Navigating Glycerol Conversion Roadmap and Heterogeneous Catalyst Selection Aided by Density Functional Theory: A Review


Abstract
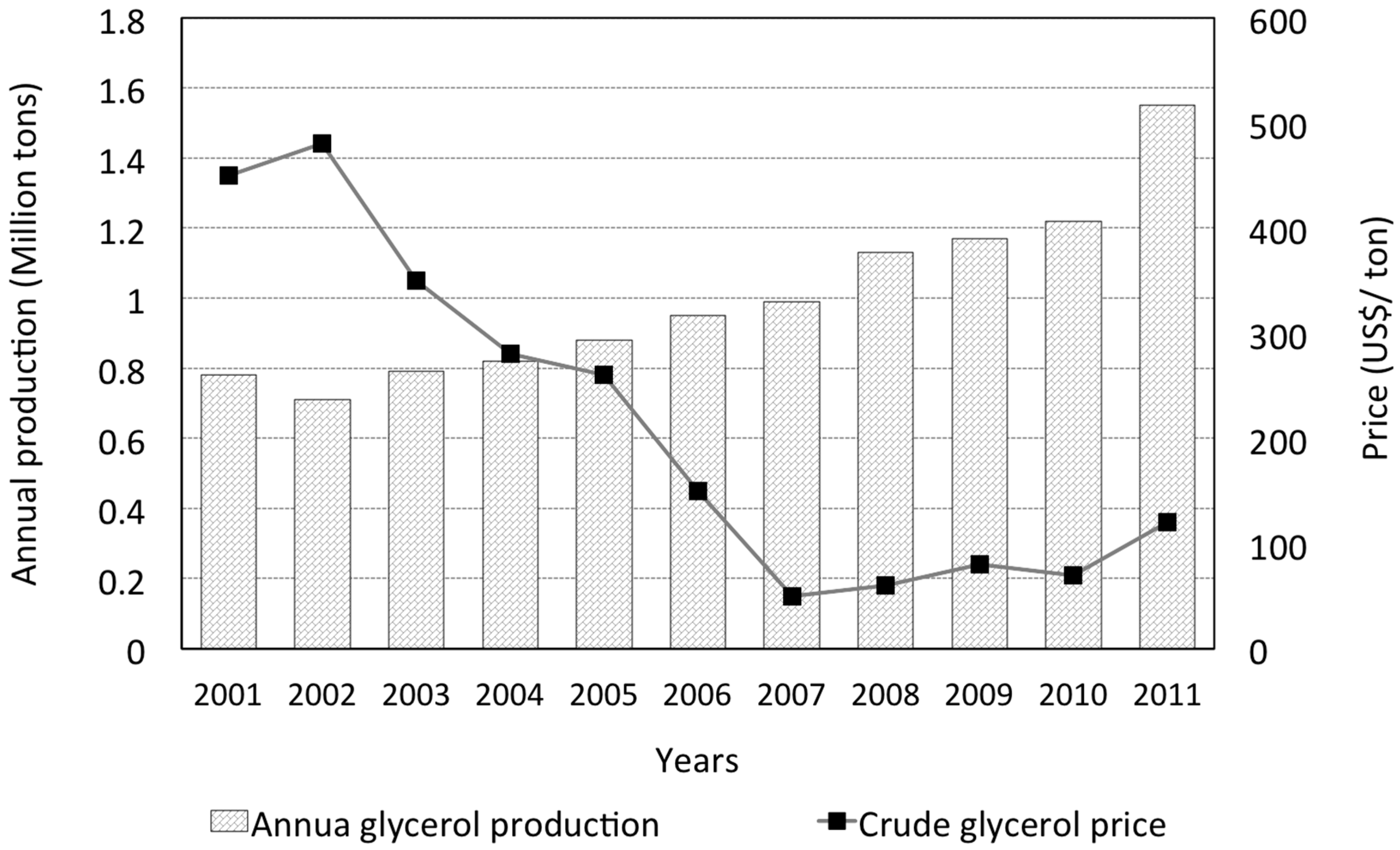
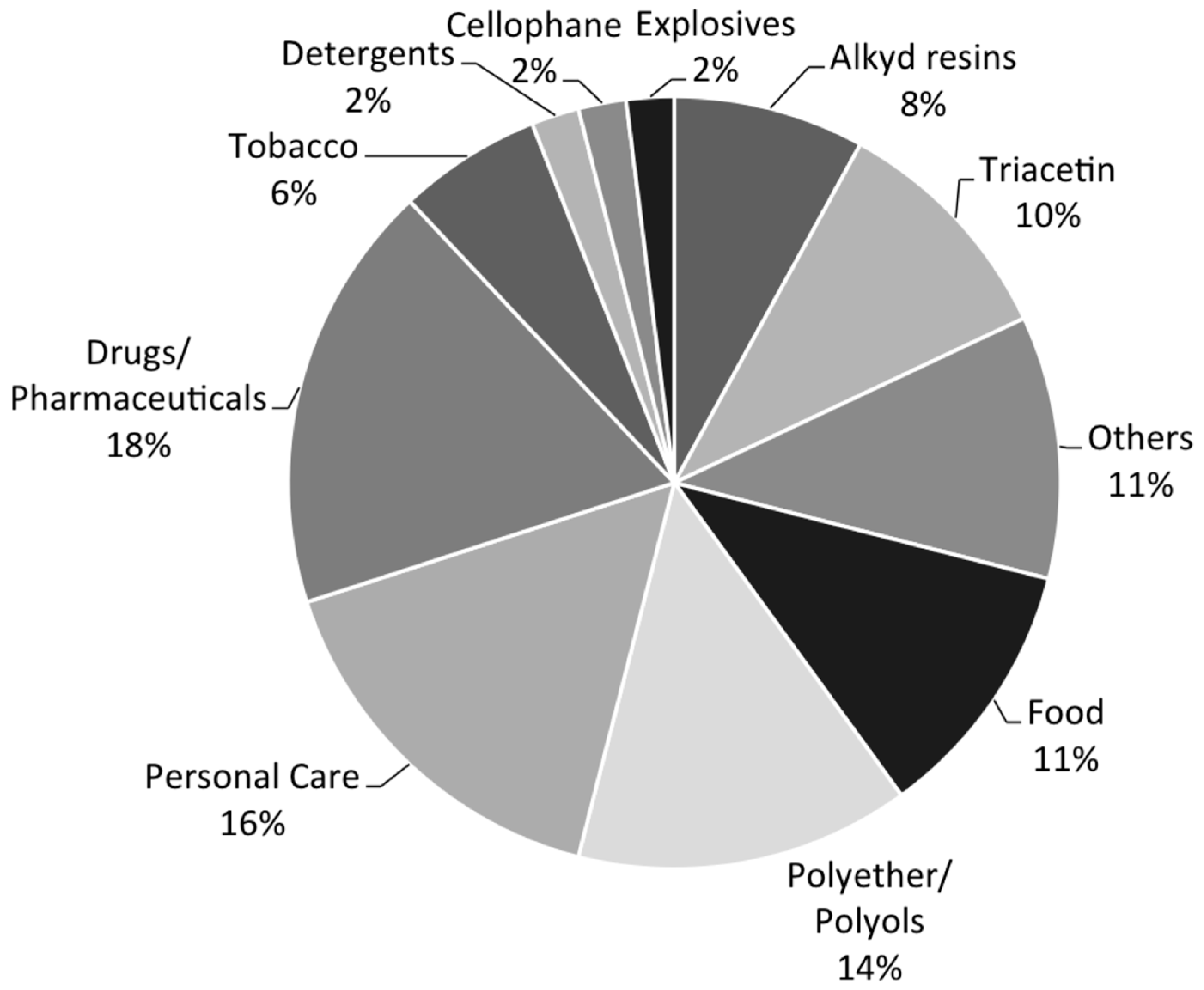
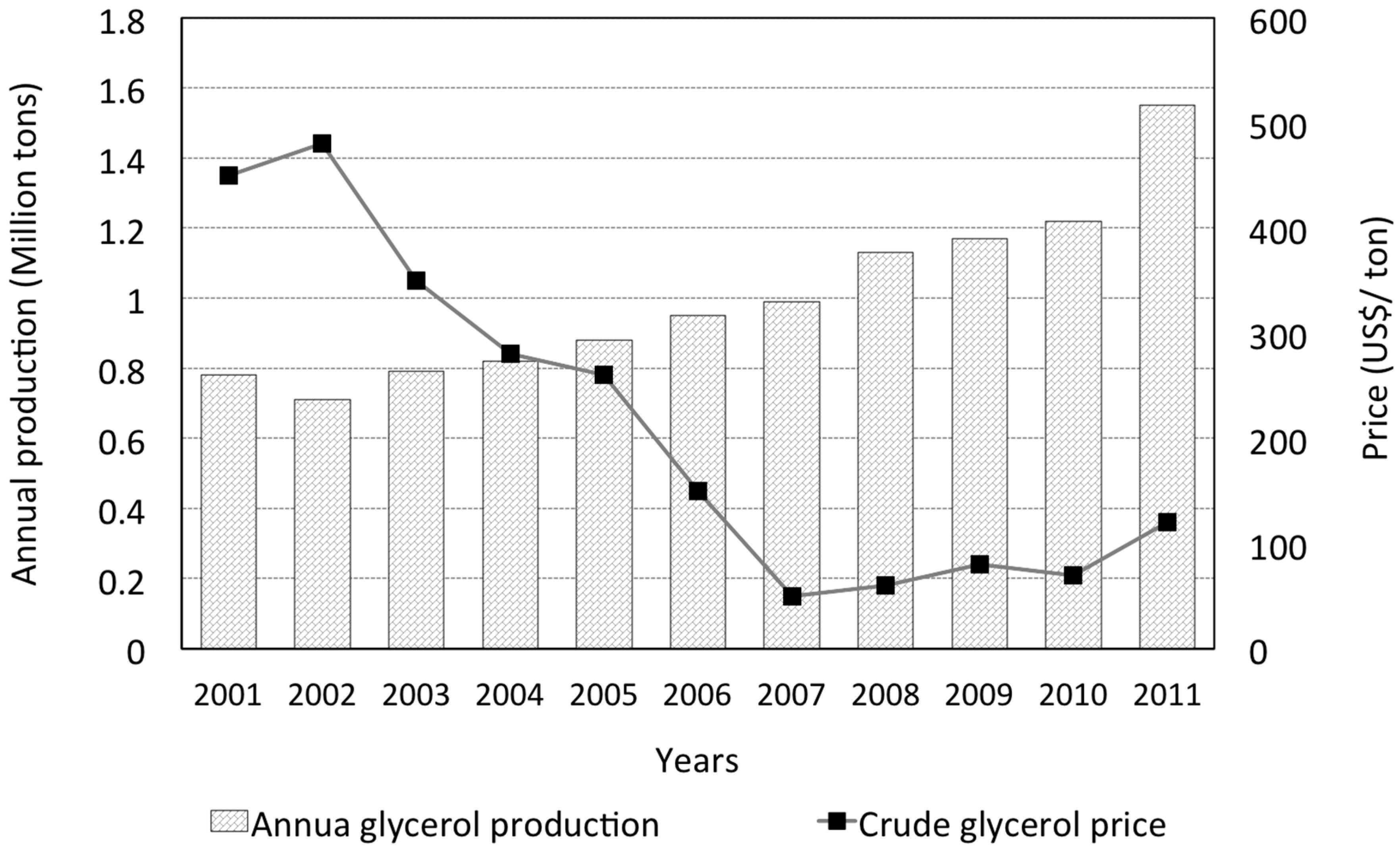
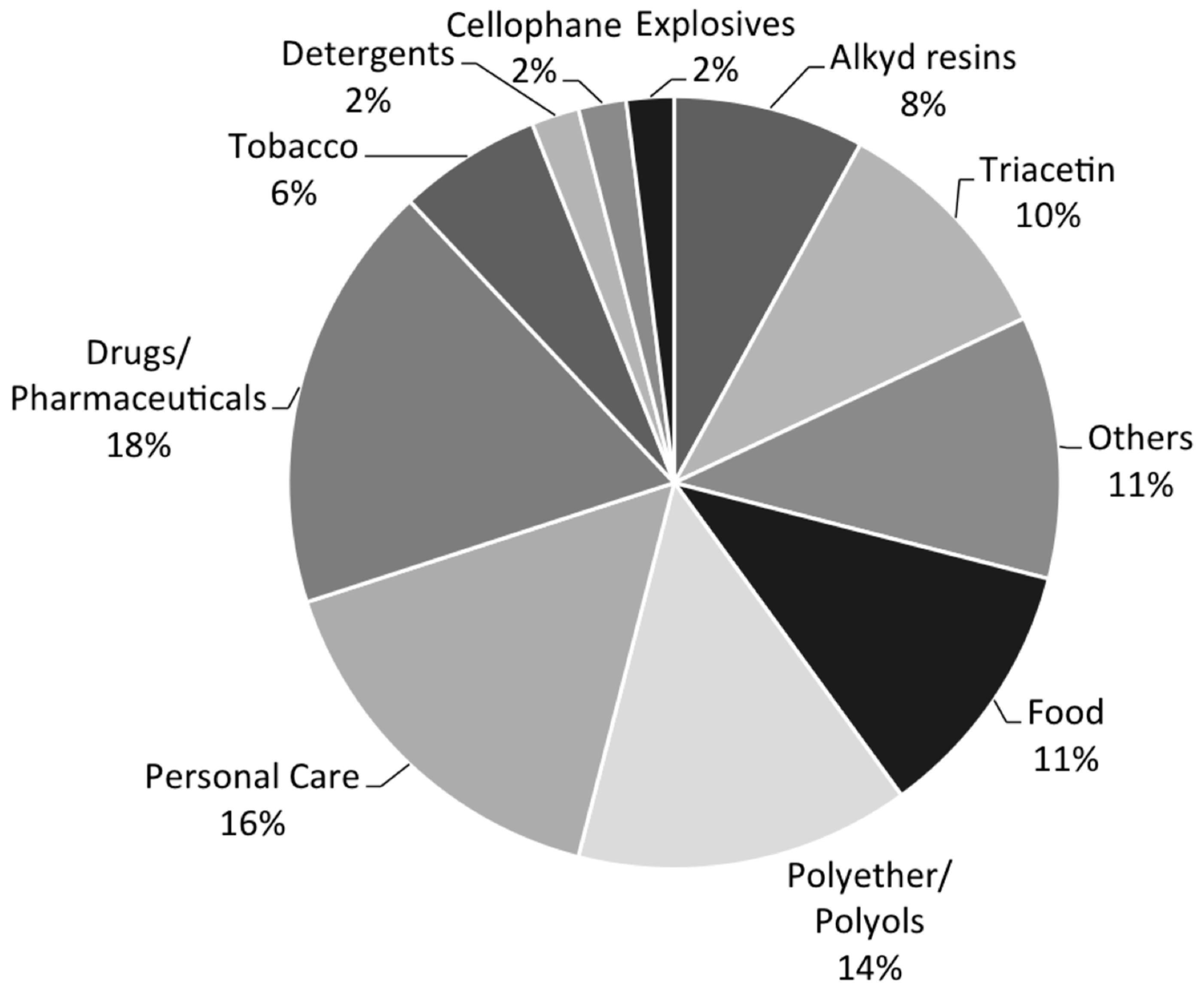
:1. Glycerol Uses and Production
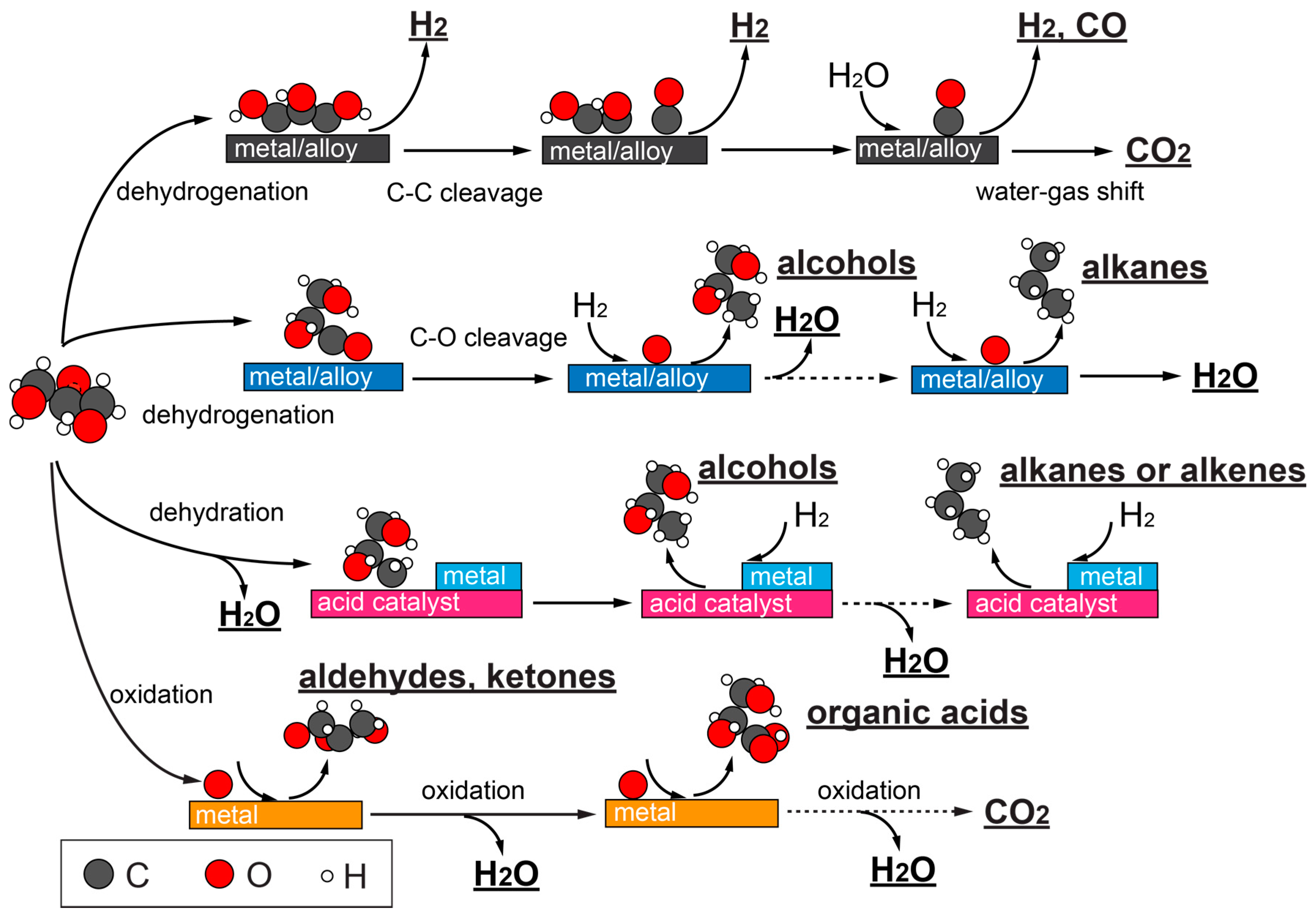
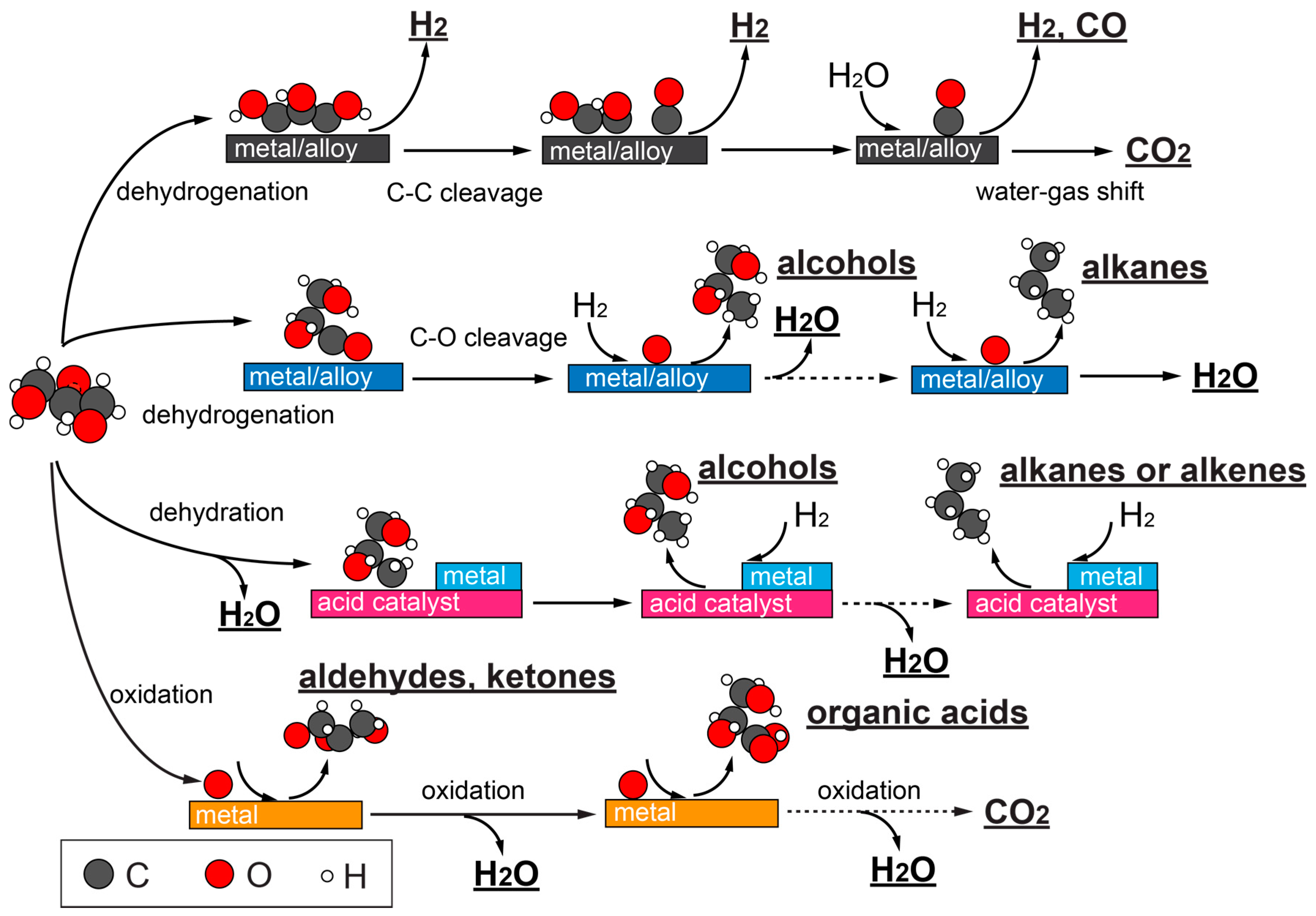
2. Chemistries in Catalytic Glycerol Conversions
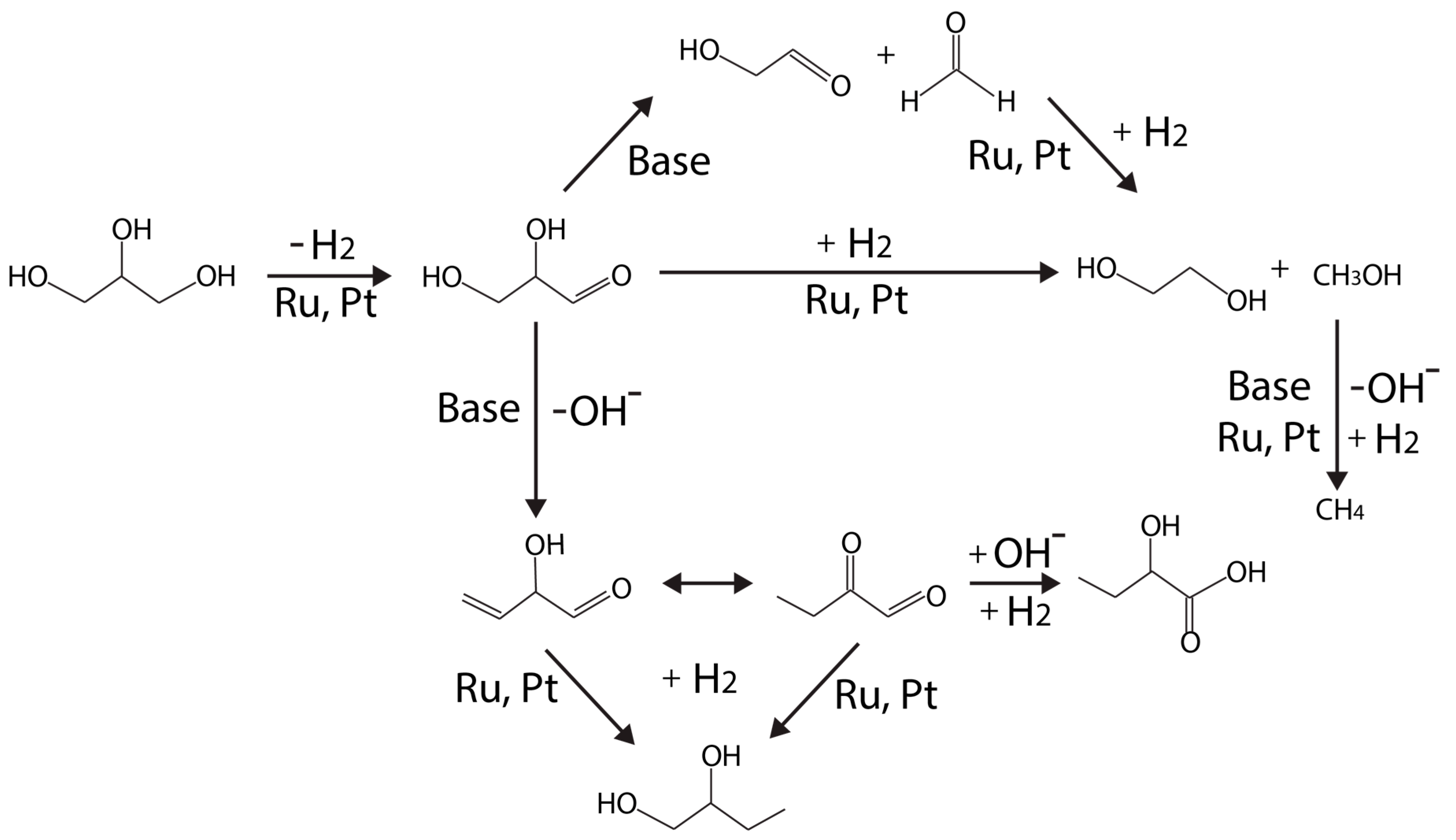
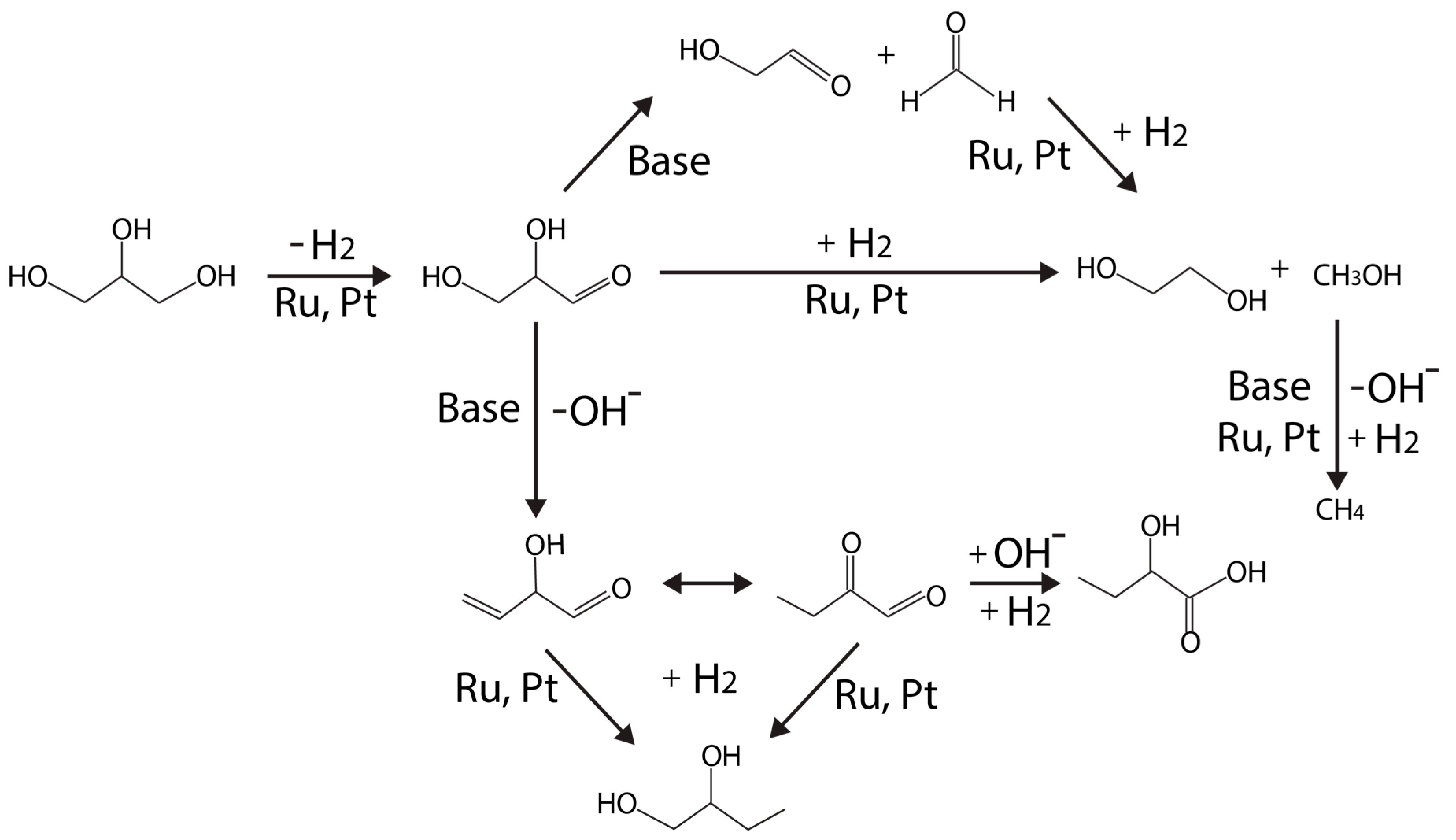
2.1. Catalytic Glycerol Reforming for Hydrogen Production
2.1.1. Aqueous and Vapor Phase Hydrogen Production
2.1.2. Catalysts for Glycerol Reforming
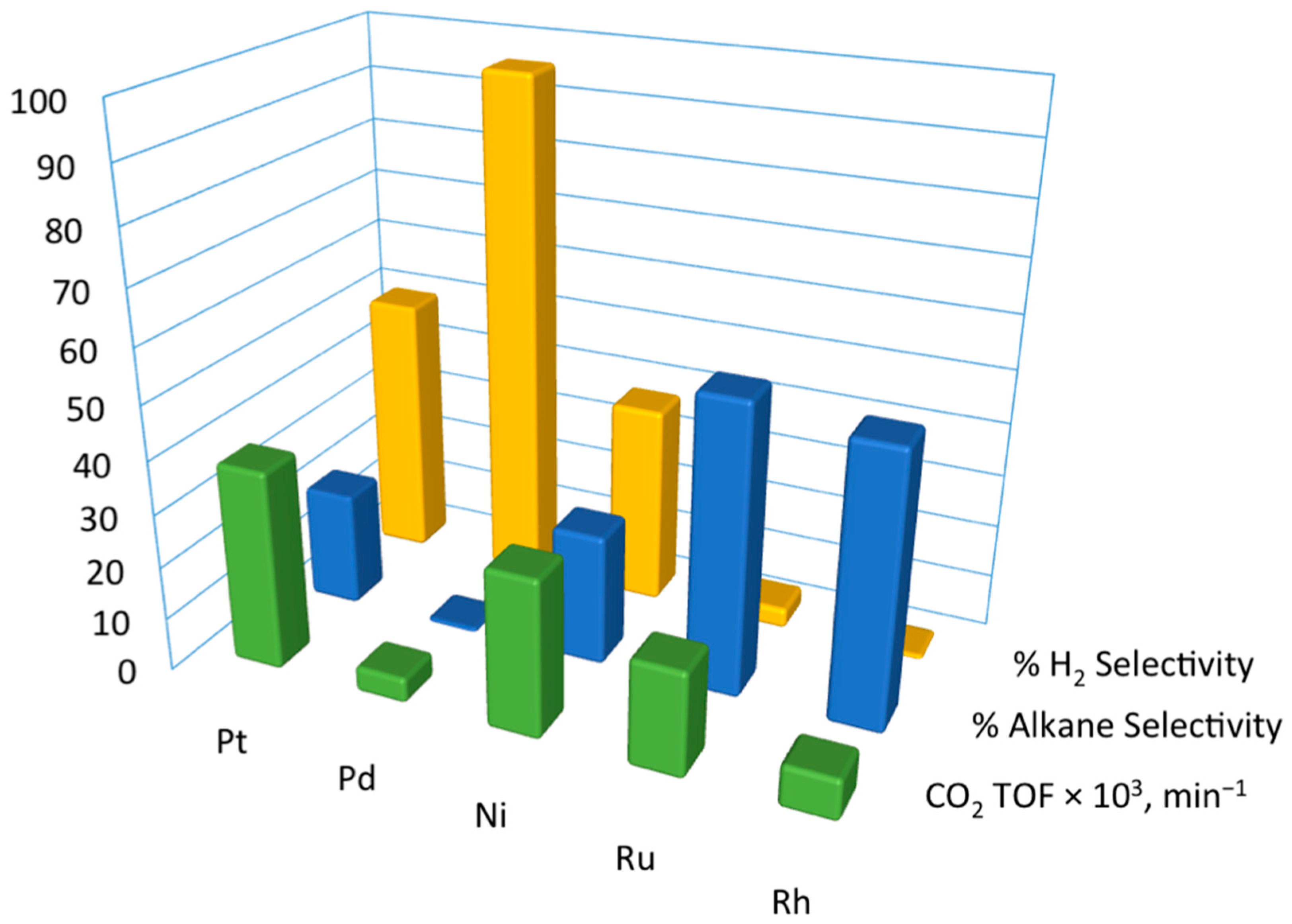
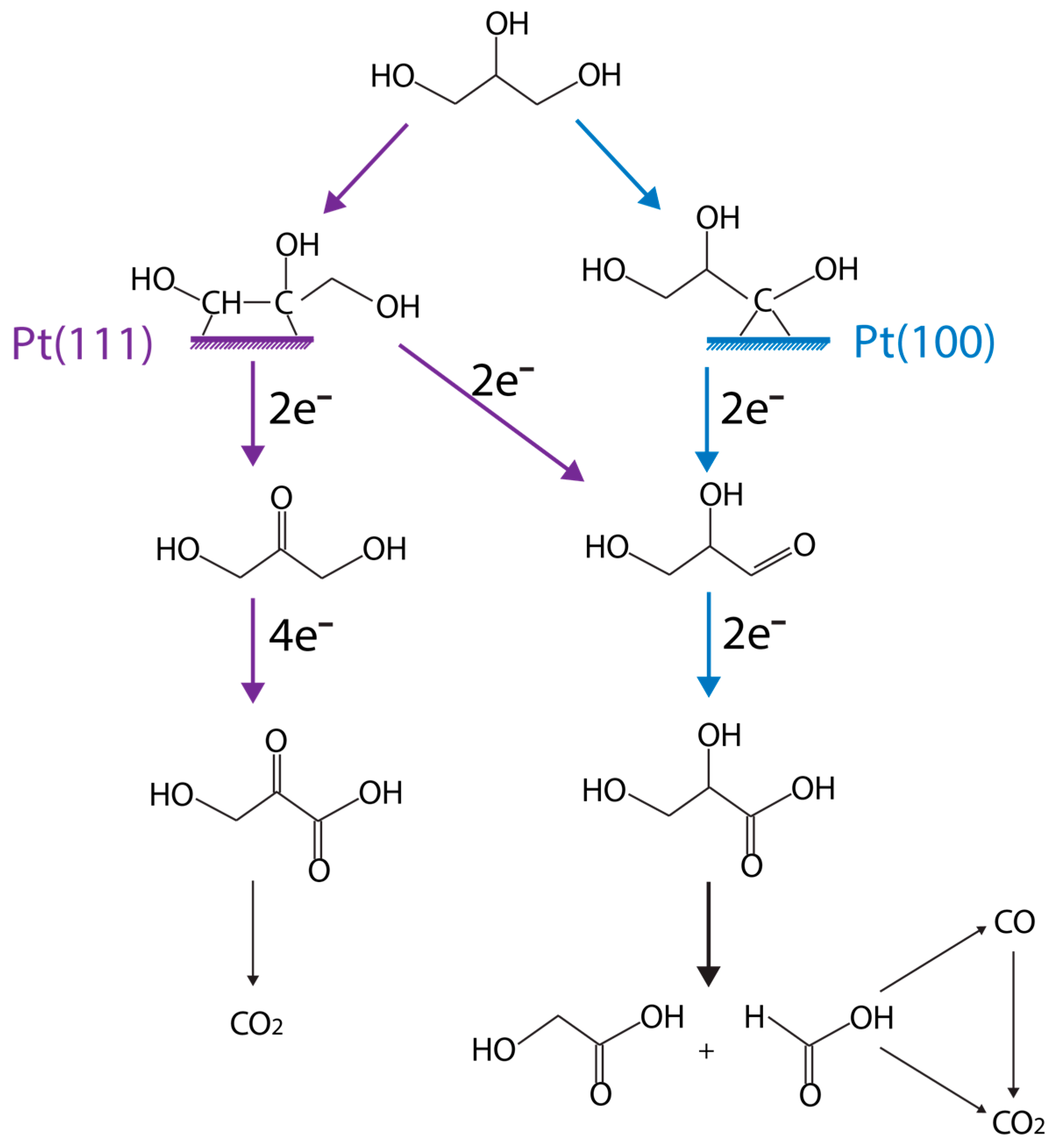
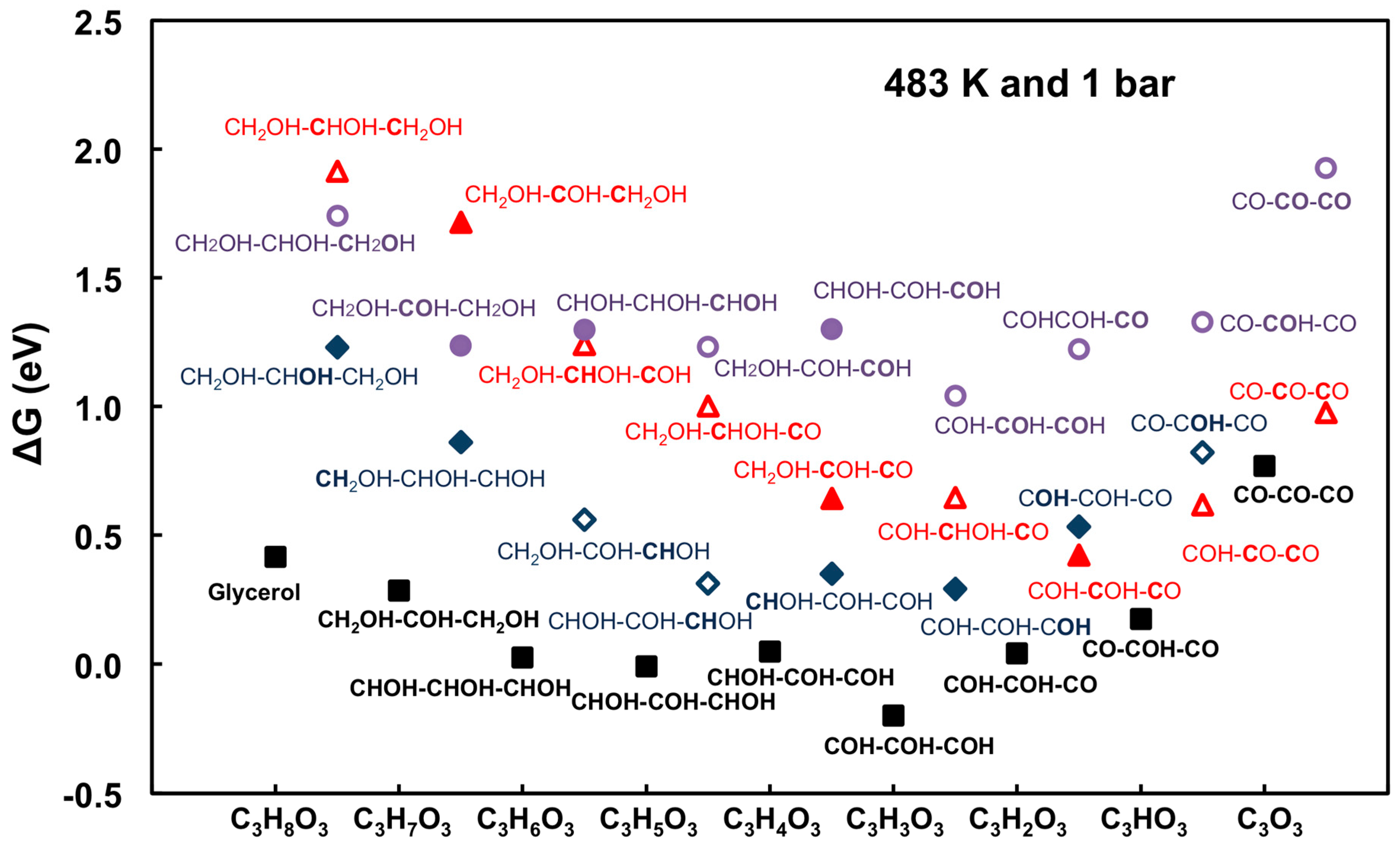
2.1.3. Understanding Molecular Behaviors for Glycerol Conversion on Transition Metals with DFT
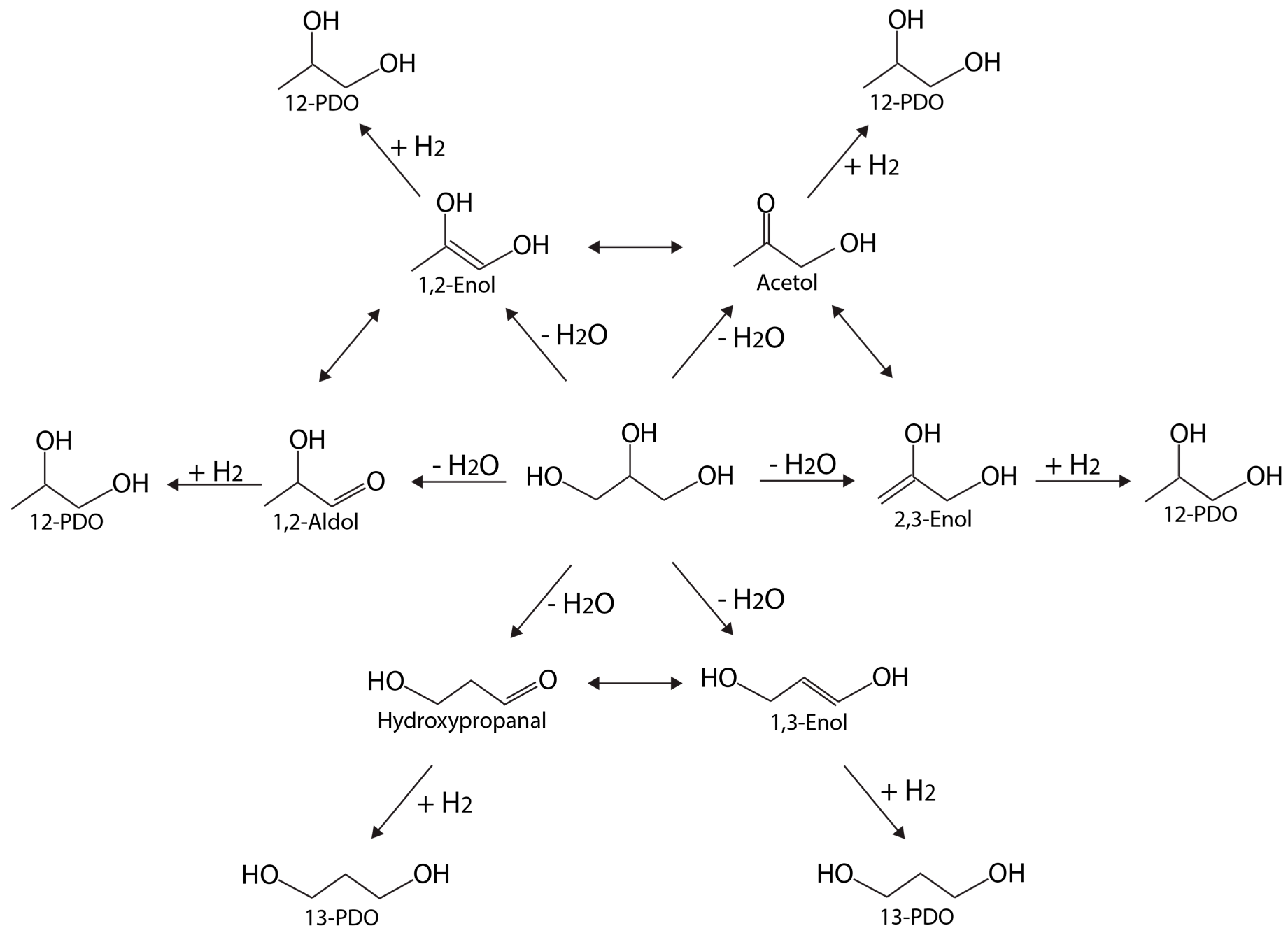
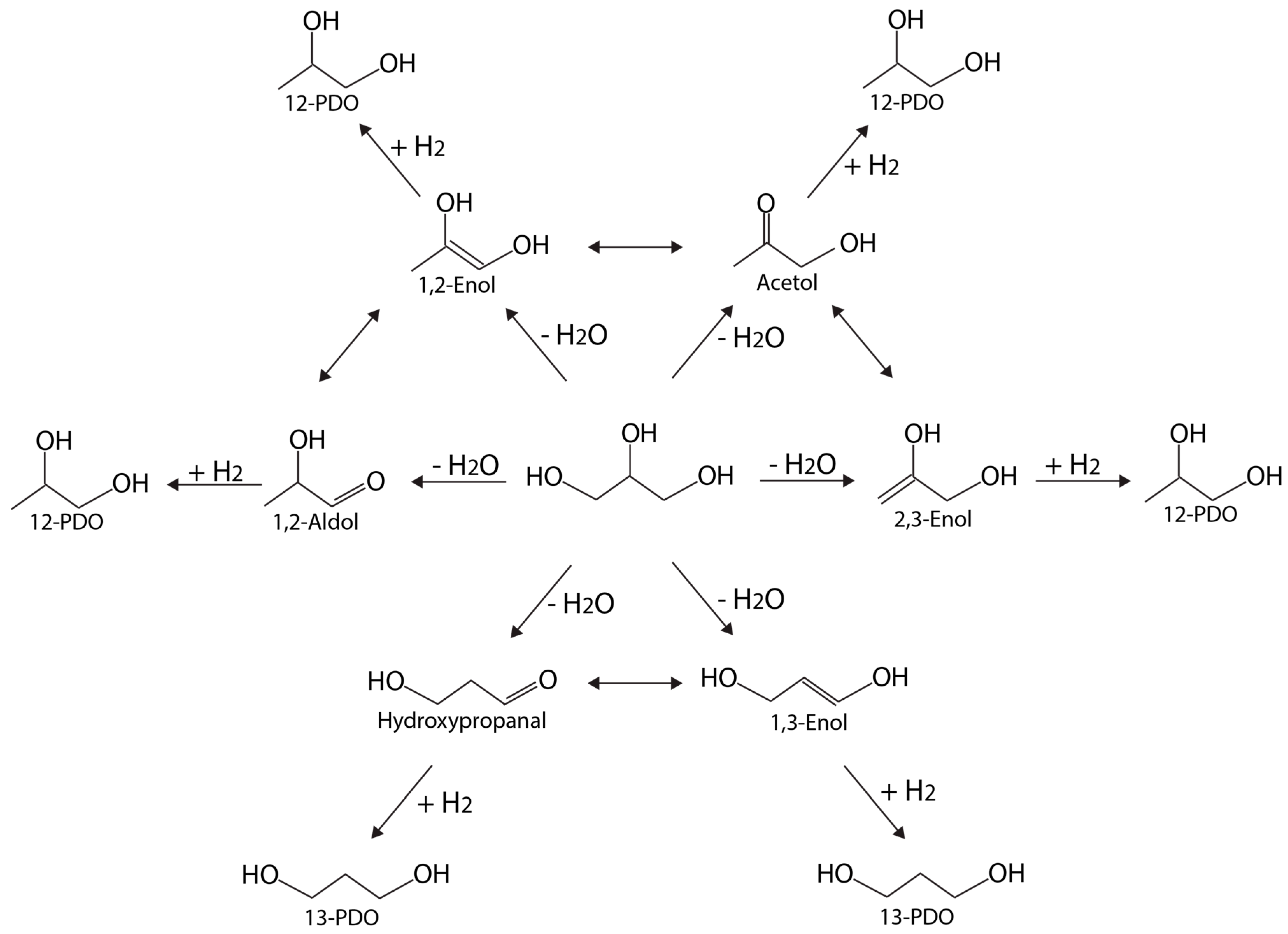
2.2. Catalysts for Glycerol Hydrogenolysis
2.3. Metal–Acid Bifunctional Catalysts for Glycerol Hydrogenolysis
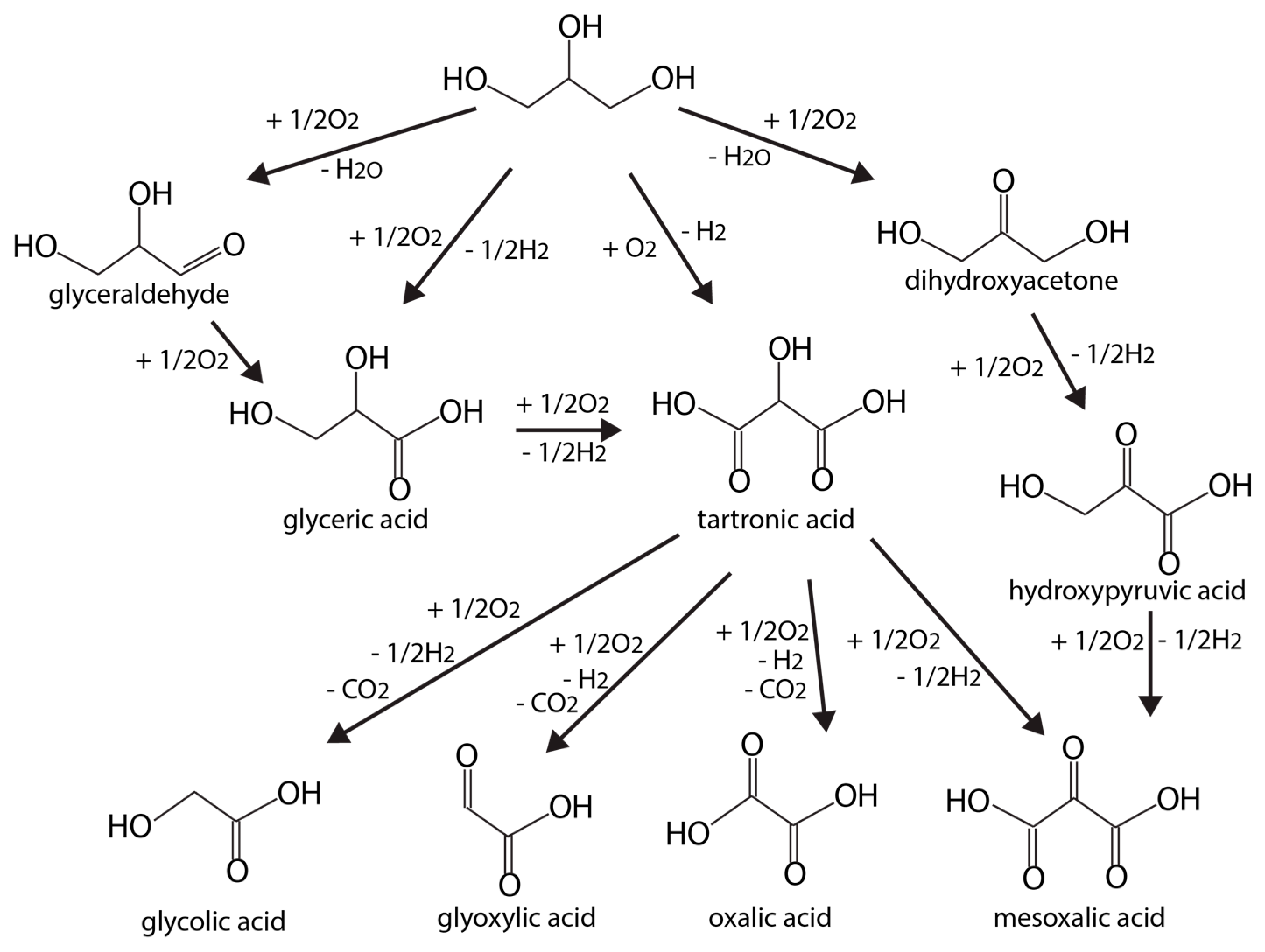
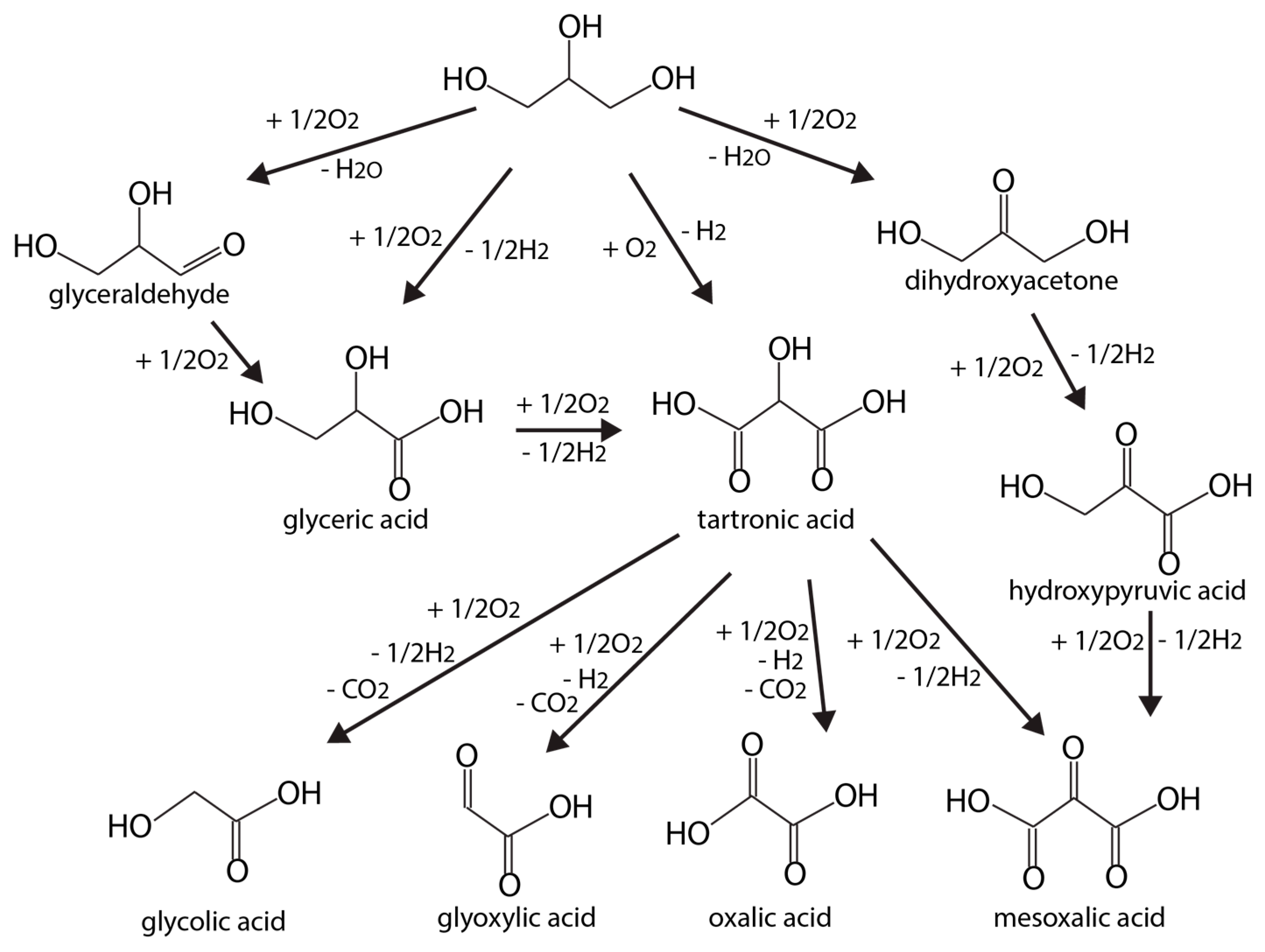
2.4. Catalysts for Glycerol Oxidation
3. Catalysts Selections Guided by First-Principles Methods
3.1. Linear Scaling Relationship to Estimate the Binding Energies of C3HxO3
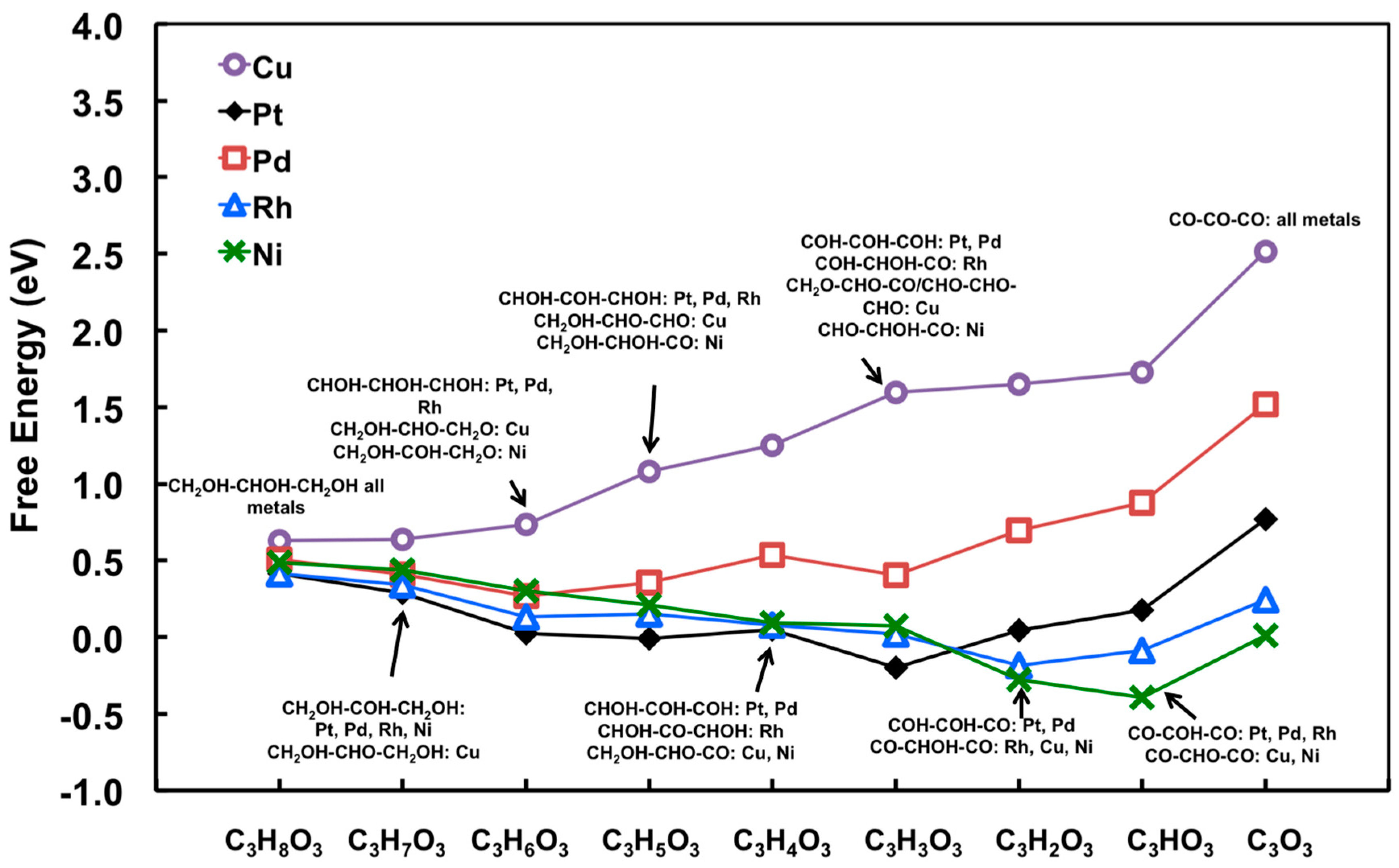
3.2. Prediction of Catalyst Activity for Glycerol Decomposition Using Scaling Relationships
4. Summary, Challenges, and Research Opportunities
4.1. Modeling Glycerol Conversion in Solutions
4.2. Modeling Synergistic Bifunctional Catalysts for Glycerol Conversion
4.3. Development Tools to Generate Reaction Pathways
Acknowledgments
Conflicts of Interest
References
- Miller, S. The Soapmaker’s Companion: A Comprehensive Guide with Recipes, Techniques & Know-How; Storey Books: North Adams, MA, USA, 1997; pp. 1–281. [Google Scholar]
- Prescott, S.C.; Dunn, C.G. Industrial Microbiology, 3rd ed.; McGraw-Hill: New York City, NY, USA, 1959; pp. 208–217. [Google Scholar]
- Wang, Z.; Zhuge, J.; Fang, H.; Prior, B.A. Glycerol production by microbial fermentation. Biotechnol. Adv. 2001, 19, 201–223. [Google Scholar] [CrossRef]
- Ciriminna, R.; Della Pina, C.; Rossi, M.; Pagliaro, M. Understanding the glycerol market, Eur. J. Lipid Sci. Technol. 2014, 116, 1432–1439. [Google Scholar] [CrossRef]
- Quispe, C.A.G.; Coronado, C.J.R.; Carvalho, J.A. Glycerol: Production, consumption, prices, characterization and new trends in combustion. Renew. Sustain. Energy Rev. 2013, 27, 475–493. [Google Scholar] [CrossRef]
- LMC International. Feedstocks for Bio-Based Chemicals; LMC International: Oxford, UK, 2013. [Google Scholar]
- Thompson, J.C.; He, B.B. Characterization of crude glycerol from biodiesel production from multiple feedstocks. Appl. Eng. Agric. 2006, 22, 261–265. [Google Scholar] [CrossRef]
- Chheda, J.N.; Huber, G.W.; Dumesic, J.A. Liquid-phase catalytic processing of biomass-derived oxygenated hydrocarbons to fuels and chemicals. Angew. Chem. Int. Ed. 2007, 46, 7164–7183. [Google Scholar] [CrossRef] [PubMed]
- Climent, M.J.; Corma, A.; De Frutos, P.; Iborra, S.; Noy, M.; Velty, A.; Concepcion, P. Chemicals from biomass: Synthesis of glycerol carbonate by transesterification and carbonylation with urea with hydrotalcite catalysts. The role of acid-base pairs. J. Catal. 2010, 269, 140–149. [Google Scholar] [CrossRef]
- Ale, C.E.; Farias, M.E.; de Saad, A.M.S.; Pasteris, S.E. Glycerol production by Oenococcus oeni during sequential and simultaneous cultures with wine yeast strains. J. Basic Microbiol. 2014, 54, S200–S209. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tao, F.; Ni, J.; Li, C.; Xu, P. Production of C3 platform chemicals from CO2 by genetically engineered cyanobacteria. Green Chem. 2015, 17, 3100–3110. [Google Scholar] [CrossRef]
- Wilson, E.K. Biodiesel revs up—Fuel made from vegetable oil leads the pack of alternatives to petroleum products. Chem. Eng. News 2002, 80, 46–49. [Google Scholar] [CrossRef]
- Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; Della Pina, C. From glycerol to value-added products. Angew. Chem. Int. Ed. 2007, 46, 4434–4440. [Google Scholar] [CrossRef] [PubMed]
- Behr, A.; Eilting, J.; Irawadi, K.; Leschinski, J.; Lindner, F. Improved utilisation of renewable resources: New important derivatives of glycerol. Green Chem. 2008, 10, 13–30. [Google Scholar] [CrossRef]
- Wen, G.D.; Xu, Y.P.; Ma, H.J.; Xu, Z.S.; Tian, Z.J. Production of hydrogen by aqueous-phase reforming of glycerol. Int. J. Hydrogen Energy 2008, 33, 6657–6666. [Google Scholar] [CrossRef]
