Effects of Synthesis on the Structural Properties and Methane Partial Oxidation Activity of Ni/CeO2 Catalyst




Abstract
:
1. Introduction
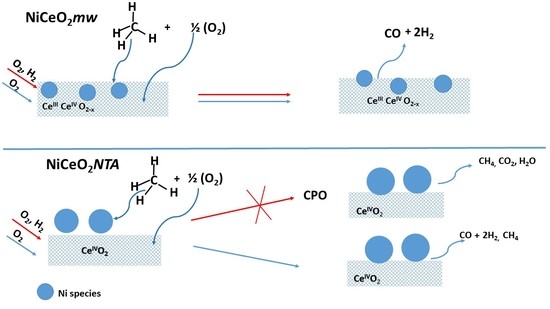
2. Results and Discussion
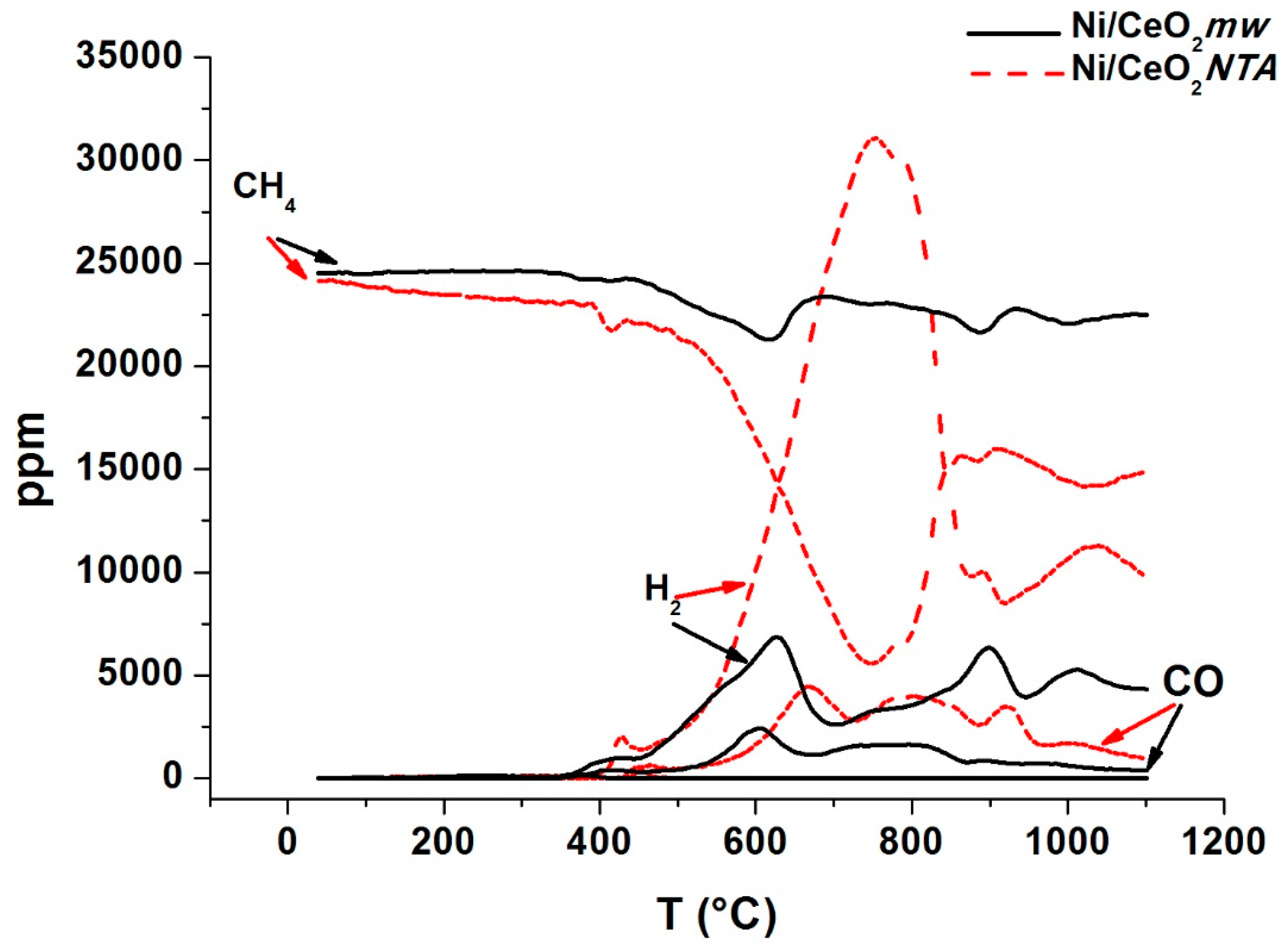
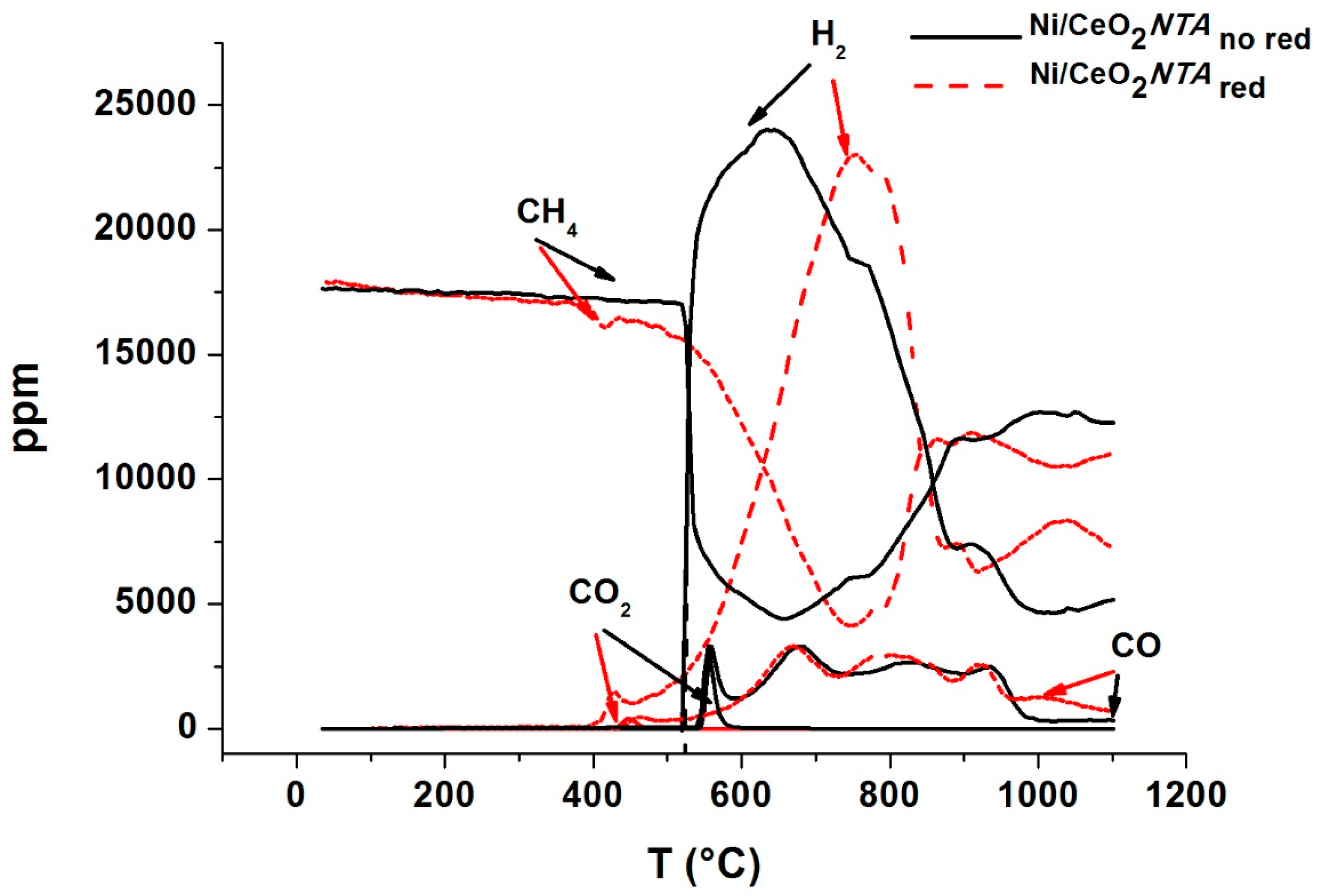
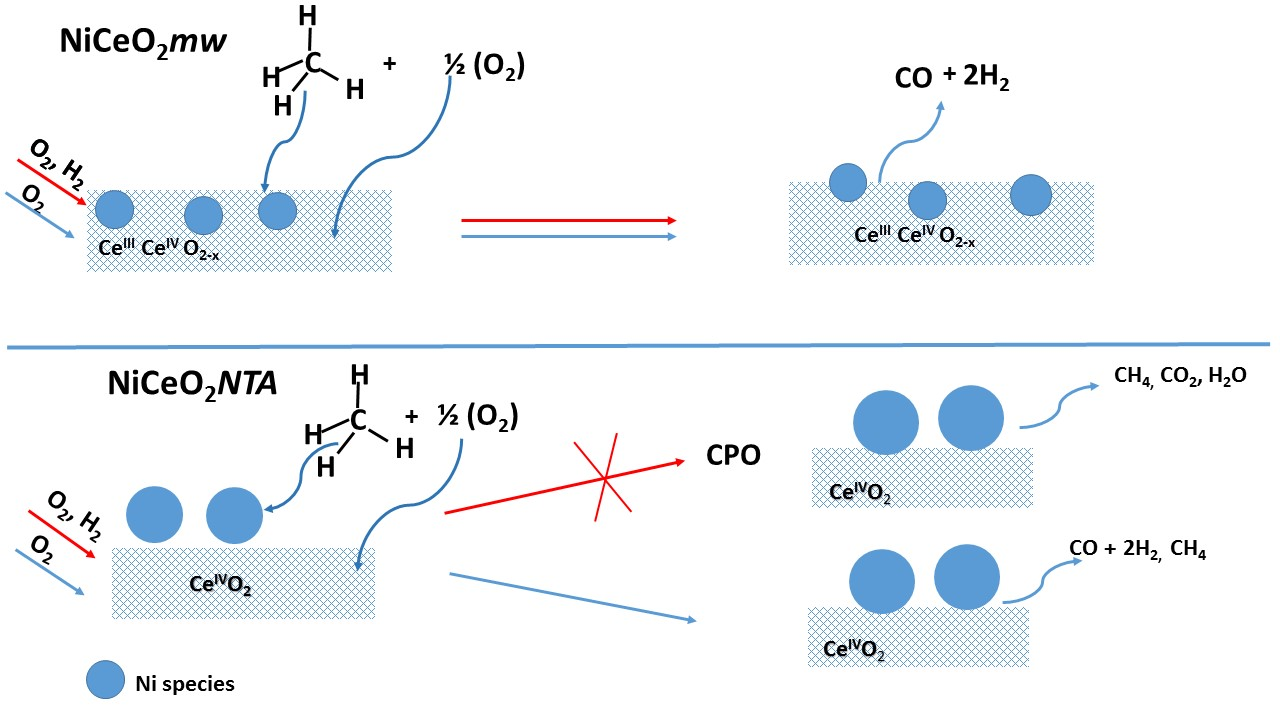
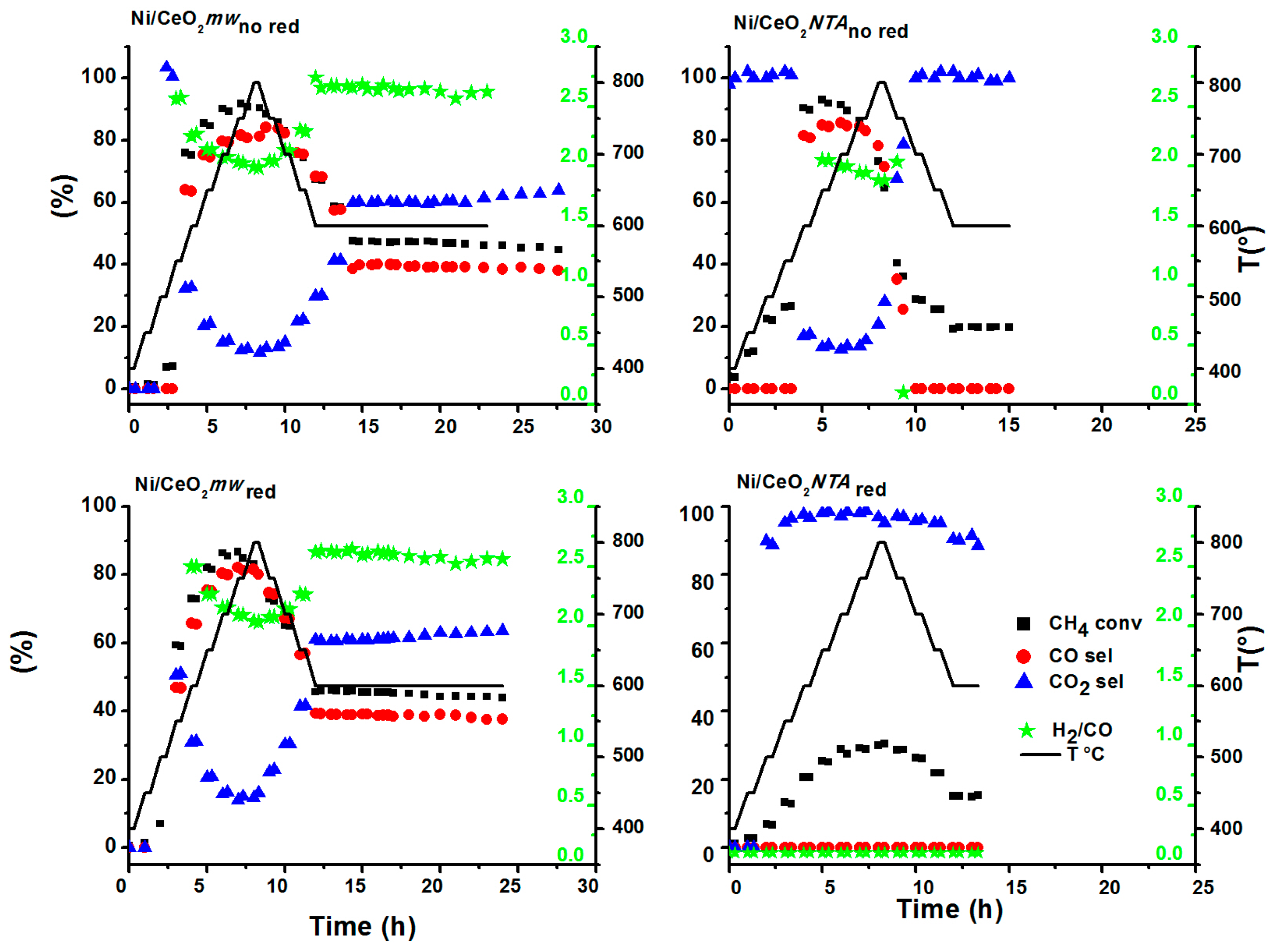
2.1. Catalytic Activity
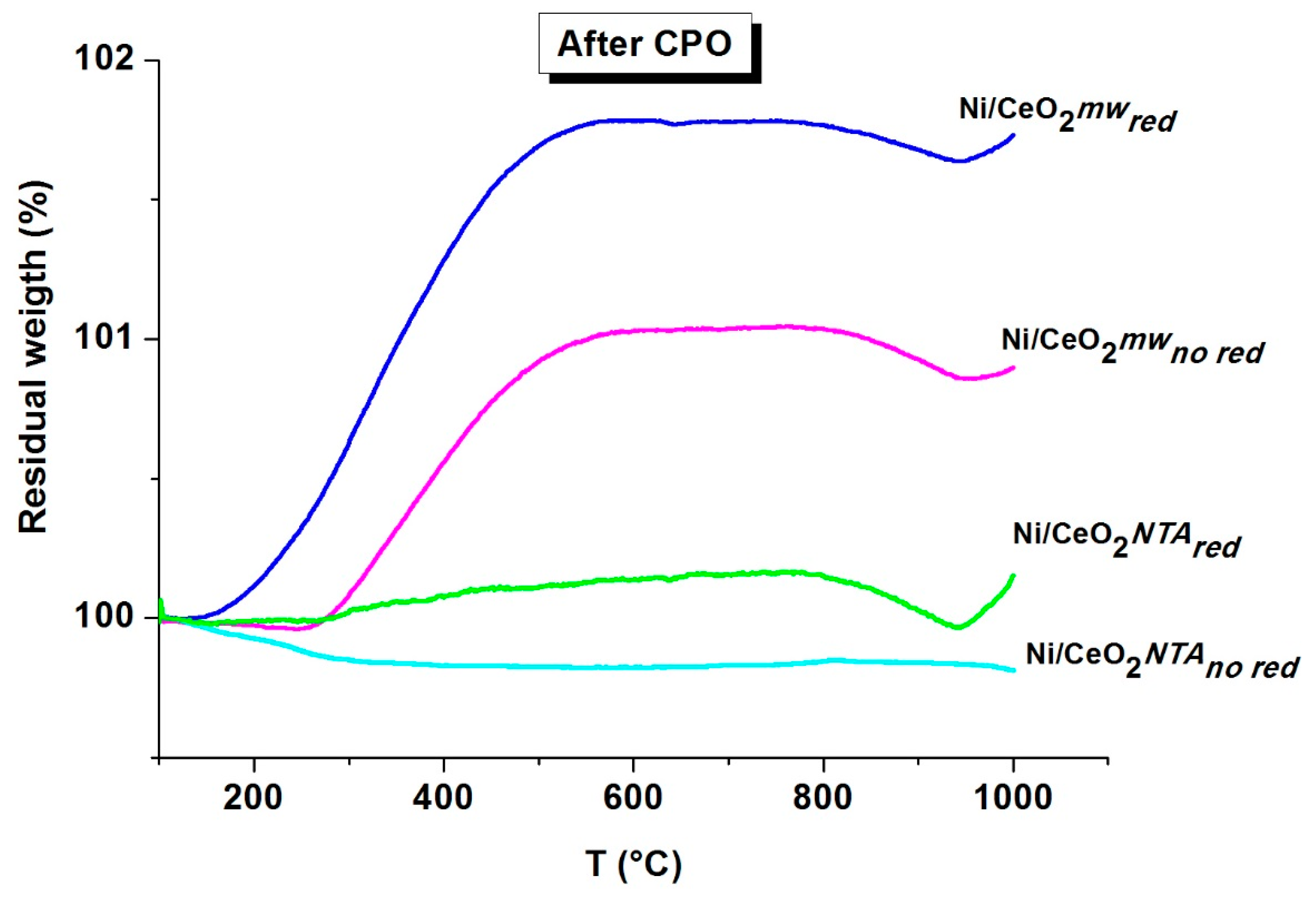
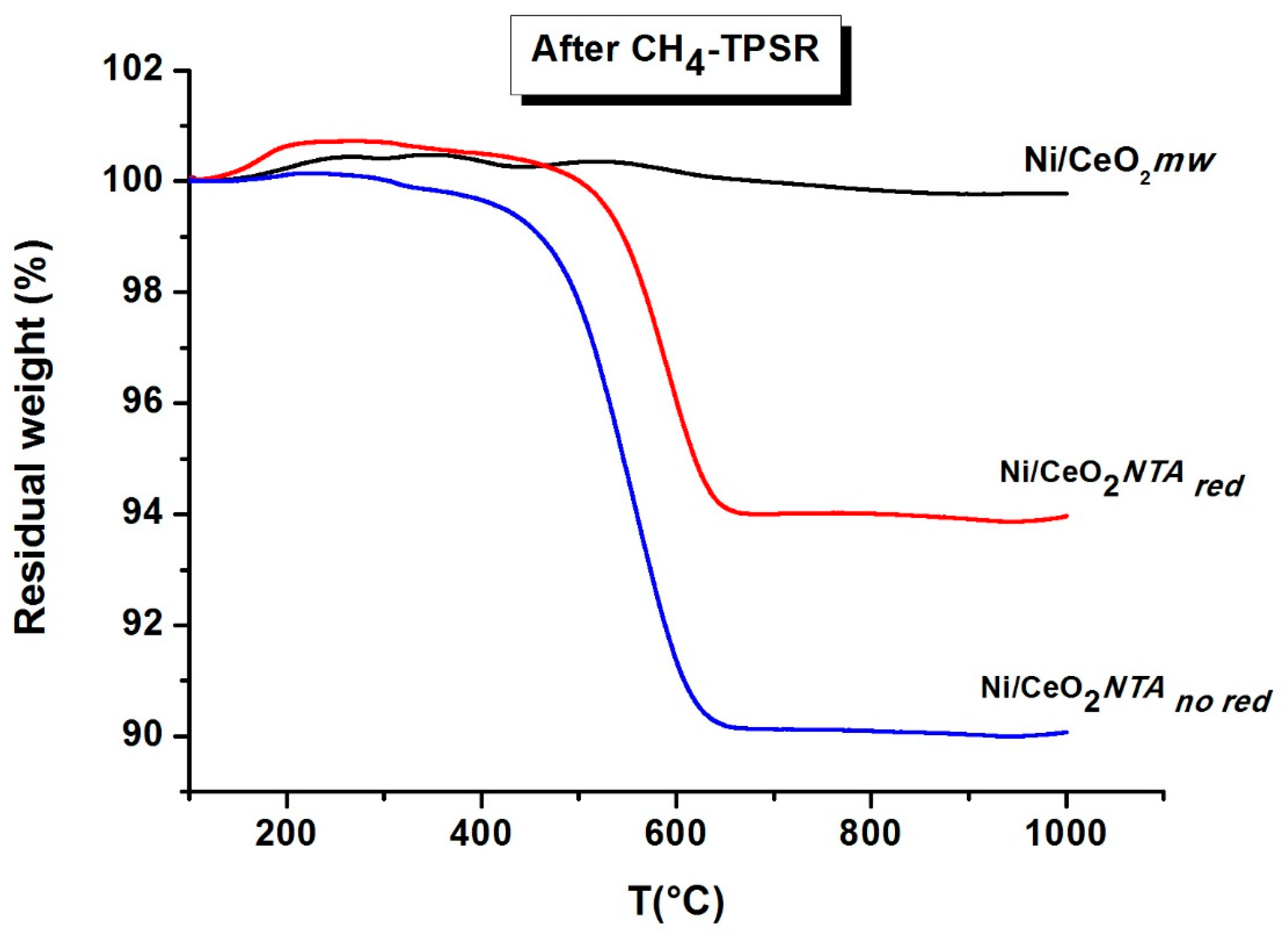
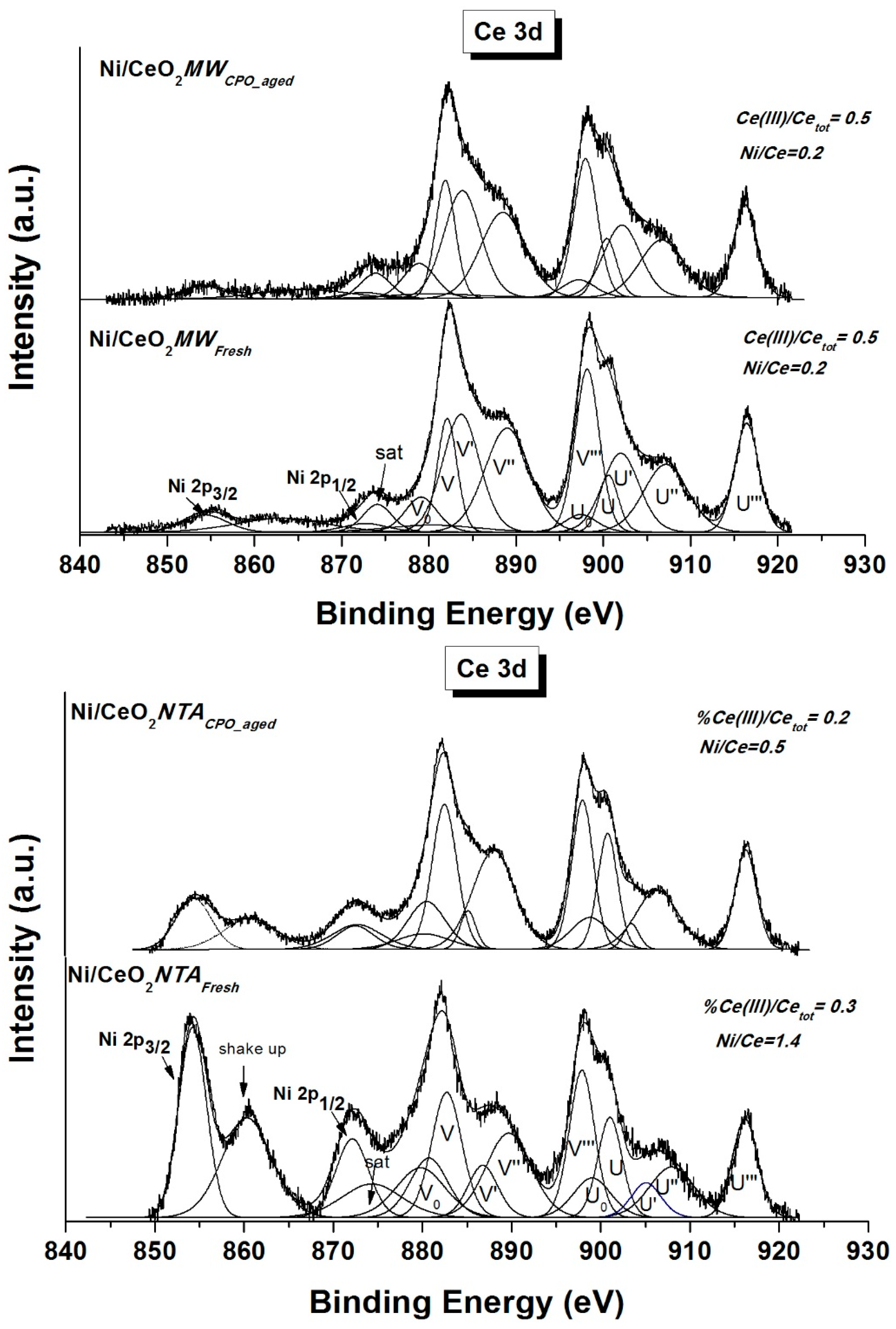
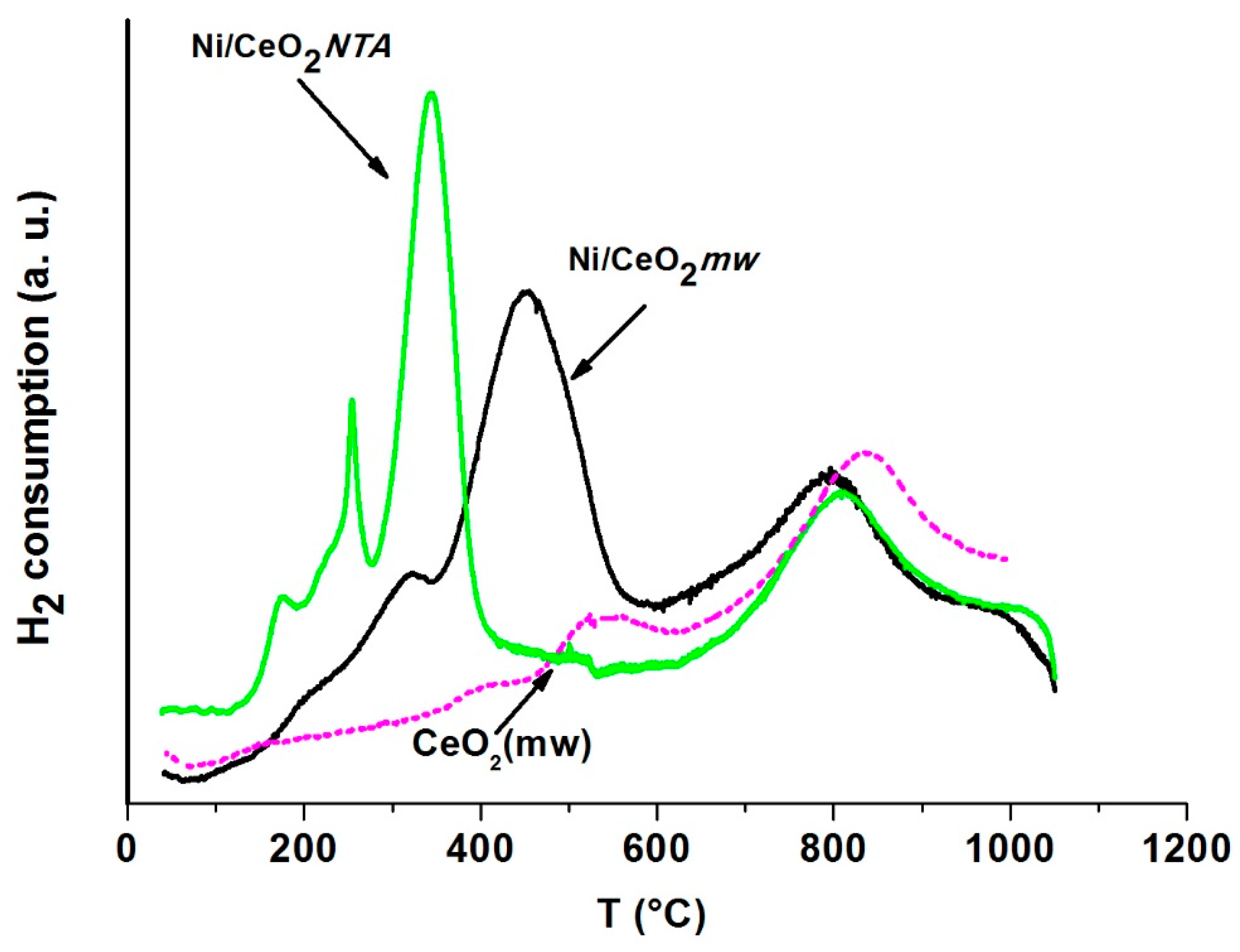
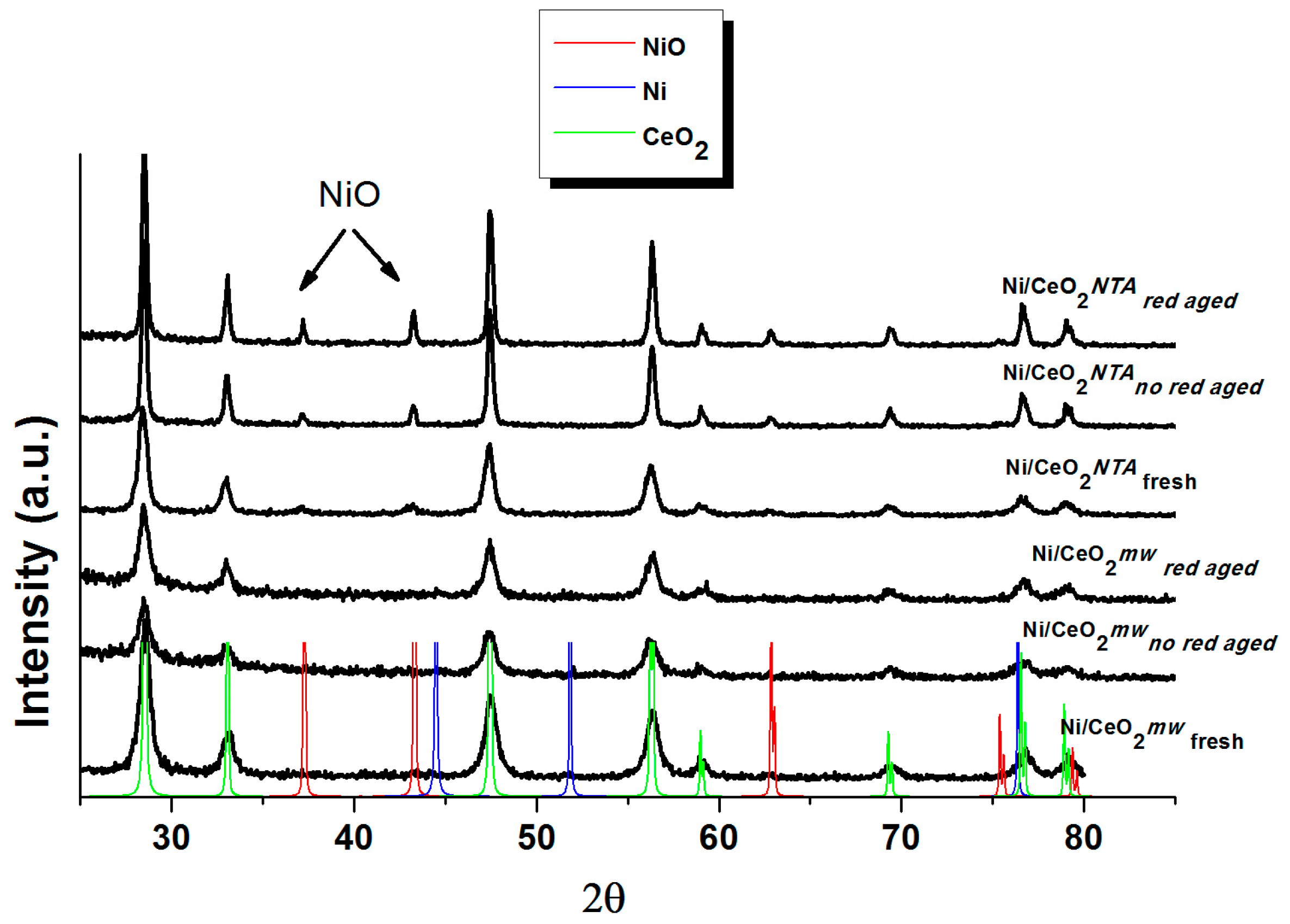
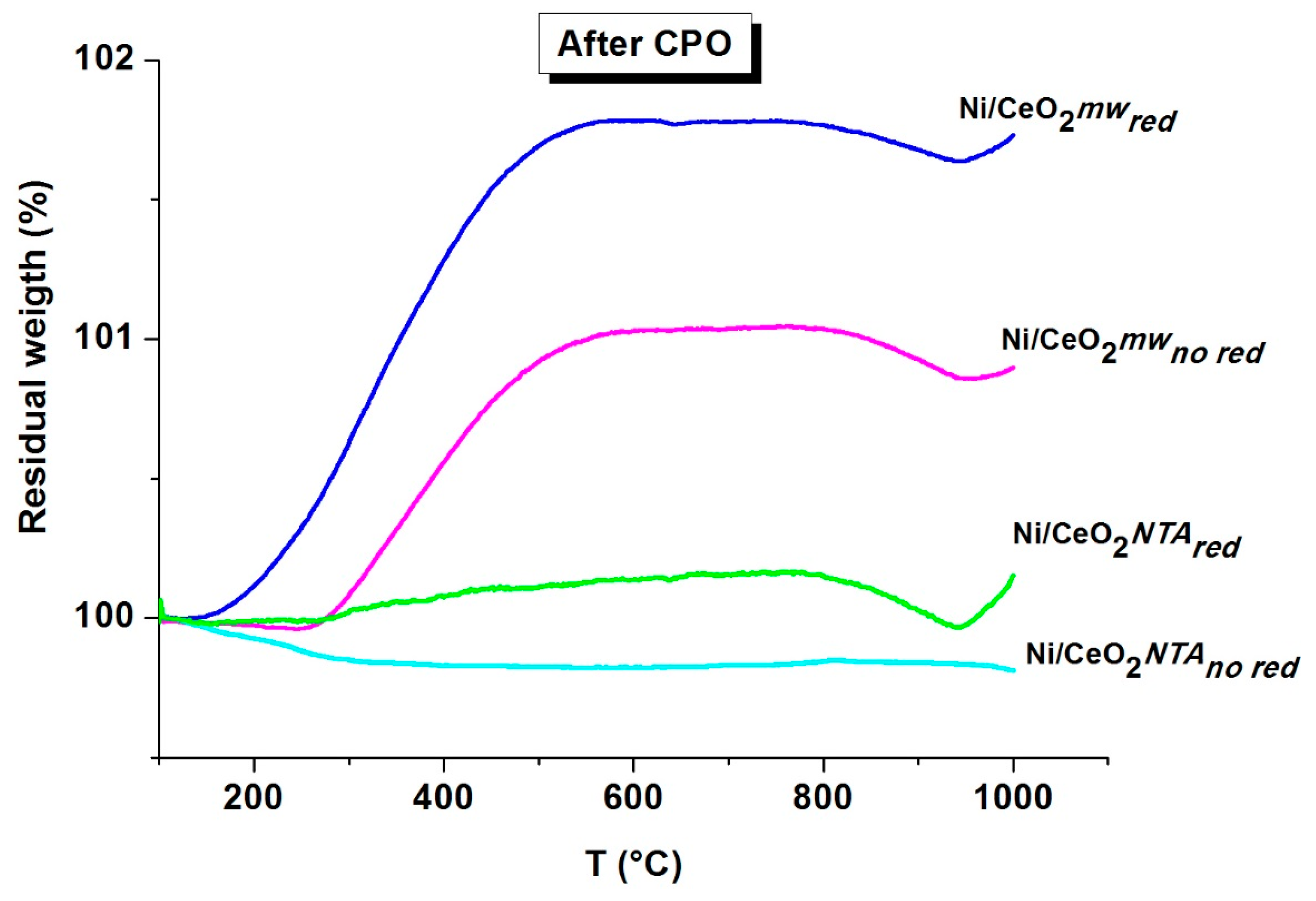
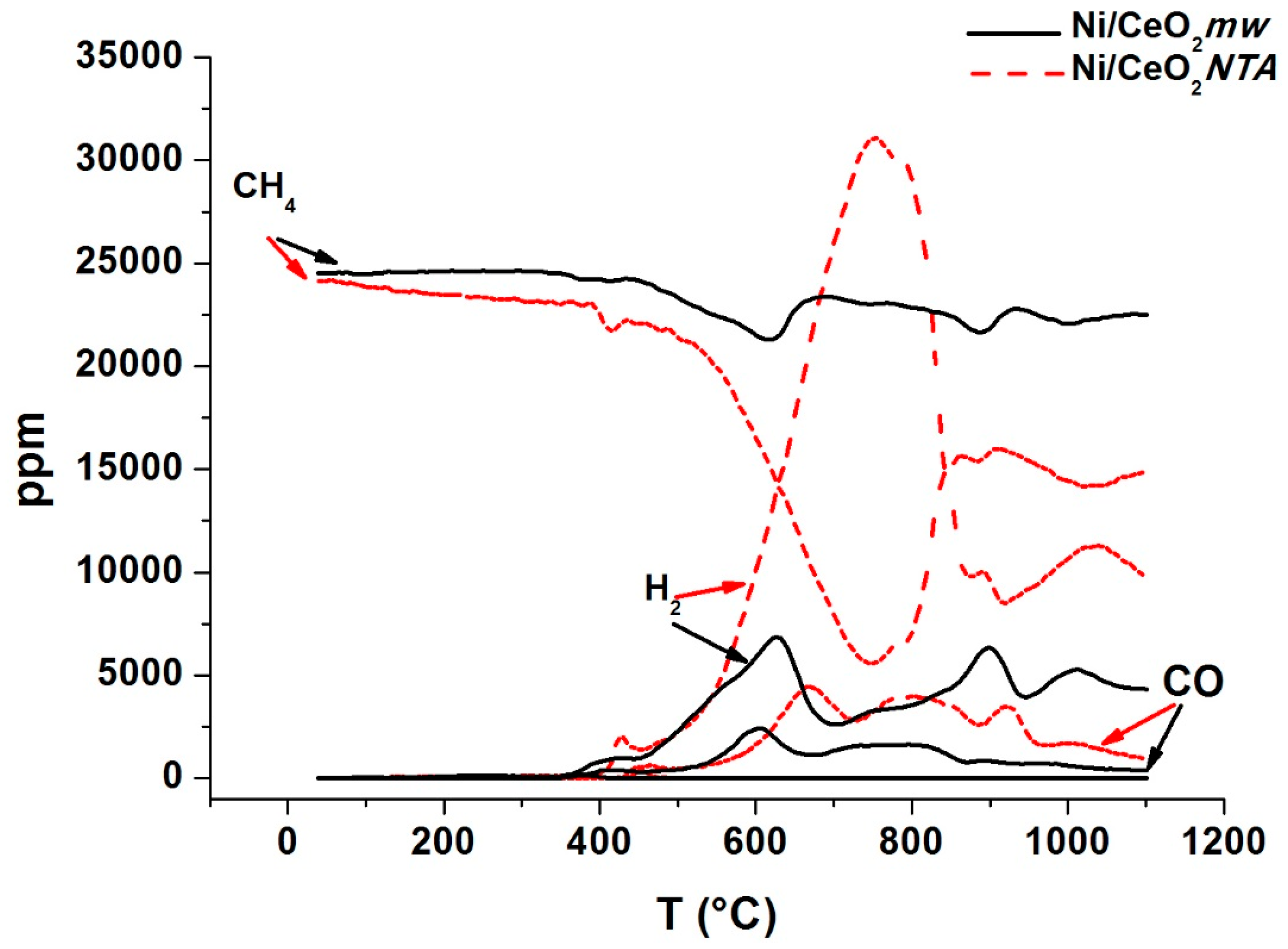
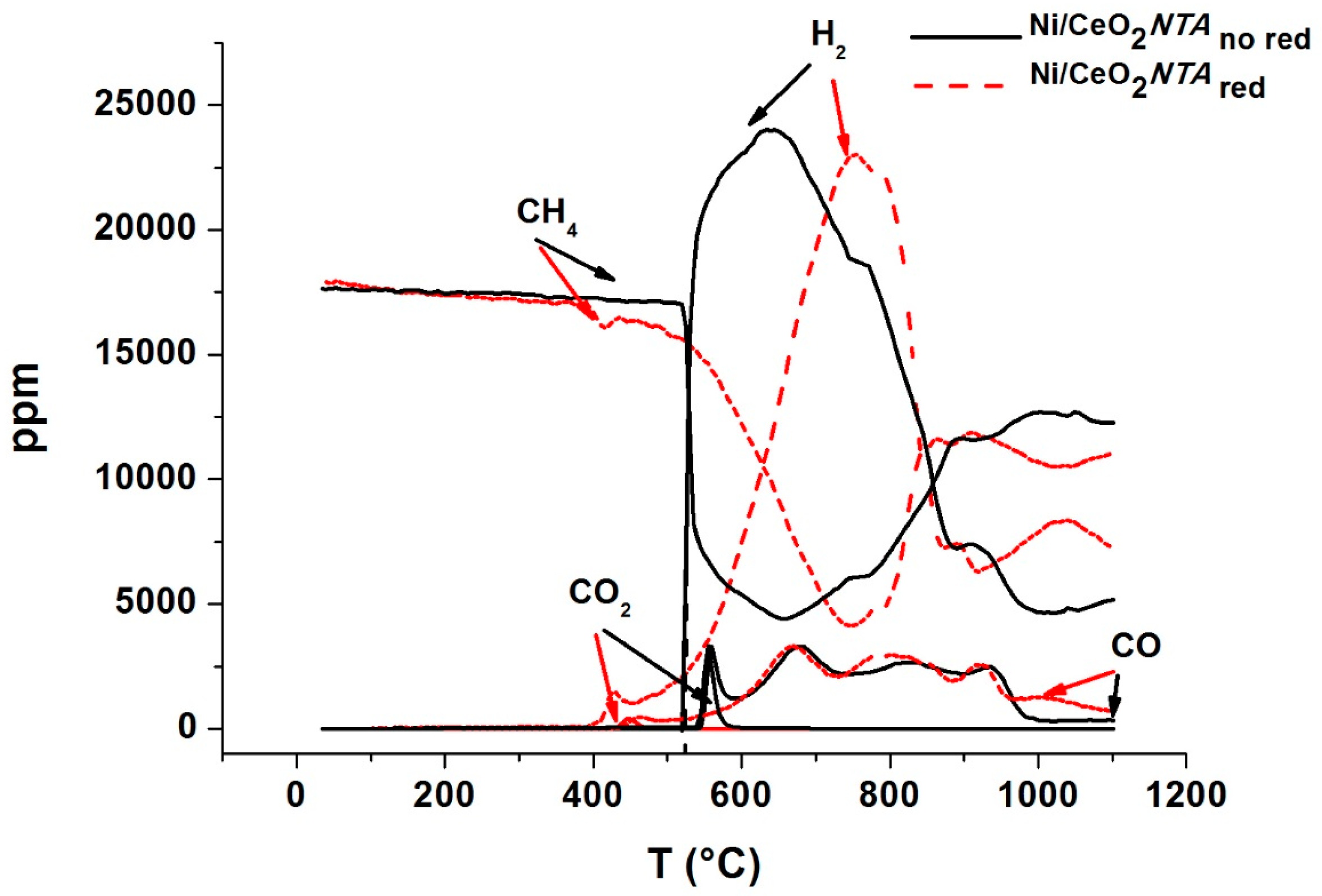
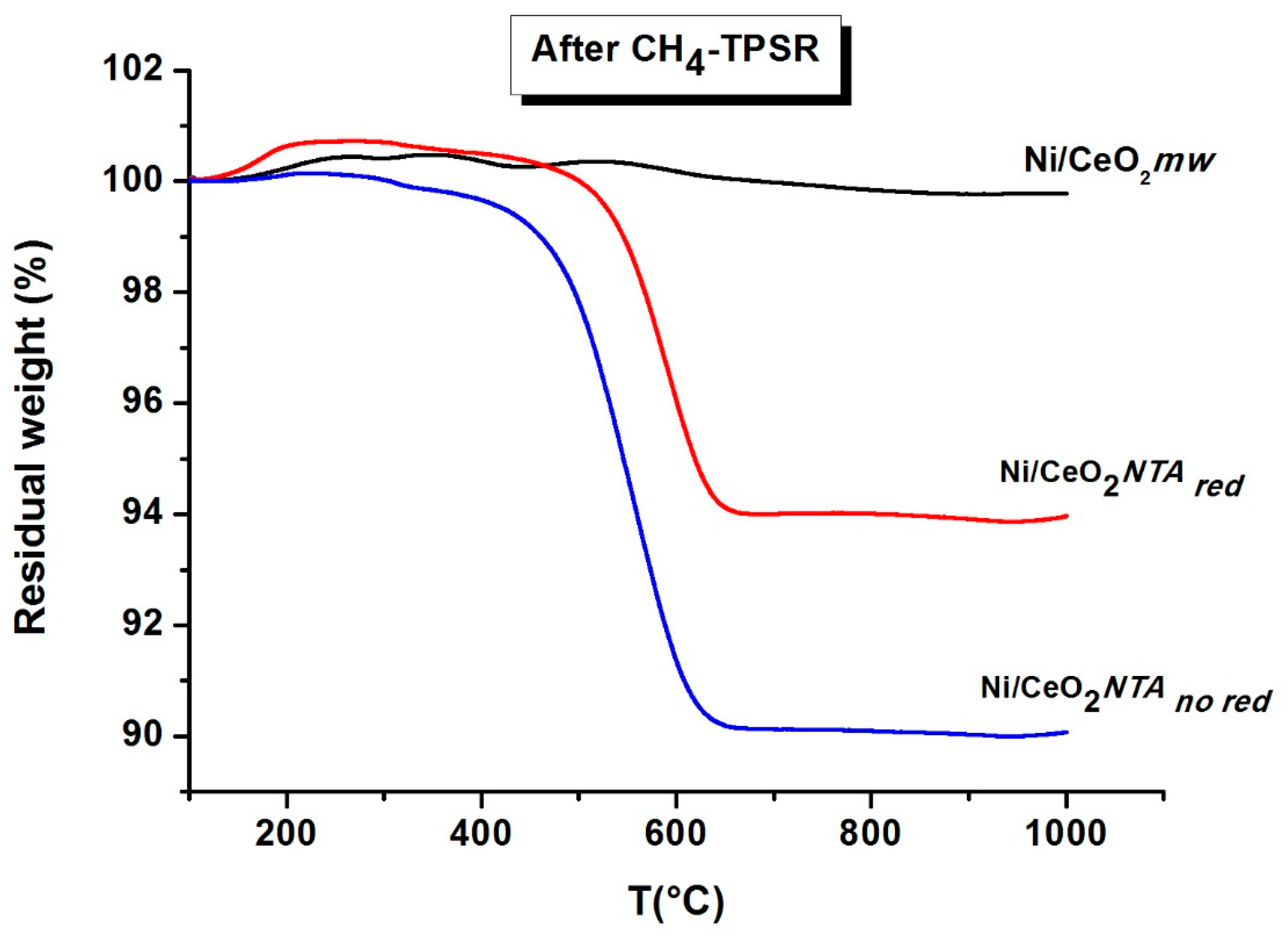
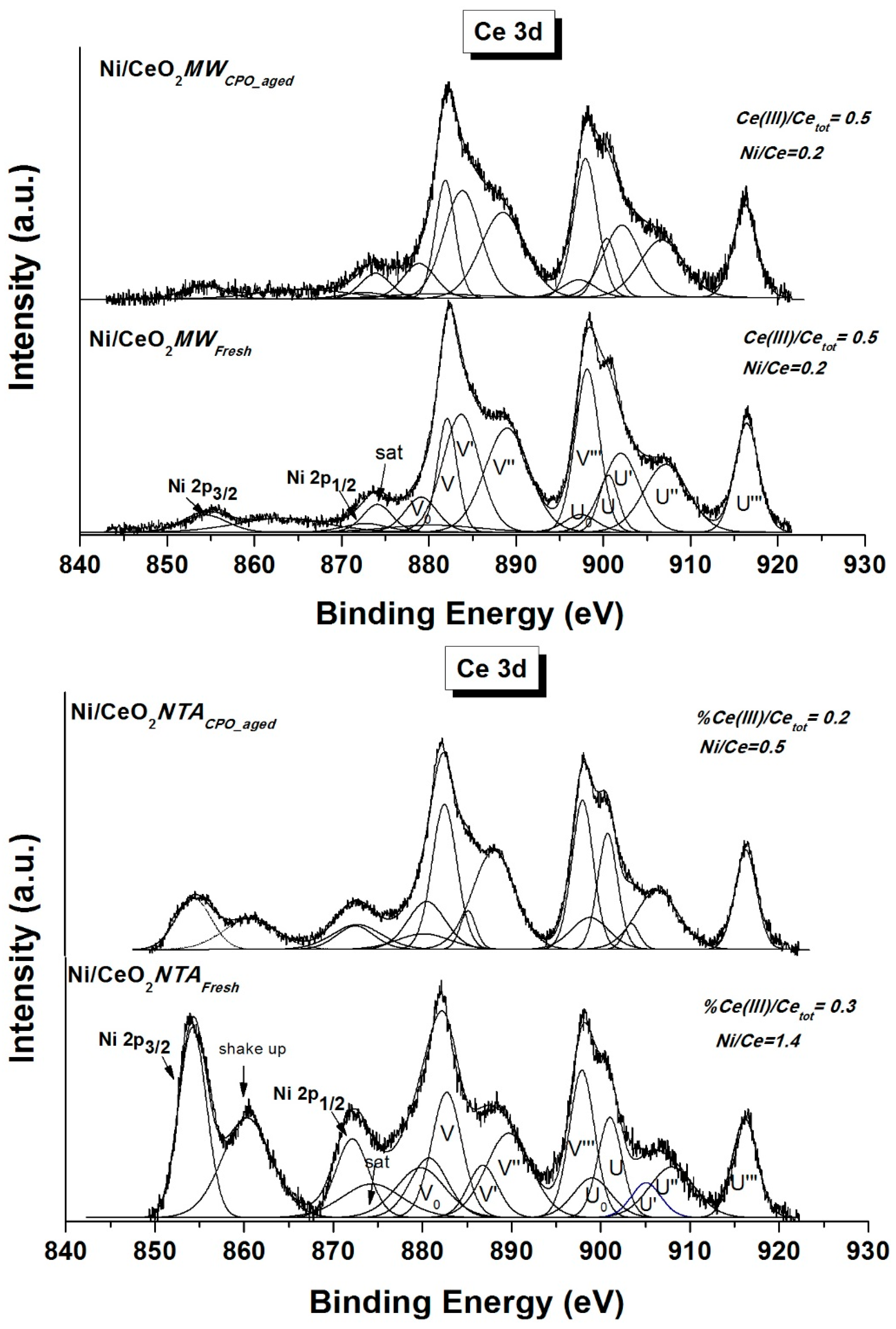
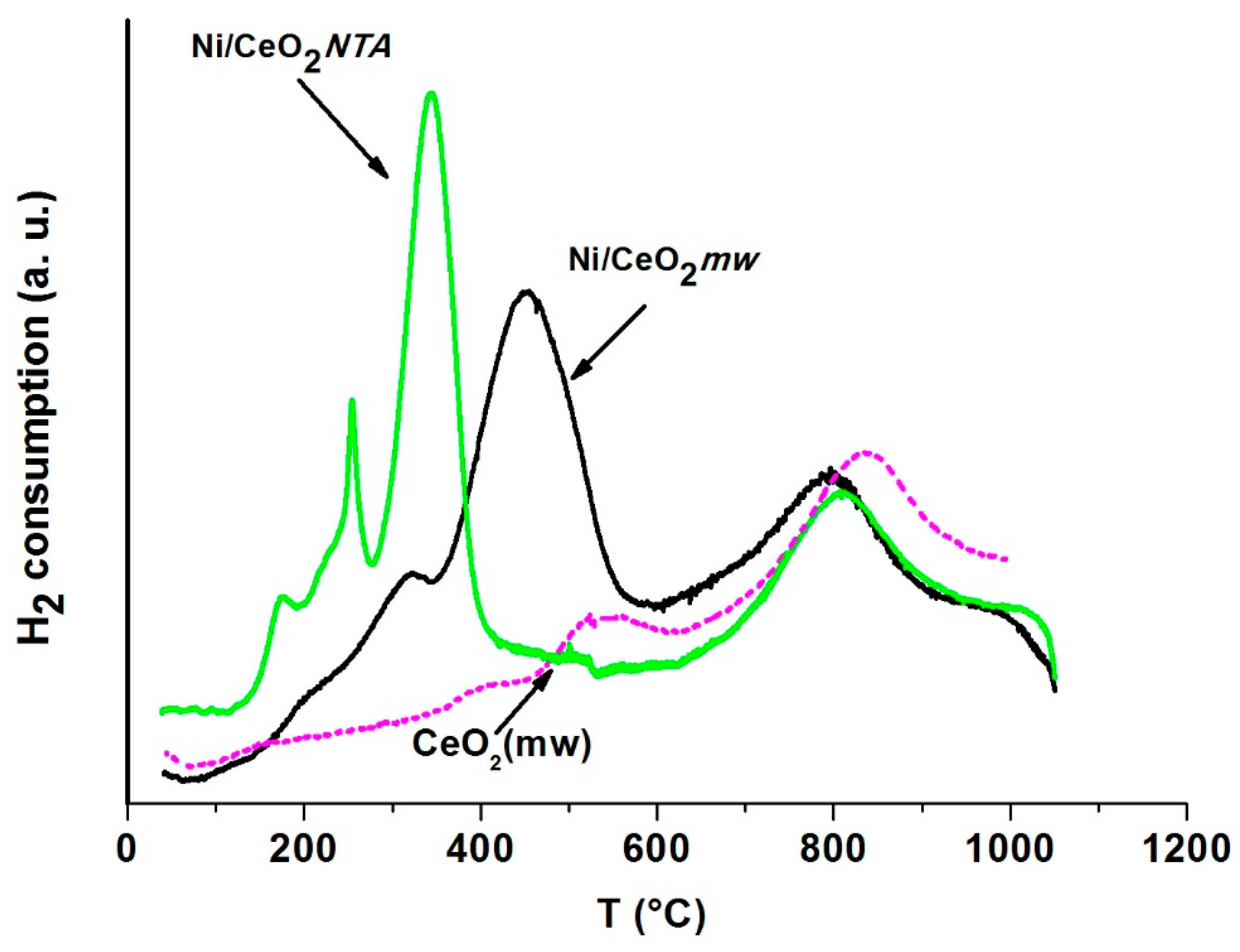
2.2. Characterization
3. Experimental
3.1. Sample Preparation
3.2. Sample Characterization
3.3. Catalytic Measurements
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Al-Sayari, S.A. Recent developments in the partial oxidation of methane to syngas. Open Catal. J. 2013, 6, 17–28. [Google Scholar] [CrossRef]
