Liquid-Phase Hydrodeoxygenation of Guaiacol over Mo2C Supported on Commercial CNF. Effects of Operating Conditions on Conversion and Product Selectivity



Abstract

1. Introduction
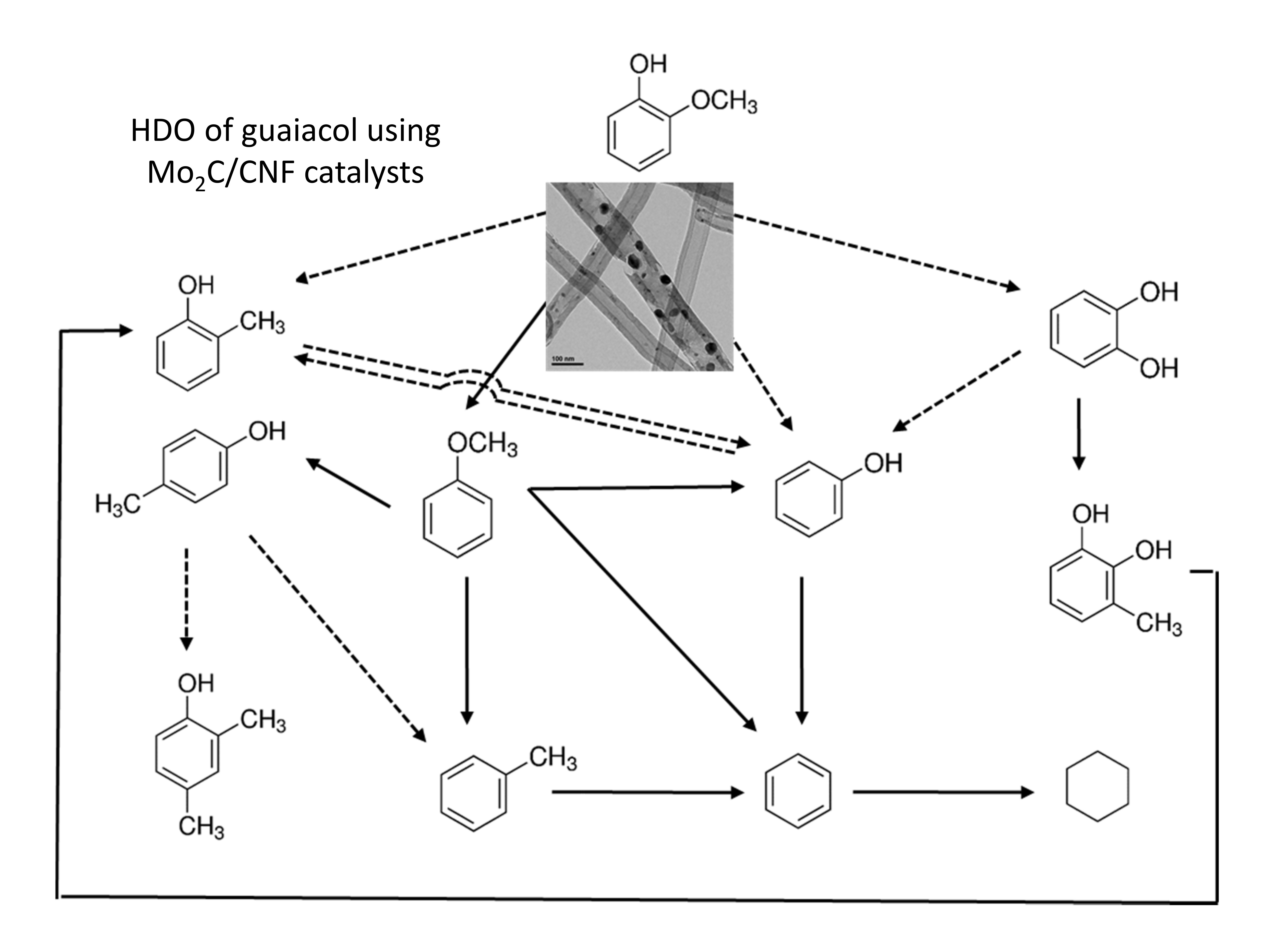
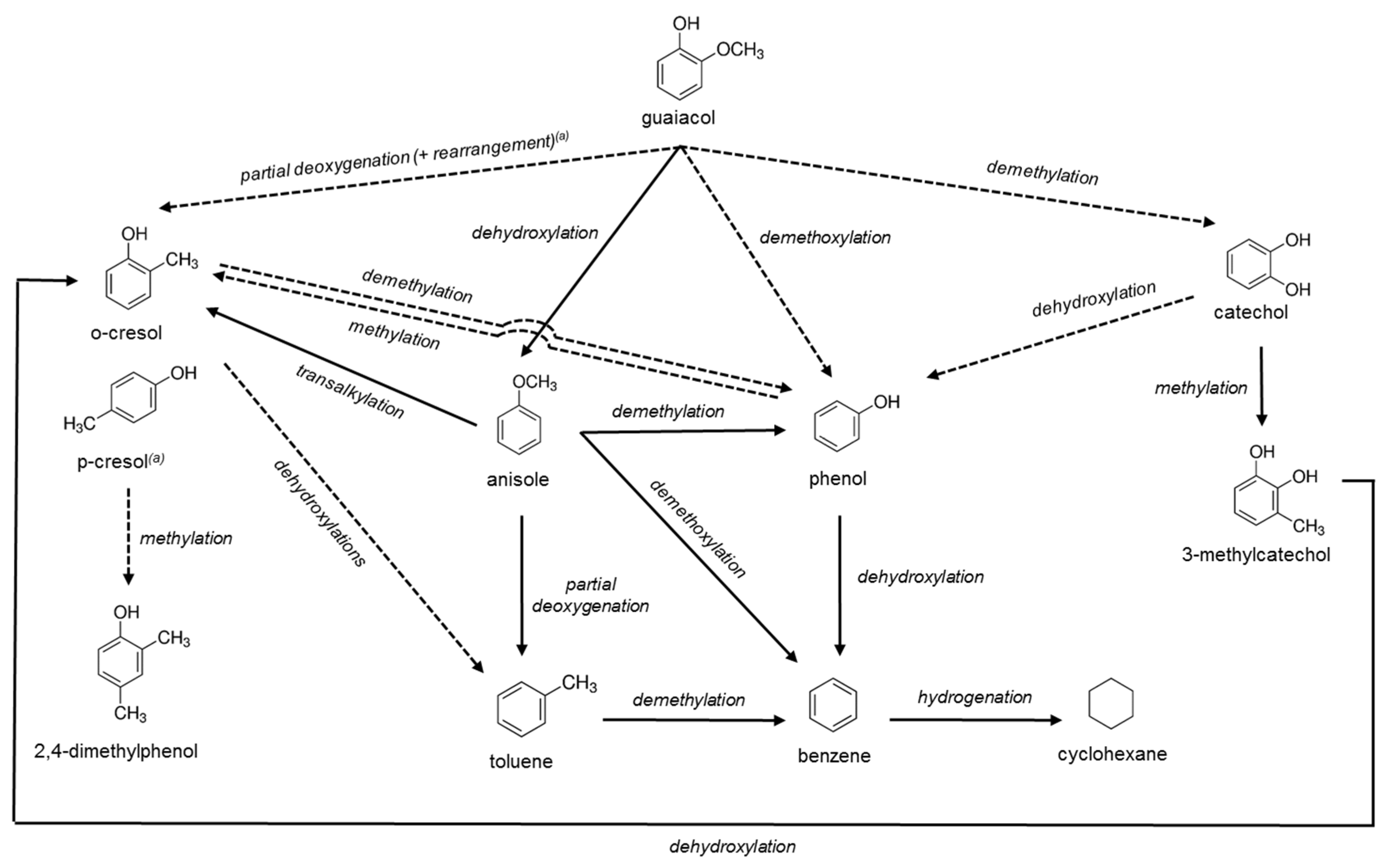
2. Results and Discussion
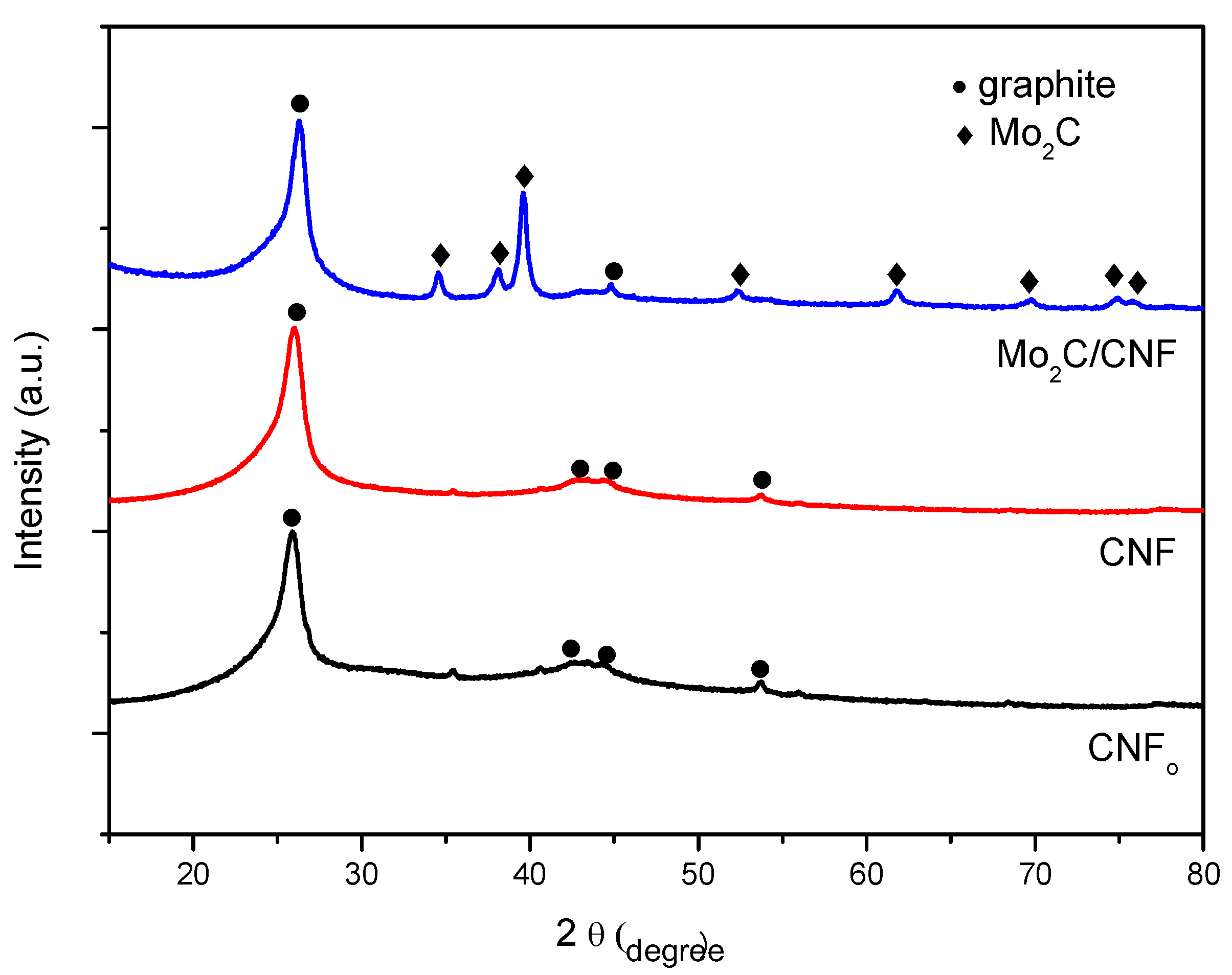
2.1. Support Characterization
2.2. Fresh Catalysts Characterization
2.3. Catalytic Activity
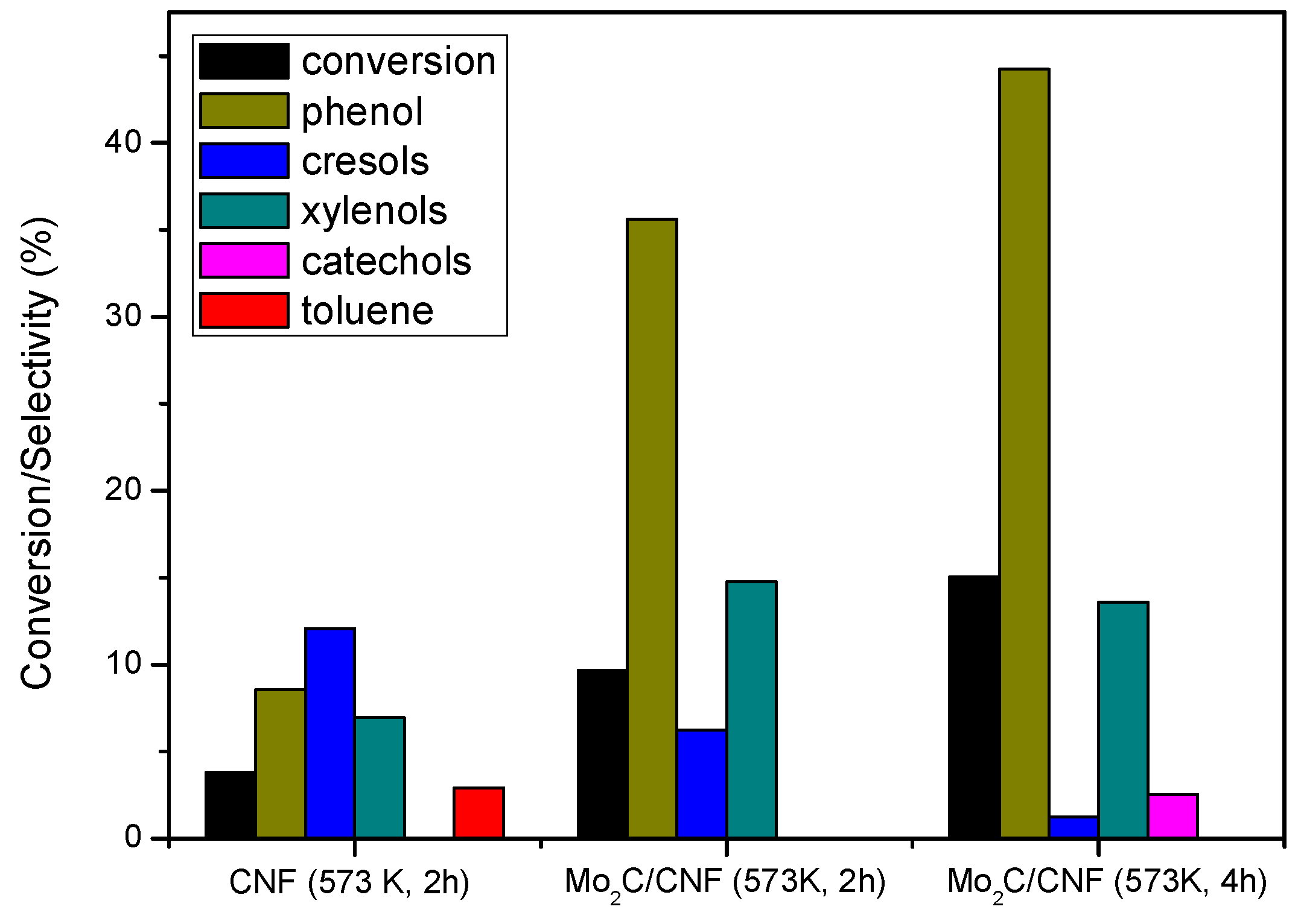
2.3.1 Catalyst Performance at 573 K
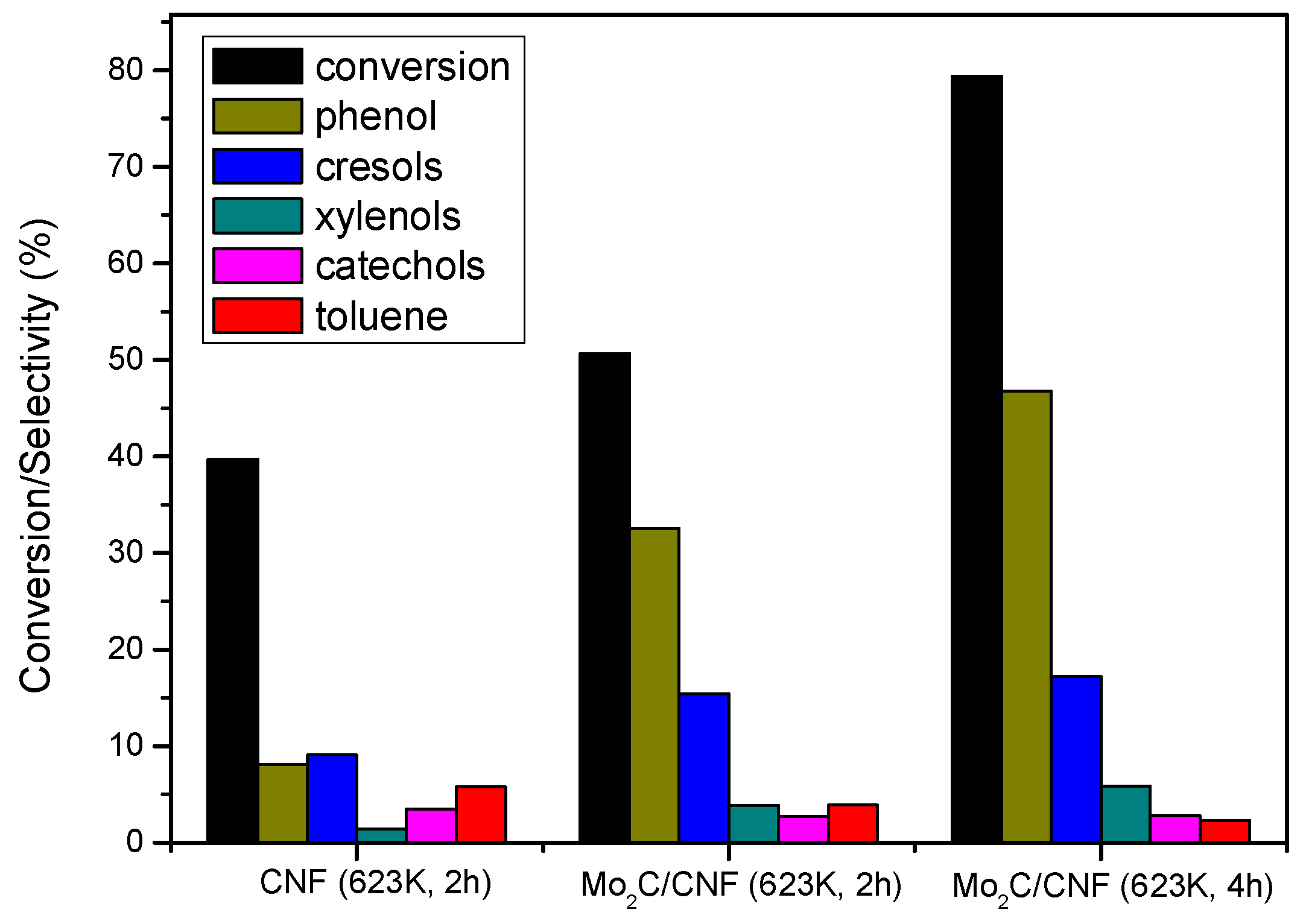
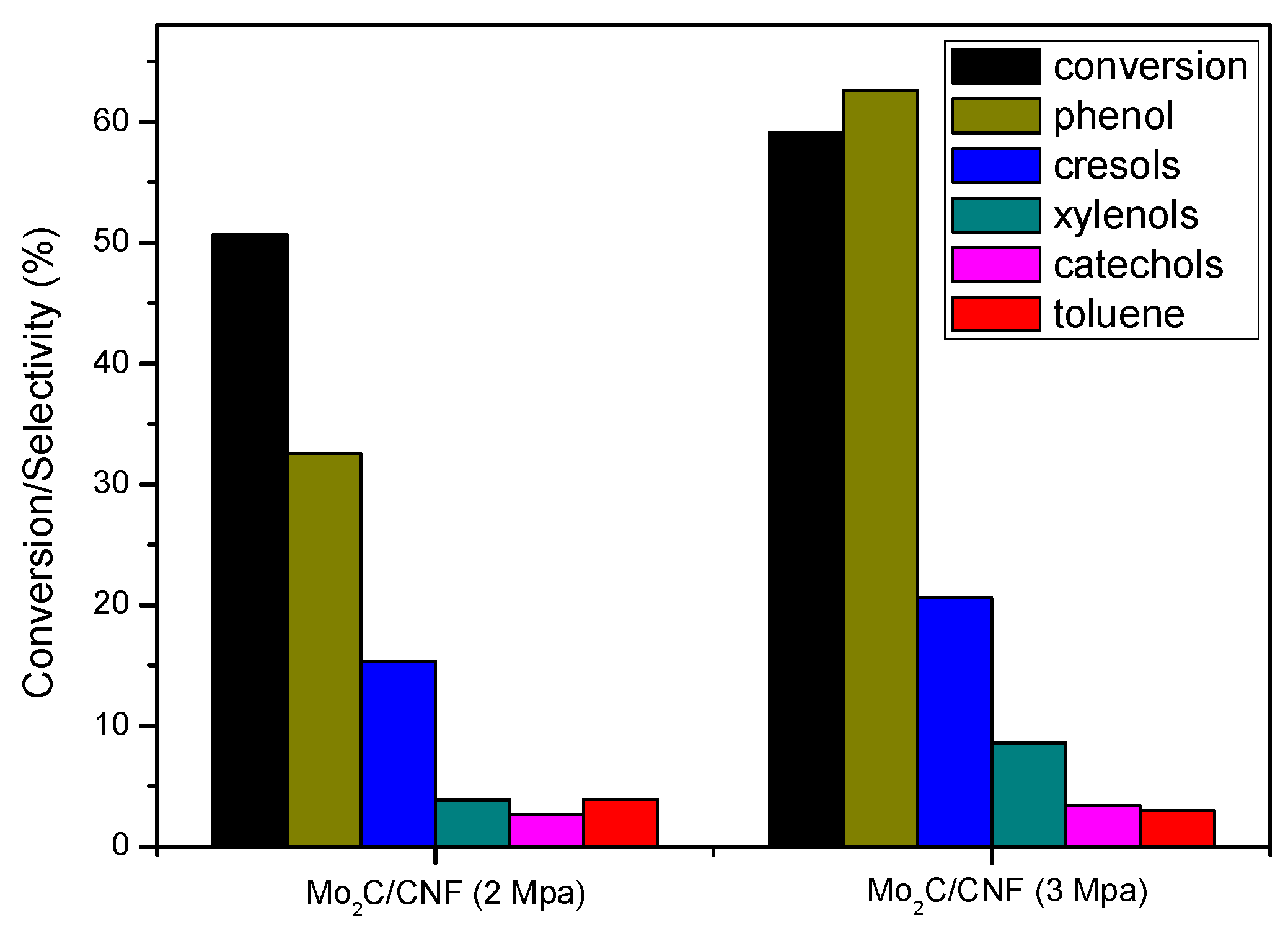
2.3.2. Catalyst Performance at 623 K
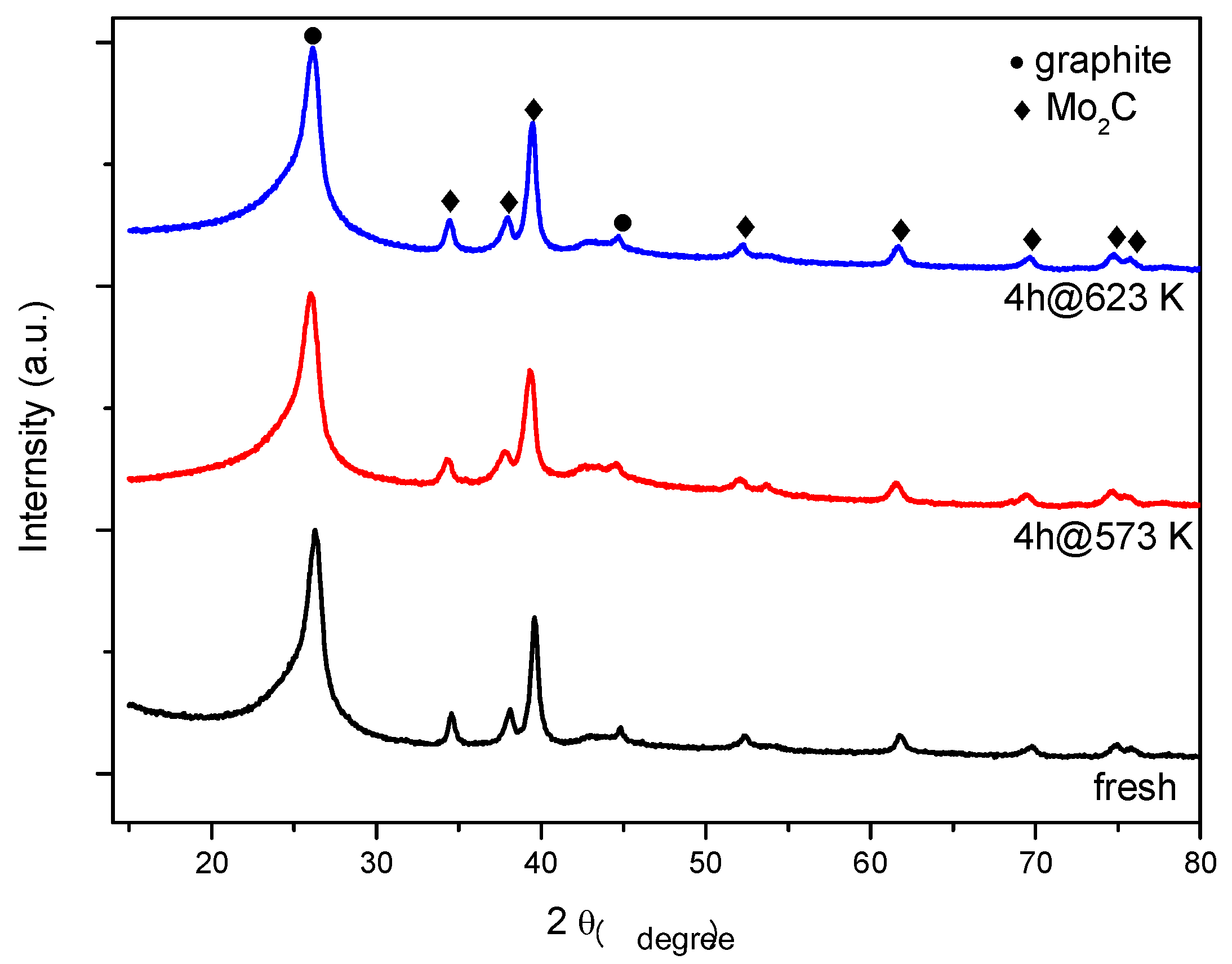
2.4. Spent Catalysts Characterization
3. Materials and Methods
3.1. Chemicals
3.2. Catalyst Preparation and Characterization
3.3. Catalytic Testing and Product Characterization
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lu, Q.; Li, W.-Z.; Zhu, X.-F. Overview of fuel properties of biomass fast pyrolysis. Energy Convers. Manag. 2009, 50, 1376–1383. [Google Scholar] [CrossRef]
- Mortensen, P.; Grunwaldt, J.; Jensen, P.; Knudsen, K.; Jensen, A. A review of catalytic upgrading of bio-oil to engine fuels. Appl. Catal. A Gen. 2011, 407, 1–19. [Google Scholar] [CrossRef]
- Butler, E.; Devlin, G.; Meier, D.; McDonnell, K. A review of recent laboratory research and commercial developments in fast pyrolysis and upgrading. Renew. Sustain. Energy Rev. 2011, 15, 4171–4186. [Google Scholar] [CrossRef]
- He, V.; Wang, X. Hydrodeoxygenation of model compounds and catalytic systems for pyrolysis bio-oils upgrading. Catal. Sustain. Energy 2013, 1, 28–52. [Google Scholar] [CrossRef]
- Venderbosch, R.; Ardiyanti, A.; Wildschut, J.; Oasmaa, A.; Heeres, H. Stabilization of biomass-derived pyrolysis oils. J. Chem. Technol. Biotechnol. 2010, 85, 674–686. [Google Scholar] [CrossRef]
