1. Introduction
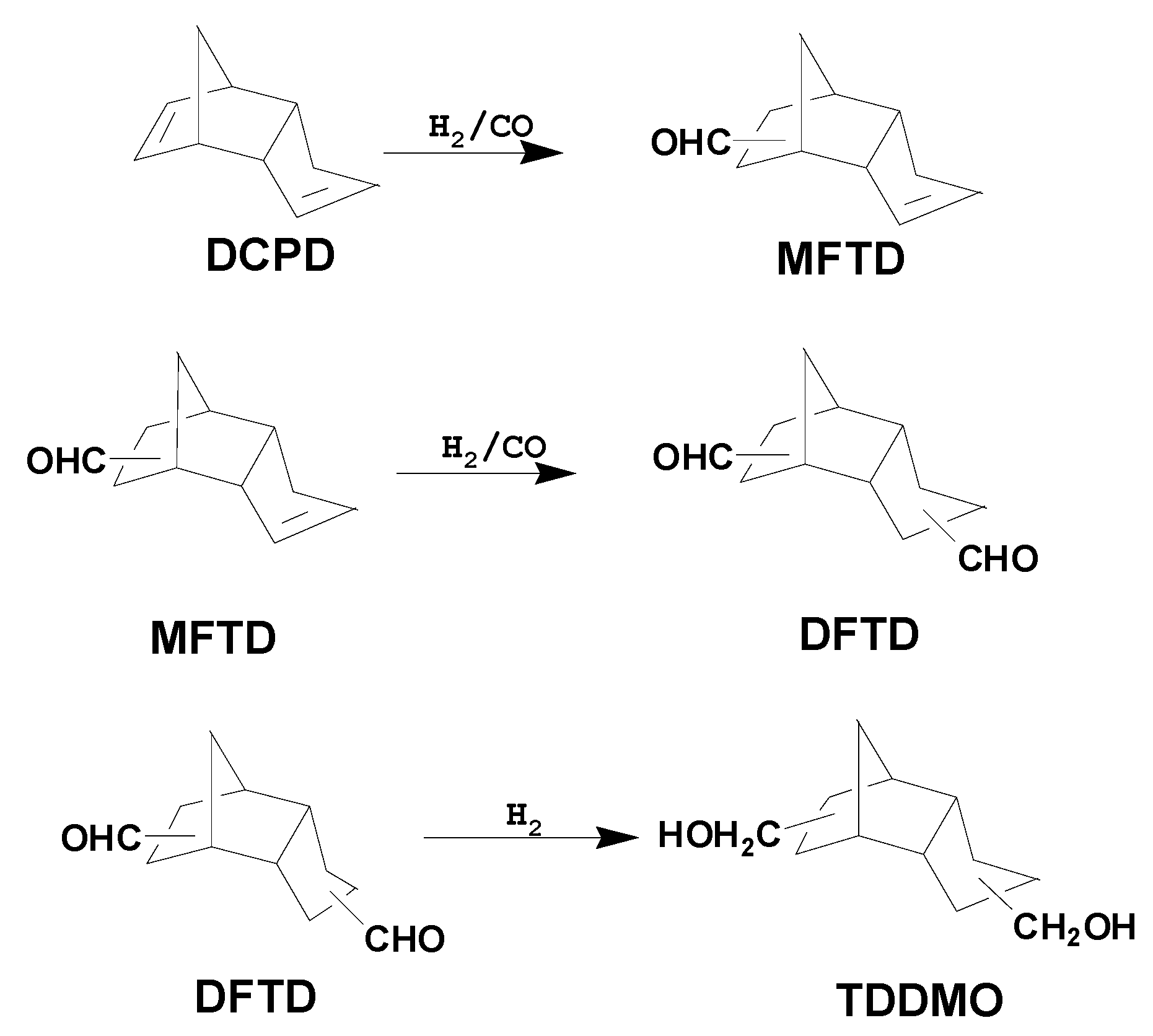
Because of the worldwide dependence on non-renewable fossil fuel reserves, society is faced with the challenge of finding alternative fuel sources for energy production [
1,
2,
3,
4,
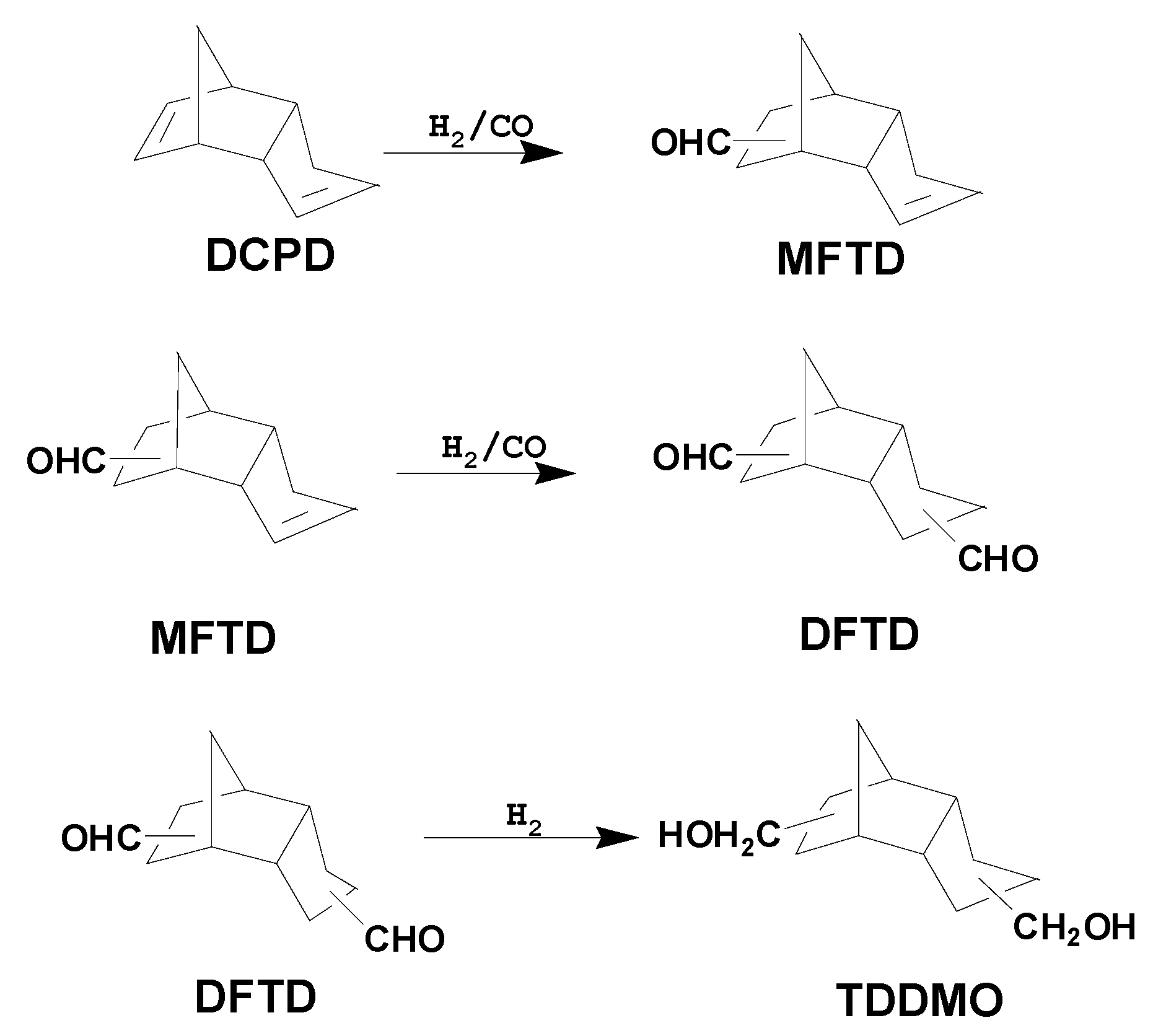
5]. However, until fossil fuels can be completely replaced, a more practical strategy is to better utilize existing fossil fuel resources. In lieu of this approach, using industrial side products to produce useful chemicals is another important strategy; for example, the hydroformylation of dicyclopentadiene (DCPD), an ethylene cracking side product, yields monoformyltricyclodecenes (MFTD) and diformyltricyclodecanes (DFTD). DCPD is currently considered to be a key intermediate that can be further derivatized to produce high-value-added fine chemicals such as DFTD and tricyclodecanedimethylol (TDDMO) [
6,
7,
8,
9,
10] (
Scheme 1). In turn, DFTD and TDDMO can be used as the starting materials in the synthesis of various agricultural chemicals, lubricating oils, plasticizers, pharmaceuticals, and perfumes [
7,
8].
