Efficient Reduction of Bromate by Iodide-Assisted UV/Sulfite Process




Abstract
1. Introduction
2. Results and Discussion
2.1. Reduction Efficiency of Using UV/Sulfite/
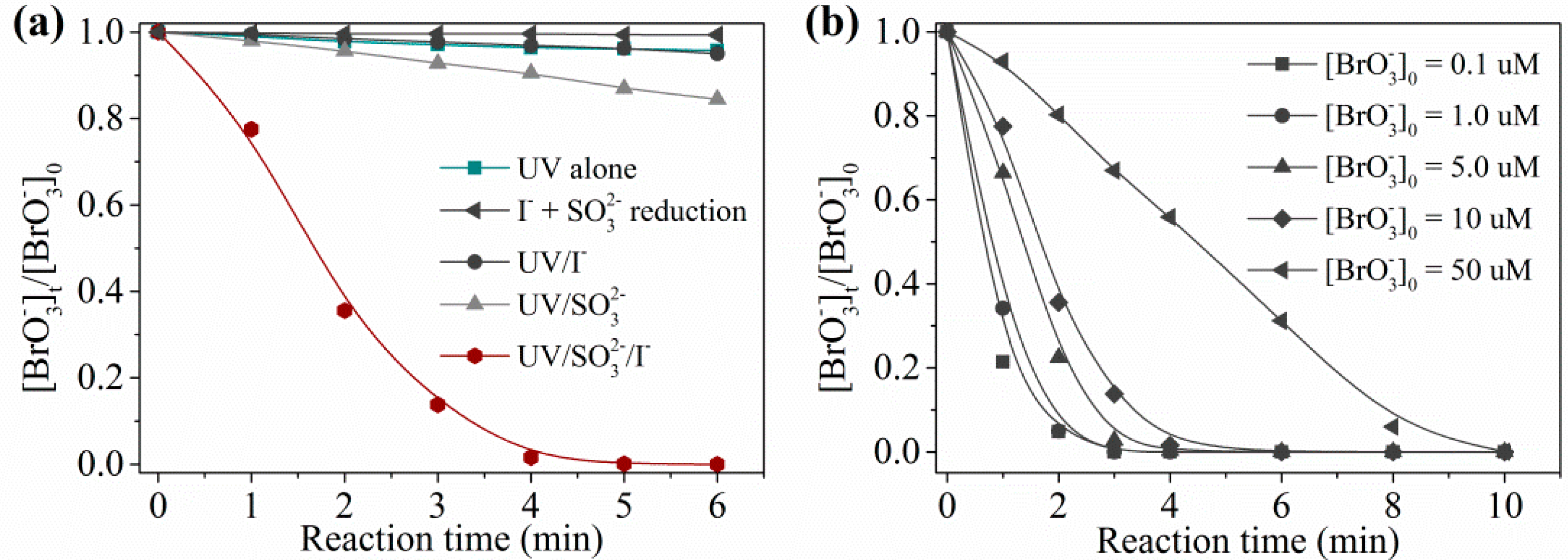
2.1.1. Degradation Efficiency in Prepared Water
2.1.2. Validation of UV/Sulfite/ Process with Real Water
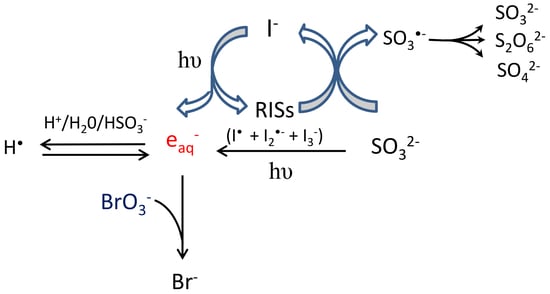
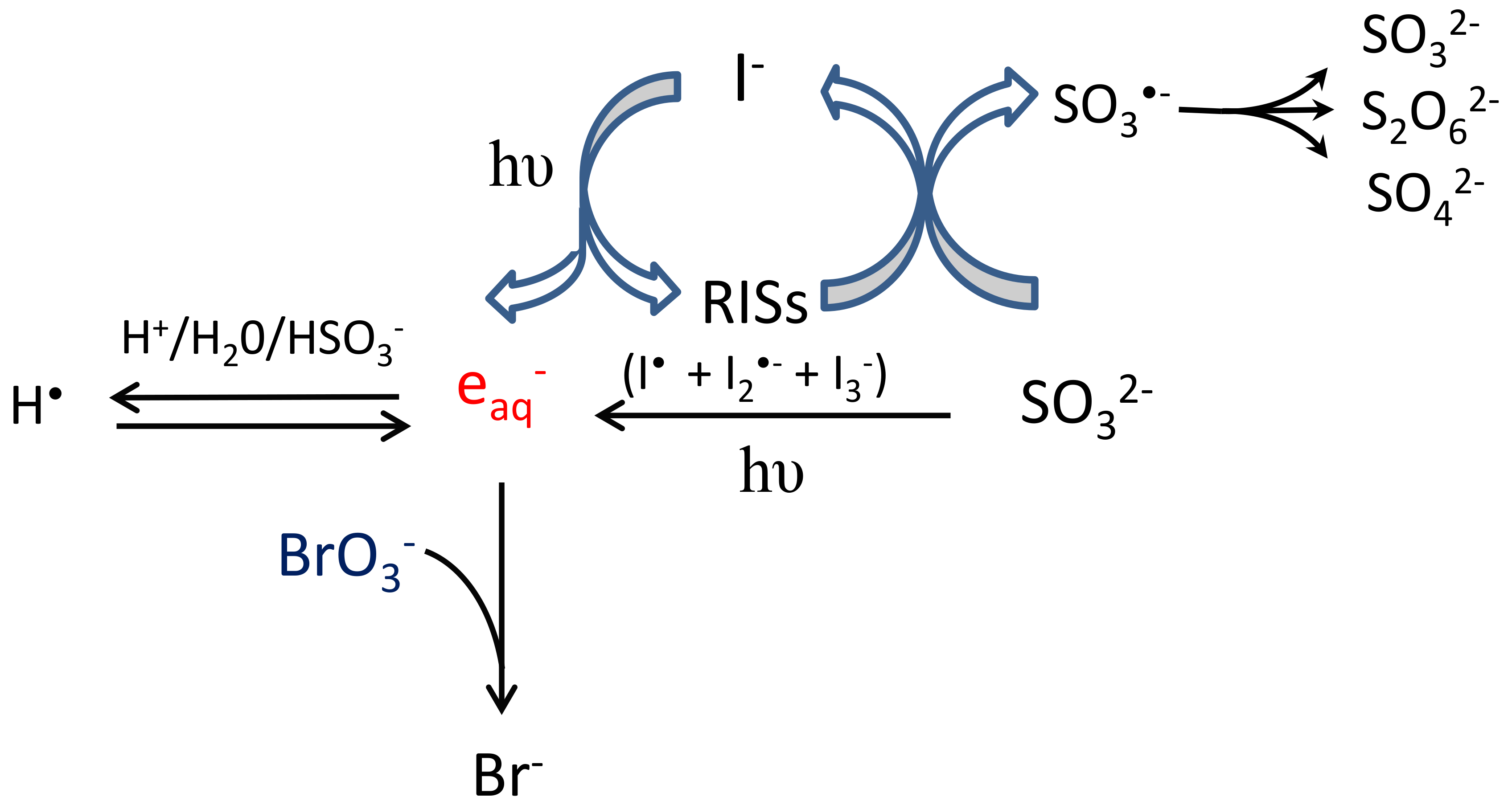
2.2. Mechanism of Reduction
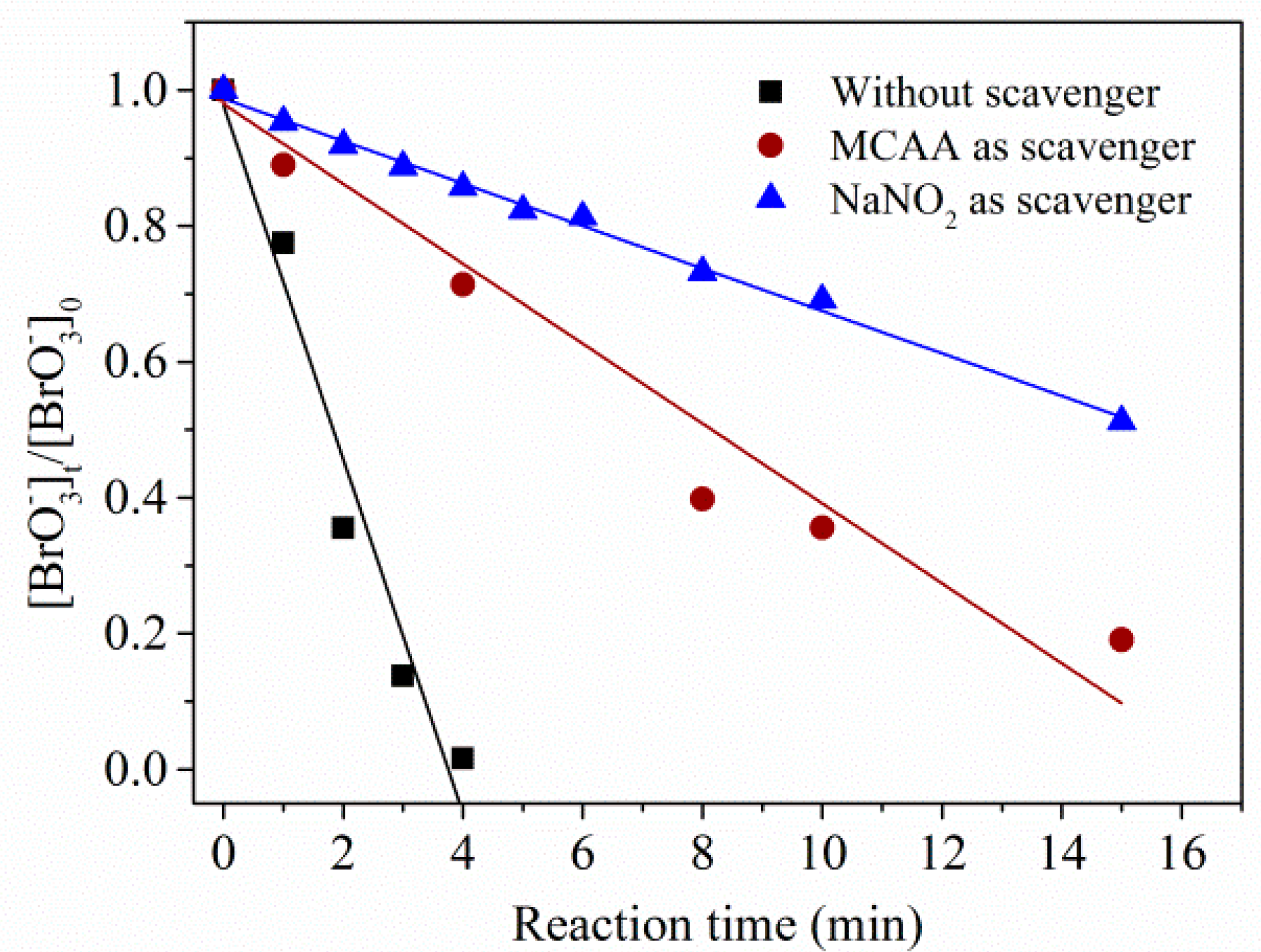
2.2.1. Main Contributor to Reduction
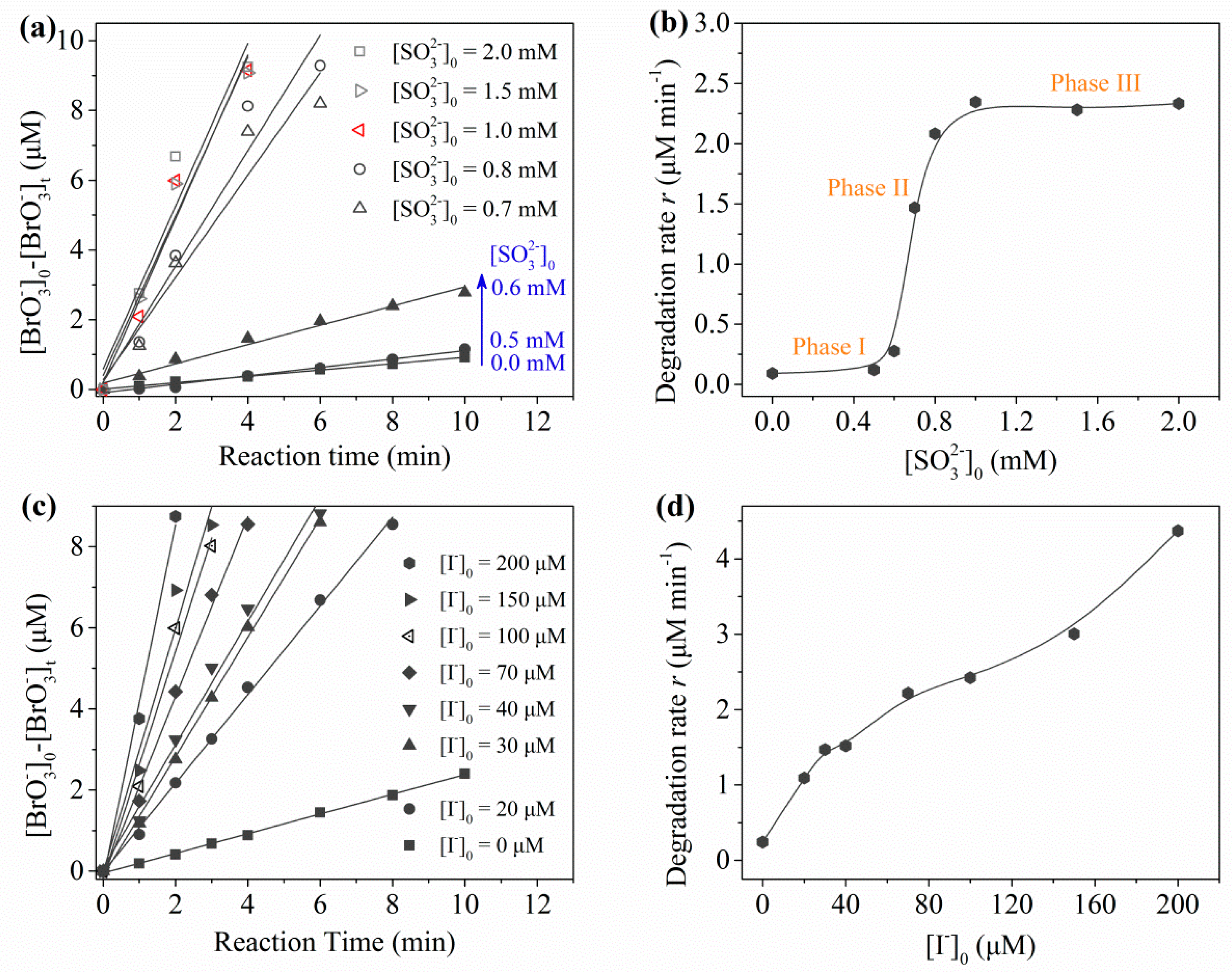
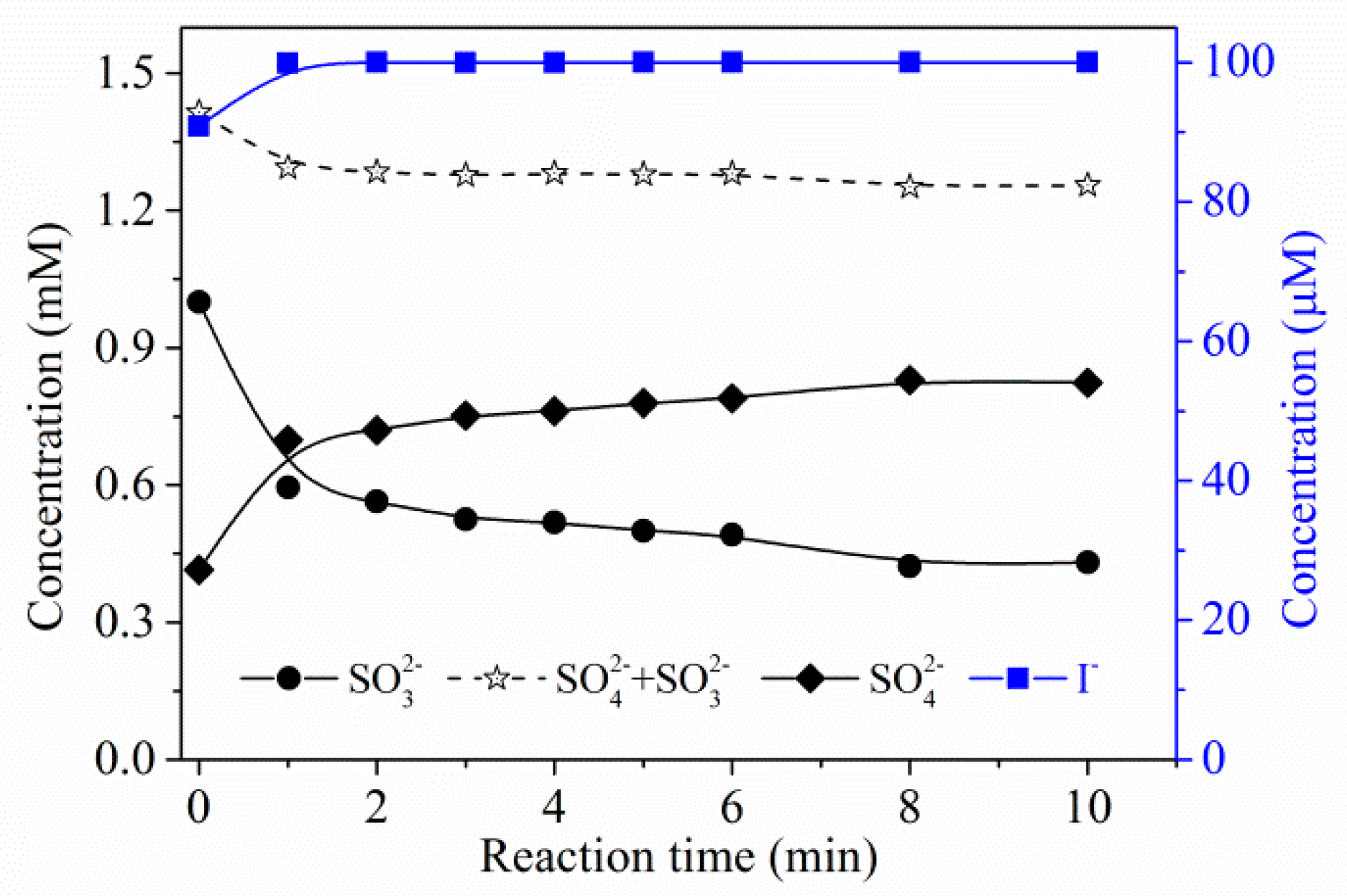
2.2.2. The Role of and Sulfite
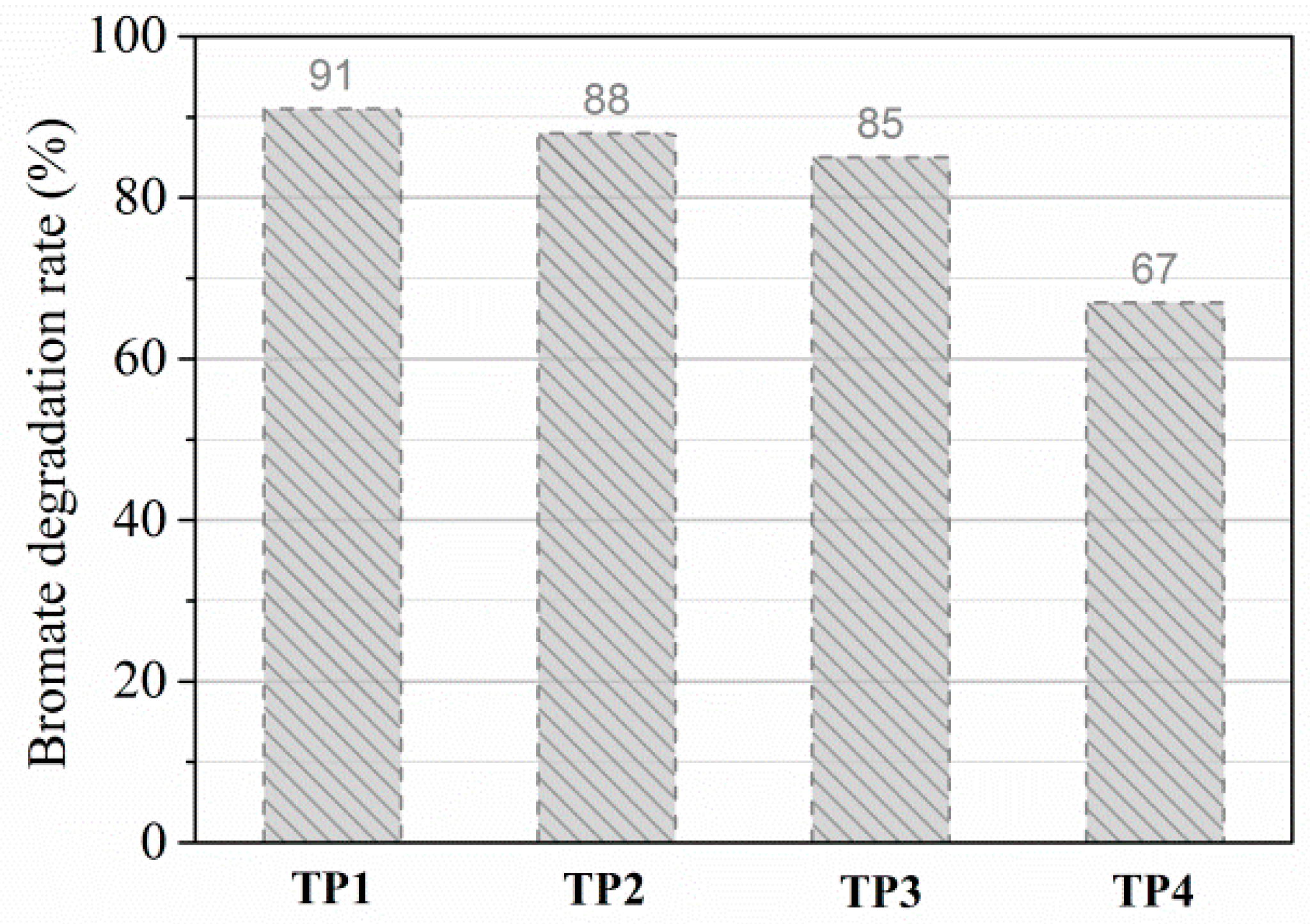
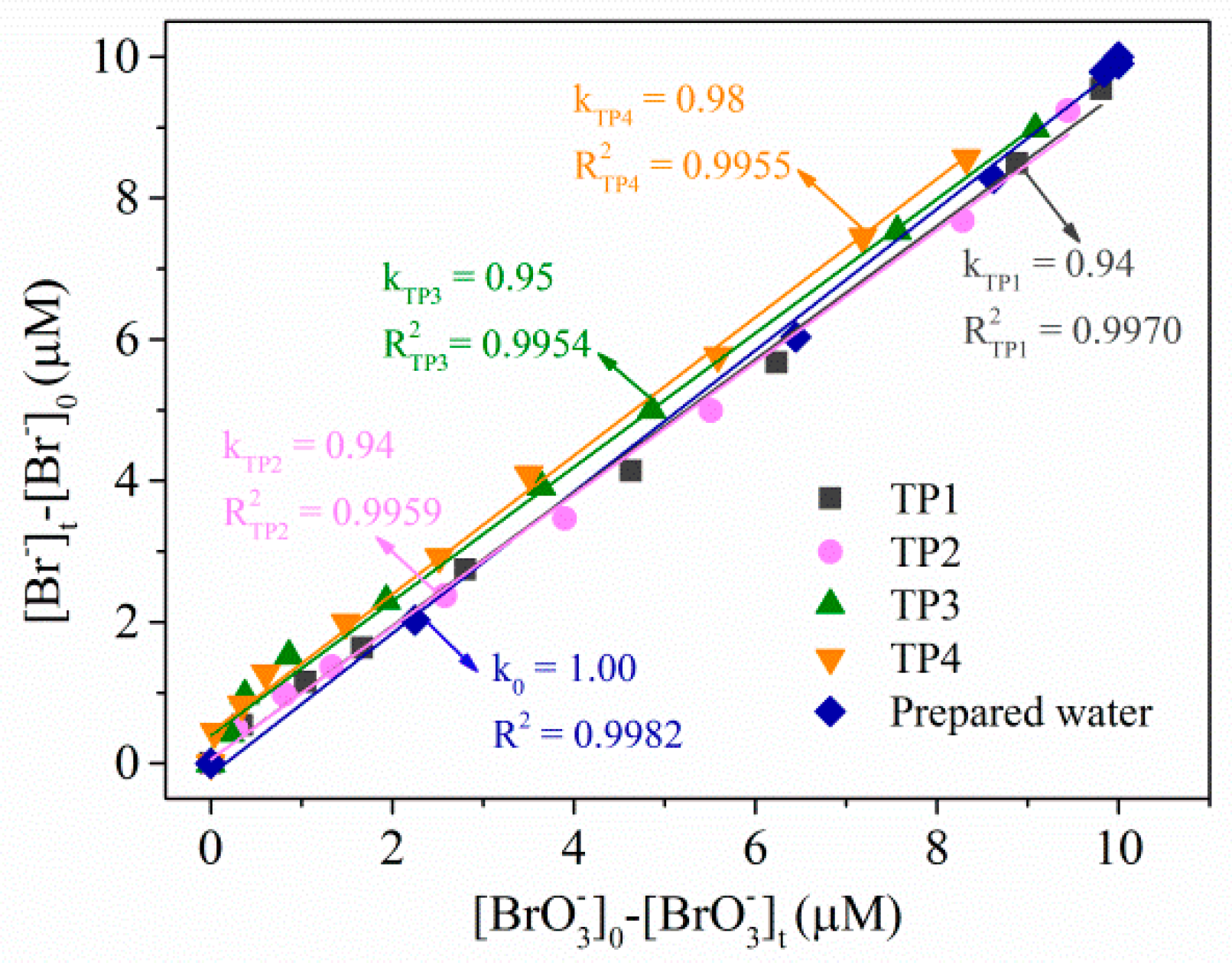
2.2.3. Transformation Products of
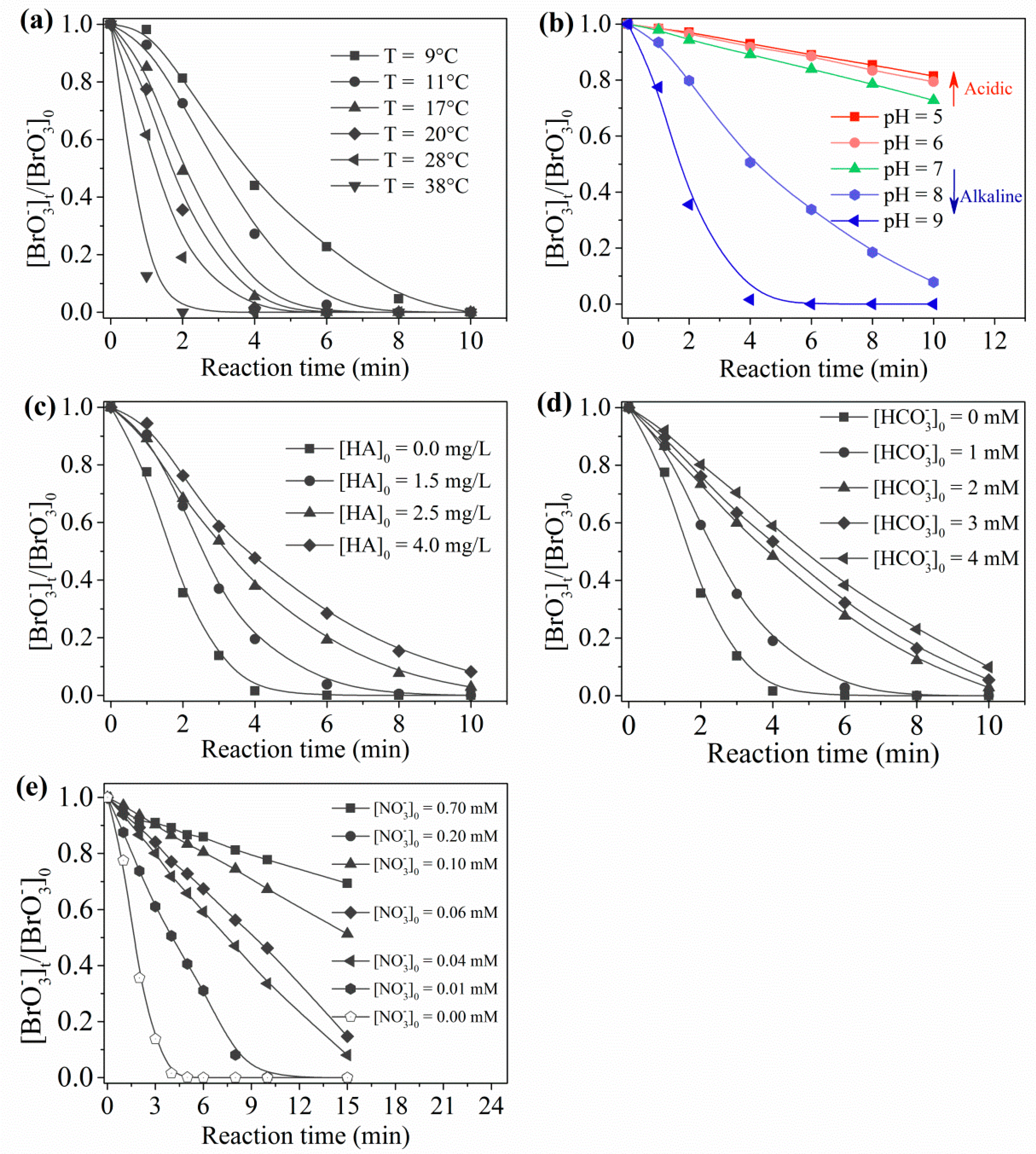
2.3. Influence of Water Quality Parameters
3. Materials and Methods
3.1. Materials
3.2. Experimental Procedure
3.3. Analytical Methods
3.4. UV Intensity and UV Fluence Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Krasner, S.W.; Glaze, W.H.; Weinberg, H.S.; Daniel, P.A.; Najm, I.N. Formation and control of bromate during ozonation of waters containing bromide. Water Res. 1993, 85, 73–81. [Google Scholar] [CrossRef]
- Huang, X.; Gao, N.; Lu, P. Bromate reduction by granular activated carbon. Environ. Sci. 2007, 28, 2264–2269. [Google Scholar] [CrossRef]
- Wang, L.; Batchelor, B.; Pillai, S.D.; Botlaguduru, V.S.V. Electron beam treatment for potable water reuse: Removal of bromate and perfluorooctanoic acid. Chem. Eng. J. 2016, 302, 58–68. [Google Scholar] [CrossRef]
- Merle, T.; Pronk, W.; Gunten, U.V. MEMBRO3X, a novel combination of a membrane contactor with advanced oxidation (O3/H2O2) for simultaneous micropollutant abatement and bromate minimization. Environ. Sci. Technol. Lett. 2017, 4, 180–185. [Google Scholar] [CrossRef]