- Choi, J.; Bugli, G.; Djéga-Mariadassou, G. Influence of the Degree of Carburization on the Density of Sites and Hydrogenating Activity of Molybdenum Carbides. J. Catal. 2000, 193, 238–247. [Google Scholar] [CrossRef]
- Nguyen, T.; Lee, T.; Safinski, T.; Adesina, A. Structural evolution of alumina supported Mo–W carbide nanoparticles synthesized by precipitation from homogeneous solution. Mater. Res. Bull. 2005, 40, 149–157. [Google Scholar] [CrossRef]
- Furimsky, E. Metal carbides and nitrides as potential catalysts for hydroprocessing. Appl. Catal. A Gen. 2003, 40, 1–28. [Google Scholar] [CrossRef]
- Boullosa-Eirasa, S.; Lødeng, R.; Bergem, H.; Stöcker, M.; Hannevold, L.; Blekkan, E. Catalytic hydrodeoxygenation (HDO) of phenol over supported molybdenum carbide, nitride, phosphide and oxide catalysts. Catal. Today 2014, 223, 44–53. [Google Scholar] [CrossRef]
- Furimsky, E. Catalytic hydrodeoxygenation. Appl. Catal. A Gen. 2000, 199, 147–190. [Google Scholar] [CrossRef]
- Politi, R.; Vines, F.; Rodriguez, J.; Illas, F. Atomic and electronic structure of molybdenum carbide phases: Bulk and low Miller-index surfaces. Phys. Chem. Chem. Phys. 2013, 15, 12617–12625. [Google Scholar] [CrossRef] [PubMed]
- Posada-Péreza, S.; Viñesa, F.; Valeroa, R.; Rodriguez, J.; Illas, F. Adsorption and dissociation of molecular hydrogen on orthorhombic β-Mo2C and cubic δ-MoC (001) surfaces. Surf. Sci. 2017, 656, 24–32. [Google Scholar] [CrossRef]
- Dongil, A.; Ghampson, I.; García, R.; Fierro, J.; Escalona, N. Hydrodeoxygenation of guaiacol over Ni/carbon catalysts: Effect of the support and Ni loading. RSC Adv. 2016, 6, 2611–2623. [Google Scholar] [CrossRef]
