Investigation of Earth-Abundant Oxygen Reduction Electrocatalysts for the Cathode of Passive Air-Breathing Direct Formate Fuel Cells


Abstract
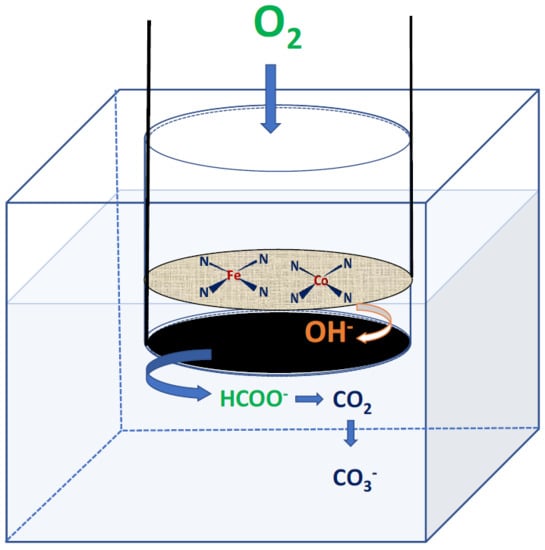
:
1. Introduction
2. Results and Discussion
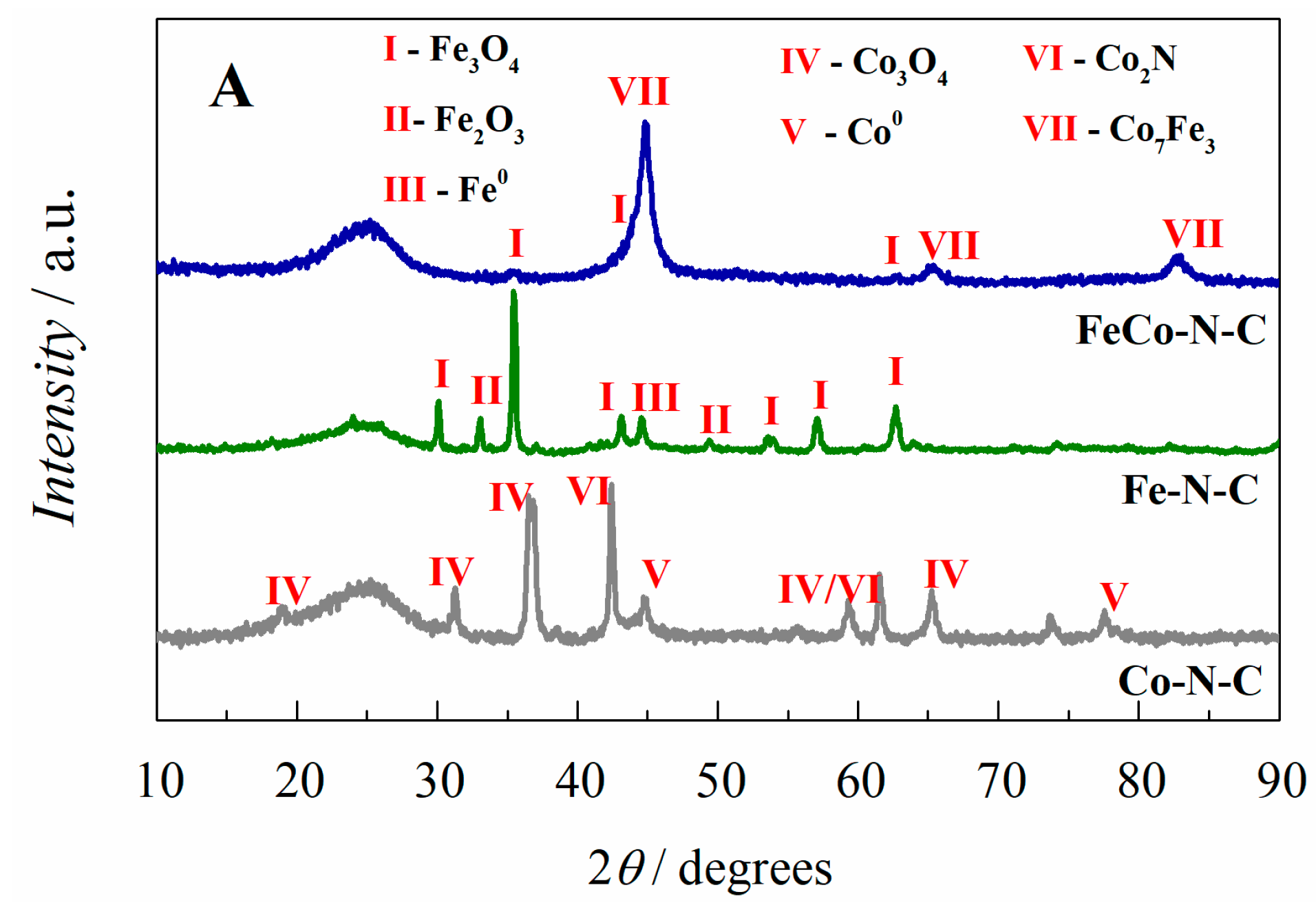
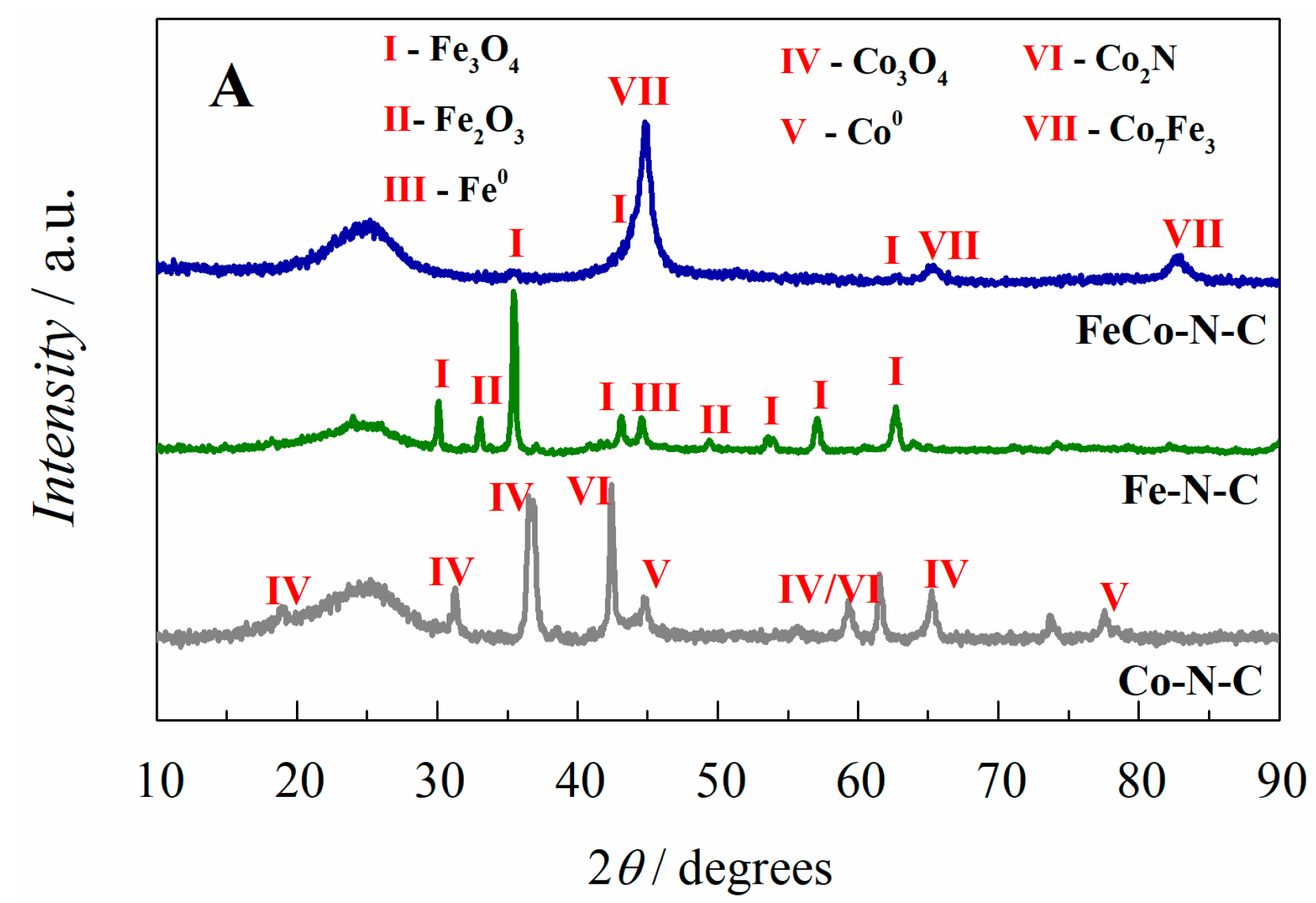
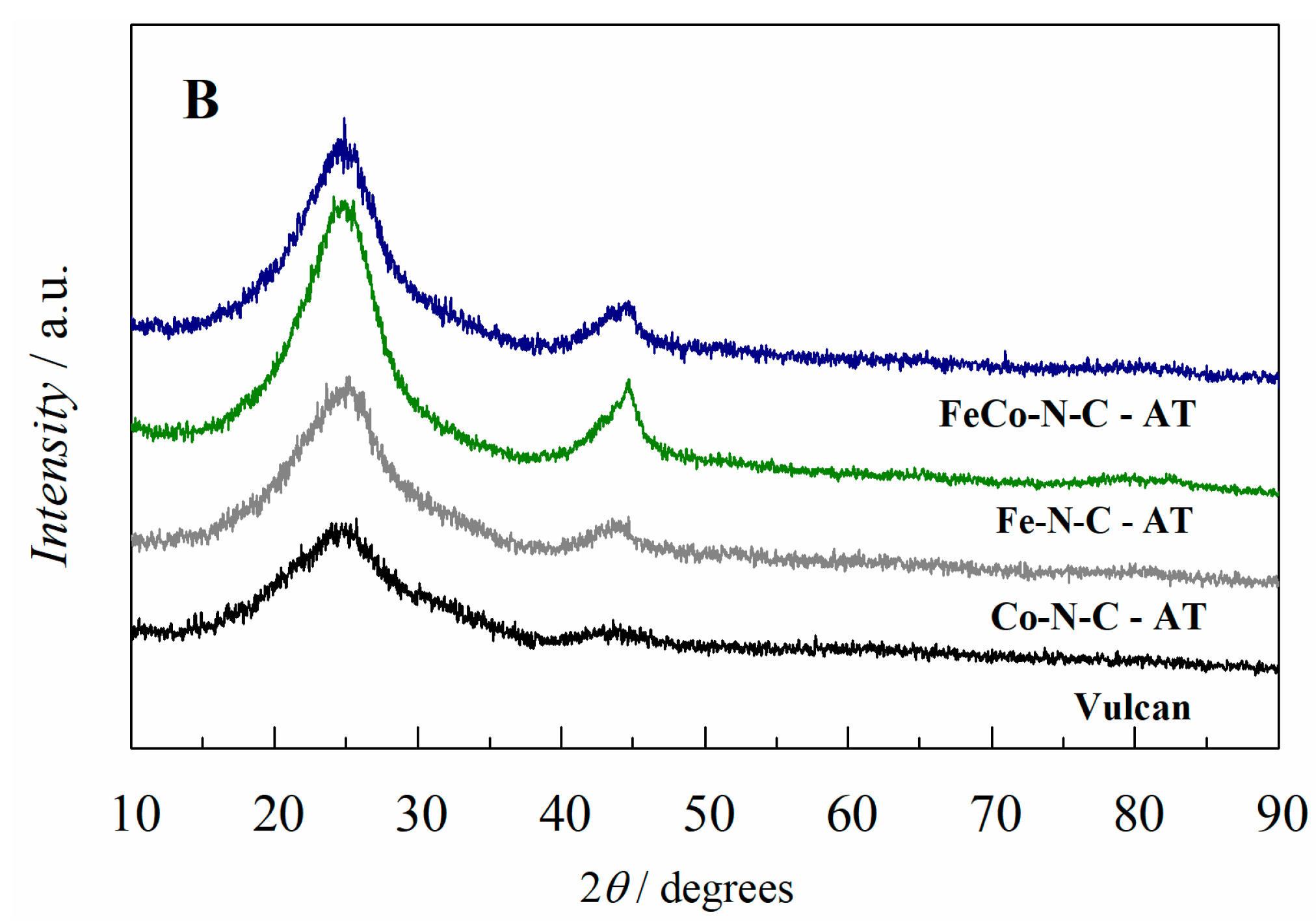
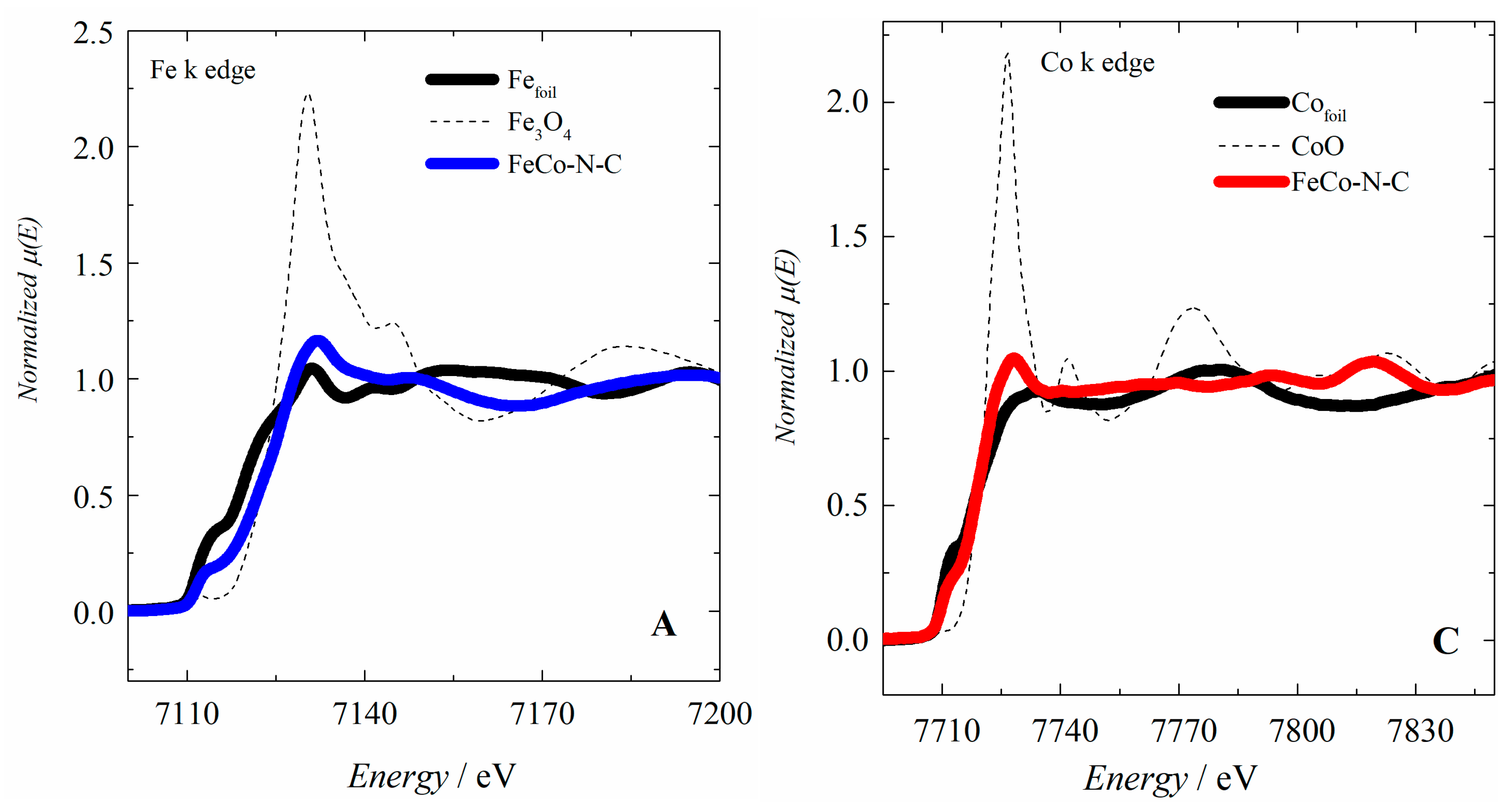
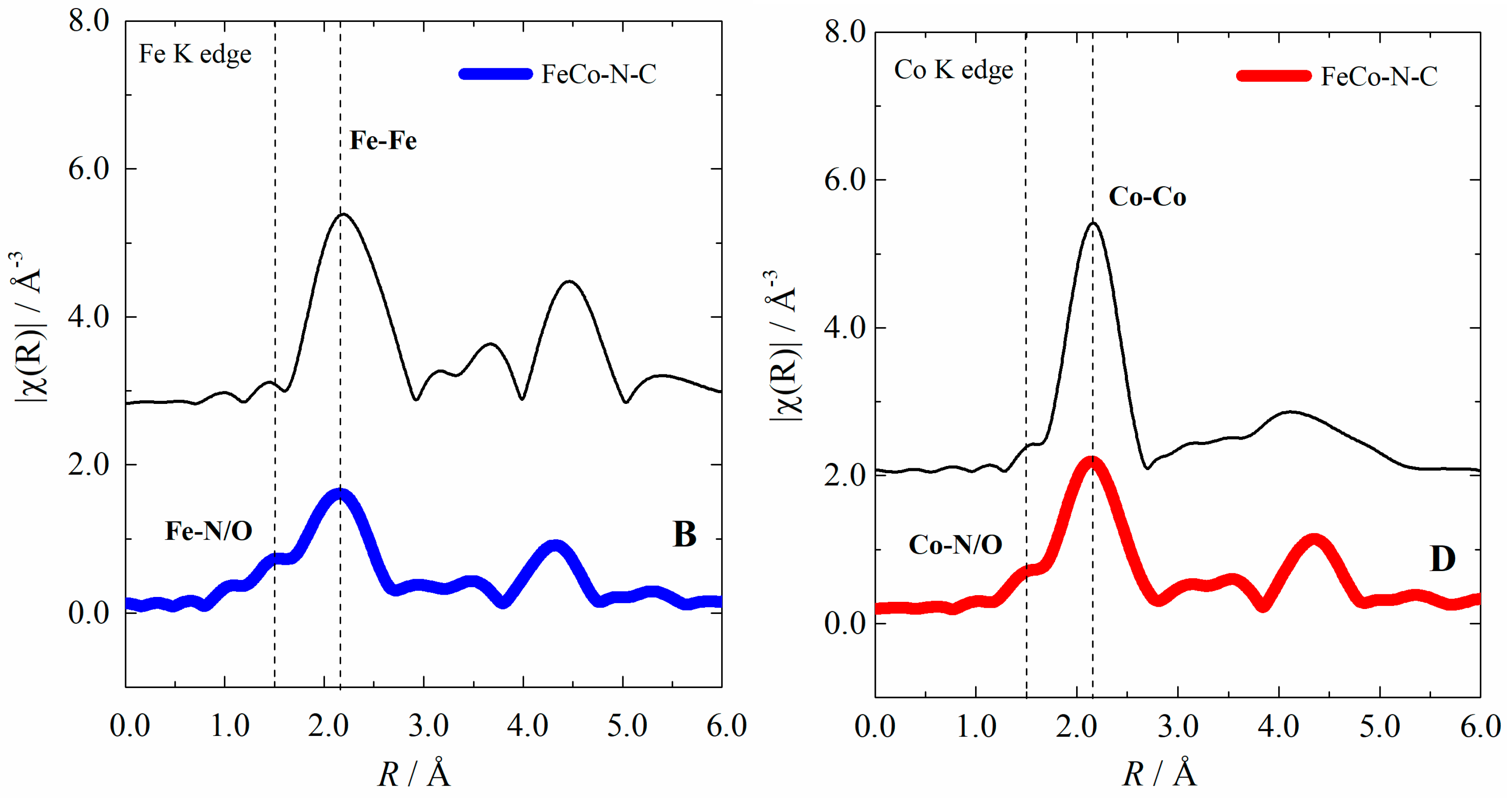
2.1. Electrocatalyst Characterization
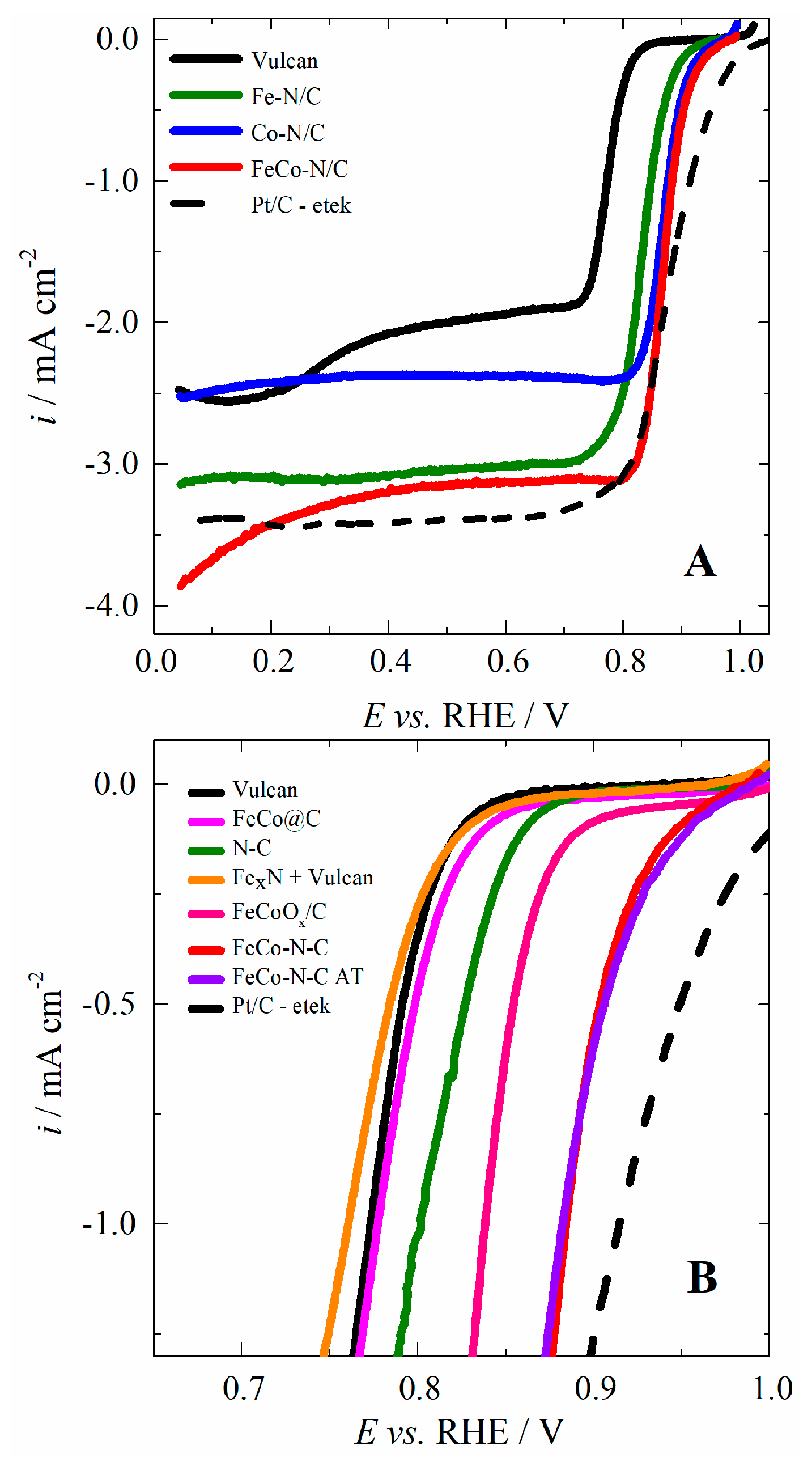
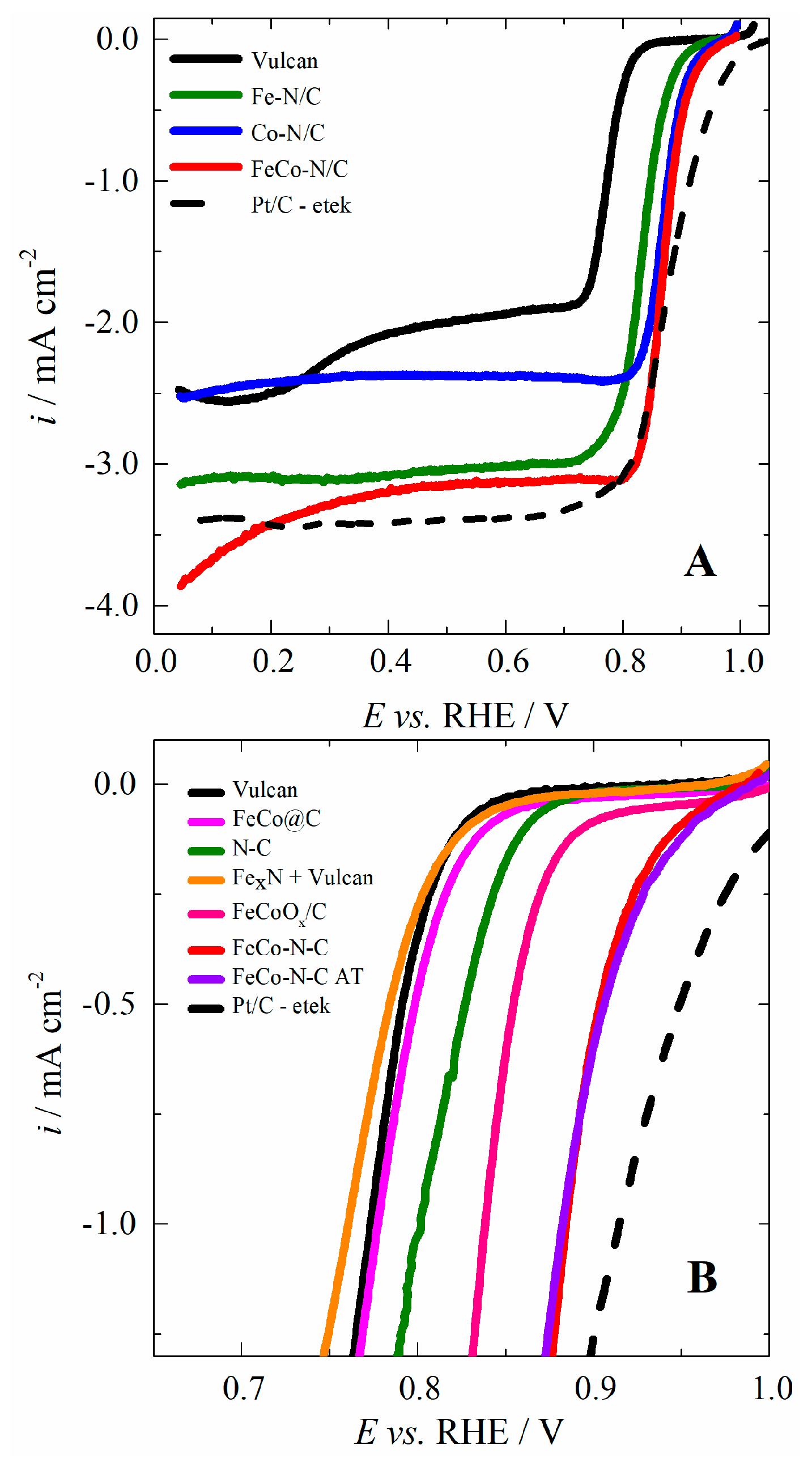
2.2. Investigation of the Electrocatalytic Activity for the ORR
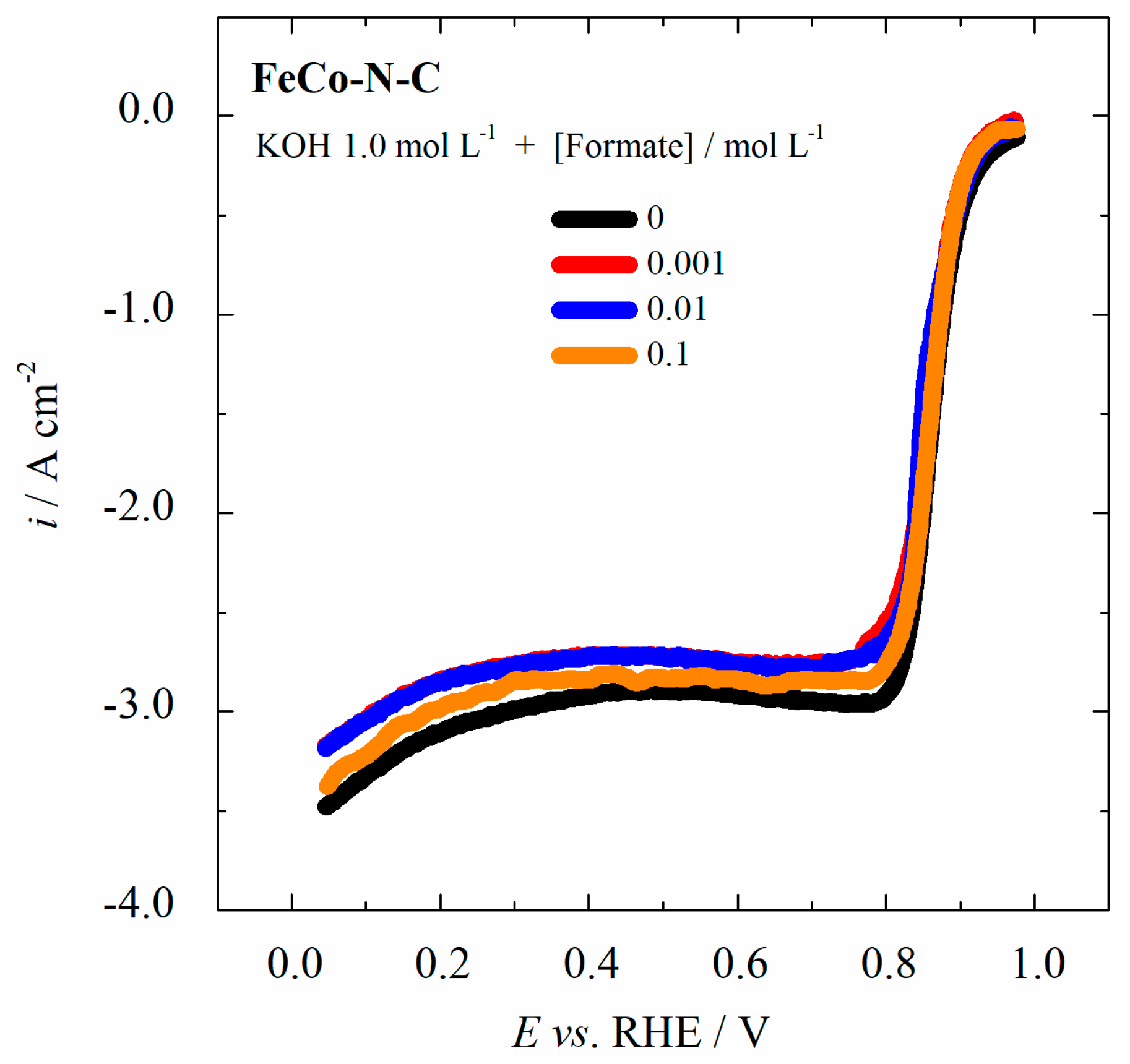
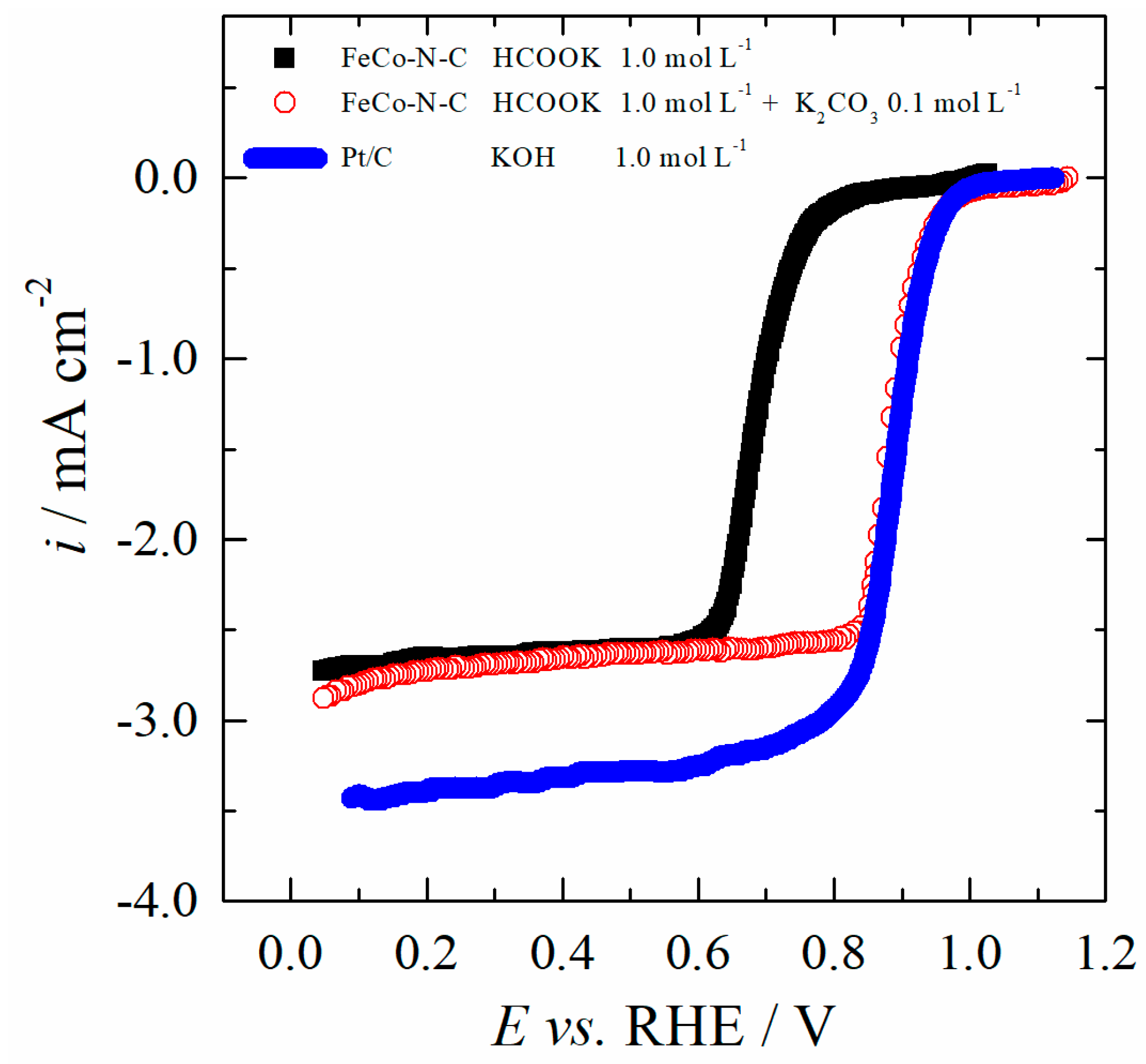
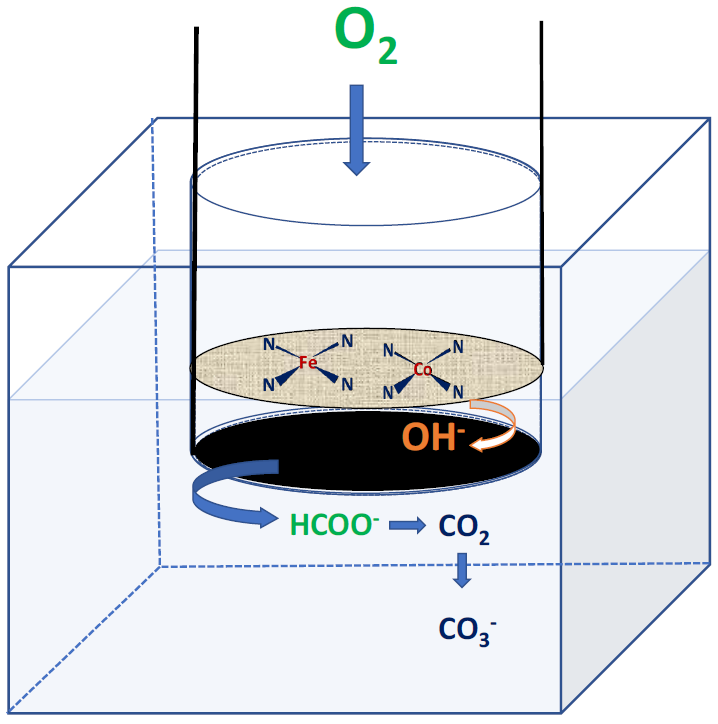
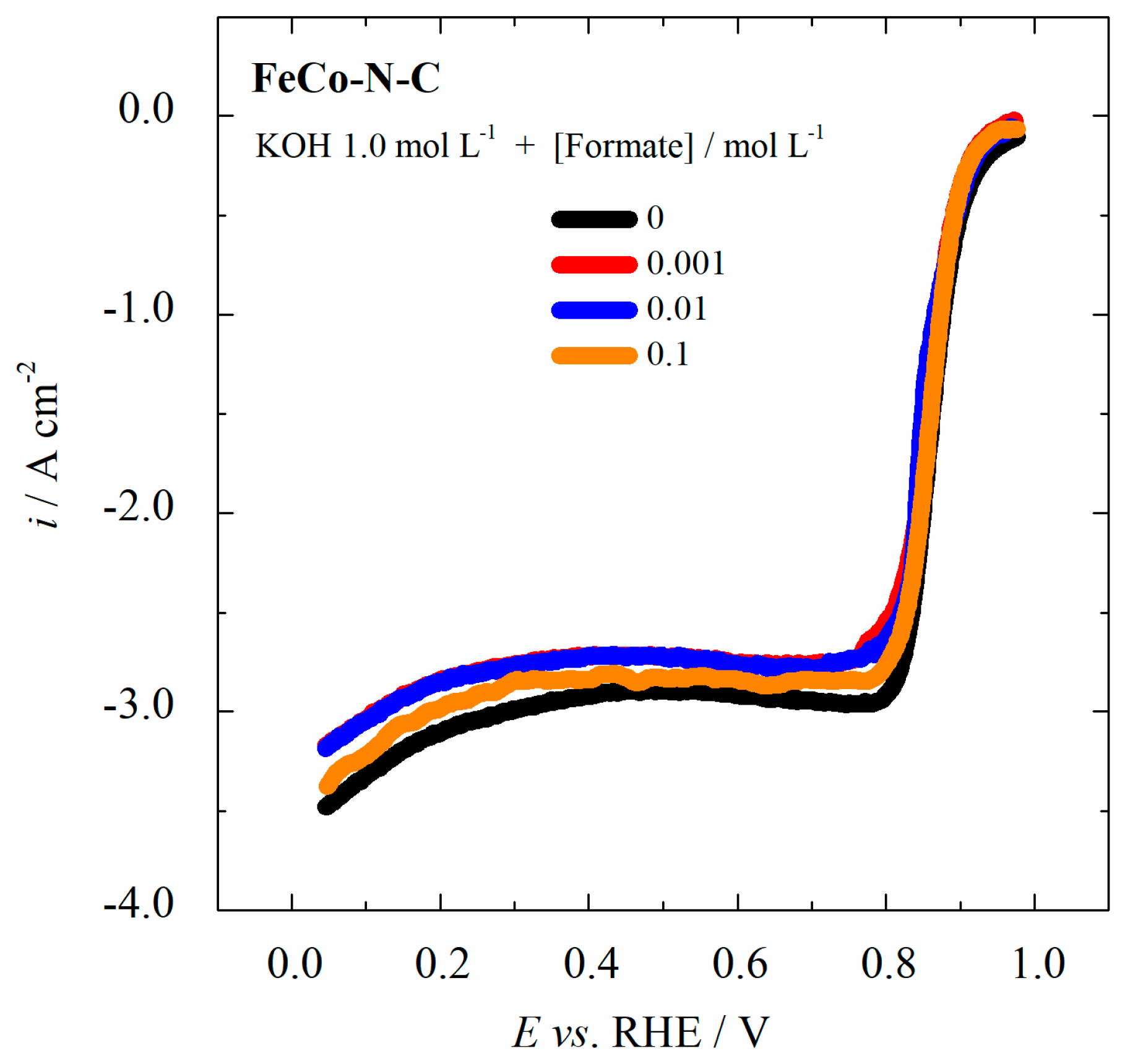
2.3. Formate Ions Tolerance of the FeCo-N-C Electrocatalyst
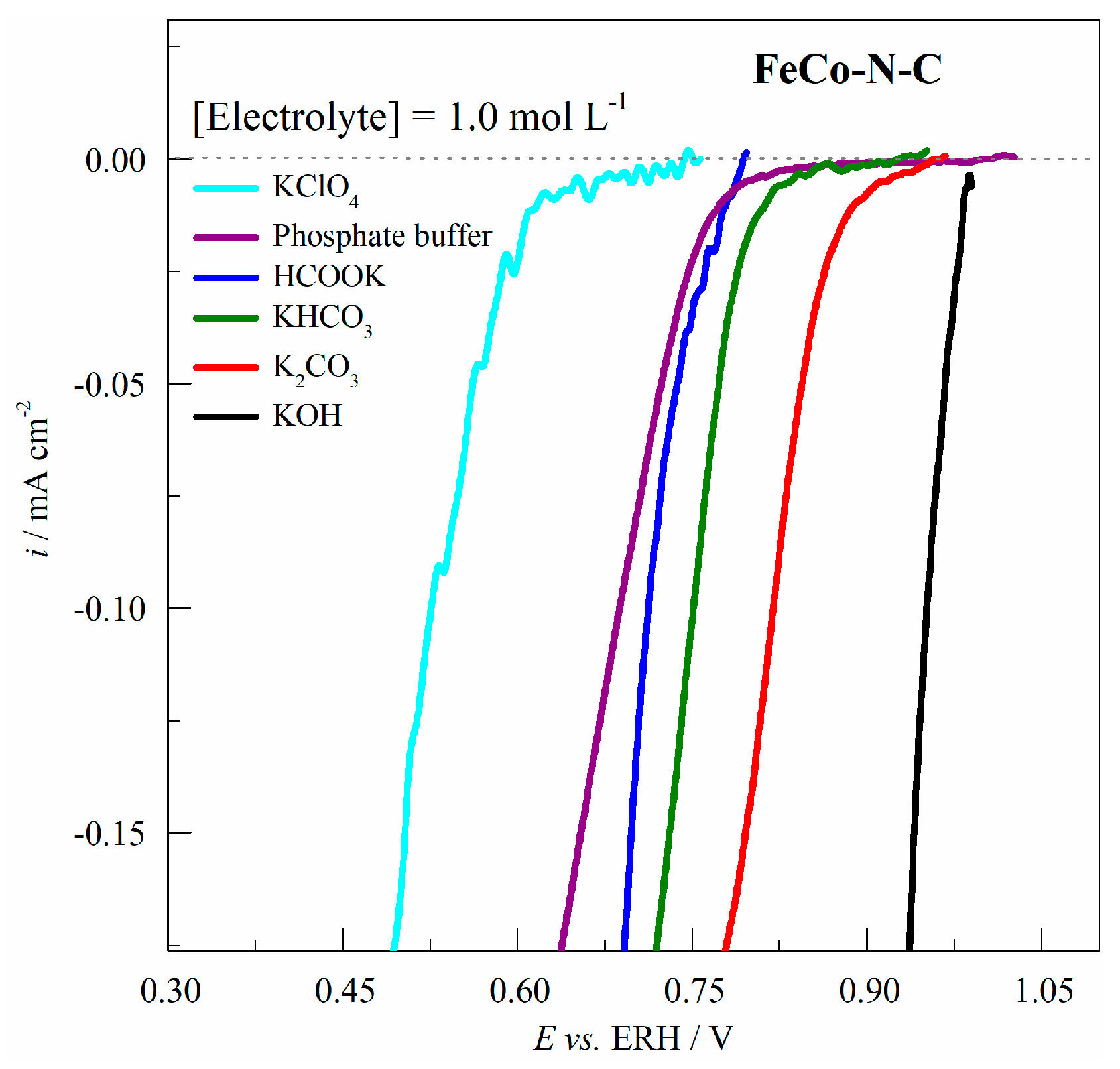
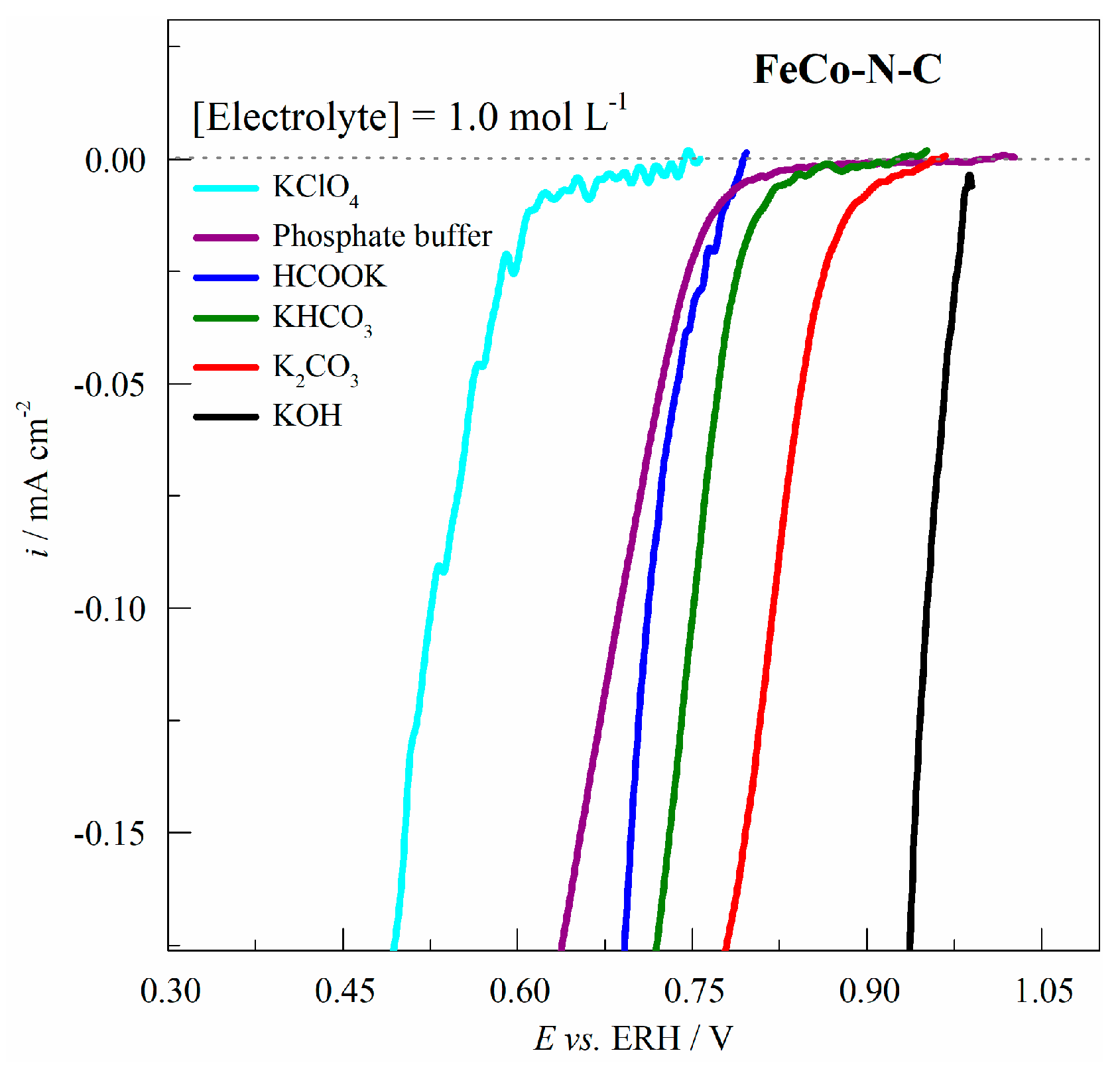
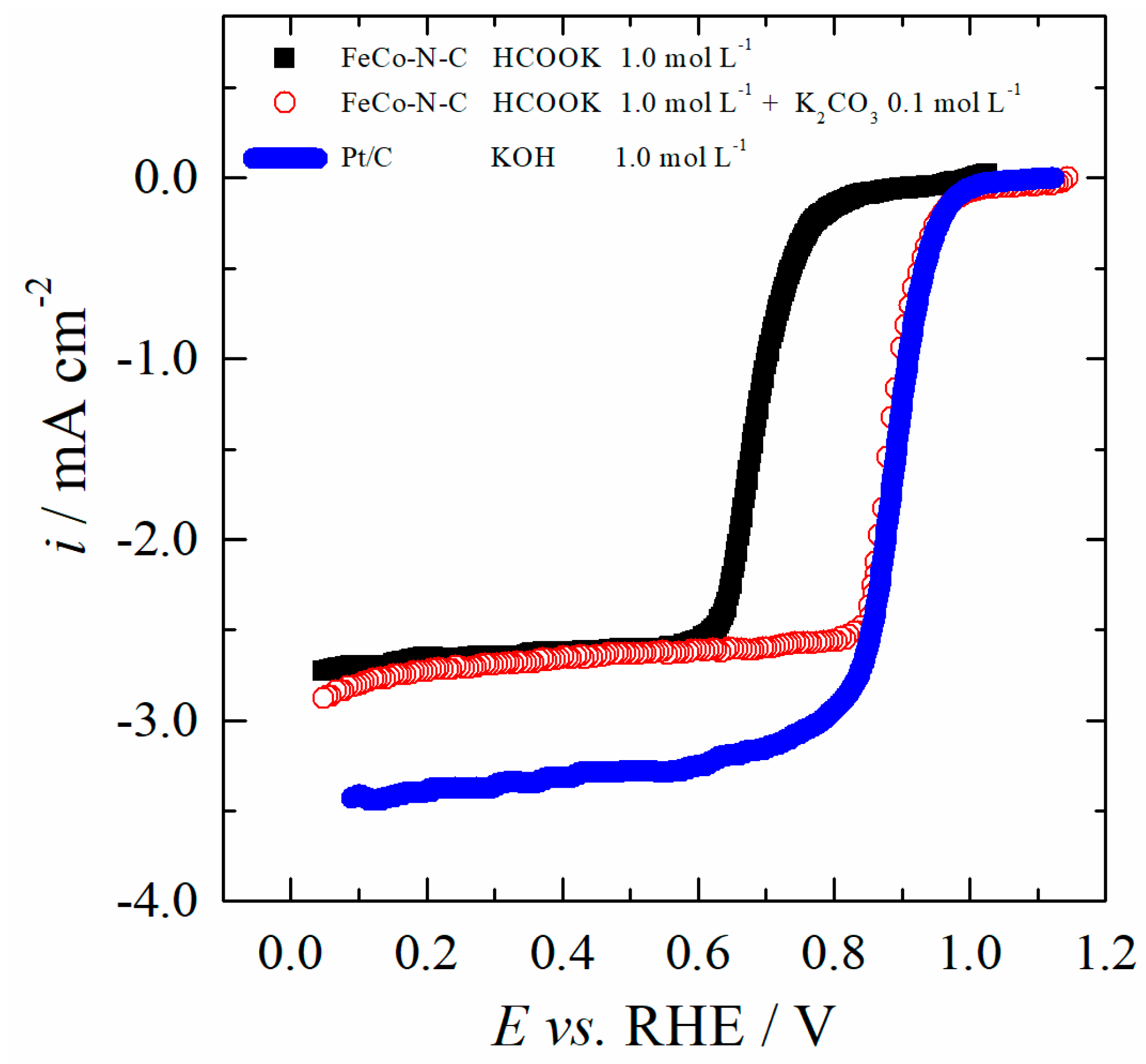
2.4. Electrocatalytic Activity of FeCo-N-C for the ORR in Different Electrolytes
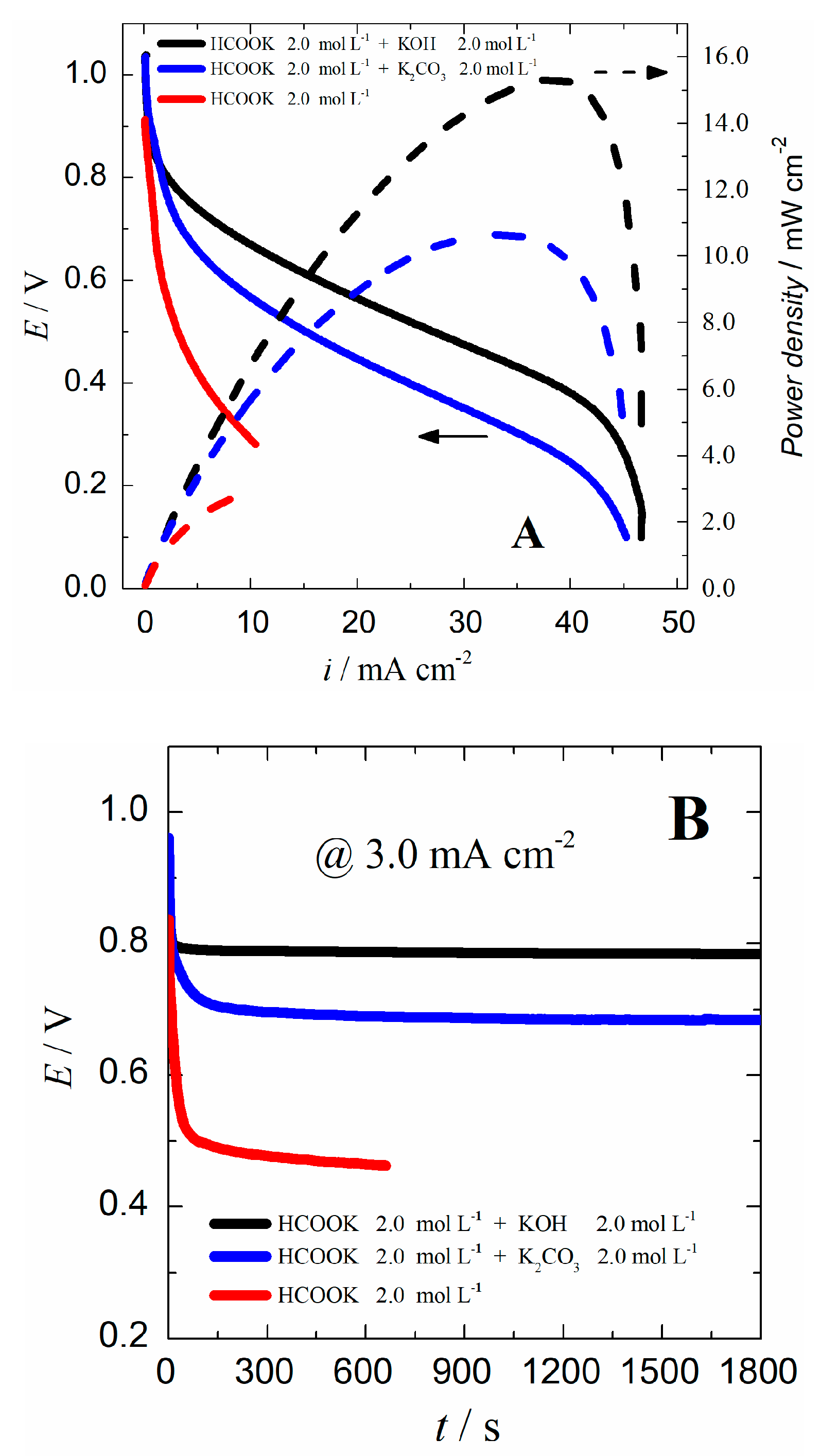
2.5. Activity and Stability in Formate/Air Single Cells
3. Experimental Section
3.1. Reactants and Materials
3.2. Electrocatalysts under the Focus of this Study—Fe-N-C, Co-N-C e FeCo-N-C
3.3. Materials Synthesized for Comparison: FeCoOx/C, FeCo@C, FexN and N-C
3.4. Synthesis of the Anode Electrocatalyst for the Single Cell Measurements
3.5. Electrocatalyst Characterization
3.6. Electrochemical Measurements
3.7. Measurements in Single Cells
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Ghoniem, A.F. Needs, resources and climate change: Clean and efficient conversion technologies. Prog. Energy Combust. Sci. 2011, 37, 15–51. [Google Scholar] [CrossRef]
- Taberner, P.; Heitbaum, J.; Vielstich, W. The influence of the electrolyte composition on the formate oxidation in alkaline formate-air fuel cells. Electrochim. Acta 1976, 21, 439–440. [Google Scholar] [CrossRef]
