Synthesis, APPI Mass-Spectrometric Characterization, and Polymerization Studies of Group 4 Dinuclear Bis(ansa-metallocene) Complexes

 , ,
, ,  ,
,  and
and 
Abstract
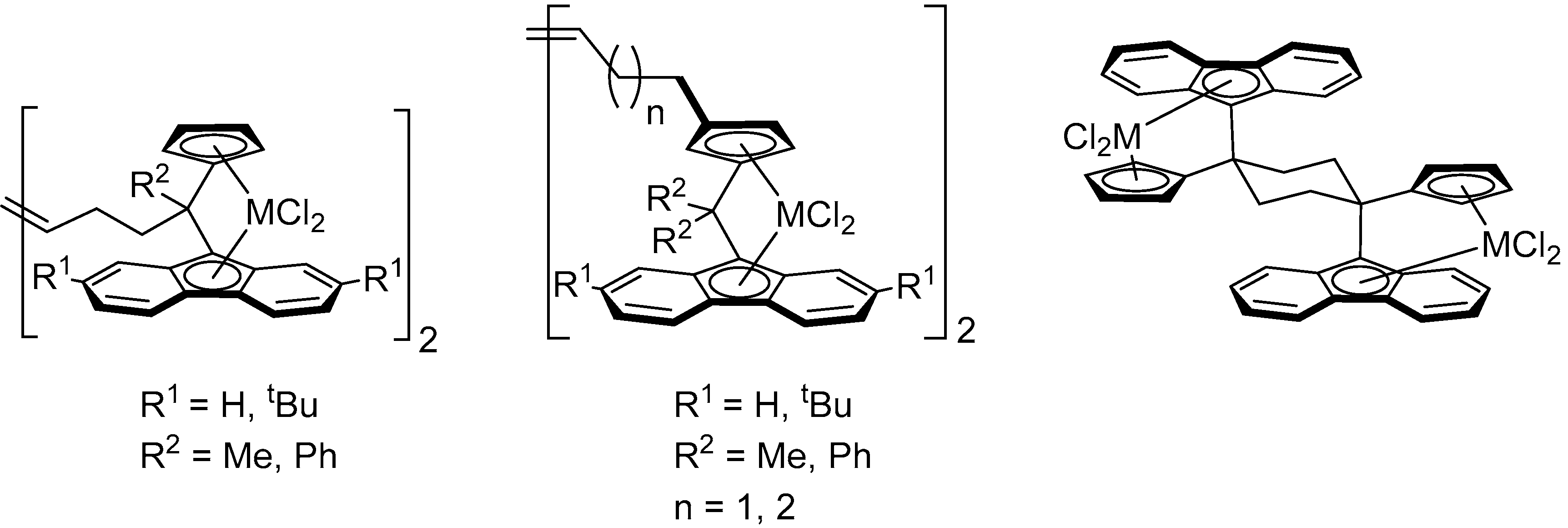
1. Introduction
2. Results and Discussion
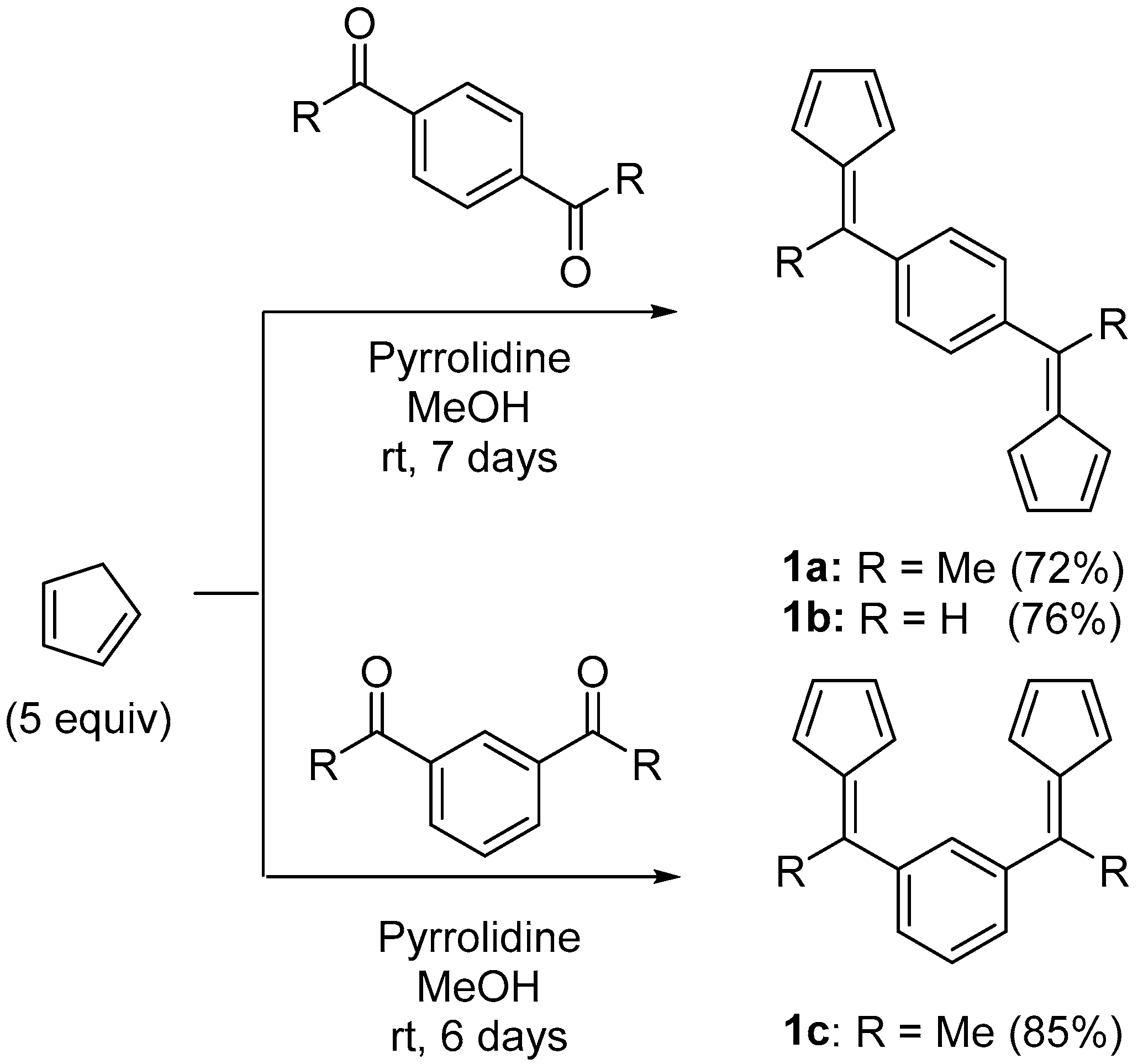
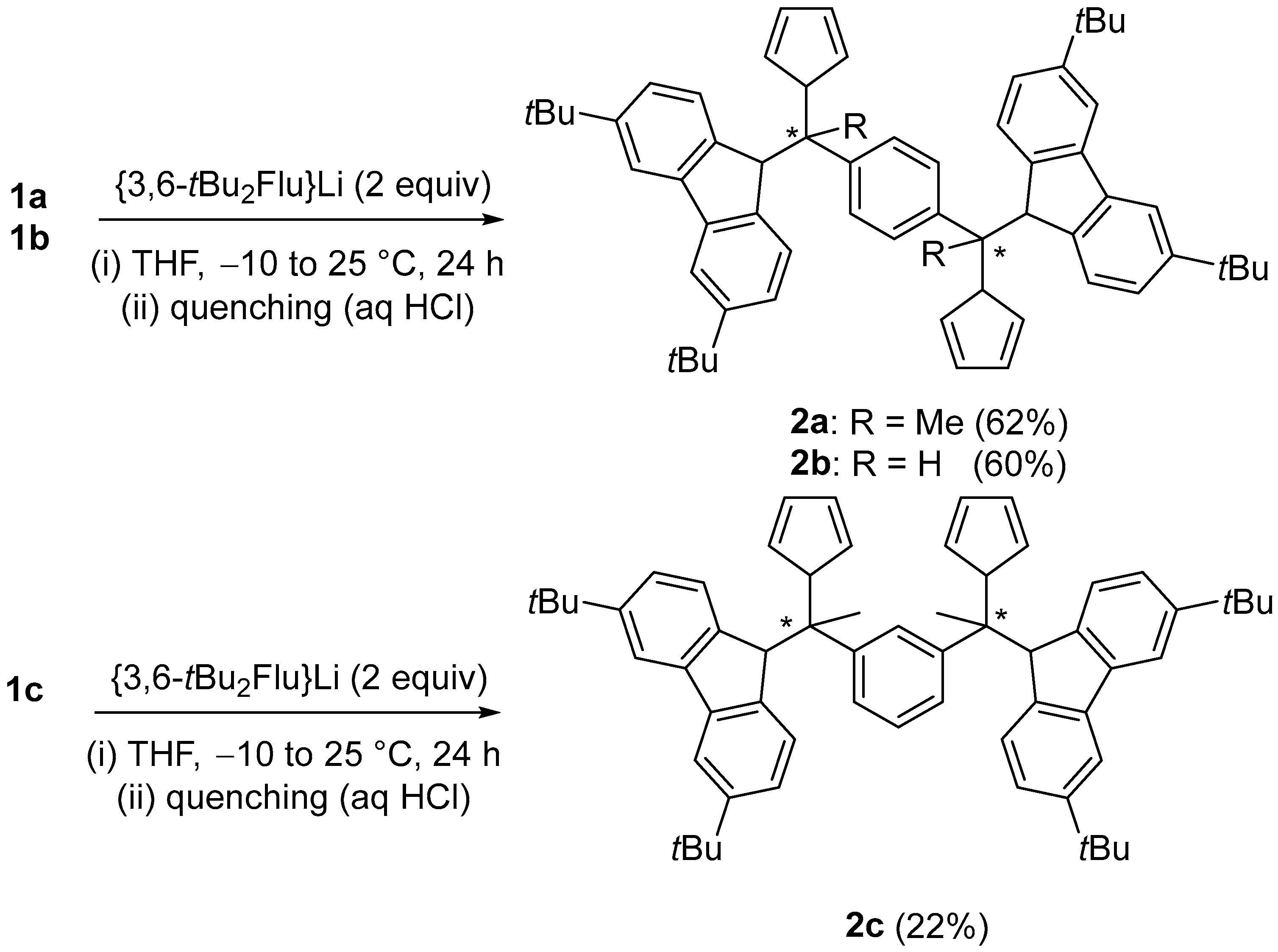
2.1. Synthesis of Proligands
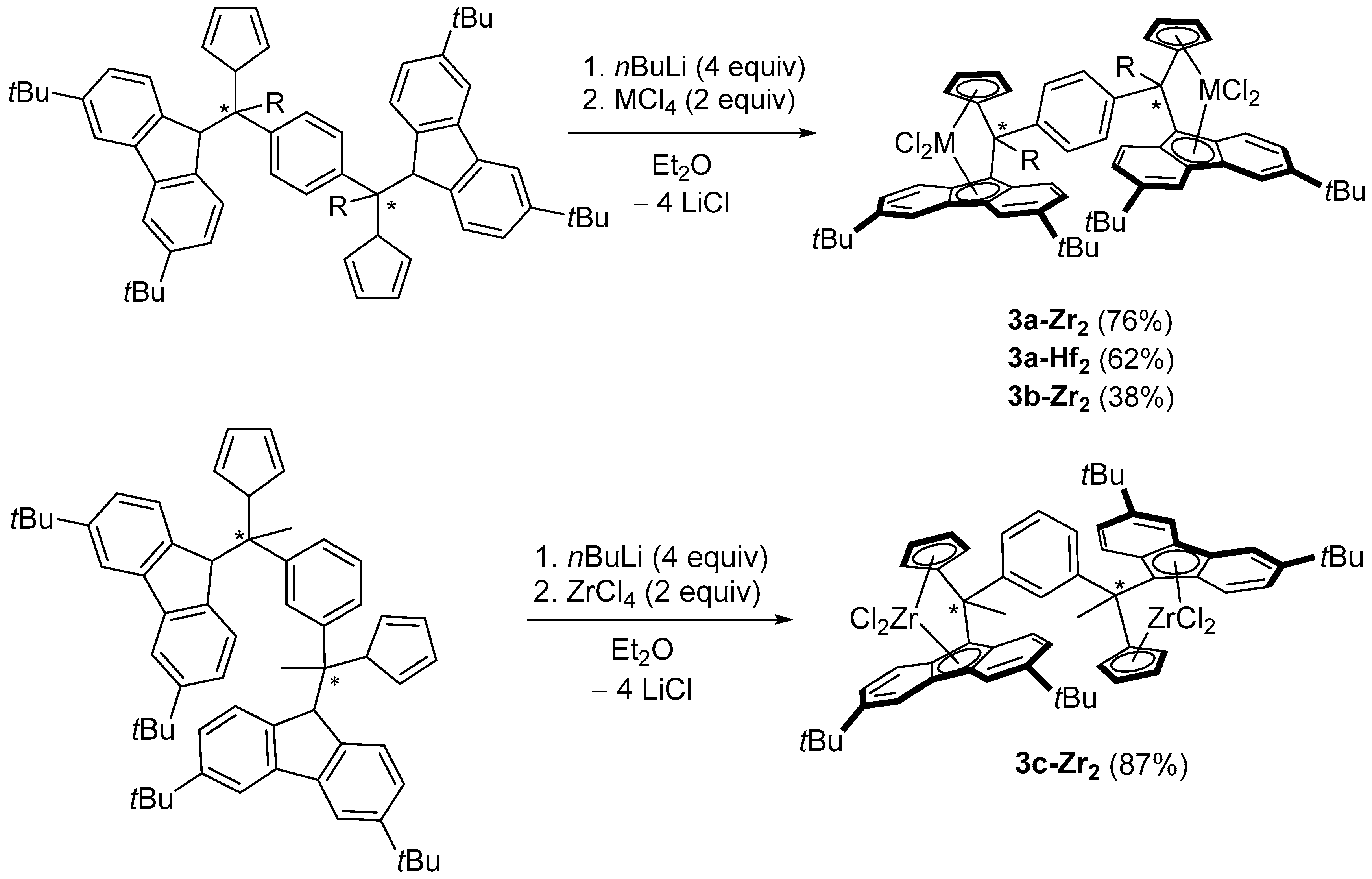
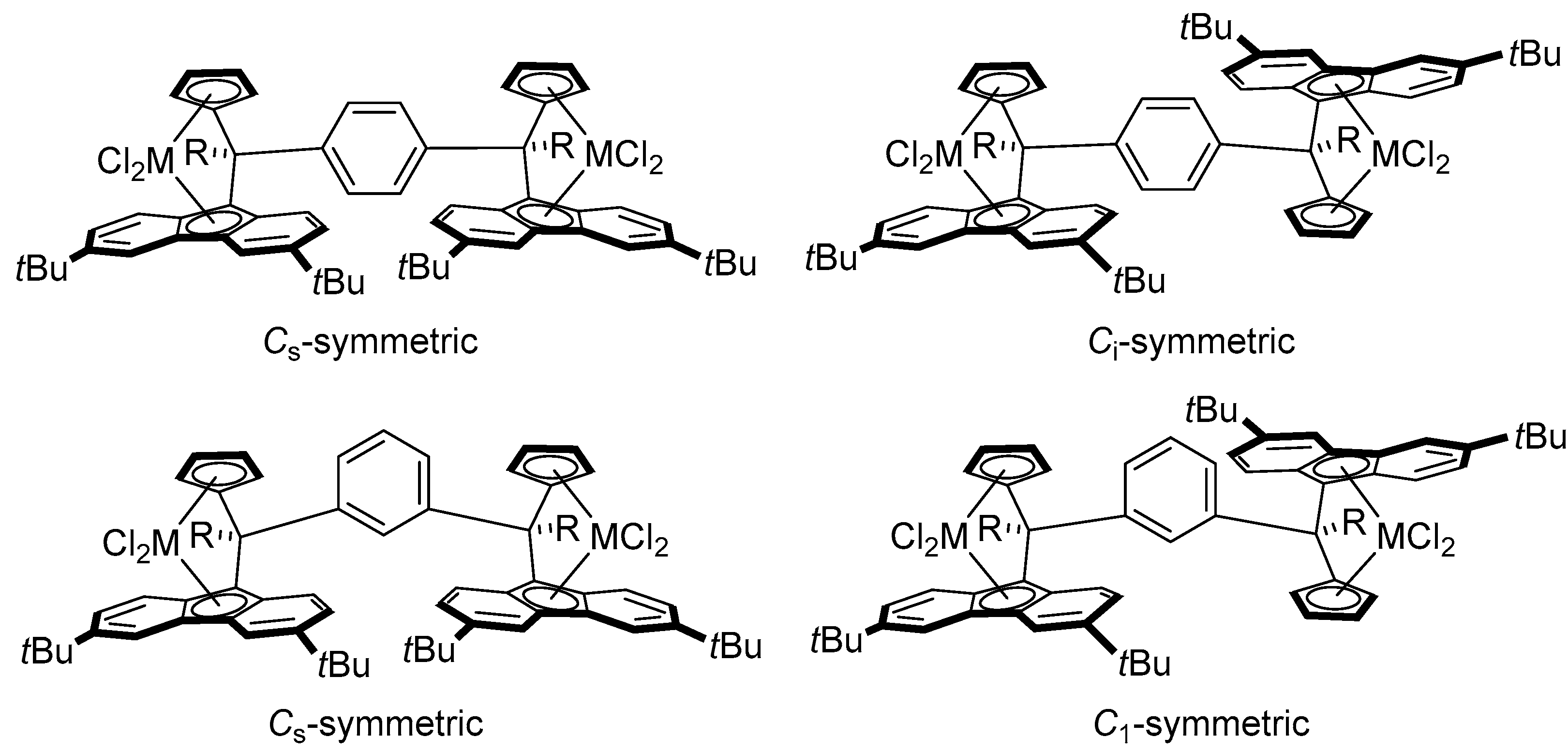
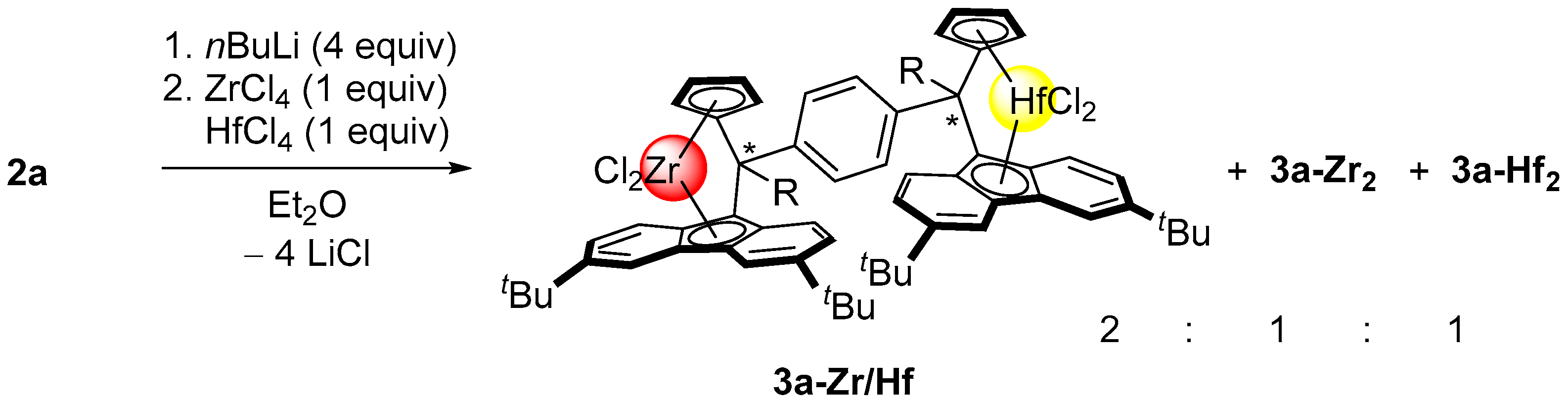
2.2. Synthesis of Group 4 Dinuclear Bis(dichloro ansa-metallocene) Complexes
2.3. Synthesis of Mononuclear Ansa-metallocene Analogues
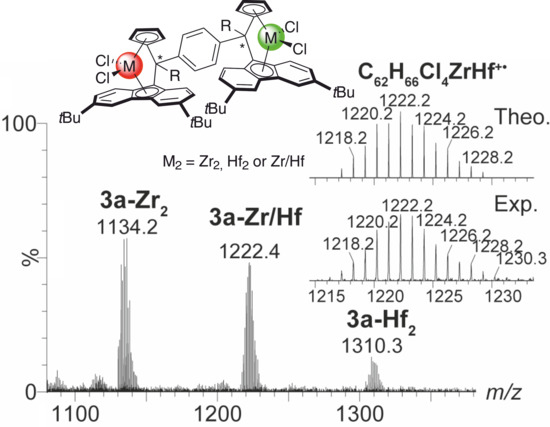
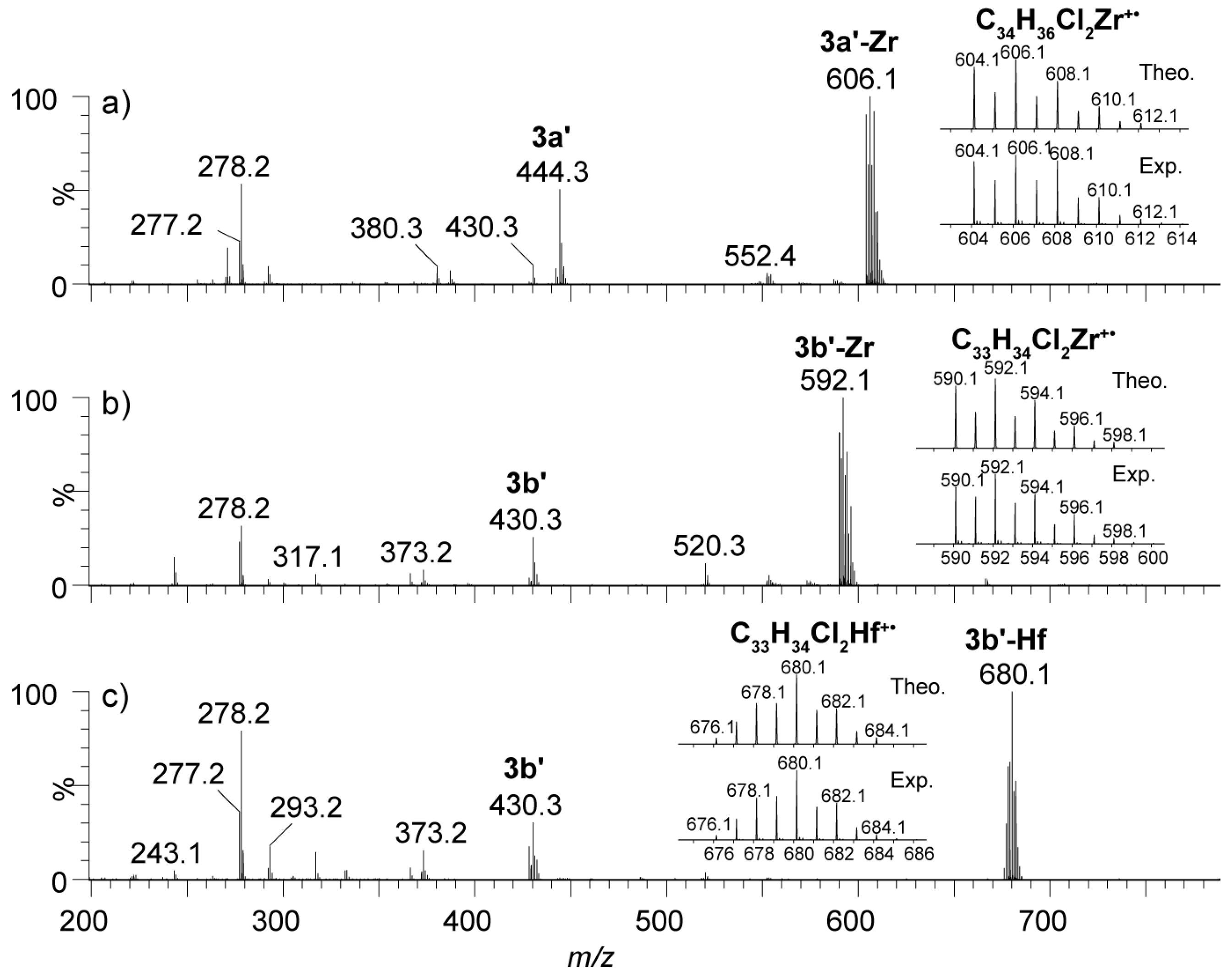
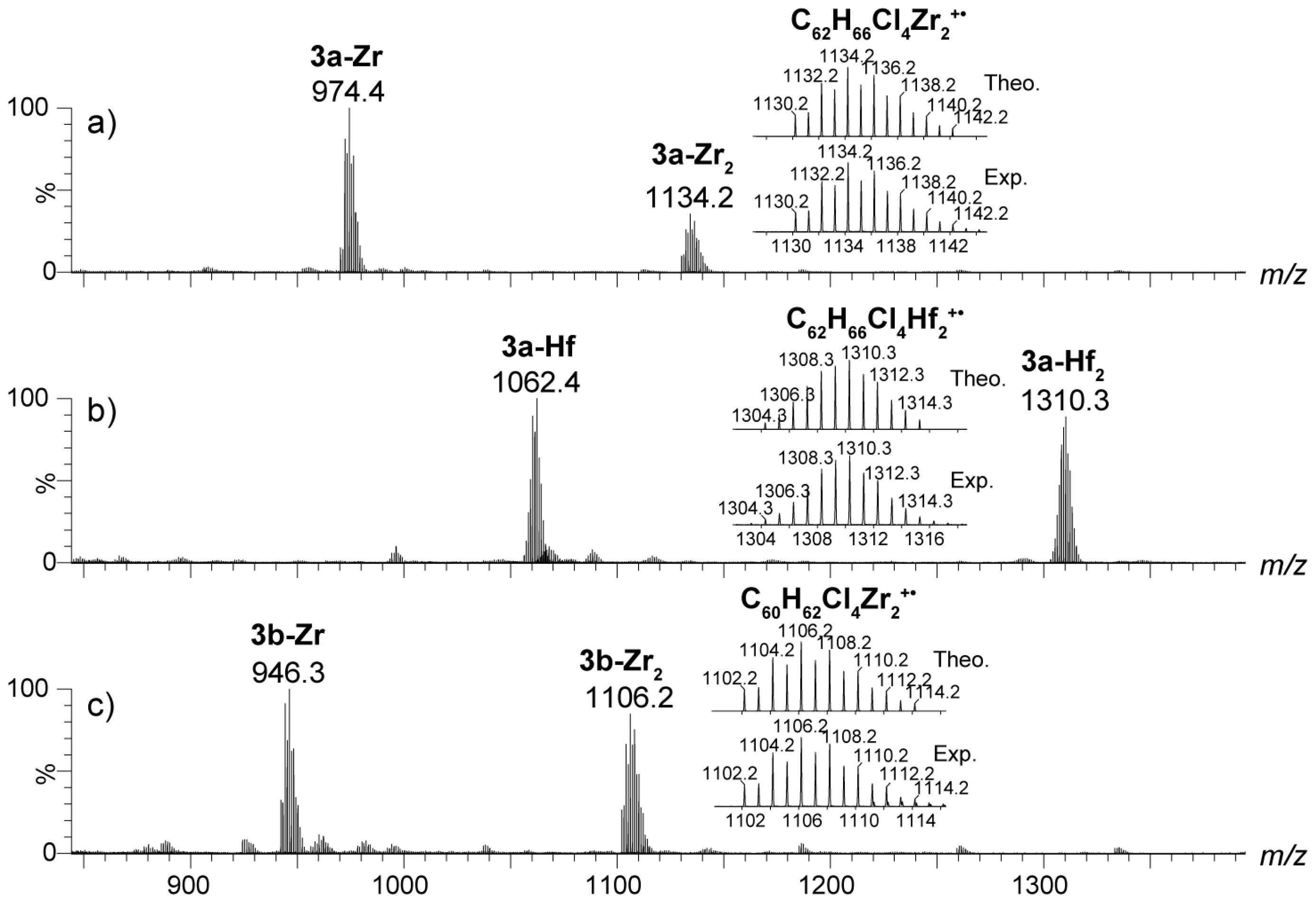
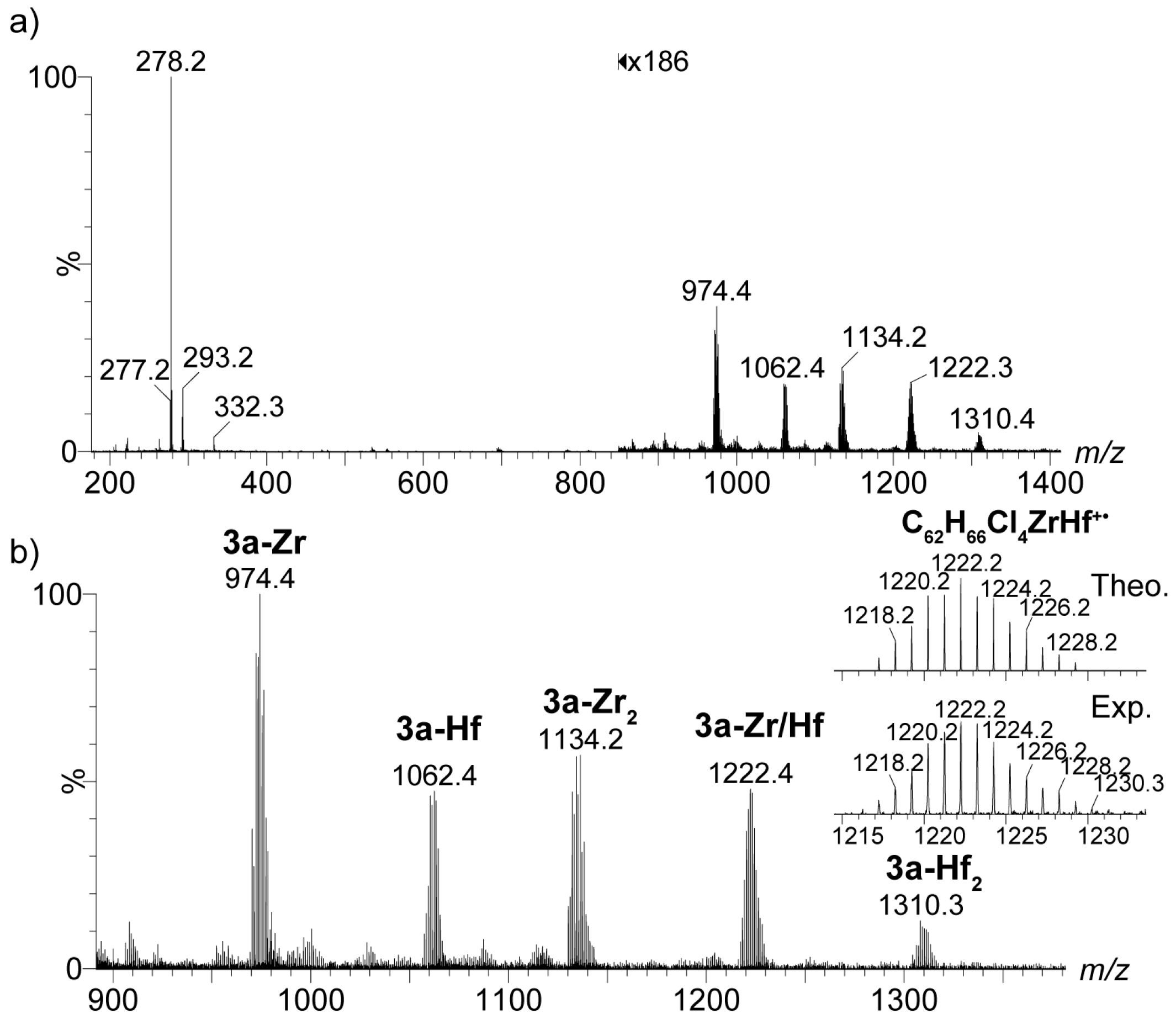
2.4. Mass Spectrometric Studies of Mononuclear and Dinuclear Bis(dichloro ansa-metallocene) Complexes
2.5. Polymerization Catalysis
3. Materials and Methods
3.1. General Considerations
3.2. Instruments and Measurements
3.3. APPI Mass Spectrometric Characterization of Metal Complexes
3.4. 1,4-Bis(1-(cyclopenta-2,4-dien-1-ylidene)ethyl)benzene (1a)
3.5. 1,4-Bis(cyclopenta-2,4-dien-1-ylidenemethyl)benzene (1b)
3.6. 1,3-Bis(1-(cyclopenta-2,4-dien-1-ylidene)ethyl)benzene (1c)
3.7. 1,4-Ph((Me)C-(3,6-tBu2FluH)(CpH))2 (2a)
3.8. 1,4-Ph((H)C-(3,6-tBu2FluH)(CpH))2 (2b)
3.9. 1,3-Ph(MeC-(3,6-tBu2FluH)(CpH))2 (2c)
3.10. PhMeC-(3,6-tBu2FluH)(CpH) (2a’)
3.11. Ph(H)C-(3,6-tBu2FluH)(CpH) (2b’)
3.12. 1,4-Ph{MeC-(3,6-tBu2Flu)(Cp)ZrCl2}2 (3a-Zr)
3.13. 1,4-Ph{MeC-(3,6-tBu2Flu)(Cp)HfCl2}2 (3a-Hf)
3.14. 1,4-Ph{(H)C-(3,6-tBu2Flu)(Cp)ZrCl2}2 (3b-Zr)
3.15. 1,3-Ph{MeC-(3,6-tBu2Flu)(Cp)ZrCl2}2 (3c-Zr)
3.16. {Ph(Me)C-(3,6-tBu2Flu)(Cp)}ZrCl2 (3a’-Zr)
3.17. {Ph(H)C-(3,6-tBu2Flu)(Cp)}ZrCl2 (3b’-Zr)
3.18. {Ph(H)C-(3,6-tBu2Flu)(Cp)}HfCl2 (3b’-Hf)