- Serp, F.; Corrias, M.; Kalck, P. Carbon nanotubes and nanofibers in catalysis. Appl. Catal. A Gen. 2003, 253, 337–358. [Google Scholar] [CrossRef]
- Vieira, R. Carbon Nanofibers as Macro-Structured Catalytic Support. In Nanofibers; InTech: Rijeka, Croatia, 2010; pp. 1–12. [Google Scholar]
- Jong, K.; Gues, J. Carbon nanofibers: Catalytic synthesis and applications. Catal. Rev. Sci. Eng. 2000, 42, 481–510. [Google Scholar] [CrossRef]
- Park, C.; Anderson, P.; Chambers, A.; Tan, C.; Hidalgo, R.; Rodriguez, N. Further Studies of the Interaction of Hydrogen with Graphite Nanofibers. J. Phys. Chem. B 1999, 103, 10572–10581. [Google Scholar] [CrossRef]
- Rodriguez-Reinoso, F. The role of carbon materials in heterogeneous catalysis. Carbon 1998, 36, 159–175. [Google Scholar] [CrossRef]
- Santillan-Jimenez, E.; Perdu, M.; Pace, R.; Morgan, T.; Crocker, M. Activated Carbon, Carbon Nanofiber and Carbon Nanotube Supported Molybdenum Carbide Catalysts for the Hydrodeoxygenation of Guaiacol. Catalysts 2015, 5, 424–441. [Google Scholar] [CrossRef]
- Jongerius, A.; Gosselink, R.; Dijkstra, J.; Bitter, J.; Bruijnincx, P.; Weckhuysen, B. Carbon Nanofiber Supported Transition-Metal Carbide Catalysts for the Hydrodeoxygenation of Guaiacol. ChemCatChem 2013, 5, 2964–2972. [Google Scholar] [CrossRef]
- Mordenti, D.; Brodzki, D.; Djéga-Mariadassou, G. New Synthesis of Mo2C 14 nm in Average Size Supported on a High Specific Surface Area Carbon Material. J. Solid State Chem. 1998, 141, 114–120. [Google Scholar] [CrossRef]
- Chaudhury, S.; Mukerjee, S.; Vaidya, V.; Venugopal, V. Kinetics and mechanism of carbothermic reduction of MoO3 to Mo2C. J. Alloys Compd. 1997, 261, 105–113. [Google Scholar] [CrossRef]
- Deutsch, K.; Shanks, B. Hydrodeoxygenation of lignin model compounds over a copper chromite catalyst. Appl. Catal. A Gen. 2012, 447, 144–150. [Google Scholar] [CrossRef]
