Hydrogenation of m-Chloronitrobenzene over Different Morphologies Ni/TiO2 without Addition of Molecular Hydrogen

Abstract
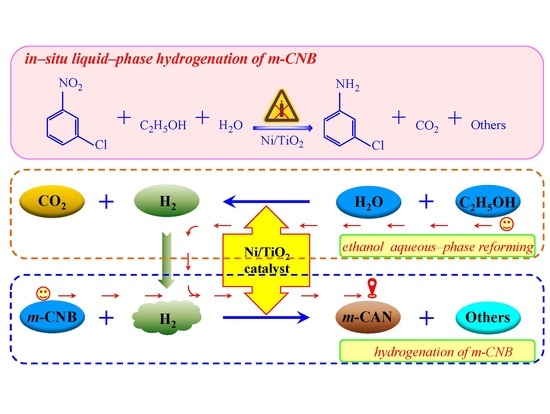
:
1. Introduction
2. Results and Discussion
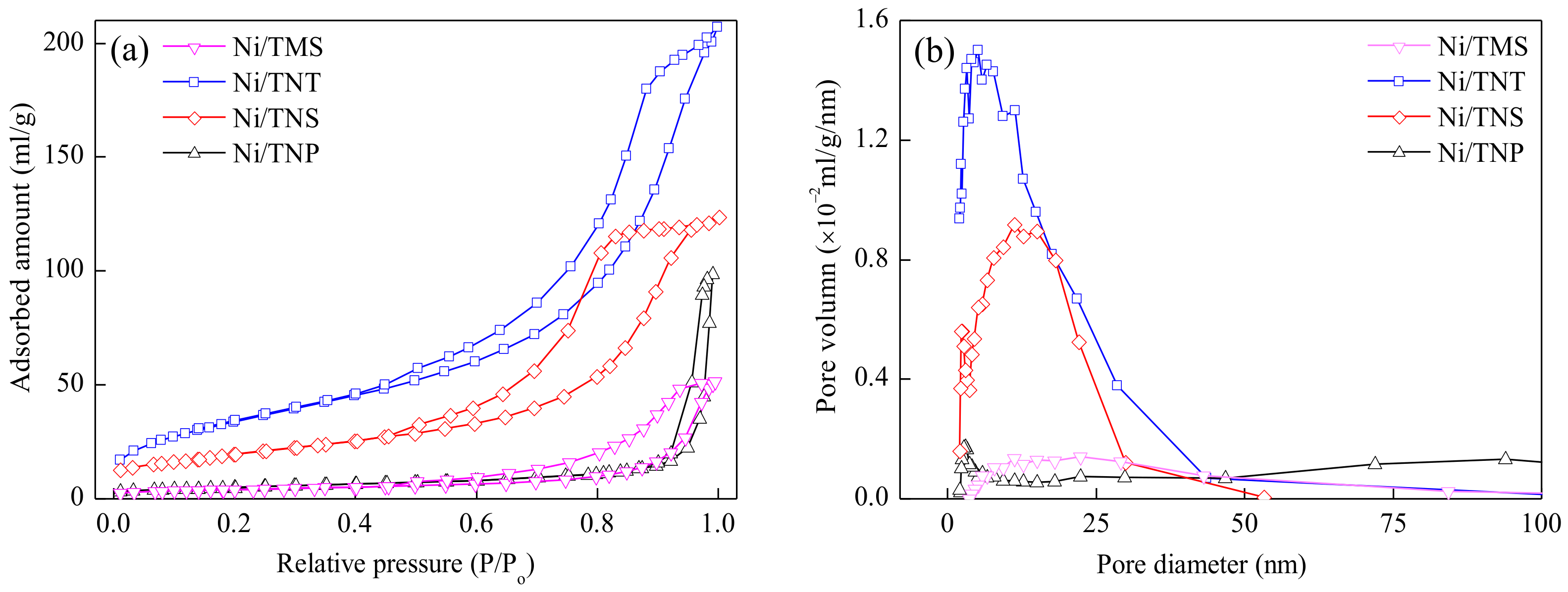
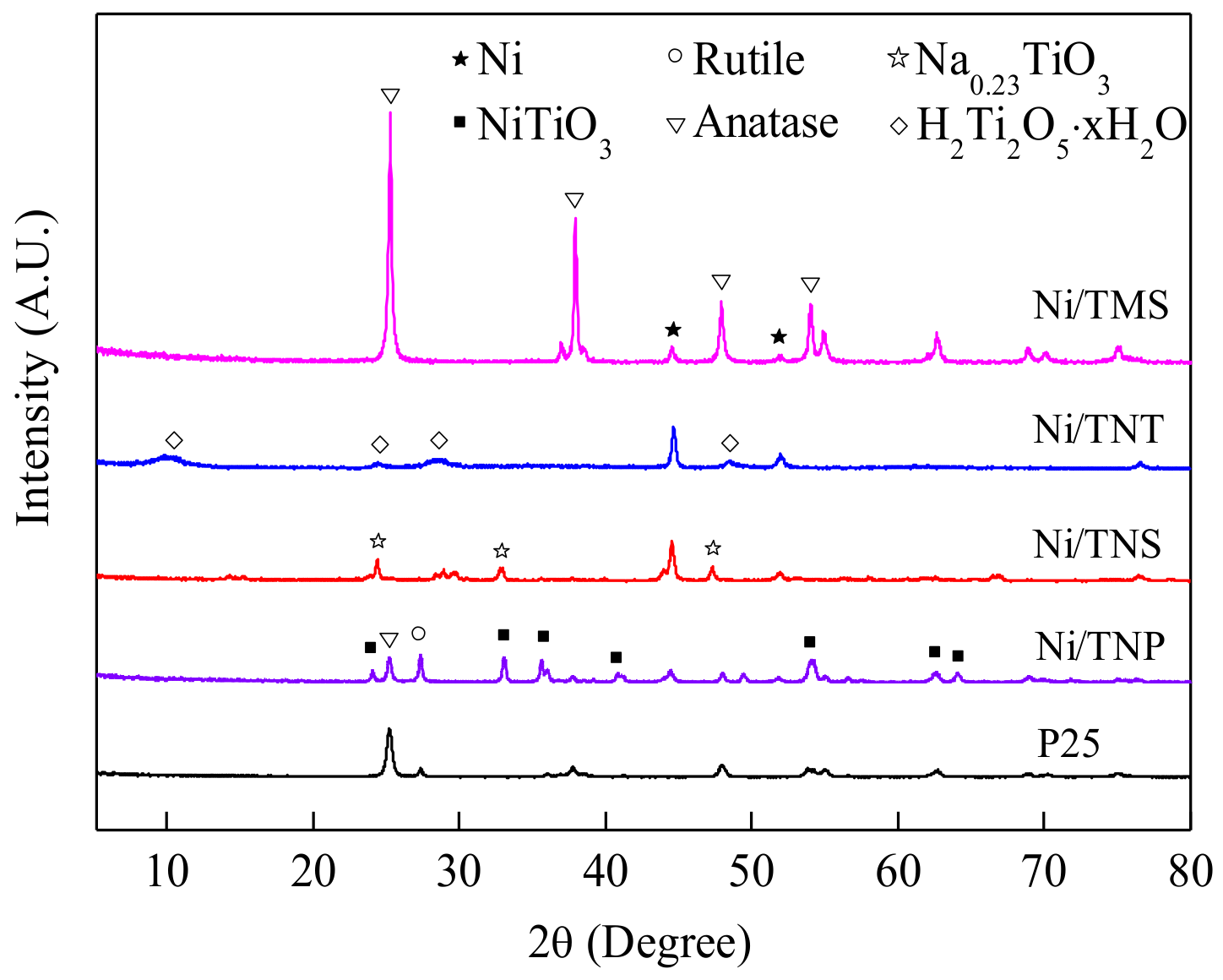
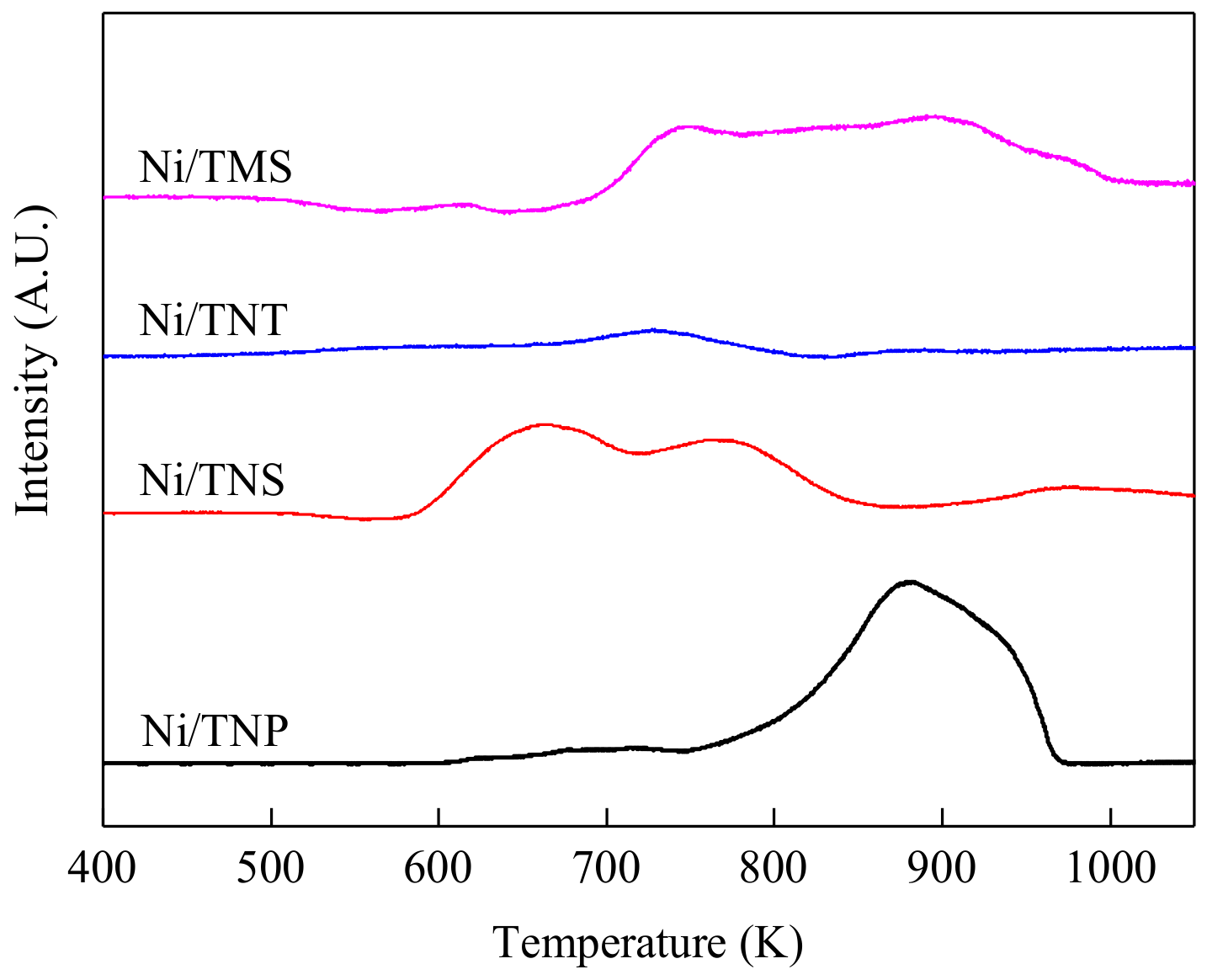
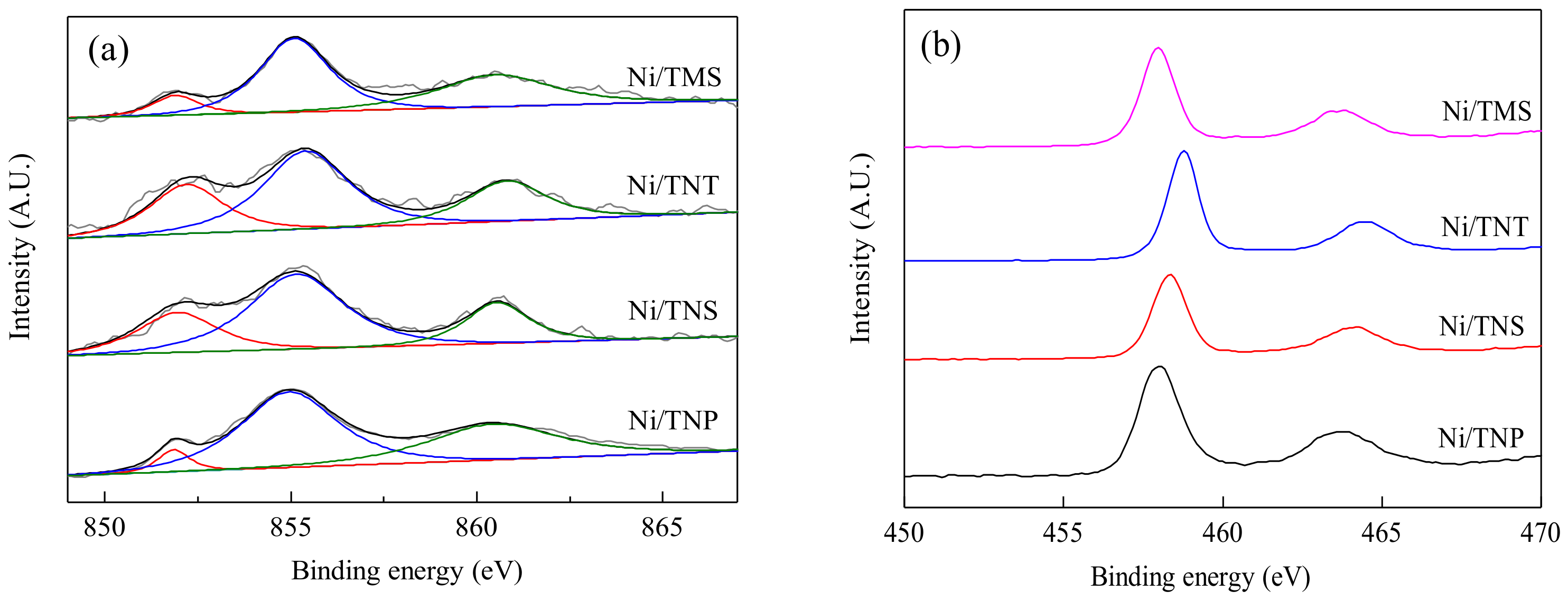
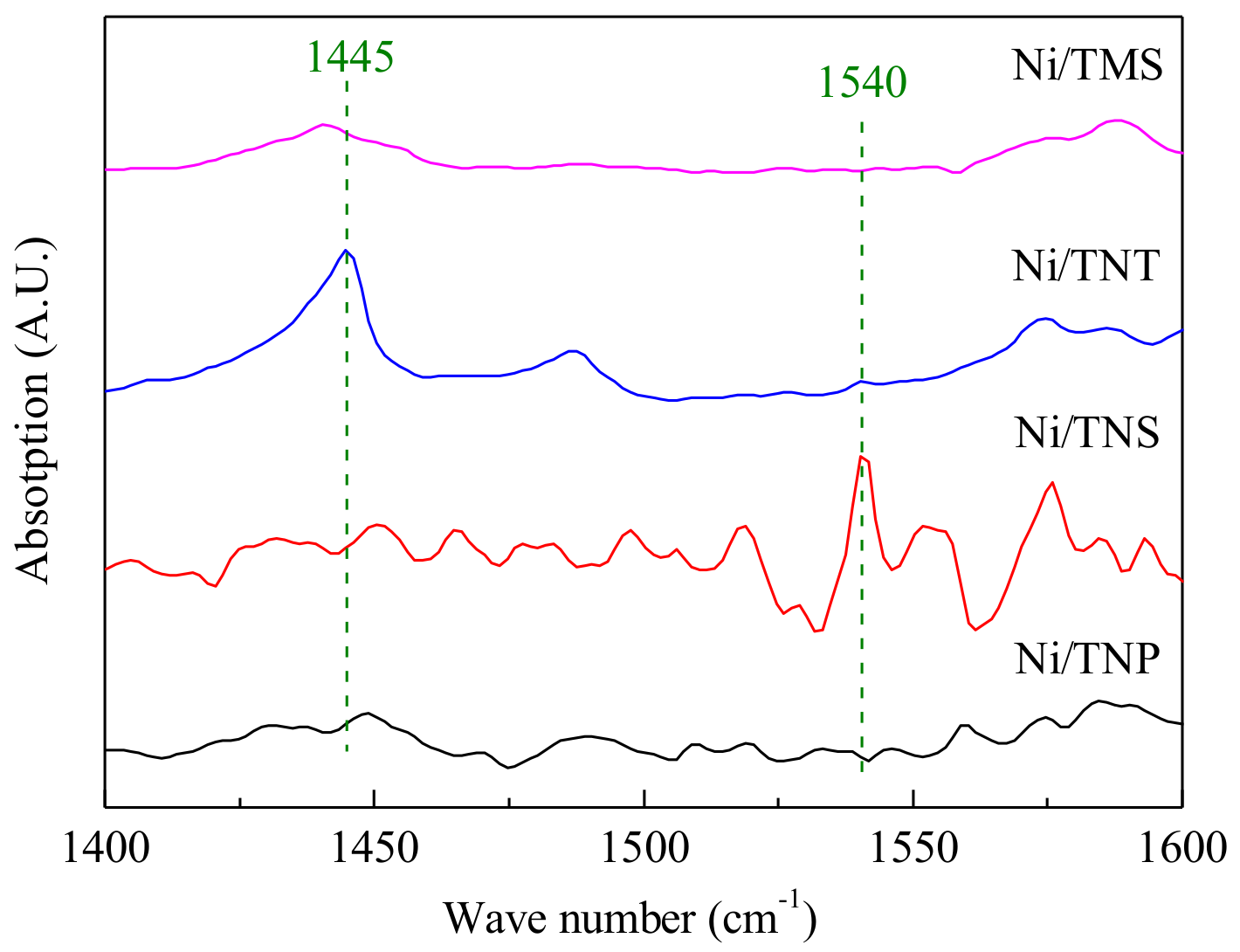
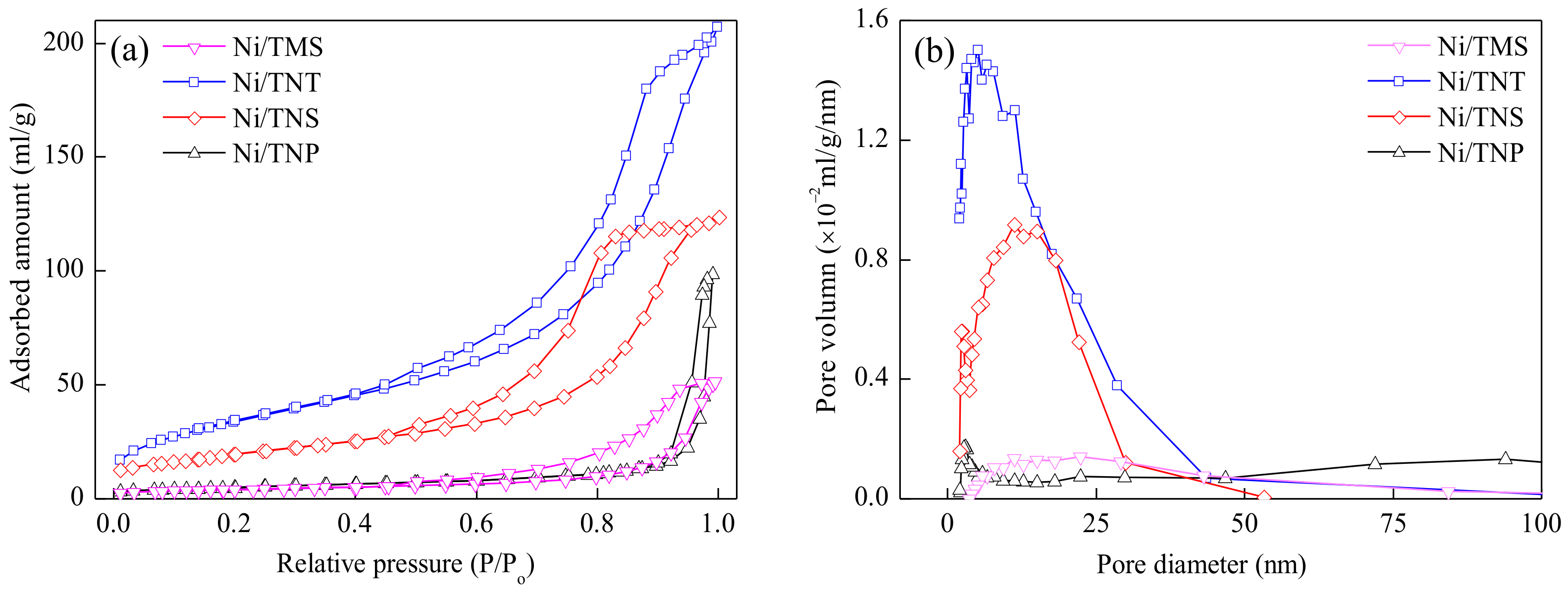
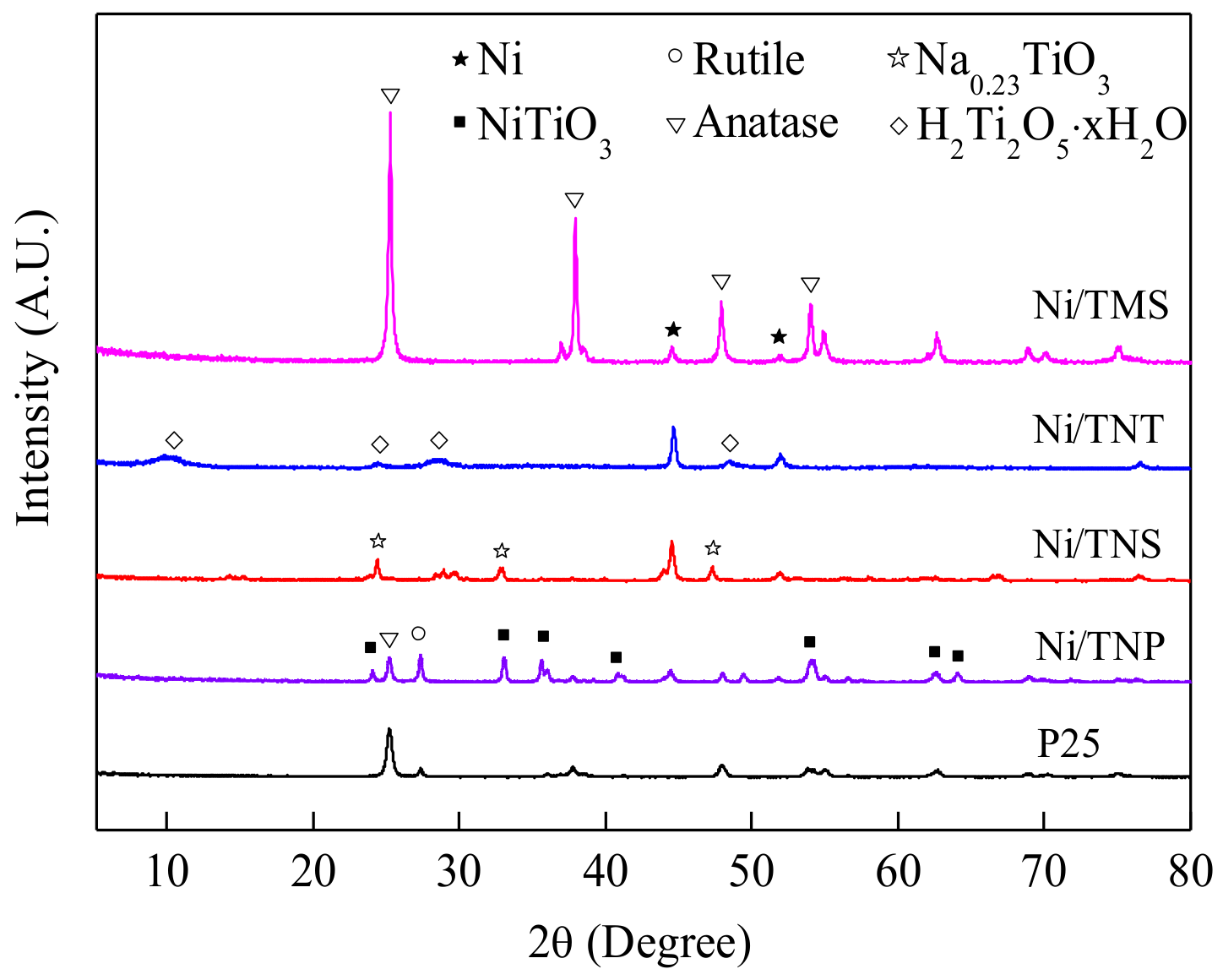
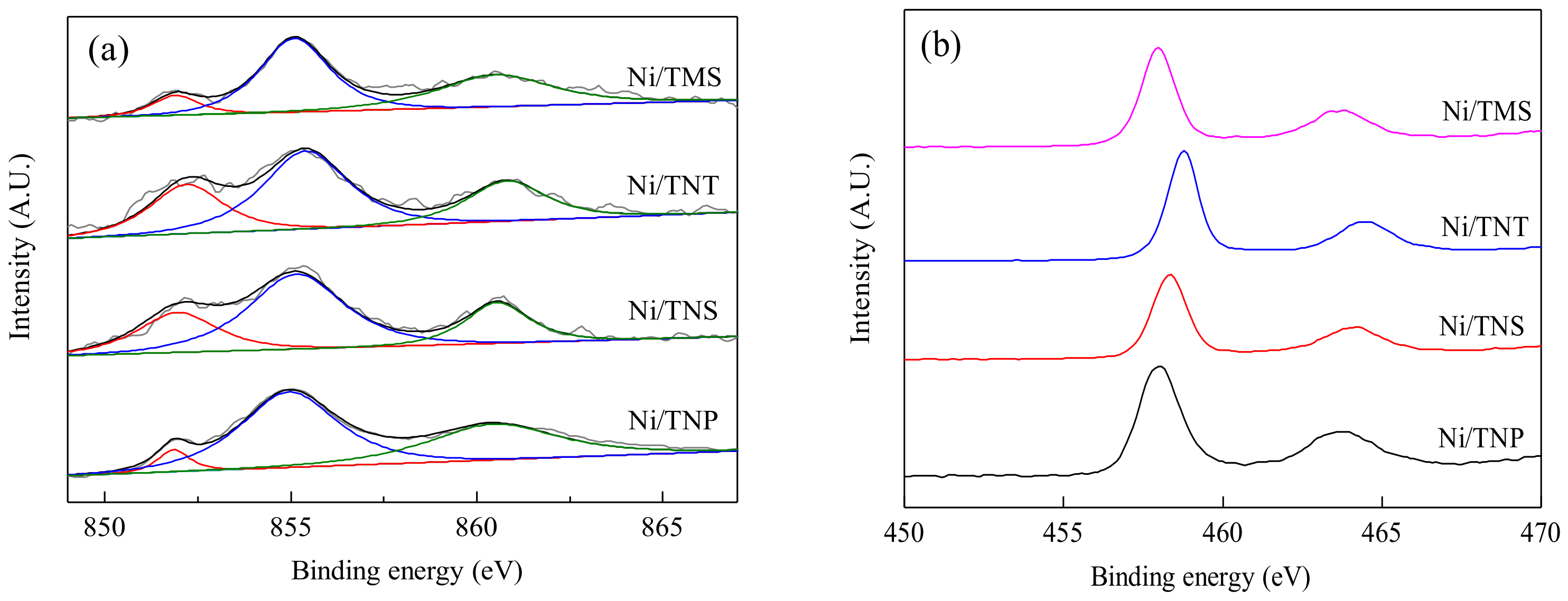
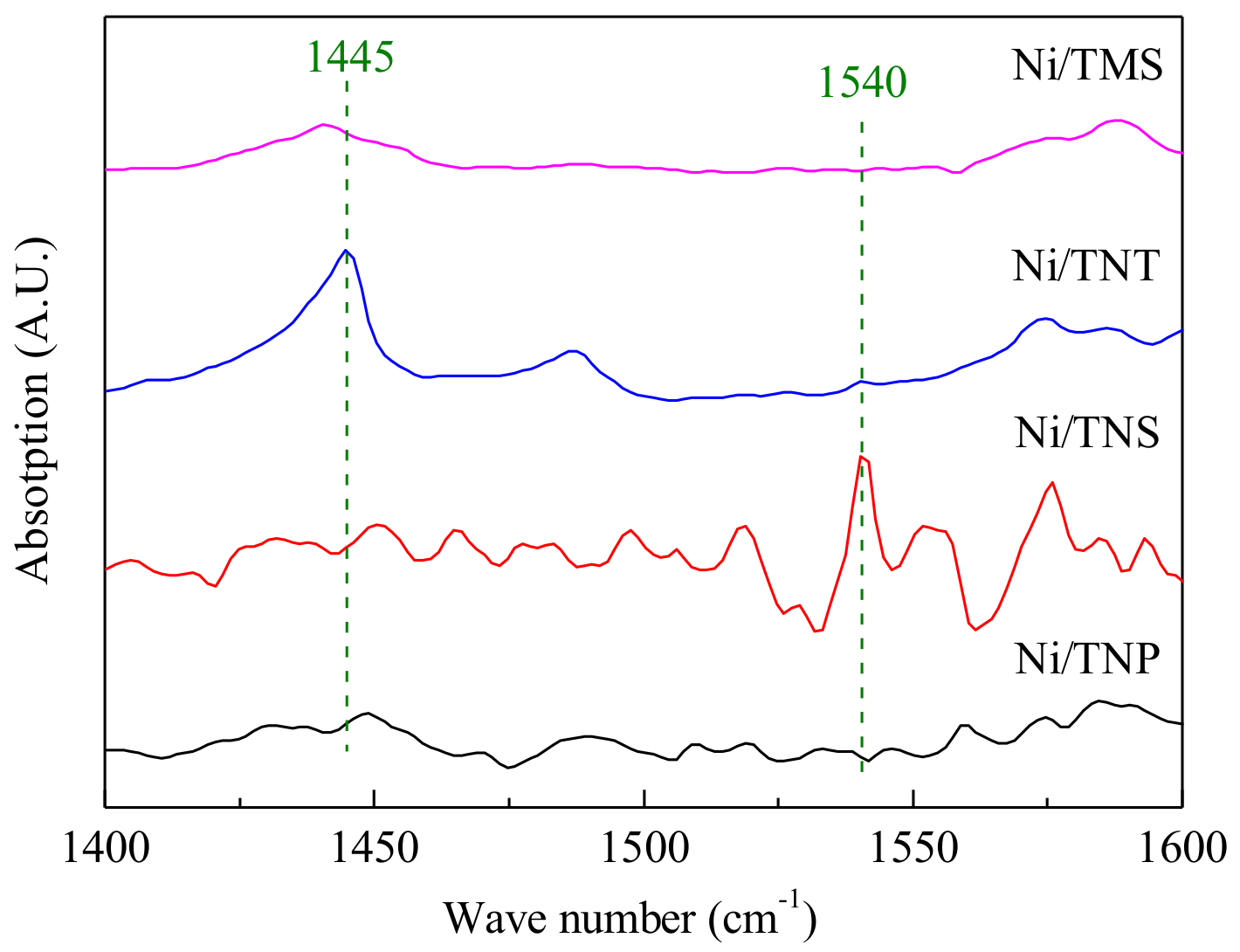
2.1. Catalyst Characterization
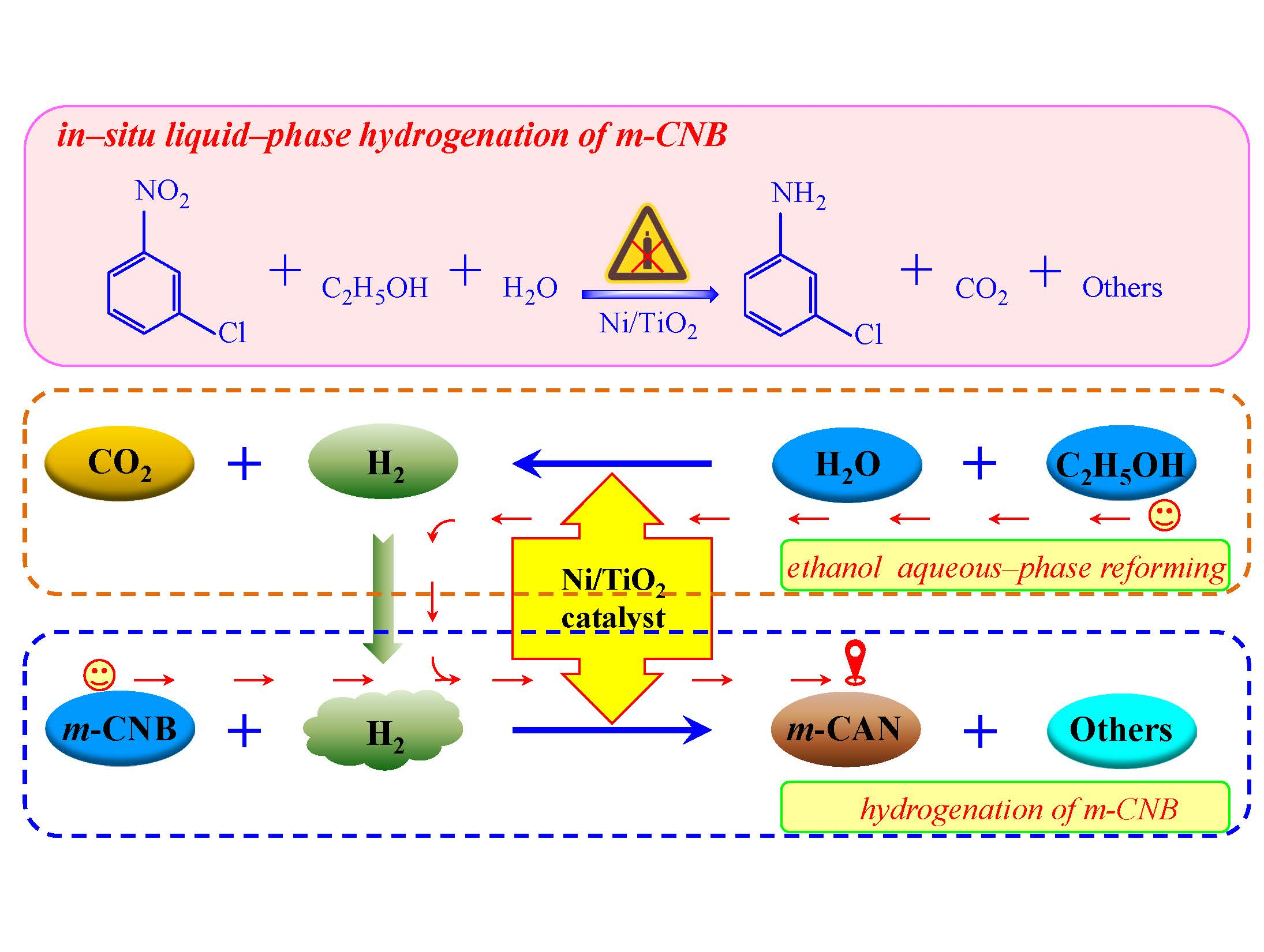
2.2. In-Situ Liquid-Phase Hydrogenation of m-CNB
3. Materials and Methods
3.1. Chemicals
3.2. Catalyst Preparation
3.3. Catalyst Characterization
3.4. Catalytic Test
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Xiong, J.; Chen, J.; Zhang, J. Liquid-phase hydrogenation of o-chloronitrobenzene over supported nickel catalysts. Catal. Commun. 2007, 8, 345–350. [Google Scholar] [CrossRef]
- Ramadas, K.; Srinivasan, N. Iron-ammonium chloride—A convenient and inexpensive reductant. Synth. Commun. 2006, 22, 3189–3195. [Google Scholar] [CrossRef]
- Pradhan, N.C.; Man, M.S. Reactions of nitrochlorobenzenes with sodium sulfide: Change in selectivity with phase-transfer catalysts. Ind. Eng. Chem. Res. 1992, 31, 1606–1609. [Google Scholar] [CrossRef]
- Lauwiner, M.; Rys, P.; Wissmann, J. Reduction of aromatic nitro compounds with hydrazine hydrate in the presence of an iron oxide hydroxide catalyst. I. The reduction of monosubstituted nitrobenzenes with hydrazine hydrate in the presence of ferrihydrite. Appl. Catal. A Gen. 1998, 172, 141–148. [Google Scholar] [CrossRef]
- Stutts, K.J.; Scortichini, C.L.; Repucci, C.M. Electrochemical reduction of nitroaromatics to anilines in basic media: Effects of positional isomerism and cathode composition. J. Org. Chem. 1989, 54, 3740–3744. [Google Scholar] [CrossRef]
- Li, F.; Liang, J.; Zhu, W.; Song, H.; Wang, K.; Li, C. In-situ liquid hydrogenation of m-chloronitrobenznen over Fe-modified Pt/carbon nanotubes catalysts. Catalysts 2018, 8, 62. [Google Scholar] [CrossRef]
