, 75–87. [Google Scholar] [CrossRef]
- Boehm, H.P. Acidic and basic properties of hydroxylated metal oxide surfaces. Discuss. Faraday Soc. 1971, 52, 264–275. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration of Main Metal Species [mg/g] | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cr | Fe | Mn | Sr | Al | Cu | Zn | Pb | Ti | |
| Biosolids-based catalyst 1 | 0.39 ± 0.02 | 26 ± 1 | 0.58 ± 0.03 | 0.37 ± 0.02 | 26 ± 1 | 0.71 ± 0.04 | 1.06 ± 0.05 | 0.10 ± 0.01 | 3.2 ± 0.2 |
| Recycled biosolids-based catalyst 2 | 0.28 ± 0.01 | 18 ± 1 | 0.39 ± 0.5 | 0.28 ± 0.01 | 20 ± 1 | 0.74 ± 0.03 | 0.72 ± 0.05 | 0.08 ± 0.01 | 0.16 ± 0.02 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartoli, M.; Zhu, C.; Chae, M.; Bressler, D.C. Value-Added Products from Urea Glycerolysis Using a Heterogeneous Biosolids-Based Catalyst. Catalysts 2018, 8, 373. https://doi.org/10.3390/catal8090373
Bartoli M, Zhu C, Chae M, Bressler DC. Value-Added Products from Urea Glycerolysis Using a Heterogeneous Biosolids-Based Catalyst. Catalysts. 2018; 8(9):373. https://doi.org/10.3390/catal8090373
Chicago/Turabian StyleBartoli, Mattia, Chengyong Zhu, Michael Chae, and David C. Bressler. 2018. "Value-Added Products from Urea Glycerolysis Using a Heterogeneous Biosolids-Based Catalyst" Catalysts 8, no. 9: 373. https://doi.org/10.3390/catal8090373
APA StyleBartoli, M., Zhu, C., Chae, M., & Bressler, D. C. (2018). Value-Added Products from Urea Glycerolysis Using a Heterogeneous Biosolids-Based Catalyst. Catalysts, 8(9), 373. https://doi.org/10.3390/catal8090373