Nucleophilic Dearomatization of Activated Pyridines



Abstract
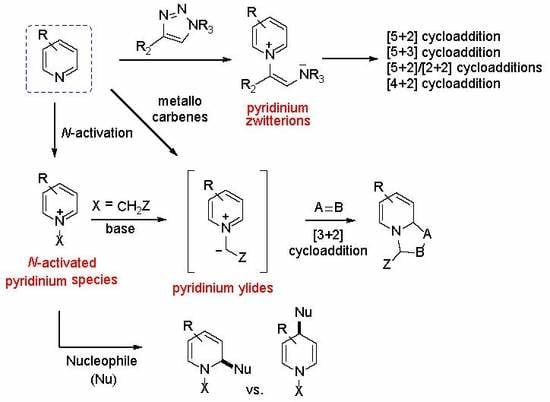
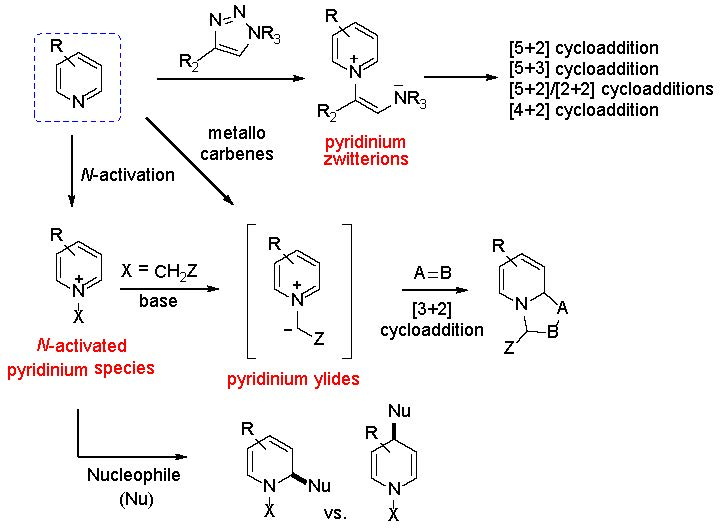{kind=link}
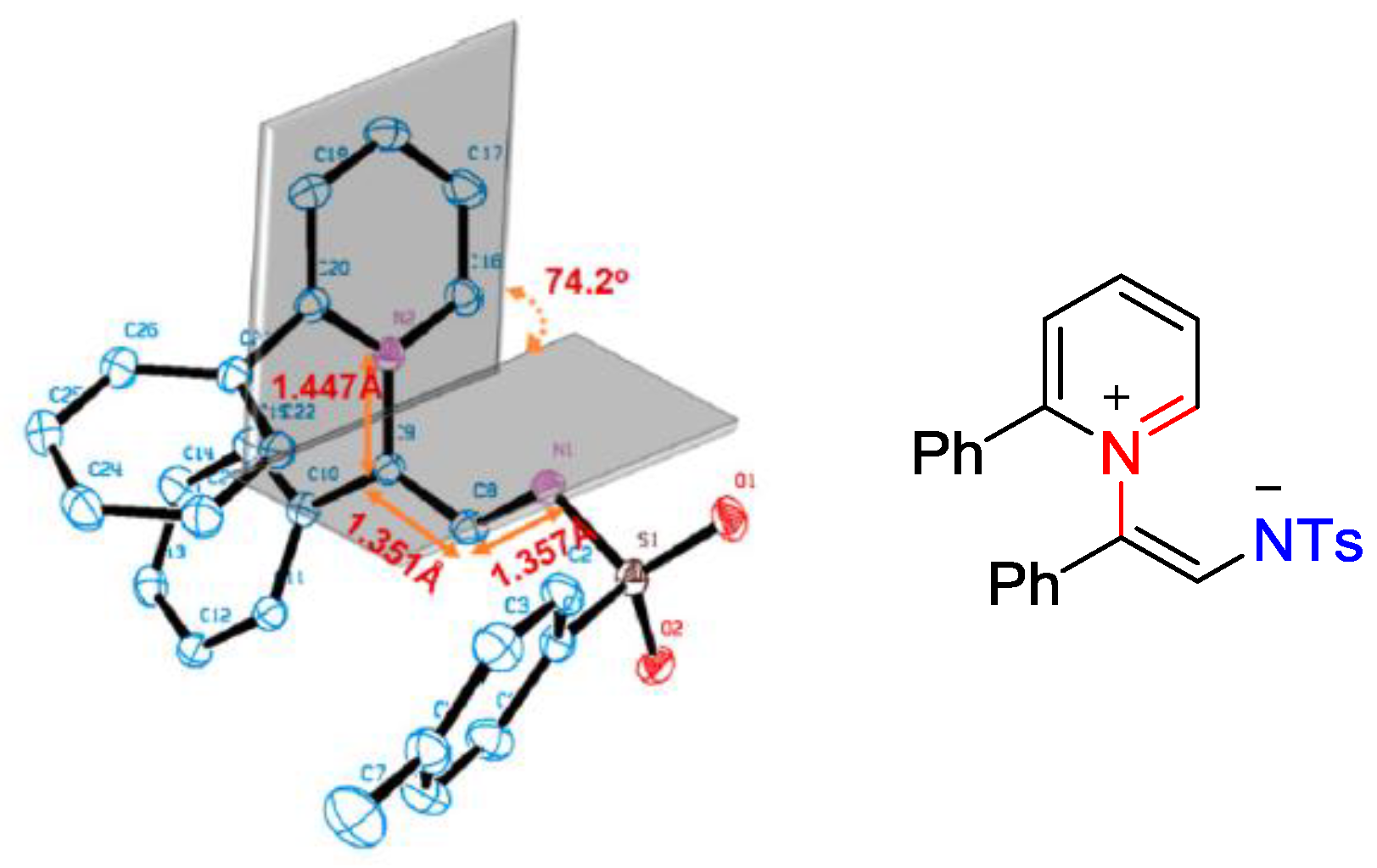{kind=link}
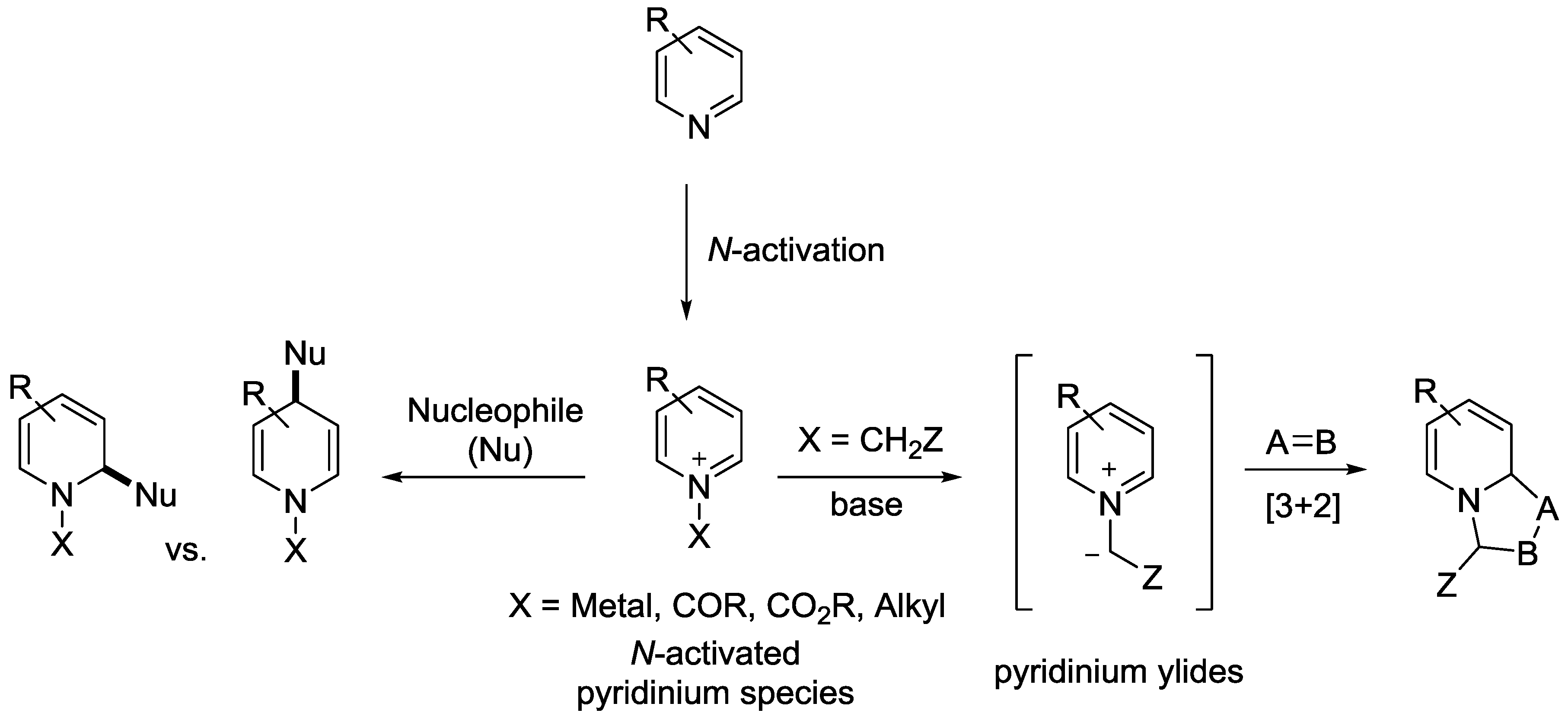{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
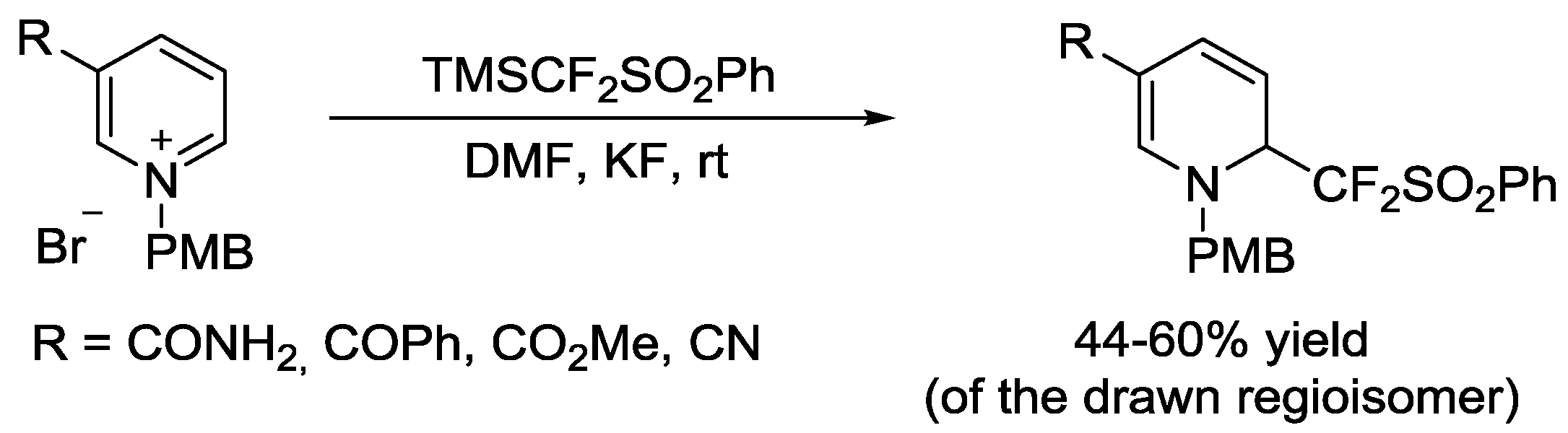{kind=link}
{kind=link}
{kind=link}
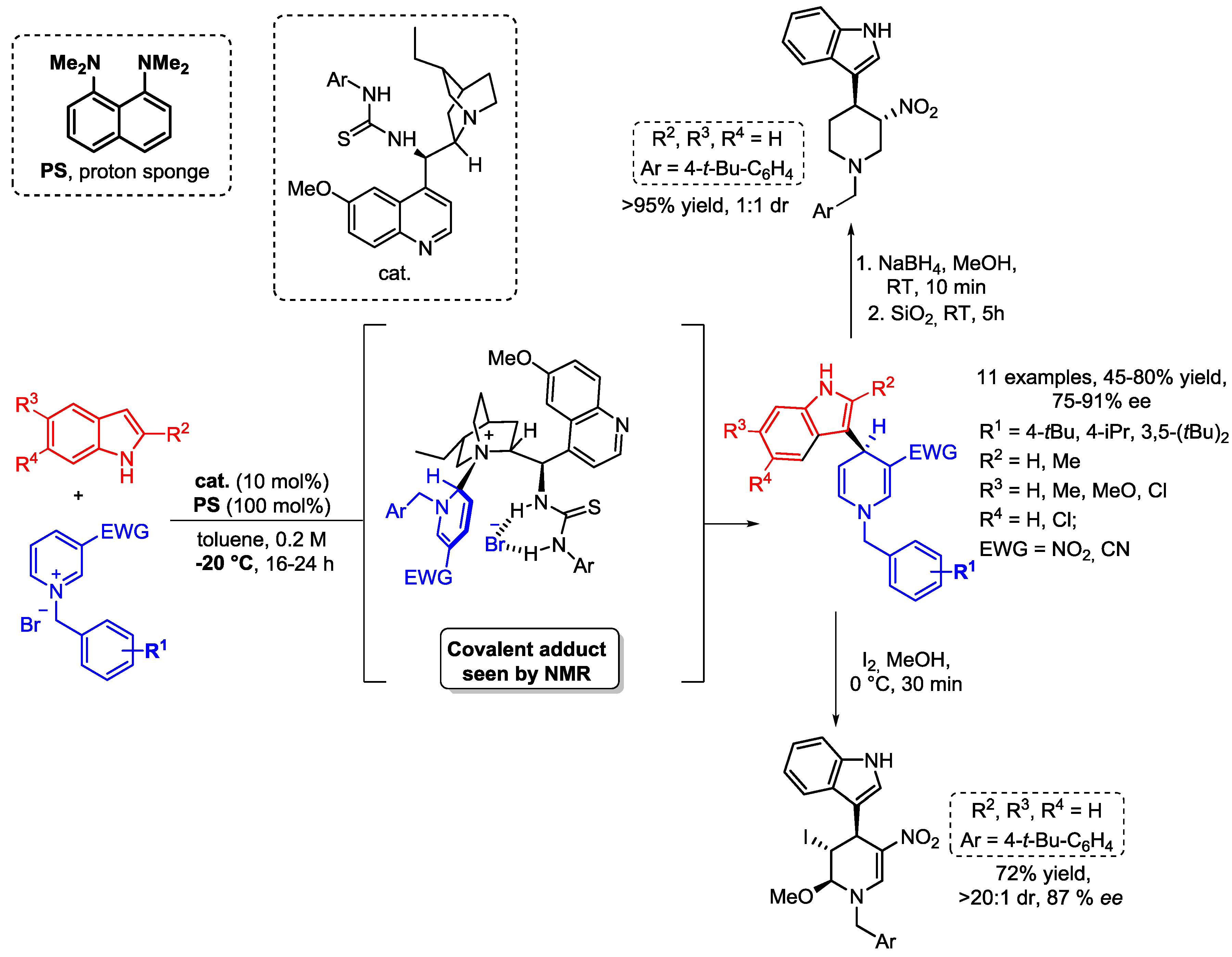{kind=link}
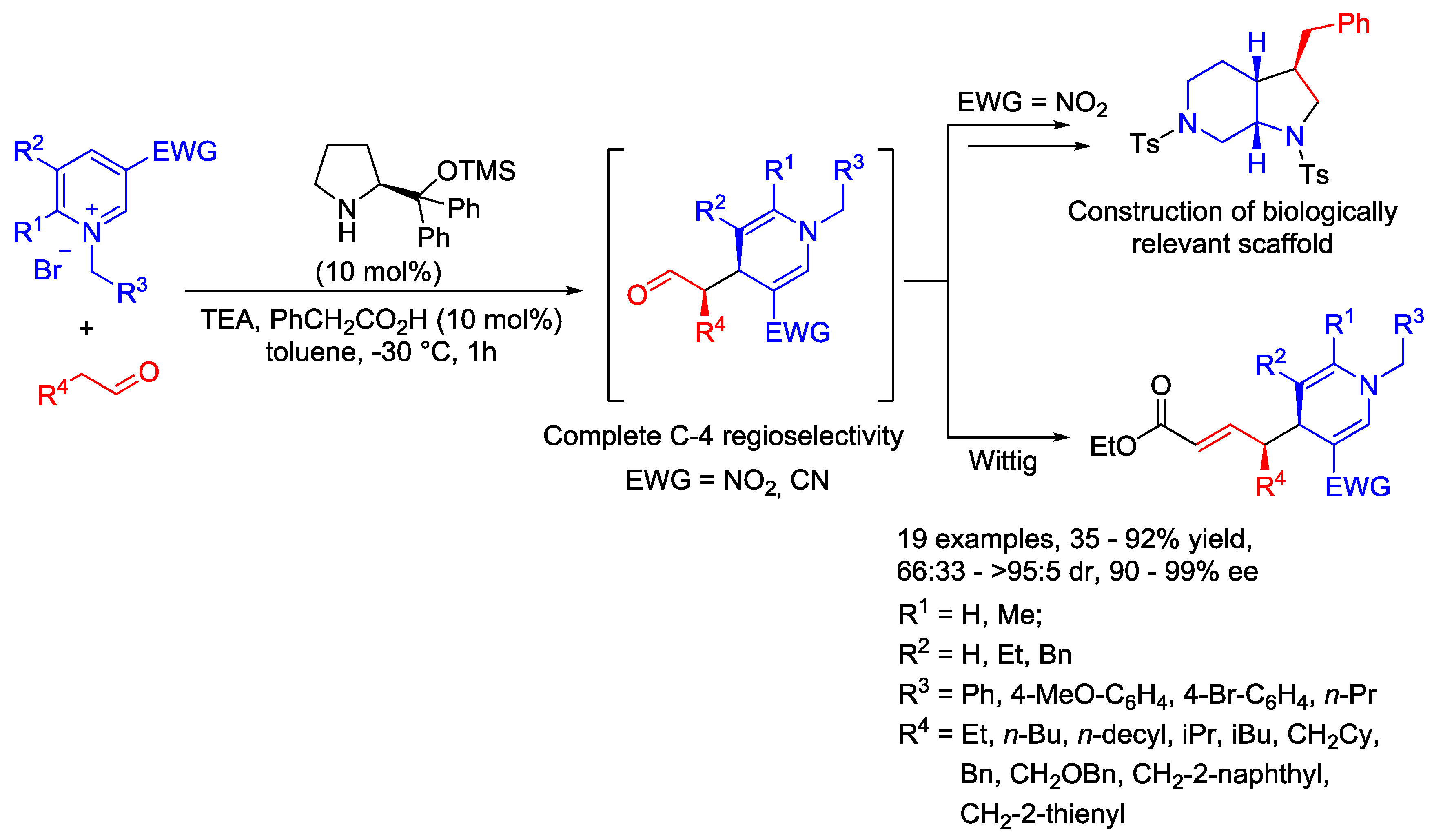{kind=link}
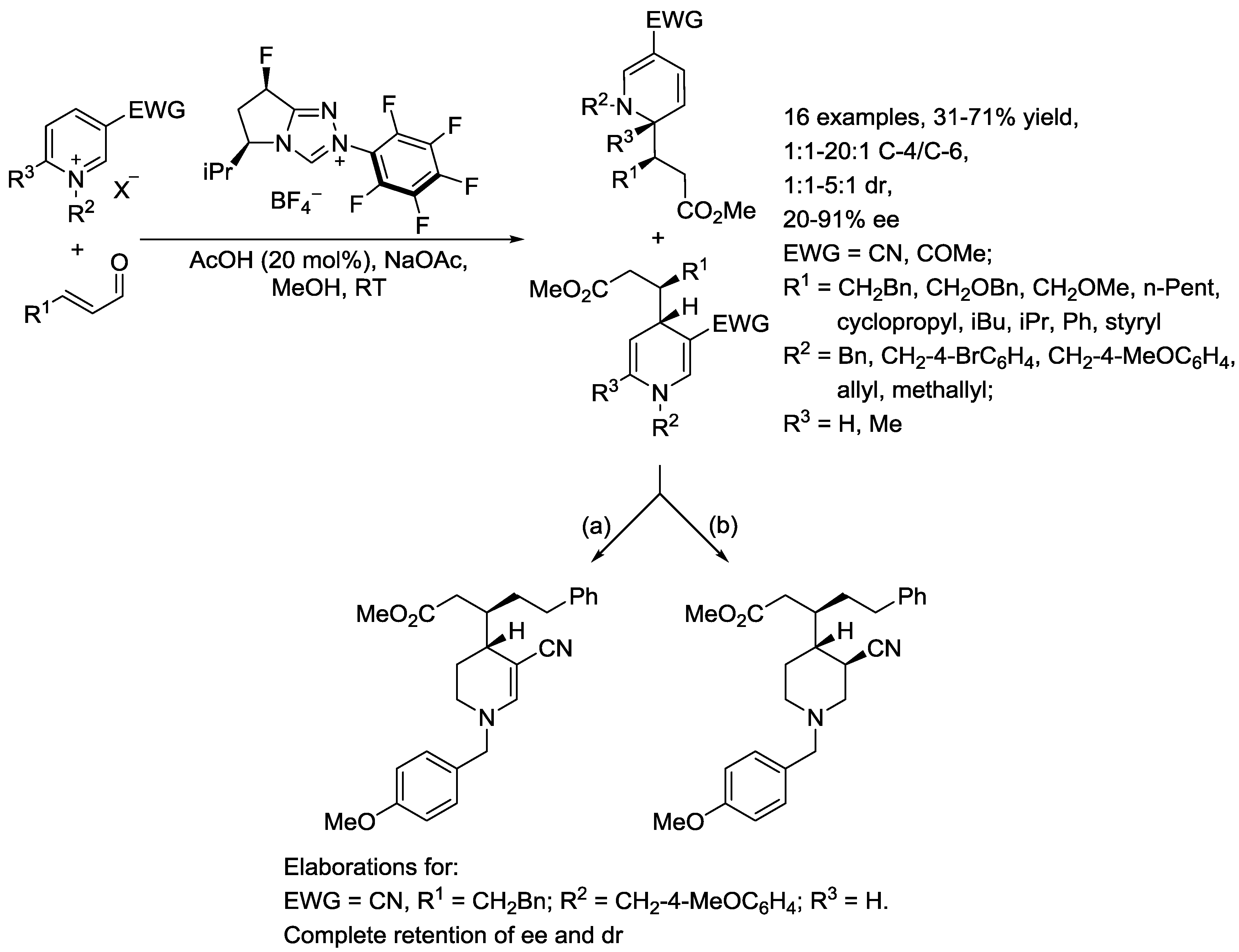{kind=link}
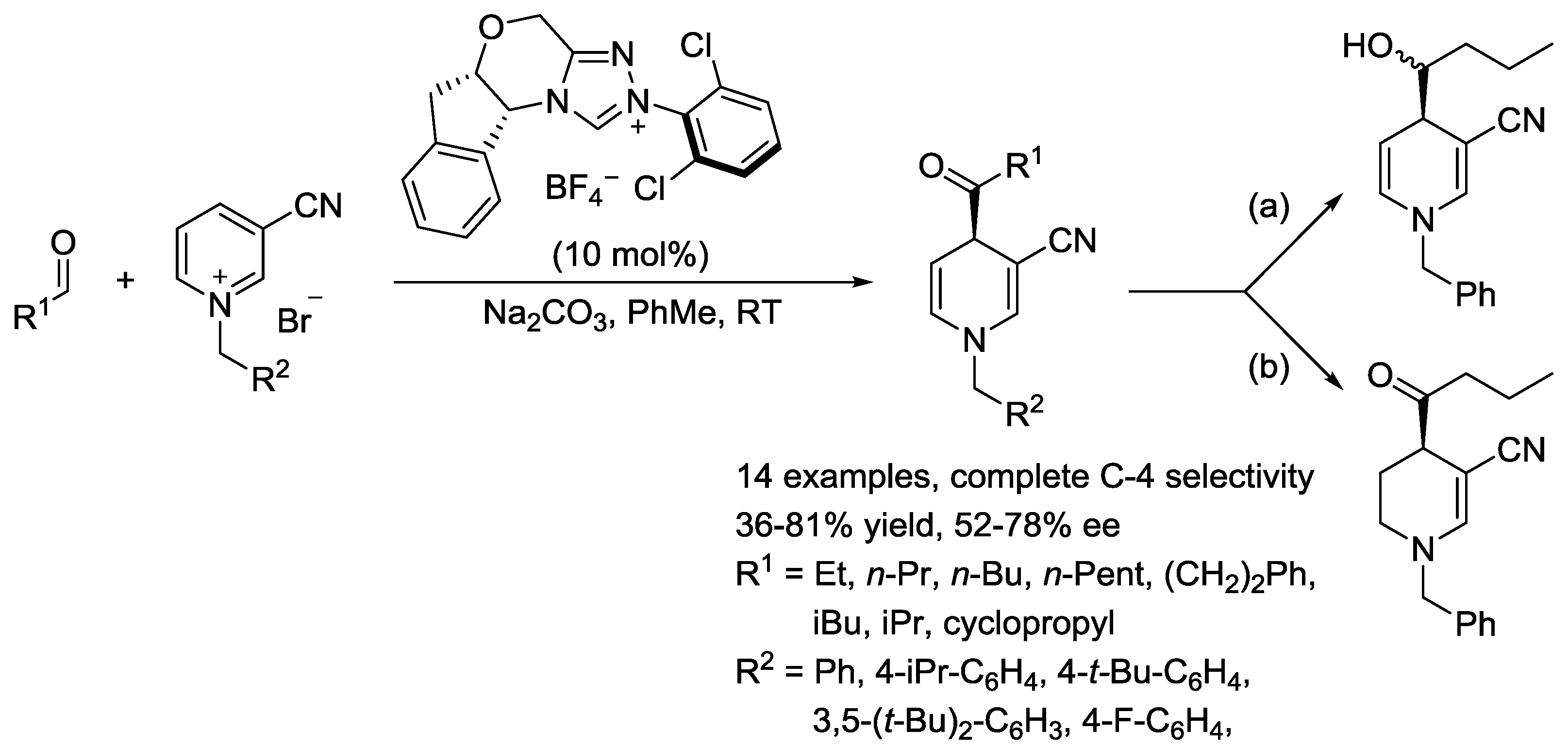{kind=link}
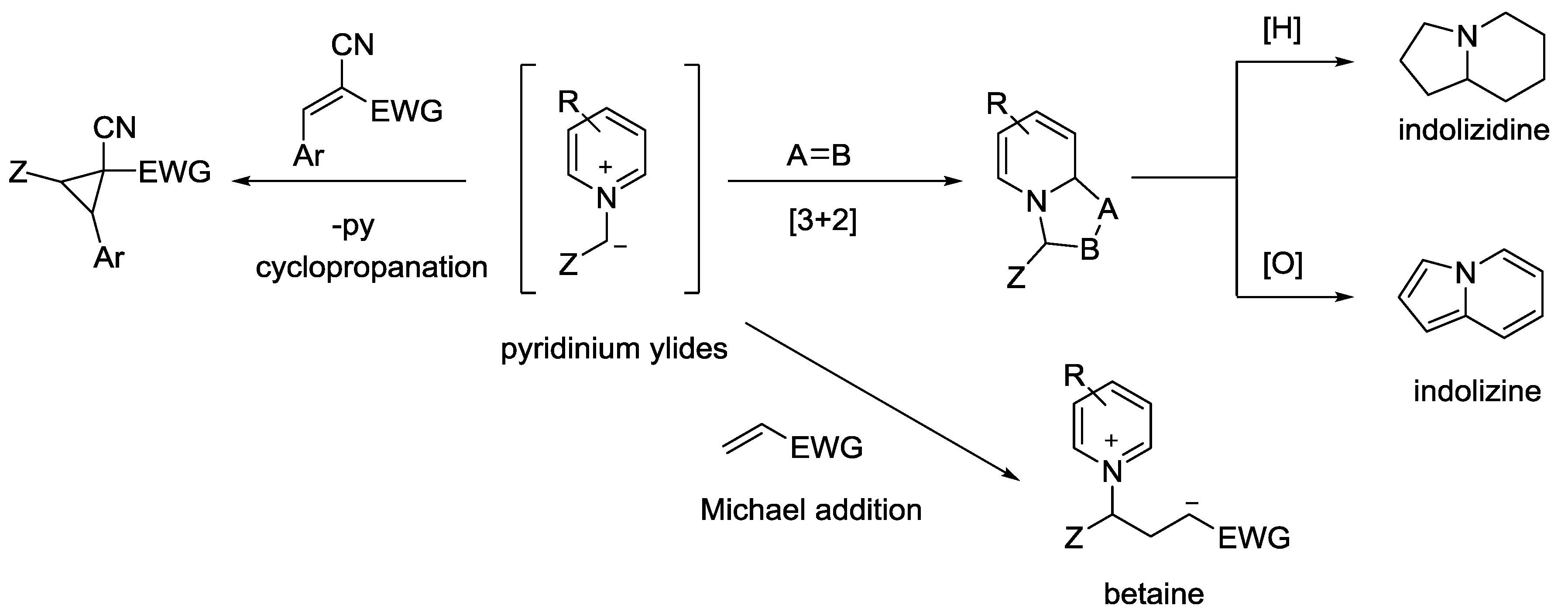{kind=link}
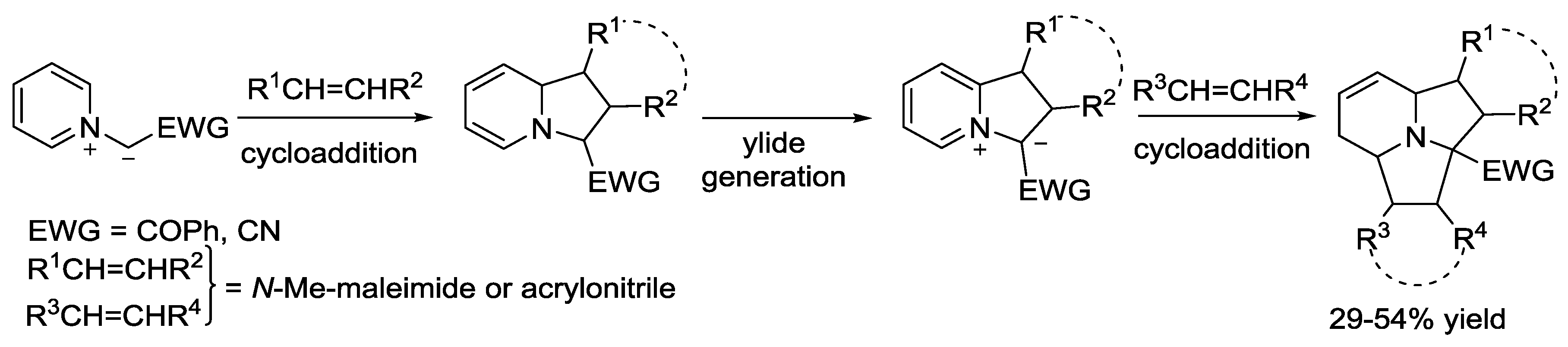{kind=link}
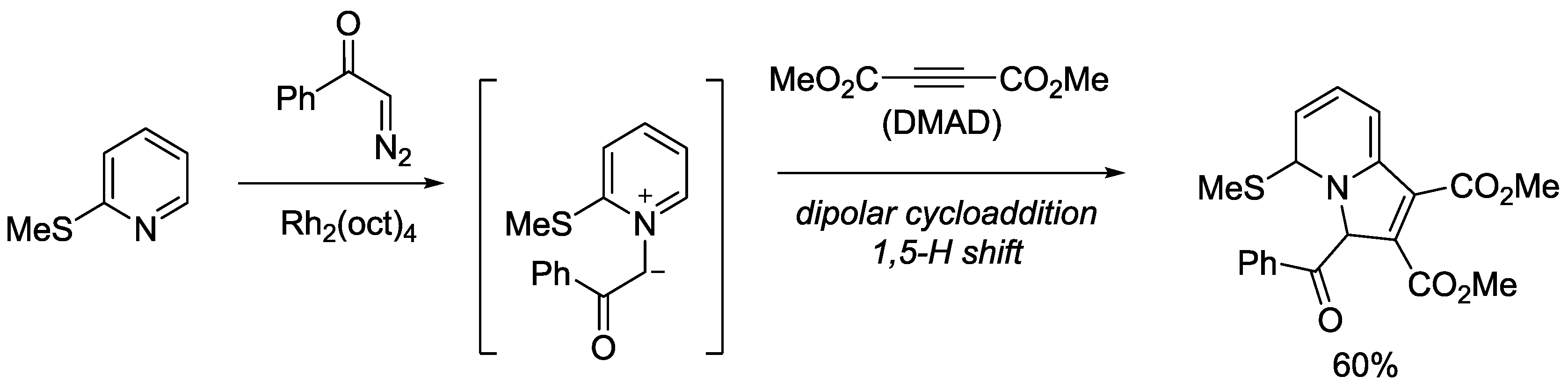{kind=link}
{kind=link}
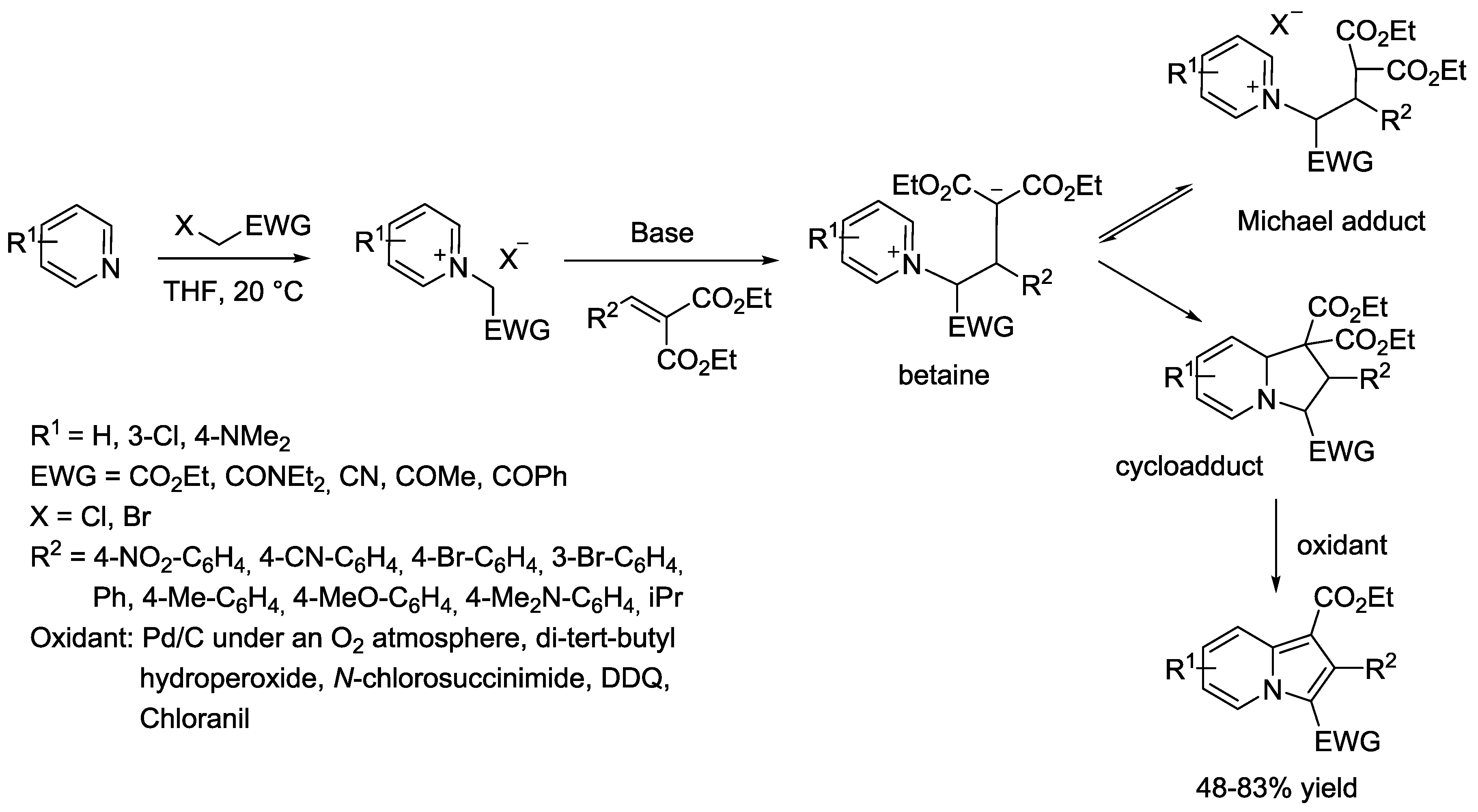{kind=link}
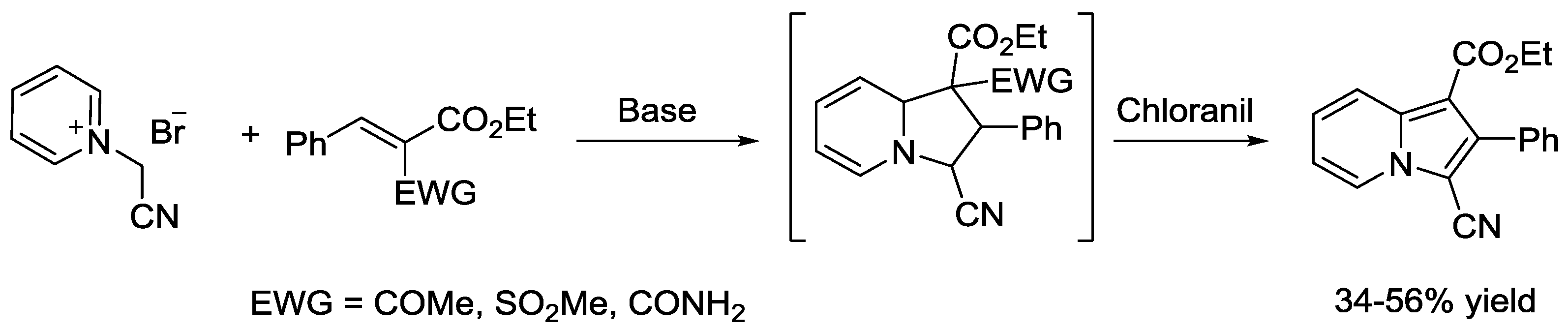{kind=link}
{kind=link}
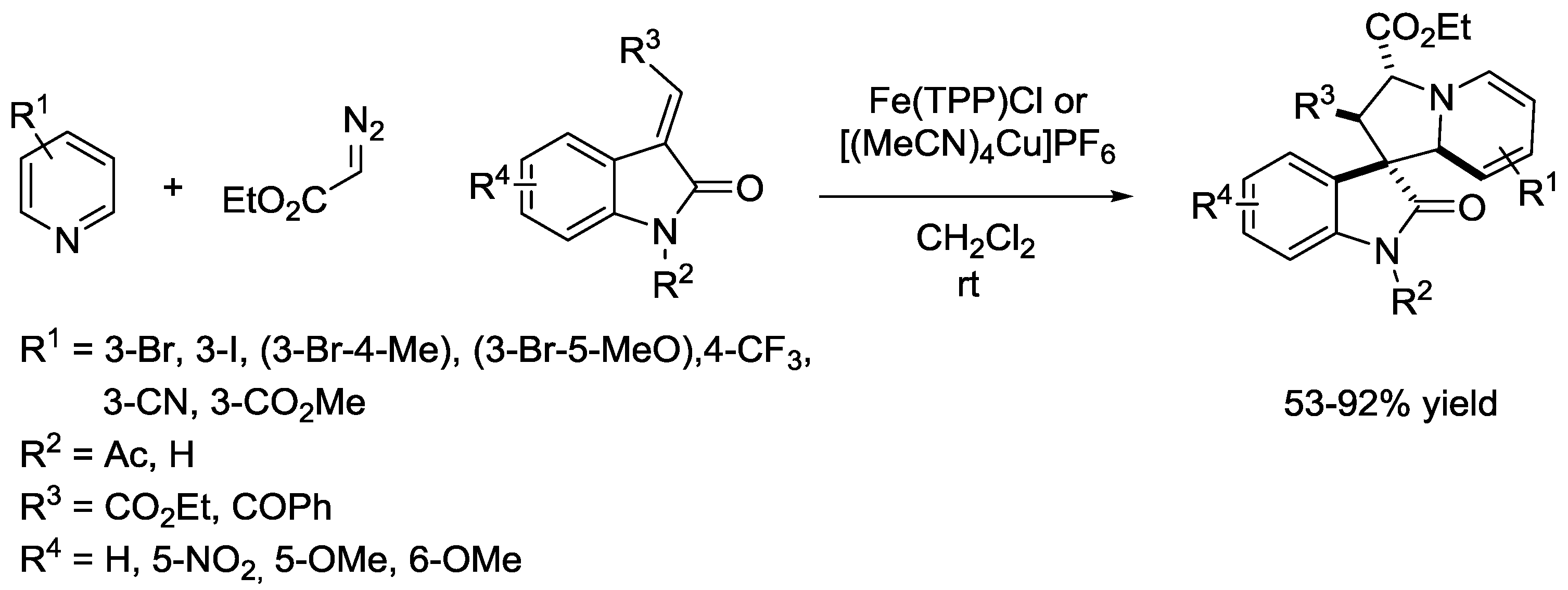{kind=link}
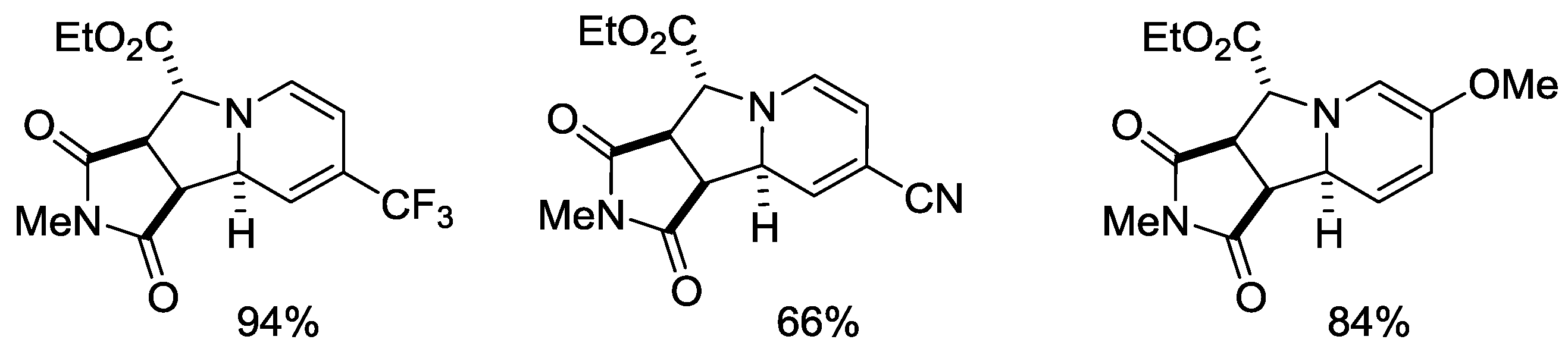{kind=link}
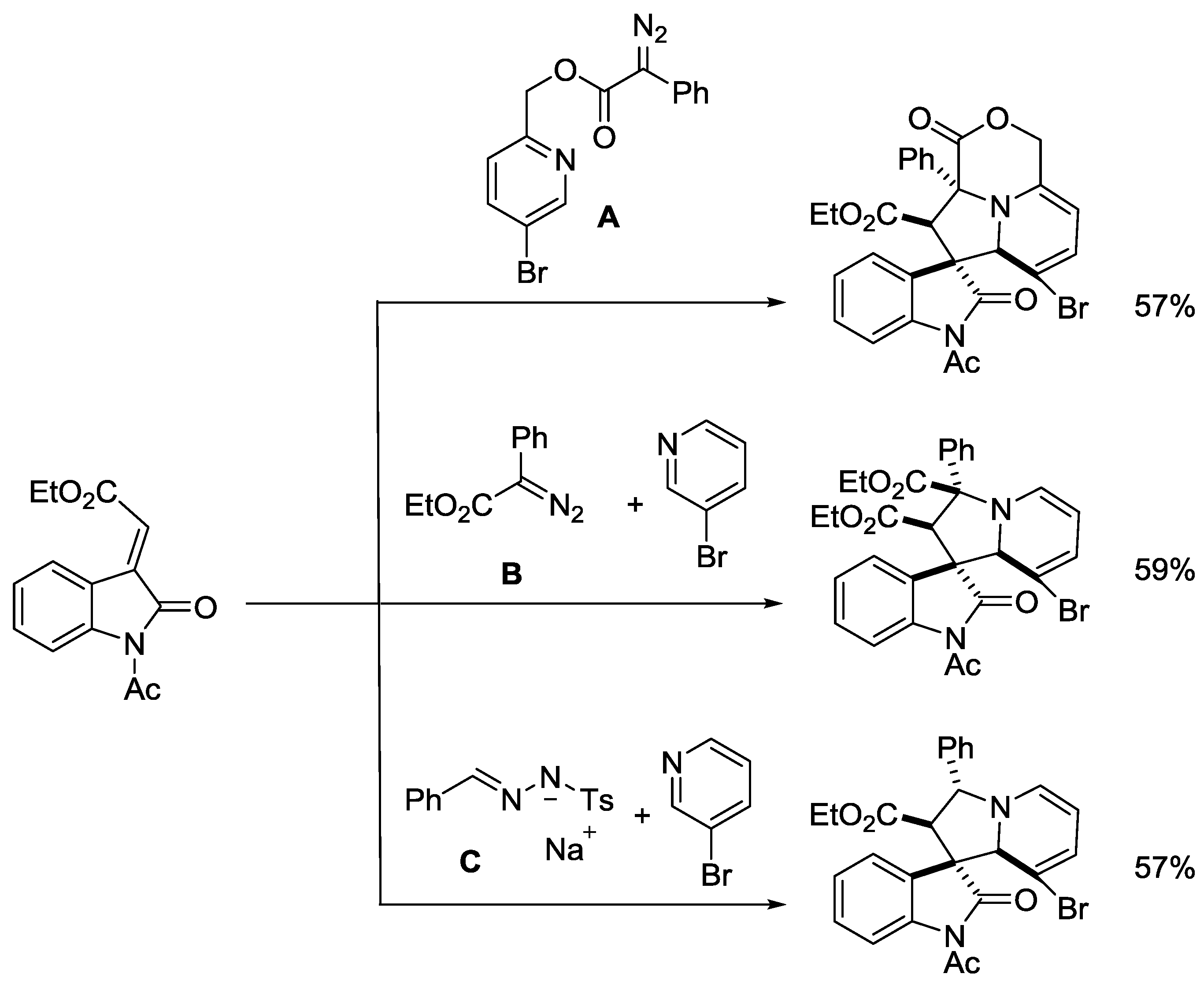{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Transient N-Metal Activation
3. N-Acylpyridinium Salts and N-Sulfonylpyridinium Salts
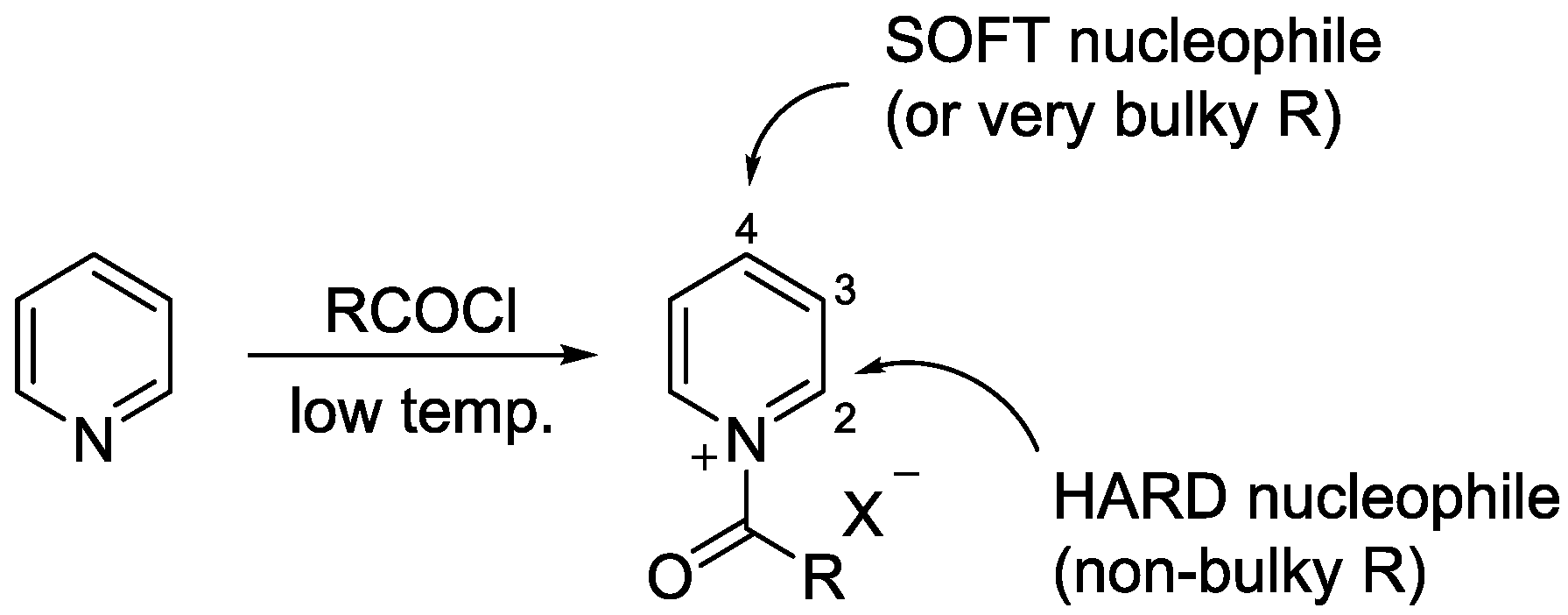
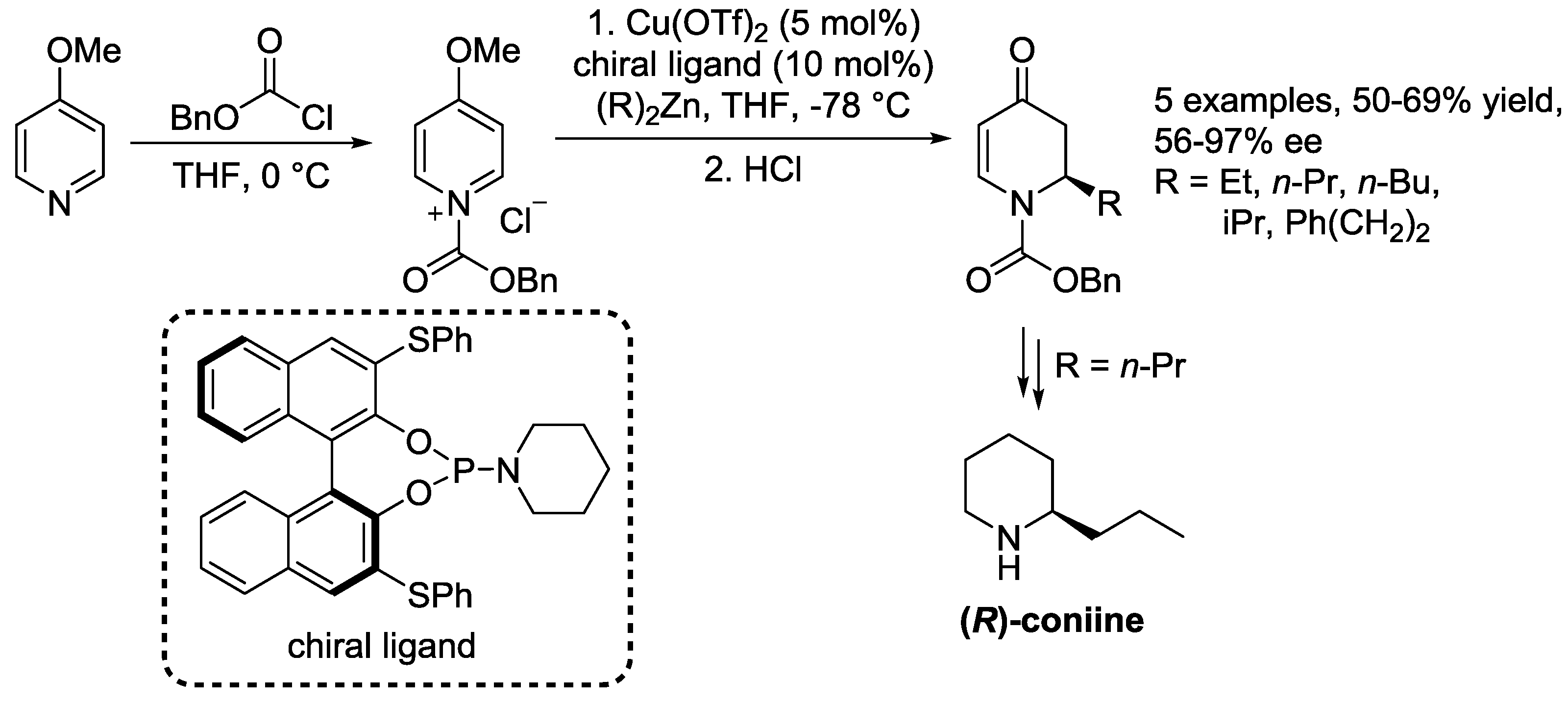
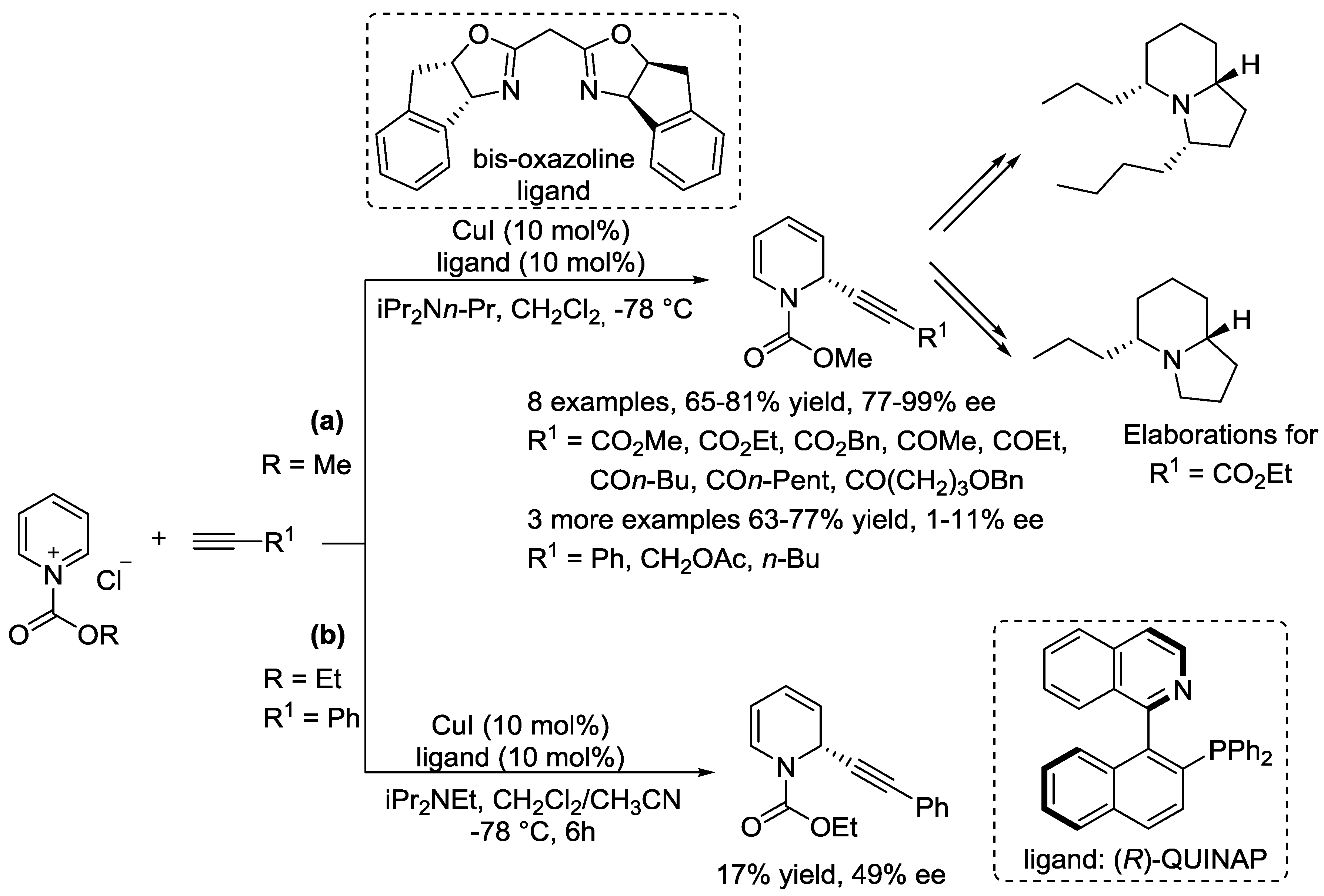
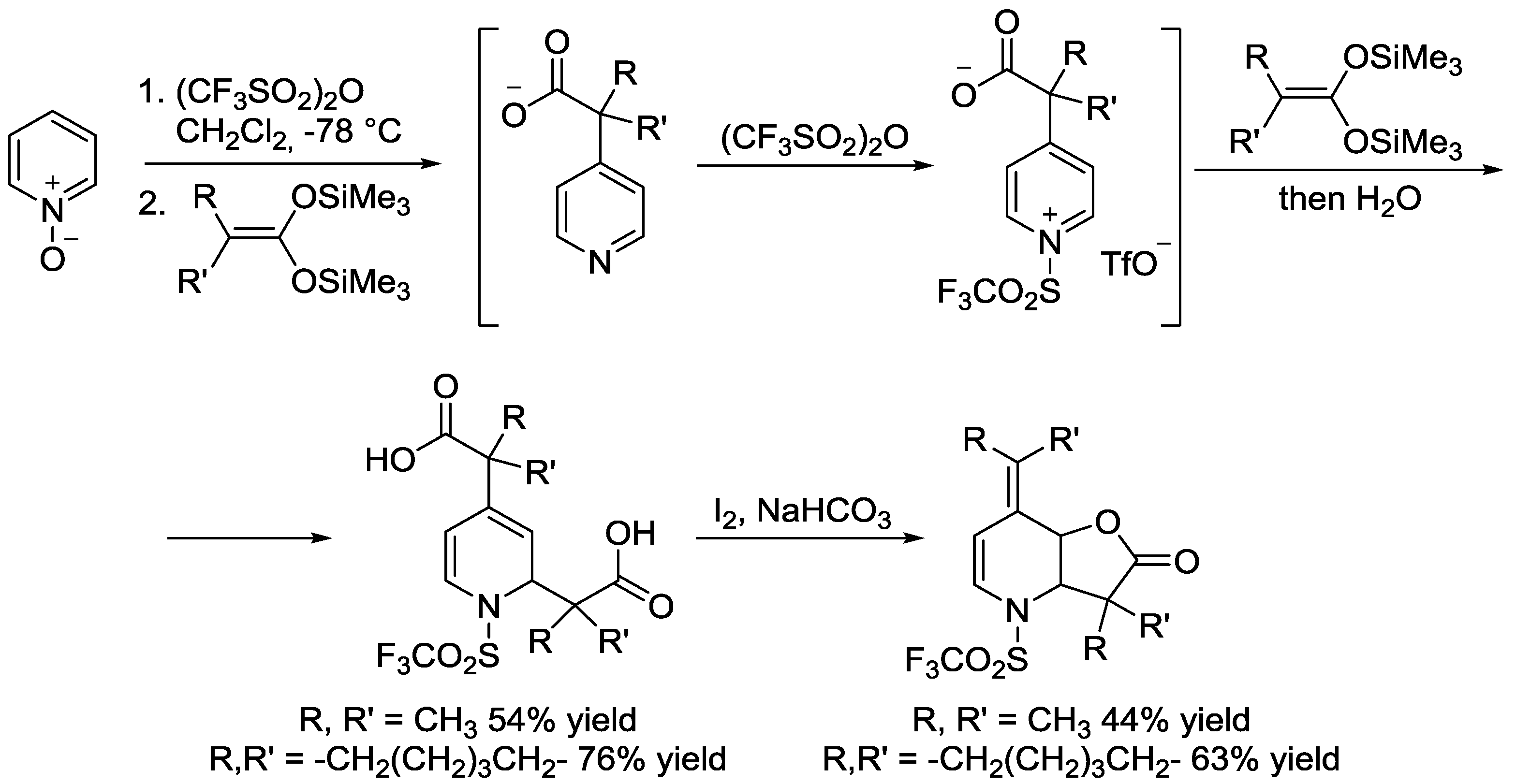
3.1. N-Acylpyridinium Salts
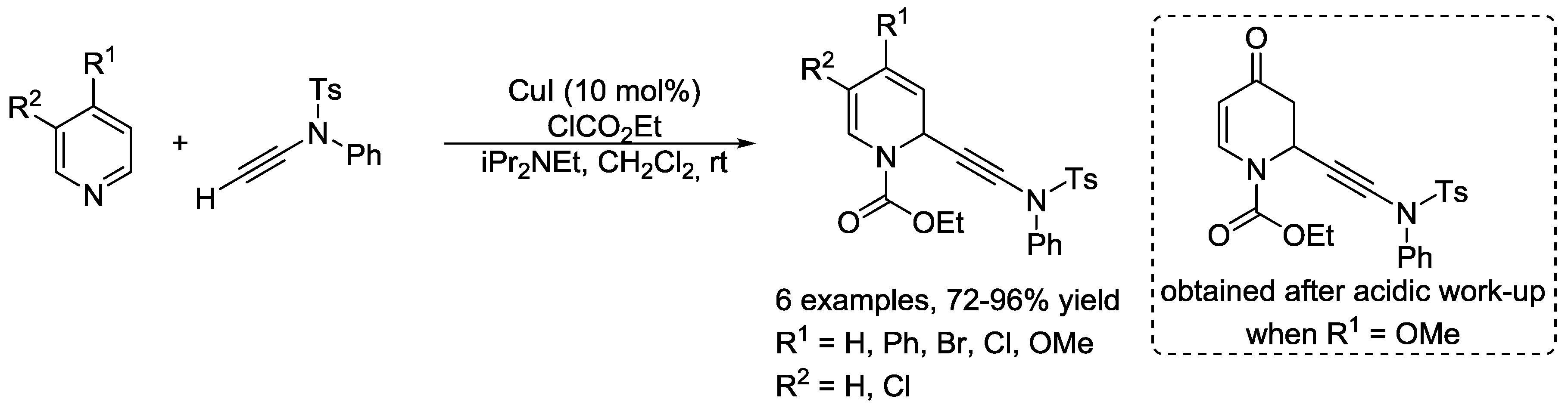
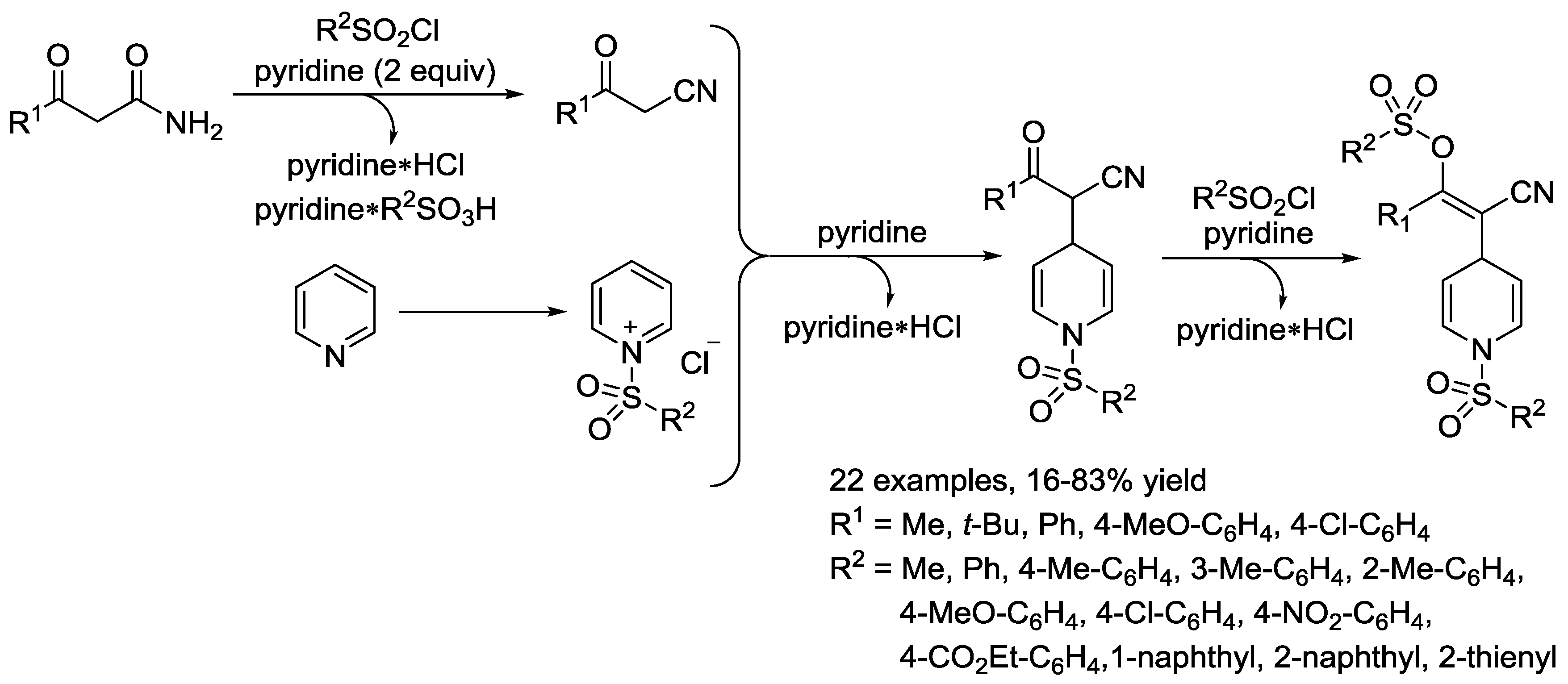
3.2. N-Sulfonylpyridinium Salts
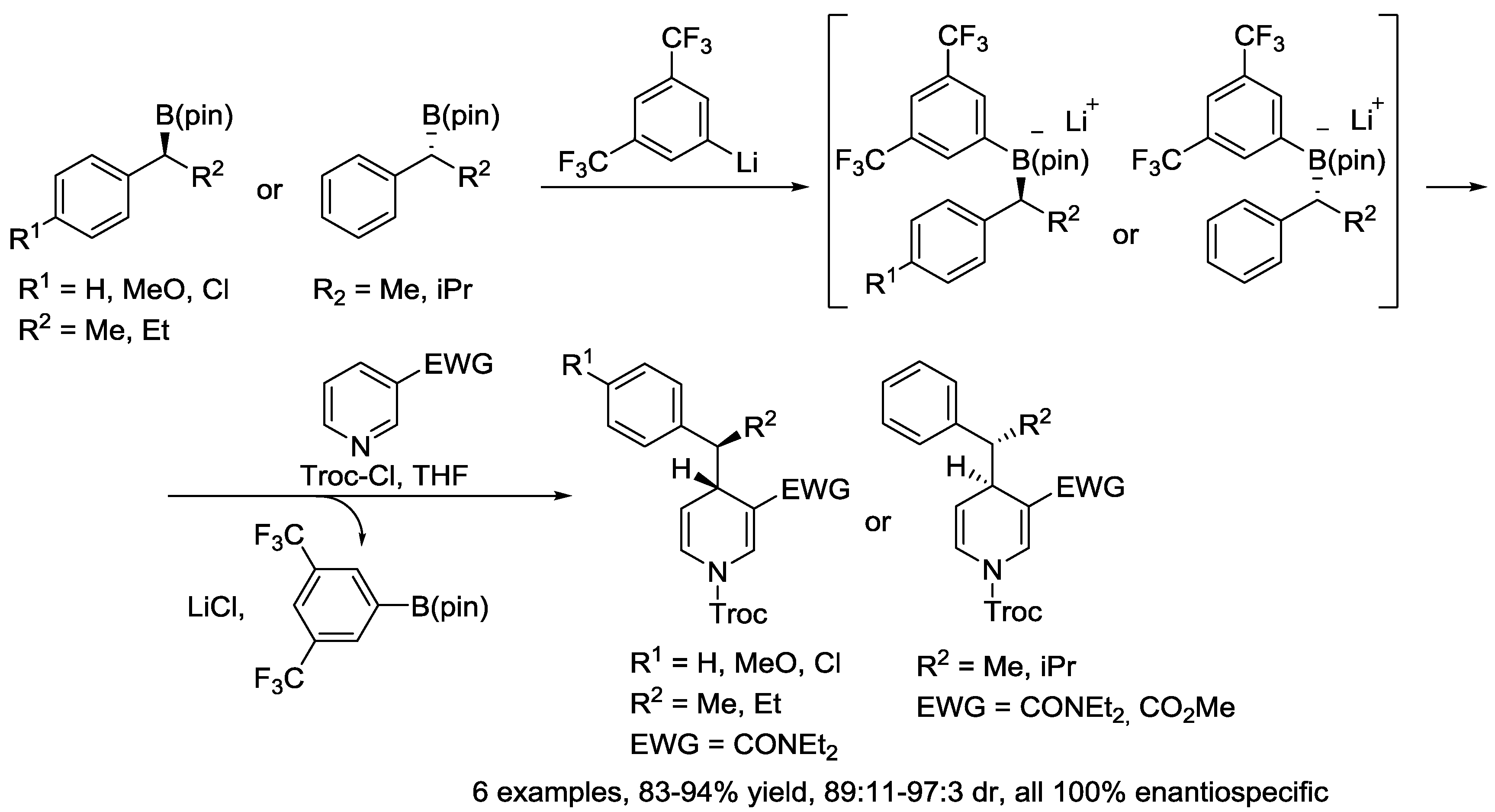
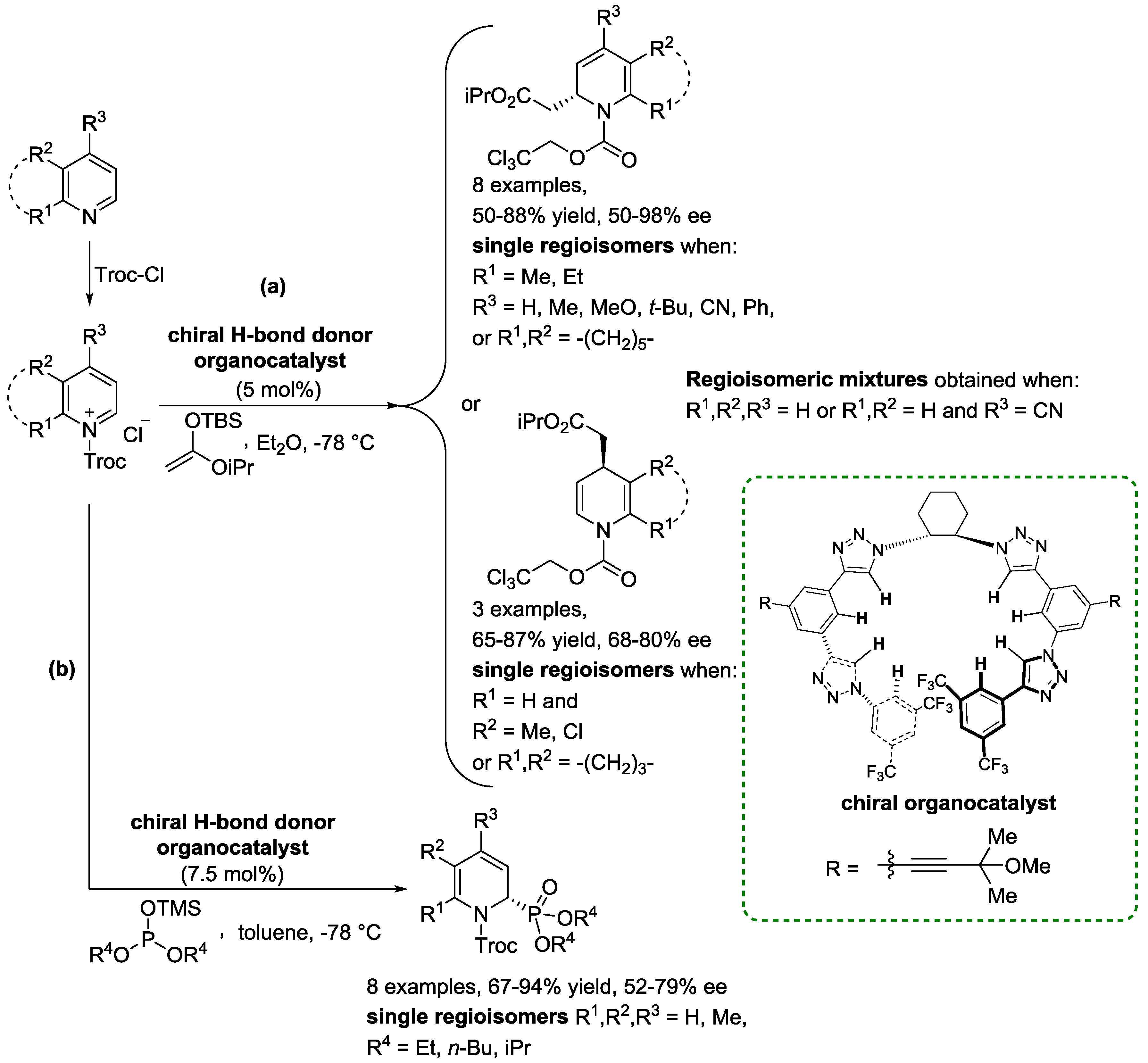
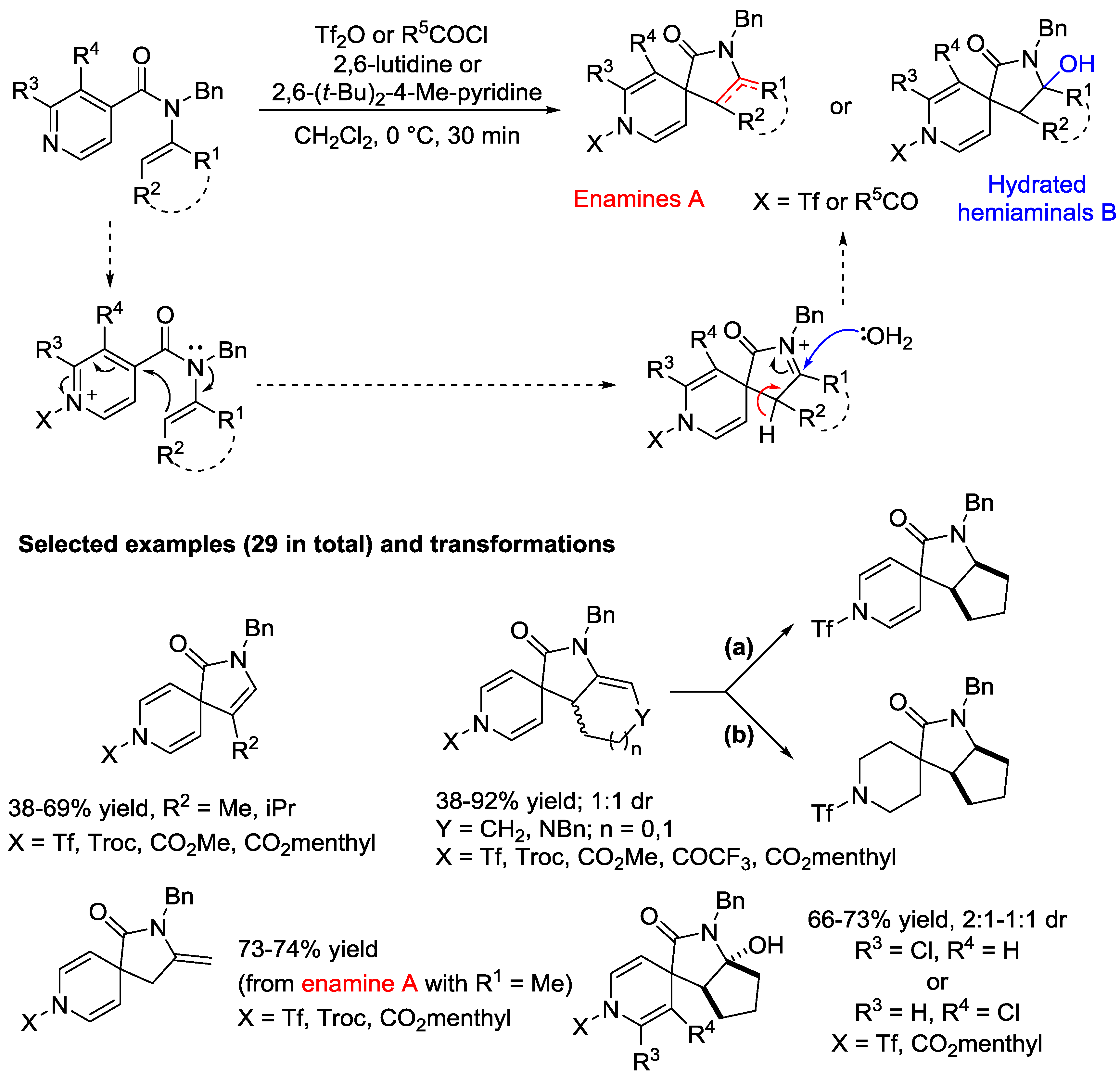
4. N-Alkylpyridinium Salts
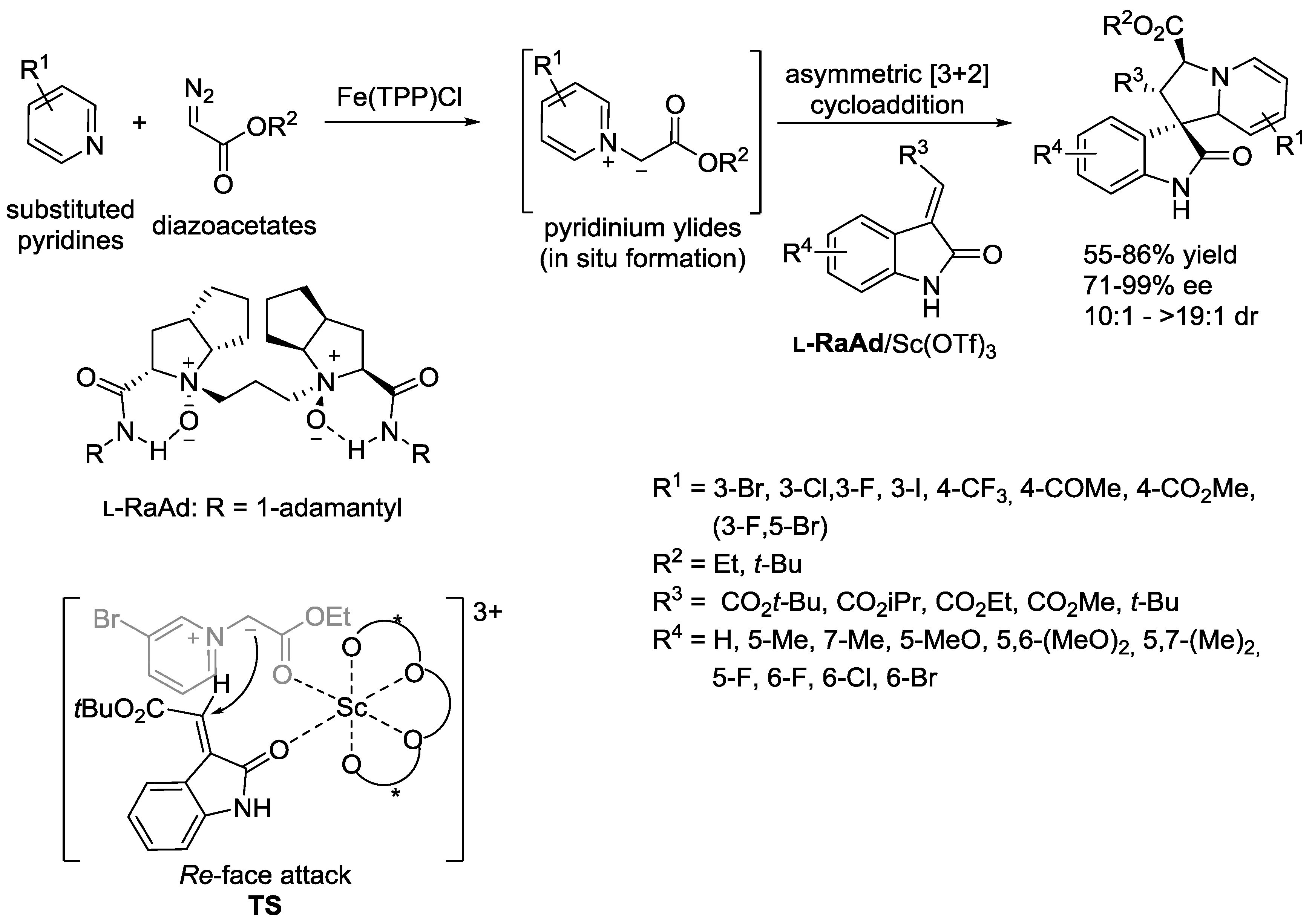
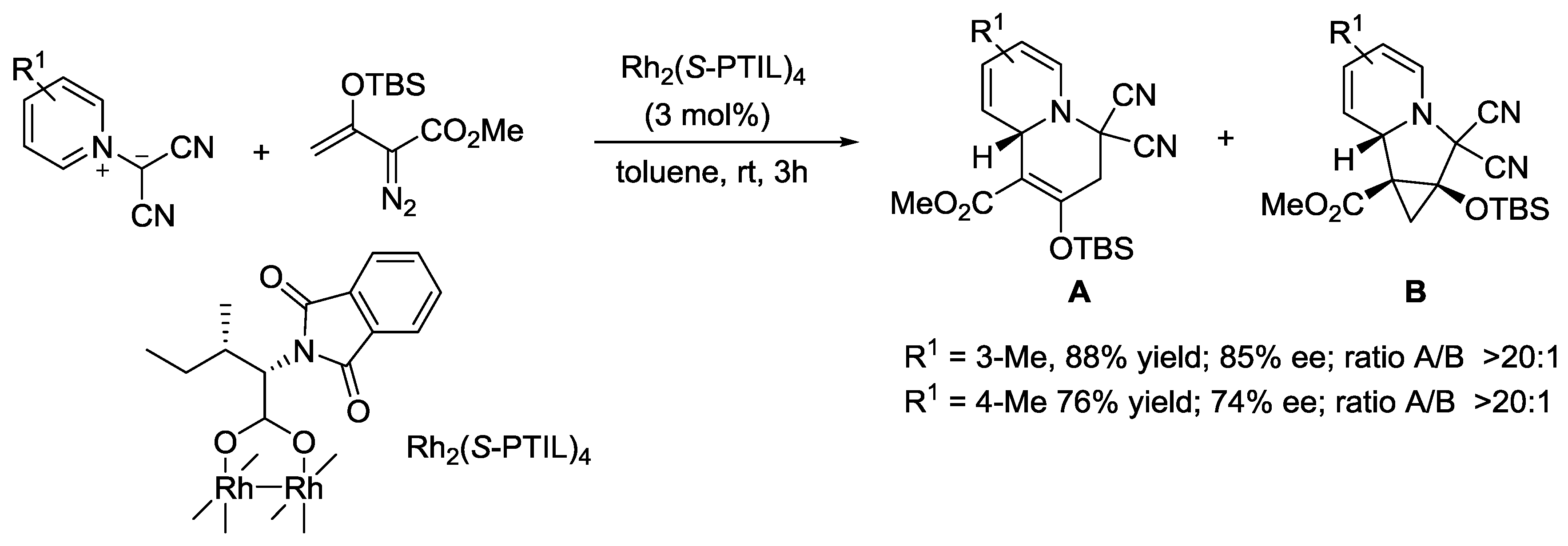
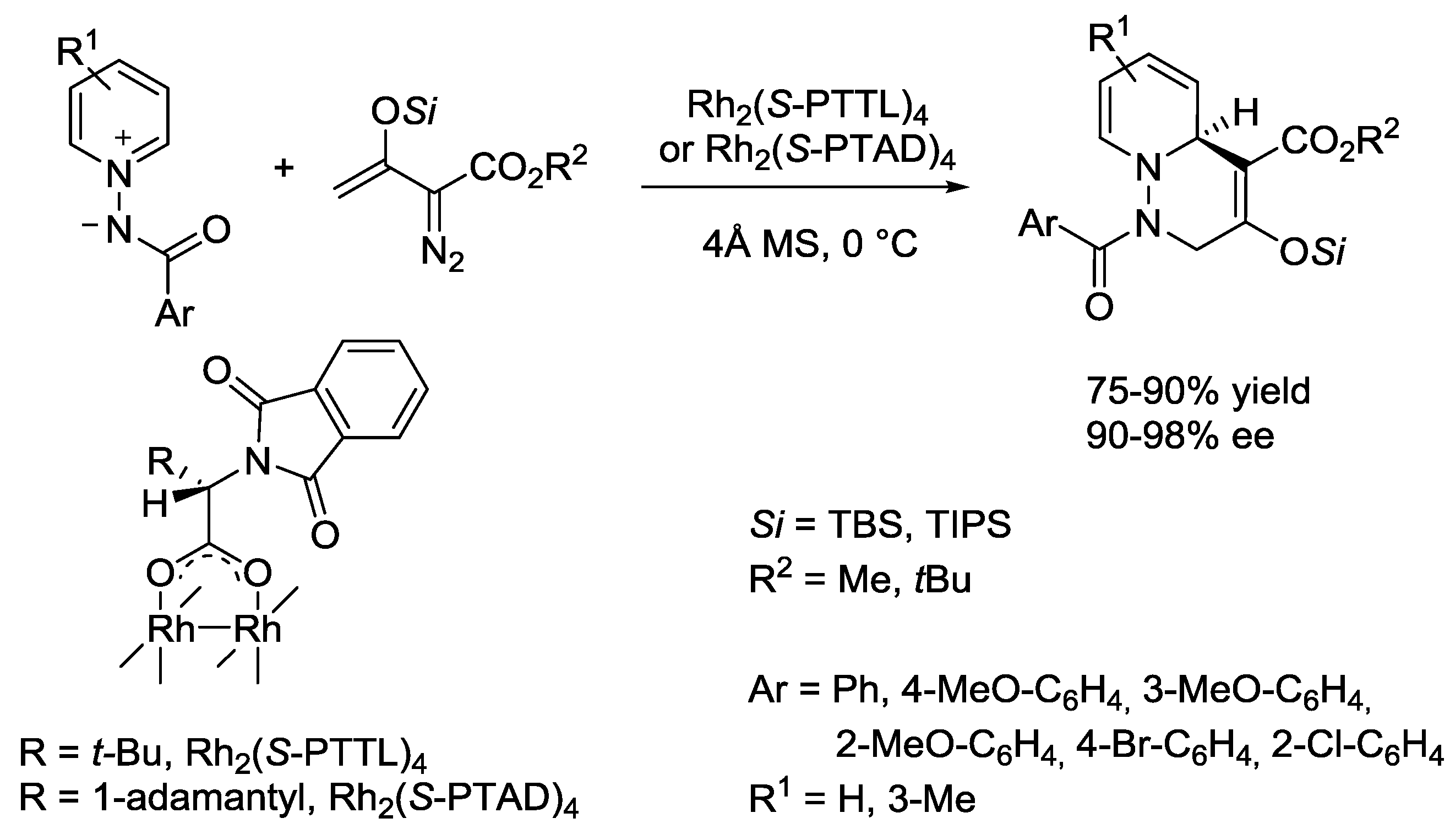
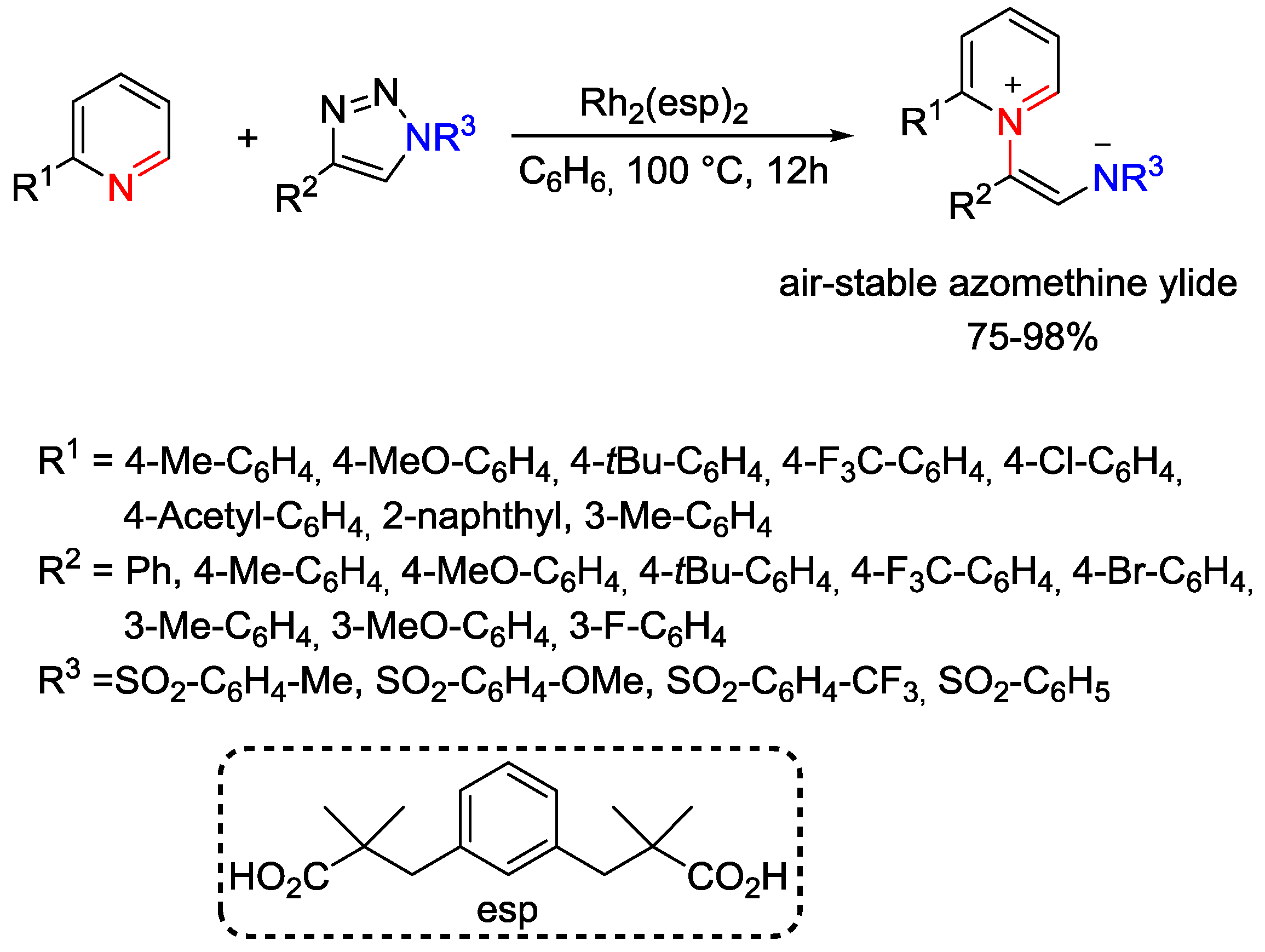
5. Pyridinium Ylides
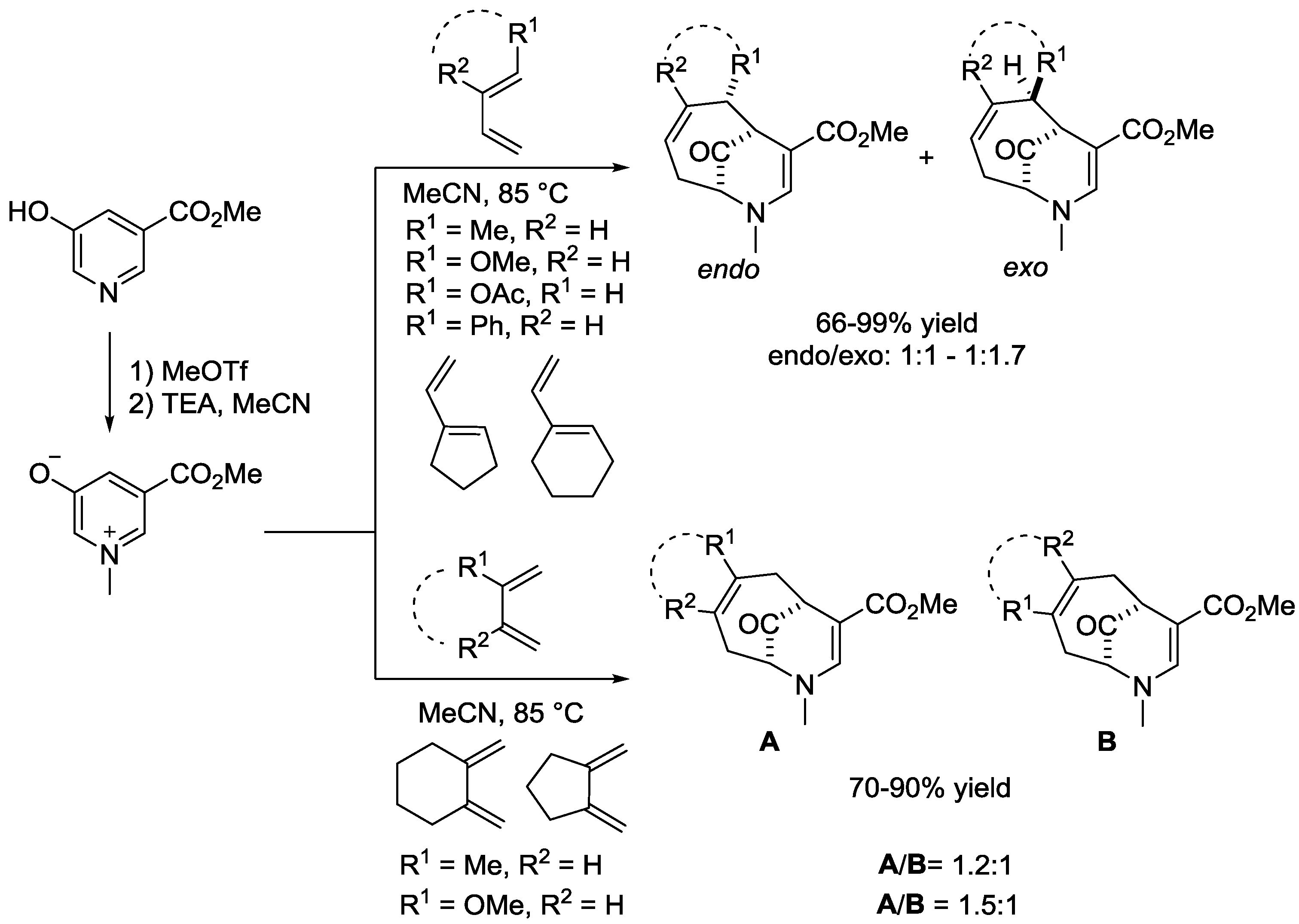
6. Pyridinium Zwitterions
7. Conclusions
Funding
Conflicts of Interest
References
- Sharma, V.K.; Singh, S.K. Synthesis, utility and medicinal importance of 1,2- & 1,4-dihydropyridines. RSC Adv. 2017, 7, 2682–2732. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Hantzsch, A. Ueber die Synthese pyridinartiger Verbindungen aus Acetessigäther und Aldehydammoniak. Justus Liebigs Ann. Chem. 1882, 215, 1–82. [Google Scholar] [CrossRef]
- Colby, D.A.; Bergman, R.G.; Ellman, J.A. Synthesis of Dihydropyridines and Pyridines from Imines and Alkynes via C−H Activation. J. Am. Chem. Soc. 2008, 130, 3645–3651. [Google Scholar] [CrossRef] [PubMed]
- Noole, A.; Borissova, M.; Lopp, M.; Kanger, T. Enantioselective Organocatalytic Aza-Ene-Type Domino Reaction Leading to 1,4-Dihydropyridines. J. Org. Chem. 2011, 76, 1538–1545. [Google Scholar] [CrossRef] [PubMed]
- Ţînţaş, M.-L.; Azzouz, R.; Peauger, L.; Gembus, V.; Petit, E.; Bailly, L.; Papamicaël, C.; Levacher, V. Access to Highly Enantioenriched Donepezil-like 1,4-Dihydropyridines as Promising Anti-Alzheimer Prodrug Candidates via Enantioselective Tsuji Allylation and Organocatalytic Aza-Ene-Type Domino Reactions. J. Org. Chem. 2018, 83, 10231–10240. [Google Scholar] [CrossRef] [PubMed]
- Eisner, U.; Kuthan, J. Chemistry of dihydropyridines. Chem. Rev. 1972, 72, 1–42. [Google Scholar] [CrossRef]
- Lavilla, R.J. Recent developments in the chemistry of dihydropyridines. J. Chem. Soc. Perkin Trans. 2002, 1, 1141–1156. [Google Scholar] [CrossRef]
- Stout, D.M.; Meyers, A.I. Recent advances in the chemistry of dihydropyridines. Chem. Rev. 1982, 82, 223–243. [Google Scholar] [CrossRef]
- Bull, J.A.; Mousseau, J.J.; Pelletier, G.; Charette, A.B. Synthesis of Pyridine and Dihydropyridine Derivatives by Regio- and Stereoselective Addition to N-Activated Pyridines. Chem. Rev. 2012, 112, 2642–2713. [Google Scholar] [CrossRef]