- Sierra-Salazar, A.F.; Hulea, V.; Ayral, A.; Chave, T.; Nikitenko, S.I.; Kooyman, P.J.; Tichelaar, F.D.; Abate, S.; Perathoner, S.; Lacroix-Desmazes, P. Engineering of silica-supported platinum catalysts with hierarchical porosity combining latex synthesis, sonochemistry and sol-gel process-II. Catalytic performance. Microporous Mesoporous Mater. 2018, 256, 227–234. [Google Scholar] [CrossRef]
- Wang, X.; Liang, M.; Zhang, J.; Wang, Y. Selective hydrogenation of aromatic chloronitro compounds. Curr. Org. Chem. 2007, 11, 299–314. [Google Scholar] [CrossRef]
- Pietrowski, M. Recent developments in heterogeneous selective hydrogenation of halogenated nitroaromatic compounds to halogenated anilines. Curr. Org. Synth. 2012, 9, 470–487. [Google Scholar] [CrossRef]
- Yu, W.; Lin, H.W.; Tan, C.S. Direct synthesis of Pd incorporated in mesoporous silica for solvent-free selective hydrogenation of chloronitrobenzenes. Chem. Eng. J. 2017, 325, 124–133. [Google Scholar] [CrossRef]
- Kothari, R.; Buddhi, D.; Sawhney, R.L. Comparison of environmental and economic aspects of various hydrogen production methods. Renew. Sustain. Energy Rev. 2008, 12, 553–563. [Google Scholar] [CrossRef]
- Zeng, K.; Zhang, D. Recent progress in alkaline water electrolysis for hydrogen production and applications. Prog. Energy Combust. Sci. 2010, 36, 307–326. [Google Scholar] [CrossRef]
- Martina, S.; Aleksi, R.P.; Reetta, K. Hydrogen production via aqueous-phase reforming of methanol over nickel modified Ce, Zr and La oxide supports. Catal. Today 2018, 304, 143–152. [Google Scholar]
- Tahay, P.; Khani, Y.; Jabari, M.; Bahadoran, F.; Safari, N. Highly porous monolith/TiO2 supported Cu, Cu-Ni, Ru, and Pt catalysts in methanol steam reforming process for H2 generation. Appl. Catal. A Gen. 2018, 554, 44–53. [Google Scholar] [CrossRef]
- Ribeirinha, P.; Mateos-Pedrero, C.; Boaventura, M.; Sousa, J.; Mendes, A. CuO/ZnO/Ga2O3 catalyst for low temperature MSR reaction: Synthesis, characterization and kinetic model. Appl. Catal. B Environ. 2018, 221, 371–379. [Google Scholar] [CrossRef]
