Plasma-Catalytic Mineralization of Toluene Adsorbed on CeO2



Abstract
1. Introduction
2. Results and Discussion
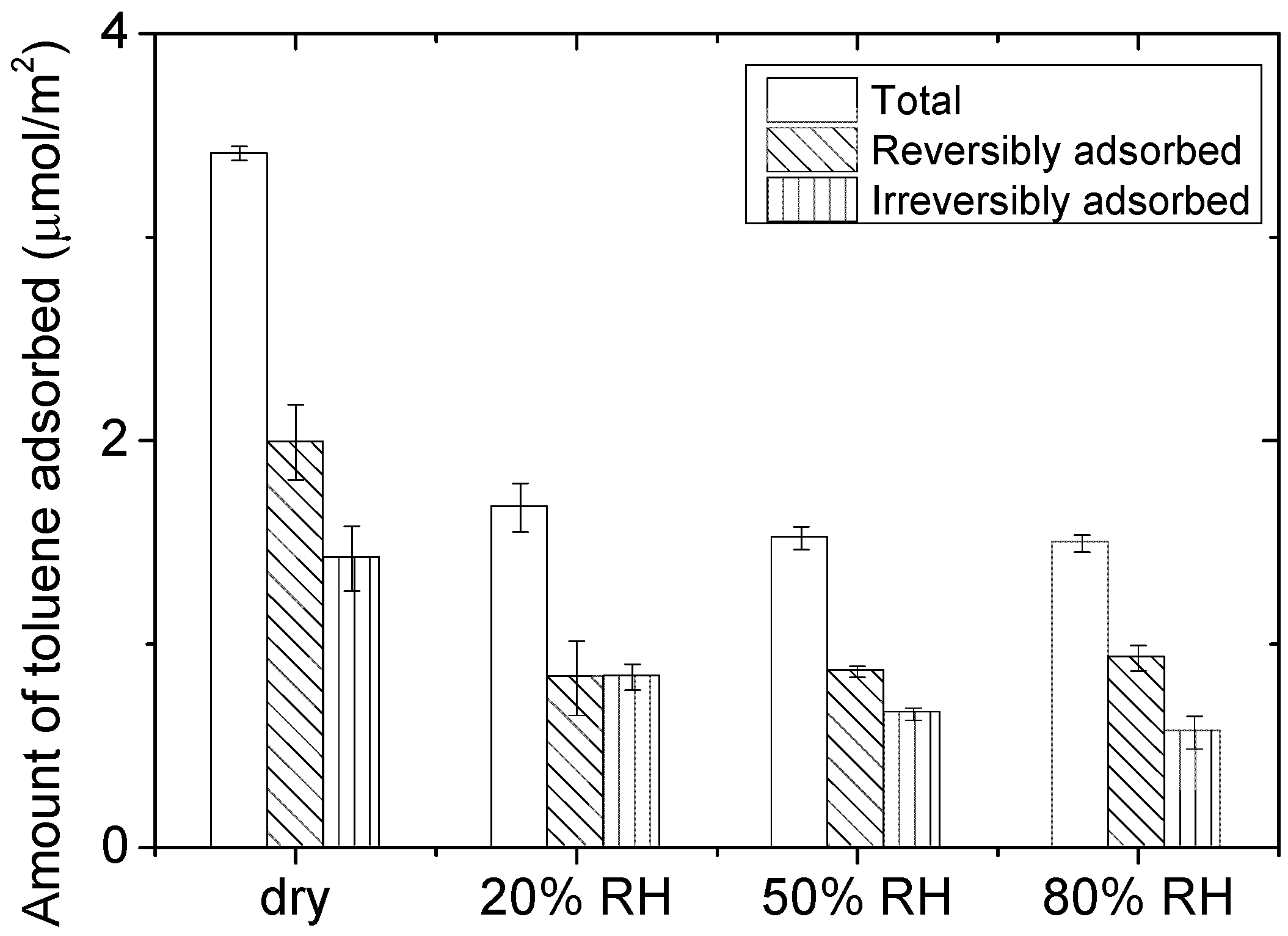
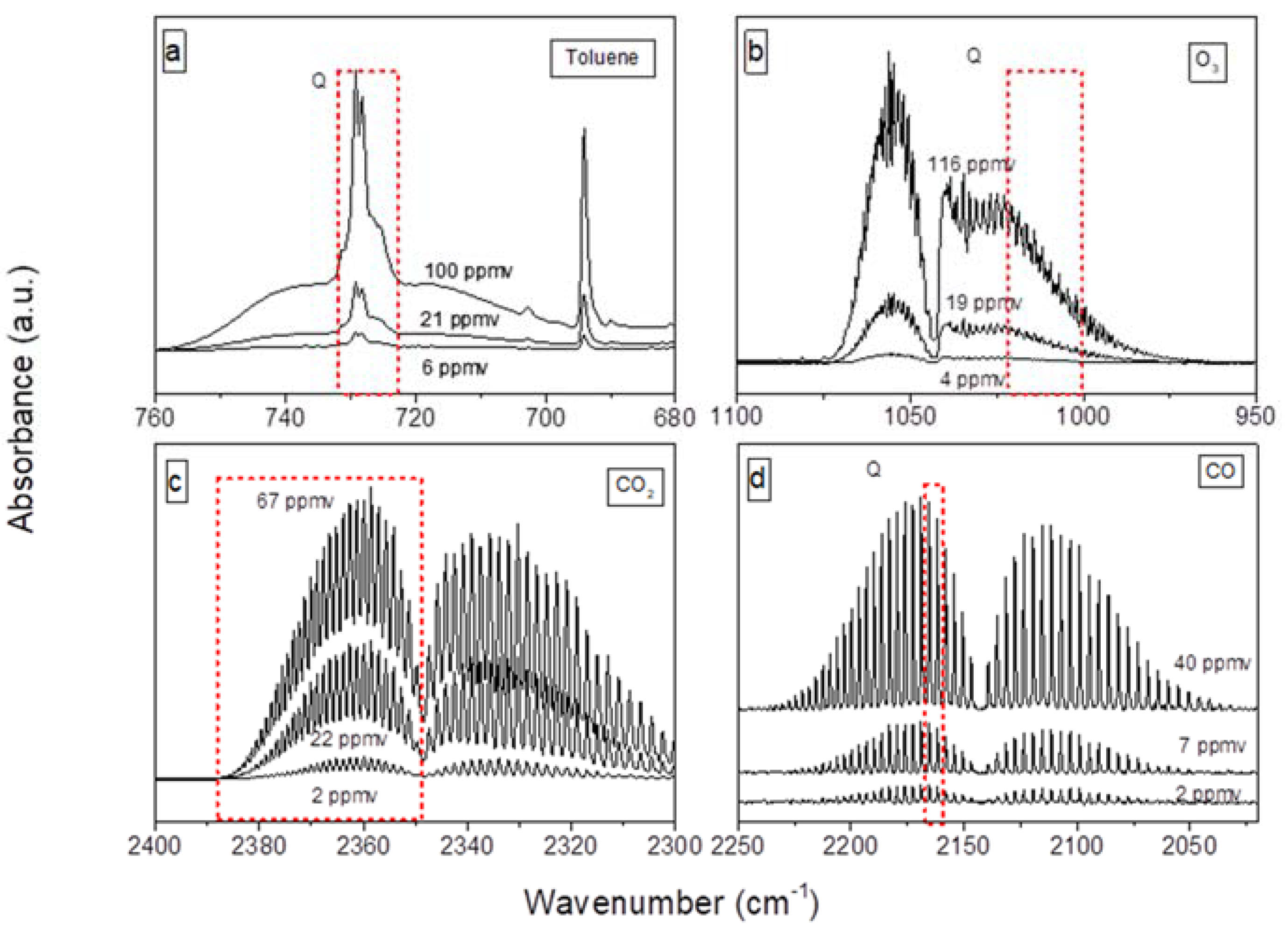
2.1. Characterization of the Adsorption of 100 ppm of Toluene on CeO2
