The Promoting Effect of Ce on the Performance of Au/CexZr1−xO2 for γ-Valerolactone Production from Biomass-Based Levulinic Acid and Formic Acid

Abstract
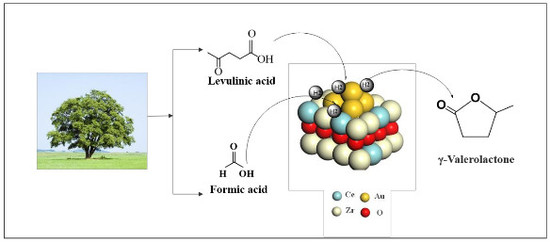
1. Introduction
2. Results and Discussion
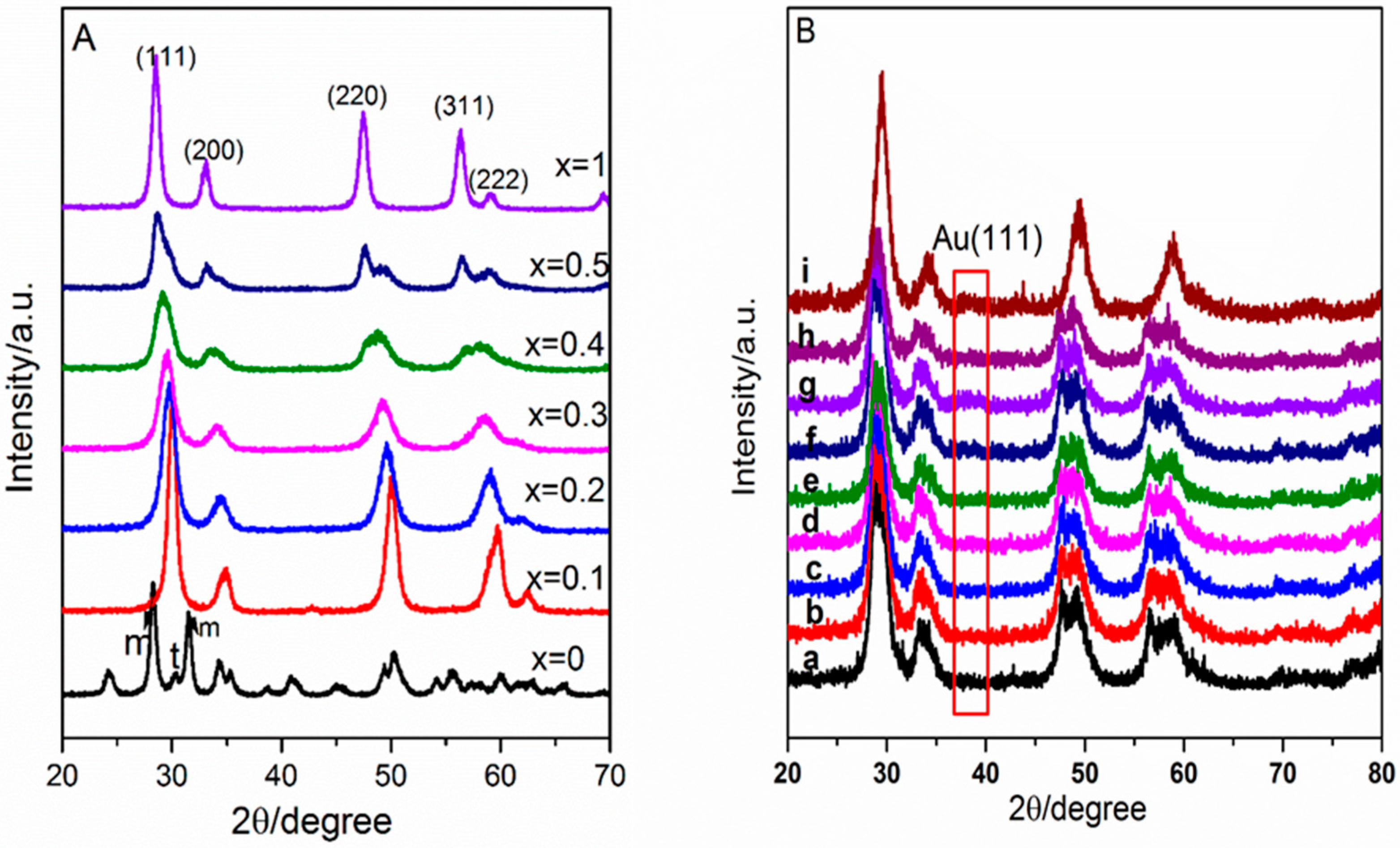
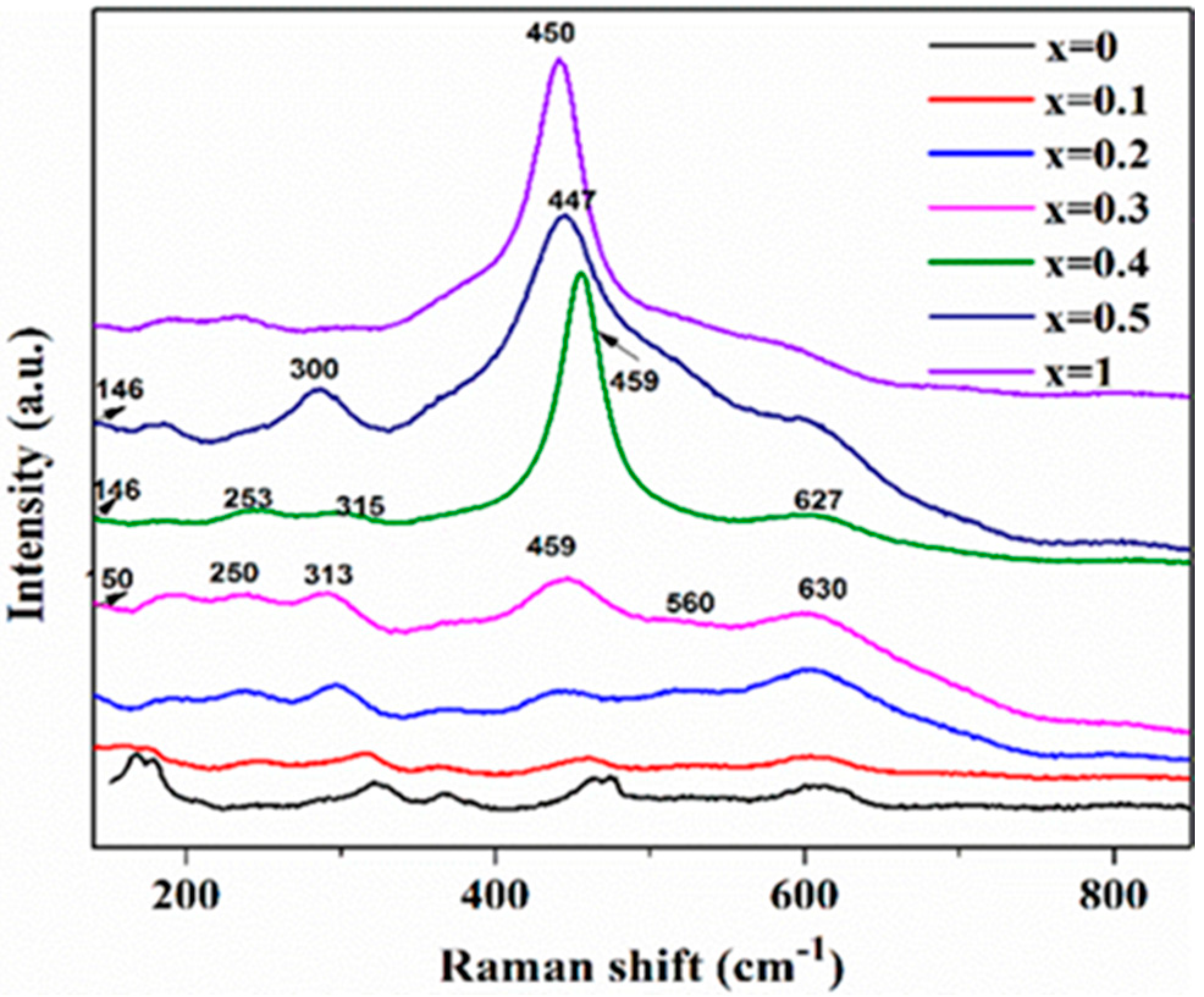
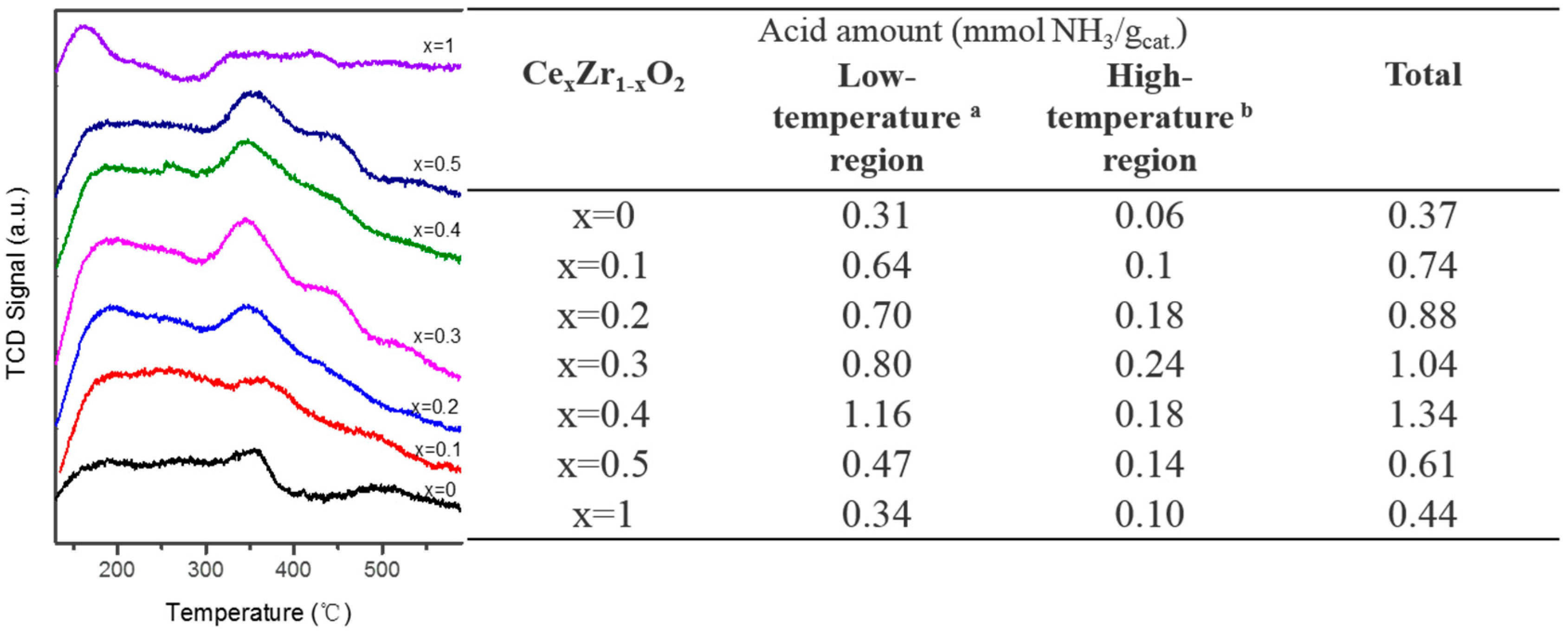
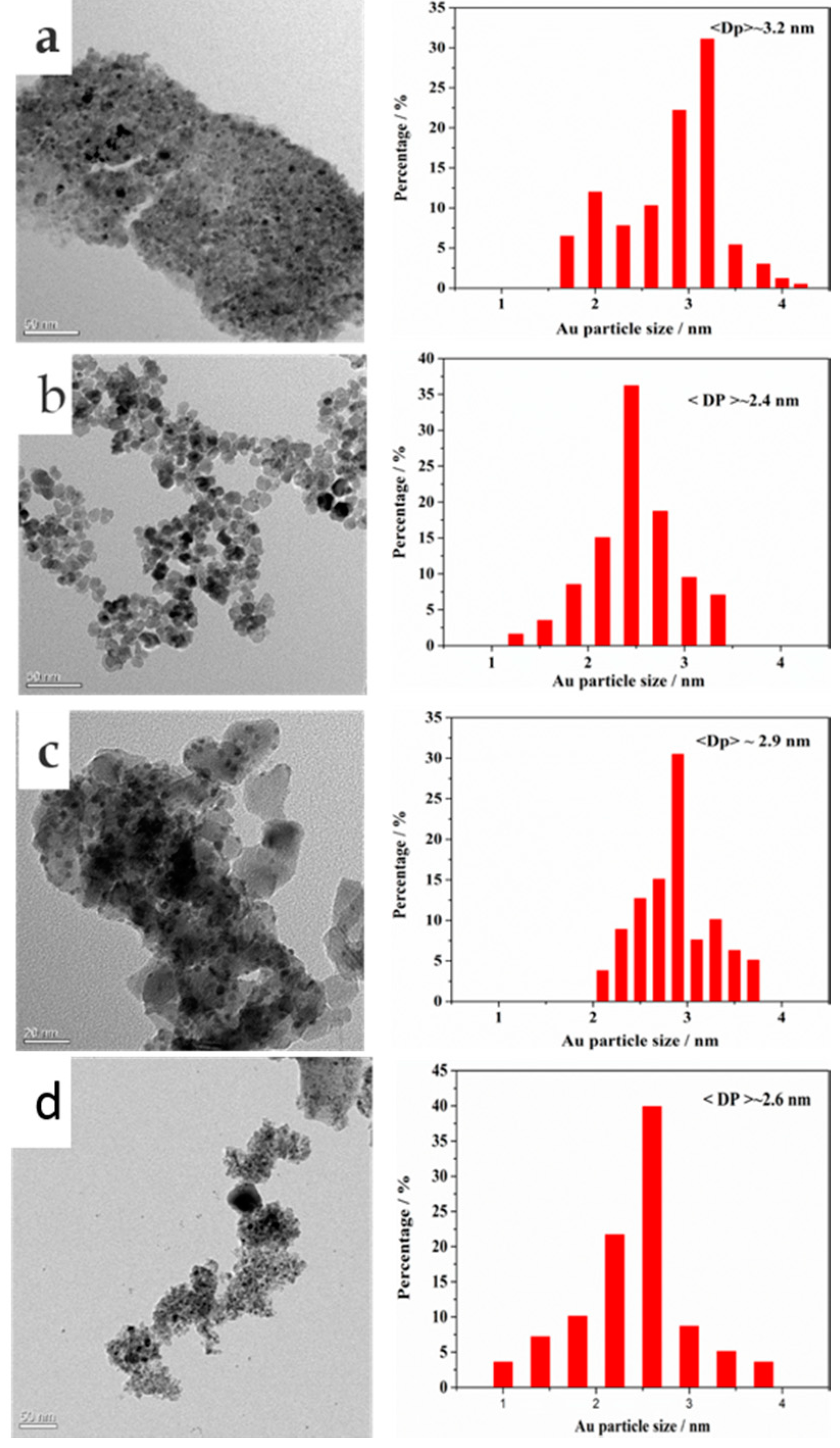
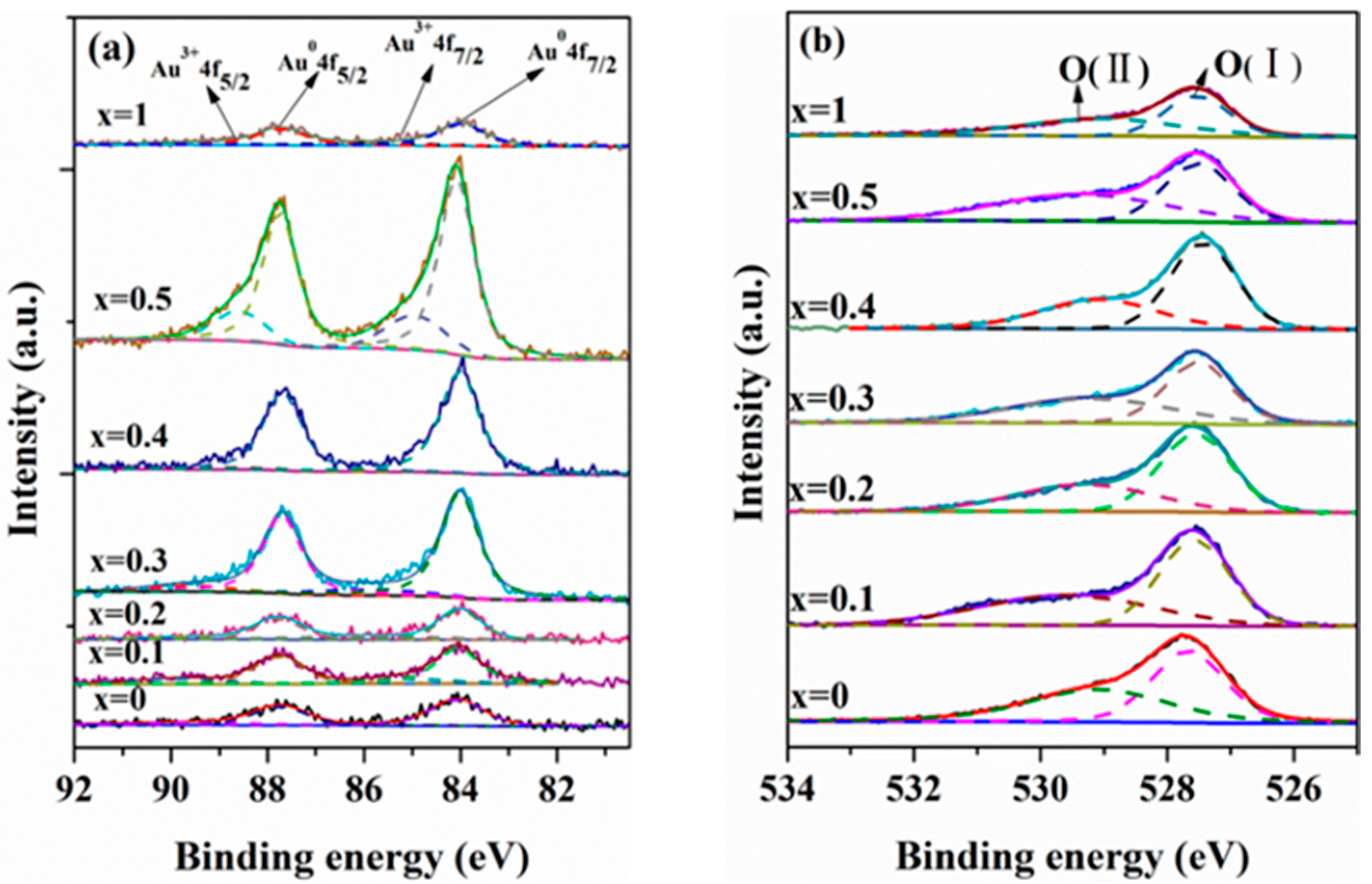
2.1. Catalyst Characterization
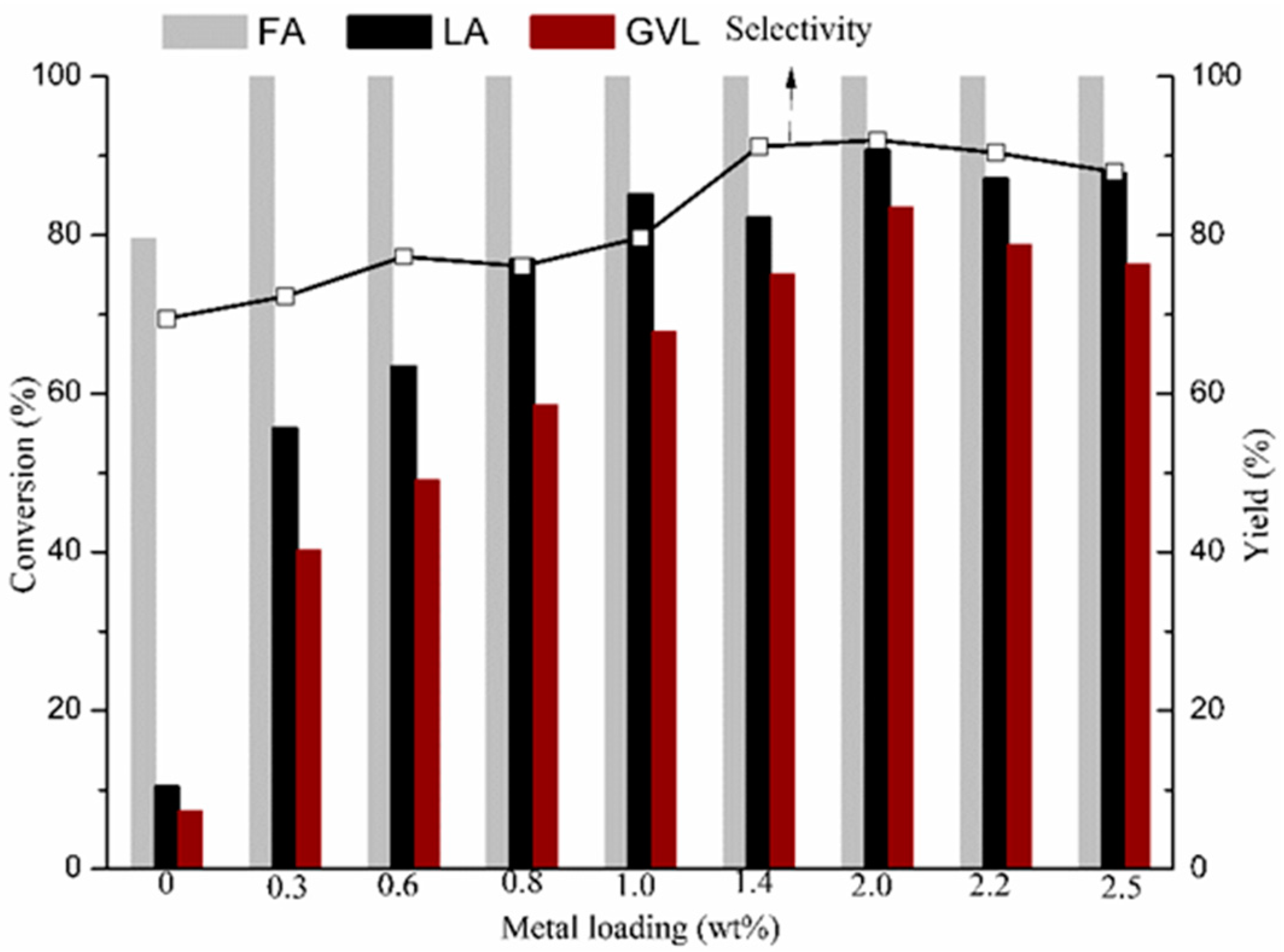
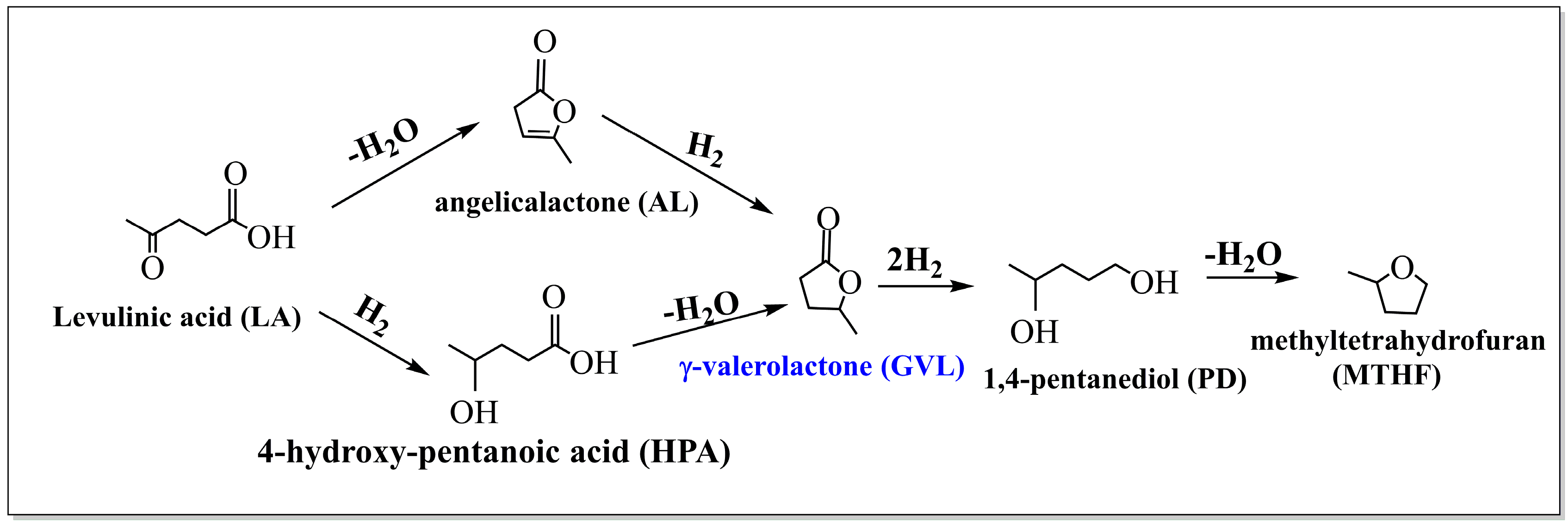
2.2. GVL Production from LA and FA
2.2.1. FA Decomposition
2.2.2. GVL Production
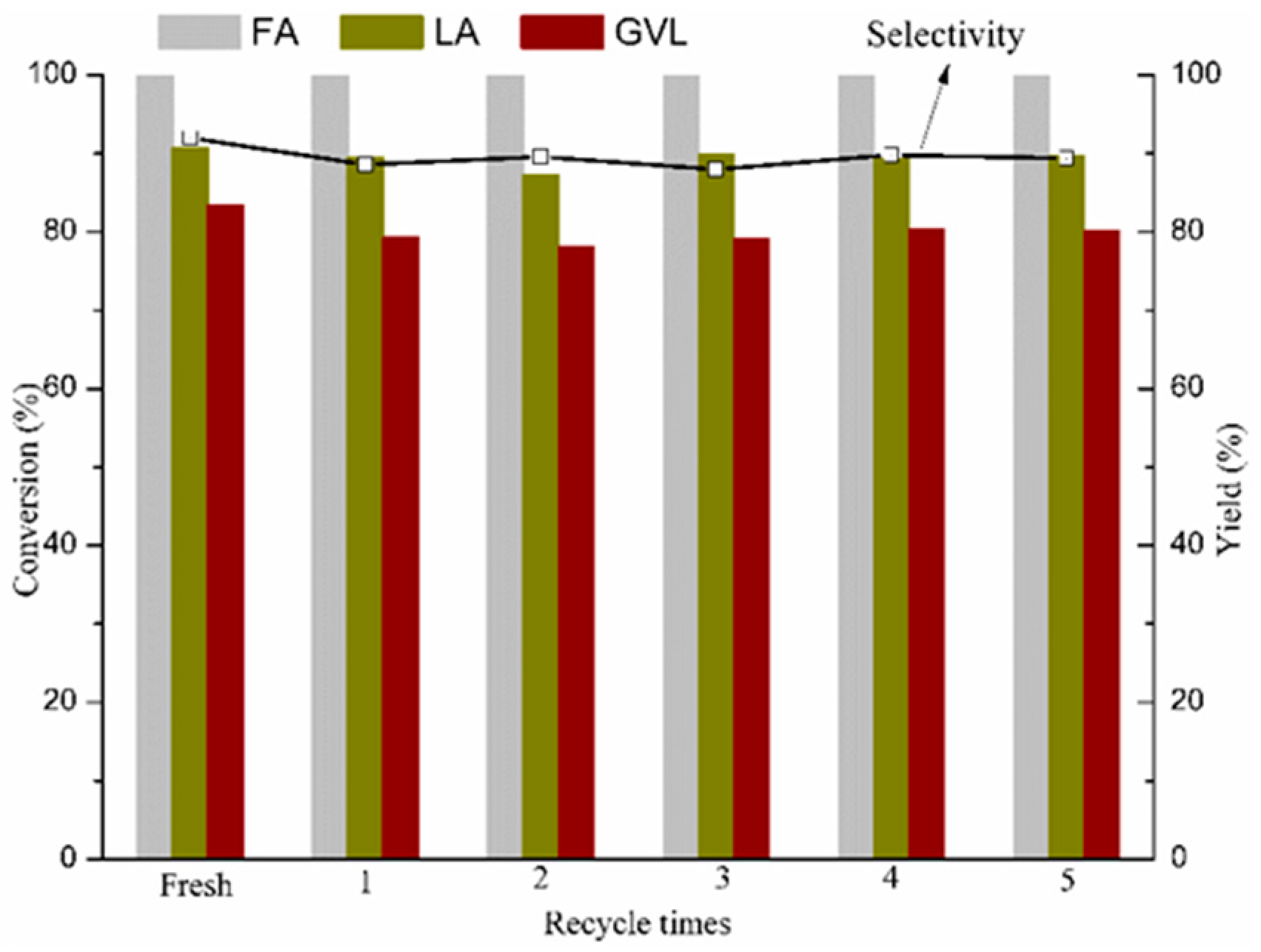
2.3. Recyclability of the Catalyst
3. Methods Experimental Section
3.1. Materials
3.2. Methods
3.2.1. Catalyst Preparation
3.2.2. Catalyst Characterization
3.2.3. Conversion of LA to GVL
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Du, X.-L.; He, L.; Zhao, S.; Liu, Y.-M.; Cao, Y.; He, H.-Y.; Fan, K.-N. Hydrogen-Independent reductive transformation of carbohydrate biomass into γ-valerlactone and pyrrolidone derivatives witn supporetd gold catalysts. Angew. Chem. 2011, 123, 7961–7965. [Google Scholar] [CrossRef]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Gamma-valerolactone, a sustainable platform molecule derived from lignocellulosic biomass. Green Chem. 2013, 15, 584–595. [Google Scholar] [CrossRef]
- Wettstein, S.G.; Alonso, D.M.; Gürbüz, E.I.; Dumesic, J.A. A roadmap for conversion of lignocellulosic biomass to chemicals and fuels. Curr. Opin. Chem. Eng. 2012, 1, 218–224. [Google Scholar] [CrossRef]
- Wright, W.R.H.; Palkovits, R. Development of heterogeneous catalysts for the conversion of levulinic acid to γ-valerolactone. ChemSusChem 2012, 5, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, S.; Li, B.; Zhang, H. Advances in the catalytic production of valuable levulinic acid derivatives. ChemCatChem 2012, 4, 1230–1237. [Google Scholar] [CrossRef]