- Ferencz, Z.; Varga, E.; Puskás, R.; Kónya, Z.; Baán, K.; Oszkó, A.; Erdőhelyi, A. Reforming of ethanol on Co./Al2O3 catalysts reduced at different temperatures. J. Catal. 2018, 358, 118–130. [Google Scholar] [CrossRef]
- Słowik, G.; Greluk, M.; Rotko, M.; Machocki, A. Evolution of the structure of unpromoted and potassium-promoted ceria-supported nickel catalysts in the steam reforming of ethanol. Appl. Catal. B Environ. 2018, 221, 490–509. [Google Scholar] [CrossRef]
- Godina, L.I.; Anton, V.T.; Simakova, I.L.; Mäki-Arvela, P.; Kortesmäki, E.; Gläsel, J.; Kronberg, L.; Etzold, B.; Murzin, Y.D. Aqueous-phase reforming of alcohols with three carbon atoms on carbon-supported Pt. Catal. Today 2018, 301, 78–89. [Google Scholar] [CrossRef]
- Cortright, R.D.; Davda, R.R.; Dumesic, J.A. Hydrogen from catalytic reforming of biomass-derived hydrocarbons in liquid water. Nature 2002, 418, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Davda, R.R.; Dumesic, J.A. Catalytic reforming of oxygenated hydrocarbons for hydrogen with low levels of carbon monoxide. Angew. Chem. Int. Ed. 2003, 42, 4068–4071. [Google Scholar] [CrossRef] [PubMed]
- Huber, G.W.; Shabaker, J.W.; Dumesic, J.A. Raney Ni-Sn catalyst for H2 production from biomass-derived hydrocarbons. Science 2003, 300, 2075–2077. [Google Scholar] [PubMed]
- Coronado, I.; Stekrova, M.; Reinikainen, M.; Simell, P.; Lefferts, L.; Lehtonen, J. A review of catalytic aqueous-phase reforming of oxygenated hydrocarbons derived from biorefinery water fractions. Int. J. Hydrog. Energy 2016, 41, 11003–11032. [Google Scholar] [CrossRef]
- Roy, B.; Martinez, U.; Loganathan, K.; Datye, A.K.; Leclerc, C.A. Effect of preparation methods on the performance of Ni/Al2O3 catalysts for aqueous-phase reforming of ethanol: Part I—catalytic activity. Int. J. Hydrog. Energy 2012, 37, 8143–8153. [Google Scholar] [CrossRef]
- Roy, B.; Artyushkova, K.; Pham, H.N.; Li, L.; Datye, A.K.; Leclerc, C.A. Effect of preparation methods on the performance of Ni/Al2O3 catalysts for aqueous-phase reforming of ethanol: Part II—characterization. Int. J. Hydrog. Energy 2012, 37, 18815–18826. [Google Scholar] [CrossRef]