- Zhou, C.H.C.; Beltramini, J.N.; Fan, Y.X.; Lu, G.Q.M. Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem. Soc. Rev. 2008, 37, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Brandner, A.; Lehnert, K.; Bienholz, A.; Lucas, M.; Claus, P. Production of Biomass-Derived Chemicals and Energy: Chemocatalytic Conversions of Glycerol. Top. Catal. 2009, 52, 278–287. [Google Scholar] [CrossRef]
- Vaidya, P.D.; Rodrigues, A.E. Glycerol reforming for hydrogen production: A review. Chem. Eng. Technol. 2009, 32, 1463–1469. [Google Scholar] [CrossRef]
- Rahmat, N.; Abdullah, A.Z.; Mohamed, A.R. Recent progress on innovative and potential technologies for glycerol transformation into fuel additives: A critical review. Renew. Sustain. Energy Rev. 2010, 14, 987–1000. [Google Scholar] [CrossRef]
- Tanksale, A.; Beltramini, J.N.; Lu, G.M. A review of catalytic hydrogen production processes from biomass. Renew. Sustain. Energy Rev. 2010, 14, 166–182. [Google Scholar] [CrossRef]
- Stelmachowski, M. Utilization of glycerol, a by-product of the transestrification process of vegetable oils: A review. Ecol. Chem. Eng. S 2011, 18, 9–30. [Google Scholar]
- Lin, Y.C. Catalytic valorization of glycerol to hydrogen and syngas. Int. J. Hydrogen Energy 2013, 38, 2678–2700. [Google Scholar] [CrossRef]
- Tran, N.H.; Kannangara, G.S.K. Conversion of glycerol to hydrogen rich gas. Chem. Soc. Rev. 2013, 42, 9454–9479. [Google Scholar] [CrossRef] [PubMed]
- Cortright, R.D.; Davda, R.R.; Dumesic, J.A. Hydrogen from catalytic reforming of biomass-derived hydrocarbons in liquid water. Nature 2002, 418, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.; Wietschel, M. The future of hydrogen—Opportunities and challenges. Int. J. Hydrogen Energy 2009, 34, 615–627. [Google Scholar] [CrossRef]
- Ewan, B.C.R.; Allen, R.W.K. A figure of merit assessment of the routes to hydrogen. Int. J. Hydrogen Energy 2005, 30, 809–819. [Google Scholar] [CrossRef]
- Alves, H.J.; Bley, C.; Niklevicz, R.R.; Frigo, E.P.; Frigo, M.S.; Coimbra-Araujo, C.H. Overview of hydrogen production technologies from biogas and the applications in fuel cells. Int. J. Hydrogen Energy 2013, 38, 5215–5225. [Google Scholar] [CrossRef]
- Schwengber, C.A.; Alves, H.J.; Schaffner, R.A.; da Silva, F.A.; Sequinel, R.; Bach, V.R.; Ferracin, R.J. Overview of glycerol reforming for hydrogen production. Renew. Sustain. Energy Rev. 2016, 58, 259–266. [Google Scholar] [CrossRef]
- Huber, G.W.; Shabaker, J.W.; Dumesic, J.A. Raney Ni-Sn catalyst for H2 production from biomass-derived hydrocarbons. Science 2003, 300, 2075–2077. [Google Scholar] [CrossRef] [PubMed]
- Shabaker, J.W.; Davda, R.R.; Huber, G.W.; Cortright, R.D.; Dumesic, J.A. Aqueous-phase reforming of methanol and ethylene glycol over alumina-supported platinum catalysts. J. Catal. 2003, 215, 344–352. [Google Scholar] [CrossRef]
- Shabaker, J.W.; Dumesic, J.A. Kinetics of aqueous-phase reforming of oxygenated hydrocarbons: Pt/Al2O3 and Sn-modified Ni catalysts. Ind. Eng. Chem. Res. 2004, 43, 3105–3112. [Google Scholar] [CrossRef]
- Shabaker, J.W.; Huber, G.W.; Dumesic, J.A. Aqueous-phase reforming of oxygenated hydrocarbons over Sn-modified Ni catalysts. J. Catal. 2004, 222, 180–191. [Google Scholar] [CrossRef]
- Huber, G.W.; Dumesic, J.A. An overview of aqueous-phase catalytic processes for production of hydrogen and alkanes in a biorefinery. Catal. Today 2006, 111, 119–132. [Google Scholar] [CrossRef]
- Kunkes, E.L.; Simonetti, D.A.; Dumesic, J.A.; Pyrz, W.D.; Murillo, L.E.; Chen, J.G.G.; Buttrey, D.J. The role of rhenium in the conversion of glycerol to synthesis gas over carbon supported platinum-rhenium catalysts. J. Catal. 2008, 260, 164–177. [Google Scholar] [CrossRef]
- Adhikari, S.; Fernando, S.; Haryanto, A. Production of hydrogen by steam reforming of glycerin over alumina-supported metal catalysts. Catal. Today 2007, 129, 355–364. [Google Scholar] [CrossRef]
- Adhikari, S.; Fernando, S.D.; Haryanto, A. Hydrogen production from glycerin by steam reforming over nickel catalysts. Renew. Energy 2008, 33, 1097–1100. [Google Scholar] [CrossRef]
- Adhikari, S.; Fernando, S.D.; To, S.D.F.; Bricka, R.M.; Steele, P.H.; Haryanto, A. Conversion of glycerol to hydrogen via a steam reforming process over nickel catalysts. Energy Fuels 2008, 22, 1220–1226. [Google Scholar] [CrossRef]
- Zhang, B.; Tang, X.; Li, Y.; Xu, Y.; Shen, W. Hydrogen production from steam reforming of ethanol and glycerol over ceria-supported metal catalysts. Int. J. Hydrogen Energy 2007, 32, 2367–2373. [Google Scholar] [CrossRef]
- Pompeo, F.; Santori, G.; Nichio, N.N. Hydrogen and/or syngas from steam reforming of glycerol. Study of platinum catalysts. Int. J. Hydrogen Energy 2010, 35, 8912–8920. [Google Scholar] [CrossRef]
