Exploring the Effect of Au/Pt Ratio on Glycerol Oxidation in Presence and Absence of a Base

 ,
, 
 and
and 
Abstract
:1. Introduction
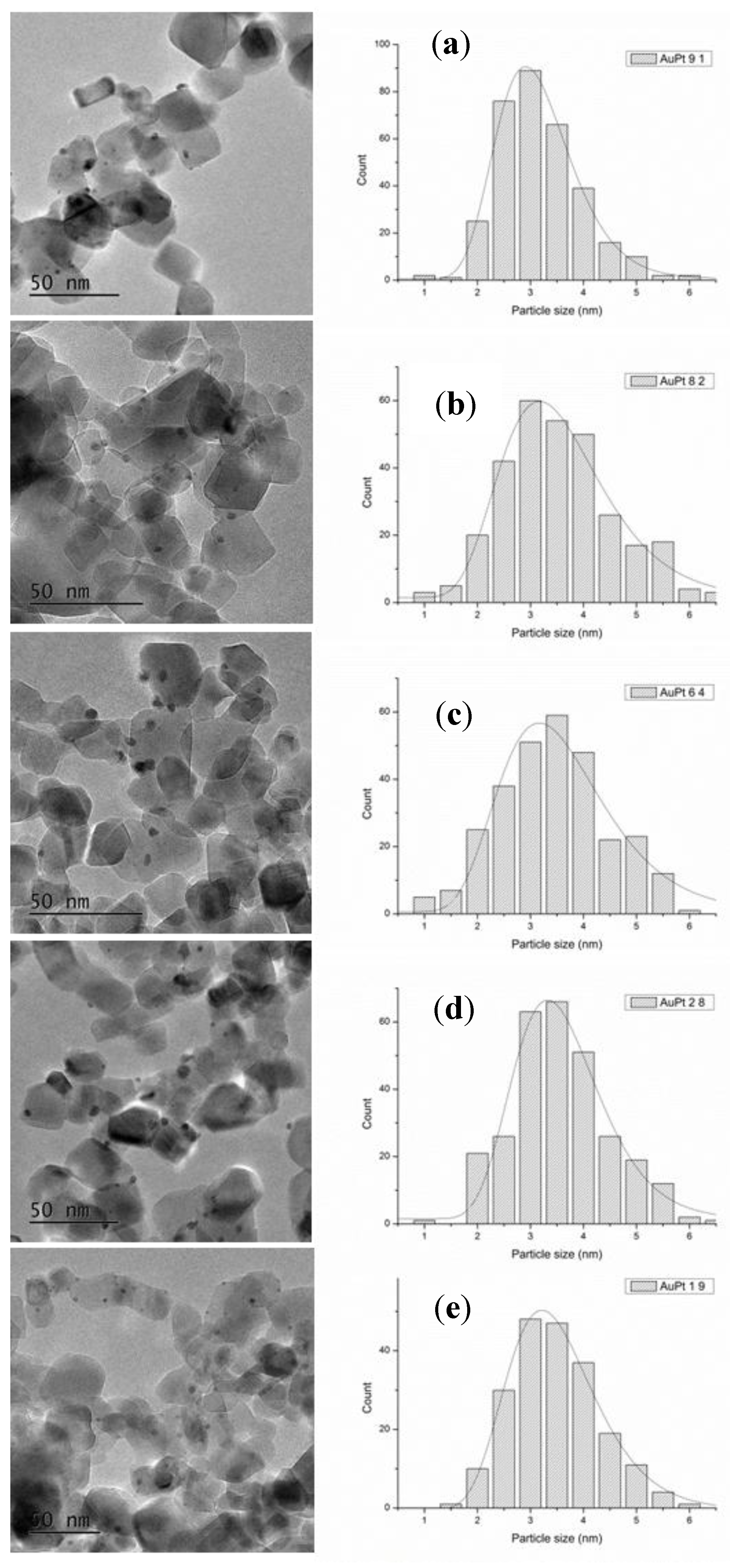
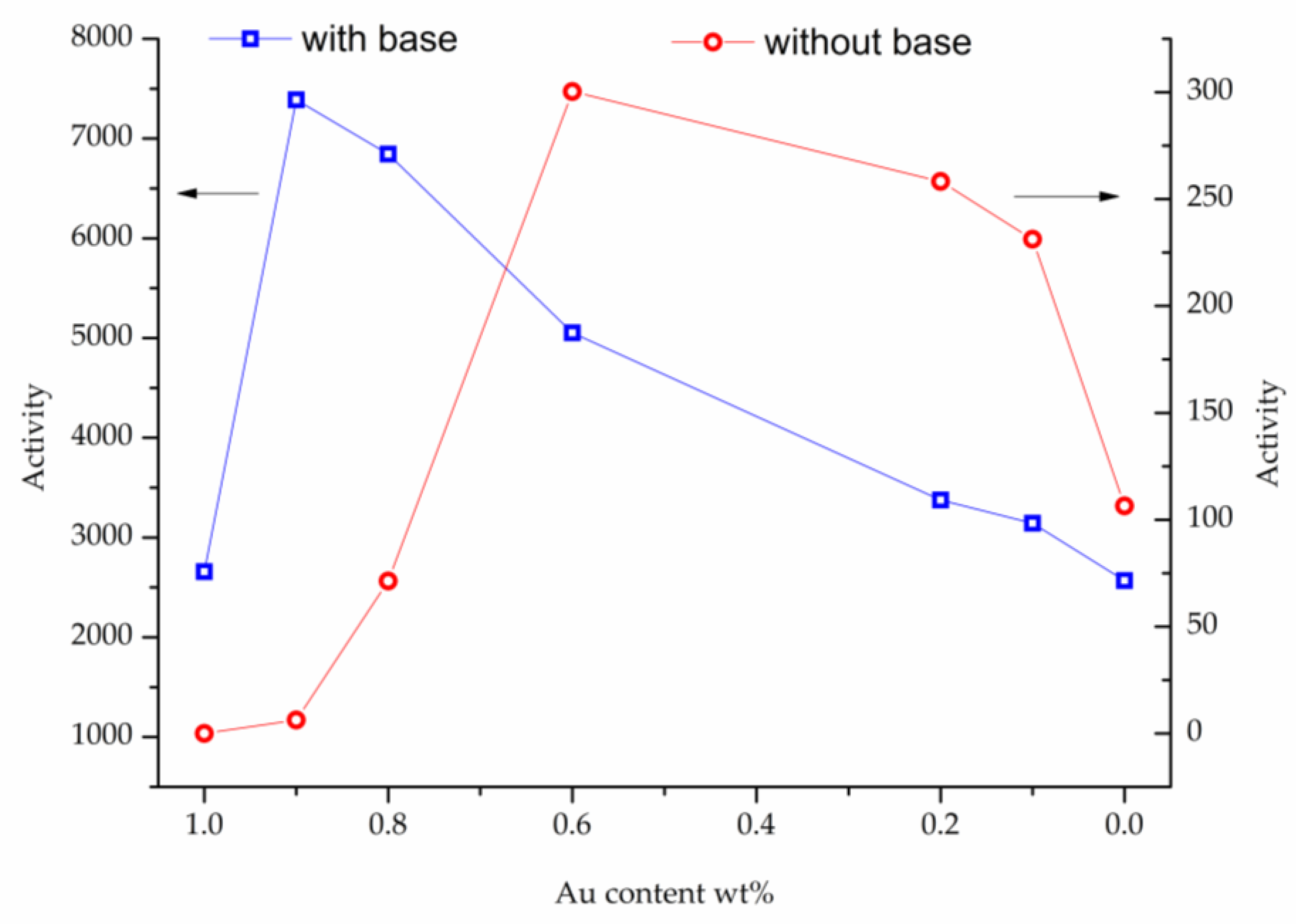
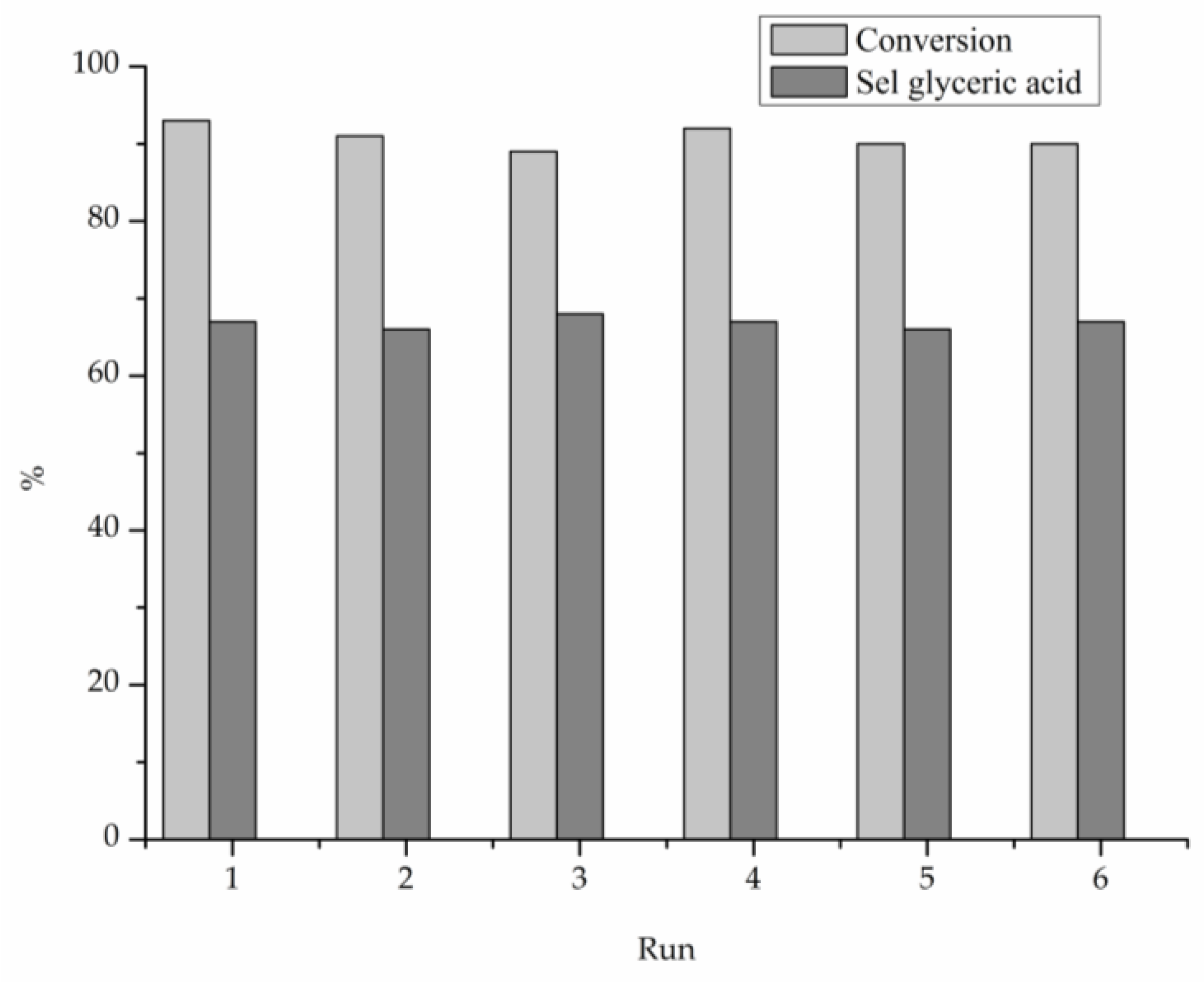
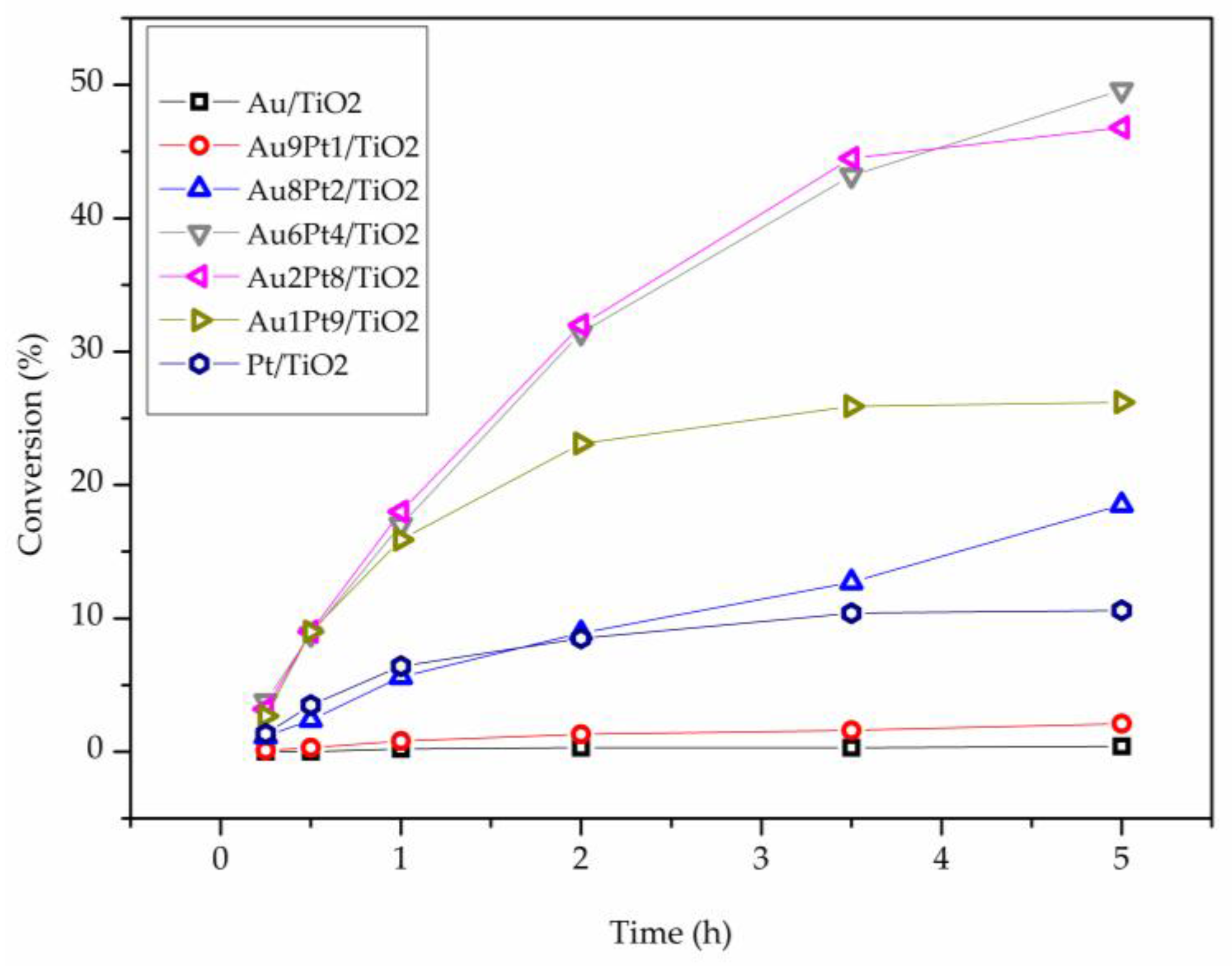
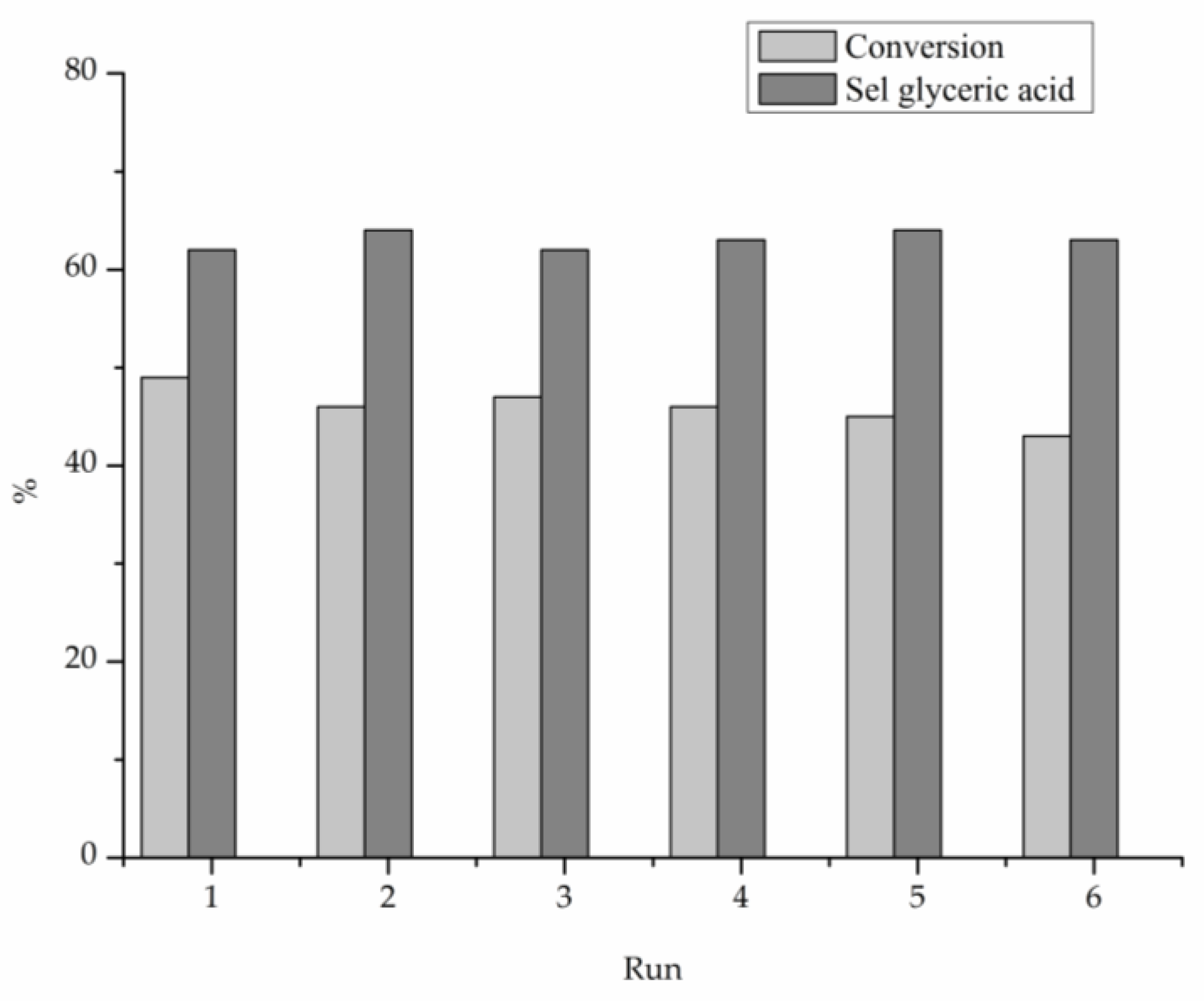
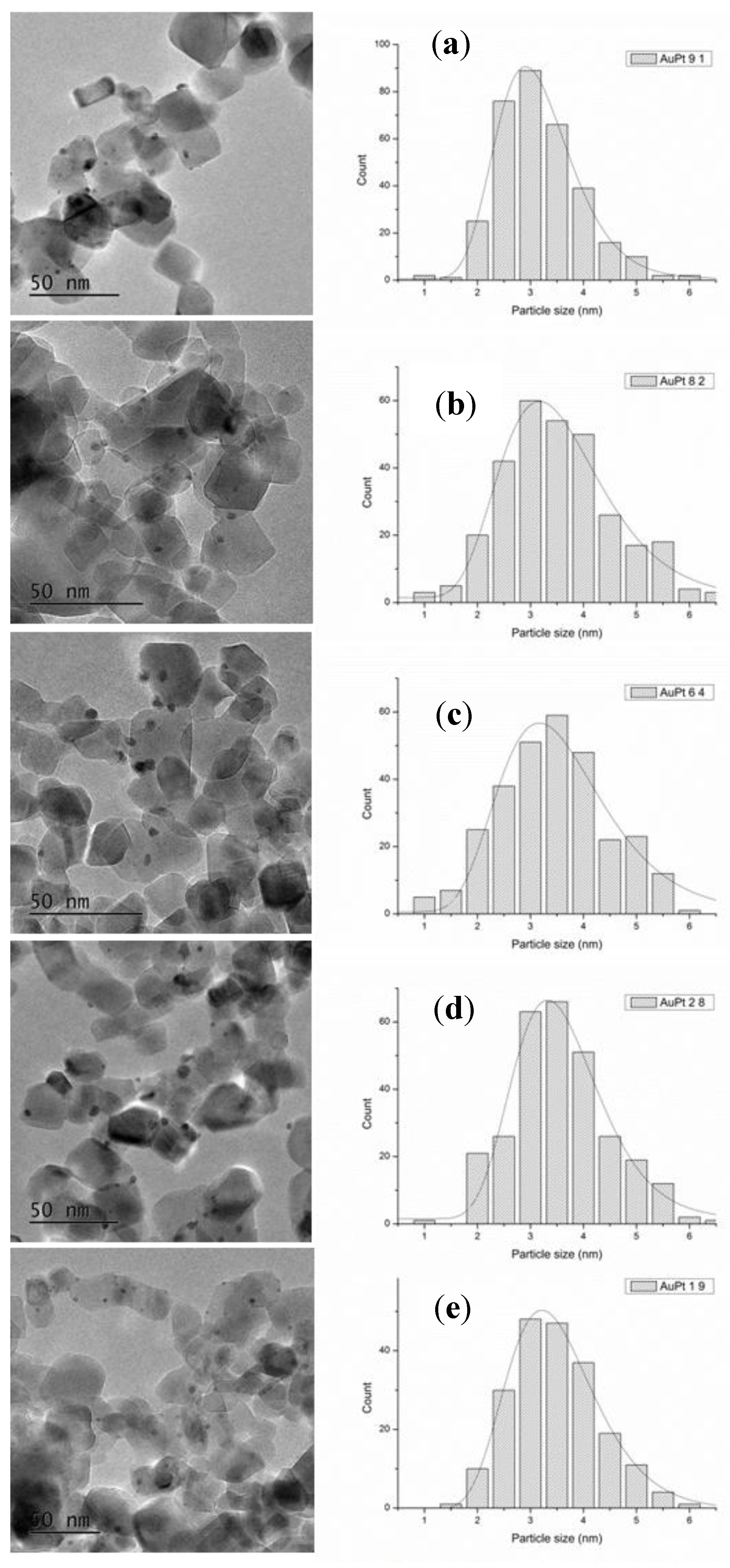
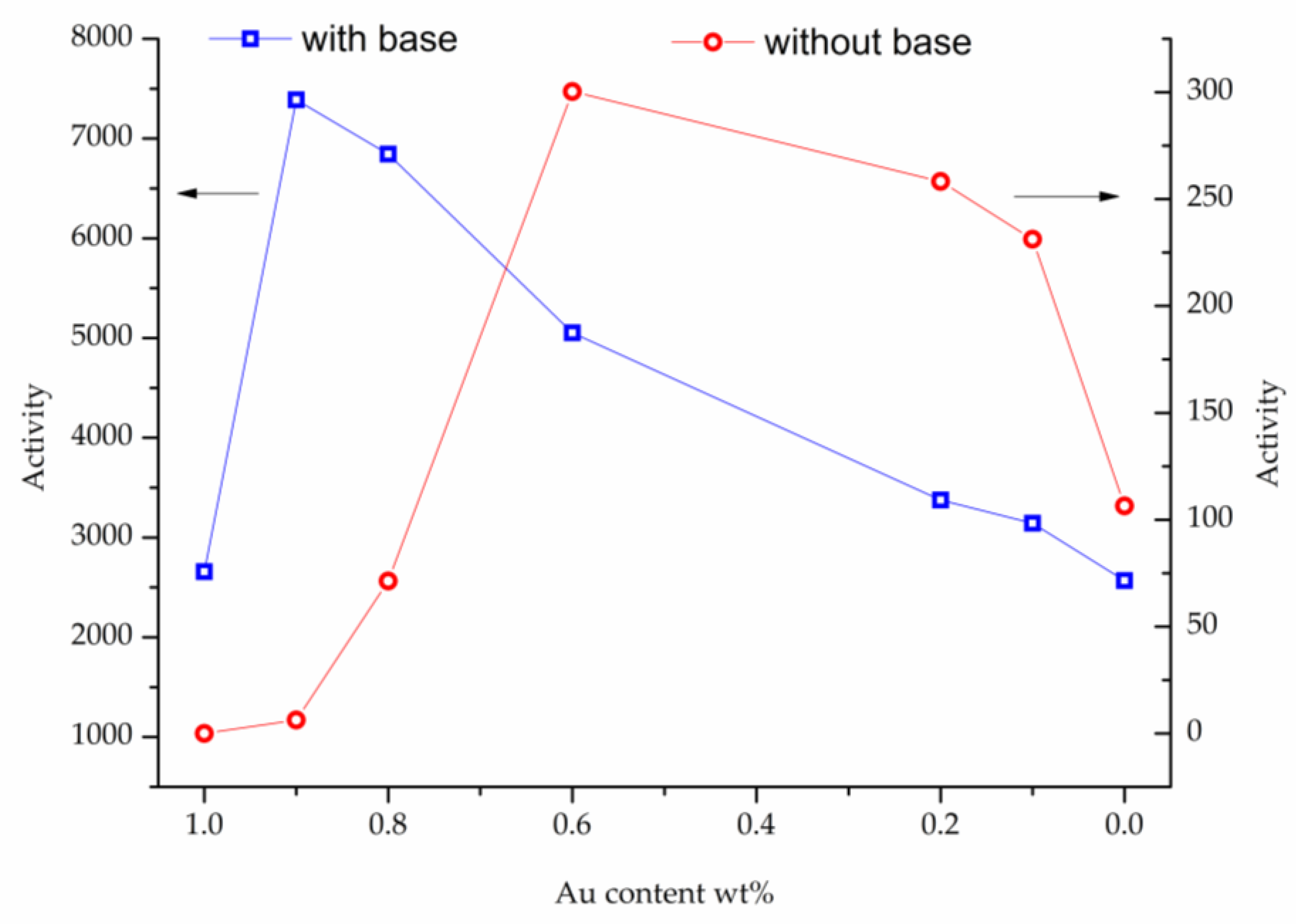
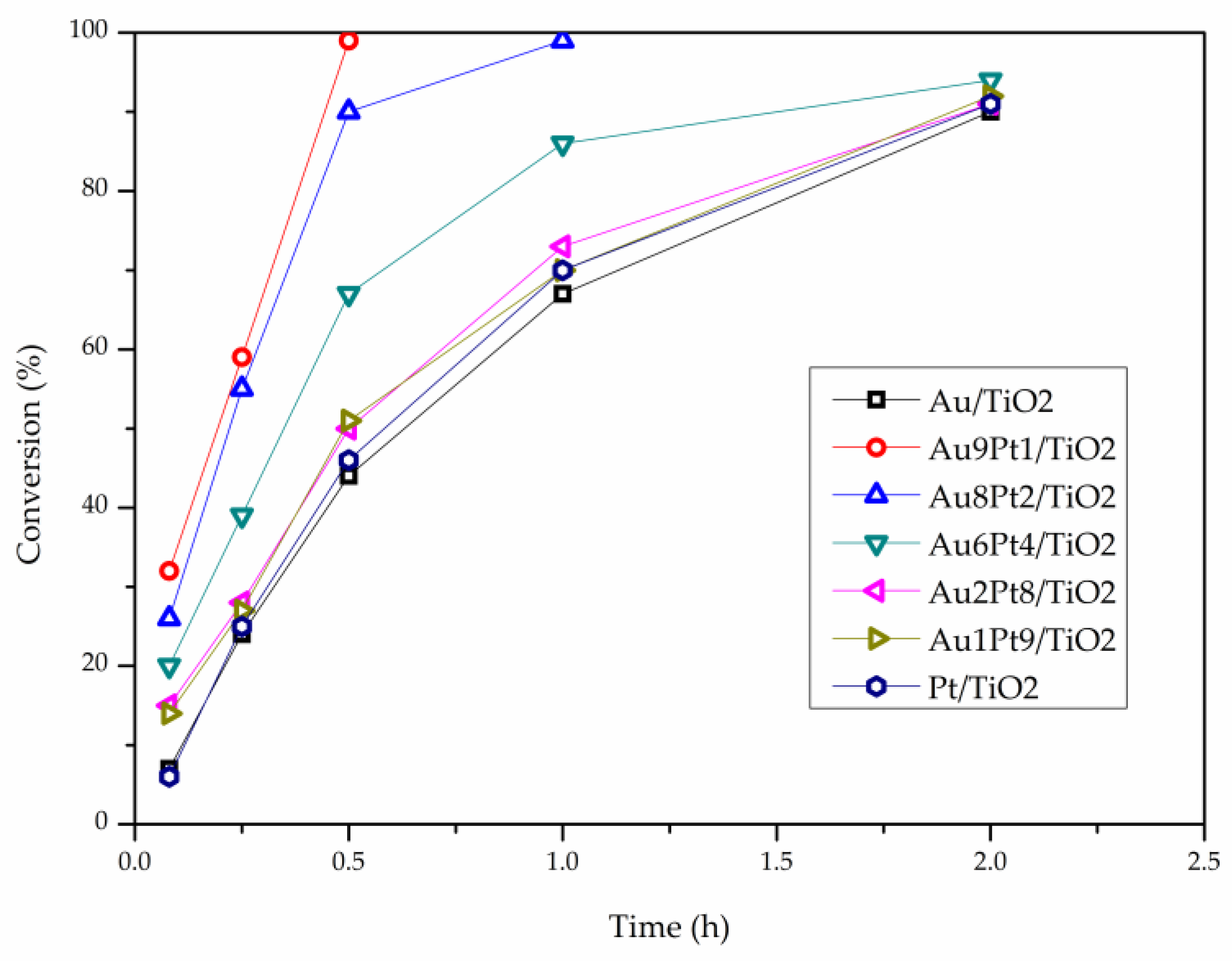
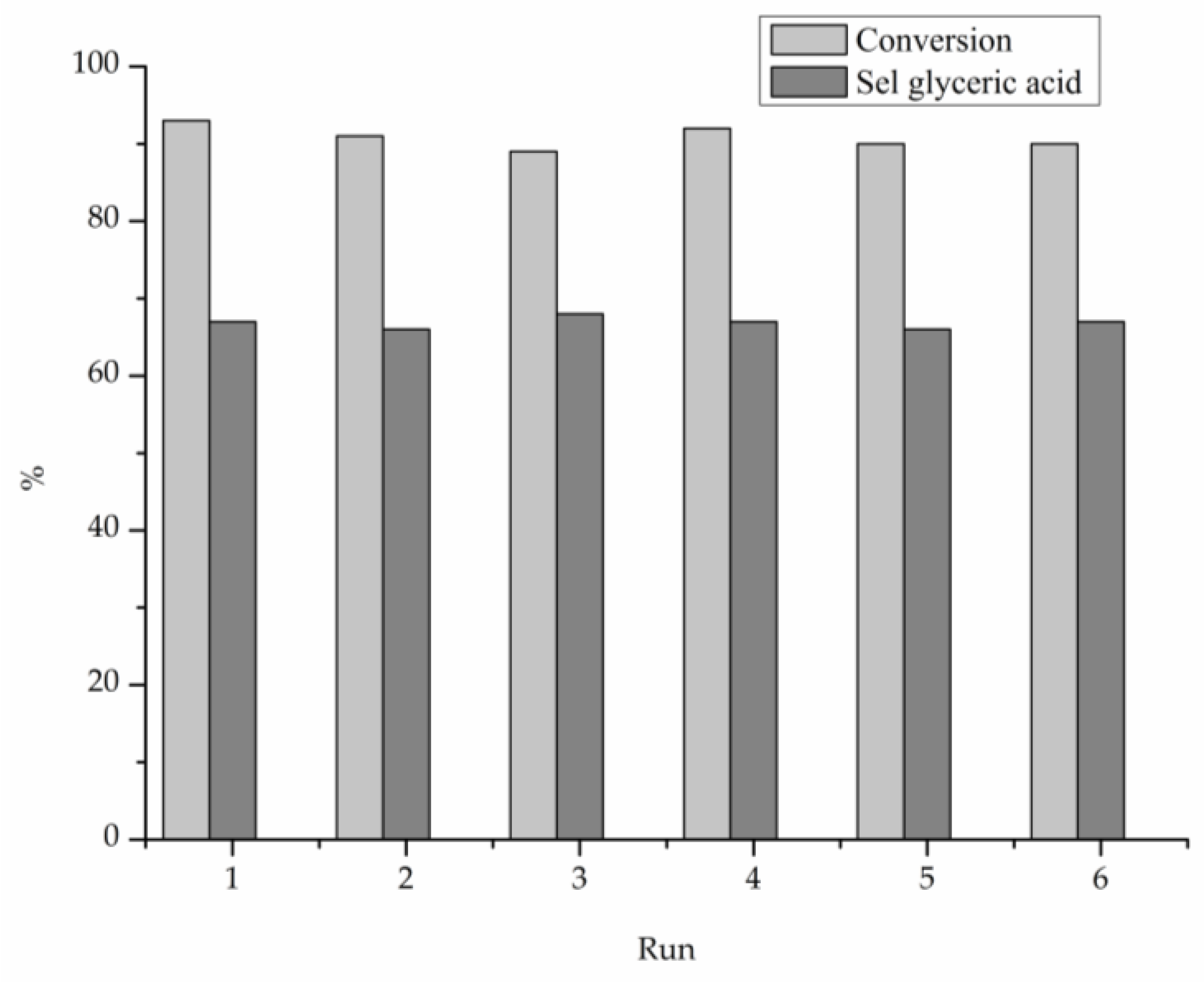
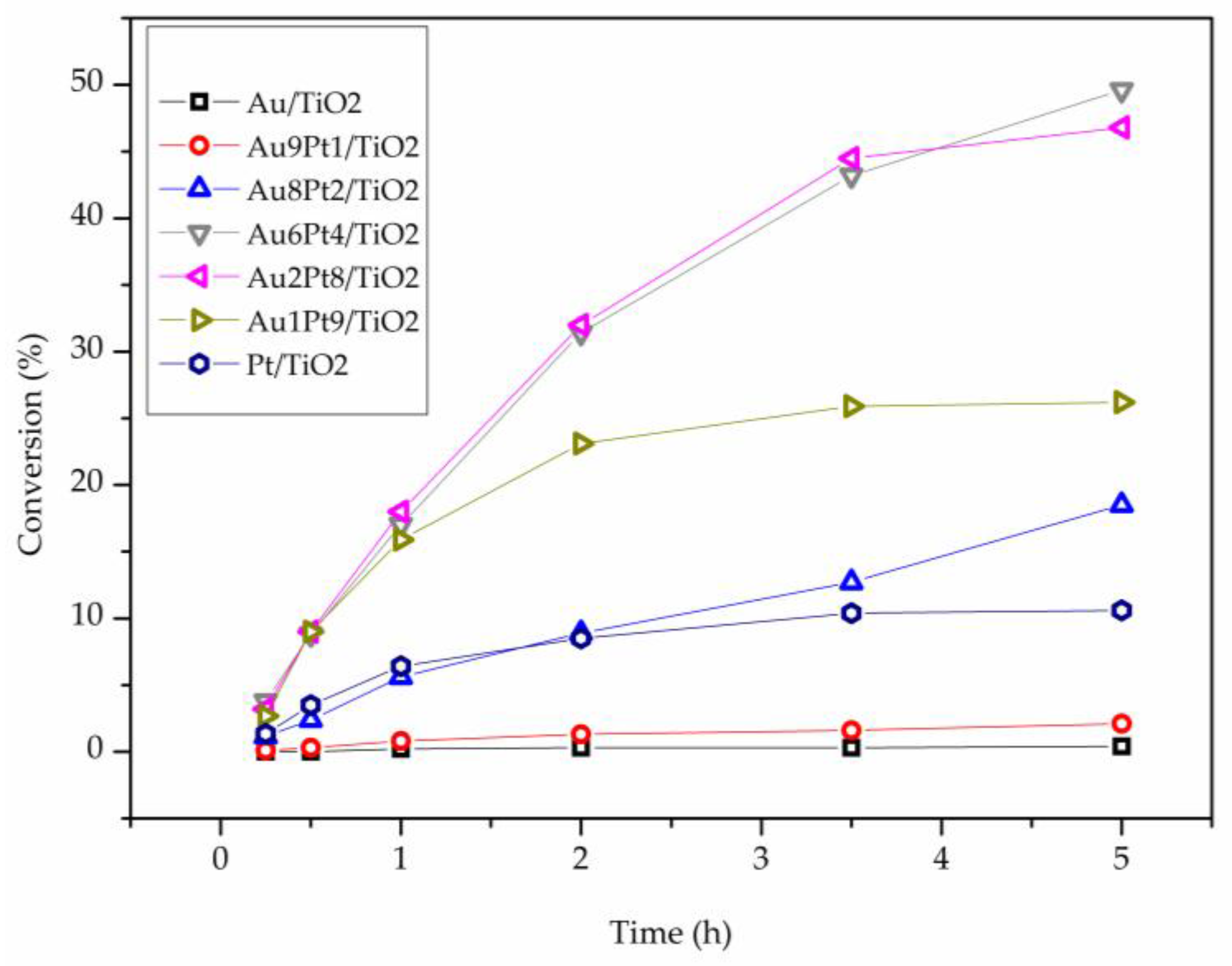
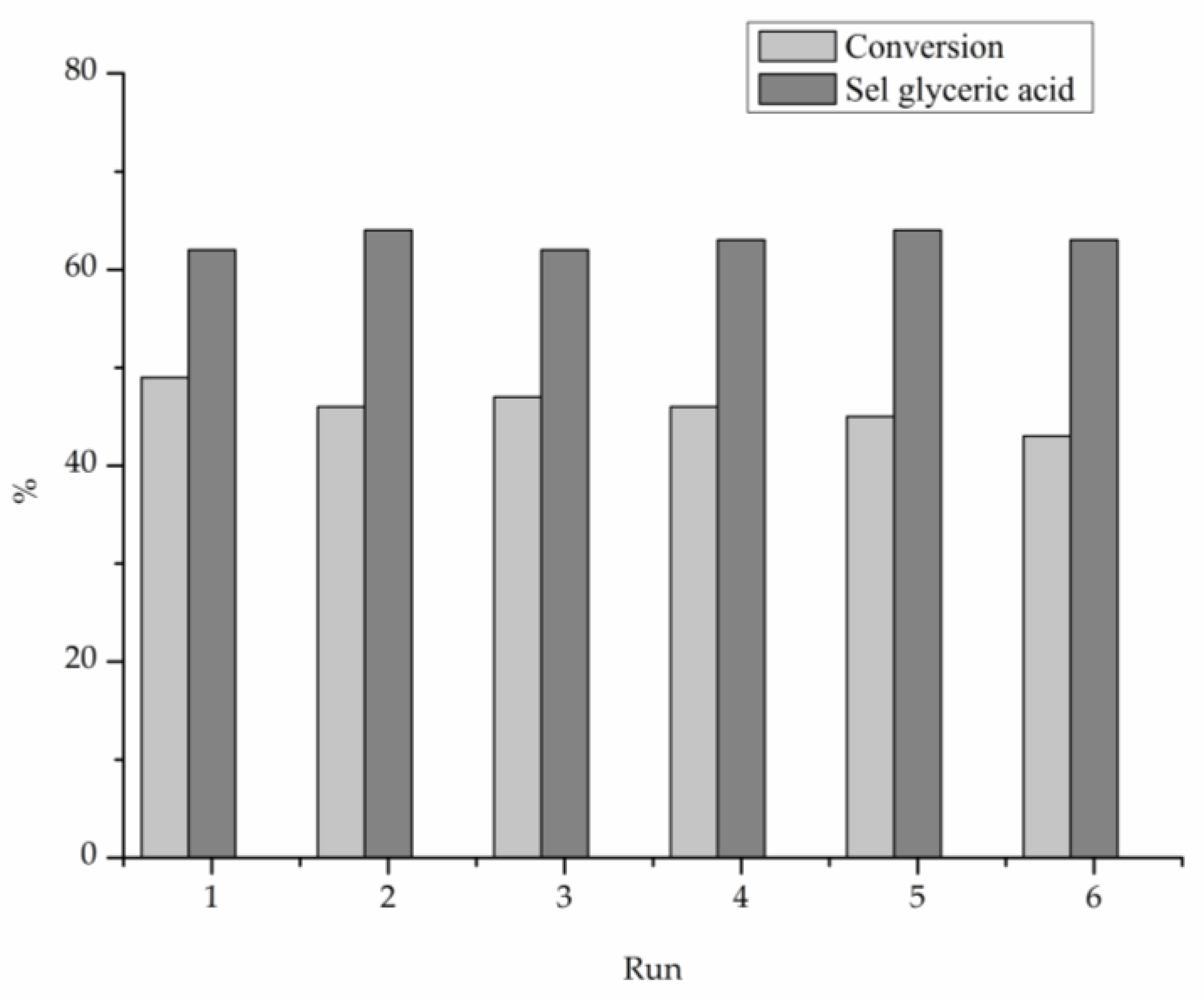
2. Results
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Materials
5.2. Catalyst Preparation
5.3. Catalytic Tests
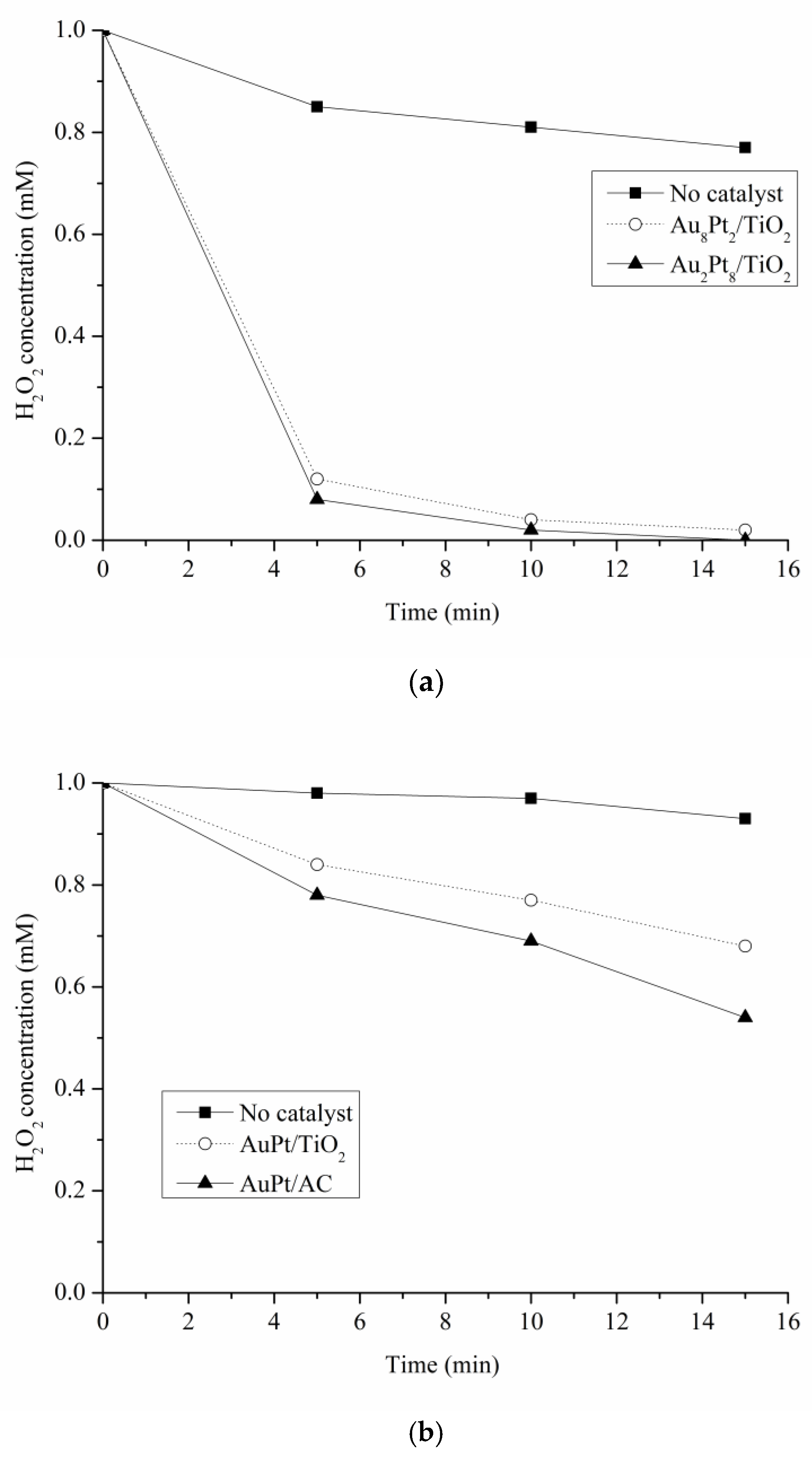
5.4. Test for Hydrogen Peroxide Detection and Degradation Test
5.5. Characterization
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Corma, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Okkerse, C.; van Bekkum, H. From fossil to green. Green Chem. 1999, 1, 107–114. [Google Scholar] [CrossRef]
- Behr, A.; Eilting, J.; Irawadi, K.; Leschinski, J.; Lindner, F. Improved utilisation of renewable resources: New important derivatives of glycerol. Green Chem. 2008, 10, 13–30. [Google Scholar] [CrossRef]
- Katryniok, B.; Kimura, H.; Skrzyńska, E.; Girardon, J.-S.; Fongarland, P.; Capron, M.; Ducoulombier, R.; Mimura, N.; Paul, S.; Dumeignil, F. Selective catalytic oxidation of glycerol: Perspectives for high value chemicals. Green Chem. 2011, 13, 1960–1979. [Google Scholar] [CrossRef]