- Enger, B.C.; Lødeng, R.; Holmen, A. A review on catalytic partial oxidation of methane to synthesis gas with emphasis on reaction mechanism over transition metal catalysts. Appl. Catal. A 2008, 346, 1–27. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, W.Z.; Jin, G.-Q.; Wang, Y.-Y.; Guo, X.-Y. Biomorphic SiC pellets as catalyst support for partial oxidation of methane to syngas. Appl. Catal. B 2008, 79, 307–312. [Google Scholar] [CrossRef]
- Pengpanich, S.; Meeyo, V.; Rirksomboon, T. Methane partial oxidation over Ni/CeO2-ZrO2 mixed oxide solid solution catalyst. Catal. Today 2004, 93–95, 95–105. [Google Scholar] [CrossRef]
- Asencios, Y.J.O.; Nascente, P.A.P.; Assaf, E.M. Partial oxidation of methane on NiO-MgO-ZrO2 catalysts. Fuel 2012, 97, 630–637. [Google Scholar] [CrossRef]
- Zhu, T.; Flytzani-Stephanopoulos, M. Catalytic partial oxidation of metahne to synthesis gas over Ni-CeO2. Appl. Catal. A 2001, 208, 403–417. [Google Scholar] [CrossRef]
- Pal, P.; Singha, R.K.; Saha, A.; Bal, R.; Panda, A.Q.B. Defect-Induced efficient partial oxidation of methane over nonstoichiometric Ni/CeO2 nanocrystals. J. Phys. Chem. C 2015, 119, 13610–13618. [Google Scholar] [CrossRef]
- Nolan, M. Charge transfer and formation of reduced Ce3+ upon adsorption of metal atoms at the ceria (110) surface. J. Chem. Phys. 2012, 136, 134703–134709. [Google Scholar] [CrossRef] [PubMed]