- King, D.L.; Zhang, L.; Xia, G.; Karim, A.M.; Heldebrant, D.J.; Wang, X.; Peterson, T.; Wang, Y. Aqueous phase reforming of glycerol for hydrogen production over Pt-Re supported on carbon. Appl. Catal. B Environ. 2010, 99, 206–213. [Google Scholar] [CrossRef]
- Luo, N.J.; Fu, X.W.; Cao, F.H.; Xiao, T.C.; Edwards, P.P. Glycerol aqueous phase reforming for hydrogen generation over Pt catalyst—Effect of catalyst composition and reaction conditions. Fuel 2008, 87, 3483–3489. [Google Scholar] [CrossRef]
- Wawrzetz, A.; Peng, B.; Hrabar, A.; Jentys, A.; Lemonidou, A.A.; Lercher, J.A. Towards understanding the bifunctional hydrodeoxygenation and aqueous phase reforming of glycerol. J. Catal. 2010, 269, 411–420. [Google Scholar] [CrossRef]
- Guo, Y.; Azmat, M.U.; Liu, X.H.; Wang, Y.Q.; Lu, G.Z. Effect of support’s basic properties on hydrogen production in aqueous-phase reforming of glycerol and correlation between WGS and APR. Appl. Energy 2012, 92, 218–223. [Google Scholar] [CrossRef]
- Iriondo, A.; Barrio, V.L.; Cambra, J.F.; Arias, P.L.; Gueemez, M.B.; Navarro, R.M.; Sanchez-Sanchez, M.C.; Fierro, J.L.G. Hydrogen production from glycerol over nickel catalysts supported on Al2O3 modified by Mg, Zr, Ce or La. Top. Catal. 2008, 49, 46–58. [Google Scholar] [CrossRef]
- Iriondo, A.; Cambra, J.F.; Barrio, V.L.; Guemez, M.B.; Arias, P.L.; Sanchez-Sanchez, M.C.; Navarro, R.M.; Fierro, J.L.G. Glycerol liquid phase conversion over monometallic and bimetallic catalysts: Effect of metal, support type and reaction temperatures. Appl. Catal. B Environ. 2011, 106, 83–93. [Google Scholar] [CrossRef]
- El Doukkali, M.; Iriondo, A.; Arias, P.L.; Requies, J.; Gandarias, I.; Jalowiecki-Duhamel, L.; Dumeignil, F. A comparison of sol-gel and impregnated Pt and/or Ni based gamma-alumina catalysts for bioglycerol aqueous phase reforming. Appl. Catal. B Environ. 2012, 125, 516–529. [Google Scholar] [CrossRef]
- He, C.; Zheng, J.; Wang, K.; Lin, H.; Wang, J.-Y.; Yang, Y. Sorption enhanced aqueous phase reforming of glycerol for hydrogen production over Pt-Ni supported on multi-walled carbon nanotubes. Appl. Catal. B Environ. 2015, 162, 401–411. [Google Scholar] [CrossRef]
- Zhang, L.; Karim, A.M.; Engelhard, M.H.; Wei, Z.; King, D.L.; Wang, Y. Correlation of Pt-Re surface properties with reaction pathways for the aqueous-phase reforming of glycerol. J. Catal. 2012, 287, 37–43. [Google Scholar] [CrossRef]
- Ciftci, A.; Ligthart, D.; Sen, A.O.; van Hoof, A.J.F.; Friedrich, H.; Hensen, E.J.M. Pt-Re synergy in aqueous-phase reforming of glycerol and the water-gas shift reaction. J. Catal. 2014, 311, 88–101. [Google Scholar] [CrossRef]
- Ciftci, A.; Eren, S.; Ligthart, D.; Hensen, E.J.M. Platinum-Rhenium Synergy on Reducible Oxide Supports in Aqueous-Phase Glycerol Reforming. ChemCatChem 2014, 6, 1260–1269. [Google Scholar] [CrossRef]
- Dietrich, P.J.; Lobo-Lapidus, R.J.; Wu, T.; Sumer, A.; Akatay, M.C.; Fingland, B.R.; Guo, N.; Dumesic, J.A.; Marshall, C.L.; Stach, E.; et al. Aqueous Phase Glycerol Reforming by PtMo Bimetallic Nano-Particle Catalyst: Product Selectivity and Structural Characterization. Top. Catal. 2012, 55, 53–69. [Google Scholar] [CrossRef]
- Dietrich, P.J.; Wu, T.; Sumer, A.; Dumesic, J.A.; Jellinek, J.; Delgass, W.N.; Ribeiro, F.H.; Miller, J.T. Aqueous Phase Glycerol Reforming with Pt and PtMo Bimetallic Nanoparticle Catalysts: The Role of the Mo Promoter. Top. Catal. 2013, 56, 1814–1828. [Google Scholar] [CrossRef]
- Tuza, P.V.; Manfro, R.L.; Ribeiro, N.F.P.; Souza, M. Production of renewable hydrogen by aqueous-phase reforming of glycerol over Ni-Cu catalysts derived from hydrotalcite precursors. Renew. Energy 2013, 50, 408–414. [Google Scholar] [CrossRef]
- Sanchez, E.A.; D’Angelo, M.A.; Comelli, R.A. Hydrogen production from glycerol on Ni/Al2O3 catalyst. Int. J. Hydrogen Energy 2010, 35, 5902–5907. [Google Scholar] [CrossRef]
- Iriondo, A.; Barrio, V.L.; Cambra, J.F.; Arias, P.L.; Guemez, M.B.; Navarro, R.M.; Sanchez-Sanchez, M.C.; Fierro, J.L.G. Influence of La2O3 modified support and Ni and Pt active phases on glycerol steam reforming to produce hydrogen. Catal. Commun. 2009, 10, 1275–1278. [Google Scholar] [CrossRef]
- Hirai, T.; Ikenaga, N.; Miyake, T.; Suzuki, T. Production of hydrogen by steam reforming of glycerin on ruthenium catalyst. Energy Fuels 2005, 19, 1761–1762. [Google Scholar] [CrossRef]
- Dou, B.L.; Wang, C.; Song, Y.C.; Chen, H.S.; Xu, Y.J. Activity of Ni-Cu-Al based catalyst for renewable hydrogen production from steam reforming of glycerol. Energy Convers. Manag. 2014, 78, 253–259. [Google Scholar] [CrossRef]
- Cheng, C.K.; Foo, S.Y.; Adesina, A.A. Glycerol Steam Reforming over Bimetallic Co-Ni/Al2O3. Ind. Eng. Chem. Res. 2010, 49, 10804–10817. [Google Scholar] [CrossRef]