- Kimura, H.; Tsuto, K.; Wakisaka, T.; Kazumi, Y.; Inaya, Y. Selective oxidation of glycerol on a platinum-bismuth catalyst. Appl. Catal. A Gen. 1993, 96, 217–228. [Google Scholar] [CrossRef]
- Garcia, R.; Besson, M.; Gallezot, P. Chemoselective catalytic oxidation of glycerol with air on platinum metals. Appl. Catal. A Gen. 1995, 127, 165–176. [Google Scholar] [CrossRef]
- Villa, A.; Dimitratos, N.; Chan-Thaw, C.E.; Hammond, C.; Prati, L.; Hutchings, G.J. Glycerol Oxidation Using Gold-Containing Catalysts. Acc. Chem. Res. 2015, 48, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Carrettin, S.; McMorn, P.; Johnston, P.; Griffin, K.; Hutchings, G.J. Selective oxidation of glycerol to glyceric acid using a gold catalyst in aqueous sodium hydroxide. Chem. Commun. 2002, 696–697. [Google Scholar] [CrossRef]
- Porta, F.; Prati, L. Selective oxidation of glycerol to sodium glycerate with gold-on-carbon catalyst: An insight into reaction selectivity. J. Catal. 2004, 224, 397–403. [Google Scholar] [CrossRef]
- Wang, D.; Villa, A.; Porta, F.; Su, D.; Prati, L. Single-phase bimetallic system for the selective oxidation of glycerol to glycerate. Chem. Commun. 2006, 1956–1958. [Google Scholar] [CrossRef] [PubMed]
- Dimitratos, N.; Lopez-Sanchez, J.A.; Anthonykutty, J.M.; Brett, G.; Carley, A.F.; Tiruvalam, R.C.; Herzing, A.A.; Kiely, C.J.; Knight, D.W.; Hutchings, G.J. Oxidation of glycerol using gold–palladium alloy-supported nanocrystals. Phys. Chem. Chem. Phys. 2009, 11, 4952–4961. [Google Scholar] [CrossRef] [PubMed]
- Chan-Thaw, C.; Campisi, S.; Wang, D.; Prati, L.; Villa, A. Selective Oxidation of Raw Glycerol Using Supported AuPd Nanoparticles. Catalysts 2015, 5, 131–144. [Google Scholar] [CrossRef]