- Kirisits, M.J.; Snoeyink, V.L.; Inan, H.; Chee-sanford, J.C.; Raskin, L.; Brown, J.C. Water quality factors affecting bromate reduction in biologically active carbon filters. Water Res. 2001, 35, 891–900. [Google Scholar] [CrossRef]
- Lin, K.A.; Lin, C.H. Enhanced reductive removal of bromate using acid-washed zero-valent iron in the presence of oxalic acid. Chem. Eng. J. 2017, 325, 144–150. [Google Scholar] [CrossRef]
- Gonzalez, M.G.; Oliveros, E.; Wörner, M.; Braun, A.M. Vacuum-ultraviolet photolysis of aqueous reaction systems. J. Photoch. Photobiol. C 2004, 5, 225–246. [Google Scholar] [CrossRef]
- Noguchi, H.; Nakajima, A.; Watanabe, T.; Hashimoto, K. Removal of bromate ion from water using TiO2 and alumina-loaded TiO2 photocatalysts. Water Sci. Technol. 2002, 46, 27–31. [Google Scholar] [CrossRef]
- Zhang, Y.; Jing, S.; Liu, H. Reactivity and mechanism of bromate reduction from aqueous solution using Zn–Fe(II)–Al layered double hydroxides. Chem. Eng. J. 2015, 266, 21–27. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, T.; Shao, Y. Aqueous bromate reduction by UV activation of sulfite. Clean-Soil Air Water 2015, 42, 1370–1375. [Google Scholar] [CrossRef]
- Xiao, Q.; Yu, S.; Li, L.; Wang, T.; Liao, X.; Ye, Y. An overview of advanced reduction processes for bromate removal from drinking water: Reducing agents, activation methods, applications and mechanisms. J. Hazard. Mater. 2017, 324, 230–240. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Jones, C.G.; Silverman, J.; Al-Sheikhly, M.; Neta, P.; Poster, D.L. Dechlorination of polychlorinated biphenyls in industrial transformer oil by radiolytic and photolytic methods. Environ. Sci. Technol. 2003, 37, 5773–5777. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, T.; Wang, L.; Shao, Y.; Fang, L. Hydrated electron-based degradation of atenolol in aqueous solution. Chem. Eng. J. 2015, 260, 740–748. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, C.; Xing, L.; Zhou, Q.; Dong, W.; Hoffmann, M.R. UV/nitrilotriacetic acid process as a novel strategy for efficient photoreductive degradation of perfluorooctanesulfonate. Environ. Sci. Technol. 2018, 52, 2953–2962. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Ma, J.; Jiang, J.; Yang, L.; Yang, J.; Zhang, J.; Chi, H.; Song, Y.; Sun, S.; Tian, W.Q. Hydrated electron (eaq−) generation from phenol/UV: Efficiency, influencing factors, and mechanism. Appl. Catal. B. Environ. 2017, 200, 585–593. [Google Scholar] [CrossRef]
- Yu, K.; Li, X.; Chen, L.; Fang, J.; Chen, H.; Li, Q.; Chi, N.; Ma, J. Mechanism and efficiency of contaminant reduction by hydrated electron in the sulfite/iodide/UV process. Water Res. 2017, 129, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, J.; Liu, G.; Fang, J.; Yue, S.; Guan, Y.; Chen, L.; Liu, X. Efficient reductive dechlorination of monochloroacetic acid by sulfite/UV process. Environ. Sci. Technol. 2012, 46, 7342–7349. [Google Scholar] [CrossRef] [PubMed]
- Bard, E.B.; Parsons, R.; Jordan, J. Standard Potentials in Aqueous Solution; Routledge: New York, NY, USA, 1985. [Google Scholar]
- Bensalah, N.; Liu, X.; Abdelwahab, A. Bromate reduction by ultraviolet light irradiation using medium pressure lamp. Int. J. Environ. Stud. 2013, 70, 566–582. [Google Scholar] [CrossRef]
- Jung, B.; Nicola, R.; Batchelor, B.; Abdel-Wahab, A. Effect of low- and medium-pressure Hg UV irradiation on bromate removal in advanced reduction process. Chemosphere 2014, 117, 663–672. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, T.; Ng, J.; Pan, J.; Sun, D. Transformation of bromine species in TiO2 photocatalytic system. Environ. Sci. Technol. 