
Treatment of Aqueous Bromate by Superparamagnetic BiOCl-Mediated Advanced Reduction Process



Abstract
:1. Introduction
2. Results and Discussion
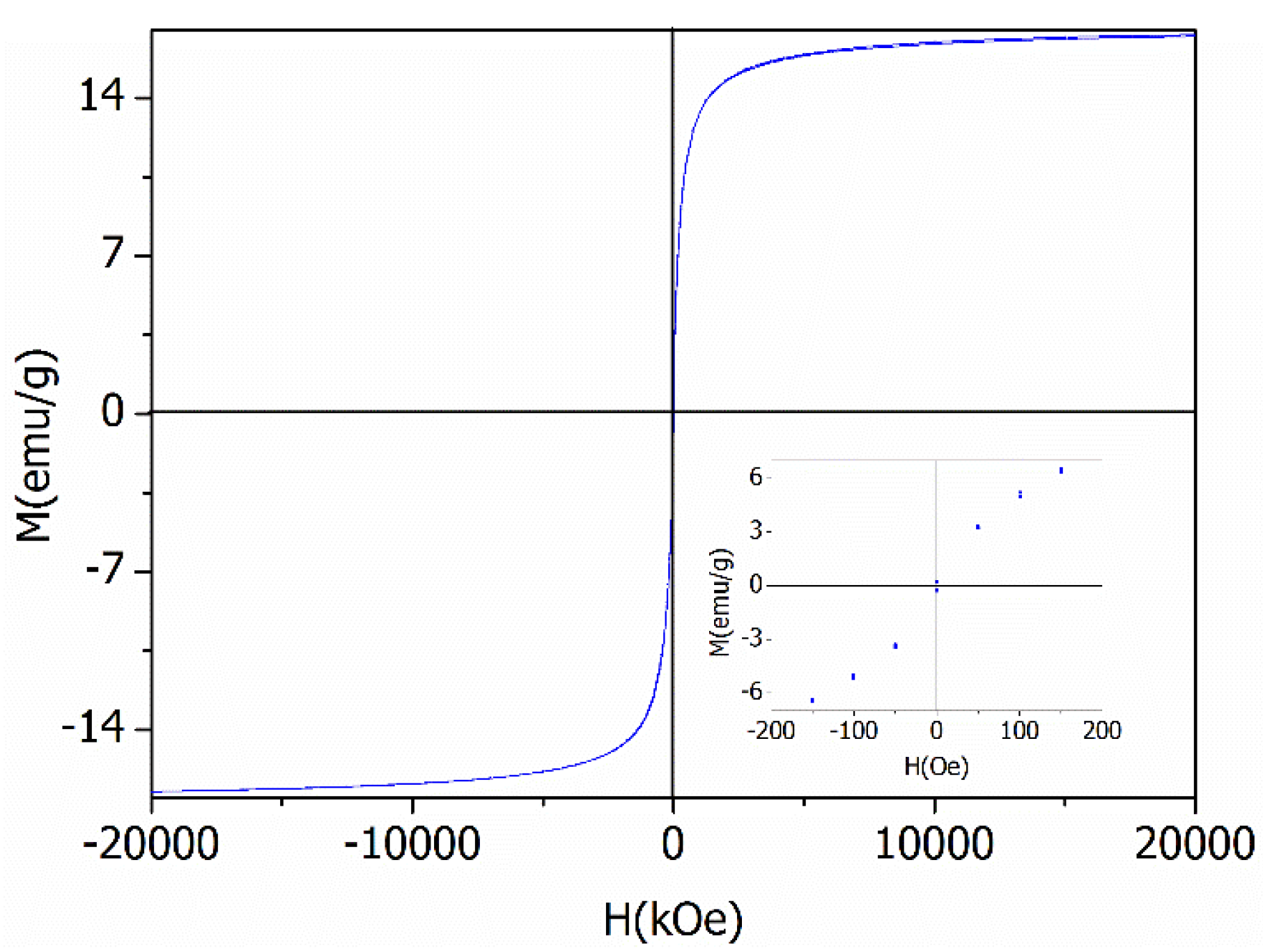
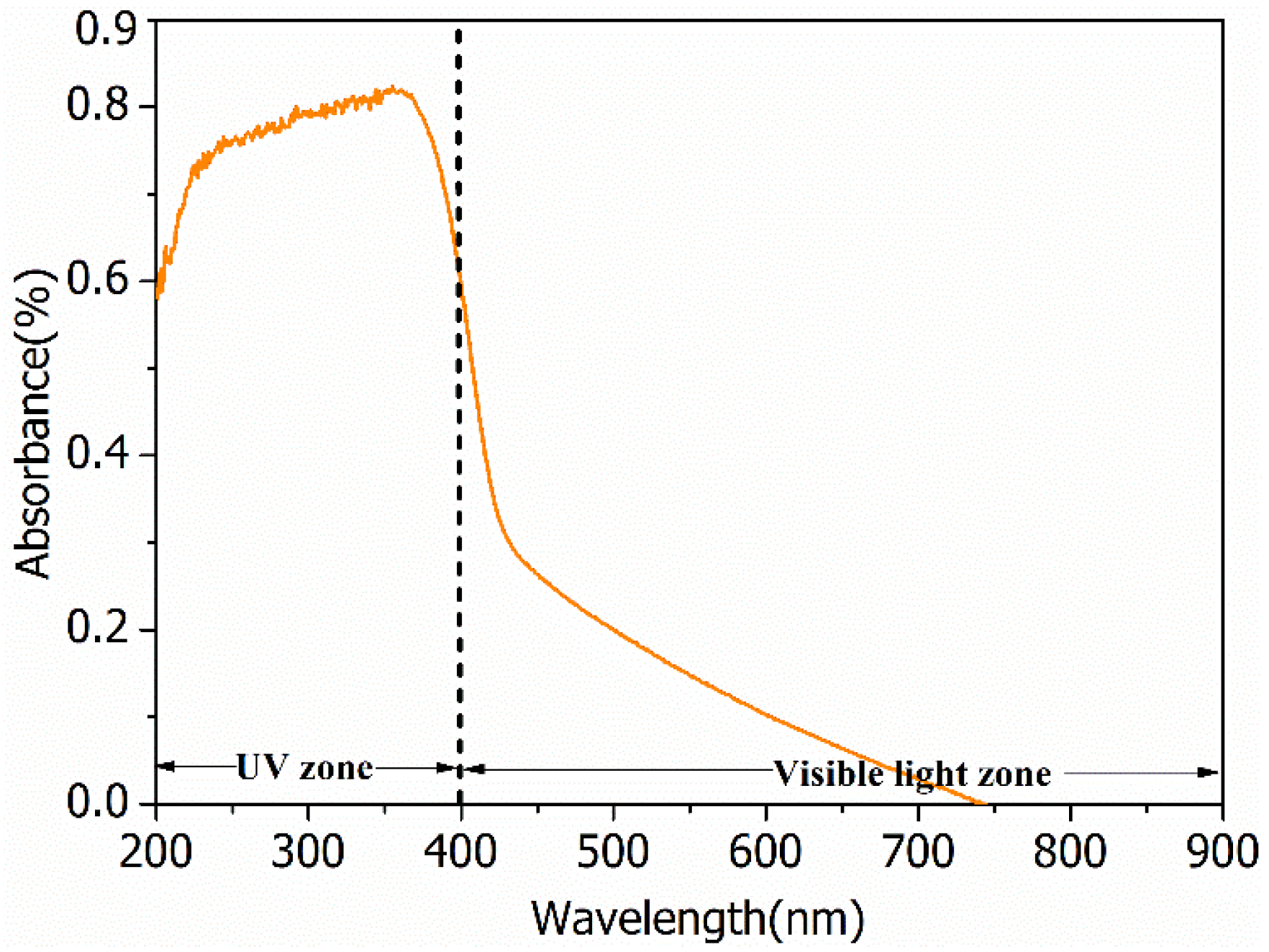
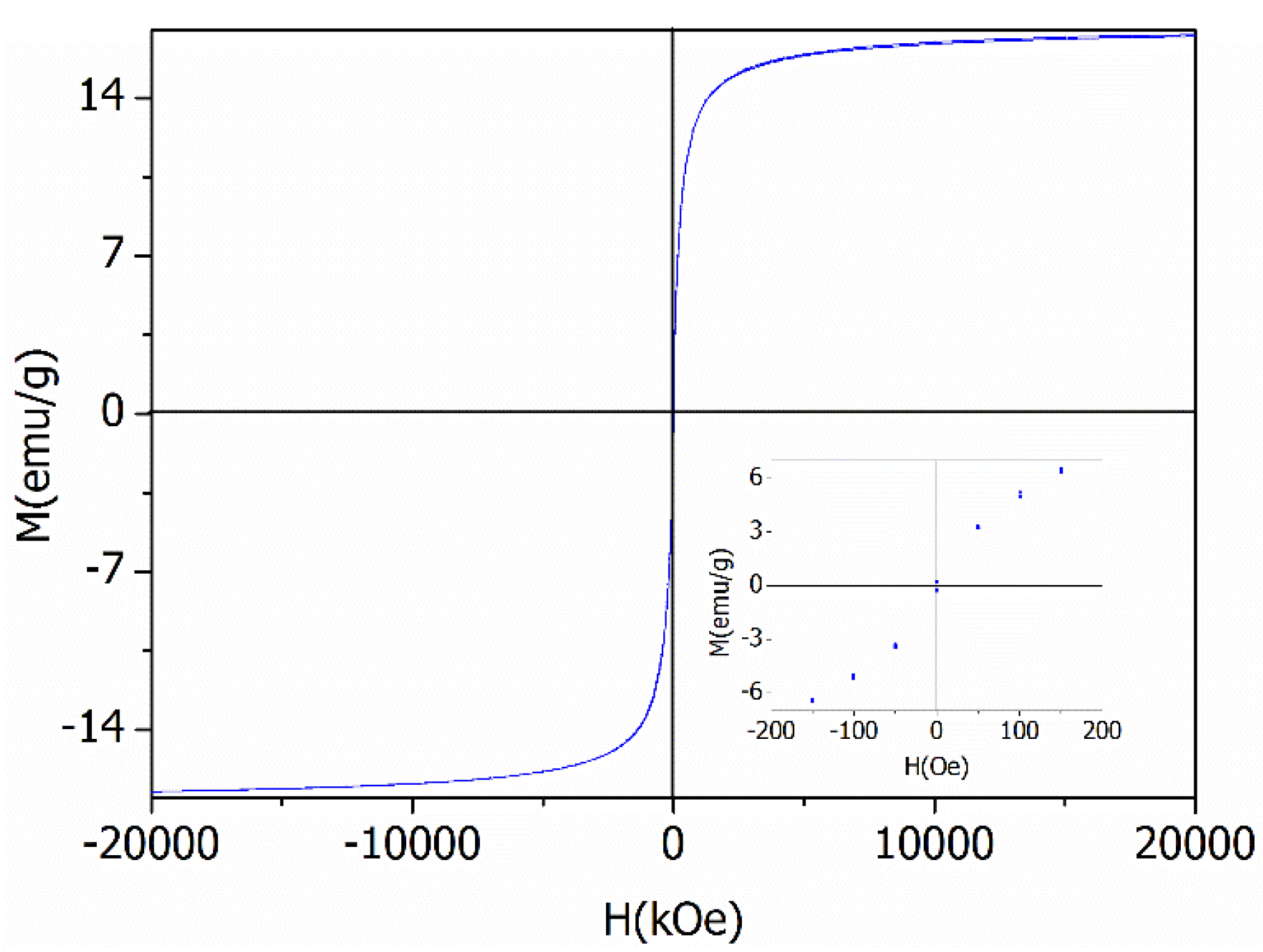
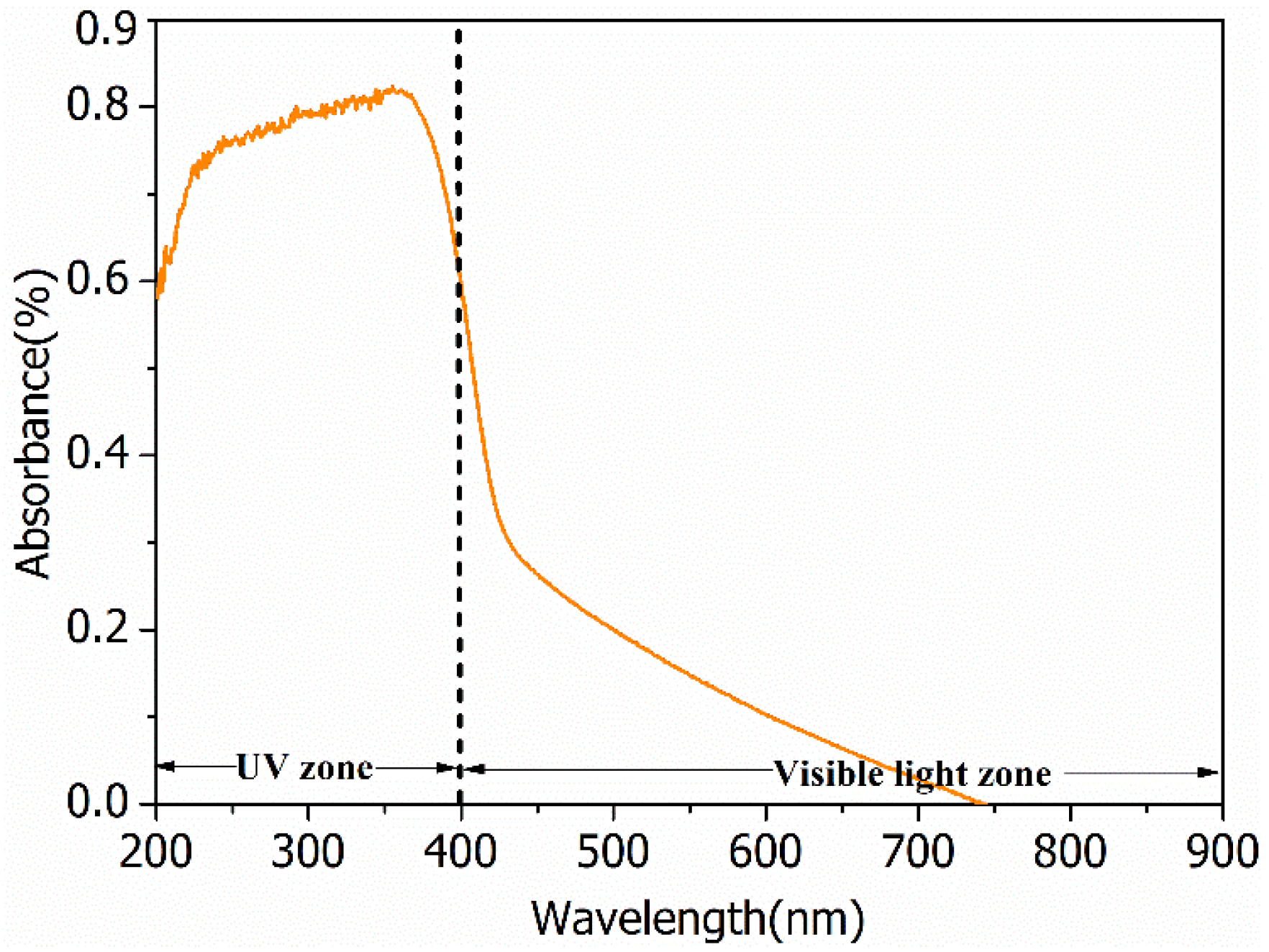
2.1. Characterization of the Photocatalyst
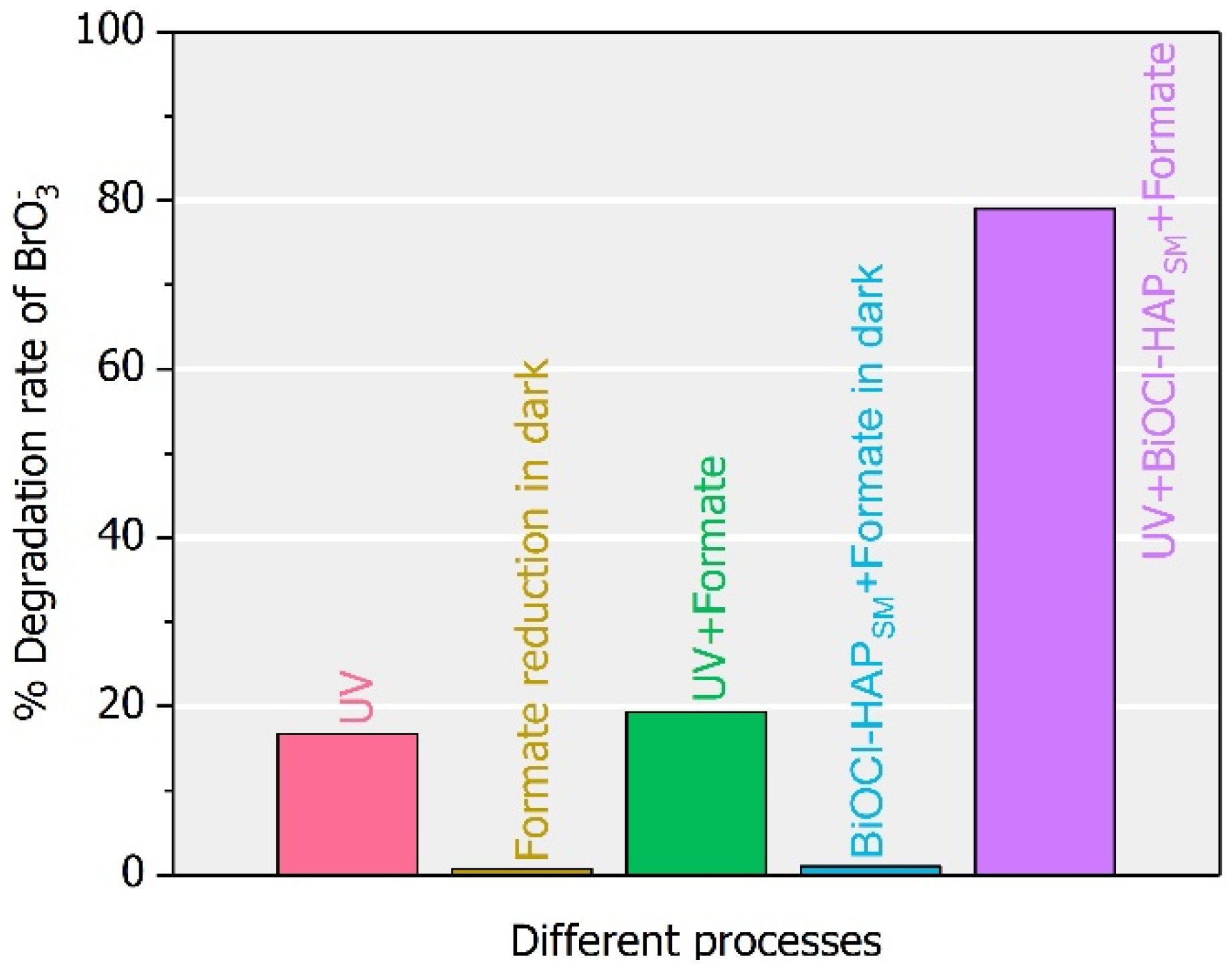
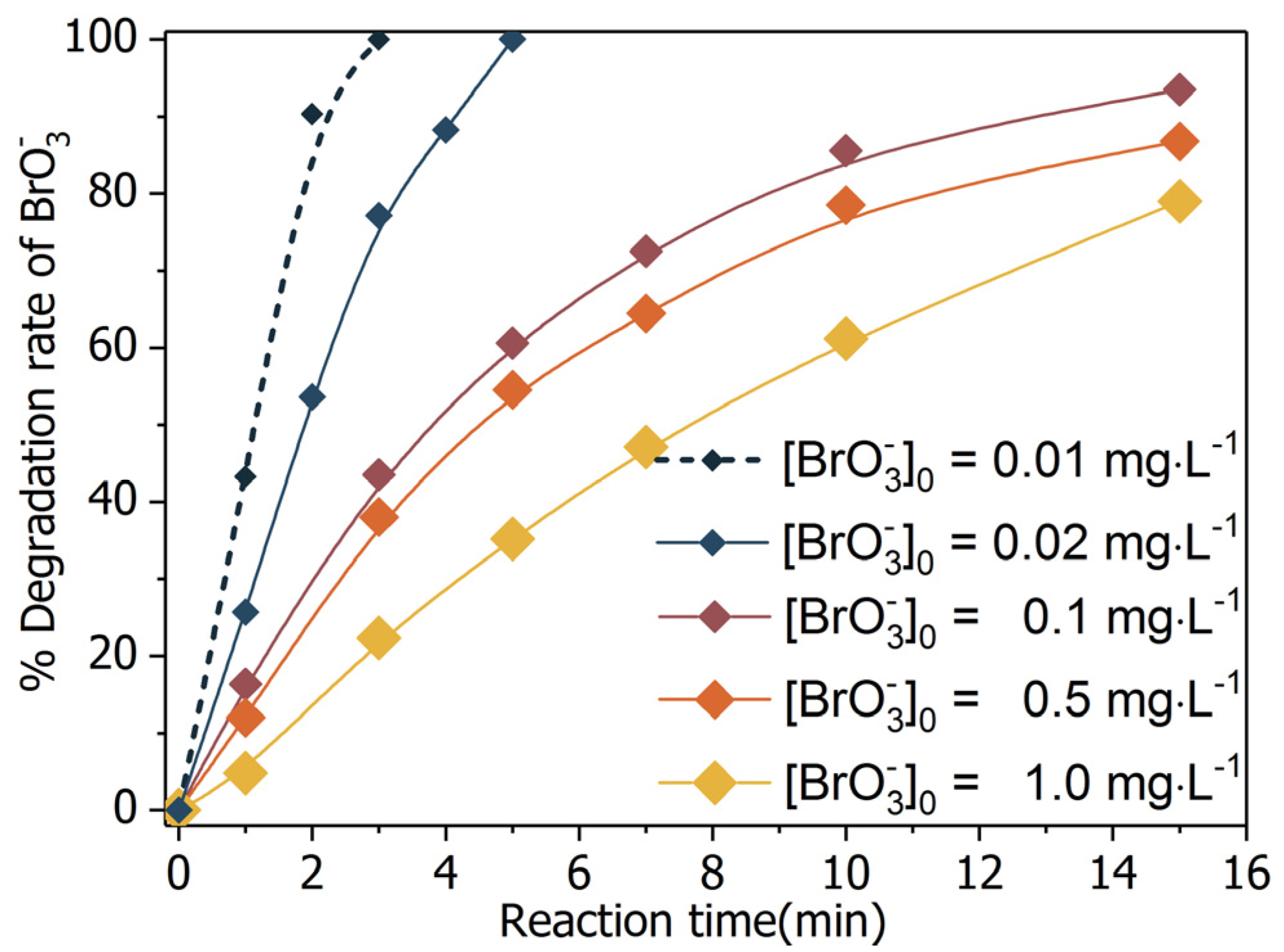
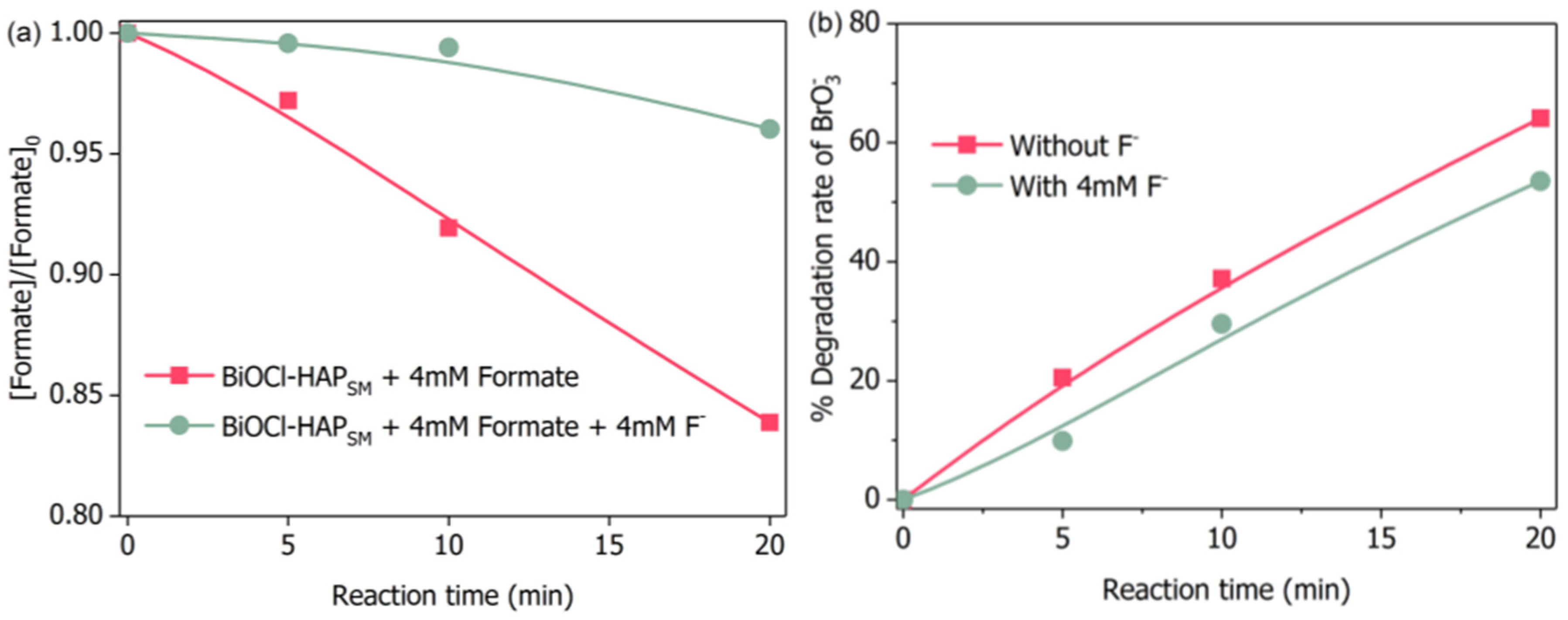
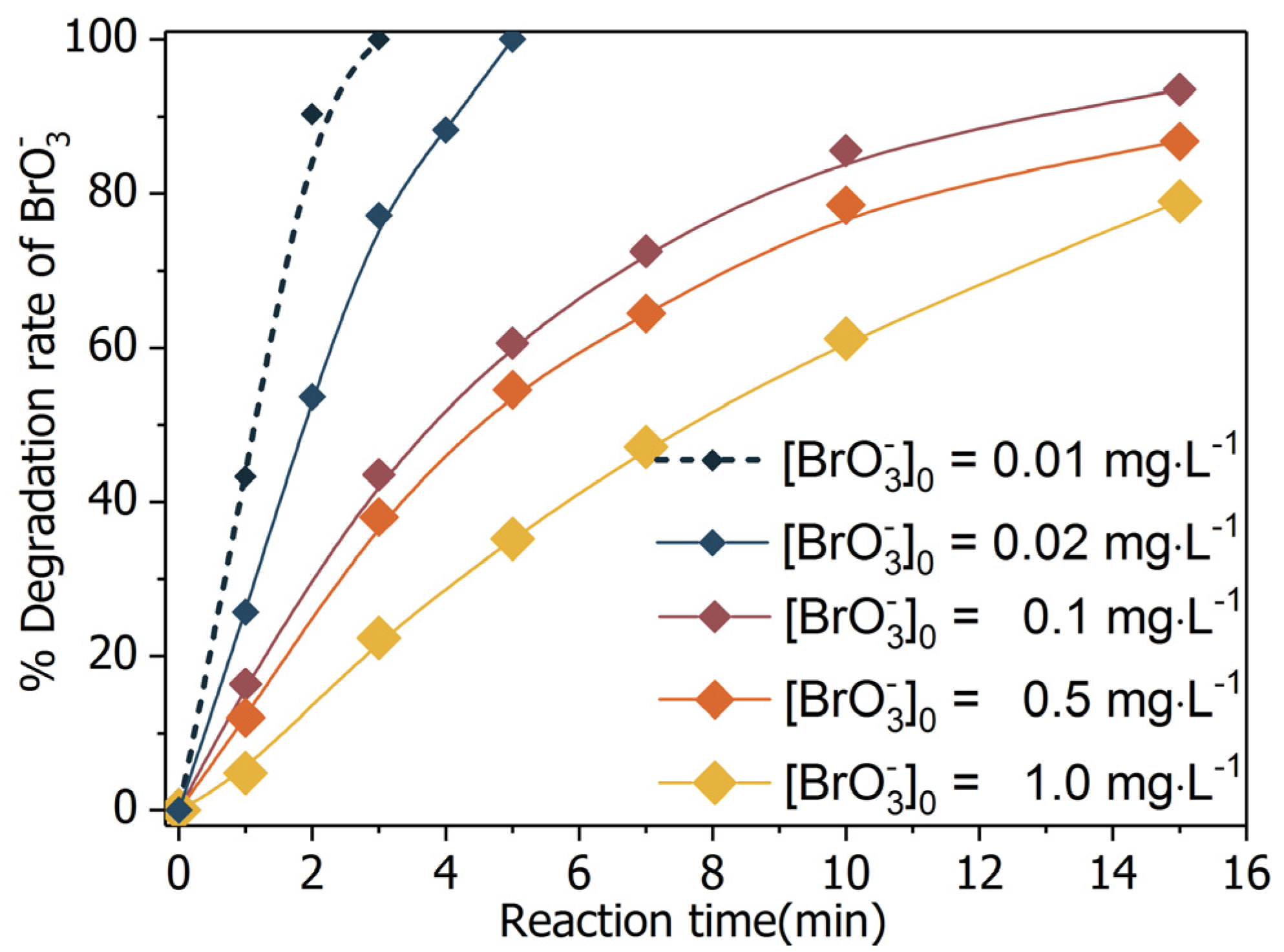
2.2. Bromate Degradation by UV/BiOCl-HAPSM in the Presence of Formate
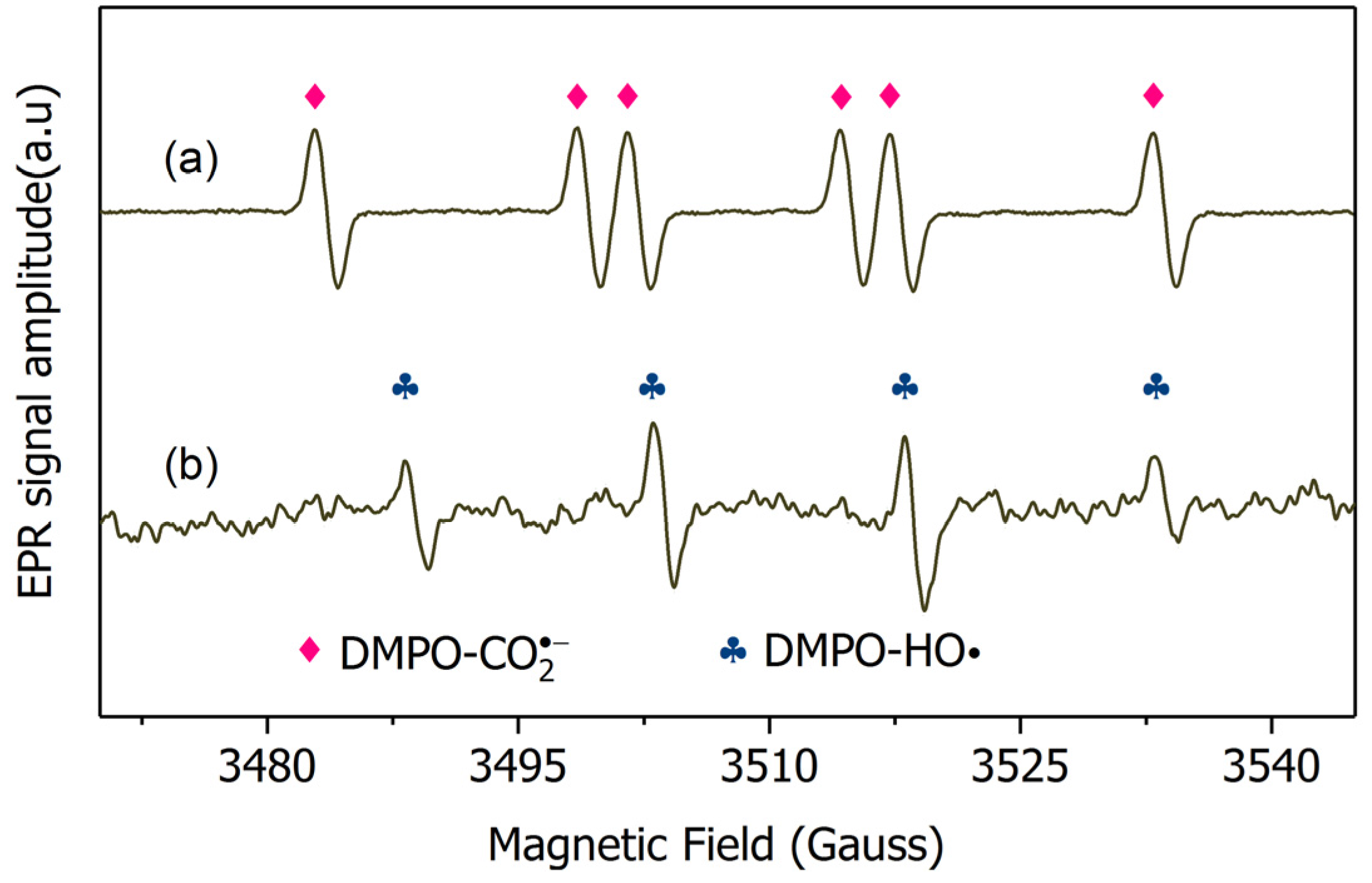
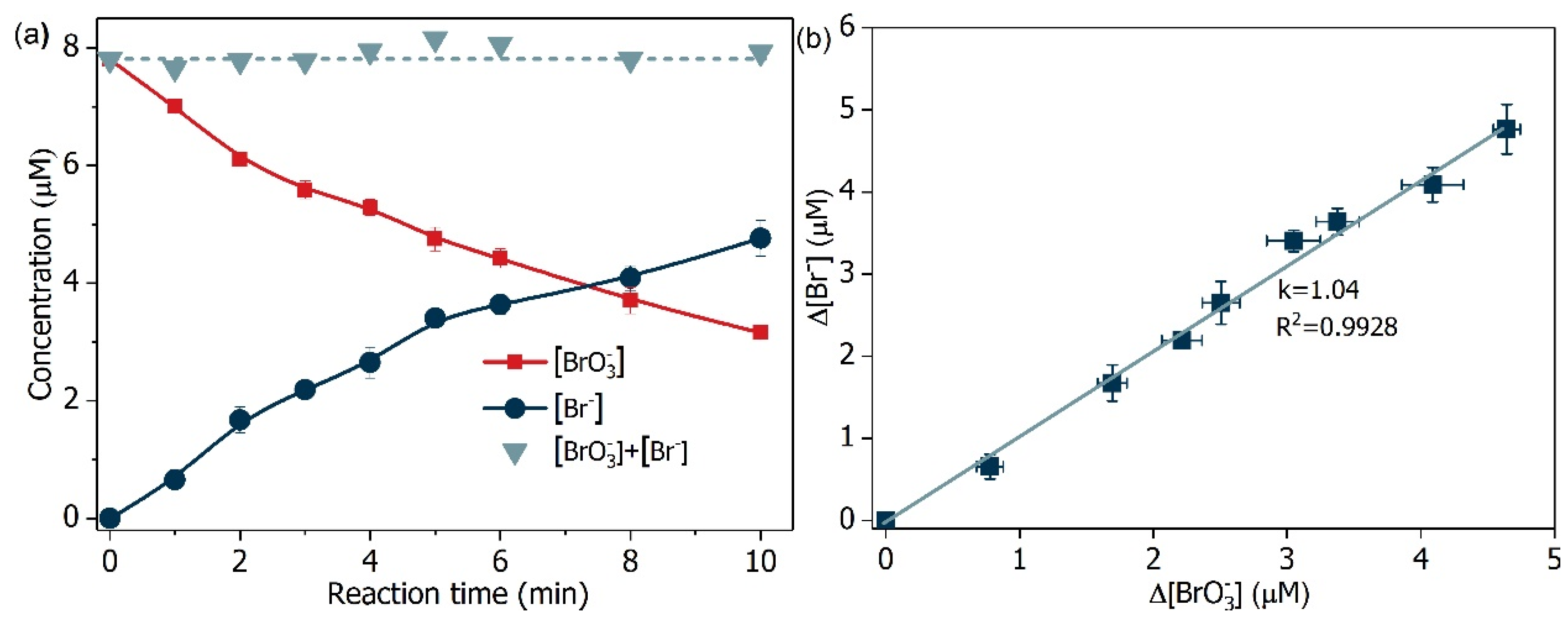
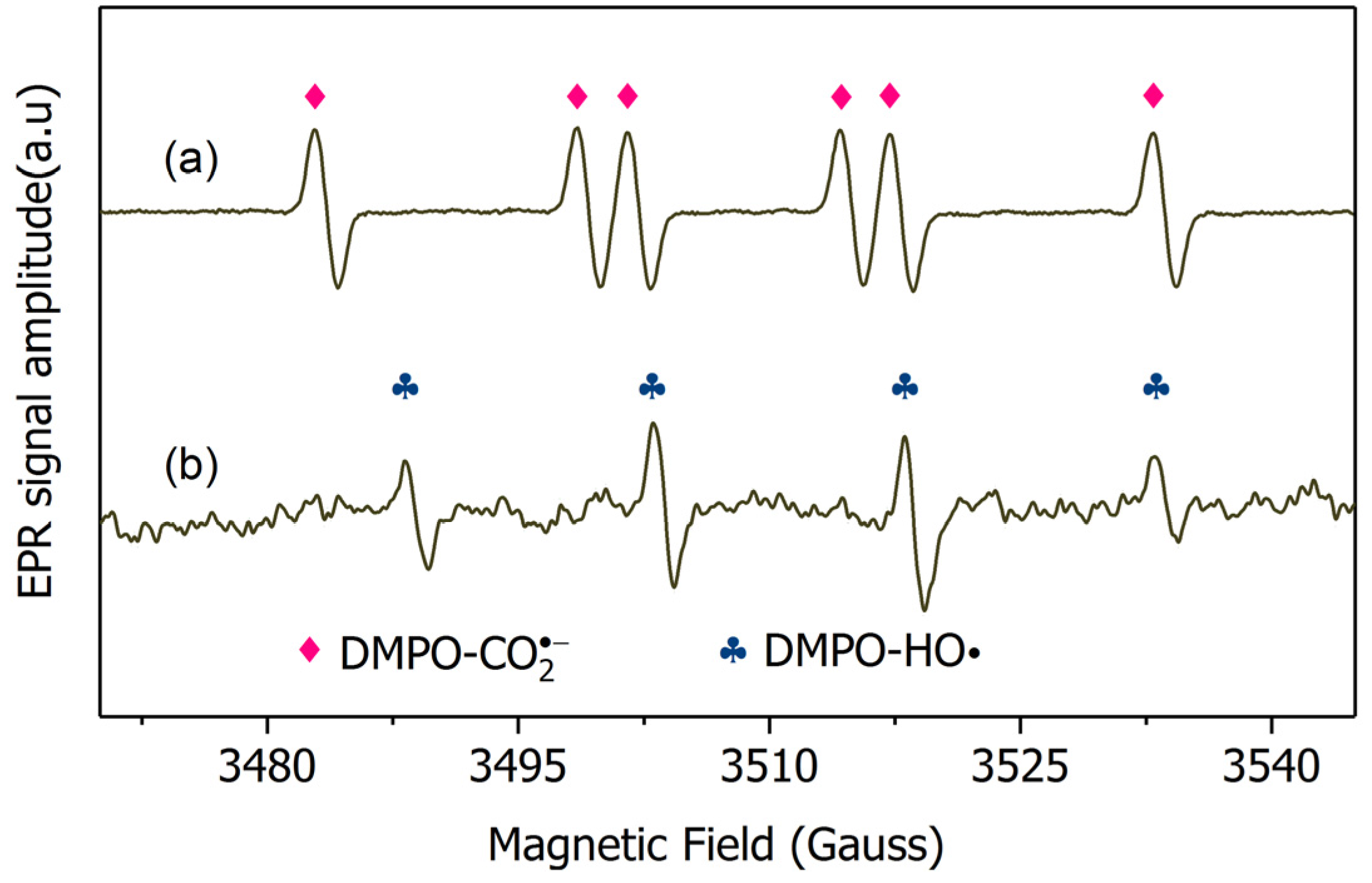
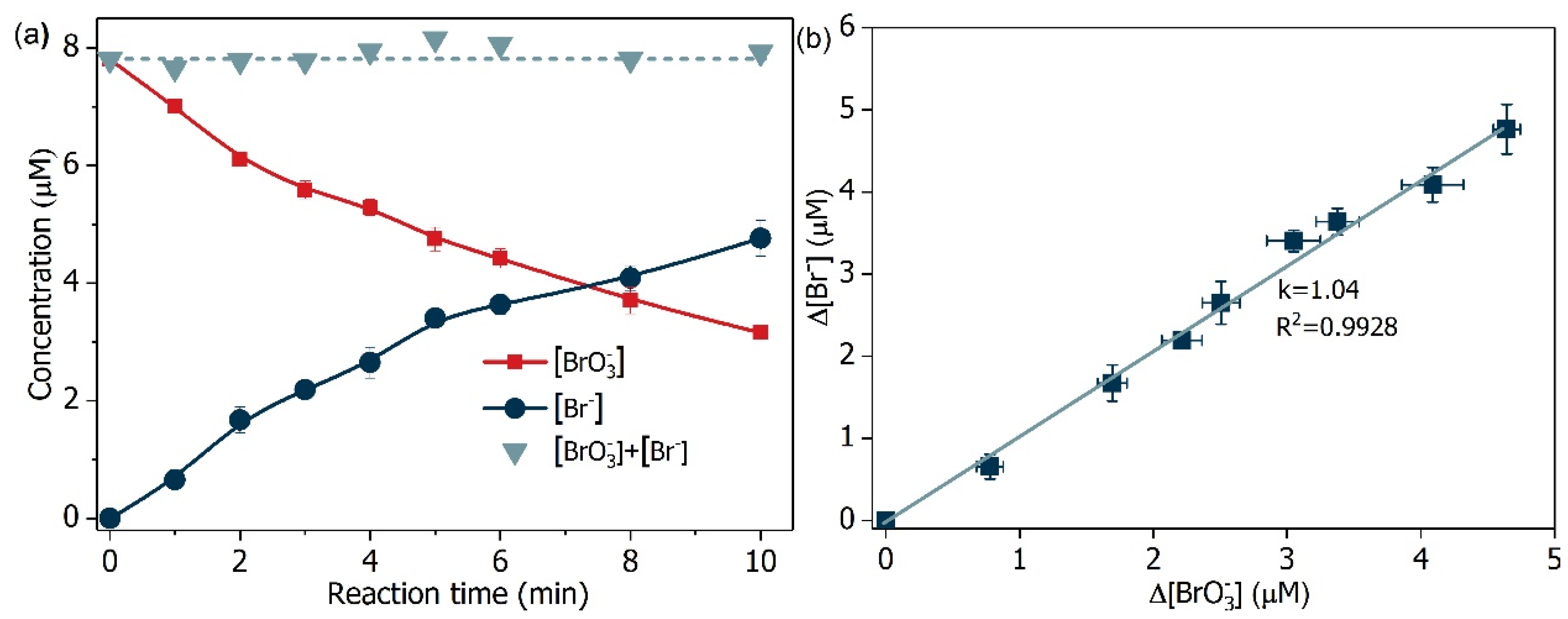
2.3. Mechanism Clarification
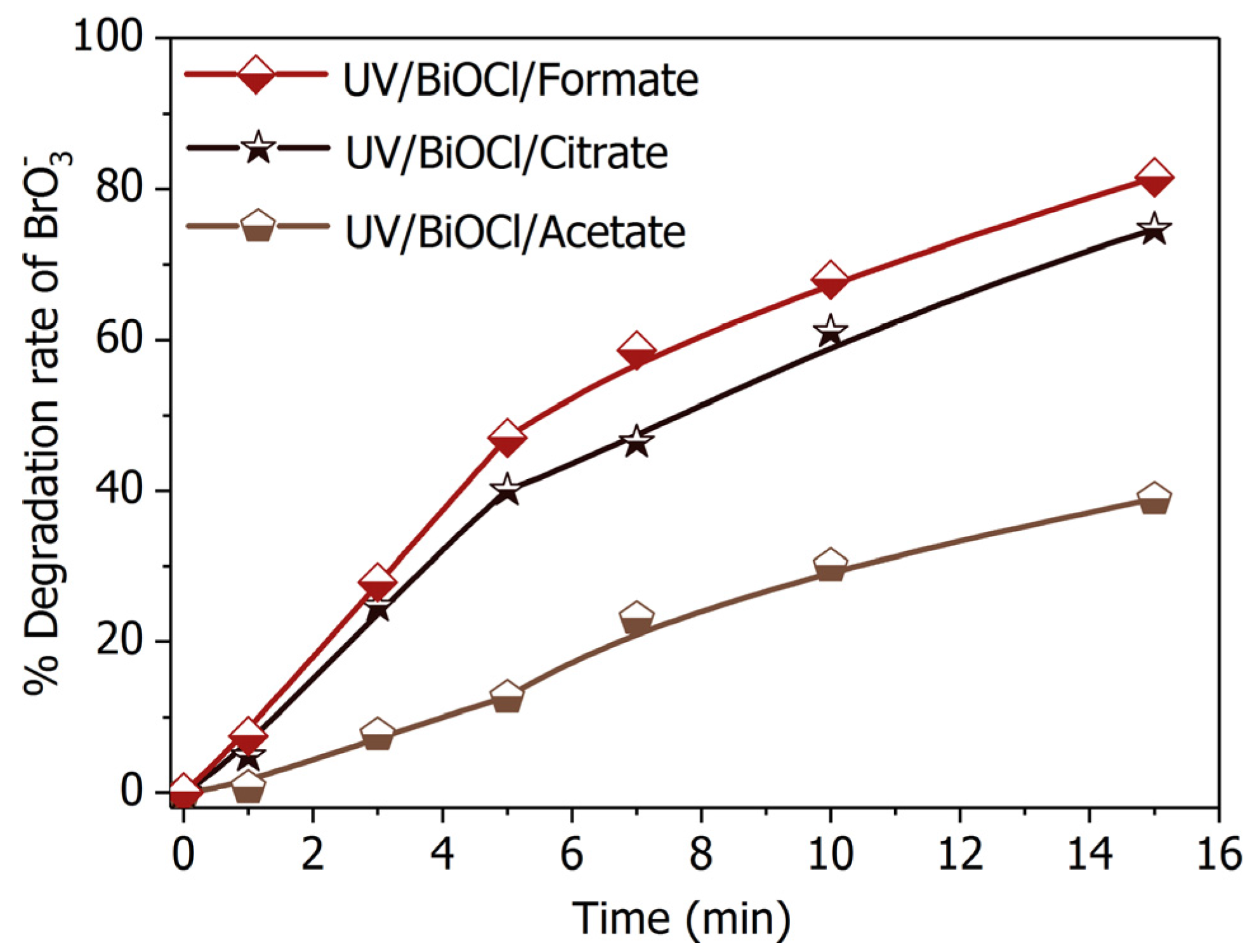
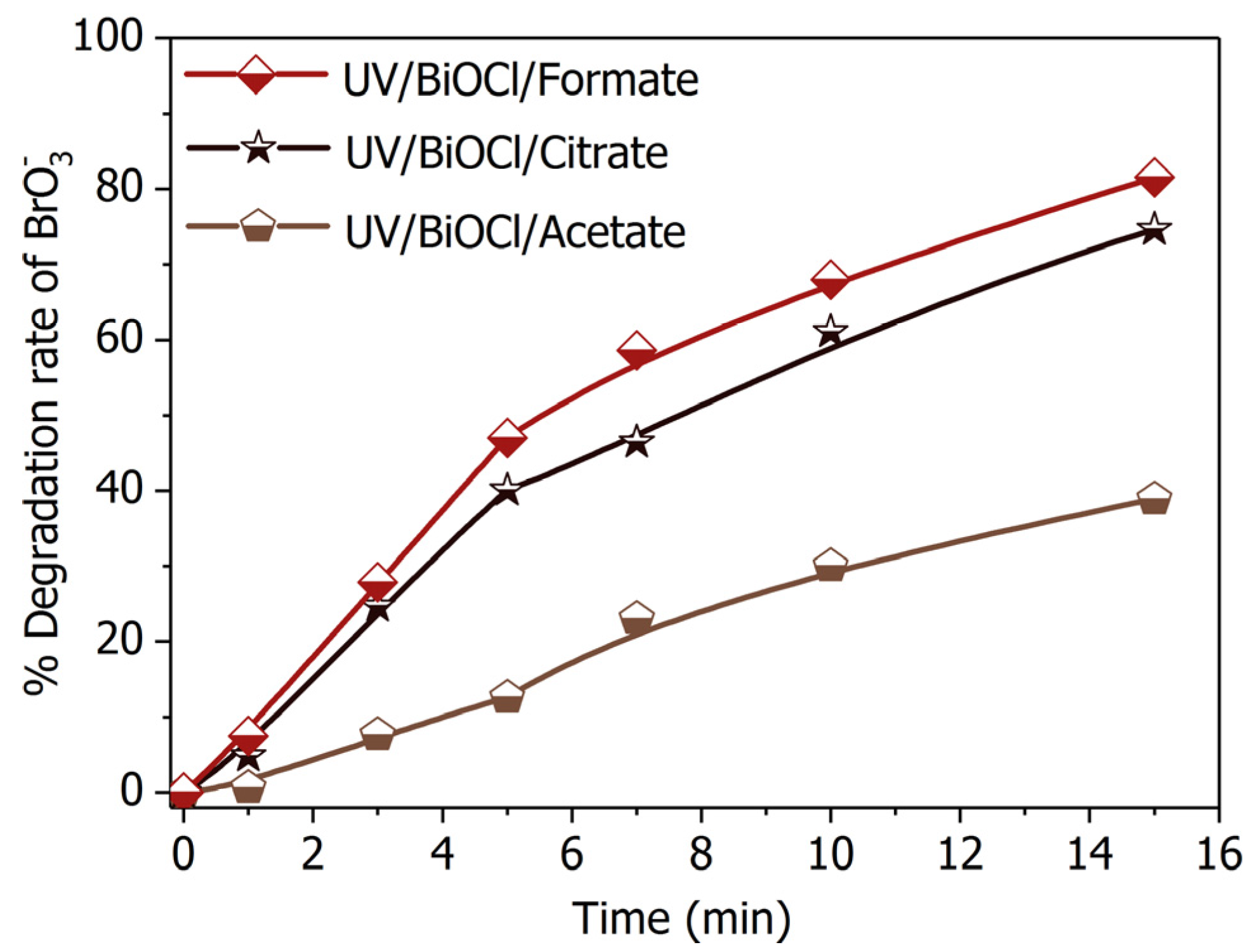
2.4. Substitutes of Formate
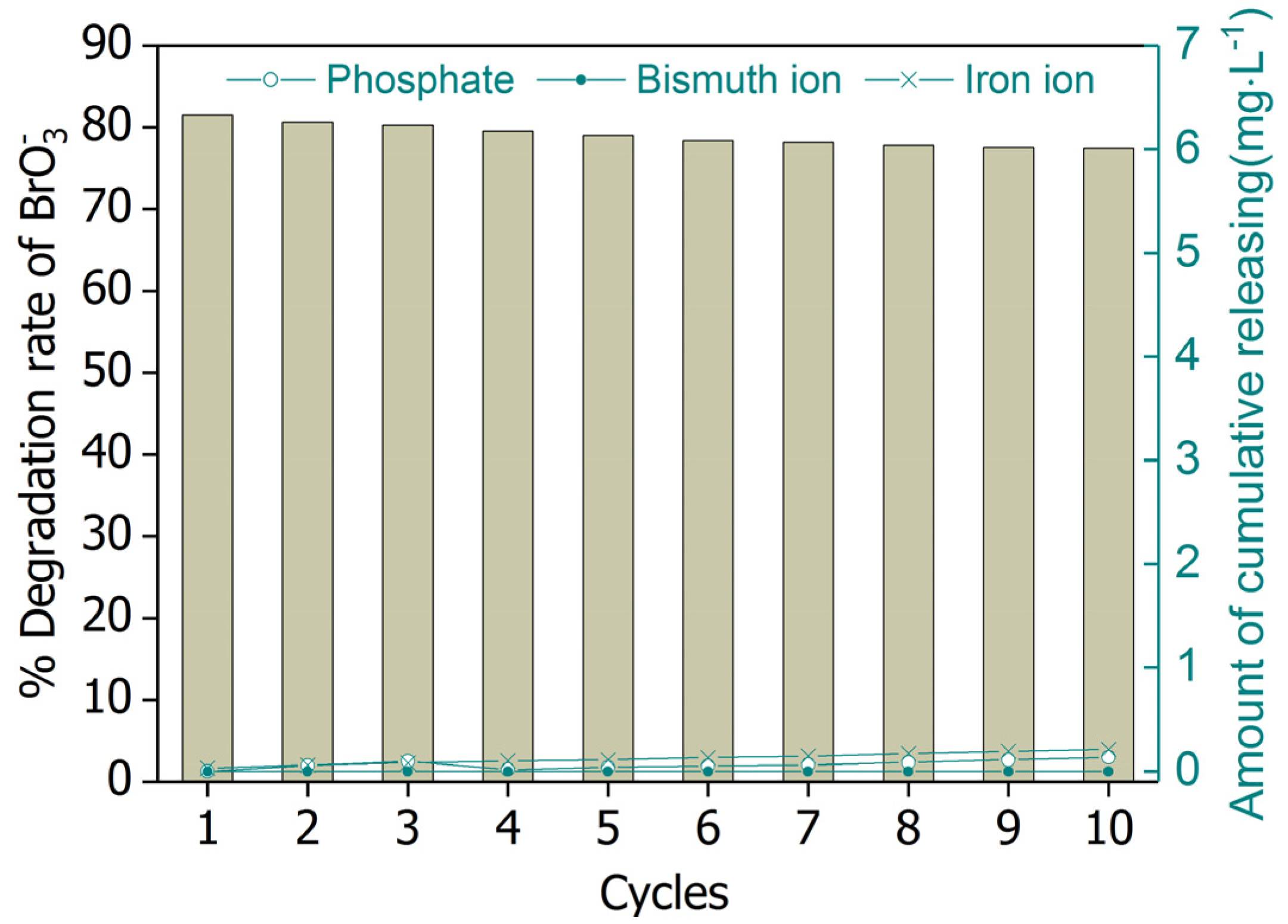
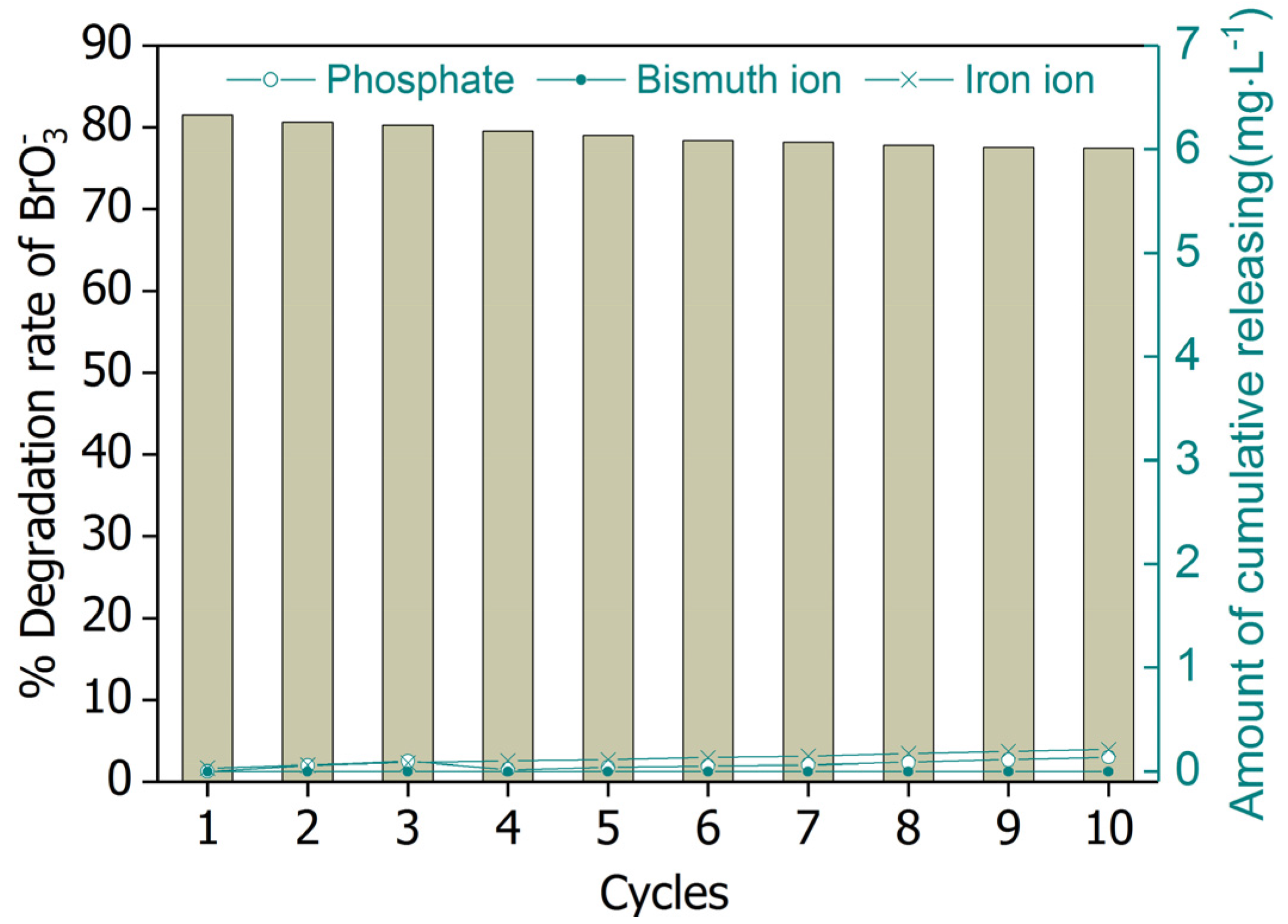
2.5. Reusability of BiOCl-HAPSM
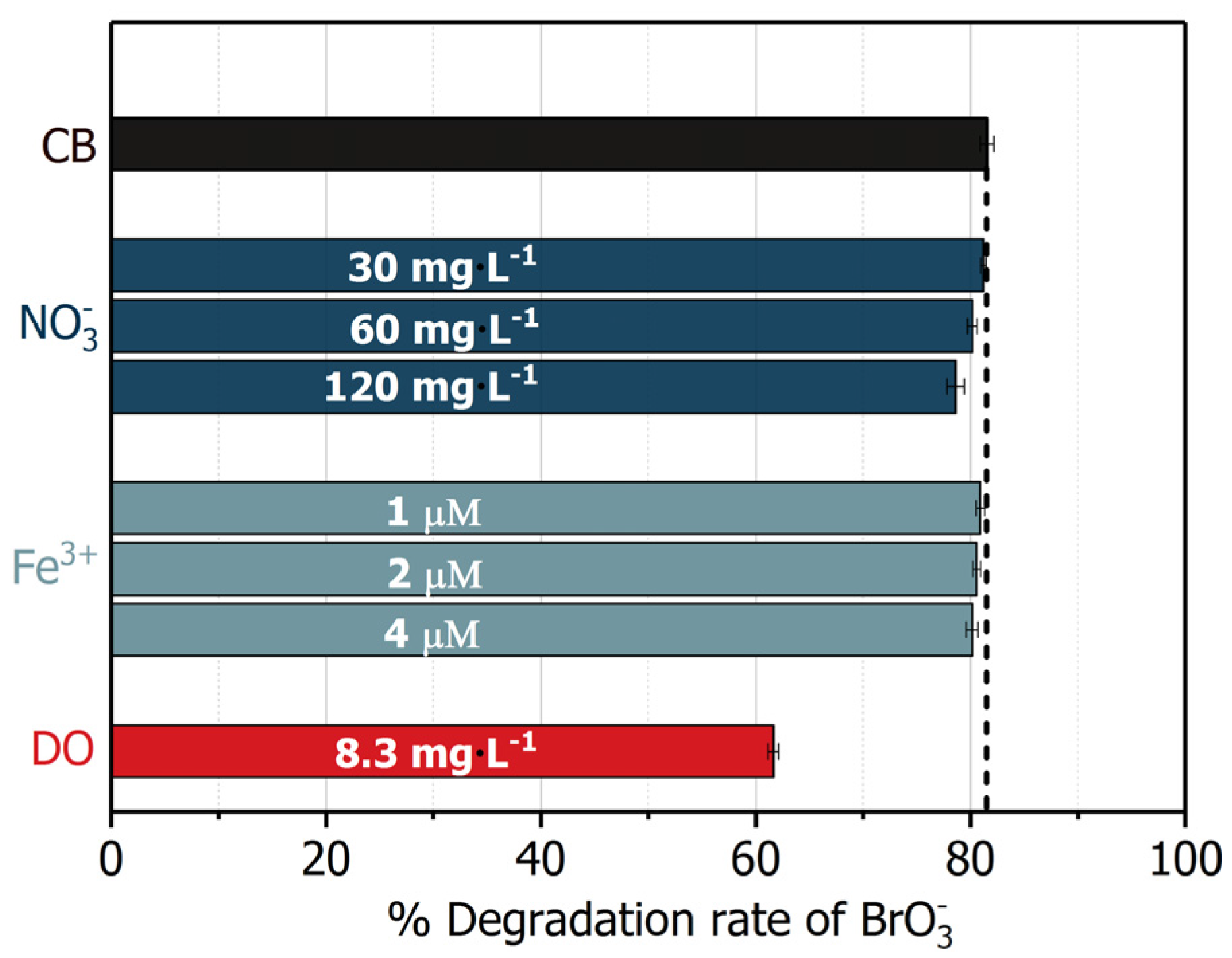
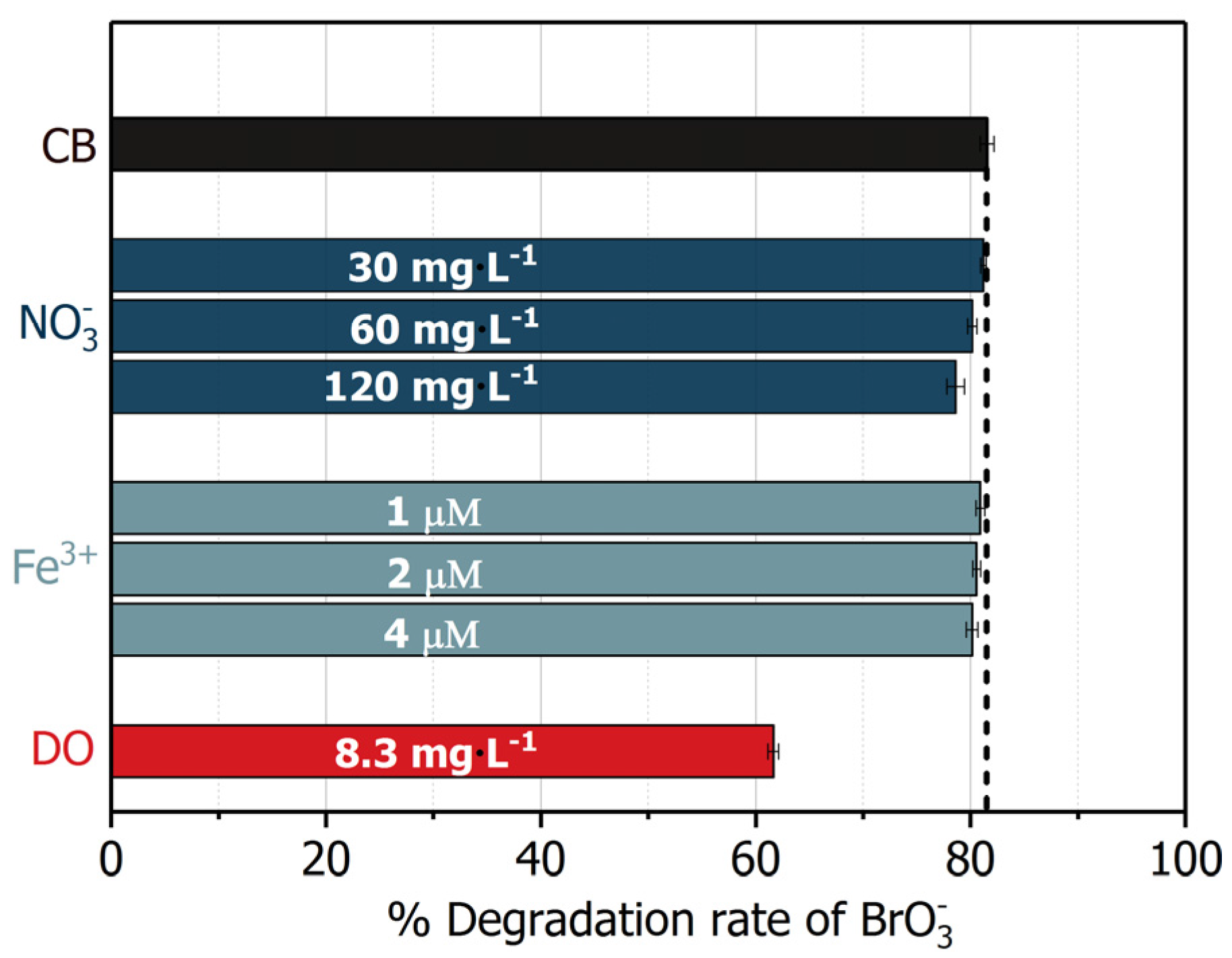
2.6. Influence of Common Electron Acceptors
2.7. Transformation Intermediates of
3. Materials and Methods
3.1. Materials and Reagents
3.2. Synthesis and Characterization of BiOCl-HAPSM
3.3. Experimental Procedures
3.4. Analysis Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
References
- Liu, C.; von Gunten, U.; Croué, J. Chlorination of bromide-containing waters: Enhanced bromate formation in the presence of synthetic metal oxides and deposits formed in drinking water distribution systems. Water Res. 2013, 47, 5307–5315. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Shang, C. Bromate formation from bromide oxidation by the UV/persulfate process. Environ. Sci. Technol. 2012, 46, 8976–8983. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, H.S.; Delcomyn, C.A.; Unnam, V. Bromate in chlorinated drinking waters: Occurrence and implications for future regulation. Environ. Sci. Technol. 2003, 37, 3104–3110. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.; Zhai, W.; Amy, G.; Mysore, C. Bromate ion removal by activated carbon. Water Res. 1996, 30, 1651–1660. [Google Scholar] [CrossRef]
- Bhatnagar, A.; Choi, Y.; Yoon, Y.; Shin, Y.; Jeon, B.; Kang, J. Bromate removal from water by granular ferric hydroxide (GFH). J. Hazard. Mater. 2009, 170, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Naushad, M.; Alothman, Z.A.; Khan, M.R.; Wabaidur, S.M. Removal of bromate from water using de-Acidite FF-IP resin and determination by ultra-performance liquid chromatography-tandem mass spectrometry. CLEAN Soil Air Water 2013, 41, 528–533. [Google Scholar] [CrossRef]
- Hatzistavros, V.S.; Koulouridakis, P.E.; Aretaki, I.I.; Kallithrakas-Kontos, N.G. Bromate determination in water after membrane complexation and total reflection X-ray fluorescence analysis. Anal. Chem. 2007, 79, 2827–2832. [Google Scholar] [CrossRef] [PubMed]
- Bensalah, N.; Liu, X.; Abdel-Wahab, A. Bromate reduction by ultraviolet light irradiation using medium pressure lamp. Int. J. Environ. Stud. 2013, 70, 566–582. [Google Scholar] [CrossRef]
- Kishimoto, N.; Matsuda, N. Bromate ion removal by electrochemical reduction using an activated carbon felt electrode. Environ. Sci. Technol. 2009, 43, 2054–2059. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Shang, C. Effects of copper and palladium on the reduction of bromate by Fe(0). Chemosphere 2006, 64, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.; Gauw, R.D.; Emmert, G.L.; Walters, B.D.; Bubnis, B. Chemical reduction methods for bromate ion removal. J. AWWA 2002, 94, 91–98. [Google Scholar]