Currently, MFTD and DFTD are primarily synthesized using Rh- and Co-based catalysts. Chen et al. [
11] reported an overall aldehyde (MFTD and DFTD) yield of up to ca. 98 mol % at nearly complete DCPD conversion using water and toluene as the solvents, and a catalyst system consisting of a homogeneous Rh-based catalyst and various phosphine ligands. Garlaschelli et al. [
9] achieved MFTD and DFTD yields of 95% and 94.5%, respectively, under different reaction conditions using a mixed cobalt-rhodium carbonyl system, PPh
3 and toluene as the catalyst, ligand and solvent, respectively. Modifying Rh-loaded mesoporous silica support with a cobalt catalyst resulted in MFTD and DFTD yields of 87% and 88.7% [
12], respectively. Chen et al. reported high conversion (99.9%) and good selectivity (95.0%) to dialdehydes in the presence of Rh/mixed monodentate and bidentate phosphorus ligand complexes under optimized reaction conditions [
13]. We recently showed that MFTD and DFTD yields of 97% and 84%, respectively, could be achieved over a cobalt-modified iron oxide-Rh/Fe
3O
4 catalyst [
14].
Recently, iron oxide-based magnetic catalysts with good activity and selectivity have been used in many reactions due to the ease of catalyst recovery [
15,
16,
17,
18,
19,
20,
21]. However, relatively few reports have dealt with their application in the hydroformylation of olefins, especially for DCPD [
17,
22]. In our previously literature, magnetically separable Fe
3O
4-supported Co-Rh binary catalysts were used to achieve 98% MFTD selectivity under mild conditions and >90% DFTD selectivity at a high reaction temperature and pressure at >99% DCPD conversion [
23]. Although a cobalt-modified Rh/Fe
3O
4 catalyst exhibited good catalytic performance in DCPD hydroformylation, the effects of the cobalt loading on the DFTD selectivity and the kinetics of DCPD hydroformylation to MFTD and DFTD are not well understood. Thus, the systematic study of the effect of the cobalt loading on DCPD hydroformylation and the underlying mechanism is still highly desirable.
In this work, six different Rh or Co-Rh bimetallic catalysts were prepared on Fe3O4 supports by the co-precipitation of RhCl3, Co(NO3)2, and Fe(NO3)3 using Na2CO3 as the precipitant. The performance of these catalysts in DCPD hydroformylation to MFTD and DFTD was evaluated. The MFTD formation rate increased with increasing cobalt loading, whereas the DFTD formation rate increased until a Co/Rh ratio of 2:1 was reached. At this Co/Rh ratio, a DFTD selectivity of 90.6% was achieved; however, it decreased to 67% when the Co/Rh ratio was further increased to 4:1. The catalysts were characterized by temperature-programmed reduction (TPR), temperature-programmed desorption (TPD), and thermogravimetric and differential thermal analyses (TG-DTA) to study the effect of the cobalt loading on the catalytic performance of the Rh/Fe3O4 catalyst. The Rh reduction and dispersion behavior varied with the cobalt loading. Therefore, the changes in the catalytic performance upon introducing cobalt into the catalytic system might be due to the formation of a more reactive Rh species with a different Rh–phosphine interaction strength on the catalyst surface.