- Silva, E.M.P.; Varandas, P.A.M.M.; Silva, A.M.S. Developments in the Synthesis of 1,2-Dihydropyridines. Synthesis 2013, 45, 3053–3089. [Google Scholar] [CrossRef]
- Balakrishna, B.; Nuñez-Rico, J.L.; Vidal-Ferran, A. Substrate Activation in the Catalytic Asymmetric Hydrogenation of N-Heteroarenes. Eur. J. Org. Chem. 2015, 2015, 5293–5303. [Google Scholar] [CrossRef]
- Zheng, C.; You, S.-L. Transfer hydrogenation with Hantzsch esters and related organic hydride donors. Chem. Soc. Rev. 2012, 41, 2498–2518. [Google Scholar] [CrossRef] [PubMed]
- Londregan, A.T.; Jennings, S.; Wei, L. Mild Addition of Nucleophiles to Pyridine-N-Oxides. Org. Lett. 2011, 13, 1840–1843. [Google Scholar] [CrossRef] [PubMed]
- Bertuzzi, G.; Pecorari, D.; Bernardi, L.; Fochi, M. An organocatalytic enantioselective direct α-heteroarylation of aldehydes with isoquinoline N-oxides. Chem. Commun. 2018, 54, 3977–3980. [Google Scholar] [CrossRef] [PubMed]
- Kakehi, A. Reactions of Pyridinium N-Ylides and Their Related Pyridinium Salts. Heterocyles 2012, 85, 1529–1577. [Google Scholar] [CrossRef]
- Yang, Z.-P.; Wu, Q.-F.; You, S.-L. Direct Asymmetric Dearomatization of Pyridines and Pyrazines by Iridium-Catalyzed Allylic Amination Reactions. Angew. Chem. Int. Ed. 2014, 53, 6986–6989. [Google Scholar] [CrossRef]
- Yang, Z.-P.; Wu, Q.-F.; Shao, W.; You, S.-L. Iridium-Catalyzed Intramolecular Asymmetric Allylic Dearomatization Reaction of Pyridines, Pyrazines, Quinolines, and Isoquinolines. J. Am. Chem. Soc. 2015, 137, 15899–15906. [Google Scholar] [CrossRef]
- Joshi, M.S.; Pigge, F.C. Sequential Pyridine Dearomatization–Mizoroki–Heck Cyclization for the Construction of Fused (Dihydropyrido)isoindolinone Ring Systems. Synthesis 2018, 50. [Google Scholar] [CrossRef]
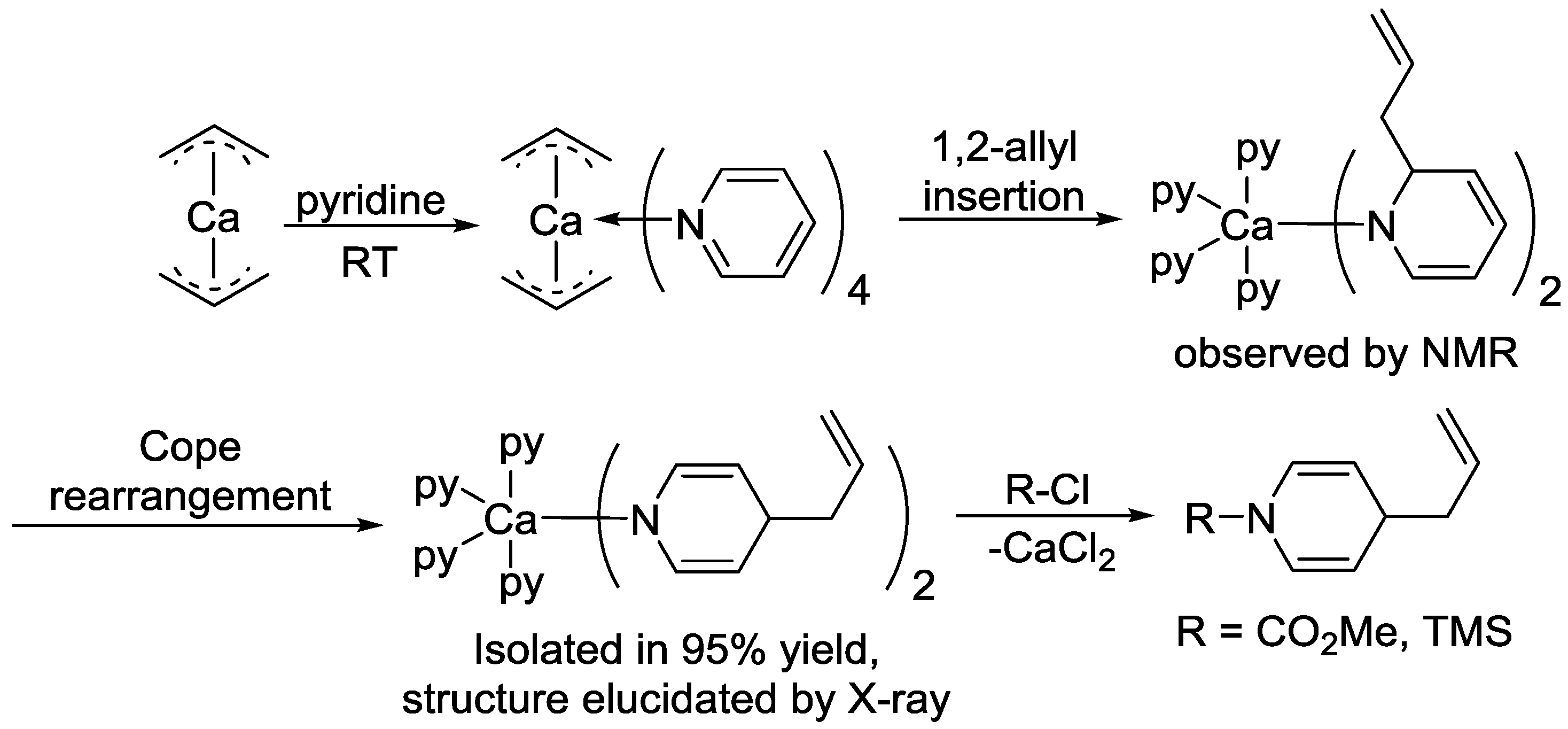
- Jochmann, P.; Dols, T.S.; Spaniol, T.P.; Perrin, L.; Maron, L.; Okuda, J. Insertion of Pyridine into the Calcium Allyl Bond: Regioselective 1,4-Dihydropyridine Formation and CH Bond Activation. Angew. Chem. Int. Ed. 2010, 49, 7795–7798. [Google Scholar] [CrossRef]
- Hao, L.; Harrod, J.F.; Lebuis, A.-M.; Mu, Y.; Shu, R.; Samuel, E.; Woo, H.-G. Homogeneous Catalytic Hydrosilylation of Pyridines. Angew. Chem. Int. Ed. 1998, 37, 3126–3129. [Google Scholar] [CrossRef]
- Gutsulyak, D.V.; van der Est, A.; Nikonov, G.I. Facile Catalytic Hydrosilylation of Pyridines. Angew. Chem. Int. Ed. 2011, 50, 1384–1387. [Google Scholar] [CrossRef] [PubMed]
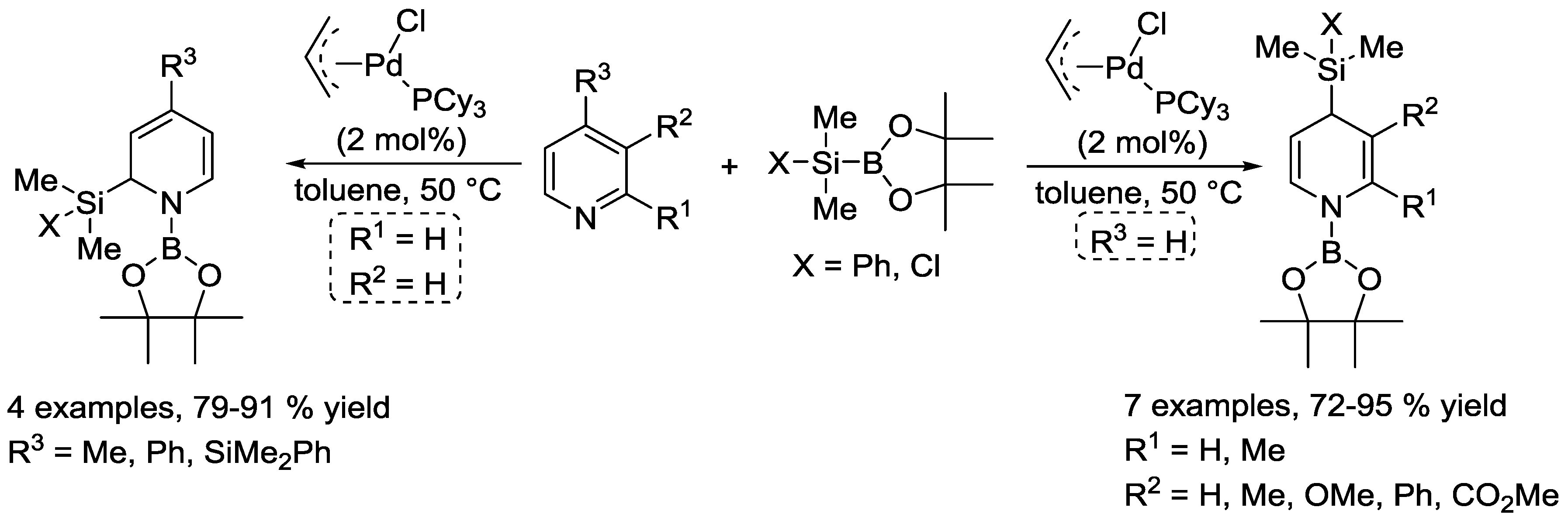
- Oshima, K.; Ohmura, T.; Suginome, M. Palladium-Catalyzed Regioselective Silaboration of Pyridines Leading to the Synthesis of Silylated Dihydropyridines. J. Am. Chem. Soc. 2011, 133, 7324–7327. [Google Scholar] [CrossRef] [PubMed]
- Gribble, M.W., Jr.; Guo, S.; Buchwald, S.L. Asymmetric Cu-Catalyzed 1,4-Dearomatization of Pyridines and Pyridazines without Preactivation of the Heterocycle or Nucleophile. J. Am. Chem. Soc. 2018, 140, 5057–5060. [Google Scholar] [CrossRef] [PubMed]
- Comins, D.-L.; Abdullah, A.H. Regioselective addition of Grignard reagents to 1-acylpyridinium salts. A convenient method for the synthesis of 4-alkyl(aryl)pyridines. J. Org. Chem. 1982, 47, 4315–4319. [Google Scholar] [CrossRef]
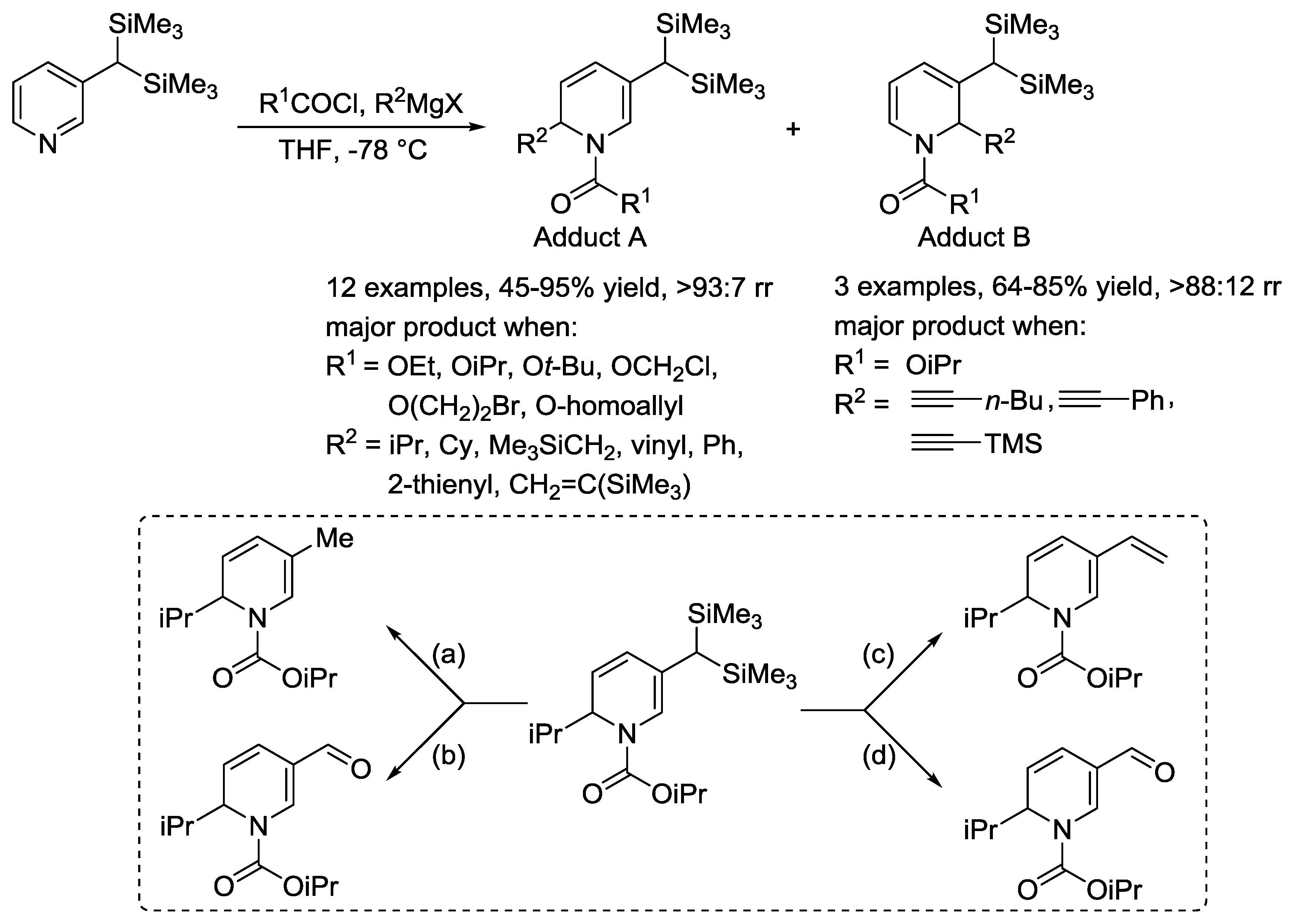
- Hoesl, C.E.; Pabel, J.; Polborn, K.; Wanner, K.T. Synthesis of Sterically Demanding 3-Silylpyridines and Their Use in Asymmetric Synthesis with Chiral N-Acyliminium Ions. Heterocycles 2002, 58, 383–392. [Google Scholar] [CrossRef]
- Wu, Y.; Li, L.; Li, H.; Gao, L.; Xie, H.; Zhang, Z.; Su, Z.; Hu, C.; Song, Z. Regioselective Nucleophilic Addition of Organometallic Reagents to 3-Geminal Bis(silyl) N-Acyl Pyridinium. Org. Lett. 2014, 16, 1880–1883. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ibáñez, M.Á.; Maciá, B.; Pizzuti, M.G.; Minnaard, A.J.; Feringa, B.L. Catalytic Enantioselective Addition of Dialkylzinc Reagents to N-Acylpyridinium Salts. Angew. Chem. Int. Ed. 2009, 48, 9339–9341. [Google Scholar] [CrossRef]