- Chen, Y.; Salciccioli, M.; Vlachos, D.G. An efficient reaction pathway search method applied to the decomposition of glycerol on platinum. J. Phys. Chem. C 2011, 115, 18707–18720. [Google Scholar] [CrossRef]
- Liu, B.; Greeley, J. Density Functional Theory Study of Selectivity Considerations for C–C versus C–O Bond Scission in Glycerol Decomposition on Pt(111). Top. Catal. 2012, 55, 280–289. [Google Scholar] [CrossRef]
- Huber, G.W.; Shabaker, J.W.; Evans, S.T.; Dumesic, J.A. Aqueous-phase reforming of ethylene glycol over supported Pt and Pd bimetallic catalysts. Appl. Catal. B Environ. 2006, 62, 226–235. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, M.; Chan, M.K.Y.; Greeley, J.P. Understanding polyol decomposition on bimetallic Pt-Mo catalysts-a DFT study of glycerol. ACS Catal. 2015, 5, 4942–4950. [Google Scholar] [CrossRef]
- Adhikari, S.; Fernando, S.; Haryanto, A. A comparative thermodynamic and experimental analysis on hydrogen production by steam reforming of glycerin. Energy Fuels 2007, 21, 2306–2310. [Google Scholar] [CrossRef]
- Davda, R.R.; Shabaker, J.W.; Huber, G.W.; Cortright, R.D.; Dumesic, J.A. Aqueous-phase reforming of ethylene glycol on silica-supported metal catalysts. Appl. Catal. B Environ. 2003, 43, 13–26. [Google Scholar] [CrossRef]
- Davda, R.R.; Alcala, R.; Shabaker, J.; Huber, G.; Cortright, R.D.; Mavrikakis, M.; Dumesic, J.A. DFT and experimental studies of C–C and C–O bond cleavage in ethanol and ethylene glycol on Pt catalysis. Stud. Surf. Sci. Catal. 2003, 145, 79–84. [Google Scholar]
- Shabaker, J.W.; Huber, G.W.; Davda, R.R.; Cortright, R.D.; Dumesic, J.A. Aqueous-phase reforming of ethylene glycol over supported platinum catalysts. Catal. Lett. 2003, 88, 1–8. [Google Scholar] [CrossRef]
- Liu, B.; Greeley, J. Decomposition pathways of glycerol via C–H, O–H and C–C bond scission on Pt(111), a density functional theory study. J. Phys. Chem. C 2011, 115, 19702–19709. [Google Scholar] [CrossRef]
- Liu, B.; Greeley, J. A Density Functional Theory Analysis of Trends in Glycerol Decomposition on Close-Packed Transition Metal Surfaces. Phys. Chem. Chem. Phys. 2013, 15, 6475–6485. [Google Scholar] [CrossRef] [PubMed]
- Sohounloue, D.K.; Montassier, C.; Barbier, J. Catalytic hydrogenolysis of sorbitol. React. Kinet. Catal. Lett. 1983, 22, 391–397. [Google Scholar] [CrossRef]
- Montassier, C.; Giraud, D.; Barbier, J. Polyol Conversion by Liquid Phase Heterogeneous Catalysis over Metals; Elsevier Science Publishers: Amsterdam, The Netherlands, 1988; p. 165. [Google Scholar]
- Montassier, C.; Menezo, J.C.; Hoang, L.C.; Renaud, C.; Barbier, J. Aqueous polyol conversions on ruthenium and on sulfur-modified ruthenium. J. Mol. Catal. 1991, 70, 99–110. [Google Scholar] [CrossRef]
- Maris, E.P.; Davis, R.J. Hydrogenolysis of glycerol over carbon-supported Ru and Pt catalysts. J. Catal. 2007, 249, 328–337. [Google Scholar] [CrossRef]
- Coll, D.; Delbecq, F.; Aray, Y.; Sautet, P. Stability of intermediates in the glycerol hydrogenolysis on transition metal catalysts from first principles. Phys. Chem. Chem. Phys. 2011, 13, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Auneau, F.; Michel, C.; Delbecq, F.; Pinel, C.; Sautet, P. Unravelling the Mechanism of Glycerol Hydrogenolysis over Rhodium Catalyst through Combined Experimental-Theoretical Investigations. Chem. A Eur. J. 2011, 17, 14288–14299. [Google Scholar] [CrossRef] [PubMed]
- Kusunoki, Y.; Miyazawa, T.; Kunimori, K.; Tomishige, K. Highly active metal-acid bifunctional catalyst system for hydrogenolysis of glycerol under mild reaction conditions. Catal. Commun. 2005, 6, 645–649. [Google Scholar] [CrossRef]
- Chaminand, J.; Djakovitch, L.; Gallezot, P.; Marion, P.; Pinel, C.; Rosier, C. Glycerol hydrogenolysis on heterogeneous catalysts. Green Chem. 2004, 6, 359–361. [Google Scholar] [CrossRef]
- Huang, L.; Zhu, Y.L.; Zheng, H.Y.; Li, Y.W.; Zeng, Z.Y. Continuous production of 1,2-propanediol by the selective hydrogenolysis of solvent-free glycerol under mild conditions. J. Chem. Technol. Biotechnol. 2008, 83, 1670–1675. [Google Scholar] [CrossRef]
- Jimenez-Morales, I.; Vila, F.; Mariscal, R.; Jimenez-Lopez, A. Hydrogenolysis of glycerol to obtain 1,2-propanediol on Ce-promoted Ni/SBA-15 catalysts. Appl. Catal. B Environ. 2012, 117, 253–259. [Google Scholar] [CrossRef]
- Balaraju, M.; Rekha, V.; Prasad, P.S.S.; Prasad, R.B.N.; Lingaiah, N. Selective Hydrogenolysis of Glycerol to 1,2-Propanediol Over Cu-ZnO Catalysts. Catal. Lett. 2008, 126, 119–124. [Google Scholar] [CrossRef]
- Gandarias, I.; Arias, P.L.; Requies, J.; Guemez, M.B.; Fierro, J.L.G. Hydrogenolysis of glycerol to propanediols over a Pt/ASA catalyst: The role of acid and metal sites on product selectivity and the reaction mechanism. Appl. Catal. B Environ. 2010, 97, 248–256. [Google Scholar] [CrossRef]