2010, 44, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Luo, Y.; Guo, Y.; Shi, W.; Wang, W.; Zhang, B.; Zhang, R.; Bao, X.; Wu, S.; Cui, F. Pd and Pt nanoparticles supported on the mesoporous silica molecular sieve SBA-15 with enhanced activity and stability in catalytic bromate reduction. Chem. Eng. J. 2018, 344, 114–123. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, H.; Li, A.; Shen, Y.; Qu, J. Bromate removal by electrochemical reduction at boron-doped diamond electrode. Electrochim. Acta 2012, 62, 181–184. [Google Scholar] [CrossRef]
- Assunção, A.; Martins, M.; Silva, G.; Lucas, H.; Coelho, M.R.; Costa, M.C. Bromate removal by anaerobic bacterial community: Mechanism and phylogenetic characterization. J. Hazard. Mater. 2011, 197, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Listiarini, K.; Tor, J.T.; Sun, D.D.; Leckie, J.O. Hybrid coagulation–nanofiltration membrane for removal of bromate and humic acid in water. J. Membr. Sci. 2010, 365, 154–159. [Google Scholar] [CrossRef]
- Fischer, M.; Warneck, P. Photodecomposition and photooxidation of hydrogen sulfite in aqueous solution. J. Phys. Chem. A 1996, 100, 15111–15117. [Google Scholar] [CrossRef]
- Lian, R.; Oulianov, D.A.; And, R.A.C.; Shkrob, I.A.; And, X.C.; Bradforth, S.E. Electron photodetachment from aqueous anions. 3. Dynamics of geminate pairs derived from photoexcitation of mono- vs polyatomic anions. J. Phys. Chem. A 2006, 110, 9071–9078. [Google Scholar] [CrossRef] [PubMed]
- Ichino, T.; Fessenden, R.W. Reactions of hydrated electron with various radicals: Spin factor in diffusion-controlled reactions. J. Phys. Chem. A 2007, 111, 2527–2541. [Google Scholar] [CrossRef] [PubMed]
- Sauer, M.C., Jr.; Crowell, R.A.; Shkrob, I.A. Electron photodetachment from aqueous anions. 1. Quantum yields for generation of hydrated electron by 193 and 248 nm laser photoexcitation of miscellaneous inorganic anions. J. Phys. Chem. A 2004, 108, 5490–5502. [Google Scholar] [CrossRef]
