
Methanation of Carbon Dioxide over Ni–Ce–Zr Oxides Prepared by One-Pot Hydrolysis of Metal Nitrates with Ammonium Carbonate

Abstract
:1. Introduction
2. Results and Discussion
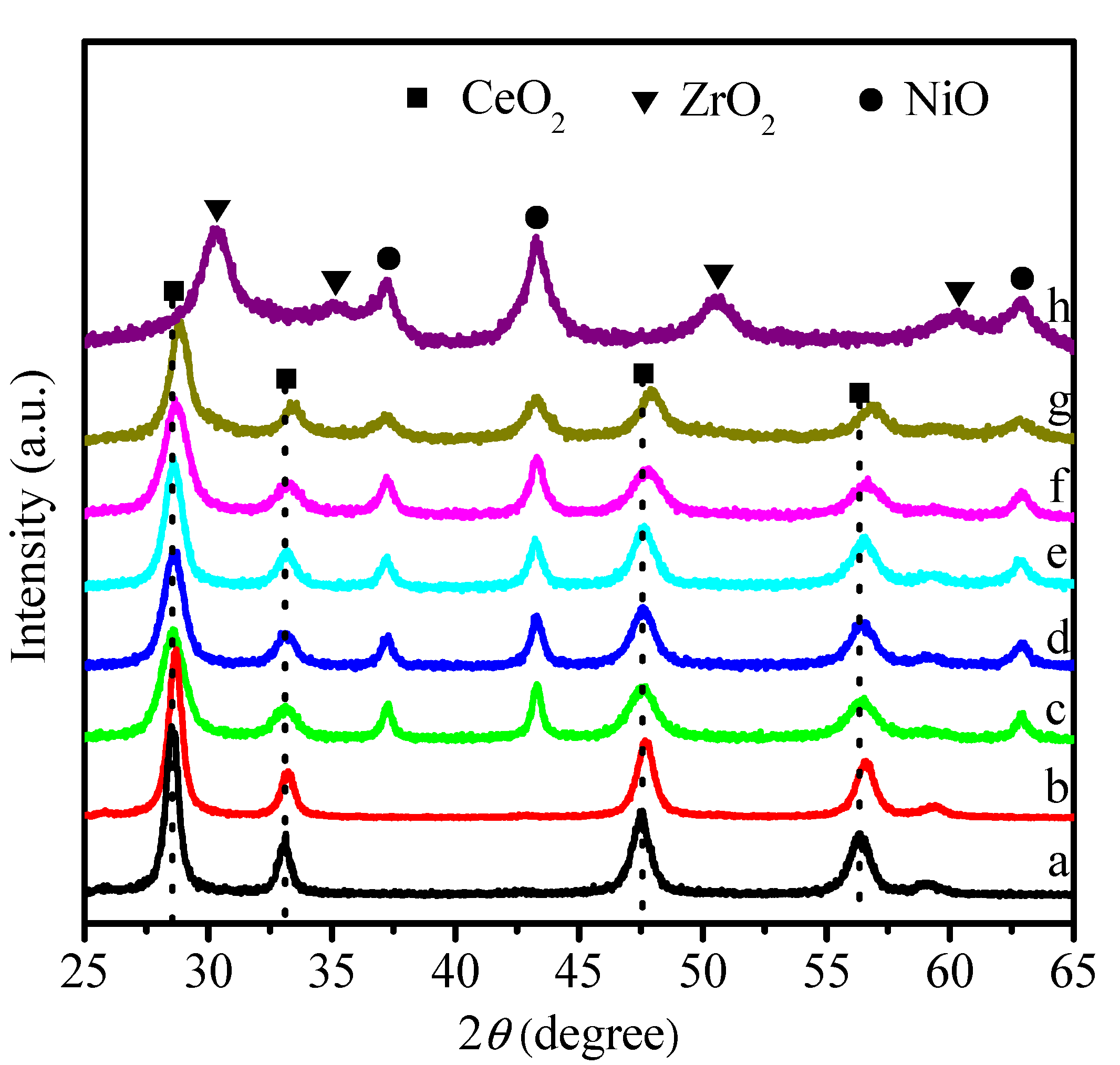
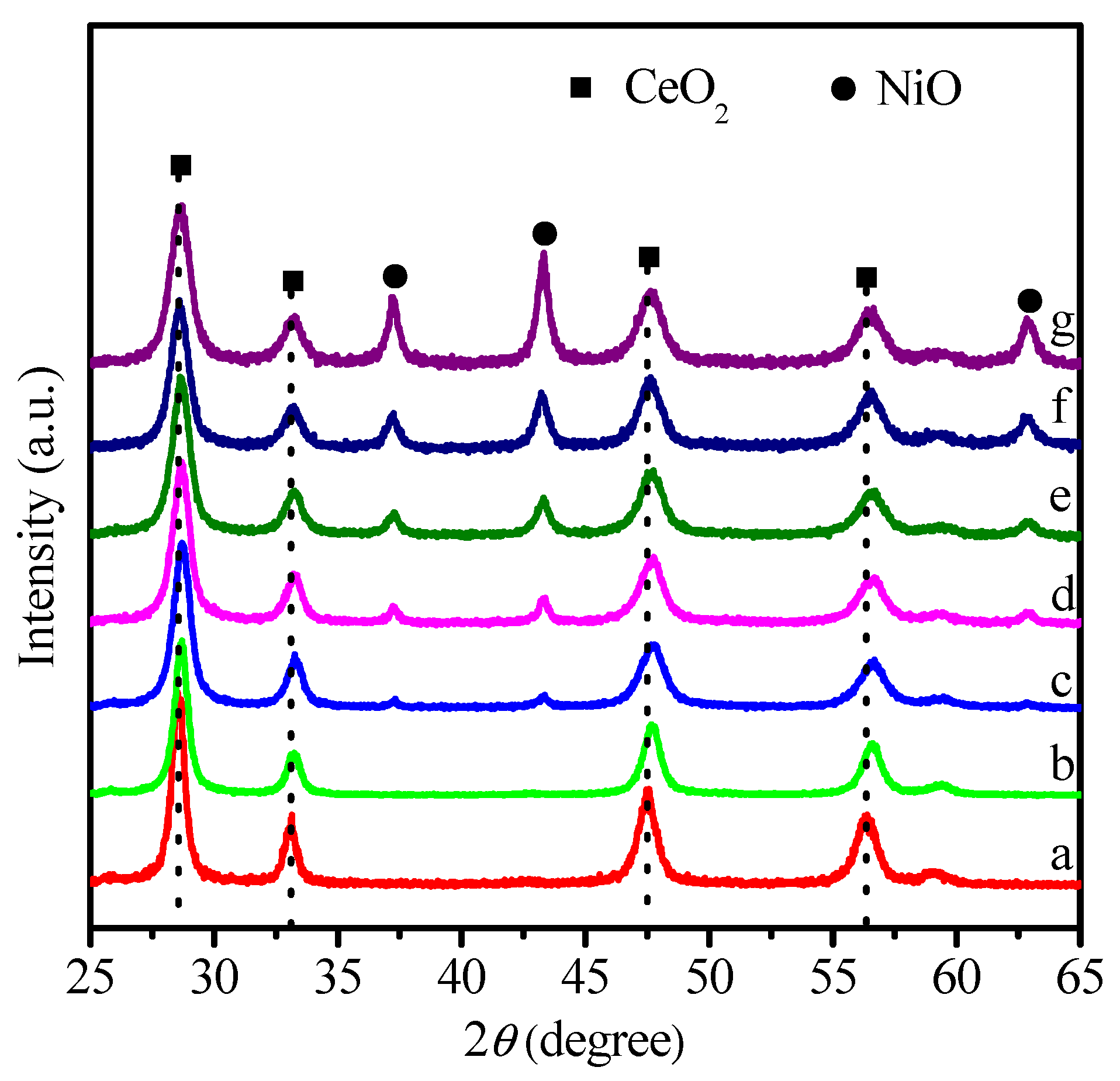
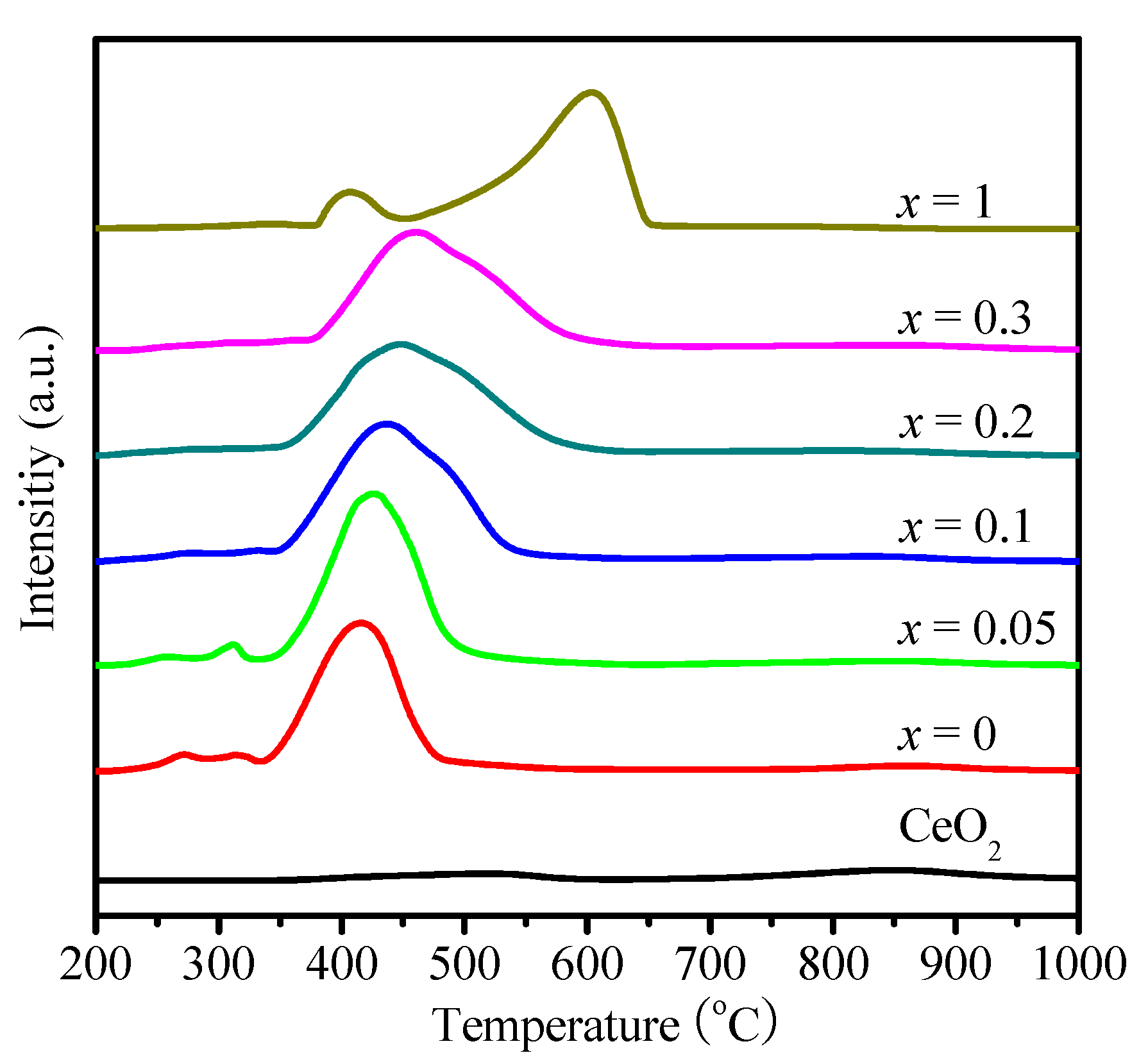
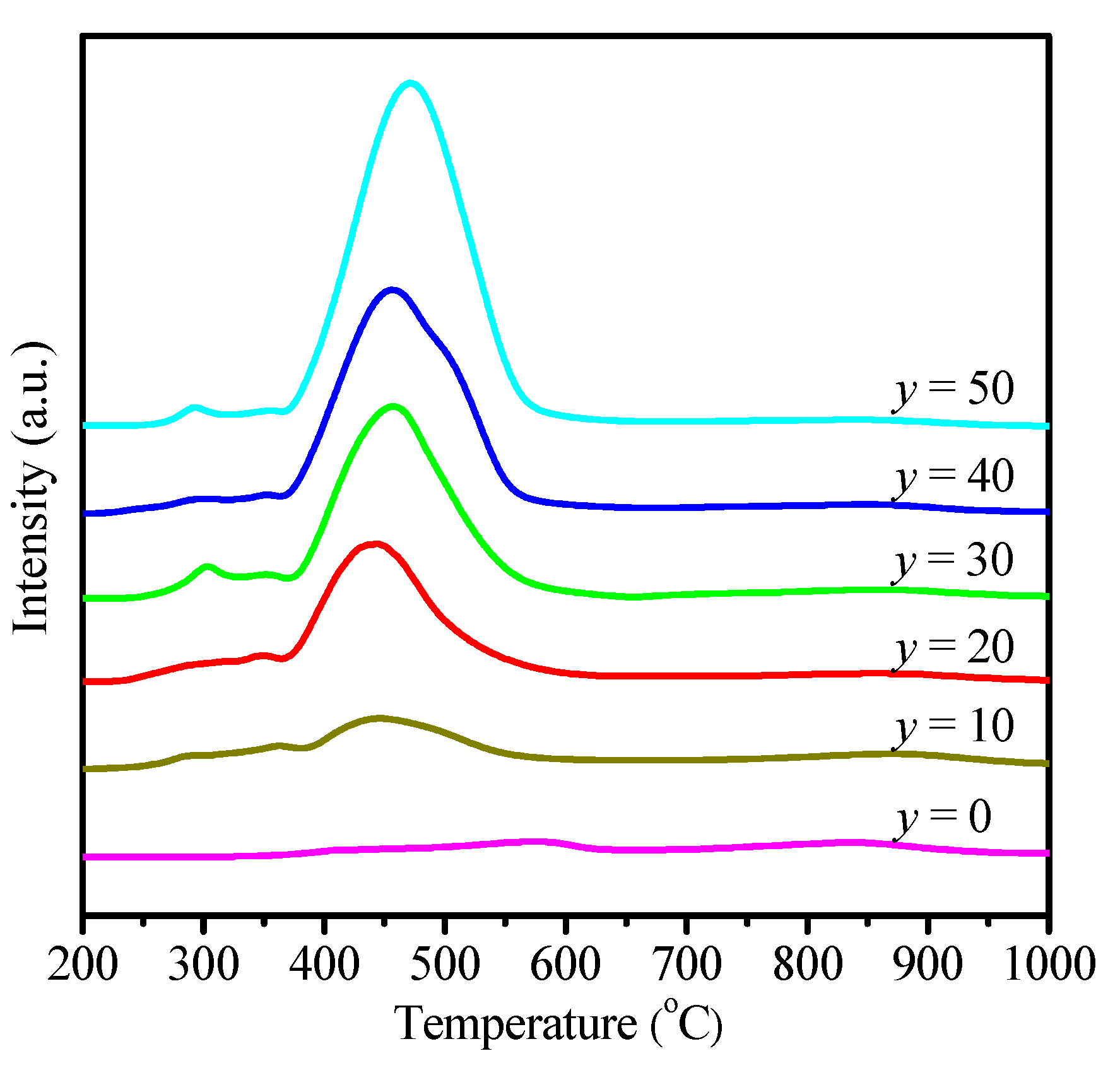
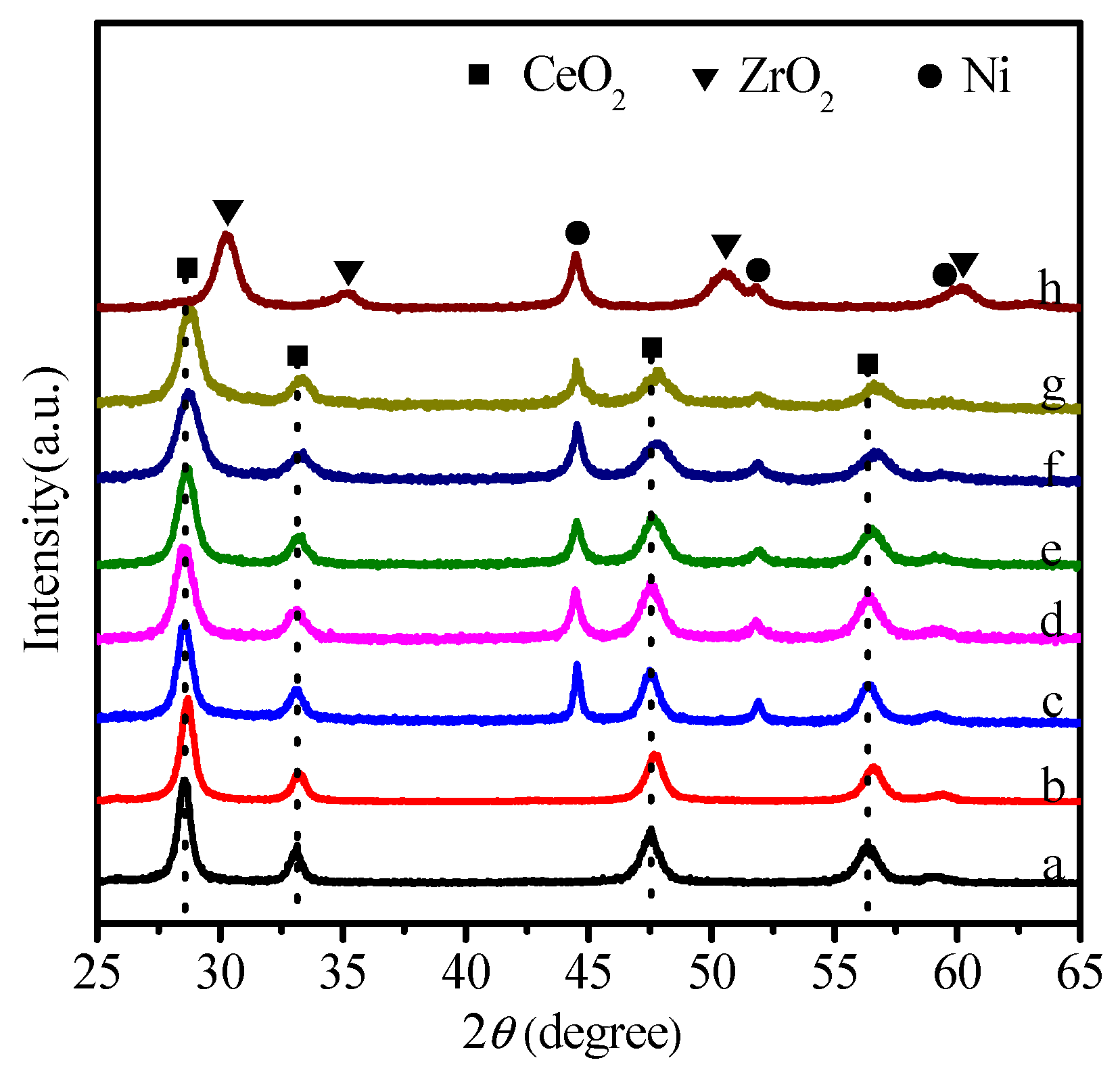
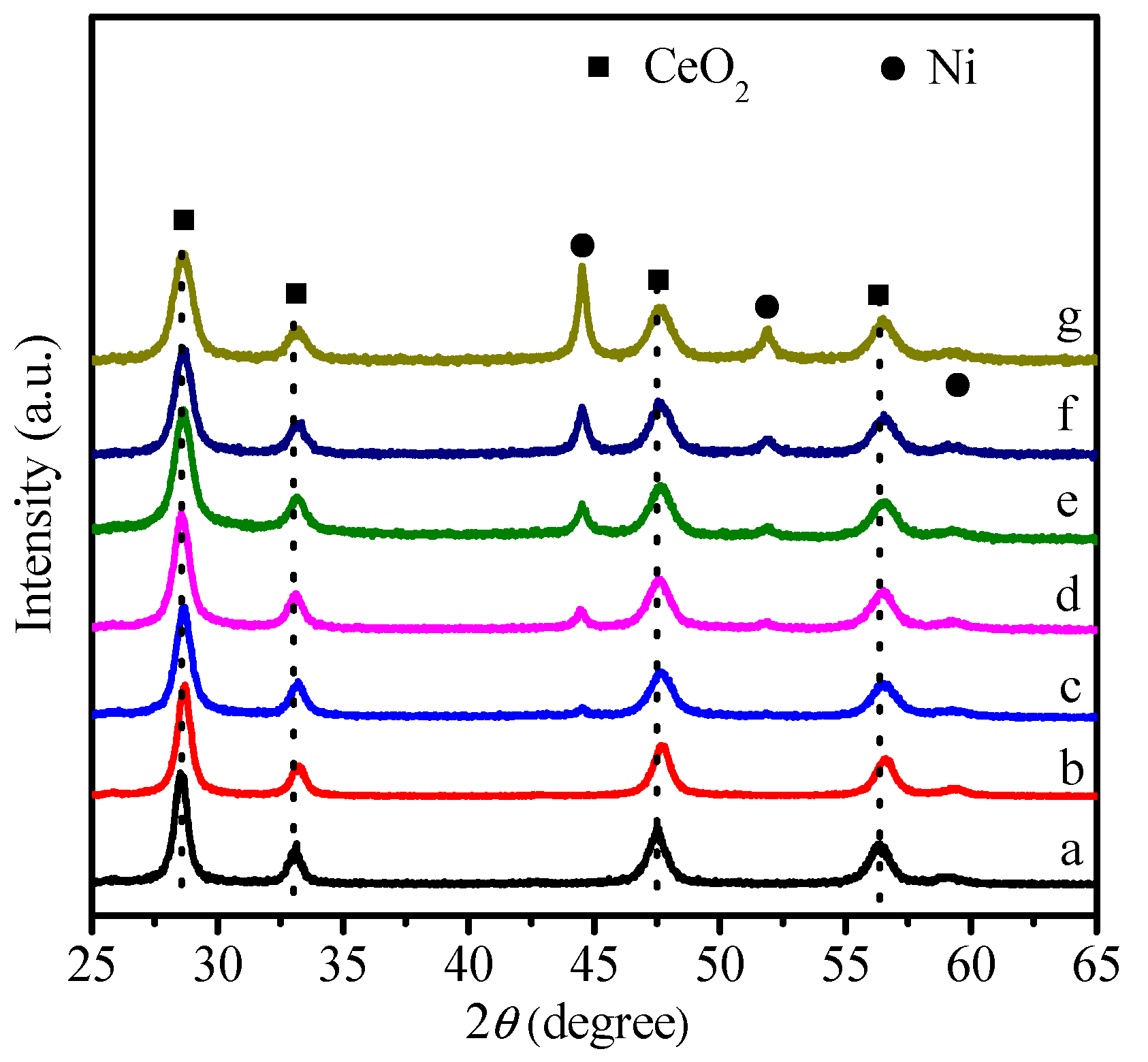
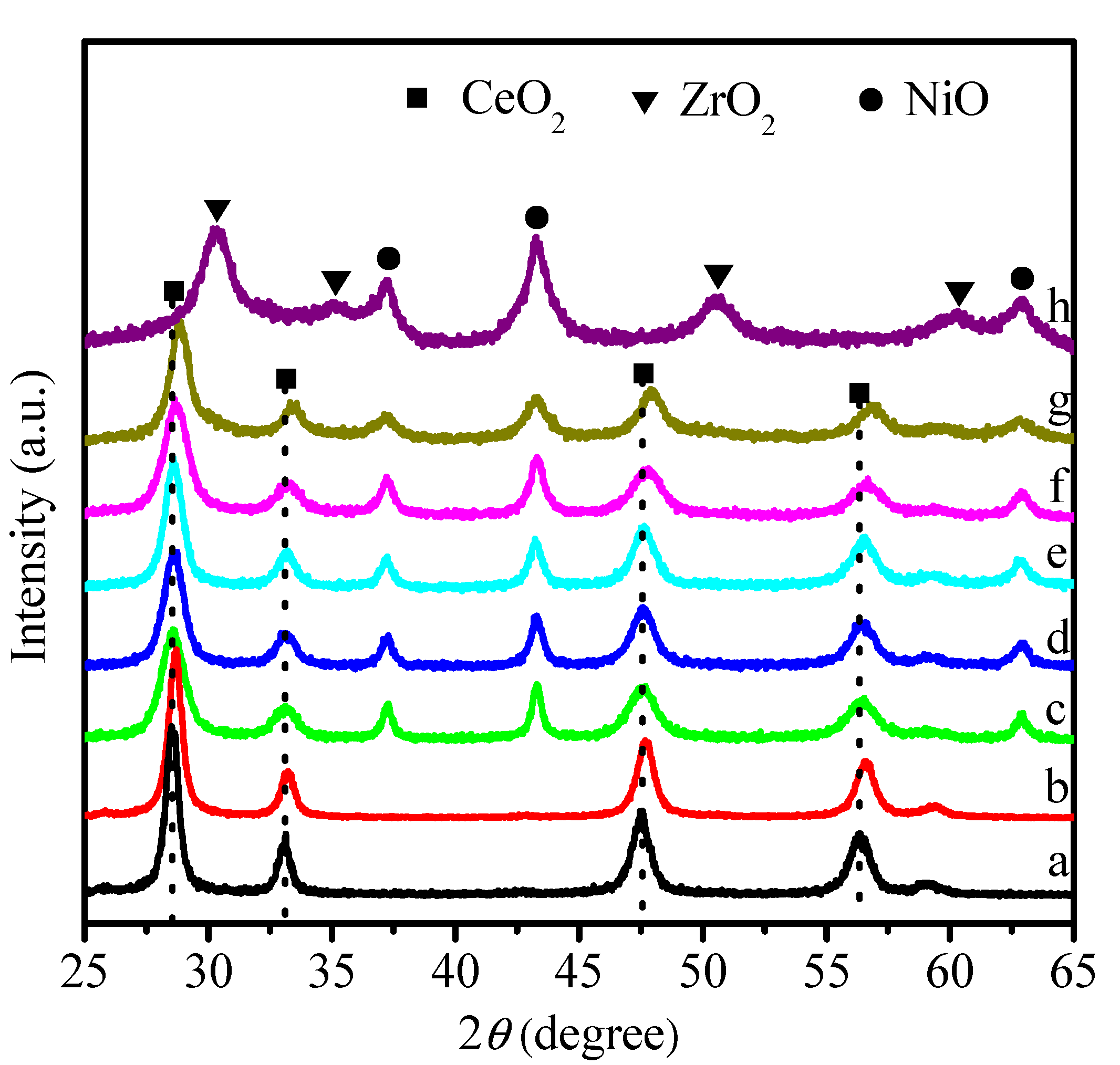
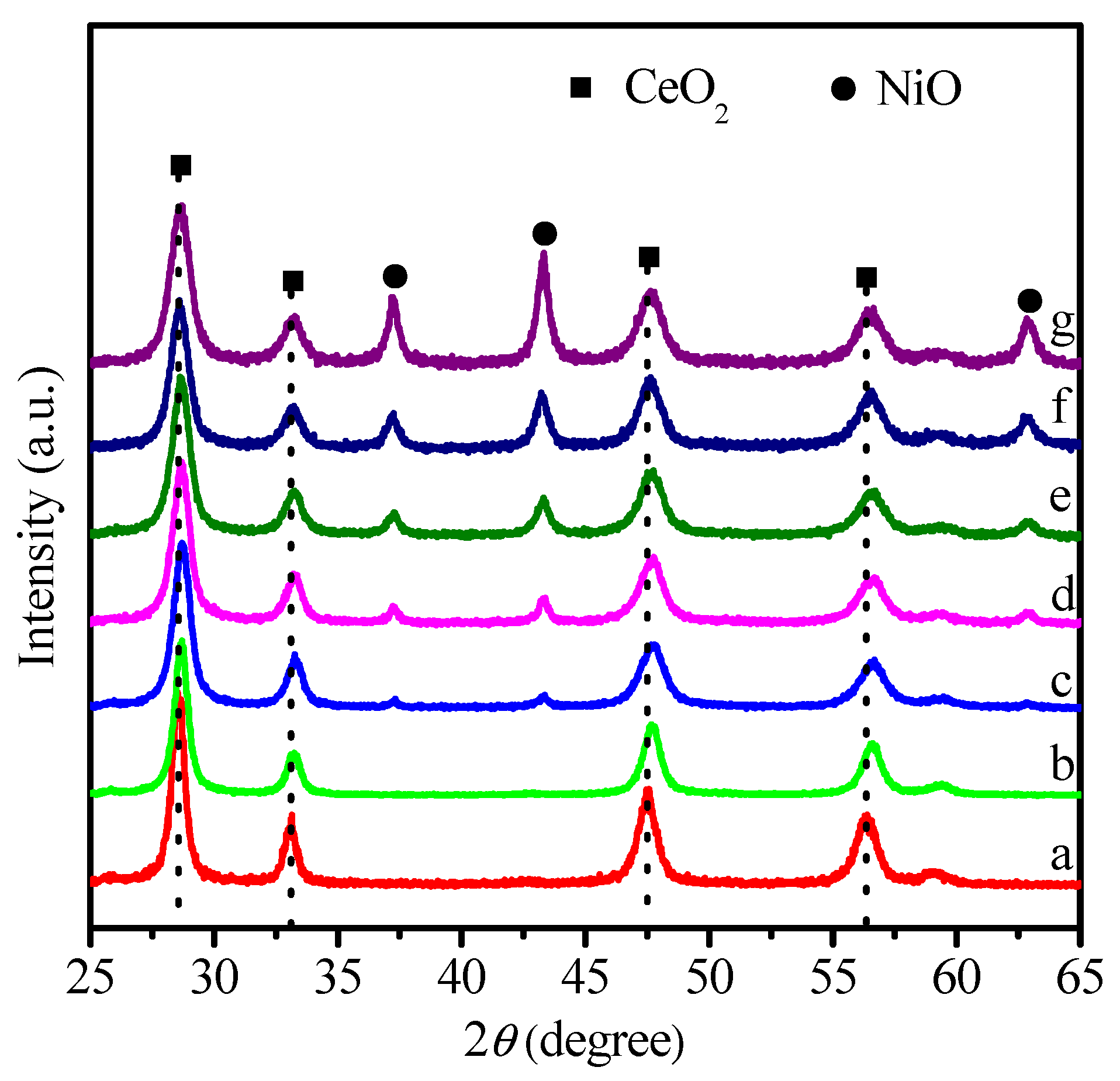
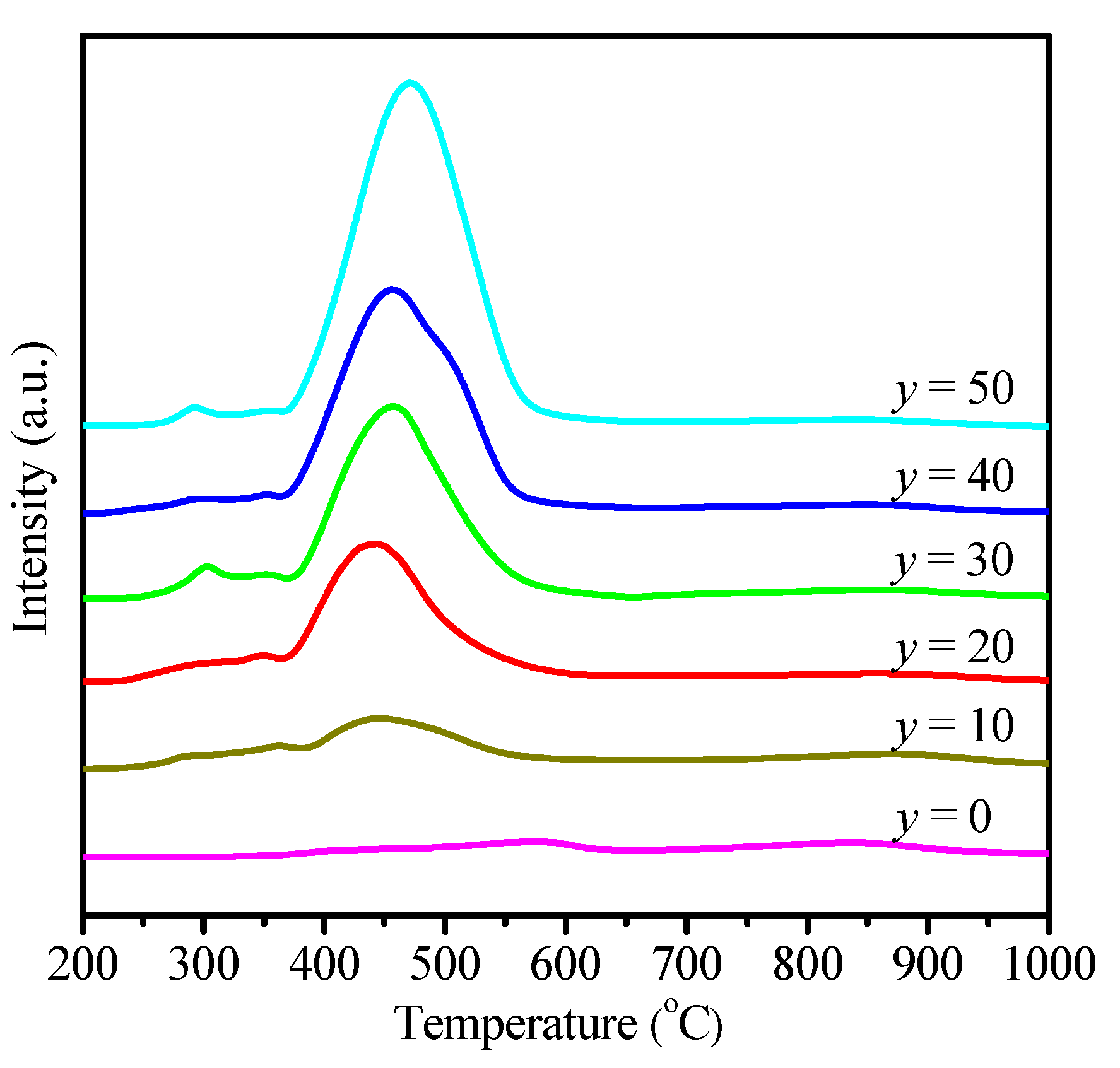
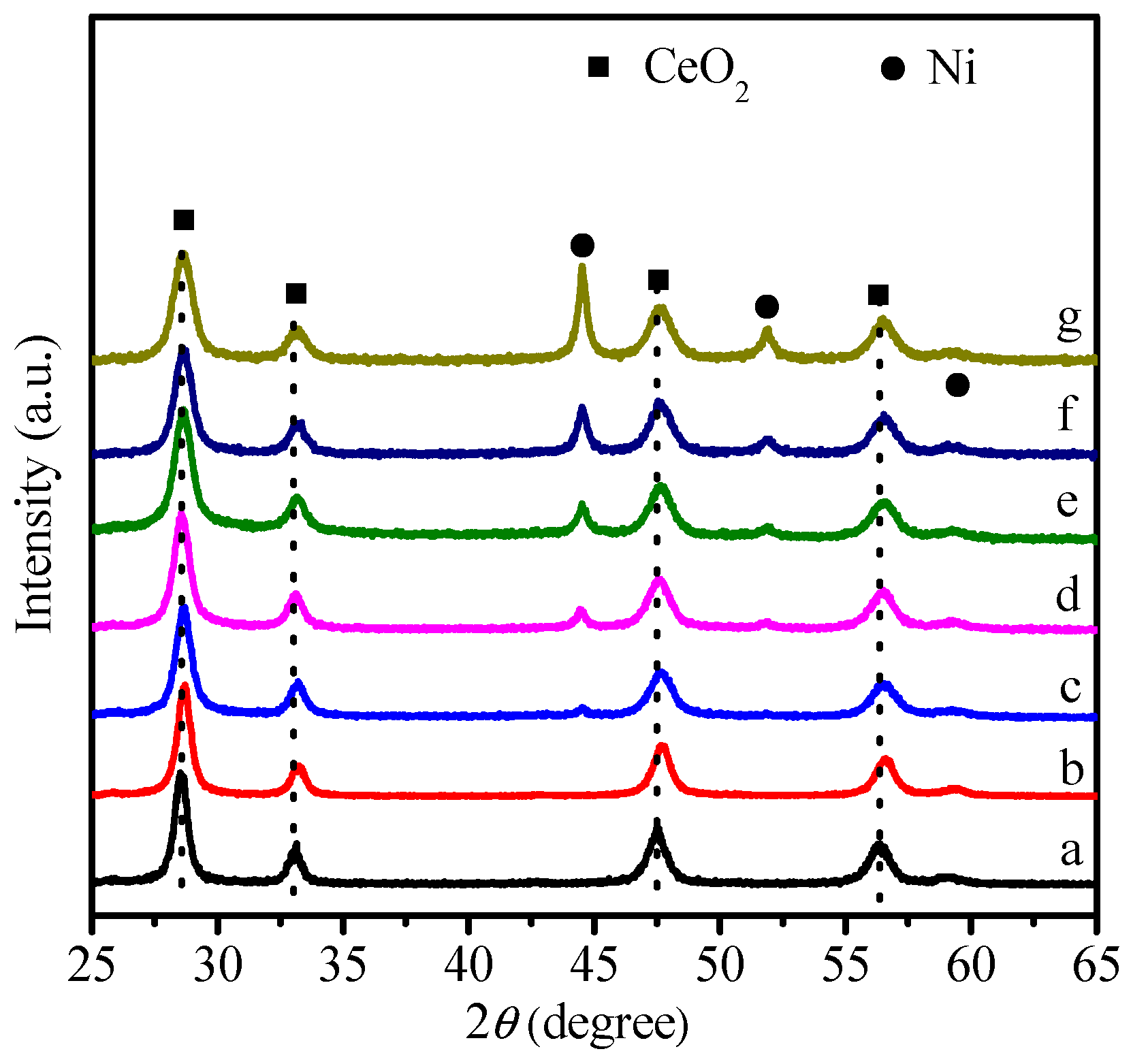
2.1. Physicochemical Properties of Ni–Ce1−xZrxO2
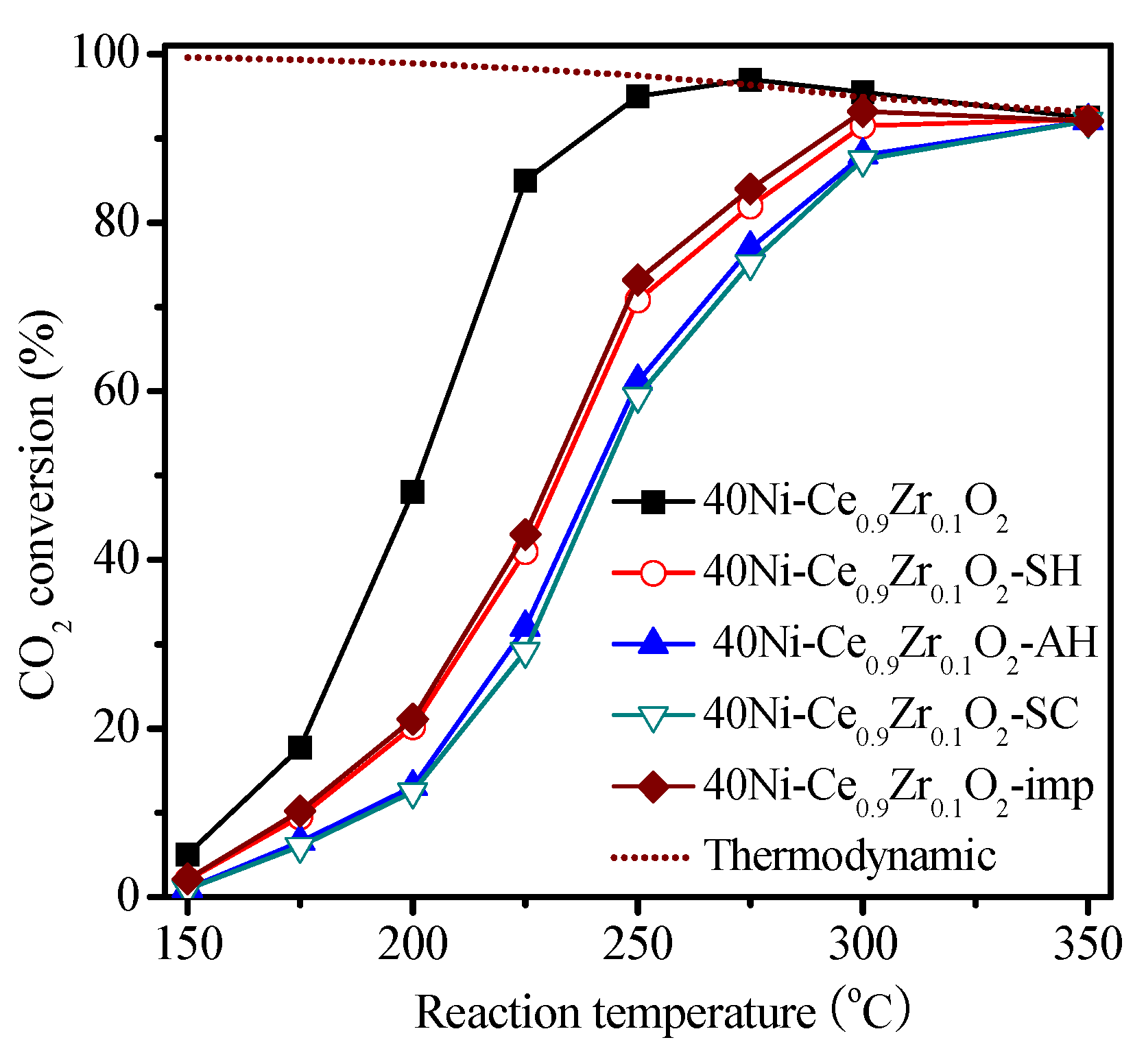
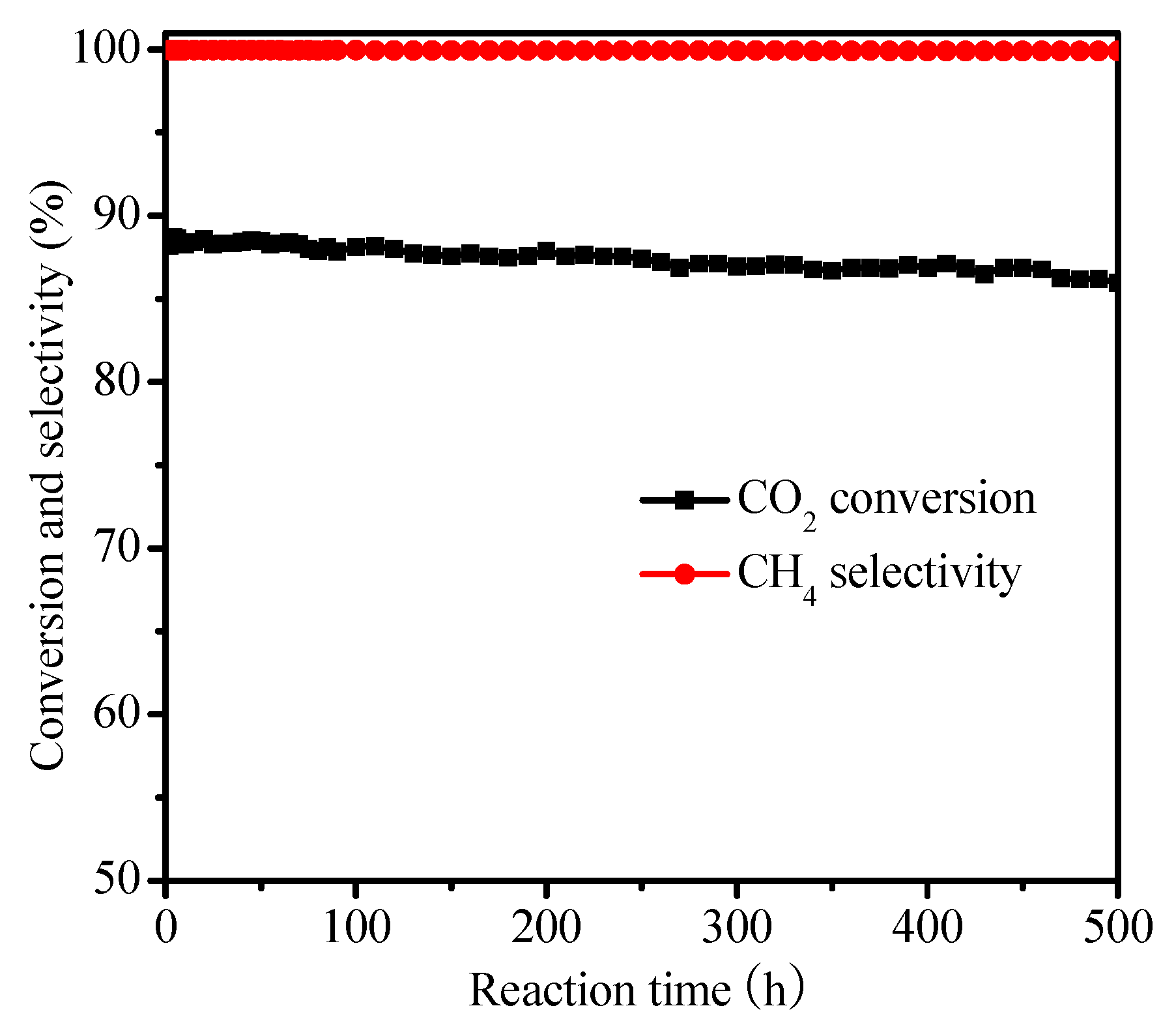
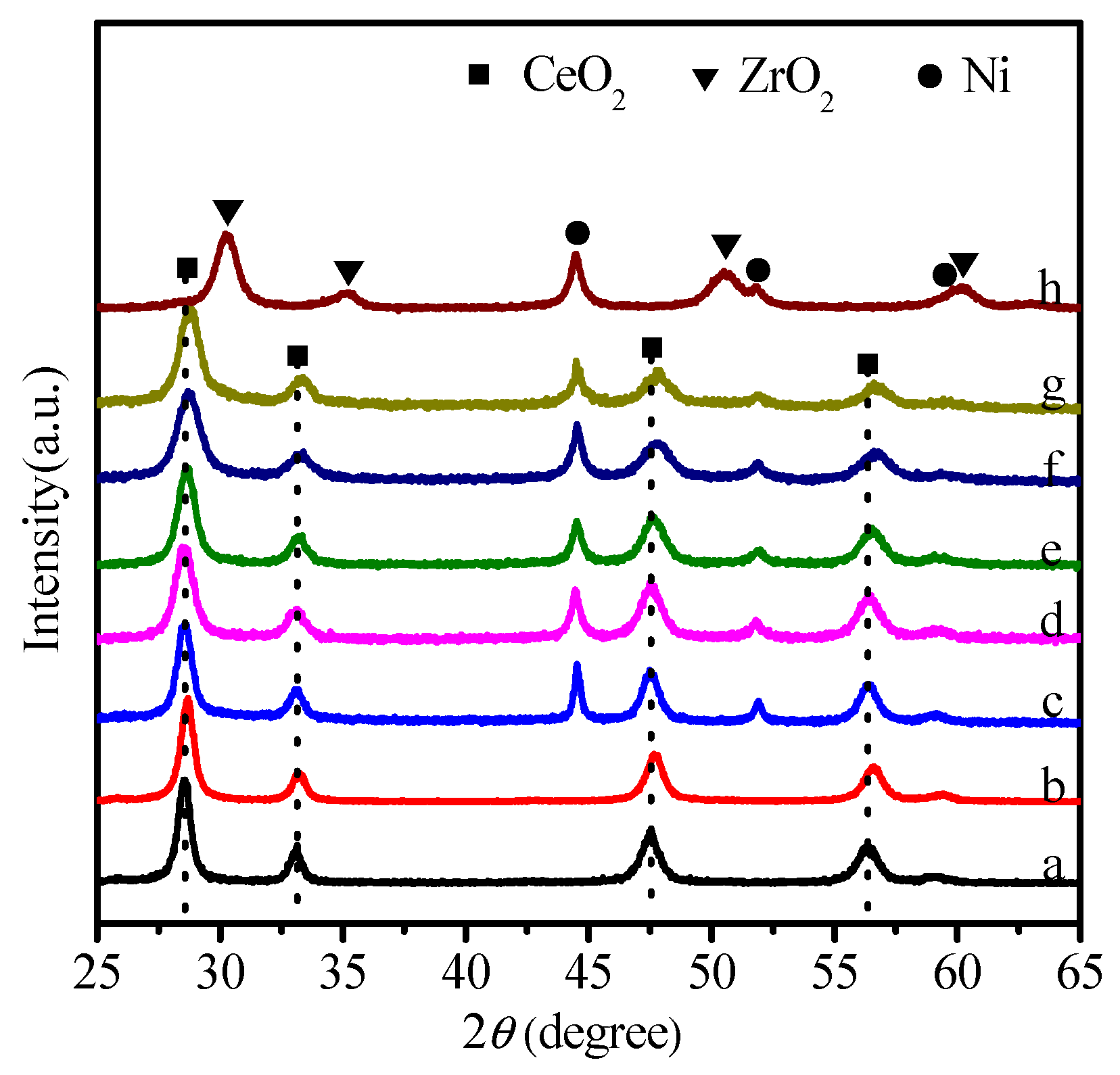
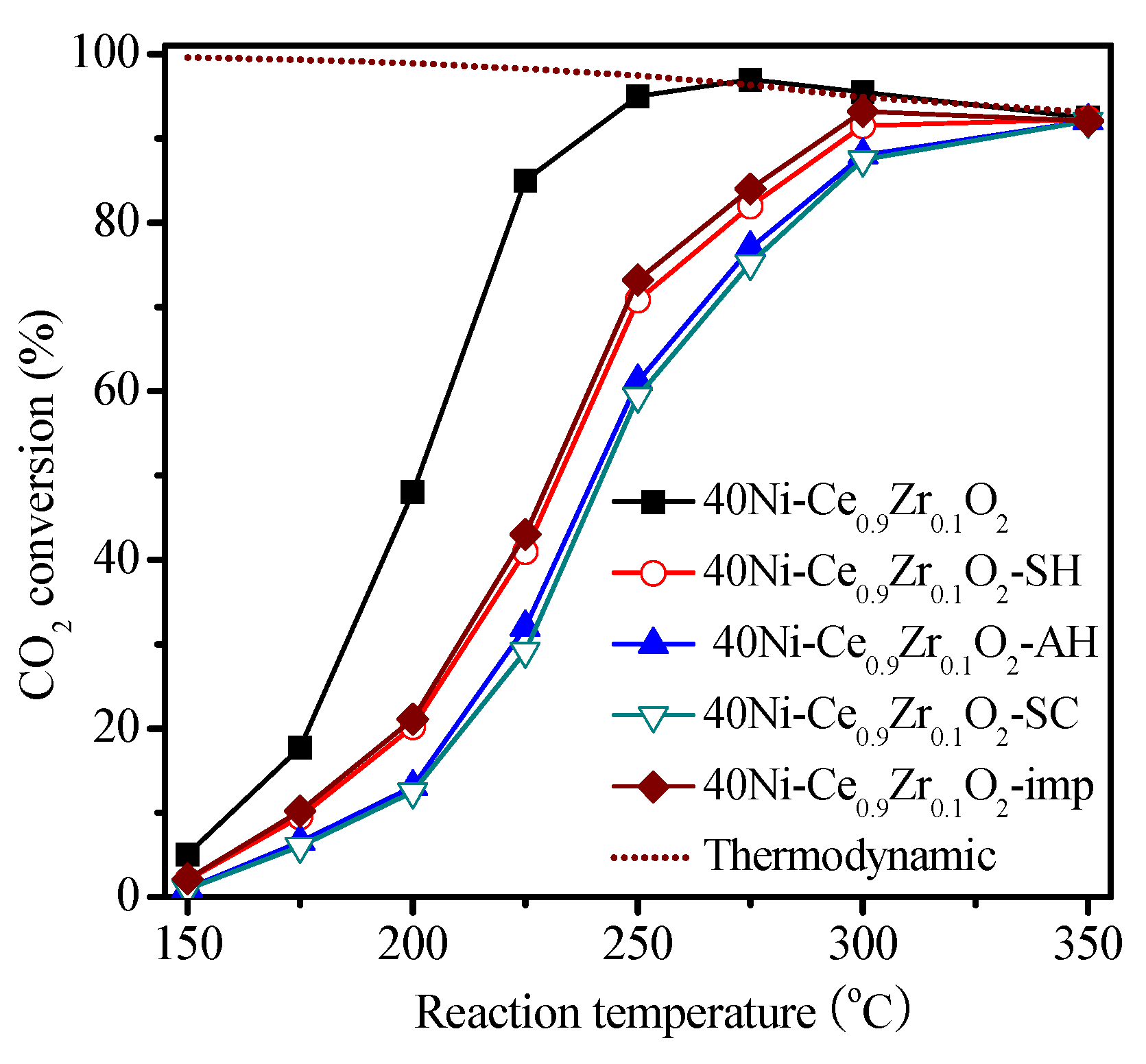
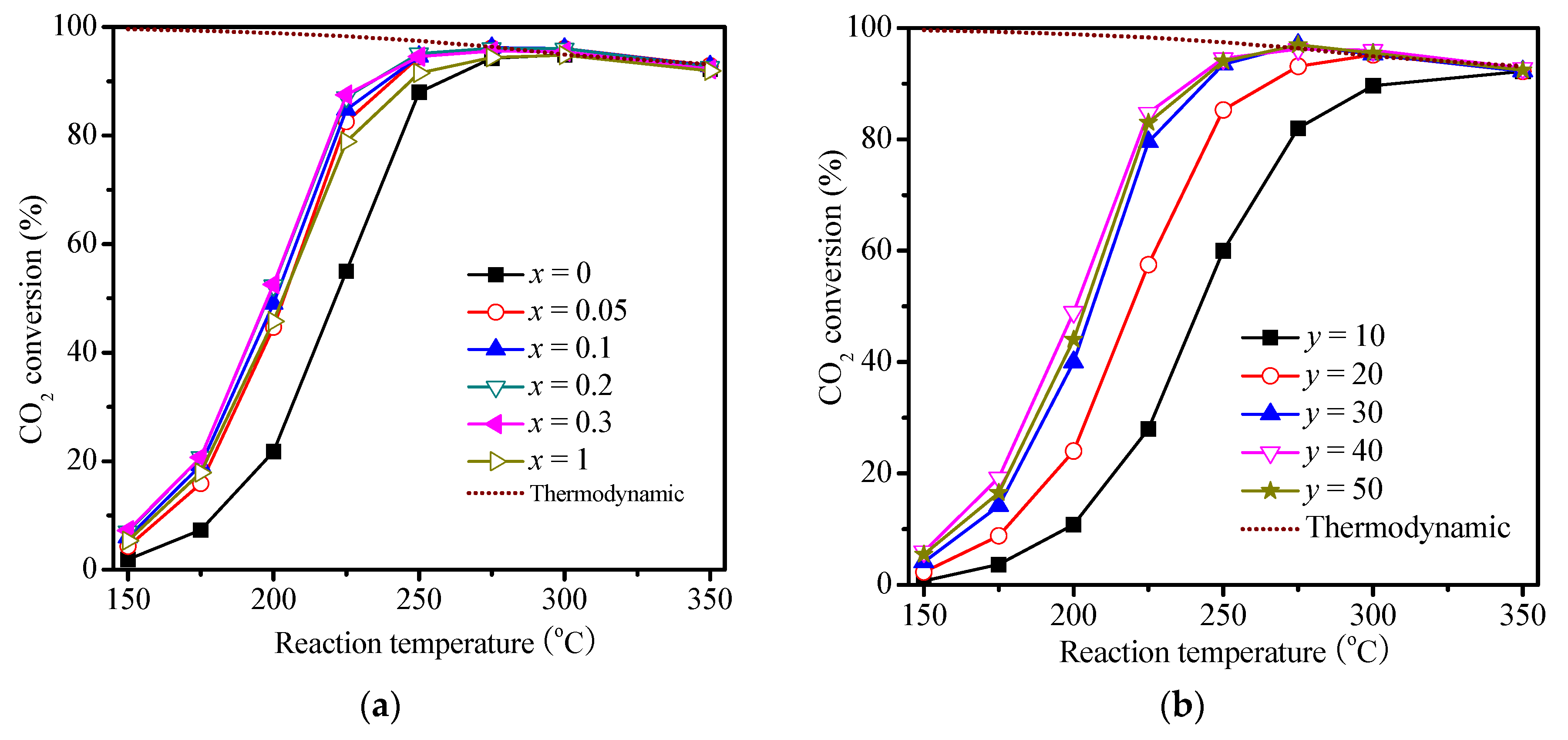
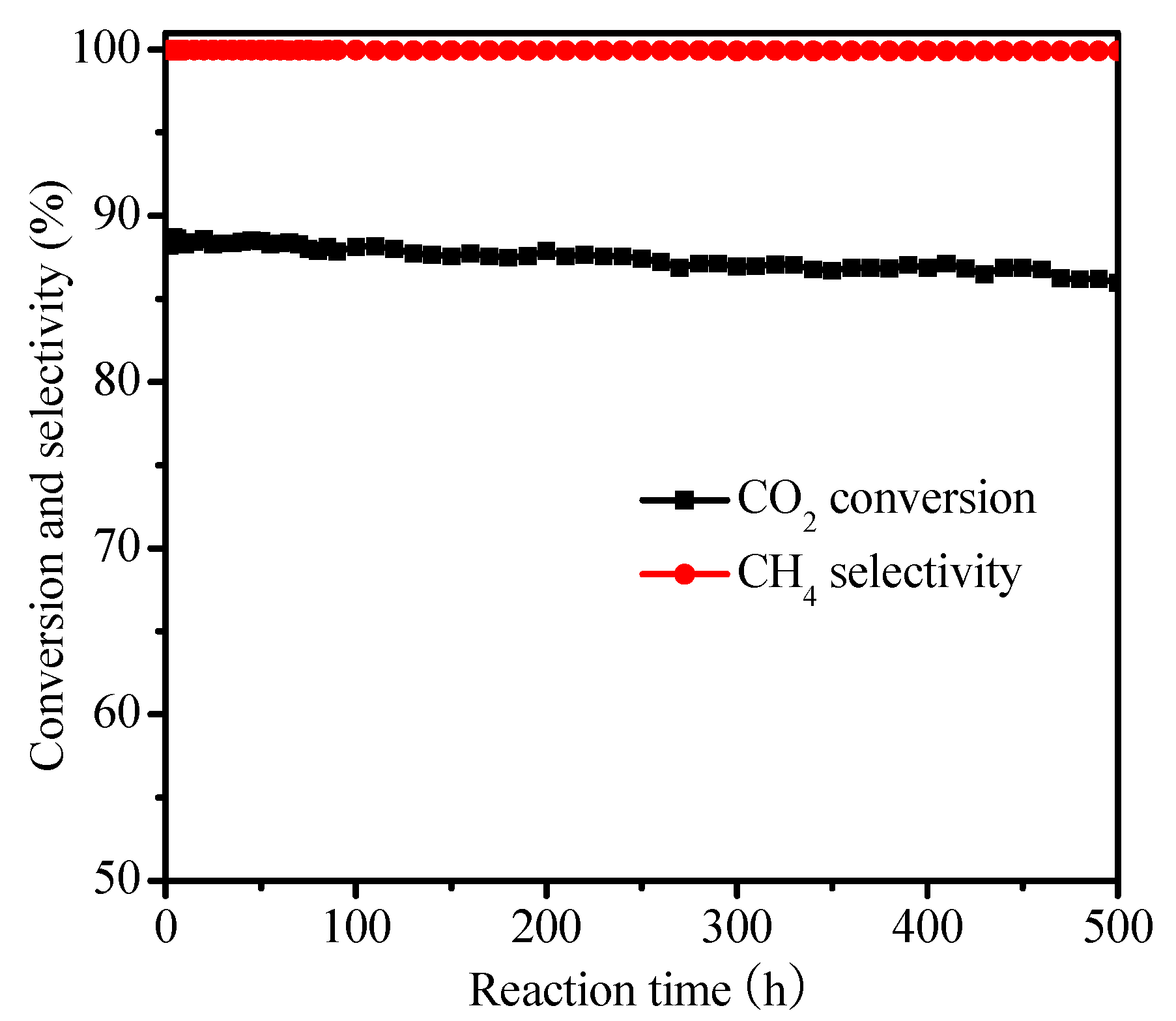
2.2. Catalysis Reaction
3. Materials and Methods
3.1. Material Preparation
3.2. Material Characterization
3.3. Catalytic Test
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferey, G.; Serr, C.; Devic, T.; Maurin, G.; Jobic, H.; Llewellyn, P.L.; De Weireld, G.; Vimont, A.; Daturi, M.; Chang, J.S. Why hybrid porous solids capture greenhouse