3.19. Ethylene Homopolymerization and Ethylene/1-hexene Copolymerization
3.20. Crystal Structure Determination of 3a’-Zr
3.21. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Delferro, M.; Marks, T.J. Multinuclear Olefin Polymerization Catalysts. Chem. Rev. 2011, 111, 2450–2485. [Google Scholar] [CrossRef] [PubMed]
- McInnis, J.P.; Delferro, M.; Marks, T.J. Multinuclear Group 4 Catalysis: Olefin Polymerization Pathways Modified by Strong Metal–Metal Cooperative Effects. Acc. Chem. Res. 2014, 47, 2545–2557. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Kim, S.K.; Do, Y. Biphenylene-Bridged Dinuclear Group 4 Metal Complexes: Enhanced Polymerization Properties in Olefin Polymerization. Organometallics 2005, 24, 3618–3620. [Google Scholar] [CrossRef]
- Soga, K.; Ban, H.T.; Uozumi, T. Synthesis of a dinuclear ansa-zirconocene catalyst having a biphenyl bridge and application to ethene polymerization. J. Mol. Catal. A Chem. 1998, 128, 273–278. [Google Scholar] [CrossRef]
- Deckers, P.J.W.; Hessen, B.; Teuben, J.H. Catalytic Trimerization of Ethene with Highly Active Cyclopentadienyl−Arene Titanium Catalysts. Organometallics 2002, 21, 5122–5135. [Google Scholar] [CrossRef]
- Lee, H.W.; Ahn, A.H.; Park, Y.H. Copolymerization characteristics of homogeneous and in situ supported [(CH2)5(C5H4)2][(C9H7)ZrCl2]2 catalyst. J. Mol. Catal. A Chem. 2003, 194, 19–28. [Google Scholar] [CrossRef]
- Noh, S.K.; Kim, J.; Jung, J.; Ra, C.S.; Lee, D.H.; Lee, H.B.; Lee, S.W.; Huh, W.S. Syntheses of polymethylene bridged dinuclear zirconocenes and investigation of their polymerisation activities. J. Organomet. Chem. 1999, 580, 90–97. [Google Scholar] [CrossRef]
- Noh, S.K.; Kim, S.; Yang, Y.; Lyoo, W.S.; Lee, D.H. Preparation of syndiotactic polystyrene using the doubly bridged dinuclear titanocenes. Eur. Polym. J. 2004, 40, 227–235. [Google Scholar] [CrossRef]