- Ketchie, W.C.; Murayama, M.; Davis, R.J. Selective oxidation of glycerol over carbon-supported AuPd catalysts. J. Catal. 2007, 250, 264–273. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, H.; Zhao, Y.; Yu, B.; Chen, S.; Li, Y.; Hao, L.; Liu, Z. Selective oxidation of glycerol to lactic acid under acidic conditions using AuPd/TiO2 catalyst. Green Chem. 2013, 15, 1520–1525. [Google Scholar] [CrossRef]
- Evans, C.D.; Kondrat, S.A.; Smith, P.J.; Manning, T.D.; Miedziak, P.J.; Brett, G.L.; Armstrong, R.D.; Bartley, J.K.; Taylor, S.H.; Rosseinsky, M.J.; et al. The preparation of large surface area lanthanum based perovskite supports for AuPt nanoparticles: Tuning the glycerol oxidation reaction pathway by switching the perovskite B site. Faraday Discuss. 2016, 188, 427–450. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Villa, A.; Porta, F.; Prati, L.; Su, D. Bimetallic Gold/Palladium Catalysts: Correlation between Nanostructure and Synergistic Effects. J. Phys. Chem. C 2008, 112, 8617–8622. [Google Scholar] [CrossRef]
- Zhao, Z.; Arentz, J.; Pretzer, L.A.; Limpornpipat, P.; Clomburg, J.M.; Gonzalez, R.; Schweitzer, N.M.; Wu, T.; Miller, J.T.; Wong, M.S. Volcano-shape glycerol oxidation activity of palladium-decorated gold nanoparticles. Chem. Sci. 2014, 5, 3715–3728. [Google Scholar] [CrossRef]
- Villa, A.; Veith, G.M.; Prati, L. Selective Oxidation of Glycerol under Acidic Conditions Using Gold Catalysts. Angew. Chem. Int. Ed. 2010, 49, 4499–4502. [Google Scholar] [CrossRef] [PubMed]