- Alfassi, Z.B.; Huie, R.E.; Marguet, S.; Natarajan, E.; Neta, P. Rate constants for reactions of iodine atoms in solution. Int. J. Chem. Kinet. 1995, 27, 181–188. [Google Scholar] [CrossRef]
- Jortner, J.; Levine, R.; Ottolenghi, M.; Stein, G. The photochemistry of the iodide ion in aqueous solution. J. Phys. Chem. 1961, 65, 1232–1238. [Google Scholar] [CrossRef]
- Park, H.; Vecitis, C.D.; Cheng, J.; Dalleska, N.F.; Mader, B.T.; Hoffmann, M.R. Reductive degradation of perfluoroalkyl compounds with aquated electrons generated from iodide photolysis at 254 nm. Photochem. Photobiol. Sci. 2011, 10, 1945–1953. [Google Scholar] [CrossRef] [PubMed]
- Neta, P.; Huie, R.E.; Ross, A.B. Rate constants for reactions of inorganic radicals in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Yiin, B.S.; Margerum, D.W. Nonmetal redox kinetics: Reactions of iodine and triiodide with sulfite and hydrogen sulfite and the hydrolysis of iodosulfate. Inorg. Chem. 1990, 29, 1559–1564. [Google Scholar] [CrossRef]
- Hayon, E.; Treinin, A.; Wilf, J. Electronic spectra, photochemistry, and autoxidation mechanism of the sulfite-bisulfite-pyrosulfite systems SO2−, SO3−, SO4−, and SO5− radicals. J. Am. Chem. Soc. 1972, 94, 47–57. [Google Scholar] [CrossRef]
- Grebel, J.E.; Pignatello, J.J.; Mitch, W.A. Effect of halide ions and carbonates on organic contaminant degradation by hydroxyl radical-based advanced oxidation processes in saline waters. Environ. Sci. Technol. 2010, 44, 6822–6828. [Google Scholar] [CrossRef]
- Ross, A.B.; Neta, P. Rate Constants for Reactions of Inorganic Radicals in Aqueous-Solution; Report NSRDS-NBS65; National Bureau of Standards: Washington, DC, USA, 1979; pp. 5–6.
- Neta, P.; Huie, R.E. Free-radical chemistry of sulfite. Environ. Health Perspect. 1985, 64, 209–217. [Google Scholar] [CrossRef]