- Roy, B.; Leclerc, C.A. Study of preparation method and oxidization/reduction effect on the performance nickel-cerium oxide catalysts for aqueous-phase reforming of ethanol. J. Power Sources 2015, 299, 114–124. [Google Scholar] [CrossRef]
- Mahata, N.; Cunha, A.F.; Órfão, J.J.M.; Figueiredo, J.L. Hydrogenation of chloronitrobenzenes over filamentous carbon stabilized nickel nanoparticles. Catal. Commun. 2009, 10, 1203–1206. [Google Scholar] [CrossRef]
- Meng, X.; Cheng, H.; Frjita, S.; Hao, Y.; Shang, Y.; Yu, Y.; Cai, S.; Zhao, F.; Arai, M. Selective hydrogenation of chloronitrobenzene to chloroaniline in supercritical carbon dioxide over Ni/TiO2: Significance of molecular interactions. J. Catal. 2010, 269, 131–139. [Google Scholar] [CrossRef]
- Fujishima, A.; Rao, T.N.; Tryk, D.A. Titanium dioxide photocatalysis. J. Photochem. Photobiol. C Photochem. Rev. 2000, 1, 1–21. [Google Scholar] [CrossRef]
- Chen, X.; Mao, S.S. Titanium dioxide nanomaterials: Synthesis, properties, modifications, and applications. Chem. Rev. 2007, 107, 2891–2959. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, H.; Lu, D.; Wu, W.H.; Ding, J.Q.; Zhao, X.N.; Xiong, R.Y.; Yang, M.C.; Wu, P.; Chen, F.T.; et al. Visible-light-driven water splitting from dyeing wastewater using Pt surface-dispersed TiO2-based nanosheets. J. Alloys Compd. 2016, 699, 183–192. [Google Scholar] [CrossRef]
- Lu, D.; Chai, W.; Yang, M.; Fang, P.; Wu, W.; Zhao, B.; Xiong, R.; Wang, H. Visible light induced photocatalytic removal of Cr(VI) over TiO2-based nanosheets loaded with surface-enriched CoOx nanoparticles and its synergism with phenol oxidation. Appl. Catal. B Environ. 2016, 190, 44–65. [Google Scholar] [CrossRef]
- Zhao, P.J.; Wu, R.; Hou, J.; Chang, A.M.; Guan, F.; Zhang, B. One-step hydrothermal synthesis and visible-light photocatalytic activity of ultrafine Cu-nanodot-modified TiO2 nanotubes. Acta Phys.-Chim. Sin. 2012, 28, 1971–1977. [Google Scholar]
- Lei, B.X.; Zhang, P.; Xie, M.L.; Li, Y.; Wang, S.N.; Yu, Y.Y.; Sun, W.; Sun, Z.F. Constructing hierarchical porous titania microspheres from titania nanosheets with exposed (001) facets for dye-sensitized solar cells. Electrochim. Acta 2015, 173, 497–505. [Google Scholar] [CrossRef]