- Daniel, O.M.; DeLaRiva, A.; Kunkes, E.L.; Datye, A.K.; Dumesic, J.A.; Davis, R.J. X-ray Absorption Spectroscopy of Bimetallic Pt-Re Catalysts for Hydrogenolysis of Glycerol to Propanediols. ChemCatChem 2010, 2, 1107–1114. [Google Scholar] [CrossRef]
- Ma, L.; He, D.H.; Li, Z.P. Promoting effect of rhenium on catalytic performance of Ru catalysts in hydrogenolysis of glycerol to propanediol. Catal. Commun. 2008, 9, 2489–2495. [Google Scholar] [CrossRef]
- Maris, E.P.; Ketchie, W.C.; Murayama, M.; Davis, R.J. Glycerol hydrogenolysis on carbon-supported PtRu and AuRu bimetallic catalysts. J. Catal. 2007, 251, 281–294. [Google Scholar] [CrossRef]
- Liu, H.Z.; Liang, S.G.; Jiang, T.; Han, B.X.; Zhou, Y.X. Hydrogenolysis of glycerol to 1,2-propanediol over Ru-Cu bimetals supported on different supports. CLEAN-Soil Air Water 2012, 40, 318–324. [Google Scholar] [CrossRef]
- Ma, L.; He, D.H. Hydrogenolysis of Glycerol to Propanediols over Highly Active Ru-Re Bimetallic Catalysts. Top. Catal. 2009, 52, 834–844. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Shinmi, Y.; Koso, S.; Tomishige, K. Direct hydrogenolysis of glycerol into 1,3-propanediol over rhenium-modified iridium catalyst. J. Catal. 2010, 272, 191–194. [Google Scholar] [CrossRef]
- Miyazawa, T.; Kusunoki, Y.; Kunimori, K.; Tomishige, K. Glycerol conversion in the aqueous solution under hydrogen over Ru/C plus an ion-exchange resin and its reaction mechanism. J. Catal. 2006, 240, 213–221. [Google Scholar] [CrossRef]
- Miyazawa, T.; Koso, S.; Kunimori, K.; Tomishige, K. Development of a Ru/C catalyst for glycerol hydrogenolysis in combination with an ion-exchange resin. Appl. Catal. Gen. 2007, 318, 244–251. [Google Scholar] [CrossRef]
- Miyazawa, T.; Koso, S.; Kunimori, K.; Tomishige, K. Glycerol hydrogenolysis to 1,2-propanediol catalyzed by a heat-resistant ion-exchange resin combined with Ru/C. Appl. Catal. Gen. 2007, 329, 30–35. [Google Scholar] [CrossRef]
- Zhu, S.H.; Qiu, Y.N.; Zhu, Y.L.; Hao, S.L.; Zheng, H.Y.; Li, Y.W. Hydrogenolysis of glycerol to 1,3-propanediol over bifunctional catalysts containing Pt and heteropolyacids. Catal. Today 2013, 212, 120–126. [Google Scholar] [CrossRef]
- Tyrlik, S.K.; Szerszen, D.; Olejnik, M.; Danikiewicz, W. Concentrated water solutions of salts as solvents for reaction of carbohydrates. Part 2. Influence of some magnesium salts and some ruthenium species on catalysis of dehydration of glucose. J. Mol. Catal. Chem. 1996, 106, 223–233. [Google Scholar] [CrossRef]
- Demirel-Gulen, S.; Lucas, M.; Claus, P. Liquid phase oxidation of glycerol over carbon supported gold catalysts. Catal. Today 2005, 102, 166–172. [Google Scholar] [CrossRef]
- Kimura, H.; Tsuto, K.; Wakisaka, T.; Kazumi, Y.; Inaya, Y. Selective oxidation of glycerol on a platinum bismuth catalyst. Appl. Catal. Gen. 1993, 96, 217–228. [Google Scholar] [CrossRef]
- Garcia, R.; Besson, M.; Gallezot, P. Chemoselective catalytic-oxidation of glycerol with air on platinum metals. Appl. Catal. Gen. 1995, 127, 165–176. [Google Scholar] [CrossRef]
- Garcia, A.C.; Birdja, Y.Y.; Tremiliosi, G.; Koper, M.T.M. Glycerol electro-oxidation on bismuth-modified platinum single crystals. J. Catal. 2017, 346, 117–124. [Google Scholar] [CrossRef]
- Xiao, Y.; Greeley, J.; Varma, A.; Zhao, Z.J.; Xiao, G.M. An experimental and theoretical study of glycerol oxidation to 1,3-dihydroxyacetone over bimetallic Pt-Bi catalysts. AIChE J. 2017, 63, 705–715. [Google Scholar] [CrossRef]
- Kwon, Y.; Koper, M.T.M. Combining voltammetry with HPLC: Application to electro-oxidation of glycerol. Anal. Chem. 2010, 82, 5420–5424. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Schouten, K.J.P.; Koper, M.T.M. Mechanism of the catalytic oxidation of glycerol on polycrystalline gold and platinum electrodes. ChemCatChem 2011, 3, 1176–1185. [Google Scholar] [CrossRef]
- Garcia, A.C.; Kolb, M.J.; Sanchez, C.V.Y.; Vos, J.; Birdja, Y.Y.; Kwon, Y.; Tremiliosi-Filho, G.; Koper, M.T.M. Strong impact of platinum surface structure on primary and secondary alcohol oxidation during electro-oxidation of glycerol. ACS Catal. 2016, 6, 4491–4500. [Google Scholar] [CrossRef]
- Kwon, Y.; Birdja, Y.; Spanos, I.; Rodriguez, P.; Koper, M.T.M. Highly selective electro-oxidation of glycerol to dihydroxyacetone on platinum in the presence of bismuth. ACS Catal. 2012, 2, 759–764. [Google Scholar] [CrossRef]