- Buchanan, W.; Roddick, F.; Porter, N.; Drikass, M. Fractionation of UV and VUV pretreated natural organic matter from drinking water. Environ. Sci. Technol. 2005, 39, 4647–4654. [Google Scholar] [CrossRef]
- Kishore, K.; Moorthy, P.N.; Rao, K.N. Use of riboflavin as a standard solute for estimating H-atom reaction rate constants by competition kinetic method. Radiat. Phys. Chem. 1984, 23, 495–497. [Google Scholar] [CrossRef]
- Zelinsky, A.G.; Pirogov, B.Y. Electrochemical oxidation of sulfite and sulfur dioxide at a renewable graphite electrode. Electrochim. Acta 2017, 231, 371–378. [Google Scholar] [CrossRef]
- Xiao, Q.; Ren, Y.; Yu, S. Pilot study on bromate reduction from drinking water by UV/sulfite systems: Economic cost comparisons, effects of environmental parameters and mechanisms. Chem. Eng. J. 2017, 330, 1203–1210. [Google Scholar] [CrossRef]
- Wenk, J.; von Gunten, U.; Canonica, S. Effect of dissolved organic matter on the transformation of contaminants induced by excited triplet states and the hydroxyl radical. Environ. Sci. Technol. 2011, 45, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Zhou, L.; Ferronato, C.; Yang, X.; Salvador, A.; Zeng, C.; Chovelon, J.M. Photocatalytic degradation of atenolol in aqueous titanium dioxide suspensions: Kinetics, intermediates and degradation pathways. J. Photochem. Photobiol. A Chem. 2013, 254, 35–44. [Google Scholar] [CrossRef]
- Fang, J.; Shang, C. Bromate formation from bromide oxidation by the UV/persulfate process. Environ. Sci. Technol. 2012, 46, 8976–8983. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, W.; Zheng, B.; Mao, S.; Jin, H. Comparative study on the determination of bicarbonate in bicarbonated ringer s injection by titrimetric method and suppressed ion chromatography. West China J. Pharm. Sci. 2016, 31, 286–288. [Google Scholar] [CrossRef]