- Jiang, J.; Wieckowski, A. Prospective direct formate fuel cell. Electrochem. Commun. 2012, 18, 41–43. [Google Scholar] [CrossRef]
- Vo, T.; Purohit, K.; Nguyen, C.; Biggs, B.; Mayoral, S.; Haan, J.L. Formate: An Energy Storage and Transport Bridge between Carbon Dioxide and a Formate Fuel Cell in a Single Device. ChemSusChem 2015, 8, 3853–3858. [Google Scholar] [CrossRef] [PubMed]
- Sandstede, G.; Cairns, E.J.; Bagotsky, V.S.; Wiesener, K. History of Low Temperature Fuel Cells. In Handbook of Fuel Cells; John Wiley & Sons: Chichester, UK, 2010; ISBN 9780470974001. [Google Scholar]
- Nishimura, K.; Machida, K.; Enyo, M. Electrooxidation of formate and formaldehyde on electrodes of alloys between Pd and Group IB metals in alkaline media Part II. The possibility of complete oxidation of formaldehyde in weak alkali. J. Electroanal. Chem. Interfacial Electrochem. 1988, 251, 117–125. [Google Scholar] [CrossRef]
- Beden, B.; Lamy, C.; Leger, J.M. Electrocatalytic Activity of Noble Metals for the Oxidation of Formate in Neutral Medium. J. Electroanal. Chem. 1979, 101, 127–131. [Google Scholar] [CrossRef]
- John, J.; Wang, H.; Rus, E.D.; Abruña, H.D. Mechanistic studies of formate oxidation on platinum in alkaline medium. J. Phys. Chem. C 2012, 116, 5810–5820. [Google Scholar] [CrossRef]
- Perales-Rondón, J.V.; Brimaud, S.; Solla-Gullón, J.; Herrero, E.; Jürgen Behm, R.; Feliu, J.M. Further Insights into the Formic Acid Oxidation Mechanism on Platinum: PH and Anion Adsorption Effects. Electrochim. Acta 2015, 180, 479–485. [Google Scholar] [CrossRef]
- Joo, J.; Uchida, T.; Cuesta, A.; Koper, M.T.M.; Osawa, M. Importance of Acid–Base Equilibrium in Electrocatalytic Oxidation of Formic Acid on Platinum. J. Am. Chem. Soc. 2013, 135, 9991–9994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banham, D.; Ye, S.; Pei, K.; Ozaki, J.I.; Kishimoto, T.; Imashiro, Y. A review of the stability and durability of non-precious metal catalysts for the oxygen reduction reaction in proton exchange membrane fuel cells. J. Power Sources 2015, 285, 334–348. [Google Scholar] [CrossRef]