- Kwon, Y.; Hersbach, T.J.P.; Koper, M.T.M. Electro-oxidation of glycerol on platinum modified by adatoms: Activity and selectivity effects. Top. Catal. 2014, 57, 1272–1276. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Xin, L.; Li, W.Z. Electrocatalytic oxidation of glycerol on Pt/C in anion-exchange membrane fuel cell: Cogeneration of electricity and valuable chemicals. Appl. Catal. B Environ. 2012, 119, 40–48. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Xin, L.; Qi, J.; Chadderdon, D.J.; Li, W.Z. Supported Pt, Pd and Au nanoparticle anode catalysts for anion-exchange membrane fuel cells with glycerol and crude glycerol fuels. Appl. Catal. B Environ. 2013, 136, 29–39. [Google Scholar] [CrossRef]
- Shustorovich, E. Activation Barrier for Adsorbate Surface Diffusion, Heat of Chemisorption, and Adsorbate Registry: Theoretical Interrelations. J. Am. Chem. Soc. 1984, 106, 6479–6481. [Google Scholar] [CrossRef]
- Van Santen, R.A. On shustorovich bond-order conservation method as applied to chemisorption. Recl. Trav. Chim. Pays-Bas 1990, 109, 59–63. [Google Scholar] [CrossRef]
- Shustorovich, E.; Sellers, H. The UBI-QEP method: A practical theoretical approach to understanding chemistry on transition metal surfaces. Surf. Sci. Rep. 1998, 31, 1–119. [Google Scholar] [CrossRef]
- Kua, J.; Faglioni, F.; Goddard, W.A. Thermochemistry for hydrocarbon intermediates chemisorbed on metal surfaces: CHn-m(CH3)m with n = 1, 2, 3 and m ≤ n on Pt, Ir, Os, Pd, Ph, and Ru. J. Am. Chem. Soc. 2000, 122, 2309–2321. [Google Scholar] [CrossRef]
- Greeley, J. Theoretical Heterogeneous Catalysis: Scaling Relationships and Computational Catalyst Design. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 605–635. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Muelas, R.; Lopez, N. Collective descriptors for the adsorption of sugar alcohols on Pt and Pd(111). J. Phys. Chem. C 2014, 118, 17531–17537. [Google Scholar] [CrossRef]
- Salciccioli, M.; Chen, Y.; Vlachos, D.G. Density Functional Theory-Derived Group Additivity and Linear Scaling Methods for Prediction of Oxygenate Stability on Metal Catalysts: Adsorption of Open-Ring Alcohol and Polyol Dehydrogenation Intermediates on Pt-Based Metals. J. Phys. Chem. C 2010, 114, 20155–20166. [Google Scholar] [CrossRef]
- Salciccioli, M.; Edie, S.M.; Vlachos, D.G. Adsorption of Acid, Ester, and Ether Functional Groups on Pt: Fast Prediction of Thermochemical Properties of Adsorbed Oxygenates via DFT-Based Group Additivity Methods. J. Phys. Chem. C 2012, 116, 1873–1886. [Google Scholar] [CrossRef]
- Benson, S.W. Thermochemical Kinetics. In Methods for the Estimation of Thermochemical Data and Rate Parameters, 2nd ed.; John Wiley and Sons, Inc.: New York, NY, USA, 1976; pp. 1–320. [Google Scholar]
- Ferrin, P.; Simonetti, D.; Kandoi, S.; Kunkes, E.; Dumesic, J.A.; Nørskov, J.K.; Mavrikakis, M. Modeling ethanol decomposition on transition metals: A combined application of scaling and Brønsted-Evans-Polanyi relations. J. Am. Chem. Soc. 2009, 131, 5809–5815. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Johannesson, G.; Jonsson, H. Methods for Finding Saddle Points and Minimum Energy Paths. In Theoretical Methods in Condensed Phase Chemistry; Springer: Dordrecht, The Netherlands, 2000; Volume 5, pp. 269–302. [Google Scholar]
- Henkelman, G.; Uberuaga, B.P.; Jonsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Henkelman, G.; Jonsson, H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
- Jacob, T.; Goddard, W.A. Water formation on Pt and Pt-based alloys: A theoretical description of a catalytic reaction. ChemPhysChem 2006, 7, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Faheem, M.; Suthirakun, S.; Heyden, A. New implicit solvation scheme for solid surfaces. J. Phys. Chem. C 2012, 116, 22458–22462. [Google Scholar] [CrossRef]
- Callam, C.S.; Singer, S.J.; Lowary, T.L.; Hadad, C.M. Computational analysis of the potential energy surfaces of glycerol in the gas and aqueous phases: Effects of level of theory, basis set, and solvation on strongly intramolecularly hydrogen-bonded systems. J. Am. Chem. Soc. 2001, 123, 11743–11754. [Google Scholar] [CrossRef] [PubMed]
- Bodenschatz, C.J.; Sarupria, S.; Getman, R.B. Molecular-level details about liquid H2O interactions with CO and sugar alcohol adsorbates on Pt(111) calculated using density functional theory and molecular dynamics. J. Phys. Chem. C 2015, 119, 13642–13651. [Google Scholar] [CrossRef]
- Mathew, K.; Sundararaman, R.; Letchworth-Weaver, K.; Arias, T.A.; Hennig, R.G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, S.N.; Sautet, P.; Michel, C. Solvation free energies for periodic surfaces: Comparison of implicit and explicit solvation models. Phys. Chem. Chem. Phys. 2016, 18, 31850–31861. [Google Scholar] [CrossRef] [PubMed]
- Plimpton, S. Fast parallel algorithms for short-range molecular-dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Yun, Y.S.; Lee, K.R.; Park, H.; Kim, T.Y.; Yun, D.; Han, J.W.; Yi, J. Rational Design of a Bifunctional Catalyst for the Oxydehydration of Glycerol: A Combined Theoretical and Experimental Study. ACS Catal. 2015, 5, 82–94. [Google Scholar] [CrossRef]