- Kuwabara, J.; Takeuchi, D.; Osakada, K. Zr/Zr and Zr/Fe Dinuclear Complexes with Flexible Bridging Ligands. Preparation by Olefin Metathesis Reaction of the Mononuclear Precursors and Properties as Polymerization Catalysts. Organometallics 2005, 24, 2705–2712. [Google Scholar] [CrossRef]
- Xiao, X.; Sun, J.; Li, X.; Li, H.; Wang, Y. Binuclear titanocenes linked by the bridge combination of rigid and flexible segment: Synthesis and their use as catalysts for ethylene polymerization. J. Mol. Catal. A Chem. 2007, 267, 86–91. [Google Scholar] [CrossRef]
- Li, H.; Stern, C.L.; Marks, T.J. Significant Proximity and Cocatalyst Effects in Binuclear Catalysis for Olefin Polymerization. Macromolecules 2005, 38, 9015–9027. [Google Scholar] [CrossRef]
- Li, L.; Metz, M.V.; Li, H.; Chen, M.-C.; Marks, T.J.; Liable Sands, L.; Rheingold, A.L. Catalyst/Cocatalyst Nuclearity Effects in Single-Site Polymerization. Enhanced Polyethylene Branching and α-Olefin Comonomer Enchainment in Polymerizations Mediated by Binuclear Catalysts and Cocatalysts via a New Enchainment Pathway. J. Am. Chem. Soc. 2002, 124, 12725–12741. [Google Scholar] [CrossRef] [PubMed]
- Salata, M.R.; Marks, T.J. Catalyst Nuclearity Effects in Olefin Polymerization. Enhanced Activity and Comonomer Enchainment in Ethylene + Olefin Copolymerizations Mediated by Bimetallic Group 4 Phenoxyiminato Catalysts. Macromolecules 2009, 42, 1920–1933. [Google Scholar] [CrossRef]
- Spaleck, W.; Küber, F.; Bachmann, B.; Fritze, C.; Winter, A. New bridged zirconocenes for olefin polymerization: Binuclear and hybrid structures. J. Mol. Catal. A Chem. 1998, 128, 279–287. [Google Scholar] [CrossRef]