- Chau, S.T.; Lutz, J.P.; Wu, K.; Doyle, A.G. Nickel-Catalyzed Enantioselective Arylation of Pyridinium Ions: Harnessing an Iminium Ion Activation Mode. Angew. Chem. Int. Ed. 2013, 52, 9153–9156. [Google Scholar] [CrossRef]
- Lutz, J.P.; Chau, S.T.; Doyle, A.G. Nickel-catalyzed enantioselective arylation of pyridine. Chem. Sci. 2016, 7, 4105–4109. [Google Scholar] [CrossRef]
- Sun, Z.; Yu, S.; Ding, Z.; Ma, D. Enantioselective Addition of Activated Terminal Alkynes to 1-Acylpyridinium Salts Catalyzed by Cu−Bis(oxazoline) Complexes. J. Am. Chem. Soc. 2007, 129, 9300–9301. [Google Scholar] [CrossRef] [PubMed]
- Black, A.D.; Beveridge, R.E.; Arndtsen, B.A. Copper-Catalyzed Coupling of Pyridines and Quinolines with Alkynes: A One-Step, Asymmetric Route to Functionalized Heterocycles. J. Org. Chem. 2008, 73, 1906–1910. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Cook, A.M.; Liu, Y.; Wolf, C. Copper(I)-Catalyzed Nucleophilic Addition of Ynamides to Acyl Chlorides and Activated N-Heterocycles. J. Org. Chem. 2014, 79, 4167–4173. [Google Scholar] [CrossRef] [PubMed]
- Mohiti, M.; Rampalakos, C.; Feeney, K.; Leonori, D.; Aggarwal, V.K. Asymmetric addition of chiral boron-ate complexes to cyclic iminium ions. Chem. Sci. 2014, 5, 602–607. [Google Scholar] [CrossRef]
- García Mancheño, O.; Asmus, S.; Zurro, M.; Fischer, T. Highly Enantioselective Nucleophilic Dearomatization of Pyridines by Anion-Binding Catalysis. Angew. Chem. Int. Ed. 2015, 54, 8823–8827. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Tokunaga, N.; Jacobsen, E.N. Enantioselective Thiourea-Catalyzed Acyl-Mannich Reactions of Isoquinolines. Angew. Chem. Int. Ed. 2005, 44, 6700–6704. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y.; Miyabe, H.; Takemoto, Y. Catalytic Enantioselective Petasis-Type Reaction of Quinolines Catalyzed by a Newly Designed Thiourea Catalyst. J. Am. Chem. Soc. 2007, 129, 6686–6687. [Google Scholar] [CrossRef] [PubMed]
- Zurro, M.; Asmus, S.; Beckendorf, S.; Mück-Lichtenfeld, C.; García Mancheño, O. Chiral Helical Oligotriazoles: New Class of Anion-Binding Catalysts for the Asymmetric Dearomatization of Electron-Deficient N-Heteroarenes. J. Am. Chem. Soc. 2014, 136, 13999–14002. [Google Scholar] [CrossRef]
- Schafer, A.G.; Wieting, J.M.; Fisher, T.J.; Mattson, A.E. Chiral Silanediols in Anion-Binding Catalysis. Angew. Chem. Int. Ed. 2013, 52, 11321–11324. [Google Scholar] [CrossRef]
- Frisch, K.; Landa, A.; Saaby, S.; Jørgensen, K.A. Organocatalytic Diastereo- and Enantioselective Annulation Reactions—Construction of Optically Active 1,2-Dihydroisoquinoline and 1,2-Dihydrophthalazine Derivatives. Angew. Chem. Int. Ed. 2005, 44, 6058–6063. [Google Scholar] [CrossRef]
- Mengozzi, L.; Gualandi, A.; Cozzi, P.G. A highly enantioselective acyl-Mannich reaction of isoquinolines with aldehydes promoted by proline derivatives: An approach to 13-alkyl-tetrahydroprotoberberine alkaloids. Chem. Sci. 2014, 5, 3915–3921. [Google Scholar] [CrossRef]
- Berti, F.; Malossi, F.; Marchetti, F.; Pineschi, M. A highly enantioselective Mannich reaction of aldehydes with cyclic N-acyliminium ions by synergistic catalysis. Chem. Commun. 2015, 51, 13694–13697. [Google Scholar] [CrossRef]