- Suleimanov, Y.V.; Green, W.H. Automated Discovery of Elementary Chemical Reaction Steps Using Freezing String and Berny Optimization Methods. J. Chem. Theory Comput. 2015, 11, 4248–4259. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Zimmerman, P.M. Reliable and Efficient Reaction Path and Transition State Finding for Surface Reactions with the Growing String Method. J. Comput. Chem. 2017, 38, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Vinu, R.; Broadbelt, L.J. Unraveling Reaction Pathways and Specifying Reaction Kinetics for Complex Systems. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 29–54. [Google Scholar] [CrossRef] [PubMed]
- Friedler, F.; Tarjan, K.; Huang, Y.W.; Fan, L.T. Graph-theoretic approach to process synthesis—Axioms and theorems. Chem. Eng. Sci. 1992, 47, 1973–1988. [Google Scholar] [CrossRef]
- Fan, L.T.; Lin, Y.C.; Shafie, S.; Hohn, K.L.; Bertok, B.; Friedler, F. Graph-theoretic and energetic exploration of catalytic pathways of the water-gas shift reaction. J. Chin. Inst. Chem. Eng. 2008, 39, 467–473. [Google Scholar] [CrossRef]
- Fan, L.T.; Lin, Y.C.; Shafie, S.; Bertok, B.; Friedler, F. Exhaustive identification of feasible pathways of the reaction catalyzed by a catalyst with multiactive sites via a highly effective graph-theoretic algorithm: Application to ethylene hydrogenation. Ind. Eng. Chem. Res. 2012, 51, 2548–2552. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
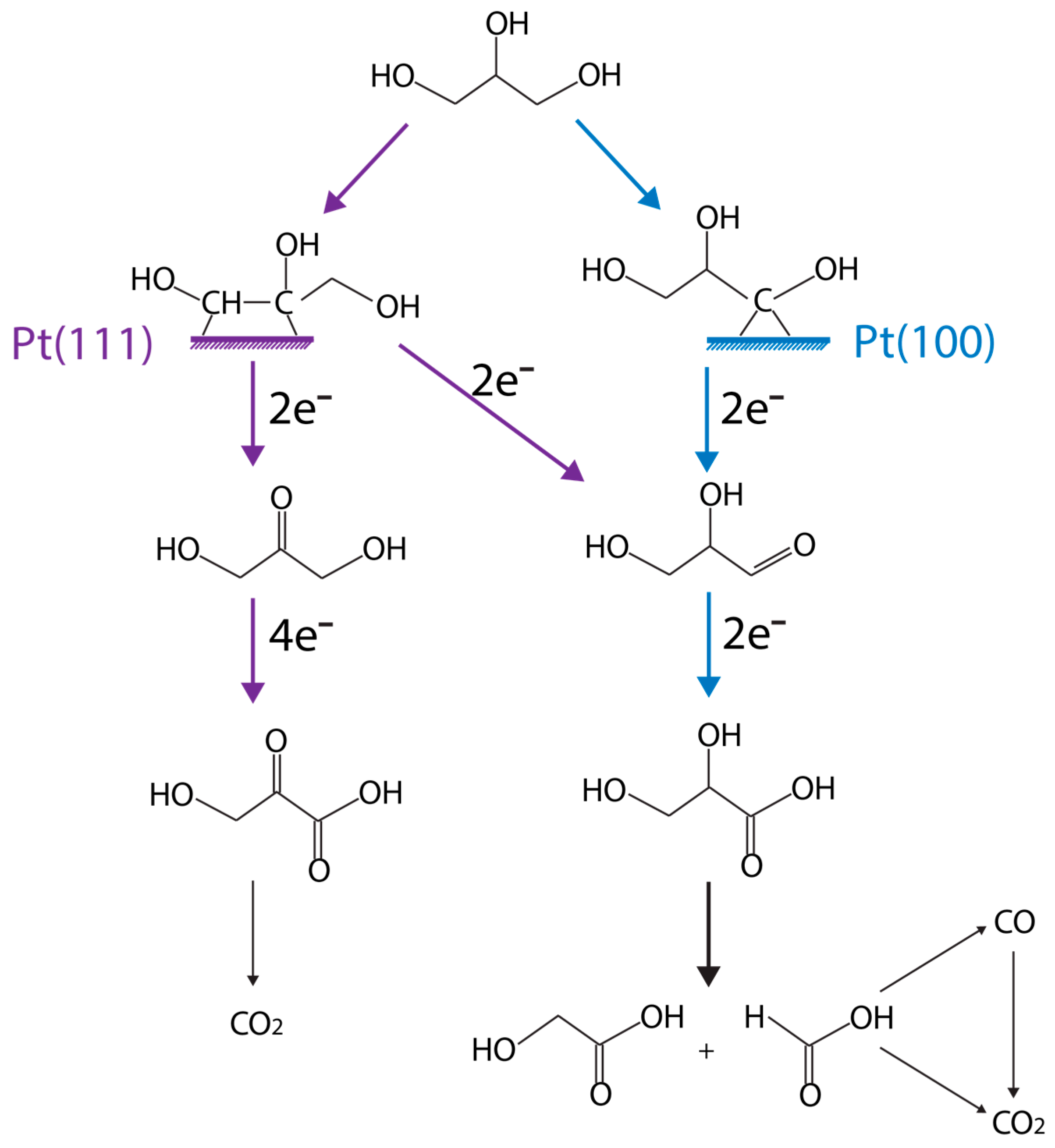{kind=link}
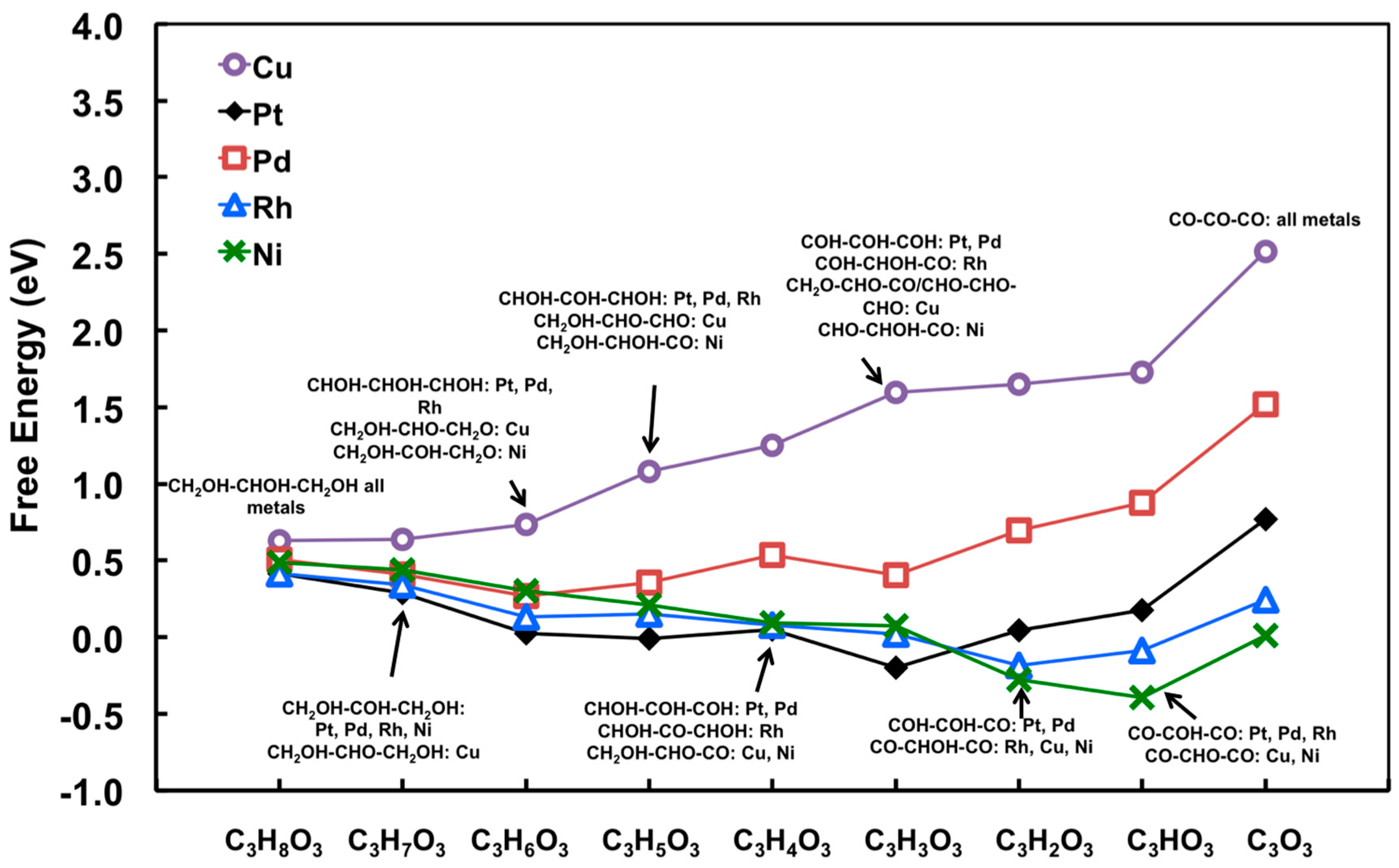{kind=link}
| Catalyst | Pressure, Temp. | Process | Gly. Conversion, % | Yield (H2) | Selectivity (H2), % | Reference |
|---|---|---|---|---|---|---|
| 3 wt.% Pt/C | 29 bar, 225 °C | APR | 5 | 68 | 57 | [40] |
| 3 wt.% Pt/γ-Al2O3 | 29–56 bar, 225–265 °C | APR | 65, 57 | 75, 51 | [24] | |
| 0.9 wt.% Pt/γ-Al2O3 | 25 bar, 220 °C | APR | 65 1 | 45 | 70 | [41] |
| 3 wt.% Pt/γ-Al2O3 | 29 bar, 225 °C | APR | ~50 | 20 2 | 20 3 | [42] |
| Pt/MgO, Al2O3, CeO2, TiO2, SiO2 | 27.6 bar, 225 °C | APR | 20–48 | 56–70 | 14–28 | [43] |
| Ni/MxOy-Al2O3 (M = Zr, Ce, La, Mg) | 30 bar, 225 °C | APR | 15–37 | 32–48 | [44] | |
| 3 wt.% Pt-10 wt.% Ni/γ-Al2O3 | 30 bar, 230 °C | APR | 75 | 54 | [45] | |
| 3.2 wt.% Pt-8 wt.% Ni/γ-Al2O3 | 50 bar, 250 °C | APR | 74 | 51 | [46] | |
| Pt-Ni/CNT | 30 bar, 230 °C | APR | 70–90 | 18–62 | [47] | |
| 3 wt.% Pt-3 wt.% Re/C | 29 bar, 225 °C, in KOH | APR | 89 | 67 | 26 | [40] |
| 3%Pt-(1–4.5%)Re/C | 29 bar, 225 °C | APR | 100 | ~70 | [48] | |
| (2.5–4 wt.%)Pt-Re (1:5 ratio)/C | 25 bar, 225 °C | APR | <45% | [49] | ||
| (2.5–3 wt.%)Pt-Re (1:2 ratio)/ZrO2, CeO2, TiO2 | 25 bar, 225 °C | APR | <35% | [50] | ||
| Pt-Mo (Pt:Mo ratio = 1:1) | 31 bar, 210–240 °C | APR | 0.5–26 | ~50 | [51,52] | |
| Ni-Sn | 25.8–51.4 bar, 225–265 °C | APR | >90 | 68–78 | [29,32] | |
| Ni-Cu | 38–52 atm, 250–270 °C | APR | 60 | >80 | [53] | |
| Pt/γ-Al2O3 | 1 bar, 350 °C | Steam | 8 | 61 | [39] | |
| Pt/SiO2, ZrO2, Ce4Zr1α | 1 bar, 350 °C | Steam | 100, 16, 78 | 69, 62, 73 | [39] | |
| 5.8 wt.% Ni/Al2O3 | 1 bar, 600–700 °C | Steam | >99 | ~100 | [54] | |
| Ni, Ir, Co./CeO2 | 1 bar, 550 °C 4 | Steam | 100 | 91, 93, 94 | [38] | |
| Ni/MxOy-Al2O3 (M = Zr, Ce, La, Mg) | 1 bar, 600 °C | Steam | 100 | 60–70 | [44] | |
| Ni/MgO | 1 bar, 550–650 °C | Steam | 100 | 43–57 | 61–66 | [36,37] |
| Ni/TiO2 | 1 bar, 550–650 °C | Steam | 96–98 | 31–47 | 44–62 | [36,37] |
| Ni/CeO2 | 1 bar, 550–650 °C | Steam | 72–98 | 31–44 | 54–67 | [36,37] |
| Pt-Ni/La2O3 | 1 bar, 600 °C | Steam | 100 | 53 | [55] | |
| 3 wt.% Ru/Y2O3 | 1 bar, 550–650 °C | Steam | 100 | ~90 | [56] | |
| Ru/Al2O3 | 900 °C | Steam | 58 | 42 | [35] | |
| Ni-Cu-Al | 1 bar, 500–600 °C | Steam | 70 | ~75 | [57] | |
| Ni-Co./Al2O3 | 1 bar, 500–550 °C | Steam | 5–6 | 65 | [58] |
| Catalyst | H2 Pressure, Temp., and Solution | Glycerol Conversion, % | Yield, % | Main Products and Selectivity, % | Reference |
|---|---|---|---|---|---|
| Ru/C | 40 bar, 200 °C, neutral | 20 | 32 (PDO) 68 (EG) | [72] | |
| Rh/C | 30 bar, 180 °C + NaOH (1 M) | 22 | 9 (12-PDO) 5 (LA) | [74] | |
| 5 wt.% Rh/C | 80 bar, 180 °C | 0.3 | 58.6 (12-PDO) 3.4 (13-PDO) | [75] | |
| Rh/C, with H2WO4 | 80 bar, 180 °C | 10 | 5.2 (12-PDO) 2.6 (13-PDO) | 52 (12-PDO) 26 (13-PDO) | [76] |
| Rh/Al2O3 | 80 bar, 180 °C | 21 | 12.4 (12-PDO) 3.1 (13-PDO) | 45 (12-PDO) 12 (13-PDO) | [76] |
| Rh/Nafion | 80 bar, 180 °C | 8 | 4.3 (12-PDO) 1.5 (13-PDO) | 54 (12-PDO) 19 (13-PDO) | [76] |
| Ni/Al2O3 | 1 bar, 190 °C | 92 | 43.6 (12-PDO) | [77] | |
| Ni/SBA-15 | 30 bar, 450 °C | 50 | 24 (12-PDO) | 30 (12-PDO) | [78] |
| Pd/C, with H2WO4 | 80 bar, 180 °C | 3 | 2 | 100 (12-PDO) | [76] |