- Salata, M.R.; Marks, T.J. Synthesis, Characterization, and Marked Polymerization Selectivity Characteristics of Binuclear Phenoxyiminato Organozirconium Catalysts. J. Am. Chem. Soc. 2008, 130, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Noh, S.K.; Byun, G.G.; Lee, C.S.; Lee, D.; Yoon, K.B.; Kang, K.S. Synthesis, characterization, and reactivities of the polysiloxane-bridged binuclear metallocenes tetramethyldisiloxanediylbis(cyclopentadienyltitanium trichloride) and hexamethyltrisiloxanediylbis(cyclopentadienyltitanium trichloride). J. Organomet. Chem. 1996, 518, 1–6. [Google Scholar] [CrossRef]
- Lee, D.H.; Yoon, K.B.; Lee, E.H.; Noh, S.K.; Byun, G.G.; Lee, C.S. Polymerizations of ethylene and styrene initiated with trisiloxane-bridged dinuclear titanium metallocene/MMAO catalyst systems. Macromol. Rapid Commun. 1995, 16, 265–268. [Google Scholar] [CrossRef]
- Linh, N.T.B.; Huyen, N.T.D.; Noh, S.K.; Lyoo, W.S.; Lee, D.H.; Kim, Y. Preparation of new dinuclear half-titanocene complexes with ortho- and meta-xylene linkages and investigation of styrene polymerization. J. Organomet. Chem. 2009, 694, 3438–3443. [Google Scholar] [CrossRef]
- Noh, S.K.; Jung, W.; Oh, H.; Lee, Y.R.; Lyoo, W.S. Synthesis and styrene polymerization properties of dinuclear half-titanocene complexes with xylene linkage. J. Organomet. Chem. 2006, 691, 5000–5006. [Google Scholar] [CrossRef]
- Liu, X.; Sun, J.; Zhang, H.; Xiao, X.; Lin, F. Ethylene polymerization by novel phenylenedimethylene bridged homobinuclear titanocene/MAO systems. Eur. Polym. J. 2005, 41, 1519–1524. [Google Scholar] [CrossRef]
- Alt, H.; Samuel, E. Fluorenyl complexes of zirconium and hafnium as catalysts for olefin polymerization. Chem. Soc. Rev. 1998, 27, 323–329. [Google Scholar] [CrossRef]
- Alt, H. From the lab bench to the plant: How to commercialize a metallocene catalyst? Macromol. Chem. Symp. 2001, 173, 65–76. [Google Scholar] [CrossRef]