- Jasinski, R. A new fuel cell cathode catalyst. Nature 1964, 201, 1212–1213. [Google Scholar] [CrossRef]
- Unni, S.M.; Devulapally, S.; Karjule, N.; Kurungot, S. Graphene enriched with pyrrolic coordination of the doped nitrogen as an efficient metal-free electrocatalyst for oxygen reduction. J. Mater. Chem. 2012, 22, 23506–23513. [Google Scholar] [CrossRef]
- Wu, G.; More, K.L.; Johnston, C.M.; Zelenay, P. High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron, and cobalt. Science 2011, 332, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Van Veen, J.A.R.; Colijn, H.A.; van Baar, J.F. On the effect of a heat treatment on the structure of carbon-supported metalloporphyrins and phthalocyanines. Electrochim. Acta 1988, 33, 801–804. [Google Scholar] [CrossRef]
- Mamtani, K.; Singh, D.; Tian, J.; Millet, J.-M.M.; Miller, J.T.; Co, A.C.; Ozkan, U.S. Evolution of N-Coordinated Iron–Carbon (FeNC) Catalysts and Their Oxygen Reduction (ORR) Performance in Acidic Media at Various Stages of Catalyst Synthesis: An Attempt at Benchmarking. Catal. Lett. 2016, 146, 1749–1770. [Google Scholar] [CrossRef]
- Bezerra, C.W.B.; Zhang, L.; Lee, K.; Liu, H.; Marques, A.L.B.; Marques, E.P.; Wang, H.; Zhang, J. A review of Fe-N/C and Co-N/C catalysts for the oxygen reduction reaction. Electrochim. Acta 2008, 53, 4937–4951. [Google Scholar] [CrossRef]
- Wu, G.; Santandreu, A.; Kellogg, W.; Gupta, S.; Ogoke, O.; Zhang, H.; Wang, H.L.; Dai, L. Carbon nanocomposite catalysts for oxygen reduction and evolution reactions: From nitrogen doping to transition-metal addition. Nano Energy 2016, 29, 83–110. [Google Scholar] [CrossRef]
- Sun, M.; Liu, H.; Qu, J.; Li, J. Earth-Rich Transition Metal Phosphide for Energy Conversion and Storage. Adv. Energy Mater. 2016, 6, 1–34. [Google Scholar] [CrossRef]