- Chen, H.; Xu, Z.; Wan, H.; Zheng, J.; Yin, D.; Zheng, S. Aqueous bromate reduction by catalytic hydrogenation over Pd/Al2O3 catalysts. Appl. Catal. B Environ. 2010, 96, 307–313. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, T.; Shao, Y. Aqueous bromate reduction by UV activation of sulfite. CLEAN Soil Air Water 2014, 42, 1370–1375. [Google Scholar] [CrossRef]
- Siddiqui, M.S.; Amy, G.L.; Cooper, W.J.; Kurucz, C.N.; Waite, T.D.; Nickelsen, M.G. Bromate ion removal by heeb irradiation. J. AWWA 1996, 88, 90–101. [Google Scholar]
- Hong, H.; Cao, H.; Wang, Y. Formation and genotoxicity of a guanine-cytosine intrastrand cross-link lesion in vivo. Nucleic Acids Res. 2007, 35, 7118–7127. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Cao, M.; Tan, Z.; Wang, L.; Yuan, S.; Chen, J. Photochemical decomposition of perfluorodecanoic acid in aqueous solution with vuv light irradiation. J. Hazard. Mater. 2010, 181, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Song, W.; Cooper, W.J.; Jung, J.; Greaves, J. Degradation of tetracycline antibiotics: Mechanisms and kinetic studies for advanced oxidation/reduction processes. Chemosphere 2010, 78, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Ma, J.; Li, X.; Fang, J.; Chen, L. Influence of pH on the formation of sulfate and hydroxyl radicals in the UV/peroxymonosulfate system. Environ. Sci. Technol. 2011, 45, 9308–9314. [Google Scholar] [CrossRef] [PubMed]
- Wenk, J.; von Gunten, U.; Canonica, S. Effect of dissolved organic matter on the transformation of contaminants induced by excited triplet states and the hydroxyl radical. Environ. Sci. Technol. 2011, 45, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Buffle, M.; Schumacher, J.; Meylan, S.; Jekel, M.; von Gunten, U. Ozonation and advanced oxidation of wastewater: Effect of O3 dose, pH, DOM and HO-scavengers on ozone decomposition and ho generation. Ozone Sci. Eng. 2006, 28, 247–259. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/O−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, T.; Wang, L.; Shao, Y.; Fang, L. Hydrated electron-based degradation of atenolol in aqueous solution. Chem. Eng. J. 2015, 260, 740–748. [Google Scholar] [CrossRef]