2.2. Comparison of PPC and IPC Performances under Dry Air Conditions
2.2.1. Performance Criteria
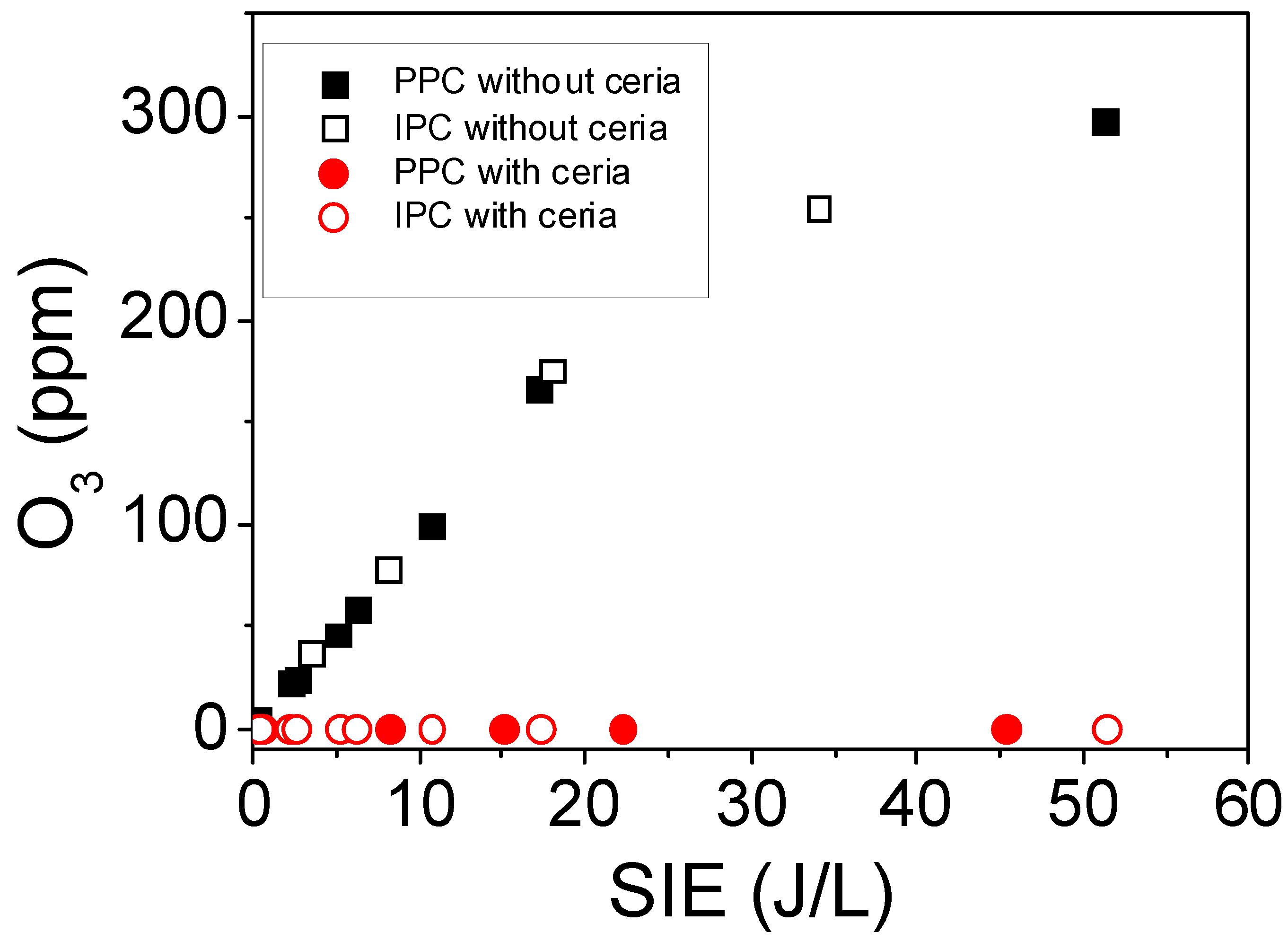
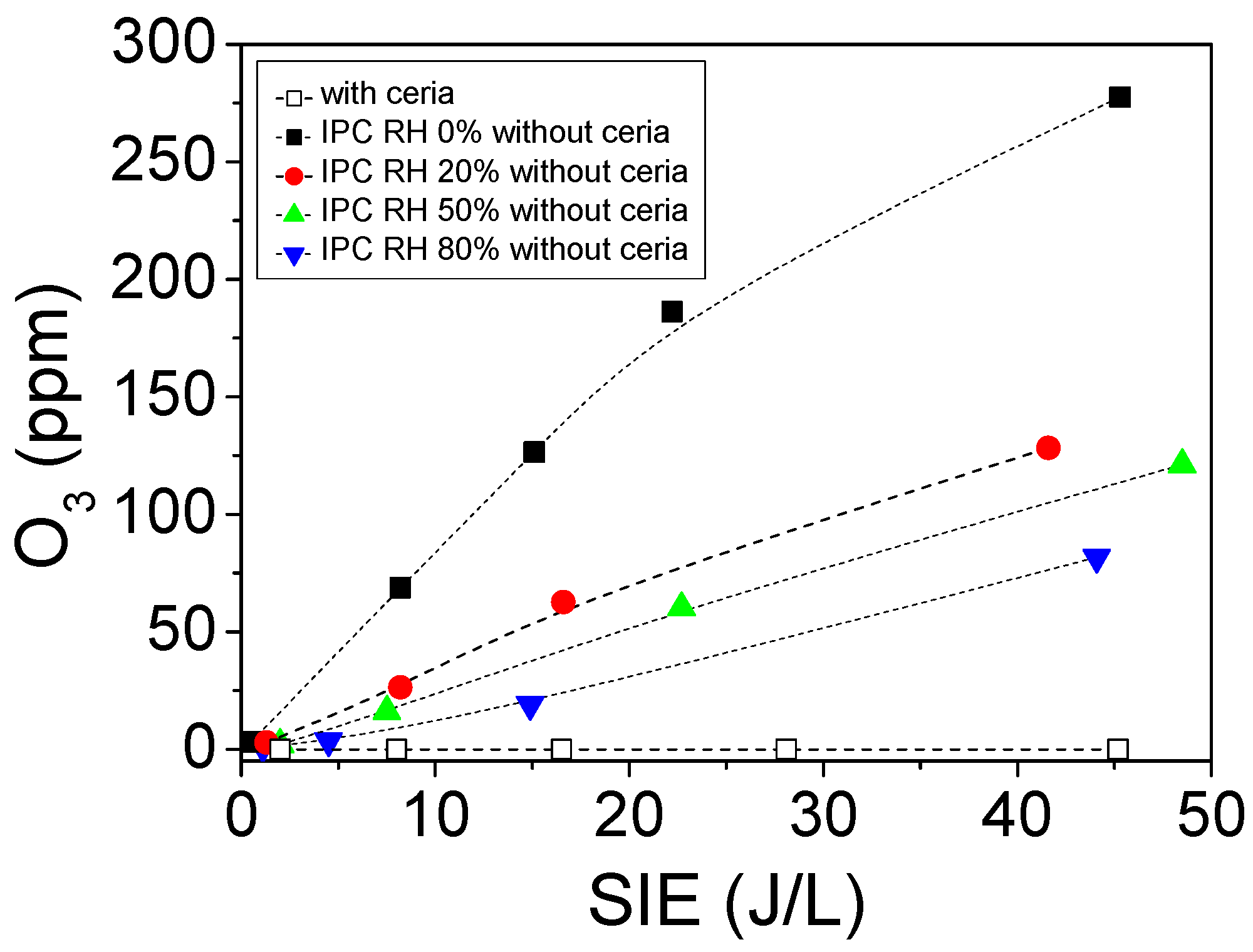
2.2.2. Formation of Ozone Using PPC and IPC Configurations
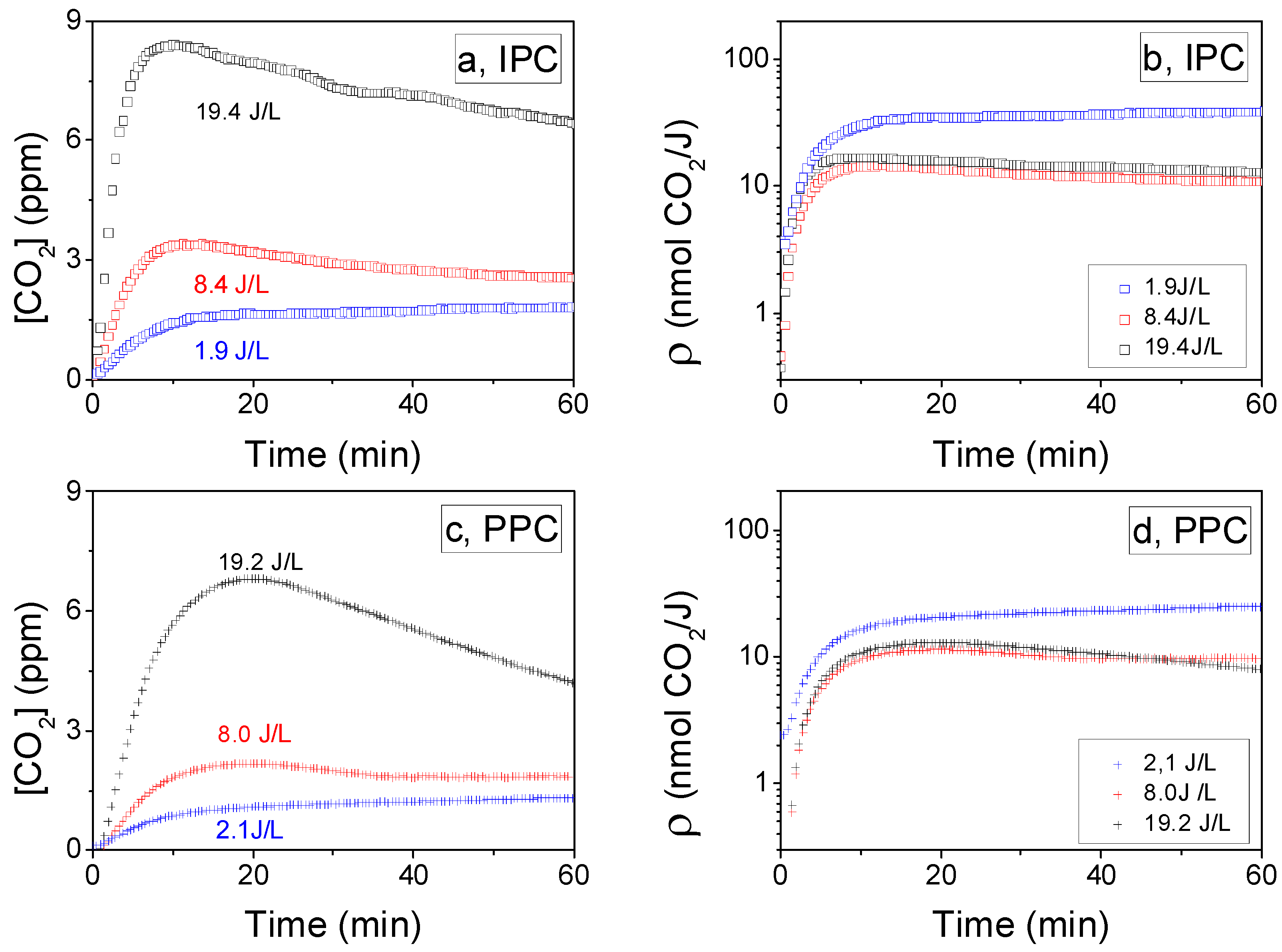