- Volla, C.M.R.; Fava, E.; Atodiresei, I.; Rueping, M. Dual metal and Lewis base catalysis approach for asymmetric synthesis of dihydroquinolines and the α-arylation of aldehydes via N-acyliminium ions. Chem. Commun. 2015, 51, 15788–15791. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Mao, Y.; Lou, H.; Liu, L. Copper(II)/amine synergistically catalyzed enantioselective alkylation of cyclic N-acyl hemiaminals with aldehydes. Chem. Commun. 2015, 51, 10691–10694. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Moquist, P.M.; Schaus, S.E. Enantioselective Boronate Additions to N-Acyl Quinoliniums Catalyzed by Tartaric Acid. Org. Lett. 2011, 13, 6316–6319. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.; Duong, Q.-N.; García Mancheño, O. Triazole-Based Anion-Binding Catalysis for the Enantioselective Dearomatization of N-Heteroarenes with Phosphorus Nucleophiles. Chem. Eur. J. 2017, 23, 5983–5987. [Google Scholar] [CrossRef] [PubMed]
- Gualo-Soberanes, N.; Ortega-Alfaro, M.C.; López-Cortés, J.G.; Toscano, R.A.; Rudler, H.; Álvarez-Toledano, C. An expedient approach to tetrahydrofuro[3,2-b]pyridine-2(3H)-ones via activation of pyridine N-oxide by triflic anhydride. Tetrahedron Lett. 2010, 51, 3186–3189. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, T.-X.; Ma, N.; Zhang, Z.; Zhang, G. Multicomponent reaction of pyridine, acetoacetamide/benzoylacetamides and sulfuryl chlorides: Regioselective construction of 4-olefinated dihydropyridines. Tetrahedron Lett. 2016, 72, 4938–4946. [Google Scholar] [CrossRef]
- Senczyszyn, J.; Brice, H.; Clayden, J. Spirocyclic Dihydropyridines by Electrophile-Induced Dearomatizing Cyclization of N-Alkenyl Pyridinecarboxamides. Org. Lett. 2013, 15, 1922–1925. [Google Scholar] [CrossRef]
- Arnott, G.; Brice, H.; Clayden, J.; Blaney, E. Electrophile-Induced Dearomatizing Spirocyclization of N-Arylisonicotinamides: A Route to Spirocyclic Piperidines. Org. Lett. 2008, 10, 3089–3092. [Google Scholar] [CrossRef]
- Brice, H.; Clayden, J. Doubly dearomatising intramolecular coupling of a nucleophilic and an electrophilicheterocycle. Chem. Commun. 2009, 1964–1966. [Google Scholar] [CrossRef] [PubMed]
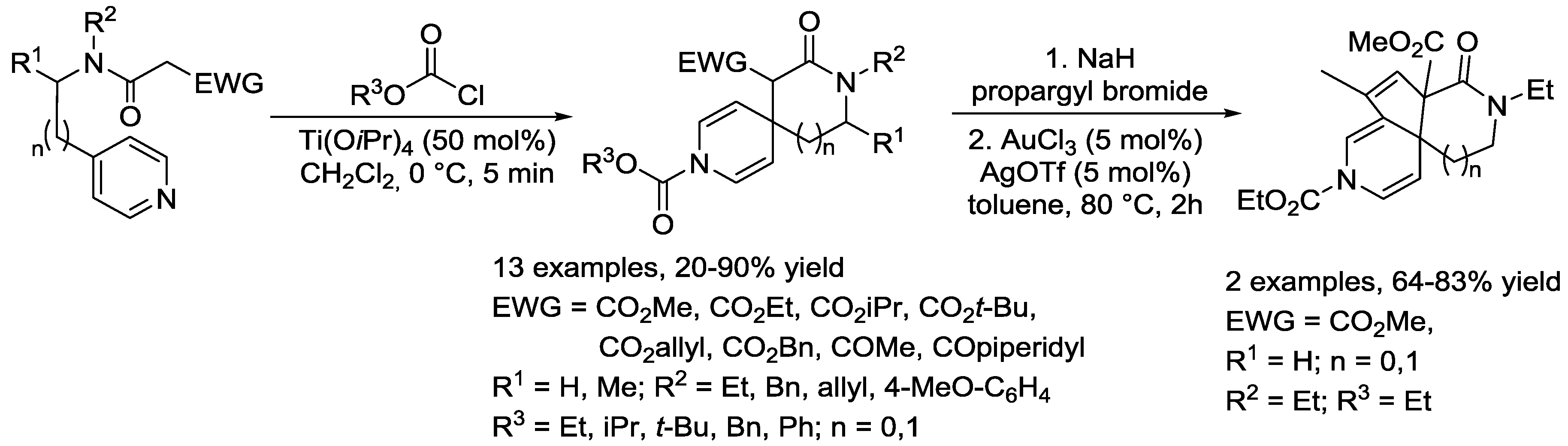
- Parameswarappa, S.G.; Pigge, F.C. Titanium-Mediated Spirocyclization Reactions of 4-Alkylpyridines. Org. Lett. 2010, 12, 3434–3437. [Google Scholar] [CrossRef] [PubMed]
- Parameswarappa, S.G.; Pigge, F.C. Synthesis of substituted 3,9-diazaspiro[5.5]undecanes via spirocyclization of pyridine substrates. Tetrahedron Lett. 2011, 52, 4357–4359. [Google Scholar] [CrossRef]
- Parameswarappa, S.G.; Pigge, F.C. Intramolecular Cyclization Manifolds of 4-Alkylpyridines Bearing Ambiphilic Side Chains: Construction of Spirodihydropyridines or Benzylic Cyclization via Anhydrobase Intermediates. J. Org. Chem. 2012, 77, 8038–8048. [Google Scholar] [CrossRef] [PubMed]
- Wenkert, E.; Reynolds, G.D. Vallesiachotamine Models. Synth. Commun. 1973, 3, 241–243. [Google Scholar] [CrossRef]
- Wang, S.-G.; Xia, Z.-L.; Xu, R.-Q.; Liu, X.-J.; Zheng, C.; You, S.-L. Construction of Chiral Tetrahydro-β-Carbolines: Asymmetric Pictet-Spengler Reaction of Indolyl Dihydropyridines. Angew. Chem. Int. Ed. 2017, 56, 7440–7443. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Ni, C.; Zhao, Y.; Zhang, W.; Dilman, A.D.; Hu, J. Nucleophilic difluoromethylation of C=N bonds in heterocycles with difluoromethyl silane reagents. Tetrahedron Lett. 2011, 68, 5137–5144. [Google Scholar] [CrossRef]
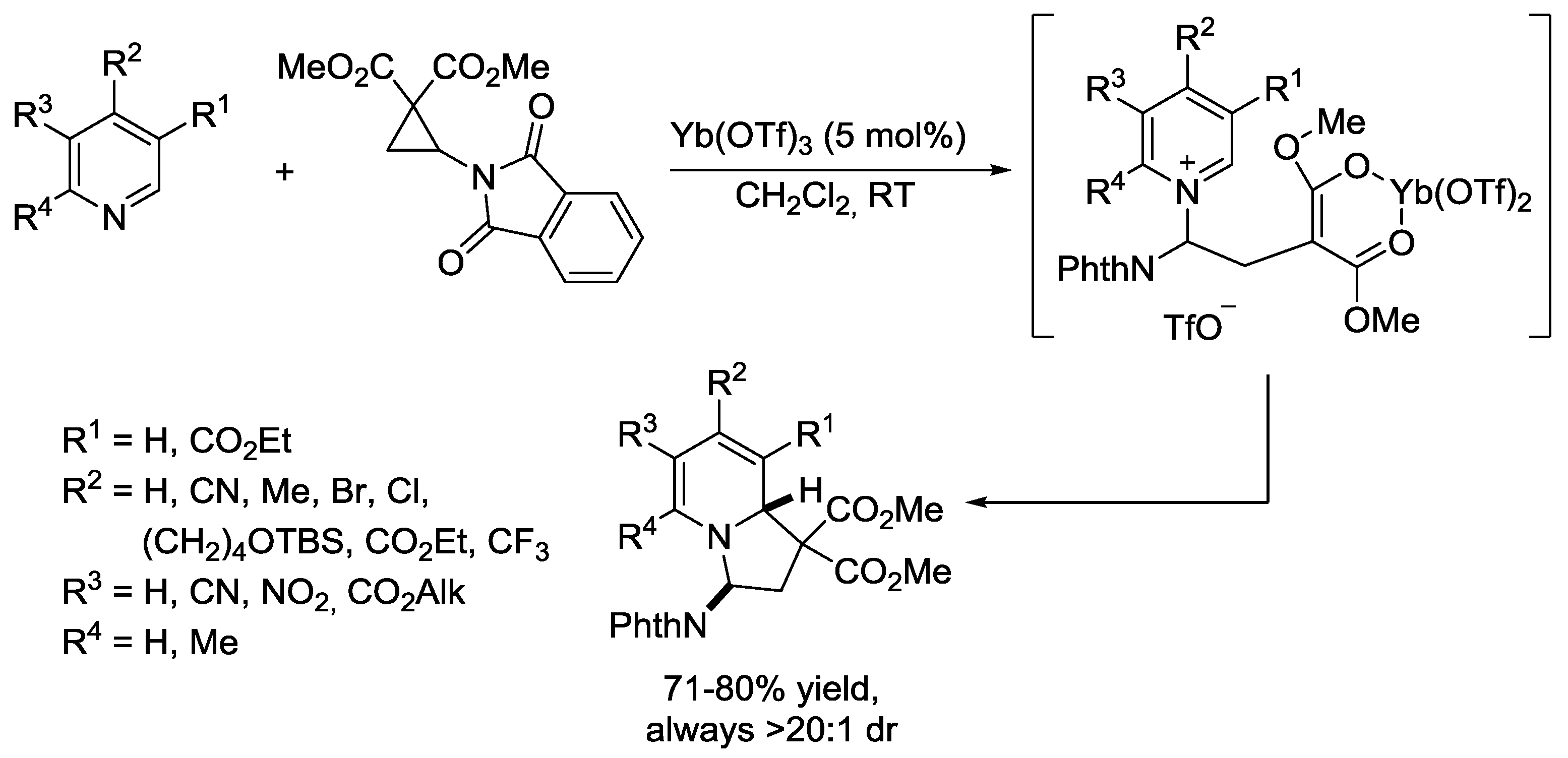
- Preindl, J.; Chakrabarty, S.; Waser, J. Dearomatization of electron poor six-membered N-heterocycles through [3+2] annulation with aminocyclopropanes. Chem. Sci. 2017, 8, 7112–7118. [Google Scholar] [CrossRef]
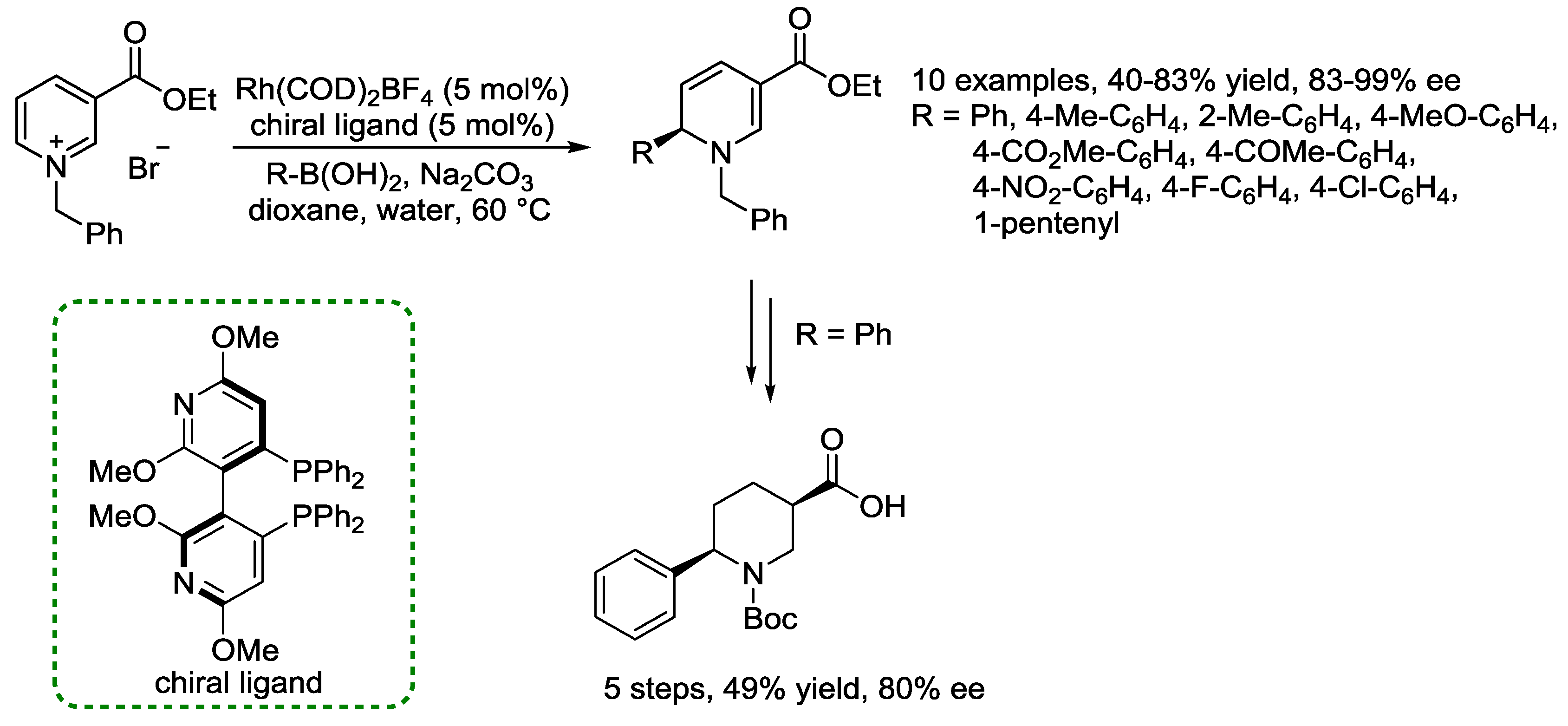
- Christian, N.; Aly, S.; Belyk, K. Rhodium-Catalyzed Enantioselective Addition of Boronic Acids to N-Benzylnicotinate Salts. J. Am. Chem. Soc. 2011, 133, 2878–2880. [Google Scholar] [CrossRef]
- Bertuzzi, G.; Sinisi, A.; Caruana, L.; Mazzanti, A.; Fochi, M.; Bernardi, L. Catalytic Enantioselective Addition of Indoles to Activated N-Benzylpyridinium Salts: Nucleophilic Dearomatization of Pyridines with Unusual C-4 Regioselectivity. ACS Catal. 2016, 6, 6473–6477. [Google Scholar] [CrossRef]