- Brett, G.L.; He, Q.; Hammond, C.; Miedziak, P.J.; Dimitratos, N.; Sankar, M.; Herzing, A.A.; Conte, M.; Lopez-Sanchez, J.A.; Kiely, C.J.; et al. Selective Oxidation of Glycerol by Highly Active Bimetallic Catalysts at Ambient Temperature under Base-Free Conditions. Angew. Chem. Int. Ed. 2011, 50, 10136–10139. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, E.G.; Pereira, M.F.R.; Chen, X.; Delgado, J.J.; Órfão, J.J.M. Selective Oxidation of Glycerol over Platinum-Based Catalysts Supported on Carbon Nanotubes. Ind. Eng. Chem. Res. 2013, 52, 17390–17398. [Google Scholar] [CrossRef]
- Villa, A.; Campisi, S.; Mohammed, K.M.H.; Dimitratos, N.; Vindigni, F.; Manzoli, M.; Jones, W.; Bowker, M.; Hutchings, G.J.; Prati, L. Tailoring the selectivity of glycerol oxidation by tuning the acid–base properties of Au catalysts. Catal. Sci. Technol. 2015, 5, 1126–1132. [Google Scholar] [CrossRef]
- Tongsakul, D.; Nishimura, S.; Ebitani, K. Platinum/Gold Alloy Nanoparticles-Supported Hydrotalcite Catalyst for Selective Aerobic Oxidation of Polyols in Base-Free Aqueous Solution at Room Temperature. ACS Catal. 2013, 3, 2199–2207. [Google Scholar] [CrossRef]
- Yang, G.-Y.; Shao, S.; Ke, Y.-H.; Liu, C.-L.; Ren, H.-F.; Dong, W.-S. PtAu alloy nanoparticles supported on thermally expanded graphene oxide as a catalyst for the selective oxidation of glycerol. RSC Adv. 2015, 5, 37112–37118. [Google Scholar] [CrossRef]
- Sánchez, B.S.; Gross, M.S.; Querini, C.A. Pt catalysts supported on ion exchange resins for selective glycerol oxidation. Effect of Au incorporation. Catal. Today 2017, 296, 35–42. [Google Scholar] [CrossRef]