- Chen, F.; Fang, P.; Gao, Y.; Liu, Z.; Yang, L.; Dai, Y. Effective removal of high-chroma crystal violet over TiO2-based nanosheet by adsorption-photocatalytic degradation. Chem. Eng. J. 2012, 204–206, 107–113. [Google Scholar] [CrossRef]
- And, M.K.; Jaroniec, M. Gas adsorption characterization of ordered organic−inorganic nanocomposite materials. Chem. Mater. 2001, 13, 3169–3183. [Google Scholar]
- Ho, S.; Chu, C.; Chen, S. Effect of thermal treatment on the nickel state and CO hydrogenation activity of titania-supported nickel catalysts. J. Catal. 1998, 178, 34–48. [Google Scholar] [CrossRef]
- Yuan, F.; Wu, C.; Cai, Y.; Zhang, L.; Wang, J.; Chen, L.; Wang, X.; Yang, S.; Wang, S. Synthesis of phytic acid-decorated titanate nanotubes for high efficient and high selective removal of U(VI). Chem. Eng. J. 2017, 322, 353–365. [Google Scholar] [CrossRef]
- Kitano, M.; Wada, E.; Nakajima, K.; Hayashi, S.; Miyazaki, S.; Kobayashi, H.; Haza, M. Protonated titanate nanotubes with lewis and brønsted acidity: Relationship between nanotube structure and catalytic activity. Chem. Mater. 2013, 25, 385–393. [Google Scholar] [CrossRef]
- Huang, J.; Cao, Y.; Huang, Q.; He, H.; Liu, Y.; Guo, W.; Hong, M. High-temperature formation of titanate nanotubes and the tranformation mechanism of nanotubes into nanowires. Cryst. Growth Des. 2009, 9, 3632–3637. [Google Scholar] [CrossRef]
- Lazaro, M.J.; Echegoyen, Y.; Alegre, C.; Suelves, I.; Moliner, R.; Palacios, J.M. TiO2 as textural promoter on high loaded Ni catalysts for methane decomposition. Int. J. Hydrog. Energy 2008, 33, 3320–3329. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Wen, X.; Liu, Y. Partial oxidation of methane over Ni/Ce-Ti-O catalysts. Chem. Eng. J. 2006, 121, 115–123. [Google Scholar] [CrossRef]
- Huo, W.; Zhang, C.; Yuan, H.; Jia, M.; Ning, C.; Tang, Y.; Zhang, Y.; Luo, J.; Wang, Z.; Zhang, W. Vapor-phase selective hydrogenation of maleic anhydride to succinic anhydride over Ni/TiO2 catalysts. J. Ind. Eng. Chem. 2014, 20, 4140–4145. [Google Scholar] [CrossRef]