- Bertuzzi, G.; Sinisi, A.; Pecorari, D.; Mazzanti, A.; Bernardi, L.; Fochi, M. Nucleophilic Dearomatization of Pyridines under Enamine Catalysis: Regio-, Diastereo-, and Enantioselective Addition of Aldehydes to Activated N-Alkylpyridinium Salts. Org. Lett. 2017, 19, 834–837. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Gotoh, H.; Hayashi, T.; Shoji, M. Diphenylprolinol Silyl Ethers as Efficient Organocatalysts for the Asymmetric Michael Reaction of Aldehydes and Nitroalkenes. Angew. Chem. Int. Ed. 2005, 44, 4212–4215. [Google Scholar] [CrossRef] [PubMed]
- Marigo, M.; Wabnitz, T.C.; Fielenbach, D.; Jørgensen, K.A. Enantioselective Organocatalyzed α Sulfenylation of Aldehydes. Angew. Chem. Int. Ed. 2005, 44, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Benfatti, E.; Benedetto, E.; Cozzi, P.G. Organocatalytic Stereoselective α-Alkylation of Aldehydes with Stable Carbocations. Chem. Asian J. 2010, 5, 2047–2052. [Google Scholar] [CrossRef]
- Mengozzi, L.; Gualandi, A.; Cozzi, P.G. Organocatalytic Stereoselective Addition of Aldehydes to Acylquinolinium Ions. Eur. J. Org. Chem. 2016, 2016, 3200–3207. [Google Scholar] [CrossRef]
- Flaningan, D.M.; Rovis, T. Enantioselective N-heterocyclic carbene-catalyzed nucleophilic dearomatization of alkyl pyridiniums. Chem. Sci. 2017, 8, 6566–6569. [Google Scholar] [CrossRef] [PubMed]
- Di Carmine, G.; Ragno, D.; Bortolini, O.; Giovannini, P.P.; Mazzanti, A.; Massi, A.; Fogagnolo, M. Enantioselective Dearomatization of Alkylpyridiniums by N-Heterocyclic Carbene-Catalyzed Nucleophilic Acylation. J. Org. Chem. 2018, 83, 2050–2057. [Google Scholar] [CrossRef]
- Padwa, A. (Ed.) 1,3-Dipolar Cycloaddition Chemistry; Wiley-Interscience: New York, NY, USA, 1984; Volumes I–II. [Google Scholar]
- Najera, C.; Sansano, J.M. Enantioselective cycloadditions of azomethine ylides. Top. Heterocycl. Chem. 2008, 12, 117–145. [Google Scholar] [CrossRef]
- Pandey, G.; Banerjee, P.; Gadre, S.R. Construction of Enantiopure Pyrrolidine Ring System via Asymmetric [3+2]-Cycloaddition of Azomethine Ylides. Chem. Rev. 2006, 106, 4484–4517. [Google Scholar] [CrossRef]
- Coldham, I.; Hufton, R. Intramolecular Dipolar Cycloaddition Reactions of Azomethine Ylides. Chem. Rev. 2005, 105, 2765–2810. [Google Scholar] [CrossRef]
- Kröhnke, F. Über Enol-Betaine (I. Mitteil.). Ber. Dtsch. Chem. Ges. 1935, 68, 1177–1195. [Google Scholar]
- Mikhailovskii, A.G.; Shklyaev, V.S. Pyrrolo[2,1-a]isoquinolines. Chem. Heterocycl. Compd. 1997, 33, 243–265. [Google Scholar] [CrossRef]
- Jacobs, J.; Van Hende, E.; Claessens, S.; De Kimpe, N. Pyridinium Ylids in Heterocyclic Synthesis. Curr. Org. Chem. 2011, 15, 1340–1362. [Google Scholar] [CrossRef]
- Allgäuer, D.S.; Mayer, P.; Mayr, H. Nucleophilicity Parameters of Pyridinium Ylides and Their Use in Mechanistic Analyses. J. Am. Chem. Soc. 2013, 135, 15216–15224. [Google Scholar] [CrossRef] [PubMed]
- For a Comprehensive Database of Nucleophilicity Parameters N, sN and Electrophilicity Parameters E. Available online: http://www.cup.lmu.de/oc/mayr (accessed on 1 November 2018).
- Kucukdisli, M.; Opatz, T. A Modular Synthesis of Polysubstituted Indolizines. Eur. J. Org. Chem. 2012, 4555–4564. [Google Scholar] [CrossRef]
- Allgäuer, D.S.; Mayr, H. One-Pot Two-Step Synthesis of 1-(Ethoxycarbonyl)indolizines via Pyridinium Ylides. Eur. J. Org. Chem. 2013, 6379–6388. [Google Scholar] [CrossRef]
- Xu, X.C.; Zavalij, P.Y.; Doyle, M.P. Catalytic Asymmetric Syntheses of Quinolizidines by Dirhodium-Catalyzed Dearomatization of Isoquinolinium/Pyridinium Methylides–The Role of Catalyst and Carbene Source. J. Am. Chem. Soc. 2013, 135, 12439–12447. [Google Scholar] [CrossRef]