- Shen, Y.; Li, Y.; Liu, H. Base-free aerobic oxidation of glycerol on TiO2-supported bimetallic Au-Pt catalysts. J. Energy Chem. 2015, 24, 669–673. [Google Scholar] [CrossRef]
- Chan-Thaw, C.E.; Chinchilla, L.E.; Campisi, S.; Botton, G.A.; Prati, L.; Dimitratos, N.; Villa, A. AuPt Alloy on TiO2 : A Selective and Durable Catalyst for l-Sorbose Oxidation to 2-Keto-Gulonic Acid. ChemSusChem 2015, 8, 4189–4194. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Manzoli, M.; Vindigni, F.; Chinchilla, L.E.; Botton, G.A.; Prati, L. Diols Production From Glycerol Over Pt-Based Catalysts: On the Role Played by the Acid Sites of the Support. Catal. Lett. 2017, 147, 2523–2533. [Google Scholar] [CrossRef]
- Radnik, J.; Mohr, C.; Claus, P. On the origin of binding energy shifts of core levels of supported gold nanoparticles and dependence of pretreatment and material synthesis. Phys. Chem. Chem. Phys. 2003, 5, 172–177. [Google Scholar] [CrossRef]
- Arrii, S.; Morfin, F.; Renouprez, A.J.; Rousset, J.L. Oxidation of CO on Gold Supported Catalysts Prepared by Laser Vaporization: Direct Evidence of Support Contribution. J. Am. Chem. Soc. 2004, 126, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Zwijnenburg, A.; Goossens, A.; Sloof, W.G.; Crajé, M.W.J.; Van der Kraan, A.M.; De Jongh, L.J.; Makkee, M.; Moulijn, J.A. XPS and Mössbauer characterization of Au/TiO2 propene epoxidation catalysts. J. Phys. Chem. B 2002, 106, 9853–9862. [Google Scholar] [CrossRef]