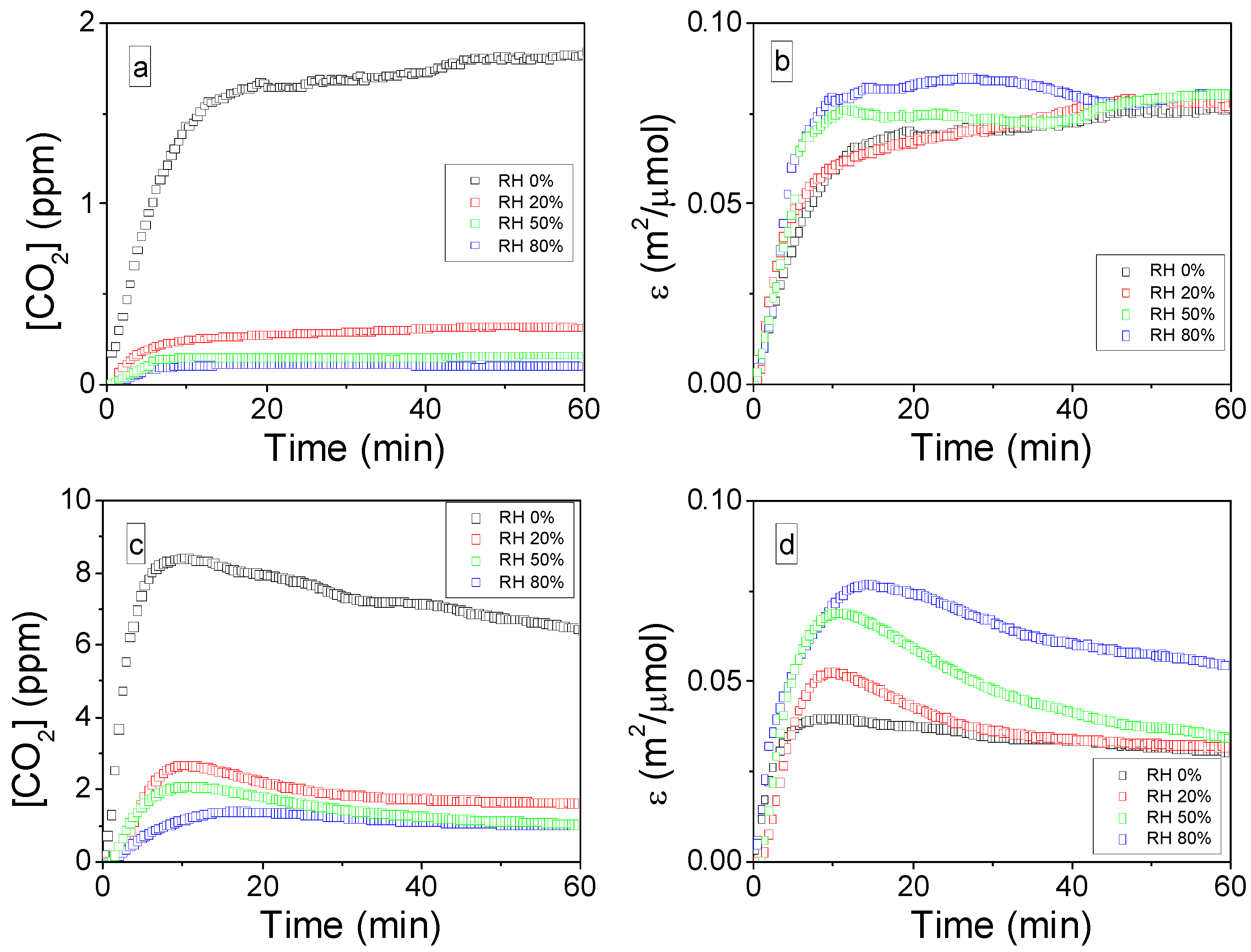
2.2.3. Influence of the SIE on CO2 Formation Yield: ρ (t)
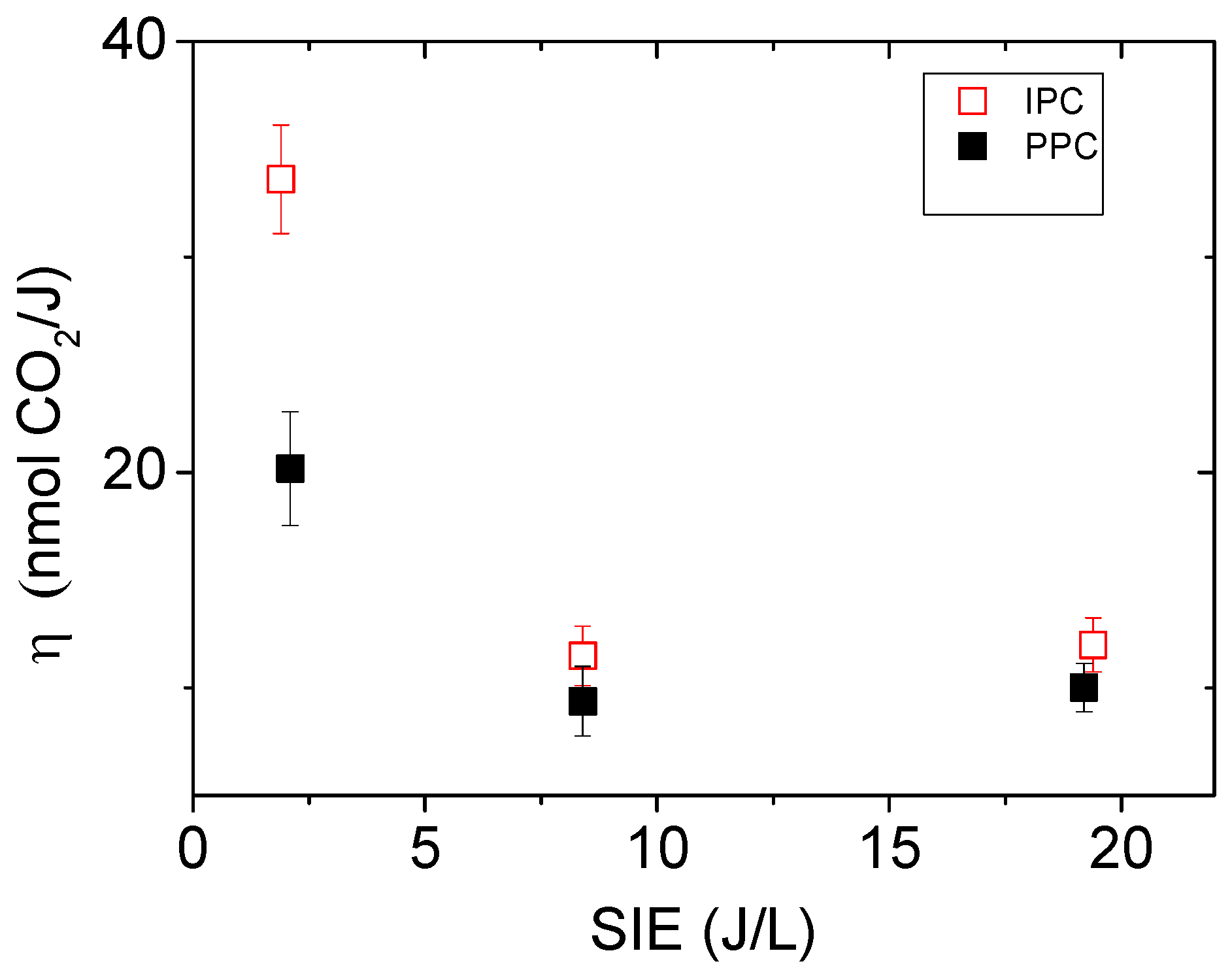
2.2.4. Influence of the SIE on the Mineralization Efficiency: η
2.3. Influence Relative Humidity on the Performances of IPC