gases? Chem. Soc. Rev. 2011, 40, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A J.; Sin, E.H.K.; Marriott, R.; Clark, J.H. Generation, capture, and utilization of industrial carbon dioxide. ChemSusChem 2010, 3, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, M.; Jorgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yan, B.H.; Chen, J.G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities. Energy Environ. Sci. 2016, 9, 62–73. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.P.; Ma, X.B.; Gong, J.L. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.J.; Liu, Q.; Gu, F.N.; Liu, B.; Zhong, Z.Y.; Su, F.B. Recent advances in methanation catalysts for the production of synthetic natural gas. RSC Adv. 2015, 5, 22759–22776. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P. Supported catalysts for CO2 methanation: A review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Fechete, I.; Vedrine, J.C. Nanoporous materials as new engineered catalysts for the synthesis of green fuels. Molecules 2015, 20, 5638–5666. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Tanizawa, M.; Watanabe, K.; Taguchi, A. CO2 methanation property of Ru nanoparticle-loaded TiO2 prepared by a polygonal barrel-sputtering method. Energy Environ. Sci. 2009, 2, 315–321. [Google Scholar] [CrossRef]
- Karelovic, A.; Ruiz, P. CO2 hydrogenation at low temperature over Rh/γ-Al2O3 catalysts: Effect of the metal particle size on catalytic performances and reaction mechanism. Appl. Catal. B 2012, 113–114, 237–249. [Google Scholar] [CrossRef]
- Jacquemin, M.; Beuls, A.; Ruiz, P. Catalytic production of methane from CO2 and H2 at low temperature: Insight on the reaction mechanism. Catal. Today 2010, 157, 462–466. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, S.; Ye, Y.; Zhang, X.; Wang, L.; Zhu, W.; Cheng, F.; Tao, F. Catalytic conversion of carbon dioxide to methane on ruthenium–cobalt bimetallic nanocatalysts and correlation between surface chemistry of catalysts under reaction conditions and catalytic performances. ACS Catal. 2012, 2, 2403–2408. [Google Scholar] [CrossRef]
- Yan, Y.; Dai, Y H.; He, H.; Yu, Y.B.; Yang, Y.H. A novel W-doped Ni-Mg mixed oxide catalyst for CO2 methanation. Appl. Catal. B Environ. 2016, 196, 108–116. [Google Scholar] [CrossRef]
- Wierzbicki, D.; Debek, R.; Motak, M.; Grzybek, T.; Gálvez, M.E.; Da Costa, P. Novel Ni-La-hydrotalcite derived catalysts for CO2 methanation. Catal. Commun. 2016, 83, 5–8. [Google Scholar] [CrossRef]