- Yan, K.; Yang, Y.; Chai, J.; Lu, Y. Catalytic reactions of gamma-valerolactone: A platform to fuels and value-added chemicals. Appl. Catal. B 2015, 179, 292–304. [Google Scholar] [CrossRef]
- Luo, W.; Sankar, M.; Beale, A.M.; He, Q.; Kiely, C.J.; Bruijnincx, P.C.A.; Weckhuysen, B.M. High performing and stable supported nano-alloys for the catalytic hydrogenation of levulinic acid to γ-valerolactone. Nat. Commun. 2015, 6, 6540–6550. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Lu, Y.; Yi, Z.; Ejaz, A.; Hu, K.; Zhang, L.; Yan, K. Selective Production of γ-Valerolactone and Valeric Acid in One-Pot Bifunctional Metal Catalysts. ChemistrySelect 2018, 3, 1097–1101. [Google Scholar] [CrossRef]
- Yan, K.; Liu, Y.; Lu, Y.; Chaib, J.; Suna, L. Catalytic application of layered double hydroxidederived catalysts for the conversion of biomass derived molecules. Catal. Sci. Technol. 2017, 7, 1622–1645. [Google Scholar] [CrossRef]
- Osatiashtiani, A.; Flee, A.; Wilson, K. Recent advances in the production of γ-valerolactone from biomass-derived feedstocks via heterogeneous catalytic transfer hydrogenation. J. Chem. Technol. Biotechnol. 2017, 92, 1125–1135. [Google Scholar] [CrossRef]
- Chia, M.; Dumesic, J.A. Liquid-phase catalytic transfer hydrogenation and cyclization of levulinic acid and its esters to γ-valerolactone over metal oxide catalysts. Chem. Commun. 2011, 47, 12233–12235. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Xue, Y.; Guo, J.; Cen, Y.; Wang, J.; Fan, W. Integrated Conversion of Hemicellulose and Furfural into γ-Valerolactone over Au/ZrO2 Catalyst Combined with ZSM-5. ACS Catal. 2016, 6, 2035–2042. [Google Scholar] [CrossRef]
- Liguori, F.; Moreno-Marrodan, C.; Barbaro, P. Environmentally Friendly Synthesis of γ-Valerolactone by Direct Catalytic Conversion of Renewable Sources. ACS Catal. 2015, 5, 1882–1894. [Google Scholar] [CrossRef]
- Fábos, V.; Mika, L.T.; Horváth, I.T. Selective Conversion of Levulinic and Formic Acids to γ-Valerolactone with the Shvo Catalyst. Organometallics 2014, 33, 181–187. [Google Scholar] [CrossRef]