- Razavi, A. Site selective ligand modification and tactic variation in polypropylene chains produced with metallocene catalysts. Coord. Chem. Rev. 2006, 250, 155–169. [Google Scholar] [CrossRef]
- Alt, H.G.; Köppl, A. Effect of the Nature of Metallocene Complexes of Group IV Metals on Their Performance in Catalytic Ethylene and Propylene Polymerization. Chem. Rev. 2000, 100, 1205–1222. [Google Scholar] [CrossRef] [PubMed]
- Coates, G.W. Precise Control of Polyolefin Stereochemistry Using Single-Site Metal Catalysts. Chem. Rev. 2000, 100, 1223–1252. [Google Scholar] [CrossRef] [PubMed]
- Resconi, L.; Cavallo, L.; Falt, A.; Piemontesi, F. Selectivity in Propene Polymerization with Metallocene Catalysts. Chem. Rev. 2000, 100, 1253–1346. [Google Scholar] [CrossRef] [PubMed]
- Gladysz, J.A. Frontiers in Metal-Catalyzed Polymerization: Designer Metallocenes, Designs on New Monomers, Demystifying MAO, Metathesis Déshabillé. Chem. Rev. 2000, 100, 1167–1168. [Google Scholar] [CrossRef] [PubMed]
- Price, C.J.; Irwin, L.J.; Aubry, D.A.; Miller, S.A. Fluorenyl Containing Catalysts for Stereoselective Propylene Polymerization. In Stereoselective Polymerization with Single Site Catalysts; Baugh, L.S., Canich, J.A.M., Eds.; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Kirillov, E.; Marquet, N.; Razavi, A.; Belia, V.; Hampel, F.; Roisnel, T.; Gladysz, J.A.; Carpentier, J.-F. New C1-Symmetric Ph2C-Bridged Multisubstituted ansa-Zirconocenes for Highly Isospecific Propylene Polymerization: Synthetic Approach via Activated Fulvenes. Organometallics 2010, 29, 5073–5082. [Google Scholar] [CrossRef]