2. Results and Discussion
In this work, the DCPD hydroformylation reaction was divided into two stages. In the first stage, the reaction was performed at 95 °C and 4 MPa for 1.5 h to convert DCPD to MFTD with a selectivity of greater than 99% (trace amount of DFTD was detected). In the second stage, the MFTD were further converted to DFTD at 140 °C and 7 MPa for additional 4 h.
2.1. Effect of the Cobalt Loading on the DFTD Selectivity
DCPD hydroformylation was performed at 95 °C and 4 MPa for 1.5 h and then at 140 °C and 7 MPa for 4 h over various Co-Rh/Fe
3O
4 catalysts with Co/Rh ratios ranging from 0:1 to 4:1, and the results are listed in
Table 1. Entry 1 showed the results of Co/Fe
3O
4, no DCPD conversion was observed. A comparison of Entries 2–7 shows that the product distribution between the MFTD (singly hydroformylated products), DFTD (doubly hydroformylated products), and other undesirable by-products was significantly affected by the cobalt loading. Furthermore, the DCPD conversion was 100% over all the Co-Rh/Fe
3O
4 catalysts. As shown in
Table 1, the DFTD selectivity increased from 21.3% to 90.6% as the Co/Rh ratio increased from 0:1 to 2:1. In addition, the DFTD selectivity gradually increased up to a Co/Rh ratio of 2:1. At a higher cobalt loading (i.e., Co/Rh ratio of 4:1,
Table 1, Entry 6), the DFTD selectivity unexpectedly decreased to 67%, showing that the catalytic performance was poorer (MFTD selectivity of 23%). These data indicated that adding an appropriate amount of cobalt to the Rh-based catalyst could greatly improve the DFTD selectivity. Moreover, within the tested range, the optimal Co/Rh ratio in the Co–Rh bimetallic catalyst was determined to be 2:1.
2.2. Effect of the Cobalt Loading on the Kinetics of DCPD Hydroformylation to MFTD
The kinetics of DCPD hydroformylation to MFTD was analyzed. The first few [DCPD]
t data points were used to construct linear ln([DCPD]
t/[DCPD]
0) versus
t plots (Equation (1)), and the observed first-order rate constants
k were obtained from the slopes of the regression lines.
In this equation, [DCPD]t and [DCPD]0 are the DCPD concentrations at time t and at t = 0 min, respectively; k is the observed rate constant; t is the time in minutes.