- Yung, T.-Y.; Liu, T.-Y.; Huang, L.-Y.; Wang, K.-S.; Tzou, H.-M.; Chen, P.-T.; Chao, C.-Y.; Liu, L.-K. Characterization of Au and Bimetallic PtAu Nanoparticles on PDDA-Graphene Sheets as Electrocatalysts for Formic Acid Oxidation. Nanoscale Res. Lett. 2015, 10, 365. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-Y.; Jeon, T.-Y.; Jang, J.H.; Yoo, S.J.; Choi, K.-H.; Jung, N.; Chung, Y.-H.; Ahn, M.; Cho, Y.-H.; Lee, K.-S.; et al. Enhancement of oxygen reduction reaction on PtAu nanoparticles via CO induced surface Pt enrichment. Appl. Catal. B Environ. 2013, 129, 375–381. [Google Scholar] [CrossRef]
- Ketchie, W.C.; Fang, Y.-L.; Wong, M.S.; Murayama, M.; Davis, R.J. Influence of gold particle size on the aqueous-phase oxidation of carbon monoxide and glycerol. J. Catal. 2007, 250, 94–101. [Google Scholar] [CrossRef]
- Zope, B.N.; Hibbitts, D.D.; Neurock, M.; Davis, R.J. Reactivity of the Gold/Water Interface during Selective Oxidation Catalysis. Science 2010, 330, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Dimitratos, N.; Villa, A.; Wang, D.; Porta, F.; Su, D.; Prati, L. Pd and Pt catalysts modified by alloying with Au in the selective oxidation of alcohols. J. Catal. 2006, 244, 113–121. [Google Scholar] [CrossRef]
- Kwon, Y.; Schouten, K.J.P.; Koper, M.T.M. Mechanism of the Catalytic Oxidation of Glycerol on Polycrystalline Gold and Platinum Electrodes. ChemCatChem 2011, 3, 1176–1185. [Google Scholar] [CrossRef]
- Zope, B.N.; Davis, R.J. Inhibition of gold and platinum catalysts by reactive intermediates produced in the selective oxidation of alcohols in liquid water. Green Chem. 2011, 13, 3484–3491. [Google Scholar] [CrossRef]