- Kirillov, E.; Marquet, N.; Bader, M.; Razavi, A.; Belia, V.; Hampel, F.; Roisnel, T.; Gladysz, J.A.; Carpentier, J.-F. Chiral-at-ansa-Bridged Group 4 Metallocene Complexes {(R1R2C)-(3,6-tBu2Flu)(3-R3-5-Me-C5H2)}MCl2: Synthesis, Structure, Stereochemistry, and Use in Highly Isoselective Propylene Polymerization. Organometallics 2011, 30, 263–272. [Google Scholar] [CrossRef]
- Bader, M.; Marquet, N.; Kirillov, E.; Roisnel, T.; Razavi, A.; Lhost, O.; Carpentier, J.-F. Old and New C1-Symmetric Group 4 Metallocenes {(R1R2C)-(R2′R3′R6′R7′-Flu)(3-R3-5-R4-C5H2)}ZrCl2: From Highly Isotactic Polypropylenes to Vinyl End-Capped Isotactic-Enriched Oligomers. Organometallics 2012, 32, 8375–8387. [Google Scholar] [CrossRef]
- Castro, L.; Kirillov, E.; Miserque, O.; Welle, A.; Haspeslagh, L.; Carpentier, J.-F.; Maron, L. Are Solvent and Dispersion Effects Crucial in Olefin Polymerization DFT Calculations? Some Insights from Propylene Coordination and Insertion Reactions with Group 3 and 4 Metallocenes. ACS Catal. 2015, 5, 416–425. [Google Scholar] [CrossRef]
- Castro, L.; Therukauff, G.; Vantomme, A.; Welle, A.; Haspeslagh, L.; Brusson, J.-M.; Maron, L.; Carpentier, J.-F.; Kirillov, E. A Theoretical Outlook on the Stereoselectivity Origins of Isoselective Zirconocene Propylene Polymerization Catalysts. Chem. Eur. J. 2018, 24, 10784–10792. [Google Scholar] [CrossRef] [PubMed]
- Murray, R.E.; Jayaratne, K.C.; Yang, Q.; Martin, J.L. Highly Soluble Olefin Polymerization Catalyst Activator. U.S. Patent 20,090,170,690 A1, 2 July 2009. [Google Scholar]
- Murray, R.E.; Martin, J.L.; Jayaratne, K.C.; Yang, Q. Nano-Linked Metallocene Catalyst Compositions and tHeir Polymer Products. European Patent WO 2,009,085,124 A1, 9 July 2009. [Google Scholar]
- Inoue, Y.; Shiomura, T.; Kuono, M.; Sonobe, Y.; Mizutani, K. Method of Polymerizing an Olefin Using a Novel Transition Metal Compound. U.S. Patent 5,439,994, 8 August 1995. [Google Scholar]