- Sayle, D.C.; Sayle, T.C.T. Atomistic modelling of ceria nanostructures: Introducing structural complexity. In Catalytic Properties of Ceria and CeO2-Containing Materials, 2nd ed.; Trovarelli, A., Fornasiero, P., Eds.; Imperial College Press: London, UK, 2013; pp. 247–290. [Google Scholar]
- Duhamel, L.J.; Zarrou, H.; D’Huysser, A. Hydrogen production at low temperature from methane on cerium and nickel based mixed oxide. Int. J. Hydrogen Energy 2008, 33, 5527–5534. [Google Scholar]
- Zou, W.; Ge, C.; Lu, M.; Wu, S.; Wang, T.; Sun, J.; Pu, Y.; Tang, C.; Gao, F.; Domg, L. Engineering the NiO/CeO2 interface to enhance the catalytic performance for CO oxidation. RSC Adv. 2015, 5, 98335–98341. [Google Scholar] [CrossRef]
- Zhang, X.; You, R.; Li, D.; Cao, T.; Huang, W. Reaction sensitivity of ceria morphology effect on Ni/CeO2 catalysis in propane oxidation reactions. Appl. Mater. Interface 2017, 9, 35897–35907. [Google Scholar] [CrossRef] [PubMed]
- Moraes, T.S.; Neto, R.C.R.; Ribeiro, M.C.; Mattos, L.V.; Kourtelesis, M.; Verykios, X.; Noronha, F.B. Effects of ceria morphology on catalytic performances of Ni/CeO2 Catalysts for low temperature steam reforming of ethanol. Top. Catal. 2015, 58, 281–294. [Google Scholar] [CrossRef]