- Prati, L.; Spontoni, P.; Gaiassi, A. From Renewable to Fine Chemicals Through Selective Oxidation: The Case of Glycerol. Top. Catal. 2009, 52, 288–296. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Au4f | Pt4f | Au-Pt Atomic Ratio | Au at % | Pt at % | Metal at % | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Au0 | Pt0 | PtO | Nominal | Bulk (AAS) | Surface (XPS) | |||||
| 1% Au/TiO2 | BE (eV) | 83.5 | - | - | - | - | - | 0.40 | - | 0.40 |
| % | 100 | |||||||||
| 1% Au9Pt1/TiO2 | BE (eV) | 83.3 | 70.3 | - | 9.0 | 9.3 | 12.0 | 0.24 | 0.02 | 0.26 |
| % | 100 | 100 | ||||||||
| 1% Au8Pt2/TiO2 | BE (eV) | 83.4 | 70.5 | - | 4.0 | 4.2 | 3.8 | 0.19 | 0.05 | 0.24 |
| % | 100 | 100 | ||||||||
| 1% Au6Pt4/TiO2 | BE (eV) | 83.4 | 70.5 | - | 1.5 | 1.4 | 1.9 | 0.14 | 0.06 | 0.20 |
| % | 100 | 100 | ||||||||
| 1% Au2Pt8/TiO2 | BE (eV) | 83.4 | 70.5 | - | 0.25 | 0.3 | 0.7 | 0.05 | 0.07 | 0.12 |
| % | 100 | 100 | ||||||||
| 1% Au1Pt9/TiO2 | BE (eV) | 83.4 | 70.6 | - | 0.1 | 0.1 | 0.14 | 0.02 | 0.14 | 0.16 |
| % | 100 | 100 | ||||||||
| 1% Pt/TiO2 | BE (eV) | - | 71.2 | 72.4 | - | - | - | 0.02 | 0.44 | 0.46 |
| % | 77.0 | 23.0 | ||||||||
| Catalyst | Initial Activity 2 | Selectivity (%) 3 | |||||
|---|---|---|---|---|---|---|---|
| Glyceric Acid | Tartronic Acid | ΣC3 Products | Formic Acid | Glycolic Acid | Oxalic Acid | ||
| Au/TiO2 | 2657 | 50 | 5 | 55 | 14 | 24 | 4 |
| Au9Pt1/TiO2 | 7389 | 58 | 7 | 65 | 10 | 23 | 1 |
| Au8Pt2/TiO2 | 6845 | 52 | 7 | 59 | 10 | 28 | 1 |
| Au6Pt4/TiO2 | 5052 | 42 | 6 | 48 | 29 | 19 | 1 |
| Au2Pt8/TiO2 | 3375 | 36 | 8 | 44 | 33 | 20 | 2 |
| Au1Pt9/TiO2 | 3142 | 31 | 6 | 37 | 37 | 16 | 4 |
| Pt/TiO2 | 2569 | 28 | 8 | 36 | 40 | 16 | 5 |