- Rosenfeldt, E.J.; Linden, K.G.; Canonica, S.; Gunten, U.V. Comparison of the efficiency of ·OH radical formation during ozonation and the advanced oxidation processes O3/H2O2 and UV/H2O2. Water Res. 2006, 40, 3695–3704. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | Predeoxygenation | (mg·L−1) | Experimental Conditions | Removal Rate (Time Needed) | Reference |
|---|---|---|---|---|---|
| UV254/sulfite/ | No | 0.01 | pH = 9.2; 20 ± 1 °C; []0 = 100 μM, [sulfite]0 = 1.0 mM, [DO]0 = 7.0 mg·L−1; 10 W | 100% (3 min) | This study |
| UV254/sulfite | Yes | 12.80 | pH = 7.0; 23 °C; []0 = 1 mM; 10 W | 61% (50 min) | [10] |
| UV-M | Yes | 0.60 | pH = 6.8; 22–23 °C; 2400 μW·cm−2 | 100% (120 min) | [20] |
| UV-L | Yes | 0.50 | pH = 6.8; 4900 μW·cm−2 | 100% (250 min) | [21] |
| UV254/TiO2 | No | 12.80 | pH = 1.5–13.5; [TiO2] = 0.5 g·L−1; 1255 mW·cm−2 | 100% (90 min) | [22] |
| UV365/TiO2 | No | 12.80 | pH = 1.5−13.5; [TiO2] = 0.5 g·L−1; 1150 mW·cm−2 | 78% (180 min) | [22] |
| Catalyst (4% Pt/SBA-15) | Yes | 100.00 | pH = 6.5; 25 °C; catalyst = 30 mg·L−1 | 80% (13 min) | [23] |
| Electrochemical reduction (BDD electrodes) | - | 20.00 | pH = 7.5; The applied bias potential of −1.0 V (vs. SCE); = 0.1 mM | 90% (2 h) | [24] |
| Zero-valent iron (ZVI) | Yes | 10.00 | pH = 7; 20 °C; ZVI = 5 g·L−1 | 100% (60 min) | [6] |
| Acid-washed Zero-valent iron | Yes | 10.00 | pH = 7; 20 °C; ZVI = 5 g·L−1, Oxalic acid = 250 μM | 93% (30 min) | [6] |
| Zn–Fe(II)–Al layered double hydroxides (LDHs) | - | 0.10 | pH = 7; 22 °C; Zn–Fe(II)–Al LDHs = 0.50 g·L−1; mixing rate = 200 rpm | 100% (30 min) | [9] |
| Biological treatment | Yes | 5.12 | pH = 7.1; 21 °C; []0 = 10 mM, with Clostridium and Citrobacter genera bacteria | 96% (5 d) | [25] |
| Hybrid coagulation–nanofiltration | - | 2.56 | pH = 7; NF-90 membrane (roughness = 388 Å, zeta potential = −26.5 mV); ionic strength 10 mM as NaCl, TMP 350 kPa | 18.9% (9 h) | [26] |
| No. | Reactions | Rate Constants (M−1∙s−1) | Reference |