- Sun, M.; Davenport, D.; Liu, H.; Qu, J.; Elimelech, M.; Li, J. Highly efficient and sustainable non-precious-metal Fe–N–C electrocatalysts for the oxygen reduction reaction. J. Mater. Chem. A 2018, 6, 2527–2539. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, L.; Liu, X.; Liu, X.; Yang, X.; Miao, S.; Wang, W.; Wang, A.; Zhang, T. Discriminating catalytically active FeNx species of atomically dispersed Fe-N-C catalyst for selective oxidation of C-H bond. J. Am. Chem. Soc. 2017, 139, 10790–10798. [Google Scholar] [CrossRef] [PubMed]
- Strickland, K.; Miner, E.; Jia, Q.; Tylus, U.; Ramaswamy, N.; Liang, W.; Sougrati, M.-T.T.; Jaouen, F.F.; Mukerjee, S. Highly active oxygen reduction non-platinum group metal electrocatalyst without direct metal-nitrogen coordination. Nat. Commun. 2015, 6, 7343. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yang, Y.; Liu, X.; Pu, Z.; Kou, Z.; Zhu, P.; Mu, S. The role of iron nitrides in the Fe-N-C catalysis system towards the oxygen reduction reaction. Nanoscale 2017, 9, 7641–7649. [Google Scholar] [CrossRef] [PubMed]
- Noh, S.H.; Seo, M.H.; Kang, J.; Okajima, T.; Han, B.; Ohsaka, T. Towards a comprehensive understanding of FeCo coated with N-doped carbon as a stable bi-functional catalyst in acidic media. NPG Asia Mater. 2016, 8. [Google Scholar] [CrossRef]
- Toh, R.J.; Sofer, Z.; Pumera, M. Transition Metal Oxides for the Oxygen Reduction Reaction: Influence of the Oxidation States of the Metal and its Position on the Periodic Table. Chemphyschem 2015, 16, 3527–3531. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Baldizzone, C.; Polymeros, G.; Pizzutilo, E.; Kasian, O.; Schuppert, A.K.; Ranjbar Sahraie, N.; Sougrati, M.-T.; Mayrhofer, K.J.J.; Jaouen, F. Minimizing Operando Demetallation of Fe-N-C Electrocatalysts in Acidic Medium. ACS Catal. 2016, 6, 3136–3146. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, H.; Zhong, H.; Xu, T.; Jin, H.; Tang, Y.; Xu, Z. Cobalt based non-precious electrocatalysts for oxygen reduction reaction in proton exchange membrane fuel cells. Electrochim. Acta 2010, 55, 7945–7950. [Google Scholar] [CrossRef]