| Catalyst | Initial Activity 2 | Selectivity (%) 3 | |||||
|---|---|---|---|---|---|---|---|
| Glyceric Acid | Glycer-Aldehyde | ΣC3 Products | Formic Acid | Glycolic Acid | Oxalic Acid | ||
| Au/TiO2 | - | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Au9Pt1/TiO2 | 7 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Au8Pt2/TiO2 | 75 | 58 | 9 | 67 | 28 | 1 | n.d. |
| Au6Pt4/TiO2 | 301 | 62 | 10 | 72 | 20 | 5 | n.d. |
| Au2Pt8/TiO2 | 263 | 66 | 12 | 88 | 14 | 6 | n.d. |
| Au1Pt9/TiO2 | 234 | 70 | 13 | 83 | 9 | 5 | 1 |
| Pt/TiO2 | 112 | 68 | 15 | 83 | 9 | 3 | 2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villa, A.; Jouve, A.; Sanchez Trujillo, F.J.; Motta, D.; Prati, L.; Dimitratos, N. Exploring the Effect of Au/Pt Ratio on Glycerol Oxidation in Presence and Absence of a Base. Catalysts 2018, 8, 54. https://doi.org/10.3390/catal8020054
Villa A, Jouve A, Sanchez Trujillo FJ, Motta D, Prati L, Dimitratos N. Exploring the Effect of Au/Pt Ratio on Glycerol Oxidation in Presence and Absence of a Base. Catalysts. 2018; 8(2):54. https://doi.org/10.3390/catal8020054
Chicago/Turabian StyleVilla, Alberto, Andrea Jouve, Felipe J. Sanchez Trujillo, Davide Motta, Laura Prati, and Nikolaos Dimitratos. 2018. "Exploring the Effect of Au/Pt Ratio on Glycerol Oxidation in Presence and Absence of a Base" Catalysts 8, no. 2: 54. https://doi.org/10.3390/catal8020054
APA StyleVilla, A., Jouve, A., Sanchez Trujillo, F. J., Motta, D., Prati, L., & Dimitratos, N. (2018). Exploring the Effect of Au/Pt Ratio on Glycerol Oxidation in Presence and Absence of a Base. Catalysts, 8(2), 54. https://doi.org/10.3390/catal8020054