- Grzechowiak, J.R.; Szyszka, I.; Masalska, A. Effect of TiO2 content and method of titania-silica preparation on the nature of oxidic nickel phases and their activity in aromatic hydrogenation. Catal. Today. 2008, 137, 433–438. [Google Scholar] [CrossRef]
- Chen, T.; Yang, B.; Li, S.; Wang, K.; Jiang, X.; Zhang, Y.; He, G. Ni2P catalysts supported on titania-modified alumina for the hydrodesulfurization of dibenzothiophene. Ind. Eng. Chem. Res. 2011, 50, 11043–11048. [Google Scholar] [CrossRef]
- Song, H.; Wang, J.; Wang, Z.; Song, H.; Jin, Z. Effect of titanium content on dibenzothiophene HDS performance over Ni2P/Ti-MCM-41 catalyst. J.Catal. 2014, 311, 257–265. [Google Scholar] [CrossRef]
- Chen, W.; Fan, Z.; Pan, X.; Bao, X. Effect of confinement in carbon nanotubes on the activity of Fischer-Tropsch iron catalyst. J. Am. Chem. Soc. 2008, 130, 9414–9419. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, X.; Long, L.; Wang, T.; Ma, L.; Wu, L.; Bai, Y.; Li, X.; Liao, S. Pt nanoparticles entrapped in titanate nanotubes (TNT) for pehnol hydrogenation: The confinement effect of TNT. Chem. Commun. 2014, 50, 2794–2796. [Google Scholar]
- Davda, R.R.; Shabaker, J.W.; Huber, G.W.; Cortright, R.D.; Dumesic, J.A. Aqueous-phase reforming of ethylene glycol on silica-supported metal catalysts. Appl. Catal. B Environ. 2003, 43, 13–26. [Google Scholar] [CrossRef]
- Shabaker, J.W.; Huber, G.W.; Dumesic, J.A. Aqueous-phase reforming of oxygenated hydrocarbons over Sn-modified Ni catalysts. J. Catal. 2004, 222, 180–191. [Google Scholar] [CrossRef]
- Shabaker, J.W.; Davda, R.R.; Huber, G.W.; Cortright, R.D.; Dumesic, J.A. Aqueous-phase reforming of methanol and ethylene glycol over alumina-supported platinum catalysts. J. Catal. 2003, 215, 344–352. [Google Scholar] [CrossRef]
- Davda, R.R.; Shabaker, J.W.; Huber, G.W.; Cortright, R.D.; Dumesic, J.A. A review of catalytic issues and process conditions for renewable hydrogen and alkanes by aqueous-phase reforming of oxygenated hydrocarbons over supported metal catalysts. Appl. Catal. B Environ. 2005, 56, 171–186. [Google Scholar] [CrossRef]