- Xu, L.L.; Wang, F.G.; Chen, M.D.; Zhang, J.K.; Yuan, D.; Wang, L.J.; Wu, K.; Xu, G.Q.; Chen, W. CO2 methanation over a Ni based ordered mesoporous catalyst for the production of synthetic natural gas. RSC Adv. 2016, 6, 28489–28499. [Google Scholar] [CrossRef]
- Du, G.A.; Lim, S.; Yang, Y.H.; Wang, C.; Pfefferle, L.; Haller, G.L. Methanation of carbon dioxide on Ni-incorporated MCM-41 catalysts: The influence of catalyst pretreatment and study of steady-state reaction. J. Catal. 2007, 249, 370–379. [Google Scholar] [CrossRef]
- Zhi, G.J.; Guo, X.N.; Wang, Y.Y.; Jin, G.Q.; Guo, X.Y. Effect of La2O3 modification on the catalytic performance of Ni/SiC for methanation of carbon dioxide. Catal. Commun. 2011, 16, 56–59. [Google Scholar] [CrossRef]
- Graca, I.; Gonzalez, L.V.; Bacariza, M.C.; Fernandes, A.; Henriques, C.; Lopes, J.M.; Ribeiro, M.F. CO2 hydrogenation into CH4 on NiHNaUSY zeolites. Appl. Catal. B Environ. 2014, 147, 101–110. [Google Scholar] [CrossRef]
- Abello, S.; Berrueco, C.; Montane, D. High-loaded nickel-alumina catalyst for direct CO2 hydrogenation into synthetic natural gas (SNG). Fuel 2013, 113, 598–609. [Google Scholar] [CrossRef]