- Zhou, J.; Sui, Z.; Zhua, J.; Li, P.; Chen, D.; Dai, Y.; Yuan, W. Characterization of surface oxygen complexes on carbon nanofibers by TPD, XPS and FT-IR. Carbon 2007, 45, 785–796. [Google Scholar] [CrossRef]
- Zhuang, Q.; Kyotani, T.; Tomita, A. DRIFT and TK/TPD Analyses of Surface Oxygen Complexes Formed during Carbon Gasification. Energy Fuels 1994, 8, 714–718. [Google Scholar] [CrossRef]
- Boehm, H. Surface oxides on carbon and their analysis: A critical assessment. Carbon 2002, 40, 145–149. [Google Scholar] [CrossRef]
- Pinilla, J.; Purón, H.; Torres, D.; Llobet, S.; Moliner, R.; Suelves, I.; Millana, M. Carbon nanofibres coated with Ni decorated MoS2 nanosheets as catalyst for vacuum residue hydroprocessing. Appl. Catal. B Environ. 2014, 148–149, 357–365. [Google Scholar] [CrossRef]
- Pinilla, J.; Purón, H.; Torres, D.; Suelves, I.; Millana, M. Ni-MoS2 supported on carbon nanofibers as hydrogenation catalysts: Effect of support functionalization. Carbon 2015, 81, 574–586. [Google Scholar] [CrossRef]
- Guil-López, R.; Nieto, E.; Botas, J.; Fierro, J. On the genesis of molybdenum carbide phases during reduction-carburization reactions. J. Solid State Chem. 2012, 190, 285–295. [Google Scholar] [CrossRef]
- Reddy, K.; Rao, T.; Revathi, J.; Joardar, J. Structural stability of α/β-Mo2C during thermochemical processing. J. Alloys Compd. 2010, 494, 386–391. [Google Scholar] [CrossRef]
- Dubois, J.; Epicier, T.; Esnouf, C.; Fantozzi, G. Neutron powder diffraction studies of transition metal hemicarbides M2C1−x—I. Motivation for a study on W2C and Mo2C and experimental background for in situ investigation at elevated temperature. Acta Metal. 1998, 36, 1891–1901. [Google Scholar] [CrossRef]