- Choi, C.H.; Baldizzone, C.; Grote, J.-P.P.; Schuppert, A.K.; Jaouen, F.; Mayrhofer, K.J.J. Stability of Fe-N-C Catalysts in Acidic Medium Studied by Operando Spectroscopy. Angew. Chemie Int. Ed. 2015, 54, 12753–12757. [Google Scholar] [CrossRef] [PubMed]
- Roncaroli, F.; Dal Molin, E.S.; Viva, F.A.; Bruno, M.M.; Halac, E.B. Cobalt and iron complexes with N-heterocyclic ligands as pyrolysis precursors for oxygen reduction catalysts. Electrochim. Acta 2015, 174, 66–77. [Google Scholar] [CrossRef]
- Armel, V.; Hindocha, S.; Salles, F.; Bennett, S.; Jones, D.; Jaouen, F. Structural descriptors of zeolitic-lmidazolate frameworks are keys to the activity of Fe-N-C catalysts. J. Am. Chem. Soc. 2017, 139, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; More, K.L.; Xu, P.; Wang, H.-L.; Ferrandon, M.; Kropf, A.J.; Myers, D.J.; Ma, S.; Johnston, C.M.; Zelenay, P. A carbon-nanotube-supported graphene-rich non-precious metal oxygen reduction catalyst with enhanced performance durability. Chem. Commun. 2013, 49, 3291–3293. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Leonard, N.; Barton, S.C. Impact of transition metal on nitrogen retention and activity of iron-nitrogen-carbon oxygen reduction catalysts. Phys. Chem. Chem. Phys. 2014, 16, 4576–4585. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Tian, J.; Mamtani, K.; King, J.; Miller, J.T.; Ozkan, U.S. A comparison of N-containing carbon nanostructures (CNx) and N-coordinated iron–carbon catalysts (FeNC) for the oxygen reduction reaction in acidic media. J. Catal. 2014, 317, 30–43. [Google Scholar] [CrossRef]
- Zhang, G.; Chenitz, R.; Lefèvre, M.; Sun, S.; Dodelet, J.-P. Is iron involved in the lack of stability of Fe/N/C electrocatalysts used to reduce oxygen at the cathode of PEM fuel cells? Nano Energy 2016, 29, 111–125. [Google Scholar] [CrossRef]
- Jia, Q.; Ramaswamy, N.; Tylus, U.; Strickland, K.; Li, J.; Serov, A.; Artyushkova, K.; Atanassov, P.; Anibal, J.; Gumeci, C.; et al. Spectroscopic insights into the nature of active sites in iron–nitrogen–carbon electrocatalysts for oxygen reduction in acid. Nano Energy 2016, 29, 65–82. [Google Scholar] [CrossRef]