- Ocampo, F.; Louis, B.; Kiwi-Minsker, L.; Roger, A.C. Effect of Ce/Zr composition and noble metal promotion on nickel based CexZr1−xO2 catalysts for carbon dioxide methanation. Appl. Catal. A Gen. 2011, 392, 36–44. [Google Scholar] [CrossRef]
- Ocampo, F.; Louis, B.; Roger, A.C. Methanation of carbon dioxide over nickel-based Ce0.72Zr0.28O2 mixed oxide catalysts prepared by sol-gel method. Appl. Catal. A Gen. 2009, 369, 90–99. [Google Scholar] [CrossRef]
- Cai, W.; Zhong, Q.; Zhao, Y.X. Fractional-hydrolysis-driven formation of non-uniform dopant concentration catalyst nanoparticles of Ni/CexZr1−xO2 and its catalysis in methanation of CO2. Catal. Commun. 2013, 39, 30–34. [Google Scholar] [CrossRef]
- Pan, Q.; Peng, J.; Sun, T.; Gao, D.; Wang, S.; Wang, S. CO2 methanation on Ni/Ce0.5Zr0.5O2 catalysts for the production of synthetic natural gas. Fuel Process. Technol. 2014, 123, 166–171. [Google Scholar] [CrossRef]
- Muroyama, H.; Tsuda, Y.; Asakoshi, T.; Masitah, H.; Okanishi, T.; Matsui, T.; Eguchi, K. Carbon dioxide methanation over Ni catalysts supported on various metal oxides. J. Catal. 2016, 343, 178–184. [Google Scholar] [CrossRef]