|---|---|---|---|
| 1 | - | [27,28] | |
| 2 | - | [27,28] | |
| 3 | [27,29] | ||
| 4 | [27,29] | ||
| 5 | [12] | ||
| 6 | - | [30] | |
| 7 | [31] | ||
| 8 | [31] | ||
| 9 | [32] | ||
| 10 | [32] | ||
| 11 | [32] | ||
| 12 | [33] | ||
| 13 | [31] | ||
| 14 | [31] | ||
| 15 | [34] | ||
| 16 | [34] | ||
| 17 | [35] | ||
| 18 | [35] | ||
| 19 | [35] | ||
| 20 | [35] | ||
| 21 | - | [35] | |
| 22 | - | [30] | |
| 23 | - | [30] | |
| 24 | - | [30] | |
| 25 | [12] | ||
| 26 | [32] | ||
| 27 | [32] | ||
| 28 | [32] | ||
| 29 | [12] | ||
| 30 | [32] | ||
| 31 | [12] | ||
| 32 | [12] | ||
| 33 | [12] | ||
| 34 | 19 | [12] | |
| 35 | + | [12] | |
| 36 | [36] | ||
| 37 | 10 | [12] | |
| 38 | [12] | ||
| 39 | [29] | ||
| 40 | slow | [27] | |
| 41 | [12] | ||
| 42 | [27] | ||
| 43 | [27] | ||
| 44 | [27] | ||
| 45 | slow | [27] | |
| 46 | [12] | ||
| 47 | [12] | ||
| 48 | <1.0 × 106 | [12] | |
| 49 | - | [37] | |
| 50 | [12] | ||
| 51 | [12] | ||
| 52 | [17] | ||
| 53 | [17] | ||
| 54 | [12] | ||
| 55 | [12] | ||
| 56 | [38] | ||
| 57 | [39] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Wang, J.; Yan, D.; Wang, L.; Liu, X. Efficient Reduction of Bromate by Iodide-Assisted UV/Sulfite Process. Catalysts 2018, 8, 652. https://doi.org/10.3390/catal8120652
Zhang T, Wang J, Yan D, Wang L, Liu X. Efficient Reduction of Bromate by Iodide-Assisted UV/Sulfite Process. Catalysts. 2018; 8(12):652. https://doi.org/10.3390/catal8120652
Chicago/Turabian StyleZhang, Tuqiao, Jiajie Wang, Dingyun Yan, Lili Wang, and Xiaowei Liu. 2018. "Efficient Reduction of Bromate by Iodide-Assisted UV/Sulfite Process" Catalysts 8, no. 12: 652. https://doi.org/10.3390/catal8120652
APA StyleZhang, T., Wang, J., Yan, D., Wang, L., & Liu, X. (2018). Efficient Reduction of Bromate by Iodide-Assisted UV/Sulfite Process. Catalysts, 8(12), 652. https://doi.org/10.3390/catal8120652