- Tylus, U.; Jia, Q.; Strickland, K.; Ramaswamy, N.; Serov, A.; Atanassov, P.; Mukerjee, S. Elucidating Oxygen Reduction Active Sites in Pyrolyzed Metal–Nitrogen Coordinated Non-Precious-Metal Electrocatalyst Systems. J. Phys. Chem. C 2014, 118, 8999–9008. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.-J.; Gu, L.; Li, L.; Zhang, Y.; Zhang, X.; Zhang, L.-J.; Wang, J.-Q.; Hu, J.-S.; Wei, Z.; Wan, L.-J. Understanding the High Activity of Fe–N–C Electrocatalysts in Oxygen Reduction: Fe/Fe3C Nanoparticles Boost the Activity of Fe–Nx. J. Am. Chem. Soc. 2016, 138, 3570–3578. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.T.; Cullen, D.A.; Higgins, D.; Sneed, B.T.; Holby, E.F.; More, K.L.; Zelenay, P. Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst. Science 2017, 357, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Tang, Z.K.; Zhao, T.S. A high-performance alkaline exchange membrane direct formate fuel cell. Appl. Energy 2014, 115, 405–410. [Google Scholar] [CrossRef]
- Sun, T.; Jiang, Y.; Wu, Q.; Du, L.; Zhang, Z.; Yang, L.; Wang, X.; Hu, Z. Is iron nitride or carbide highly active for oxygen reduction reaction in acidic medium? Catal. Sci. Technol. 2017, 7, 51–55. [Google Scholar] [CrossRef]
- Yu, X.; Manthiram, A. Catalyst-selective, scalable membraneless alkaline direct formate fuel cells. Appl. Catal. B Environ. 2015, 165, 63–67. [Google Scholar] [CrossRef]
- Yuan, W.; Xu, X.; Han, F.; Ye, G.; Tang, Y. Developing a passive air-breathing tubular direct methanol fuel cell fed with concentrated methanol. Int. J. Green Energy 2017, 13, 1100–1109. [Google Scholar] [CrossRef]
- Chen, C.Y.; Yang, P. Performance of an air-breathing direct methanol fuel cell. J. Power Sources 2003, 123, 37–42. [Google Scholar] [CrossRef]