- Du, Y.L.; Wu, X.; Cheng, Q.; Huang, Y.L.; Huang, W. Development of Ni-based catalysts derived from hydrotalcite-like compounds precursors for synthesis gas production via methane or ethanol reforming. Catalysts 2017, 7, 70. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, R.; Lin, X.; Wang, Z.; Liu, J.; Zhou, J.; Cen, K. Effects of Ce/Zr composition on nickel based Ce(1−x)ZrxO2 catalysts for hydrogen production in sulfur-iodine cycle. Int. J. Hydrog. Energy 2014, 39, 10853–10860. [Google Scholar] [CrossRef]
- Kramer, M.; Stowe, K.; Duisberg, M.; Muller, F.; Reiser, M.; Sticher, S.; Maier, W.F. The impact of dopants on the activity and selectivity of a Ni-based methanation catalyst. Appl. Catal. A Gen. 2009, 369, 42–52. [Google Scholar] [CrossRef]
- Feng, S.; Pan, D.; Wang, Z. Facile synthesis of cubic fluorite nano-Ce1-xZrxO2 via hydrothermal crystallization method. Adv. Powder Technol. 2011, 22, 678–681. [Google Scholar] [CrossRef]
- Xu, S.; Wang, X. Highly active and coking resistant Ni/CeO2–ZrO2 catalyst for partial oxidation of methane. Fuel 2005, 84, 563–567. [Google Scholar] [CrossRef]
- Roh, H.S.; Eum, I.H.; Jeong, D.W. Low temperature steam reforming of methane over Ni–Ce(1−x)ZrxO2 catalysts under severe conditions. Renew. Energy 2012, 42, 212–216. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, M.C.; Navarro, R.M.; Fierro, J.L.G. Ethanol steam reforming over Ni/MxOy–Al2O3 (M = Ce, La, Zr and Mg) catalysts: Influence of support on the hydrogen production. Int. J. Hydrog. Energy 2010, 32, 1462–1471. [Google Scholar] [CrossRef]
- Tan, M.W.; Wang, X.G.; Shang, X.F.; Zou, X.J.; Lu, X.G.; Ding, W.Z. Template-free synthesis of mesoporous γ-alumina-supported Ni-Mg oxides and their catalytic properties for pre-reforming liquefied petroleum gas. J. Catal. 2014, 314, 117–131. [Google Scholar] [CrossRef]
- Zou, X.J.; Wang, X.G.; Li, L.; Shen, K.; Lu, X.G.; Ding, W.Z. Development of highly effective supported nickel catalysts for pre-reforming of liquefied petroleum gas under low steam to carbon molar ratios. Int. J. Hydrog. Energy 2010, 35, 13191–13200. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | SBET (m2 g−1) | Ni Crystallite Size (nm) a | XCO2 (%) b | TOF (h−1) |
|---|---|---|---|---|
| CeO2 | 27 | - | - | - |
| 40Ni–CeO2 | 64 | 27 | 4.2 | 0.08 |
| 40Ni–Ce0.95Zr0.05O2 | 53 | 21 | 7.1 | 0.18 |
| 40Ni–Ce0.9Zr0.1O2 | 46 | 18 | 10.3 | 0.31 |
| 40Ni–Ce0.8Zr0.2O2 | 68 | 18 | 10.5 | 0.30 |
| 40Ni–Ce0.7Zr0.3O2 | 56 | 19 | 10.8 | 0.28 |
| 40Ni–ZrO2 | 78 | 16 | 8.2 | 0.27 |
| Ce0.9Zr0.1O2 | 43 | - | - | - |
| 10Ni–Ce0.9Zr0.1O2 | 50 | 23 | 3.3 | 0.08 |