- Liu, X.; Zhong, J.; Fang, L.; Wang, L.; Ye, M.; Shao, Y.; Li, J.; Zhang, T. Trichloroacetic acid reduction by an advanced reduction process based on carboxyl anion radical. Chem. Eng. J. 2016, 303, 56–63. [Google Scholar] [CrossRef]
- Ibhadon, A.; Fitzpatrick, P. Heterogeneous photocatalysis: Recent advances and applications. Catalysts 2013, 3, 189–218. [Google Scholar] [CrossRef]
- Li, J.; Yu, Y.; Zhang, L. Bismuth oxyhalide nanomaterials: Layered structures meet photocatalysis. Nanoscale 2014, 6, 8473–8488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liu, C.; Huang, F.; Zheng, C.; Wang, W. Study of the electronic structure and photocatalytic activity of the biocl photocatalyst. Appl. Catal. B Environ. 2006, 68, 125–129. [Google Scholar] [CrossRef]
- Deng, H.; Wang, J.; Peng, Q.; Wang, X.; Li, Y. Controlled hydrothermal synthesis of bismuth oxyhalide nanobelts and nanotubes. Chem. Eur. J. 2005, 11, 6519–6524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ai, Z.; Jia, F.; Zhang, L. Generalized one-pot synthesis, characterization, and photocatalytic activity of hierarchical biox (X = Cl, Br, I) nanoplate microspheres. J. Phys. Chem. C 2008, 112, 747–753. [Google Scholar] [CrossRef]
- Xia, J.; Di, J.; Li, H.; Xu, H.; Li, H.; Guo, S. Ionic liquid-induced strategy for carbon quantum dots/biox (X = Cl, Br, I) hybrid nanosheets with superior visible light-driven photocatalysis. Appl. Catal. B Environ. 2016, 181, 260–269. [Google Scholar] [CrossRef]