- Queiroz, A.C.; Lima, F.H.B. Electrocatalytic activity and stability of Co and Mn-based oxides for the oxygen reduction reaction in alkaline electrolyte. J. Electroanal. Chem. 2013, 707, 142–150. [Google Scholar] [CrossRef]
- Fu, X.; Liu, Y.; Cao, X.; Jin, J.; Liu, Q.; Zhang, J. FeCo-Nxembedded graphene as high performance catalysts for oxygen reduction reaction. Appl. Catal. B Environ. 2013, 130–131, 143–151. [Google Scholar] [CrossRef]
- Fang, X.; Jiao, L.; Yu, S.H.; Jiang, H.L. Metal–Organic Framework-Derived FeCo-N-Doped Hollow Porous Carbon Nanocubes for Electrocatalysis in Acidic and Alkaline Media. ChemSusChem 2017, 10, 3019–3024. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yang, D.S.; Kweun, J.M.; Li, C.; Tan, K.; Kong, F.; Liang, C.; Chabal, Y.J.; Kim, Y.Y.; Cho, M.; et al. Rational design of common transition metal-nitrogen-carbon catalysts for oxygen reduction reaction in fuel cells. Nano Energy 2016, 30, 443–449. [Google Scholar] [CrossRef]
- Yang, H.B.; Miao, J.; Hung, S.-F.; Chen, J.; Tao, H.B.; Wang, X.; Zhang, L.; Chen, R.; Gao, J.; Chen, H.M.; et al. Identification of catalytic sites for oxygen reduction and oxygen evolution in N-doped graphene materials: Development of highly efficient metal-free bifunctional electrocatalyst. Sci. Adv. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, N.; Mukerjee, S. Influence of Inner-and Outer-Sphere Electron Transfer Mechanisms during Electrocatalysis of Oxygen Reduction in Alkaline Media. J. Phys. Chem. C 2011, 115, 18015–18026. [Google Scholar] [CrossRef]
- Singh, M.R.; Papadantonakis, K.; Xiang, C.; Lewis, N.S. An electrochemical engineering assessment of the operational conditions and constraints for solar-driven water-splitting systems at near-neutral pH. Energy Environ. Sci. 2015, 8, 2760–2767. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.; Vielstich, W. Einfluß von Edelmetall-Mischkatalysatoren auf die anodische Oxydation von Methanol und Formiat. Fresenius Z. Anal. Chem. 1967, 224, 84–93. [Google Scholar] [CrossRef]