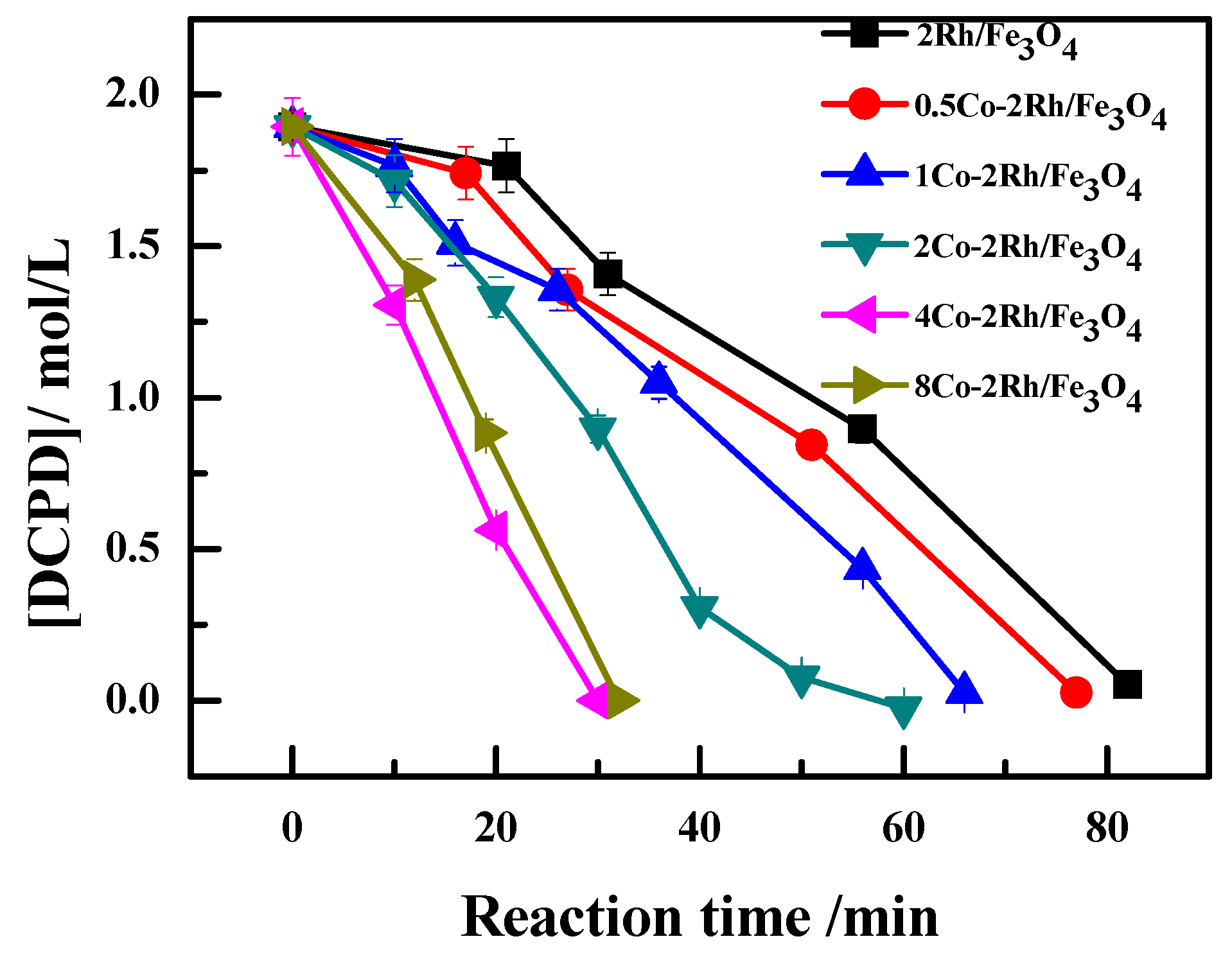
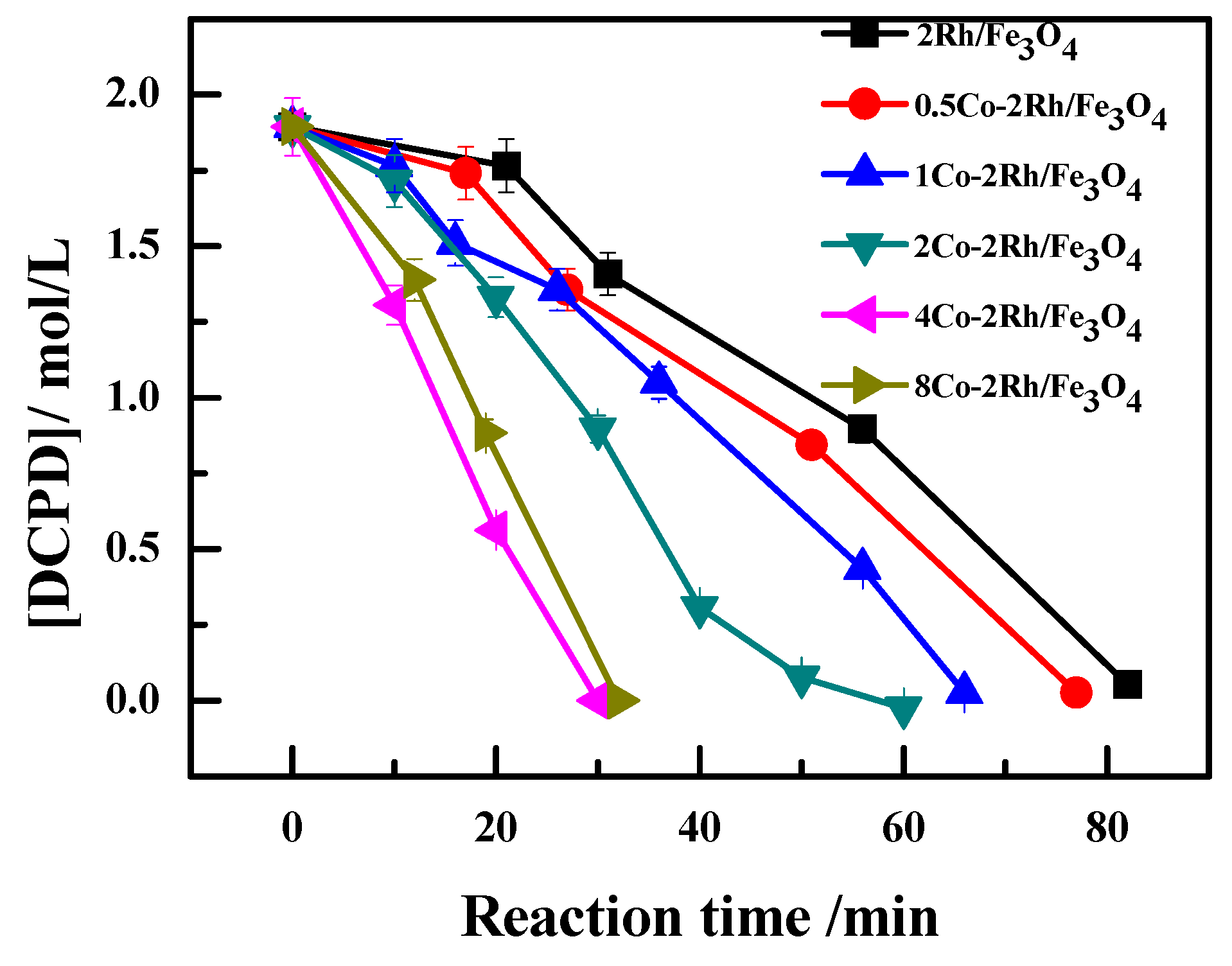
The kinetic parameters for DCPD hydroformylation to MFTD over the studied Co-Rh/Fe
3O
4 catalysts are summarized in
Table 2. As shown in
Figure 1 (Reaction conditions: 95 °C and 4 MPa for 1.5 h, 5 g DCPD, 0.2 g catalyst, 0.2 g PPh
3, 20 mL solvent) and
Table 2, the rate constant increased as the Co/Rh ratio increased from 0:1 to 2:1. However, the rate constant decreased slightly when the Co/Rh ratio was further increased to 4:1, indicating that the DCPD hydroformylation reaction rate could be accelerated by increasing the cobalt loading.
2.3. Effect of the Cobalt Loading on the Kinetics of MFTD HydroformSylation to DFTD
The kinetics of MFTD hydroformylation to DFTD was analyzed. The first few [MFTD]t data points were used to construct linear ln([MFTD]t/[MFTD]0) versus t plots, and the observed first-order rate constants k were obtained from the slopes of the regression lines. Here, [MFTD]t and [MFTD]0 are the MFTD concentrations at time t and at t = 0 min, respectively; k is the observed rate constant; t is the time in minutes.