- Kong, L.; Jiang, Z.; Xiao, T.; Lu, L.; Jones, M.O.; Edwards, P.P. Exceptional visible-light-driven photocatalytic activity over BiOBr-ZnFe2O4 heterojunctions. Chem. Commun. 2011, 47, 5512–5514. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Huang, W.; Zhou, H.; Yin, H.; Zheng, Y.; Song, X. A novel Fe3O4@SiO2@BiOBr photocatalyst with highly active visible light photocatalytic properties. Mater. Chem. Phys. 2014, 148, 896–902. [Google Scholar] [CrossRef]
- Choi, Y.; Kim, Y.; Cho, D.; Kang, J.; Leung, K.; Sohn, Y. Recyclable magnetic CoFe2O4/ BiOX (X = Cl, Br and I) microflowers for photocatalytic treatment of water contaminated with methyl orange, rhodamine B, methylene blue, and a mixed dye. RSC Adv. 2015, 5, 79624–79634. [Google Scholar] [CrossRef]
- Jiang, W.; Qiu, Z.; Yao, W.; Zhu, Y.; Cui, W. TiO2/Al(H2PO4)3 composite film as separation-free and washing-resistance photocatalyst. Appl. Catal. B Environ. 2017, 204, 43–48. [Google Scholar] [CrossRef]
- Wang, N.; Zhu, L.; Deng, K.; She, Y.; Yu, Y.; Tang, H. Visible light photocatalytic reduction of Cr(VI) on TiO2 in situ modified with small molecular weight organic acids. Appl. Catal. B Environ. 2010, 95, 400–407. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, P. Photocatalytic decomposition of perfluorooctanoic acid (PFOA) by TiO2 in the presence of oxalic acid. J. Hazard. Mater. 2011, 192, 1869–1875. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.; Sin, J.; Abdullah, A.Z.; Mohamed, A.R. Degradation of wastewaters containing organic dyes photocatalysed by zinc oxide: A review. Desalin. Water Treat. 2012, 41, 131–169. [Google Scholar] [CrossRef]
- Doudrick, K.; Yang, T.; Hristovski, K.; Westerhoff, P. Photocatalytic nitrate reduction in water: Managing the hole scavenger and reaction by-product selectivity. Appl. Catal. B Environ. 2013, 136–137, 40–47. [Google Scholar] [CrossRef]
- Zhang, J.; Nosaka, Y. Mechanism of the OH radical generation in photocatalysis with TiO2 of different crystalline types. J. Phys. Chem. C 2014, 118, 10824–10832. [Google Scholar] [CrossRef]