- Jin, R.; Chen, Y.; Li, W.; Cui, W.; Ji, Y.; Yu, C.; Jiang, Y. Mechanism for catalytic partial oxidation of methane to syngas over a Ni/Al2O3 catalyst. Appl. Catal. A 2000, 201, 71–80. [Google Scholar] [CrossRef]
- Shan, W.; Fleys, M.; Lapicque, F.; Swierczynski, D.; Kiennemann, A.; Simon, Y.; Marquaire, P.-M. Syngas production from partial oxidation of methane over Ce1−XNiXOY catalysts prepared by complexation-combustion method. Appl. Catal. A 2006, 311, 24–33. [Google Scholar] [CrossRef]
- Singha, R.K.; Shukla, A.; Yadav, A.; Konathala, L.N.S.; Bal, R. Effect of metal-support interaction on activity and stability of Ni-CeO2 catalyst for partial oxidation of methane. Appl. Catal. B 2017, 201, 473–488. [Google Scholar] [CrossRef]
- Pantaleo, G.; la Parola, V.; Deganello, F.; Singha, R.K.; Bal, R.; Venezia, A.M. Ni/CeO2 catalysts for methane partial oxidation: Synthesis driven structural and catalytic effects. Appl. Catal. B 2016, 189, 233–241. [Google Scholar] [CrossRef]
- Pantaleo, G.; la Parola, V.; Deganello, F.; Calatozzo, P.; Bal, R.; Venezia, A.M. Synthesis and support composition effects on CH4 partial oxidation over Ni-CeLa oxides. Appl. Catal. B 2015, 164, 135–141. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, B.; Tang, X.; Xu, Y.; Shen, W. Hydrogen production from methane decomposition over Ni/CeO2 catalysts. Catal. Commun. 2006, 7, 380–386. [Google Scholar] [CrossRef]
- Klug, H.P.; Alexander, L.E. X-ray Diffraction Procedures for Polycrystalline and Amorphous Materials, 2nd ed.; John Wiley and Sons: New York, NY, USA, 1974. [Google Scholar]
- Biesinger, M.C.; Payen, B.P.; Lau, L.W.M.; Gerson, A.; Smart, R.S.C. X-ray photoelectron spectroscopic chemical state quantification of mixed nickel metal, oxide and hydroxide systems. Surf. Interface Anal. 2009, 41, 324–332. [Google Scholar] [CrossRef]
- Renuka, N.K.; Harsha, N.; Divya, T. Supercharged ceria quantum dots with exceptionally high oxygen buffer action. RSC Adv. 2015, 5, 38837–38841. [Google Scholar] [CrossRef]
- Rãduţoiu, N.; Teodorescu, C.M. Satellites in Ce 3d X-ray photoelectron spectroscopy of ceria. Dig. J. Nanomater. Biostruct. 2013, 8, 1535–1549. [Google Scholar]
- Choudhury, B.; Choudhury, A. Ce3+ and oxygen vacancy mediated tuning of structural and optical properties of CeO2 nanoparticles. Mater. Chem. Phys. 2012, 131, 666–671. [Google Scholar] [CrossRef]