- Deuss, P.J.; Barta, K.; de Vries, J.G. Homogeneous catalysis for the conversion of biomass and biomass-derived platform chemicals. Catal. Sci. Technol. 2014, 4, 1174–1196. [Google Scholar] [CrossRef]
- Horváth, I.T.; Mehdi, H.; Fabos, V.; Boda, L.; Mika, L.T. γ-Valerolactone—A sustainable liquid for energy and carbon-based chemicals. Green Chem. 2008, 10, 238–242. [Google Scholar] [CrossRef]
- Deng, L.; Zhao, Y.; Li, J.; Fu, Y.; Liao, B.; Guo, Q.X. Conversion of Levulinic Acid and Formic Acid into γ-Valerolactone over Heterogeneous Catalysts. ChemSusChem 2010, 3, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Luo, W.; Ge, H.; Su, Y.; Wang, A.; Zhang, T. UiO-66 derived Ru/ZrO2@C as a highly stable catalyst for hydrogenation of levulinic acid to γ-valerolactone. Green Chem. 2017, 19, 2201–2211. [Google Scholar] [CrossRef]
- Alonso, D.M.; Wettstein, S.G.; Bond, J.Q.; Root, T.W.; Dumesic, J.A. Production of Biofuels from Cellulose and Corn Stover Using Alkylphenol Solvents. ChemSusChem 2011, 4, 1078–1081. [Google Scholar] [CrossRef] [PubMed]
- Braden, D.J.; Henao, C.A.; Heltzel, J.; Maravelias, C.C.; Dumesic, J.A. Production of liquid hydrocarbon fuels by catalytic conversion of biomass-derived levulinic acid. Green Chem. 2011, 13, 1755–1765. [Google Scholar] [CrossRef]
- Sen, S.M.; Henao, C.A.; Braden, D.J.; Dumesic, J.A.; Maravelias, C.T. Catalytic conversion of lignocellulosic biomass to fuels: Process development and technoeconomic evaluation. Chem. Eng. Sci. 2012, 67, 57–67. [Google Scholar]
- Serrano-Ruiz, J.C.; Wang, D.; Dumesic, J.A. Catalytic upgrading of levulinic acid to 5-nonanone. Green Chem. 2010, 12, 574–577. [Google Scholar] [CrossRef]
- Yan, Z.P.; Lin, L.; Liu, S. Synthesis of γ-Valerolactone by Hydrogenation of Biomass-derived Levulinic Acid over Ru/C Catalyst. Energy Fuels 2009, 23, 3853–3858. [Google Scholar] [CrossRef]
- Zacharska, M.; Chuvilin, A.L.; Kriventsov, V.V.; Beloshapkin, S.; Estrada, M.; Simakov, A.; Bulushev, D.A. Support effect for nanosized Au catalysts in hydrogen production from formic acid decomposition. Catal. Sci. Technol. 2016, 6, 6853–6860. [Google Scholar] [CrossRef]