- Perissinotti, L.L.; Brusa, M.A.; Grela, M.A. Yield of carboxyl anion radicals in the photocatalytic degradation of formate over TiO2 particles. Langmuir 2001, 17, 8422–8427. [Google Scholar] [CrossRef]
- Chawla, O.P.; Fessenden, R.W. Electron spin resonance and pulse radiolysis studies of some reactions of SO4−. J. Phys. Chem. 1975, 79, 2693–2700. [Google Scholar] [CrossRef]
- Lai, Y.; Liu, F.; Zhang, Z.; Liu, J.; Li, Y.; Kuang, S.; Li, J.; Liu, Y. Cyclic voltammetry study of electrodeposition of Cu(In,Ga)Se2 thin films. Electrochim. Acta 2009, 54, 3004–3010. [Google Scholar] [CrossRef]
- Hotten, P.; Marotta, F.; Naito, Y.; Minelli, E.; Helmy, A.; Lighthouse, J.; Fuji, H.; Fesce, E. Effects of probiotics, lactitol and rifaximin on intestinal flora and fecal excretion of organic acids in cirrhotic patients. Chin. J. Digest. Dis. 2003, 4, 13–18. [Google Scholar] [CrossRef]
- Hu, P.; Long, M. Cobalt-catalyzed sulfate radical-based advanced oxidation: A review on heterogeneous catalysts and applications. Appl. Catal. B Environ. 2016, 181, 103–117. [Google Scholar] [CrossRef]
- Marinho, B.A.; Cristóvão, R.O.; Djellabi, R.; Loureiro, J.M.; Boaventura, R.A.R.; Vilar, V.J.P. Photocatalytic reduction of Cr(VI) over TiO2-coated cellulose acetate monolithic structures using solar light. Appl. Catal. B Environ. 2017, 203, 18–30. [Google Scholar] [CrossRef]
- Tan, T.; Beydoun, D.; Amal, R. Effects of organic hole scavengers on the photocatalytic reduction of selenium anions. J. Photochem. Photobiol. A 2003, 159, 273–280. [Google Scholar] [CrossRef]