| Pd/C | 80 bar, 180 °C | 0.7 | 93.1 (12-PDO) 1.4 (13-PDO) | [75] | |
| Cu-ZnO (Cu:Zn ratio 50:50) | 20 bar, 120–220 °C | 37 | >93 (12-PDO), 3.4 (Acetol), 3 (EG) | [79] | |
| Cu/ZnO/Al2O3 | 6.4 bar, 190 °C | 96 | 92.2 (12-PDO) | [77] | |
| CuO/ZnO, with H2WO4 | 80 bar, 180 °C | 21 | 17 | 100 (12-PDO) | [76] |
| Pt/amorphous silico alumina | 45 bar, 220 °C | 20 | 6.3 (12-PDO) | 35.3 (12-PDO), 1.2 (13-PDO) | [80] |
| Pt/C | 40 bar, 200 °C, neutral | 13 | 79 (PDO) 17 (EG) | [72] | |
| Pt/C | 40 bar, 200 °C, with 0.8 M NaOH | 20 | 30 (PDO) 62 (LA) 2 (EG) | [72] | |
| Pt/C | 80–90 bar, 160 °C | 20 | 43 (12-PDO) | [81] | |
| Pt/C | 80 bar, 180 °C | 1.1 | 87.6 (12-PDO) 1.9 (13-PDO) | [75] | |
| Ru/C | 40 bar, 200 °C, with 0.8 M NaOH | 20 | 37 (PDO) 47 (LA) 12 (EG) | [72] | |
| Ru/C | 80 bar, 160 °C | 29.7 1 | 50.9 (12-PDO) 0.8 (13-PDO) 22.9 (1-PO) 3.2 (2-PO) | [82] | |
| Ru/C | 80 bar, 180°C | 6.3 | 17.9 (12-PDO) 0.5 (13-PDO) | [75] | |
| Pt-Ru/C | 40 bar, 200 °C, with 0.8 M NaOH | 22 | 37 (PDO) 41 (LA) 15 (EG) | [83] | |
| Au-Ru/C | 40 bar, 200 °C, with 0.8 M NaOH | 21 | 25 (PDO) 60 (LA) 10 (EG) | [83] | |
| Pt-Re/C sintered | 80–90 bar, 200 °C | 20 | 33 (12-PDO) 34 (13-PDO) | [81] | |
| Cu-Ru/ZrO2 (Cu:Ru ratio = 1:10) | 80 bar, 180 °C | 100 | 78.5 (12-PDO) | 84 (12-PDO) 6.4 (1-PO) 9.3 (EG) | [84] |
| Ru-Re/ZrO2 | 80 bar, 160 °C | 57 | 47.2 (12-PDO) 5.5 (13-PDO) 27.2 (1-PO) 8.1 (2-PO) | [85] | |
| Ir-ReOx/SiO2 | 80 bar, 160 °C | 60 2 | 5 (12-PDO) 54 (13-PDO) 31 (1-PO) 4 (2-PO) | [86] |
| H2 Pressure, and Temp. | Glycerol Conversion, % | Main Product Selectivity, % | Reference | |
|---|---|---|---|---|
| Ru/C + Amberlyst | 80 bar, 140 °C | 41 | 43.1 (12-PDO) 1.0 (13-PDO) 18.2 (1-PO) 2.9 (2-PO) | [75] |
| Ru/C + H2SO4 | 80 bar, 140 °C | 3.2 | 47.4 (12-PDO) 5.4 (13-PDO) 19.6 (1-PO) 1.6 (2-PO) | [75] |
| Pt/amorphous silico alumina | 45 bar, 220 °C | 20 | 35.3 (12-PDO) 1.2 (13-PDO) | [80] |
| Pt/C + Amberlyst | 80 bar, 140 °C | 0.5 | 4.4 (12-PDO) 4.4 (13-PDO) 52.7 (1-PO) 14.7 (2-PO) | [75] |
| Pd/C + Amberlyst | 80 bar, 140 °C | 0.3 | 6.1 (12-PDO) 51.5 (1-PO) 15.2 (2-PO) | [75] |
| Rh/C + Amberlyst | 80 bar, 140 °C | 6.4 | 19.5 (12-PDO) 7.2 (13-PDO) 53.2 (1-PO) 14.7 (2-PO) | [75] |
| Ru/C +Amberlyst | 80 bar, 120 °C | 13 | 55.4 (12-PDO) 4.9 (13-PDO) 14.1 (1-PO) 0.9 (2-PO) | [87] |
| Ru5/C(I) | 80 bar, 120 °C | 21 | 76.7 (12-PDO) 1.5 (13-PDO) 2.5 (1-PO) 0.5 (2-PO) | [88] |
| h-Ru/C + A70 | 80 bar, 180 °C | 49 | 70.2 (12-PDO) 1.3 (13-PDO) 7.1 (1-PO) 1.0 (2-PO) | [89] |
| Pt-HSiW/ZrO2 | 50 bar, 180 °C | 24 | 16.5 (12-PDO) 48.1 (13-PDO) 21.8 (1-PO) 4.5 (2-PO) | [90] |
| Pt-HPW/ZrO2 | 50 bar, 180 °C | 25.5 | 10.9 (12-PDO) 32.9 (13-PDO) 37.9 (1-PO) 5.2 (2-PO) | [90] |
| Pt-HPMo/ZrO2 | 50 bar, 180 °C | 27 | 39.2 (12-PDO) 7.8 (13-PDO) 30.4 (1-PO) 3.2 (2-PO) | [90] |
| Catalyst | Experimental Conditions | Selectivity, % | Reference |
|---|---|---|---|
| 5 wt.% Pt-1 wt.% Bi/C | With air in acidic media at 323 K and normal pressure | 20 (DHA) | [93] |
| Pd/C | pH = 11 in air | 70 (Glyceric acid) 1 8 (DHA)) | [94] |
| Pt/C | pH = 7 in air | 55 (Glyceric acid) 1 12 (DHA) | [94] |
| Pt-Bi/C | pH = 11 in air | 50 (DHA) 1 | [94] |
| Au/AC | (10 bar), 60 °C in NaOH solution | 75 (Glyceric acid) 2 15 (Glycolic acid) 2 | [92] |
| Pt(111), Pt(100) | 0.5 M HClO4, CV performed at 293 K in the range of 0.0–1.0 V | 80 3 (Glyceraldehyde) ~20 3 (DHA) | [95] |
| Pt(111)/Bi, Pt(100)/Bi | 0.5 M HClO4, CV performed at 293 K in the range of 0.0–1.0 V | 80 4 (Glyceraldehyde) ~20 4 (DHA) 90 4 (Glyceraldehyde) ~10 4 (DHA) | [95] |
| Pt/MCM-41 | (30 psi), 75 °C | 15.25 (DHA) 5 | [96] |
| Pt-Bi/AC | (30 psi), 75 °C | 77.12 (DHA) 5 | [96] |
| Pt-Bi/ZSM-5 | (30 psi), 75 °C | 41.07 (DHA) 5 | [96] |
| Pt-Bi/MCM-41 | (30 psi), 75 °C | 65.26 (DHA) 5 | [96] |
| Pt/Bi-MCM-41 | (30 psi), 75 °C | 33.73 (DHA) 5 | [96] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Gao, F. Navigating Glycerol Conversion Roadmap and Heterogeneous Catalyst Selection Aided by Density Functional Theory: A Review. Catalysts 2018, 8, 44. https://doi.org/10.3390/catal8020044
Liu B, Gao F. Navigating Glycerol Conversion Roadmap and Heterogeneous Catalyst Selection Aided by Density Functional Theory: A Review. Catalysts. 2018; 8(2):44. https://doi.org/10.3390/catal8020044
Chicago/Turabian StyleLiu, Bin, and Feng Gao. 2018. "Navigating Glycerol Conversion Roadmap and Heterogeneous Catalyst Selection Aided by Density Functional Theory: A Review" Catalysts 8, no. 2: 44. https://doi.org/10.3390/catal8020044
APA StyleLiu, B., & Gao, F. (2018). Navigating Glycerol Conversion Roadmap and Heterogeneous Catalyst Selection Aided by Density Functional Theory: A Review. Catalysts, 8(2), 44. https://doi.org/10.3390/catal8020044