- Yuan, J.; Li, S.; Yu, L.; Liu, Y.; Cao, Y.; He, H.; Fan, K. Copper-based catalysts for the efficient conversion of carbohydrate biomass into γ-valerolactone in the absence of externally added hydrogen. Energy Environ. Sci. 2013, 6, 3308–3313. [Google Scholar] [CrossRef]
- Kumar, V.V.; Naresh, G.; Sudhakar, M.; Anjaneyulu, C.; Bhargava, S.K.; Tardio, J.; Reddy, V.K.; Padmasri, A.H.; Venugopal, A. An investigation on the influence of support type for Ni catalysed vapour phase hydrogenation of aqueous levulinic acid to γ-valerolactone. RSC Adv. 2016, 6, 9872–9879. [Google Scholar] [CrossRef]
- Lomate, S.; Sultana, A.; Fujitani, T. Effect of SiO2 support properties on the performance of Cu–SiO2 catalysts for the hydrogenation of levulinic acid to gamma valerolactone using formic acid as a hydrogen source. Catal. Sci. Technol. 2017, 7, 3073–3083. [Google Scholar] [CrossRef]
- Ruppert, A.M.; Grams, J.; Jędrzejczyk, M.; Matras-Michalska, J.; Keller, N.; Ostojska, K.; Sautet, P. Titania-Supported Catalysts for Levulinic Acid Hydrogenation: Influence of Support and its Impact on γ-Valerolactone Yield. ChemSusChem 2015, 8, 1538–1547. [Google Scholar] [CrossRef] [PubMed]
- Valekar, A.H.; Cho, K.; Chitale, S.K.; Hong, D.; Cha, G.; Lee, U.; Hwang, D.W.; Serre, C.; Chang, J.; Hwang, Y.K. Catalytic transfer hydrogenation of ethyl levulinate to γ-valerolactone over zirconium-based metal–organic frameworks. Green Chem. 2016, 18, 4542–4552. [Google Scholar] [CrossRef]
- Lange, J.P.; Price, R.; Ayoub, P.; Louis, J.; Petrus, L.; Clarke, L.; Gosselink, H. Valeric Biofuels: A Platform of Cellulosic Transportation Fuels. Angew. Chem. Int. Ed. 2010, 49, 4479–4483. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Deka, U.; Beale, A.M.; Eck, E.R.H.V.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Ruthenium-catalyzed hydrogenation of levulinic acid: Influence of the support and solvent on catalyst selectivity and stability. J. Catal. 2013, 301, 175–186. [Google Scholar] [CrossRef]