- Carraway, E.R.; Hoffman, A.J.; Hoffmann, M.R. Photocatalytic oxidation of organic acids on quantum-sized semiconductor colloids. Environ. Sci. Technol. 1994, 28, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Cooper, W.J.; Cramer, C.J.; Martin, N.H.; Mezyk, S.P.; O’Shea, K.E.; Sonntag, C.V. Free radical mechanisms for the treatment of methyl tert-butyl ether (MTBE) via advanced oxidation/reductive processes in aqueous solutions. Chem. Rev. 2009, 109, 1302–1345. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J. Heterogeneous photocatalysis: Fundamentals and applications to the removal of various types of aqueous pollutants. Catal. Today 1999, 53, 115–129. [Google Scholar] [CrossRef]
- Meichtry, J.M.; Quici, N.; Mailhot, G.; Litter, M.I. Heterogeneous photocatalytic degradation of citric acid over TiO2: II. Mechanism of citric acid degradation. Appl. Catal. B Environ. 2011, 102, 555–562. [Google Scholar] [CrossRef]
- Salvador, P. On the nature of photogenerated radical species active in the oxidative degradation of dissolved pollutants with TiO2 aqueous suspensions: A revision in the light of the electronic structure of adsorbed water. J. Phys. Chem. C 2007, 111, 17038–17043. [Google Scholar] [CrossRef]
- Li, X.; Ma, J.; Liu, G.; Fang, J.; Yue, S.; Guan, Y.; Chen, L.; Liu, X. Efficient reductive dechlorination of monochloroacetic acid by sulfite/UV process. Environ. Sci. Technol. 2012, 46, 7342–7349. [Google Scholar] [CrossRef] [PubMed]
- Neta, P.; Huie, R.E.; Ross, A.B. Rate constants for reactions of inorganic radicals in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Bard, A.J.; Parsons, R.; Jordan, J. Standard Potentials in Aqueous Solution, 1st ed.; IUPAC: New York, NY, USA, 1985; pp. 78–83. [Google Scholar]
- Huff Hartz, K.E.; Nicoson, J.S.; Wang, L.; Margerum, D.W. Kinetics and mechanisms of S(IV) reductions of bromite and chlorite ions. Inorg. Chem. 2003, 42, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Kilduff, J.; Weber, W.J. Transport and separation of organic macromolecules in ultrafiltration processes. Environ. Sci. Technol. 1992, 26, 569–577. [Google Scholar] [CrossRef]