- Dubois, J.; Epicier, T.; Esnouf, C.; Fantozzi, G. Neutron powder diffraction studies of transition metal hemicarbides M2C1−x—II. In situ high temperature study on W2C1−x and Mo2C1−x. Acta Metal. 1998, 36, 1903–1921. [Google Scholar]
- Araujo, C.; Souza, C.; Maia, L.; Souto, M.; Barbosa, C. On the synthesis of molybdenum carbide with cobalt addition via gas-solid reactions in a CH4/H2 atmosphere. Braz. J. Chem. Eng. 2016, 3, 577–588. [Google Scholar] [CrossRef][Green Version]
- Stellwagen, D.; Bitter, J. Structure–performance relations of molybdenum and tungsten carbide catalysts for deoxygenation. Green Chem. 2015, 17, 582–593. [Google Scholar] [CrossRef]
- Baaziz, W.; Melinte, G.; Ersen, O.; Pham-Huua, C.; Janowska, I. Effect of nitriding/nanostructuration of few layer graphene supported iron-based particles; catalyst in graphene etching and carbon nanofilament growth. Phys. Chem. Chem. Phys. 2014, 16, 15988–15993. [Google Scholar] [CrossRef] [PubMed]
- Papaefthimiou, V.; Florea, I.; Baaziz, W.; Janowska, I.; Doh, W.; Begin, D.; Blume, R.; Knop-Gericke, A.; Ersen, O.; Pham-Huu, C.; et al. Effect of the Specific Surface Sites on the Reducibility of α-Fe2O3/Graphene Composites by Hydrogen. J. Phys. Chem. Chem. Phys. 2013, 117, 20313–20319. [Google Scholar] [CrossRef]
- Datta, S.; Strachan, D.; Khamis, S.; Johnson, A. Crystallographic Etching of Few-Layer Graphene. Nano Lett. 2008, 7, 1912–1915. [Google Scholar] [CrossRef] [PubMed]
- Ramasse, Q.; Zan, R.; Ursel Bangert, U.; Boukhvalov, D.; Son, Y.; Novoselov, K. Direct Experimental Evidence of Metal-Mediated Etching of Suspended Graphene. ACS Nano 2012, 6, 4063–4071. [Google Scholar] [CrossRef] [PubMed]
- Schäffel, F.; Warner, J.; Bachmatiuk, A.; Rellinghaus, B.; Büchner, B.; Schultz, L.; Rümmeli, M. Shedding Light on the Crystallographic Etching of Multi-Layer Graphene at the Atomic Scale. Nano Res. 2009, 2, 695–705. [Google Scholar] [CrossRef]