2.3.1. Influence of Relative Humidity on O3 Formation
2.3.2. Influence of Relative Humidity on the Mineralization of Adsorbed Toluene
3. Materials and Methods
3.1. Gas Flow Preparation
3.2. Materials
3.3. FTIR Monitoring
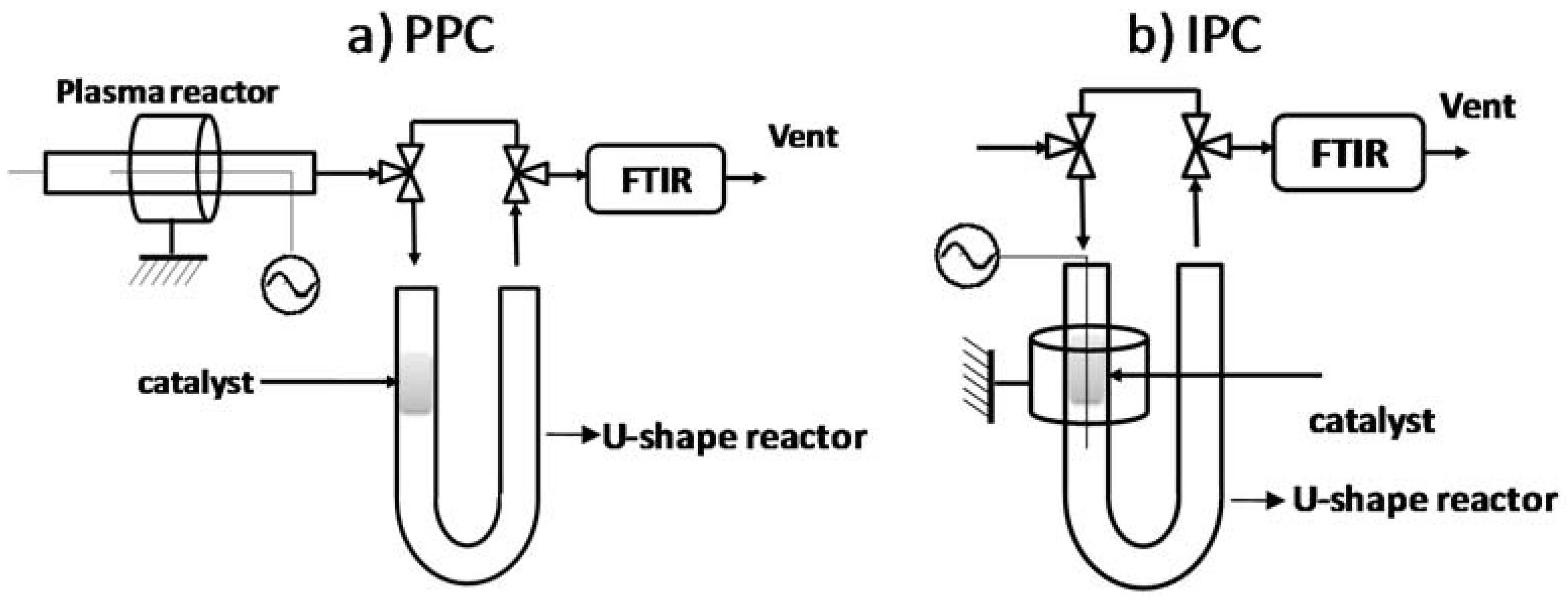
3.4. Post vs in Plasma–Catalytic Reactors
3.4.1. Post Plasma Reactor–PPC
3.4.2. In Plasma Reactor—IPC
3.5. Typical Experimental Protocole
- Pretreatment of CeO2 sample under dry air at 400 °C to remove water and other adsorbed species and ensure repeatability of experiments.