- Liao, J.; Sihler, H.; Huey, L.G.; Neuman, J.A.; Tanner, D.J.; Friess, U.; Platt, U.; Flocke, F.M.; Orlando, J.J.; Shepson, P.B.; et al. A comparison of arctic BrO measurements by chemical ionization mass spectrometry and long path-differential optical absorption spectroscopy. J. Geophys. Res. Atmos. 2011, 116, 1–14. [Google Scholar] [CrossRef]
- Orban, M.; Epstein, I.R. Simple and complex pH oscillations and bistability in the phenol-perturbed bromite-hydroxylamine reaction. J. Phys. Chem. 1994, 98, 2930–2935. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electron Acceptors e | Concentration | Reaction Rate Constant with | Scavenging Rate |
|---|---|---|---|
| 6.6 μM | 1.4 × 109 M−1·s−1 | 9.24 × 103 s−1 | |
| 30–120 mg·L−1 | 6.5 × 105 M−1·s−1 [52] | (0.3–1.3) × 103 s−1 | |
| DO | 8.1 mg·L−1 | 2.4 × 109 M−1·s−1 [52] | 6.0 × 105 s−1 |
| 1–4 μM | 5.0 × 107 M−1·s−1 [52] | (0.1–2.0) × 102 s−1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Wang, L.; Sun, Z.; Shao, Y.; Yu, T. Treatment of Aqueous Bromate by Superparamagnetic BiOCl-Mediated Advanced Reduction Process. Catalysts 2017, 7, 131. https://doi.org/10.3390/catal7050131
Liu X, Wang L, Sun Z, Shao Y, Yu T. Treatment of Aqueous Bromate by Superparamagnetic BiOCl-Mediated Advanced Reduction Process. Catalysts. 2017; 7(5):131. https://doi.org/10.3390/catal7050131
Chicago/Turabian StyleLiu, Xiaowei, Lili Wang, Zhe Sun, Yu Shao, and Tingchao Yu. 2017. "Treatment of Aqueous Bromate by Superparamagnetic BiOCl-Mediated Advanced Reduction Process" Catalysts 7, no. 5: 131. https://doi.org/10.3390/catal7050131
APA StyleLiu, X., Wang, L., Sun, Z., Shao, Y., & Yu, T. (2017). Treatment of Aqueous Bromate by Superparamagnetic BiOCl-Mediated Advanced Reduction Process. Catalysts, 7(5), 131. https://doi.org/10.3390/catal7050131