- Schäffel, F.; Warner, J.; Bachmatiuk, A.; Rellinghaus, B.; Büchner, B.; Schultz, L.; Rümmeli, M. On the catalytic hydrogenation of graphite for graphene nanoribbon fabrication. Phys. Status Solidi B 2009, 246, 2540–2544. [Google Scholar] [CrossRef]
- Wang, R.; Wang, J.; Gong, H.; Luo, Z.; Zhan, D.; Shen, Z.; Thong, J. Cobalt-Mediated Crystallographic Etching of Graphite from Defects. Small 2012, 8, 2515–2523. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.; Manfrinato, V.; Sanchez-Yamagishi, J.; Kong, J.; Jarillo-Herrero, P. Anisotropic Etching and Nanoribbon Formation in Single-Layer Graphene. Nano Lett. 2009, 9, 2600–2604. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Motchelaho, M.; Moyo, M.; Jewell, L.; Coville, N. Cobalt catalysts supported on a micro-coil carbon in Fischer–Tropsch synthesis: A comparison with CNTs and CNFs. Catal. Today 2013, 214, 50–60. [Google Scholar] [CrossRef]
- Chen, W.; Fan, Z.; Pan, X.; Bao, X. Effect of Confinement in Carbon Nanotubes on the Activity of Fischer-Tropsch Iron Catalyst. J. Am. Chem. Soc. 2008, 130, 9414–9419. [Google Scholar] [CrossRef] [PubMed]
- Abbaslou, R.; Tavassoli, A.; Soltan, J.; Dalai, A. Iron catalysts supported on carbon nanotubes for Fischer–Tropsch synthesis: Effect of catalytic site position. Appl. Catal. A Gen. 2009, 367, 47–52. [Google Scholar] [CrossRef]
- Wandas, R.; Surygata, J.; Sliwka, E. Conversion of cresols and naphthalene in the hydroprocessing of three-component model mixtures simulating fast pyrolysis tars. Fuel 1996, 75, 687–694. [Google Scholar] [CrossRef]