- Abdelrahman, O.A.; Luo, H.Y.; Heyden, A.; Román-Leshkov, Y.; Bond, J.Q. Toward rational design of stable supported metal catalysts for aqueous-phase processing: Insights from the hydrogenation of levulinic acid. J. Catal. 2015, 329, 10–21. [Google Scholar] [CrossRef]
- Ftouni, J.; Muñoz-Murillo, A.; Goryachev, A.; Hofmann, J.P.; Hensen, E.J.M.; Lu, L.; Kiely, C.J.; Bruijnincx, P.C.A.; Weckhuysen, B.M. ZrO2 is Preferred over TiO2 as Support for the Ru-Catalyzed Hydrogenation of Levulinic Acid to γ-Valerolactone. ACS Catal. 2016, 6, 5462–5472. [Google Scholar] [CrossRef]
- Deng, L.; Li, J.; Lai, D.M.; Fu, Y.; Guo, Q.X. Catalytic Conversion of Biomass-Derived Carbohydrates into γ-Valerolactone without Using an External H2 Supply. Angew. Chem. 2009, 121, 6651–6654. [Google Scholar] [CrossRef]
- Luo, W.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Selective, one-pot catalytic conversion of levulinic acid to pentanoic acid over Ru/H-ZSM5. J. Catal. 2014, 320, 33–41. [Google Scholar] [CrossRef]
- Luo, Y.; Yi, J.; Tong, D.; Hu, C. Production of γ-valerolactone via selective catalytic conversion of hemicellulose in pubescens without addition of external hydrogen. Green Chem. 2016, 18, 848–857. [Google Scholar] [CrossRef]
- Pengpanich, S.; Meeyoo, V.; Rirksomboon, T.; Bunyakiat, K. Catalytic oxidation of methane over CeO2-ZrO2 mixed oxide solid solution catalysts prepared via urea hydrolysis. Appl. Catal. A Gen. 2002, 234, 221–233. [Google Scholar] [CrossRef]
- Roh, H.S.; Potdar, H.S.; Jun, K.W.; Kim, J.W.; Oh, Y.S. Carbon dioxide reforming of methane over Ni incorporated into Ce–ZrO2 catalysts. Appl. Catal. A Gen. 2004, 276, 231–239. [Google Scholar] [CrossRef]
- Gregg, S.J.; Sing, K.S.W. Adsorption Surface Area and Porosity. J. Electrochem. Soc. 1967, 114, 279. [Google Scholar] [CrossRef]
- Menegazzo, F.; Signoretto, M.; Marchese, D.; Pinna, F.; Manzoli, M. Structure–activity relationships of Au/ZrO2 catalysts for 5-hydroxymethylfurfural oxidative esterification: Effects of zirconia sulphation on gold dispersion, position and shape. J. Catal. 2015, 326, 1–8. [Google Scholar] [CrossRef]