- Adsorption of 100 ppm toluene on CeO2 until breakthrough: the air flow with different RH levels (0–80%) containing toluene breaks through the CeO2 bed and is gradually adsorbed on the catalyst surface until equilibration of the sorption sites.
- Flushing of CeO2 under synthetic dry air flow to remove the reversibly adsorbed fraction of toluene. Flushing the sorbent bed under air desorbs the molecules with the weakest heats of adsorption, that is, physisorbed species, leaving only the irreversibly adsorbed toluene molecules on CeO2 surface.
- Surface exposure under air flow with different RH levels (0–80%) by switching on the nonthermal plasma for 60 minutes for IPC or PPC. Once the plasma is turned off, the system is purged with synthetic air flow until ozone concentration returns to zero.
- Temperature programmed oxidation (TPO) under synthetic air flow with different RH levels (0–80%) at 400 °C is performed in order to remove the remaining adsorbed species and regenerate the CeO2 surface.
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Theloke, J.; Friedrich, R. Compilation of a database on the composition of anthropogenic VOC emissions for atmospheric modeling in Europe. Atmos. Environ. 2007, 41, 4148–4160. [Google Scholar] [CrossRef]
- Coates, J.D.; Chakraborty, R.; Lack, J.G.; O’Connor, S.M.; Cole, K.A.; Bender, K.S.; Achenbach, L.A. Anaerobic benzene oxidation coupled to nitrate reduction in pure culture by two strains of Dechloromonas. Nature 2001, 411, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Benignus, V.A. Health effects of toluene: A review. Neurotoxicology 1981, 2, 567–588. [Google Scholar] [PubMed]
- Fishbein, L. An overview of environmental and toxicological aspects of aromatic hydrocarbons II. Toluene. Sci. Total Environ. 1985, 42, 267–288. [Google Scholar] [CrossRef]
- Lebret, E.; Van de Wiel, H.J.; Bos, H.P.; Noij, D.; Boleij, J.S.M. Volatile organic compounds in Dutch homes. Environ. Int. 1986, 12, 323–332. [Google Scholar] [CrossRef]
- Feng, X.; Liu, H.; He, C.; Shen, Z.; Wang, T. Synergistic effects and mechanism of a non-thermal plasma catalysis system in volatile organic compound removal: A review. Catal. Sci. Technol. 2018, 8, 936–954. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, S.; Yang, Y.; Zheng, C.; Zhou, J.; Gao, X.; Tu, X. Enhanced performance for plasma-catalytic oxidation of ethyl acetate over La1−xCexCoO3+δ catalysts. Appl. Catal. B Environ. 2017, 213, 97–105. [Google Scholar] [CrossRef]
- Wang, W.; Kim, H.-H.; Van Laer, K.; Bogaerts, A. Streamer propagation in a packed bed plasma reactor for plasma catalysis applications. Chem. Eng. J. 2018, 334, 2467–2479. [Google Scholar] [CrossRef]
- Veerapandian, S.; Leys, C.; De Geyter, N.; Morent, R. Abatement of VOCs Using Packed Bed Non-Thermal Plasma Reactors: A Review. Catalysts 2017, 7, 113. [Google Scholar] [CrossRef]
- Ye, Z.; Giraudon, J.-M.; De Geyter, N.; Morent, R.; Lamonier, J.-F. The Design of MnOx Based Catalyst in Post-Plasma Catalysis Configuration for Toluene Abatement. Catalysts 2018, 8. [Google Scholar] [CrossRef]
- Xu, X.; Wang, P.; Xu, W.; Wu, J.; Chen, L.; Fu, M.; Ye, D. Plasma-catalysis of metal loaded SBA-15 for toluene removal: Comparison of continuously introduced and adsorption-discharge plasma system. Chem. Eng. J. 2016, 283, 276–284. [Google Scholar] [CrossRef]
- Thevenet, F.; Sivachandiran, L.; Guaitella, O.; Barakat, C.; Rousseau, A. Plasma-catalyst coupling for volatile organic compound removal and indoor air treatment: A review. J. Phys. D Appl. Phys. 2014, 47. [Google Scholar] [CrossRef]
- Jia, Z.; Ben Amar, M.; Yang, D.; Brinza, O.; Kanaev, A.; Duten, X.; Vega-González, A. Plasma catalysis application of gold nanoparticles for acetaldehyde decomposition. Chem. Eng. J. 2018, 347, 913–922. [Google Scholar] [CrossRef]
- Jia, Z.; Vega-Gonzalez, A.; Amar, M.B.; Hassouni, K.; Tieng, S.; Touchard, S.; Kanaev, A.; Duten, X. Acetaldehyde removal using a diphasic process coupling a silver-based nano-structured catalyst and a plasma at atmospheric pressure. Catal. Today 2013, 208, 82–89. [Google Scholar] [CrossRef]
- Jia, Z.; Barakat, C.; Dong, B.; Rousseau, A. VOCs Destruction by Plasma Catalyst Coupling Using AL-KO PURE Air Purifier on Industrial Scale. J. Mater. Sci. Chem. Eng. 2015, 3, 19–26. [Google Scholar] [CrossRef]