- Saidi, M.; Samimi, F.; Karimipourfard, D.; Nimmanwudipong, T.; Gates, B.; Rahimpour, M. Upgrading of lignin-derived bio-oils by catalytic hydrodeoxygenation. Energy Environ. Science 2014, 7, 103–129. [Google Scholar]
- Nimmanwudipong, T.; Aydin, C.; Lu, J.; Ron, C.; Runnebaum, R.; Brodwater, K.; Browning, N.; Block, D.; Gates, B. Selective Hydrodeoxygenation of Guaiacol Catalyzed by Platinum Supported on Magnesium Oxide. Catal. Lett. 2012, 142, 1190–1196. [Google Scholar] [CrossRef]
- Lee, H.; Kim, H.; Yu, M.; Ko, C.; Jeon, J.; Jae, J.; Park, S.; Jung, S.; Park, Y. Catalytic Hydrodeoxygenation of Bio-oil Model Compounds over Pt/HY Catalyst. Nat. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; He, J.; Lemonidou, A.; Li, X.; Lercher, J. Aqueous-phase hydrodeoxygenation of bio-derived phenols to cycloalkanes. J. Catal. 2011, 280, 8–16. [Google Scholar] [CrossRef]
- Zhao, H.; Li, D.; Bui, P.; Oyama, S. Hydrodeoxygenation of guaiacol as model compound for pyrolysis oil on transition metal phosphide hydroprocessing catalysts. Appl. Catal. A Gen. 2011, 391, 305–310. [Google Scholar] [CrossRef]
- Zhao, C.; Camaioni, D.; Lercher, J. Selective catalytic hydroalkylation and deoxygenation of substituted phenols to bicycloalkanes. J. Catal. 2012, 288, 92–103. [Google Scholar] [CrossRef]
- Furimsky, E. Carbons and Carbon-Supported Catalysts in Hydroprocessing; The Royal Society of Chemistry, IMAF Group: Ottawa, ON, Canada, 2008; pp. 12–126. [Google Scholar]
- Ma, R.; Cui, K.; Yang, L.; Ma, X.; Li, Y. Selective catalytic conversion of guaiacol to phenols over a molybdenum carbide catalyst. Chem. Commun. 2015, 51, 10299–10301. [Google Scholar] [CrossRef] [PubMed]
- Kwon, K.; Mayfield, H.; Marolla, T.; Nichols, B.; Mashburn, M. Catalytic Deoxygenation of Liquid Biomass for Hydrocarbon Fuels. Renew. Energy 2011, 36, 907–1015. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Surface Area (m2/g) | Pore Volume (cm3/g) | Pore Size (nm) | CO (mmol/g) | CO2 (mmol/g) | Oxygen Content (wt %) |