- Ma, Z.Y.; Yang, C.; Wei, W.; Li, W.H.; Sun, Y.H. Surface properties and CO adsorption on zirconia polymorphs. J. Mol. Catal. A Chem. 2005, 227, 119–124. [Google Scholar] [CrossRef]
- Guo, R.; Zhou, Y.; Pan, W.; Hong, J.; Zhen, W.; Jin, Q.; Ding, C.; Guo, S. Effect of preparation methods on the performance of CeO2/Al2O3 catalysts for selective catalytic reduction of NO with NH3. J. Ind. Eng. Chem. 2013, 19, 2022–2025. [Google Scholar] [CrossRef]
- Khaodee, W.; Tangchupong, N.; Jongsomjit, B.; Praserthdam, P.; Assabumrungrat, S. A study on isosynthesis via CO hydrogenation over ZrO2–CeO2 mixed oxide catalysts. Catal. Commun. 2009, 10, 494–501. [Google Scholar] [CrossRef]
- Harrison, B.; Diwell, A.F.; Hallett, C. Promoting Platinum Metals by Ceria. Platin. Met. Rev. 1988, 32, 73–83. [Google Scholar]
- Eder, D.; Kramer, R. The stoichiometry of hydrogen reduced zirconia and its influence on catalytic activity Part 1: Volumetric and conductivity studies. Phys. Chem. Chem. Phys. 2002, 4, 795–801. [Google Scholar] [CrossRef]
- Trovarelli, A.; Leitenburg, C.; Dolcetti, G. Design better cerium-based oxidation catalysts. Chemtech 1997, 27, 32–37. [Google Scholar]
- Damyanova, S.; Pawelec, B.; Arishtirova, K.; Huerta, M.V.M.; Fierro, J.L.G. Study of the surface and redox properties of ceria–zirconia oxides. Appl. Catal. A Gen. 2008, 337, 86–96. [Google Scholar] [CrossRef]
- Li, M.; Wang, X.; Cárdenas-Lizana, F.; Keane, M.A. Effect of support redox character on catalytic performance in the gas phase hydrogenation of benzaldehyde and nitrobenzene over supported gold. Catal. Today 2017, 279, 19–28. [Google Scholar] [CrossRef]
- Tedsree, K.; Li, T.; Jones, S.; Chan, C.W.; Yu, K.M.; Bagot, P.A.; Marquis, E.A.; Smith, G.D.; Tsang, S.C. Hydrogen production from formic acid decomposition at room temperature using a Ag–Pd core–shell nanocatalyst. Nat. Nanotechnol. 2011, 6, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Ojeda, M.; Iglesia, E. Formic Acid Dehydrogenation on Au-Based Catalysts at Near-Ambient Temperatures. Angew. Chem. 2009, 121, 4894–4897. [Google Scholar] [CrossRef]