- Peifer, B.; Milius, W.; Alt, H.G. Selbstimmobilisierende Metallocenkatalysatoren. J. Organomet. Chem. 1998, 553, 205–220. [Google Scholar] [CrossRef]
- Jüngling, S.; Mülhaupt, R. Cooperative effects in binuclear zirconocenes: Their synthesis and use as catalyst in propene polymerization. J. Organomet. Chem. 1993, 460, 191–195. [Google Scholar] [CrossRef]
- Farenc, M.; Assumani, B.; Afonso, C.; Giusti, P. Analysis of Metallocene using Atmospheric Pressure Chemical Ionization and Atmospheric Pressure Photo Ionization Coupled with Ion Mobility Mass Spectrometry. In Proceedings of the 64th ASMS conference on Mass Spectrometry and Allied Topics, San Atonio, TX, USA, 5–9 June 2016. [Google Scholar]
- Farenc, M.; Corilo, Y.E.; Lalli, P.M.; Riches, E.; Rodgers, R.P.; Afonso, C.; Giusti, P. Comparison of Atmospheric Pressure Ionization for the Analysis of Heavy Petroleum Fractions with Ion Mobility-Mass Spectrometry. Energy Fuels 2016, 30, 8896–8903. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R.; Pellechia, R.; Talarico, G.; Razavi, A. Hafnocenes and MAO: Beware of Trimethylaluminum! Macromolecules 2009, 42, 1789–1791. [Google Scholar] [CrossRef]
- Coşkun, N.; Erden, I. An efficient catalytic method for fulvene synthesis. Tetrahedron 2011, 67, 8607–8614. [Google Scholar] [CrossRef] [PubMed]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Cryst. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Treutler, O.; Ahlrichs, R. Efficient molecular numerical integration schemes. J. Chem. Phys. 1995, 102, 346–354. [Google Scholar] [CrossRef]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary basis sets to approximate coulomb potentials. Chem. Phys. Lett. 1995, 242, 652–660. [Google Scholar] [CrossRef]
- TURBOMOLE V6.2 2010, A Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 1 July 2010).