|---|---|---|---|---|---|---|
| CNFo | 39.6 | 0.156 | 15.5 | 0.256 | 0.079 | 0.7 |
| CNF | 37.8 | 0.110 | 13.7 | 0.410 | 0.398 | 1.9 |
| Mo2C/CNF | 44.2 | 0.147 | 13.3 | n.d. | n.d. | n.d. |
| Temperature (K) | Reaction Time (h) | Pressure (Mpa) | Mo2C Crystallite Size (nm) |
|---|---|---|---|
| fresh catalyst | 11.2 | ||
| 573 | 2 | 2 | 9.7 |
| 573 | 4 | 2 | 10.2 |
| 623 | 2 | 2 | 7.8 |
| 623 | 4 | 2 | 8.6 |
| 623 | 2 | 3 | 7.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreira, R.; Ochoa, E.; Pinilla, J.L.; Portugal, A.; Suelves, I. Liquid-Phase Hydrodeoxygenation of Guaiacol over Mo2C Supported on Commercial CNF. Effects of Operating Conditions on Conversion and Product Selectivity. Catalysts 2018, 8, 127. https://doi.org/10.3390/catal8040127
Moreira R, Ochoa E, Pinilla JL, Portugal A, Suelves I. Liquid-Phase Hydrodeoxygenation of Guaiacol over Mo2C Supported on Commercial CNF. Effects of Operating Conditions on Conversion and Product Selectivity. Catalysts. 2018; 8(4):127. https://doi.org/10.3390/catal8040127
Chicago/Turabian StyleMoreira, Rui, Elba Ochoa, José Luis Pinilla, António Portugal, and Isabel Suelves. 2018. "Liquid-Phase Hydrodeoxygenation of Guaiacol over Mo2C Supported on Commercial CNF. Effects of Operating Conditions on Conversion and Product Selectivity" Catalysts 8, no. 4: 127. https://doi.org/10.3390/catal8040127
APA StyleMoreira, R., Ochoa, E., Pinilla, J. L., Portugal, A., & Suelves, I. (2018). Liquid-Phase Hydrodeoxygenation of Guaiacol over Mo2C Supported on Commercial CNF. Effects of Operating Conditions on Conversion and Product Selectivity. Catalysts, 8(4), 127. https://doi.org/10.3390/catal8040127