- Brioche, J.; Meyer, C.; Cossy, J. Synthesis of 2-Aminoindolizines by 1,3-Dipolar Cycloaddition of Pyridinium Ylides with Electron-Deficient Ynamides. Org. Lett. 2015, 17, 2800–2803. [Google Scholar] [CrossRef]
- Wang, W.H.; Han, J.W.; Sun, J.W.; Liu, Y. CuBr-Catalyzed Aerobic Decarboxylative Cycloaddition for the Synthesis of Indolizines under Solvent-Free Conditions. J. Org. Chem. 2017, 82, 2835–2842. [Google Scholar] [CrossRef]
- Douglas, T.; Pordea, A.; Dowden, J. Iron-Catalyzed Indolizine Synthesis from Pyridines, Diazo Compounds, and Alkynes. Org. Lett. 2017, 19, 6396–6399. [Google Scholar] [CrossRef]
- Bermudez, J.; Fake, C.S.; Joiner, G.F.; Joiner, K.A.; King, F.D.; Miner, W.D.; Sanger, G.J. 5-Hydroxytryptamine (5-HT3) receptor antagonists. Indazole and indolizine-3-carboxylic acid derivatives. J. Med. Chem. 1990, 33, 1924–1929. [Google Scholar] [CrossRef]
- Hagishita, S.; Yamada, M.; Shirahase, K.; Okada, T.; Murakami, Y.; Ito, Y.; Matsuura, T.; Wada, M.; Kato, T.; Ueno, M.; et al. Potent Inhibitors of Secretory Phospholipase A2: Synthesis and Inhibitory Activities of Indolizine and Indene Derivatives. J. Med. Chem. 1996, 39, 3636–3658. [Google Scholar] [CrossRef]
- Sonnet, P.; Dallemagne, P.; Guillon, J.; Enguehard, C.; Stiebing, S.; Tanguy, J.; Bureau, R.; Rault, S.; Auvray, P.; Moslemi, S.; et al. New aromatase inhibitors. Synthesis and biological activity of aryl-substituted pyrrolizine and indolizine derivatives. Bioorg. Med. Chem. 2000, 8, 945–955. [Google Scholar] [CrossRef]
- Teklu, S.; Gundersen, L.L.; Larsen, T.; Malterud, K.E.; Rise, F. Indolizine 1-sulfonates as potent inhibitors of 15-lipoxygenase from soybeans. Bioorg. Med. Chem. 2005, 13, 3127–3139. [Google Scholar] [CrossRef] [PubMed]
- James, D.A.; Koya, K.; Li, H.; Liang, G.-Q.; Xia, Z.-Q.; Ying, W.-W.; Wu, Y.-M.; Sun, L.-J. Indole- and indolizine-glyoxylamides displaying cytotoxicity against multidrug resistant cancer cell lines. Bioorg. Med. Chem. Lett. 2008, 18, 1784–1787. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.-M.; Lv, P.-C.; Chen, W.; Liu, P.-G.; Zhang, M.-Z.; Zhu, H.-L. Synthesis and antiproliferative activity of indolizine derivatives incorporating a cyclopropylcarbonyl group against Hep-G2 cancer cell line. Eur. J. Med. Chem. 2010, 45, 3184–3190. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.S.; Mmatli, E.E. Recent progress in synthesis and bioactivity studies of indolizines. Eur. J. Med. Chem. 2011, 46, 5237–5257. [Google Scholar] [CrossRef]
- Sharma, V.; Kumar, V. Indolizine: A biologically active moiety. Med. Chem. Res. 2014, 23, 3593–3606. [Google Scholar] [CrossRef]
- Hay, D.A.; Rogers, C.M.; Fedorov, O.; Tallant, C.; Martin, S.; Monteiro, O.P.; Muller, S.; Knapp, S.; Schofield, C.J.; Brennan, P.E. Design and synthesis of potent and selective inhibitors of BRD7 and BRD9 bromodomains. MedChemComm. 2015, 6, 1381–1386. [Google Scholar] [CrossRef]
- Ortega, N.; Tang, D.-T.D.; Urban, S.; Zhao, D.; Glorius, F. Ruthenium–NHC-Catalyzed Asymmetric Hydrogenation of Indolizines: Access to Indolizidine Alkaloids. Angew. Chem. Int. Ed. 2013, 52, 9500–9503. [Google Scholar] [CrossRef]
- Teodoro, B.V.M.; Correia, J.T.M.; Coelho, F. Selective Hydrogenation of Indolizines: An Expeditious Approach To Derive Tetrahydroindolizines and Indolizidines from Morita–Baylis–Hillman Adducts. J. Org. Chem. 2015, 80, 2529–2538. [Google Scholar] [CrossRef] [PubMed]
- Rotaru, A.V.; Druta, I.D.; Oeser, T.; Müller, T.J.J. A Novel Coupling 1,3-Dipolar Cycloaddition Sequence as a Three-Component Approach to Highly Fluorescent Indolizines. Helv. Chim. Acta 2005, 88, 1798–1812. [Google Scholar] [CrossRef]
- Kim, E.; Koh, M.; Ryu, J.; Park, S.B. Combinatorial Discovery of Full-Color-Tunable Emissive Fluorescent Probes Using a Single Core Skeleton, 1,2-Dihydropyrrolo[3,4-β]indolizin-3-one. J. Am. Chem. Soc. 2008, 130, 12206–12207. [Google Scholar] [CrossRef] [PubMed]