- Vandenbroucke, A.M.; Morent, R.; De Geyter, N.; Leys, C. Non-thermal plasmas for non-catalytic and catalytic VOC abatement. J. Hazard. Mater. 2011, 195, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Christensen, P.A.; Mashhadani, Z.T.A.W.; Md Ali, A.H.B.; Carroll, M.A.; Martin, P.A. The Production of Methane, Acetone, “Cold” CO and Oxygenated Species from IsoPropyl Alcohol in a Non-Thermal Plasma: An In-Situ FTIR Study. J. Phys. Chem. A 2018, 122, 4273–4284. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.L.; Lee, H.M.; Chen, S.H.; Chang, M.B.; Yu, S.J.; Li, S.N. Removal of Volatile Organic Compounds by Single-Stage and Two-Stage Plasma Catalysis Systems: A Review of the Performance Enhancement Mechanisms, Current Status, and Suitable Applications. Environ. Sci. Technol. 2009, 43, 2216–2227. [Google Scholar] [CrossRef] [PubMed]
- Norsic, C.; Tatibouët, J.-M.; Batiot-Dupeyrat, C.; Fourré, E. Methanol oxidation in dry and humid air by dielectric barrier discharge plasma combined with MnO2-CuO based catalysts. Chem. Eng. J. 2018, 347, 944–952. [Google Scholar] [CrossRef]
- Ogata, A.; Einaga, H.; Kabashima, H.; Futamura, S.; Kushiyama, S.; Kim, H.-H. Effective Combination of Nonthermal Plasma and Catalysts for Decomposition of Benzene in Air. Appl. Catal. B Environ. 2003, 46, 87–95. [Google Scholar] [CrossRef]
- Hammer, T.; Kappes, T.; Baldauf, M. Plasma catalytic hybrid processes: Gas discharge initiation and plasma activation of catalytic processes. Catal. Today 2004, 89, 5–14. [Google Scholar] [CrossRef]
- Kirkpatrick, M.J.; Finney, W.C.; Locke, B.R. Plasma-Catalyst Interactions in the Treatment of Volatile Organic Compounds and NOx with Pulsed Corona Discharge and Reticulated Vitreous Carbon Pt/Rh-Coated Electrodes. Catal. Today 2004, 89, 117–126. [Google Scholar] [CrossRef]
- Ayrault, C.; Barrault, J.; Blin-Simiand, N.; Jorand, F.; Pasquiers, S.; Rousseau, A.; Tatibouët, J.M. Oxidation of 2-heptanone in air by a DBD-type plasma generated within a honeycomb monolith supported Pt-based catalyst. Catal. Today 2004, 89, 75–81. [Google Scholar] [CrossRef]
- Du, C.M.; Yan, J.H.; Cheron, B. Decomposition of toluene in a gliding arc discharge plasma reactor. Plasma Sources Sci. Technol. 2007, 16, 791–797. [Google Scholar] [CrossRef]
- Guo, Y.; Ye, D.; Tian, Y.; Chen, K. Humidity Effect on Toluene Decomposition in a Wire-plate Dielectric Barrier Discharge Reactor. Plasma Chem. Plasma Process. 2006, 26, 237–249. [Google Scholar] [CrossRef]
- Van Durme, J.; Dewulf, J.; Demeestere, K.; Leys, C.; Van Langenhove, H. Post-plasma catalytic technology for the removal of toluene from indoor air: Effect of humidity. Appl. Catal. B Environ. 2009, 87, 78–83. [Google Scholar] [CrossRef]
- Thevenet, F.; Guaitella, O.; Puzenat, E.; Guillard, C.; Rousseau, A. Influence of water vapour on plasma/photocatalytic oxidation efficiency of acetylene. Appl. Catal. B Environ. 2008, 84, 813–820. [Google Scholar] [CrossRef]
- Chang, T.; Shen, Z.; Huang, Y.; Lu, J.; Ren, D.; Sun, J.; Cao, J.; Liu, H. Post-plasma-catalytic removal of toluene using MnO2–Co3O4 catalysts and their synergistic mechanism. Chem. Eng. J. 2018, 348, 15–25. [Google Scholar] [CrossRef]
- Batault, F.; Thevenet, F.; Hequet, V.; Rillard, C.; Le Coq, L.; Locoge, N. Acetaldehyde and acetic acid adsorption on TiO2 under dry and humid conditions. Chem. Eng. J. 2015, 264, 197–210. [Google Scholar] [CrossRef]
- Huang, H.; Ye, D. Combination of photocatalysis downstream the non-thermal plasma reactor for oxidation of gas-phase toluene. J. Hazard. Mater. 2009, 171, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Wang, X.; Thevenet, F.; Rousseau, A. Dynamic probing of plasma-catalytic surface processes: Oxidation of toluene on CeO2. Plasma Process. Polym. 2017, 14. [Google Scholar] [CrossRef]
- Jia, Z.; Rousseau, A. Sorbent track: Quantitative monitoring of adsorbed VOCs under in-situ plasma exposure. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Stere, C.E.; Anderson, J.A.; Chansai, S.; Delgado, J.J.; Goguet, A.; Graham, W.G.; Hardacre, C.; Taylor, S.F.R.; Tu, X.; Wang, Z.; et al. Non-Thermal Plasma Activation of Gold-Based Catalysts for Low-Temperature Water–Gas Shift Catalysis. Angew. Chem. Int. Ed. Engl. 2017, 56, 5579–5583. [Google Scholar] [CrossRef] [PubMed]
- Barakat, C.; Gravejat, P.; Guaitella, O.; Thevenet, F.; Rousseau, A. Oxidation of isopropanol and acetone adsorbed on TiO2 under plasma generated ozone flow: Gas phase and adsorbed species monitoring. Appl. Catal. B Environ. 2014, 147, 302–313. [Google Scholar] [CrossRef]