- Wang, X.; Mi, J.; Lin, Z.; Lin, Y.; Jiang, L.; Cao, Y. Effect fabrication of mesoporous active Cu-Co.-CeO2 catalysts for water-gas shift. Mater. Lett. 2016, 162, 214–217. [Google Scholar] [CrossRef]
- Zhao, B.; Chen, Y.W. Hydrogenation of p-chloronitrobenzene on Mo, La, Fe, and W-modified NiCoB nanoalloy catalysts. J. Non-Cryst. Solids 2010, 356, 839–847. [Google Scholar] [CrossRef]
- Chen, J.; Yao, N.; Wang, R.; Zhang, J. Hydrogenation of chloronitrobenzene to chloroaniline over Ni/TiO2 catalysts prepared by sol-gel method. Chem. Eng. J. 2009, 148, 164–172. [Google Scholar] [CrossRef]
- Pan, C.; Tsai, M.; Su, W.; Rick, J.; Akalework, N.G.; Agegnehu, A.K.; Cheng, S.; Hwang, B. Tuning/exploiting strong metal-support interaction (SMSI) in heterogeneous catalysis. J. Taiwan Inst. Chem. Eng. 2017, 74, 154–186. [Google Scholar] [CrossRef]
- Englisch, M.; Jentys, A.; Lercher, J.A. Structure sensitivity of the hydrogenation of crotonaldehyde over Pt/SiO2 and Pt/TiO2. J. Catal. 1997, 166, 25–35. [Google Scholar] [CrossRef]
- Yin, A.; Guo, X.; Dai, W.; Fan, K. The synthesis of propylene glycol and ethylene glycol from glycerol using Raney Ni as a versatile catalyst. Green Chem. 2009, 11, 1514–1516. [Google Scholar] [CrossRef]
- Kong, D.; Jiang, T.; Zhang, Y.; Cao, F. Catalytic performance of activated carbon supported Pt-Ni bimetallic catalyst for glycerol in situ hydrogenolysis. Chem. J. Chin. Univ. Chin. 2016, 37, 1140–1147. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | SBET (m²/g) | Vpore (cm³/g) | Dpore (nm) | Ni Loading (wt.%) | H2 Consumption 1 (mmol/g-cat) | Surface Atomic Ratio 2 | |
|---|---|---|---|---|---|---|---|
| Ni/Ti | Ni0/Ni2+ | ||||||
| Ni/TNP | 17.9 | 0.15 | 34.1 | 20.7 | 3.37 | 18.1/81.9 | 11.3/88.7 |
| Ni/TNS | 71.3 | 0.20 | 11.0 | 19.1 | 3.07 | 17.1/82.9 | 44.1/55.9 |
| Ni/TNT | 126.4 | 0.34 | 10.3 | 18.6 | 0.88 | 16.6/83.4 | 53.7/46.3 |
| Ni/TMS | 14.5 | 0.08 | 21.9 | 19.5 | 3.17 | 17.8/82.2 | 39.3/60.7 |
| Catalysts | Conversion (%) | Selectivity (%) | TOF 2 (h−1) | ||
|---|---|---|---|---|---|
| m-CAN | AN | Others | |||
| Blank 3 | - | - | - | - | - |