- Paparazzo, E.; Ingo, G.M.; Zacchetti, N. X-ray induced reduction effects at CeO2 surfaces: An X-ray photoelectron spectroscopy study. J. Vac. Sci. Technol. A 1991, 9, 1416–1419. [Google Scholar] [CrossRef]
- Diskin, A.M.; Cunningham, R.H.; Ormerod, R.M. The oxidative chemistry of methane over supported nickel catalysts. Catal. Today 1998, 46, 147–154. [Google Scholar] [CrossRef]
- Qin, W.; Xie, K.; Liu, M.; Wiu, G.; Wang, Y.; Zhang, Y. Single phase nickel-doped ceria cathode with in situ grown nickel nanocatalyst for direct high temperature carbon dioxide electrolysis. RSC Adv. 2014, 4, 40494–40504. [Google Scholar]
- Liu, Z.; Grinter, D.C.; Lustemberg, P.G.; Nguyen-Phan, T.; Zhou, Y.; Luo, S.; Waluyo, I.; Crumlin, E.J.; Stacchiola, D.J.; Zhou, J.; et al. Dry reforming of methane on a highly active Ni-CeO2 catalyst: Effects of metal-support interactions on C-H bond breaking. Angew. Chem. Int. Ed. Engl. 2016, 55, 7455–7459. [Google Scholar] [CrossRef] [PubMed]
- Van Dillen, A.J.; Terorde, R.J.A.M.; Lensveld, D.J.; Geus, J.W.; de Jong, K.P. Synthesis of supported catalysts by impregnation and drying using aqueous chelated metal complexes. J. Catal. 2003, 216, 257–264. [Google Scholar] [CrossRef]