- Sierka, M.; Hogekamp, A.; Ahlrichs, R. Fast evaluation of the coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J. Chem. Phys. 2003, 118, 9136–9148. [Google Scholar] [CrossRef]
- Deglmann, P.; May, K.; Furche, F.; Ahlrichs, R. Nuclear second analytical derivative calculations using auxiliary basis set expansions. Chem. Phys. Lett. 2004, 384, 103–107. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Comp. | mPE (g) | Prod. (kg·mol−1·h−1) | Mwb (kg·mol−1) | Mw/Mnb | Tmc (°C) | %Me d (wt%) | %Et d (wt%) | %nBu d (wt%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3a-Zr2 | 6.20 | 24,800 | 175.1 | 3.2 | 132.3 | 0.0 | 0.1 | 0.0 |
| 2 | 3a-Zr2 | 5.62 | 22,500 | 196.6 | 3.4 | 132.2 | 0.0 | 0.2 | 0.0 |
| 3 | 3a’-Zr | 5.64 | 22,600 | 260.4 | 3.6 | 132.1 | 0.0 | 0.1 | 0.0 |
| 4 | 3a’-Zr | 6.14 | 24,600 | 307.9 | 4.1 | 131.8 | 0.0 | 0.0 | 0.0 |
| 5 | 3b-Zr2 | 5.82 | 23,280 | 134.6 | 3.1 | 127.2 | 1.5 | 0.2 | 0.0 |
| 6 | 3b-Zr2 | 5.20 | 20,800 | 189.2 | 3.5 | 129.0 | 0.9 | 0.2 | 0.0 |
| 7 | 3b’-Zr | 6.79 | 27,160 | 180.9 | 3.4 | 132.1 | 0.0 | 0.1 | 0.0 |
| 8 | 3b’-Zr | 5.27 | 21,080 | 272.6 | 4.3 | 133.2 | 0.0 | 0.1 | 0.0 |
| 9 | 3c-Zr2 | 4.96 | 19,800 | 85.0 | 3.3 | nd | 0.0 | 0.2 | 0.0 |
| 10 | 3a-Hf2 | 1.53 | 6100 | - f | - f | 132.7 | 0.0 | 0.0 | 0.0 |
| 11 e | 3a-Hf2 | 1.63 | 6500 | - f | - f | 133.6 | 0.0 | 0.0 | 0.0 |
| 12 | 3b’-Hf | 1.38 | 5500 | 1146.3 | 4,1 | nd | 0.0 | 0.0 | 0.0 |
| Entry | Comp. | mPE (g) | Prod. (kg·mol−1·h−1) | Mwb (kg·mol−1) | Mw/Mnb | Tmc (°C) | %Me d (wt%) | %Et d (wt%) | %nBu d (wt%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3a-Zr2 | 6.20 | 24,800 | 70.6 | 2.6 | 122.5 | 0.0 | 0.1 | 15.7 |
| 2 | 3a-Zr2 | 5.62 | 22,500 | 84.3 | 3.0 | 123.8 | 0.0 | 0.0 | 23.2 |
| 3 | 3a’-Zr | 7.22 | 28,900 | 129.3 | 3.4 | 117.7 | 0.0 | 0.0 | 22.6 |
| 4 | 3a’-Zr | 6.77 | 27,100 | 132.1 | 3.3 | 118.2 | 0.0 | 0.0 | 21.3 |
| 5 | 3b-Zr2 | 7.74 | 31,000 | 75.8 | 2.8 | 111.4 | 0.9 | 0.1 | 22.5 |
| 6 | 3b-Zr2 | 6.70 | 26,800 | 64.0 | 2.7 | 112.2 | 0.4 | 0.1 | 19.4 |
| 7 | 3b’-Zr | 7.57 | 30,300 | 89.9 | 2.7 | 113.9 | 0.0 | 0.0 | 21.1 |
| 8 | 3b’-Zr | 7.30 | 29,200 | 96.9 | 2.9 | 108.2 | 0.0 | 0.0 | 22.7 |
| 9 | 3c-Zr2 | 7.75 | 31,000 | 190.6 | 3.5 | 116.0 | 0.0 | 0.0 | 21.3 |
| 10 | 3a-Hf2 | 3.78 | 15,100 | 447.1 | 3.1 | nd | 0.0 | 0.0 | 28.1 |
| 11 e | 3a-Hf2 | 4.34 | 17,400 | 581.5 | 4.0 | nd | 0.0 | 0.0 | 29.6 |
| 12 | 3b’-Hf | 1.66 | 6600 | - f | - f | nd | 0.0 | 0.0 | 28.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schnee, G.; Farenc, M.; Bitard, L.; Vantomme, A.; Welle, A.; Brusson, J.-M.; Afonso, C.; Giusti, P.; Carpentier, J.-F.; Kirillov, E. Synthesis, APPI Mass-Spectrometric Characterization, and Polymerization Studies of Group 4 Dinuclear Bis(ansa-metallocene) Complexes. Catalysts 2018, 8, 558. https://doi.org/10.3390/catal8110558
Schnee G, Farenc M, Bitard L, Vantomme A, Welle A, Brusson J-M, Afonso C, Giusti P, Carpentier J-F, Kirillov E. Synthesis, APPI Mass-Spectrometric Characterization, and Polymerization Studies of Group 4 Dinuclear Bis(ansa-metallocene) Complexes. Catalysts. 2018; 8(11):558. https://doi.org/10.3390/catal8110558
Chicago/Turabian StyleSchnee, Gilles, Mathilde Farenc, Leslie Bitard, Aurelien Vantomme, Alexandre Welle, Jean-Michel Brusson, Carlos Afonso, Pierre Giusti, Jean-François Carpentier, and Evgueni Kirillov. 2018. "Synthesis, APPI Mass-Spectrometric Characterization, and Polymerization Studies of Group 4 Dinuclear Bis(ansa-metallocene) Complexes" Catalysts 8, no. 11: 558. https://doi.org/10.3390/catal8110558
APA StyleSchnee, G., Farenc, M., Bitard, L., Vantomme, A., Welle, A., Brusson, J.-M., Afonso, C., Giusti, P., Carpentier, J.-F., & Kirillov, E. (2018). Synthesis, APPI Mass-Spectrometric Characterization, and Polymerization Studies of Group 4 Dinuclear Bis(ansa-metallocene) Complexes. Catalysts, 8(11), 558. https://doi.org/10.3390/catal8110558