| Ni/TNP | 20.1 | 97.9 | 1.9 | 0.2 | 0.07 |
| Ni/TNT | 97.5 | 96.2 | 3.5 | 0.3 | 0.36 |
| Ni/TNS | 67.7 | 97.2 | 2.7 | 0.1 | 0.25 |
| Ni/TMS | 41.1 | 97.6 | 2.2 | 0.2 | 0.15 |
| H2O/C2H5OH 2 | T (K) | P (MPa) | Conversion (%) | Selectivity (%) | TOF 3 (h−1) | ||
|---|---|---|---|---|---|---|---|
| m-CAN | AN | Others | |||||
| 1/9 | 473 | 3 | 69.7 | 97.7 | 2.1 | 0.2 | 0.26 |
| 2/8 | 473 | 3 | 88.1 | 96.5 | 3.4 | 0.1 | 0.32 |
| 3/7 | 473 | 3 | 97.5 | 96.2 | 3.5 | 0.3 | 0.36 |
| 4/6 | 473 | 3 | 78.3 | 97.5 | 2.1 | 0.4 | 0.29 |
| 3/7 | 493 | 3 | 98.2 | 94.7 | 4.5 | 0.8 | 0.36 |
| 3/7 | 453 | 3 | 91.3 | 95.8 | 3.7 | 0.5 | 0.33 |
| 3/7 | 433 | 3 | 66.9 | 96.8 | 3.0 | 0.2 | 0.25 |
| 3/7 | 473 | 4 | 95.2 | 96.1 | 3.6 | 0.3 | 0.35 |
| 3/7 | 473 | 2 | 94.6 | 96.6 | 3.3 | 0.1 | 0.35 |
| 3/7 | 473 | 1 | 93.9 | 97.2 | 2.6 | 0.2 | 0.34 |
| Run | Conversion (%) | Selectivity (%) | TOF 2 (h−1) | ||
|---|---|---|---|---|---|
| m-CAN | AN | Others | |||
| 1 | 97.5 | 96.2 | 3.5 | 0.3 | 0.36 |
| 2 | 92.6 | 96.5 | 3.3 | 0.2 | 0.34 |
| 3 | 89.1 | 97.0 | 2.9 | 0.1 | 0.33 |
| 4 | 85.2 | 97.2 | 2.6 | 0.2 | 0.31 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Liang, J.; Zhu, W.; Song, H.; Wang, K.; Li, C. Hydrogenation of m-Chloronitrobenzene over Different Morphologies Ni/TiO2 without Addition of Molecular Hydrogen. Catalysts 2018, 8, 182. https://doi.org/10.3390/catal8050182
Li F, Liang J, Zhu W, Song H, Wang K, Li C. Hydrogenation of m-Chloronitrobenzene over Different Morphologies Ni/TiO2 without Addition of Molecular Hydrogen. Catalysts. 2018; 8(5):182. https://doi.org/10.3390/catal8050182
Chicago/Turabian StyleLi, Feng, Jinrong Liang, Wenxi Zhu, Hua Song, Keliang Wang, and Cuiqin Li. 2018. "Hydrogenation of m-Chloronitrobenzene over Different Morphologies Ni/TiO2 without Addition of Molecular Hydrogen" Catalysts 8, no. 5: 182. https://doi.org/10.3390/catal8050182
APA StyleLi, F., Liang, J., Zhu, W., Song, H., Wang, K., & Li, C. (2018). Hydrogenation of m-Chloronitrobenzene over Different Morphologies Ni/TiO2 without Addition of Molecular Hydrogen. Catalysts, 8(5), 182. https://doi.org/10.3390/catal8050182