- Batault, F. Effect of Adsorption and Operating Parameters on Volatile Organic Compounds Mixture Photocatalytic Treatment in Indoor Air Conditions. Ph.D. Thesis, University of Lille, Lille, France, 2014. [Google Scholar]
- Bouzaza, A.; Laplanche, A. Photocatalytic degradation of toluene in the gas phase: Comparative study of some TiO2 supports. J. Photochem. Photobiol. A Chem. 2002, 150, 207–212. [Google Scholar] [CrossRef]
- Takeuchi, M.; Hidaka, M.; Anpo, M. Efficient removal of toluene and benzene in gas phase by the TiO2/Y-zeolite hybrid photocatalyst. J. Hazard. Mater. 2012, 237–238, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Goss, K.-U. The Air/Surface Adsorption Equilibrium of Organic Compounds Under Ambient Conditions. Crit. Rev. Environ. Sci. Technol. 2004, 34, 339–389. [Google Scholar] [CrossRef]
- Fowkes, F.M. Attractive Forces at Interfaces. Ind. Eng. Chem. Res. 1964, 56, 40–52. [Google Scholar] [CrossRef]
- Goss, K.-U.; Eisenreich, S.J. Adsorption of VOCs from the Gas Phase to Different Minerals and a Mineral Mixture. Environ. Sci. Technol. 1996, 30, 2135–2142. [Google Scholar] [CrossRef]
- Mao, L.; Chen, Z.; Wu, X.; Tang, X.; Yao, S.; Zhang, X.; Jiang, B.; Han, J.; Wu, Z.; Lu, H.; et al. Plasma-catalyst hybrid reactor with CeO2/Γ-Al2O3 for benzene decomposition with synergetic effect and nano particle by-product reduction. J. Hazard. Mater. 2018, 347, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Blount, M.C.; Falconer, J.L. Steady-state surface species during toluene photocatalysis. Appl. Catal. B Environ. 2002, 39, 39–50. [Google Scholar] [CrossRef]
- Augugliaro, V.; Coluccia, S.; Loddo, V.; Marchese, L.; Martra, G.; Palmisano, L.; Schiavello, M. Photocatalytic oxidation of gaseous toluene on anatase TiO2 catalyst: Mechanistic aspects and FT-IR investigation. Appl. Catal. B Environ. 1999, 20, 15–27. [Google Scholar] [CrossRef]
- Harling, A.M.; Demidyuk, V.; Fischer, S.J.; Whitehead, J.C. Plasma-catalysis destruction of aromatics for environmental clean-up: Effect of temperature and configuration. Appl. Catal. B Environ. 2008, 82, 180–189. [Google Scholar] [CrossRef]
- Van Durme, J.; Dewulf, J.; Sysmans, W.; Leys, C.; Van Langenhove, H. Efficient toluene abatement in indoor air by a plasma catalytic hybrid system. Appl. Catal. B Environ. 2007, 74, 161–169. [Google Scholar] [CrossRef]
- Wu, J.; Huang, Y.; Xia, Q.; Li, Z. Decomposition of Toluene in a Plasma Catalysis System with NiO, MnO2, CeO2, Fe2O3, and CuO Catalysts. Plasma Chem. Plasma Process. 2013, 33, 1073–1082. [Google Scholar] [CrossRef]
- Orge, C.A.; Órfão, J.J.M.; Pereira, M.F.R.; Duarte de Farias, A.M.; Fraga, M.A. Ceria and cerium-based mixed oxides as ozonation catalysts. Chem. Eng. J. 2012, 200–202, 499–505. [Google Scholar] [CrossRef]
- Diehl, J.W.; Finkbeiner, J.W.; DiSanzo, F.P. Determination of benzene, toluene, ethylbenzene, and xylenes in gasolines by gas chromatography/deuterium isotope dilution Fourier transform infrared spectroscopy. Anal. Chem. 1993, 65, 2493–2496. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SIE (J/L) | Ozone Produced During 60 min (μmol) | Ratio of Ozone to Toluene |
|---|---|---|
| 2 | 24 | 3:1 |
| 8 | 120 | 17:1 |
| 19 | 204 | 29:1 |
| RH (%) | Amount of Irreversibly Adsorbed Toluene (µmol/m2) |
|---|---|
| 0 | 1.40 |
| 20 | 0.80 |
| 50 | 0.66 |
| 80 | 0.56 |
| Compound | Detection Limit (ppbv) |
|---|---|
| Toluene | 130 |
| CO2 | 20 |
| CO | 10 |
| O3 | 15 |
| H2O | 2800 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, Z.; Wang, X.; Foucher, E.; Thevenet, F.; Rousseau, A. Plasma-Catalytic Mineralization of Toluene Adsorbed on CeO2. Catalysts 2018, 8, 303. https://doi.org/10.3390/catal8080303
Jia Z, Wang X, Foucher E, Thevenet F, Rousseau A. Plasma-Catalytic Mineralization of Toluene Adsorbed on CeO2. Catalysts. 2018; 8(8):303. https://doi.org/10.3390/catal8080303
Chicago/Turabian StyleJia, Zixian, Xianjie Wang, Emeric Foucher, Frederic Thevenet, and Antoine Rousseau. 2018. "Plasma-Catalytic Mineralization of Toluene Adsorbed on CeO2" Catalysts 8, no. 8: 303. https://doi.org/10.3390/catal8080303
APA StyleJia, Z., Wang, X., Foucher, E., Thevenet, F., & Rousseau, A. (2018). Plasma-Catalytic Mineralization of Toluene Adsorbed on CeO2. Catalysts, 8(8), 303. https://doi.org/10.3390/catal8080303