- Aittala, P.J.; Cramariuc, O.; Hukka, T.I.; Vasilescu, M.; Bandula, R.; Lemmetyinen, H. A TDDFT Study of the Fluorescence Properties of Three Alkoxypyridylindolizine Derivatives. J. Phys. Chem. A 2010, 114, 7094–7101. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Koh, M.; Lim, B.J.; Park, S.B. Emission Wavelength Prediction of a Full-Color-Tunable Fluorescent Core Skeleton, 9-Aryl-1,2-dihydropyrrolo [3,4-b]indolizin-3-one. J. Am. Chem. Soc. 2011, 133, 6642–6649. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Lee, S.; Park, S.B. A Seoul-fluor-based bioprobe for lipid droplets and its application in image-based high throughput screening. Chem. Commun. 2012, 48, 2331–2333. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Lee, Y.; Lee, S.; Park, S.B. Discovery, Understanding, and Bioapplication of Organic Fluorophore: A Case Study with an Indolizine-Based Novel Fluorophore, Seoul-Fluor. Acc. Chem. Res. 2015, 48, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Huckaba, A.J.; Yella, A.; Brogdon, P.; Scott Murphy, J.; Nazeeruddin, M.K.; Grätzel, M.; Delcamp, J.H. A low recombination rate indolizine sensitizer for dye-sensitized solar cells. Chem. Commun. 2016, 52, 8424–8427. [Google Scholar] [CrossRef]
- Huckaba, A.J.; Yella, A.; McNamara, L.E.; Steen, A.E.; Murphy, J.S.; Carpenter, C.A.; Puneky, G.D.; Hammer, N.I.; Nazeeruddin, M.K.; Grätzel, M.; et al. Molecular Design Principles for Near-Infrared Absorbing and Emitting Indolizine Dyes. Chem. Eur. J. 2016, 22, 15536–15542. [Google Scholar] [CrossRef]
- Song, Y.R.; Lim, C.W.; Kim, T.W. Synthesis and photophysical properties of 1,2-diphenylindolizine derivatives: Fluorescent blue-emitting materials for organic light-emitting device. Luminescence 2016, 31, 364–371. [Google Scholar] [CrossRef]
- Radzhabov, M.R.; Tsyganov, D.V.; Pivina, T.S.; Krayushkin, M.M.; Seeboth, N.; Ivanovc, S.; Salitc, A.-F. Synthesis of pyridinium ylides and simulation of their 1,3-dipolar cycloaddition mechanism. Mendeleev Commun. 2017, 27, 503–505. [Google Scholar] [CrossRef]
- Kanemasa, S.; Takenaka, S.; Watanabe, H.; Tsuge, O. Tandem 1,3-Dipolar Cycloadditions of Pyridinium or Isoquinolinium Methylides with Olefinic Dipolarophiles Leading to Cycl[3.2.2Jazines. “Enamine Route” as a New Generation Method of Azomethine Ylides. J. Org. Chem. 1989, 54, 420–424. [Google Scholar] [CrossRef]
- Padwa, A.; Austin, D.J.; Precedo, L.; Zhi, L. Cycloaddition Reactions of Pyridinium and Related Azomethine Ylides. J. Org. Chem. 1993, 58, 1144–1150. [Google Scholar] [CrossRef]
- Wang, Q.-F.; Song, X.-K.; Chen, J.; Yan, C.-G. Pyridinium Ylide-Assisted One-Pot Two-Step Tandem Synthesis of Polysubstituted Cyclopropanes. J. Comb. Chem. 2009, 11, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Day, J.; Uroos, M.; Castledine, R.A.; Lewis, W.; McKeever-Abbas, B.; Dowden, J. Alkaloid inspired spirocyclic oxindoles from 1,3-dipolar cycloaddition of pyridinium ylides. Org. Biomol. Chem. 2013, 11, 6502–6509. [Google Scholar] [CrossRef] [PubMed]
- Day, J.; McKeever-Abbas, B.; Dowden, J. Stereoselective Synthesis of Tetrahydroindolizines through the Catalytic Formation of Pyridinium Ylides from Diazo Compounds. Angew. Chem. Int. Ed. 2016, 55, 5809–5813. [Google Scholar] [CrossRef] [PubMed]
- Fulton, J.R.; Aggarwal, V.K.; de Vicente, J. The Use of Tosylhydrazone Salts as a Safe Alternative for Handling Diazo Compounds and Their Applications in Organic Synthesis. Eur. J. Org. Chem. 2005, 1479–1492. [Google Scholar] [CrossRef]
- Zhang, D.; Lin, L.; Yang, J.; Liu, X.H.; Feng, X.M. Asymmetric Synthesis of Tetrahydroindolizines by Bimetallic Relay Catalyzed Cycloaddition of Pyridinium Ylides. Angew. Chem. Int. Ed. 2018, 57, 12323–12327. [Google Scholar] [CrossRef]