- Venezia, A.M.; la Parola, V.; Deganello, G.; Cauzzi, D.; Leonardi, G.; Predieri, G. Influence of the prepration method on the thiophene HDS activbity of silica supported CoMo catalysts. Appl. Catal. A 2002, 229, 261–271. [Google Scholar] [CrossRef]
- Inorganic Crystal Structure Database (ICSD). Fachinformationszentrum Karlsruhe; Inorganic Crystal Structure Database: Karlsruhe, Germany, 2014. [Google Scholar]
- Lødeng, R.; Bjørgum, E.; Enger, B.C.; Eilertsen, J.L.; Holmen, A.; Krogh, B.; Rønnekleiv, M.; Rytter, E. Catalytic partial oxidation of CH4 to H2 over cobalt catalysts at moderate temperatures. Appl. Catal. A 2007, 333, 11–23. [Google Scholar] [CrossRef]
- Boucouvalas, Y.; Zhang, Z.; Verykios, X.E. Heat transport limitations and reaction scheme of partial oxidation of methane to synthesis gas over rhodium catalysts. Catal. Lett. 1994, 27, 131–142. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | BET (m2/g) | NiO d (nm) | CeO2 d (nm) | Lattice Parameter a (Å) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Fresh | Aged | Fresh | Aged | Fresh CeO2 | |||||
| No Red | Red | No Red | Red | ||||||
| Support | CeO2 | 30 | - | - | - | 18 | - | - | 5.411 |
| Catalyst | Ni/CeO2mw | 26 | nd | nd | nd | 13 | 15 | 17 | 5.412 |
| Ni/CeO2NTA | 28 | 13 | 49 | 60 | 16 | 30 | 34 | 5.410 | |
| Sample | Tmax (°C) | V (mL/gcatal) | |||||