| 20Ni–Ce0.9Zr0.1O2 | 46 | 22 | 4.6 | 0.11 |
| 30Ni–Ce0.9Zr0.1O2 | 55 | 19 | 7.8 | 0.22 |
| 50Ni–Ce0.9Zr0.1O2 | 52 | 21 | 10.1 | 0.26 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nie, W.; Zou, X.; Chen, C.; Wang, X.; Ding, W.; Lu, X. Methanation of Carbon Dioxide over Ni–Ce–Zr Oxides Prepared by One-Pot Hydrolysis of Metal Nitrates with Ammonium Carbonate. Catalysts 2017, 7, 104. https://doi.org/10.3390/catal7040104
Nie W, Zou X, Chen C, Wang X, Ding W, Lu X. Methanation of Carbon Dioxide over Ni–Ce–Zr Oxides Prepared by One-Pot Hydrolysis of Metal Nitrates with Ammonium Carbonate. Catalysts. 2017; 7(4):104. https://doi.org/10.3390/catal7040104
Chicago/Turabian StyleNie, Wangxin, Xiujing Zou, Chenju Chen, Xueguang Wang, Weizhong Ding, and Xionggang Lu. 2017. "Methanation of Carbon Dioxide over Ni–Ce–Zr Oxides Prepared by One-Pot Hydrolysis of Metal Nitrates with Ammonium Carbonate" Catalysts 7, no. 4: 104. https://doi.org/10.3390/catal7040104
APA StyleNie, W., Zou, X., Chen, C., Wang, X., Ding, W., & Lu, X. (2017). Methanation of Carbon Dioxide over Ni–Ce–Zr Oxides Prepared by One-Pot Hydrolysis of Metal Nitrates with Ammonium Carbonate. Catalysts, 7(4), 104. https://doi.org/10.3390/catal7040104