- Hengne, A.M.; Kadu, B.S.; Biradar, N.S.; Chikate, R.C.; Rode, C.V. Transfer hydrogenation of biomass-derived levulinic acid to γ-valerolactone over supported Ni catalysts. RSC Adv. 2016, 6, 59753–59761. [Google Scholar] [CrossRef]
- Li, C.; Xu, G.; Zhai, Y.; Liu, X.; Ma, Y.; Zhang, Y. Hydrogenation of biomass-derived ethyl levulinate into γ-valerolactone by activated carbon supported bimetallic Ni and Fe catalysts. Fuel 2017, 203, 23–31. [Google Scholar] [CrossRef]
- Wąchała, M.; Grams, J.; Kwapiński, W.; Ruppert, A.M. Influence of ZrO2 on catalytic performance of Ru catalyst in hydrolytic hydrogenation of cellulose towards γ-valerolactone. Int. J. Hydrogen Energy 2016, 41, 8688–8695. [Google Scholar] [CrossRef]
- Wang, N.; Chua, W.; Zhang, T.; Zhao, X. Manganese promoting effects on the Co–Ce–Zr–Ox nano catalysts for methane dry reforming with carbon dioxide to hydrogen and carbon monoxide. Chem. Eng. J. 2011, 170, 457–463. [Google Scholar] [CrossRef]
- Oemar, U.; Hidajat, K.; Kawi, S. Pd–Ni catalyst over spherical nanostructured Y2O3 support for oxy-CO2 reforming of methane: Role of surface oxygen mobility. Int. J. Hydrogen Energy 2015, 40, 12227–12238. [Google Scholar] [CrossRef]
- He, J.; Li, H.; Lu, Y.; Liu, Y.; Wu, Z.; Hu, D.; Yang, S. Cascade catalytic transfer hydrogenation–cyclization of ethyl levulinate to γ-valerolactone with Al–Zr mixed oxides. Appl. Catal. A Gen. 2016, 510, 11–19. [Google Scholar] [CrossRef]
- Li, M.; Collado, L.; Cárdenas-Lizana, F.; Keane, M.A. Role of Support Oxygen Vacancies in the Gas Phase Hydrogenation of Furfural over Gold. Catal. Lett. 2018, 148, 90–96. [Google Scholar] [CrossRef]
- Bond, G.C. The origins of particle size effects in heterogeneous catalysis. Surf. Sci. 1985, 156, 966–981. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Percentage (%) | (Ce/Zr)bulk a | (Ce/Zr)XPS | Au/(Ce + Zr)XPS | |