|---|---|---|---|---|---|---|---|
| 1st Peak | 2nd Peak | 3rd Peak | 1st Peak | 2nd Peak | 3rd Peak | ||
| Support | CeO2 | - | 538 | 835 | - | 6 | 27 |
| Catalyst | Ni/CeO2mw | 320 | 450 | 796 | 5 | 28 | 27 |
| Ni/CeO2NTA | 250, 170 | 343 | 807 | 6 | 22 | 28 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Parola, V.; Pantaleo, G.; Venezia, A.M. Effects of Synthesis on the Structural Properties and Methane Partial Oxidation Activity of Ni/CeO2 Catalyst. Catalysts 2018, 8, 220. https://doi.org/10.3390/catal8050220
La Parola V, Pantaleo G, Venezia AM. Effects of Synthesis on the Structural Properties and Methane Partial Oxidation Activity of Ni/CeO2 Catalyst. Catalysts. 2018; 8(5):220. https://doi.org/10.3390/catal8050220
Chicago/Turabian StyleLa Parola, Valeria, Giuseppe Pantaleo, and Anna Maria Venezia. 2018. "Effects of Synthesis on the Structural Properties and Methane Partial Oxidation Activity of Ni/CeO2 Catalyst" Catalysts 8, no. 5: 220. https://doi.org/10.3390/catal8050220
APA StyleLa Parola, V., Pantaleo, G., & Venezia, A. M. (2018). Effects of Synthesis on the Structural Properties and Methane Partial Oxidation Activity of Ni/CeO2 Catalyst. Catalysts, 8(5), 220. https://doi.org/10.3390/catal8050220