- Lima, F.H.B.; Profeti, D.; Chatenet, M.; Riello, D.; Ticianelli, E.A.; Gonzalez, E.R. Electro-oxidation of Ethanol on Rh/Pt and Ru/Rh/Pt Sub-monolayers Deposited on Au/C Nanoparticles. Electrocatalysis 2010, 1, 72–82. [Google Scholar] [CrossRef]
- Kristian, N.; Wang, X. Ptshell-Aucore/C electrocatalyst with a controlled shell thickness and improved Pt utilization for fuel cell reactions. Electrochem. Commun. 2008, 10, 12–15. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Gasteiger, H.A.; Stäb, G.D.; Urban, P.M.; Kolb, D.M.; Behm, R.J. Characterization of High-Surface-Area Electrocatalysts Using a Rotating Disk Electrode Configuration. J. Electrochem. Soc. 1998, 145, 2354–2358. [Google Scholar] [CrossRef]
- Gulzow, E.; Giilzow, E. Alkaline fuel cells: A critical view. J. Power Sources 1996, 61, 99–104. [Google Scholar] [CrossRef]
- Paganin, V.A.; Ticianelli, E.A.; Gonzalez, E.R. Development and electrochemical studies of gas diffusion electrodes for polymer electrolyte fuel cells. J. Appl. Electrochem. 1996, 26, 297–304. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrocatalyst | Composition (wt.%) | ||
|---|---|---|---|
| Fe | Co | C | |
| FeCo-N-C | 5.0 | 4.6 | 90.4 |
| FeCo-N-C AT | 3.1 | 2.8 | 94.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
E. R. Oliveira, F.; A. Galiote, N.; H. B. Lima, F. Investigation of Earth-Abundant Oxygen Reduction Electrocatalysts for the Cathode of Passive Air-Breathing Direct Formate Fuel Cells. Catalysts 2018, 8, 320. https://doi.org/10.3390/catal8080320
E. R. Oliveira F, A. Galiote N, H. B. Lima F. Investigation of Earth-Abundant Oxygen Reduction Electrocatalysts for the Cathode of Passive Air-Breathing Direct Formate Fuel Cells. Catalysts. 2018; 8(8):320. https://doi.org/10.3390/catal8080320
Chicago/Turabian StyleE. R. Oliveira, Francisca, Nelson A. Galiote, and Fabio H. B. Lima. 2018. "Investigation of Earth-Abundant Oxygen Reduction Electrocatalysts for the Cathode of Passive Air-Breathing Direct Formate Fuel Cells" Catalysts 8, no. 8: 320. https://doi.org/10.3390/catal8080320