|---|---|---|---|---|---|
| Aun+ | Au0 | ||||
| Au/ZrO2 | 32.9 | 67.1 | — | — | 0.007 |
| Au/Ce0.1Zr0.9O2 | 23.7 | 76.3 | 0.07 | 0.11 | 0.010 |
| Au/Ce0.2Zr0.8O2 | 27.7 | 72.3 | 0.19 | 0.24 | 0.009 |
| Au/Ce0.3Zr0.7O2 | 11.1 | 88.9 | 0.37 | 0.47 | 0.020 |
| Au/Ce0.4Zr0.6O2 | 10.6 | 89.4 | 0.45 | 0.53 | 0.022 |
| Au/Ce0.5Zr0.5O2 | 20.9 | 79.3 | 0.67 | 0.88 | 0.011 |
| Au/CeO2 | 18.4 | 81.6 | — | — | 0.008 |
| Catalyst b | CFA(%) c | CLA(%) d | YGVL(%) e | SGVL(%) f | CPD g |
|---|---|---|---|---|---|
| Au/ZrO2 | 99.9 | 40.7 | 36.4 | 89.5 | trace |
| Au/Ce0.1Zr0.9O2 | 100 | 47.0 | 33.6 | 82.6 | trace |
| Au/Ce0.2Zr0.8O2 | 100 | 51.7 | 43.1 | 83.4 | trace |
| Au/Ce0.3Zr0.7O2 | 100 | 60.3 | 46.8 | 77.6 | trace |
| Au/Ce0.4Zr0.6O2 | 100 | 63.5 | 49.7 | 77.3 | trace |
| Au/Ce0.5Zr0.5O2 | 100 | 55.4 | 37.1 | 67.0 | trace |
| Au/CeO2 | 95.6 | 13.1 | 9.2 | 70.2 | trace |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Li, J.; Liu, X.; Tian, Q.; Hu, C. The Promoting Effect of Ce on the Performance of Au/CexZr1−xO2 for γ-Valerolactone Production from Biomass-Based Levulinic Acid and Formic Acid. Catalysts 2018, 8, 241. https://doi.org/10.3390/catal8060241
Li X, Li J, Liu X, Tian Q, Hu C. The Promoting Effect of Ce on the Performance of Au/CexZr1−xO2 for γ-Valerolactone Production from Biomass-Based Levulinic Acid and Formic Acid. Catalysts. 2018; 8(6):241. https://doi.org/10.3390/catal8060241
Chicago/Turabian StyleLi, Xiaoling, Jianmei Li, Xudong Liu, Qi Tian, and Changwei Hu. 2018. "The Promoting Effect of Ce on the Performance of Au/CexZr1−xO2 for γ-Valerolactone Production from Biomass-Based Levulinic Acid and Formic Acid" Catalysts 8, no. 6: 241. https://doi.org/10.3390/catal8060241
APA StyleLi, X., Li, J., Liu, X., Tian, Q., & Hu, C. (2018). The Promoting Effect of Ce on the Performance of Au/CexZr1−xO2 for γ-Valerolactone Production from Biomass-Based Levulinic Acid and Formic Acid. Catalysts, 8(6), 241. https://doi.org/10.3390/catal8060241