- Xu, X.; Zavalij, P.Y.; Doyle, M.P. Highly Enantioselective Dearomatizing Formal [3+3]-Cycloaddition Reactions of N-Acyliminopyridinium Ylides with Electrophilic Enol carbene Intermediates. Angew. Chem. Int. Ed. 2013, 52, 12664–12668. [Google Scholar] [CrossRef]
- Xu, X.; Doyle, M.P. The [3+3]-Cycloaddition Alternative for Heterocycle Syntheses: Catalytically Generated Metalloenolcarbenes as Dipolar Adducts. Acc. Chem. Res. 2014, 47, 1396–1405. [Google Scholar] [CrossRef]
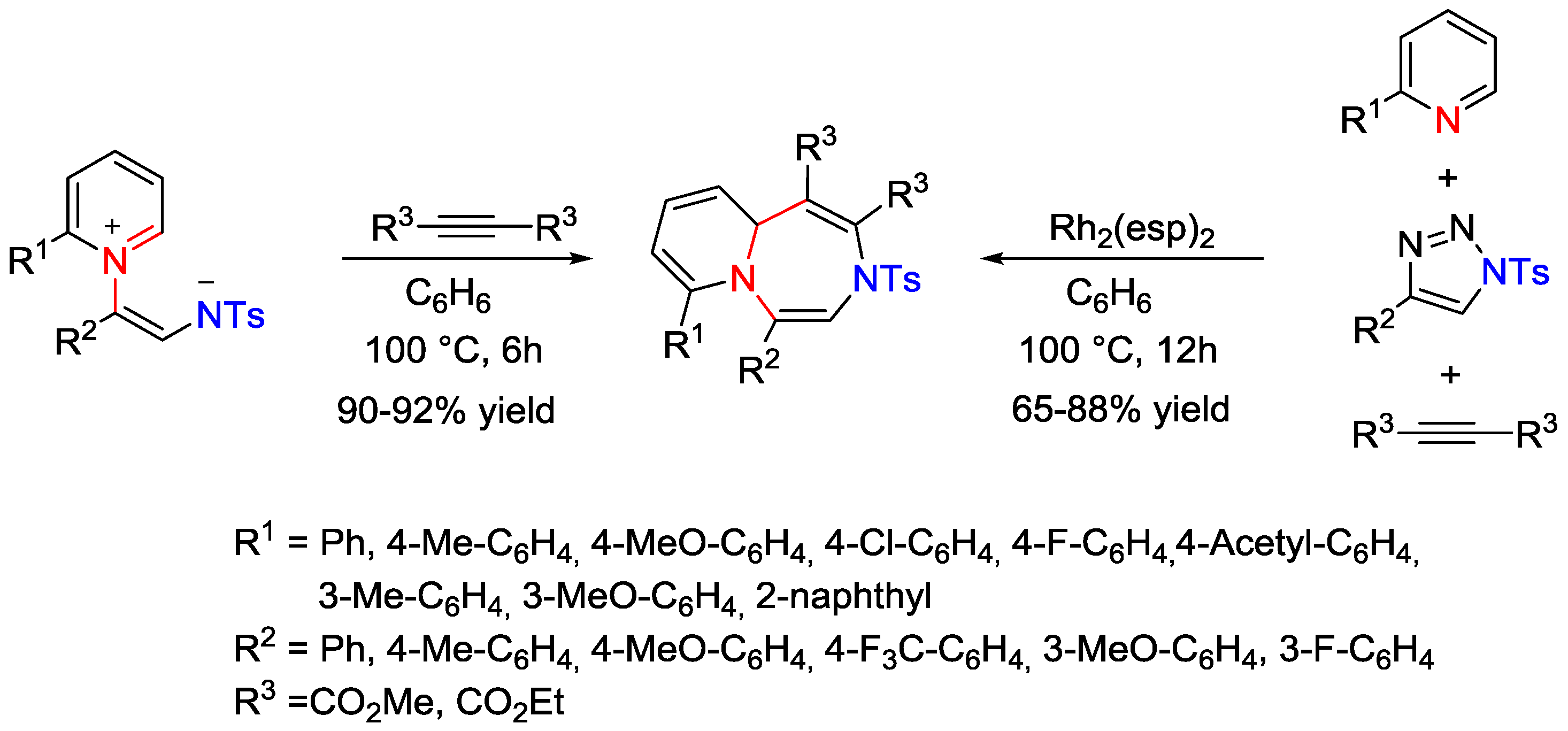
- Lee, D.J.; Han, H.S.; Shin, J.; Yoo, E.J. Multicomponent [5+2] Cycloaddition Reaction for the Synthesis of 1,4-Diazepines: Isolation and Reactivity of Azomethine Ylides. J. Am. Chem. Soc. 2014, 136, 11606–11609. [Google Scholar] [CrossRef] [PubMed]
- Yoo, E.J. Azomethine Ylide: An Isolable 1,5-Dipole for Affecting [5+2] Cycloaddition Reactions. Synlett 2015, 26, 2189–2193. [Google Scholar] [CrossRef]
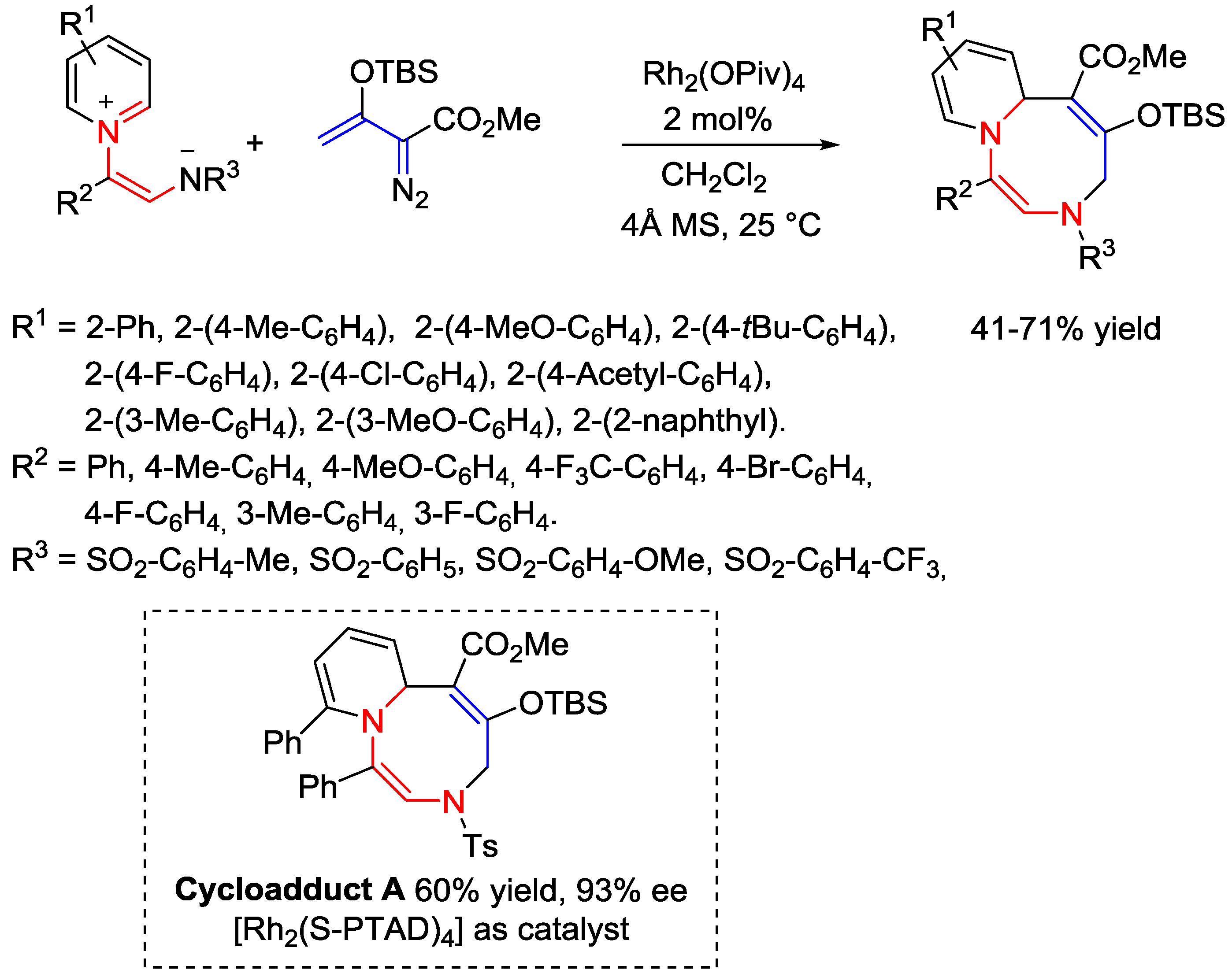
- Lee, D.J.; Ko, D.; Yoo, E.J. Rhodium(II)-Catalyzed Cycloaddition Reactions of Non-classical 1,5-Dipoles for the Formation of Eight-Membered Heterocycles. Angew. Chem. Int. Ed. 2015, 54, 13715–13718. [Google Scholar] [CrossRef] [PubMed]
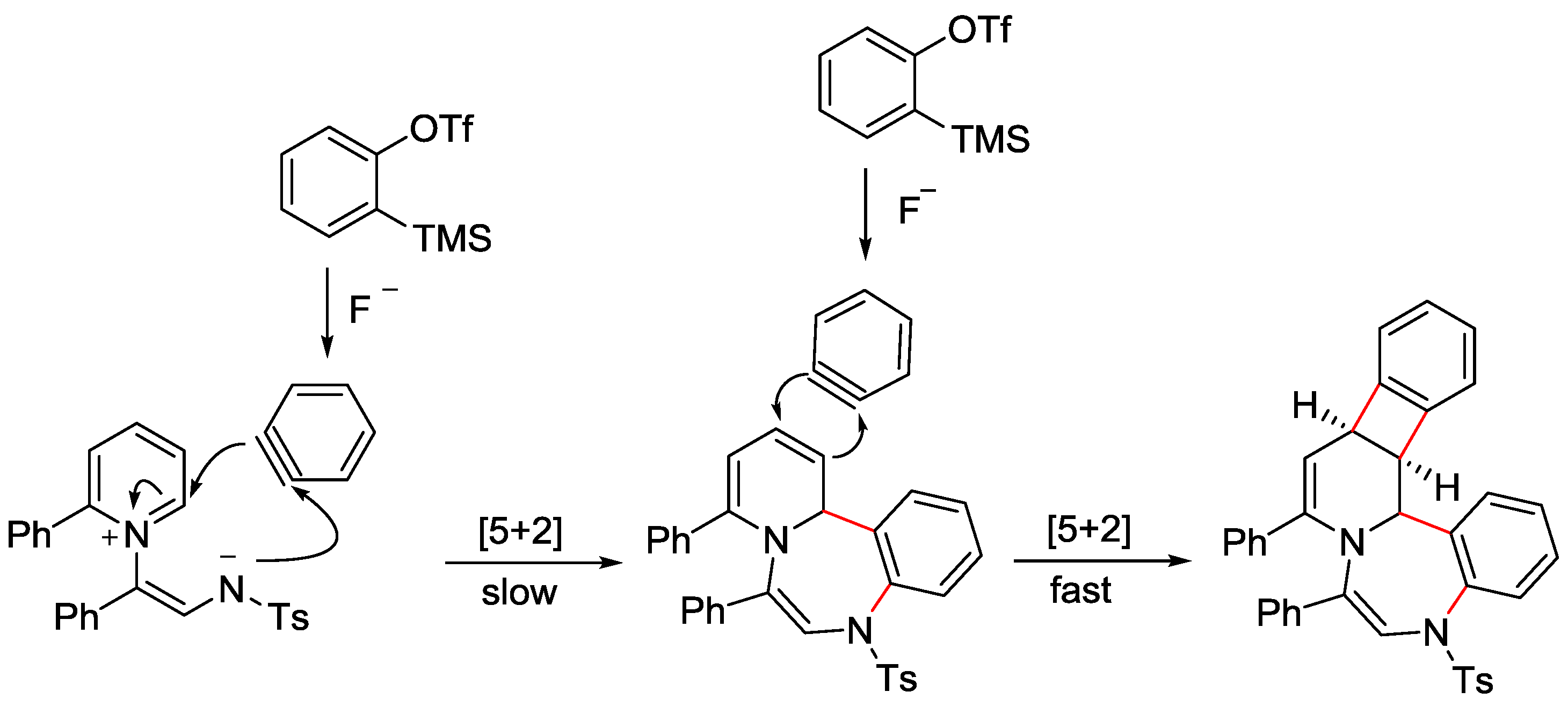
- Shin, J.; Lee, J.; Ko, D.; De, N.; Yoo, E.J. Synthesis of Fused Polycyclic 1,4-Benzodiazepines via Metal-Free Cascade [5+2]/[2+2] Cycloadditions. Org. Lett. 2017, 19, 2901–2904. [Google Scholar] [CrossRef]
- Jeganmohan, M.; Bhuvaneswari, S.; Cheng, C.-H. Synthesis of N-arylated 1,2-dihydroheteroaromatics Through the Three-Component Reaction of Arynes with N-Heteroaromatics and Terminal Alkynes or Ketones. Chem. Asian J. 2010, 5, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Jeganmohan, M.; Cheng, C.-H. Reaction of arynes, N-heteroaromatics and nitriles. Chem. Commun. 2006, 2454–2456. [Google Scholar] [CrossRef] [PubMed]
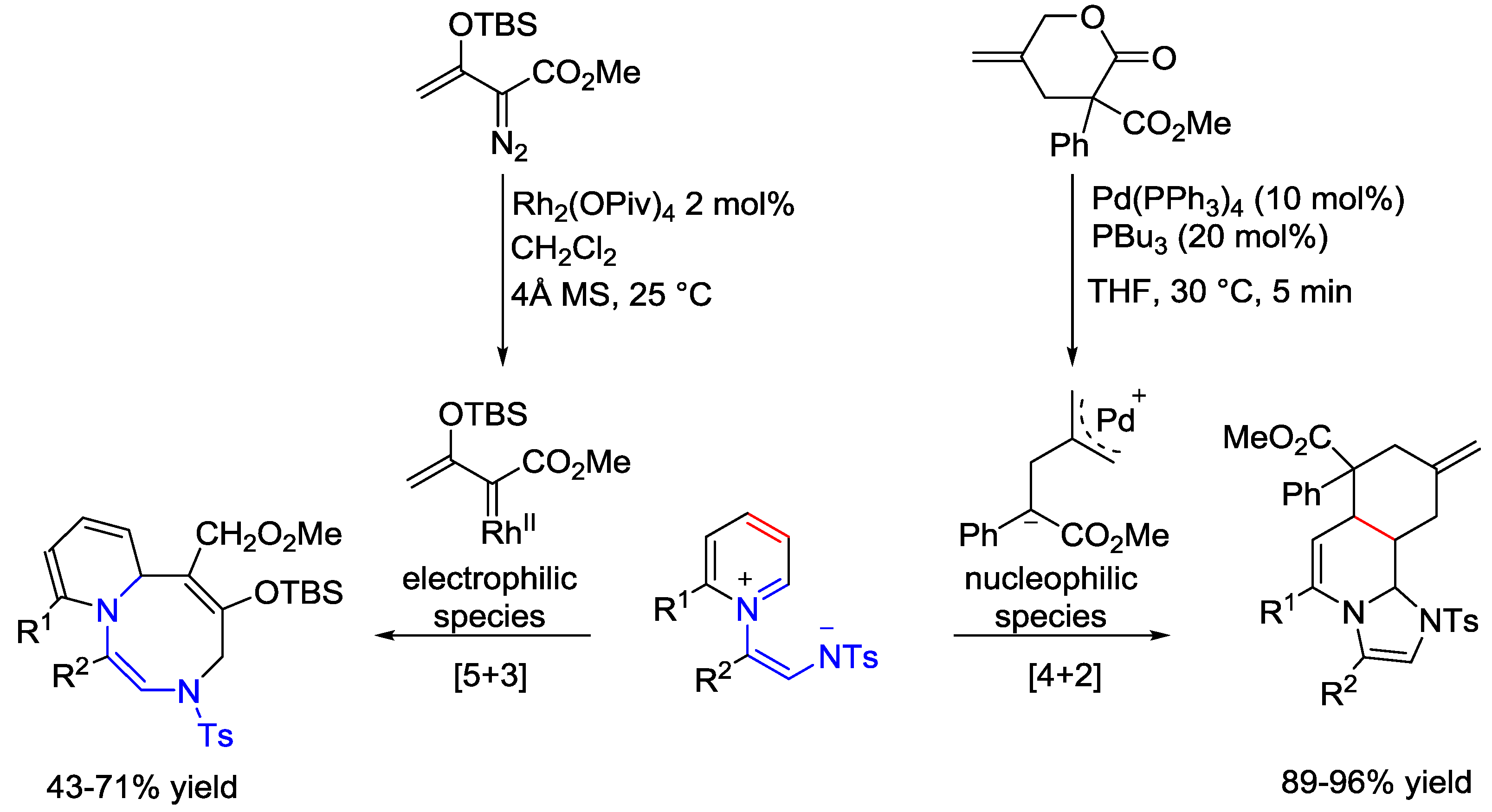
- Baek, S.-Y.; Lee, J.Y.; Ko, D.; Baik, M.-H.; Yoo, E.J. Rationally Designing Regiodivergent Dipolar Cycloadditions: Frontier Orbitals Show How To Switch between [5+3] and [4+2] Cycloadditions. ACS Catal. 2018, 8, 6353–6361. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Dennis, N. Cycloaddition Reactions of Heteroaromatic Six-Membered Rings. Chem. Rev. 1989, 89, 827–861. [Google Scholar] [CrossRef]
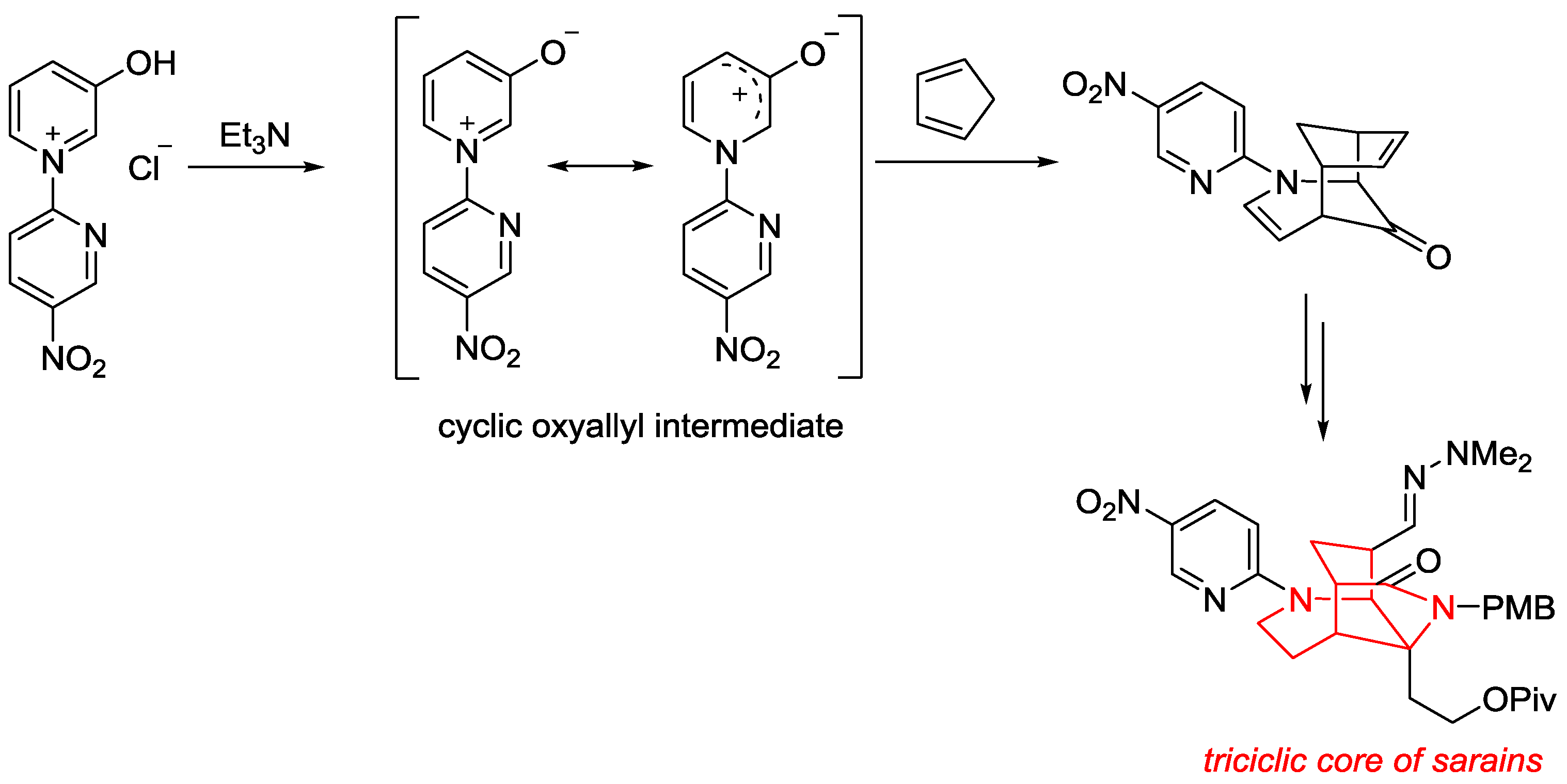
- Sung, M.J.; Lee, H.I.; Chong, Y.; Cha, J.K. Facile Synthesis of the Tricyclic Core of Sarain A. 3-Oxidopyridinium Betaine Cycloaddition Approach. Org. Lett. 1999, 1, 2017–2019. [Google Scholar] [CrossRef]
- Lee, H.I.; Sung, M.J.; Lee, H.B.; Cha, J.K. An Improved Synthesis of the Tricyclic Core of Sarains by a 3-Oxidopyridinium Betaine Cycloaddition. Heterocycles 2004, 62, 407–422. [Google Scholar] [CrossRef]
- Fu, C.; Lora, N.; Kirchhoefer, P.L.; Lee, D.R.; Altenhofer, E.; Barnes, C.L.; Hungerford, N.L.; Krenske, E.H.; Harmata, M. (4+3) Cycloaddition Reactions of N-Alkyl Oxidopyridinium Ions. Angew. Chem. Int. Ed. 2017, 56, 14682–14687. [Google Scholar] [CrossRef] [PubMed]













































© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertuzzi, G.; Bernardi, L.; Fochi, M. Nucleophilic Dearomatization of Activated Pyridines. Catalysts 2018, 8, 632. https://doi.org/10.3390/catal8120632
Bertuzzi G, Bernardi L, Fochi M. Nucleophilic Dearomatization of Activated Pyridines. Catalysts. 2018; 8(12):632. https://doi.org/10.3390/catal8120632
Chicago/Turabian StyleBertuzzi, Giulio, Luca Bernardi, and Mariafrancesca Fochi. 2018. "Nucleophilic Dearomatization of Activated Pyridines" Catalysts 8, no. 12: 632. https://doi.org/10.3390/catal8120632
APA StyleBertuzzi, G., Bernardi, L., & Fochi, M. (2018). Nucleophilic Dearomatization of Activated Pyridines. Catalysts, 8(12), 632. https://doi.org/10.3390/catal8120632