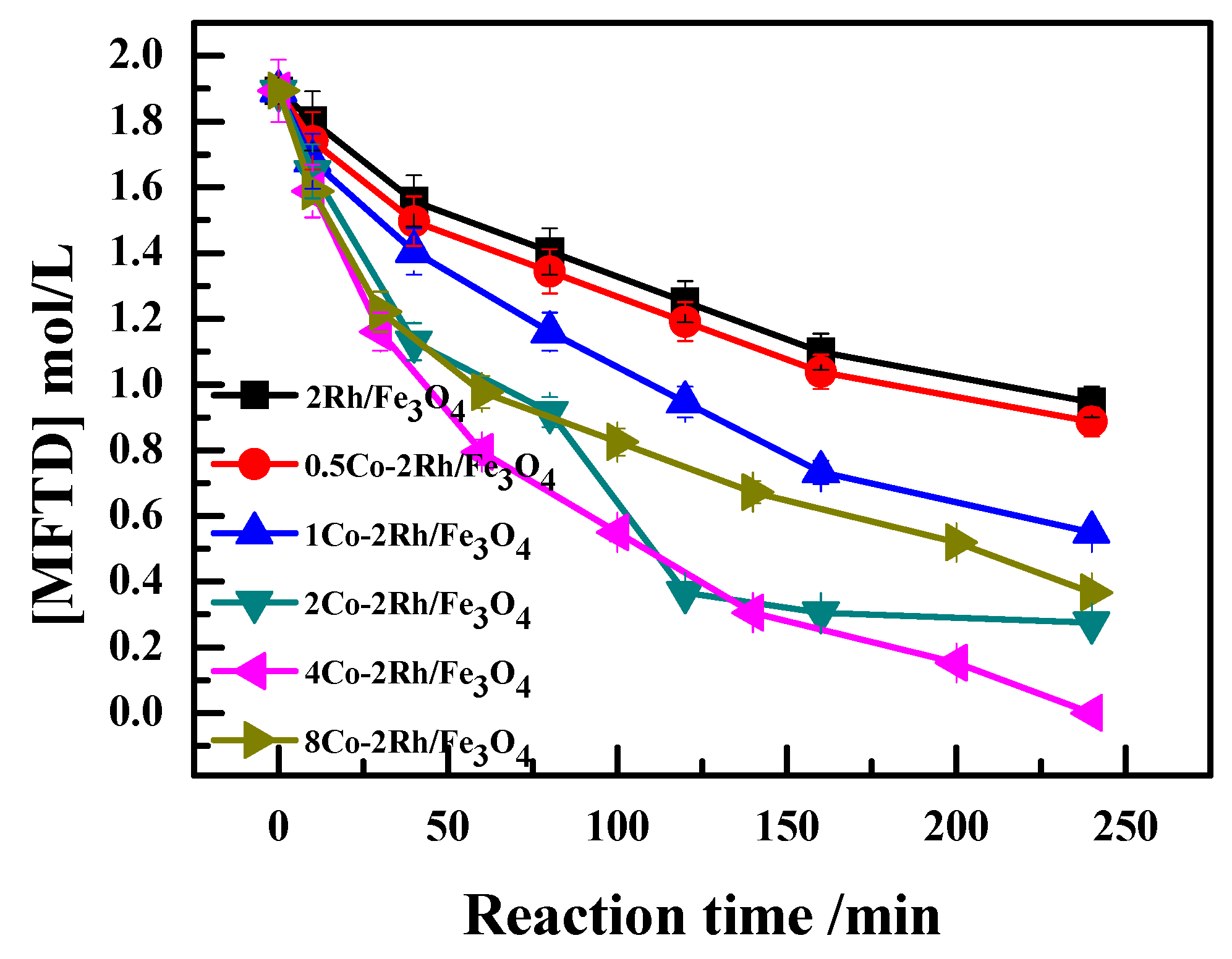
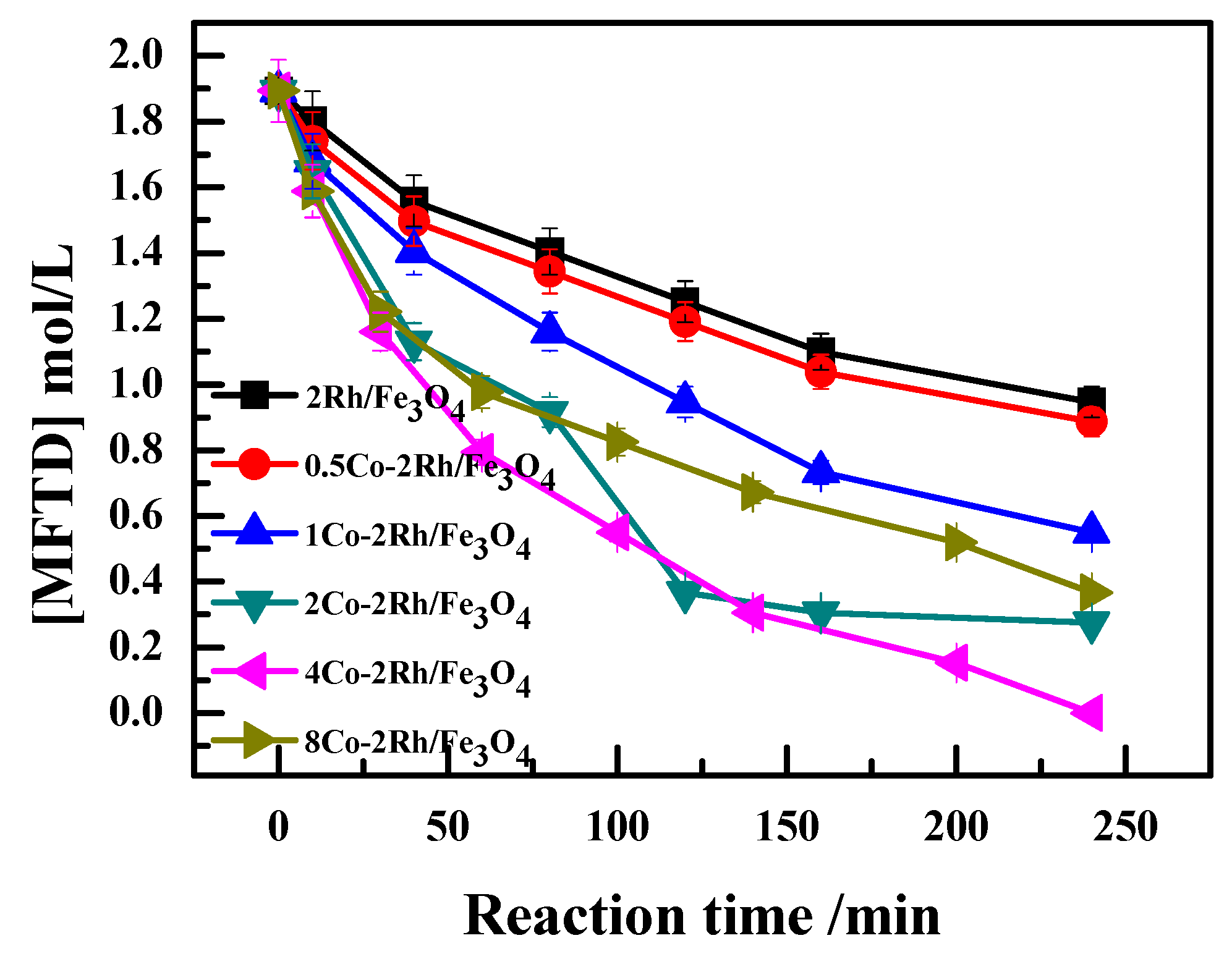
As shown in
Figure 2 (Reaction conditions: 140 °C and 7 MPa for 4 h, 5 g DCPD, 0.2 g catalyst, 0.2 g PPh
3, 20 mL solvent.), the MFTD consumption rate over the different Co–Rh/Fe
3O
4 catalysts decreased in the following order: 4Co-2Rh/Fe
3O
4 > 2Co-2Rh/Fe
3O
4 > 8Co-2Rh/Fe
3O
4 > 1Co-2Rh/Fe
3O
4 > 0.5Co-2Rh/Fe
3O
4 > 2Rh/Fe
3O
4. These results indicated that the reaction rate increased with increasing Co/Rh ratio up to a ratio of 2:1 and decreased as the ratio was further increased.
Table 3 lists the kinetic parameters for MFTD hydroformylation to DFTD over the tested Co-Rh/Fe
3O
4 catalysts. The rate constant increased as the Co/Rh ratio increased from 0:1 to 2:1 and then decreased with a further increase in the ratio from 2:1 to 4:1. The highest rate constant was observed for the 4Co-2Rh/Fe
3O
4. Therefore, 4Co-2Rh/Fe
3O
4 might be a suitable catalyst for DFTD synthesis by DCPD hydroformylation.
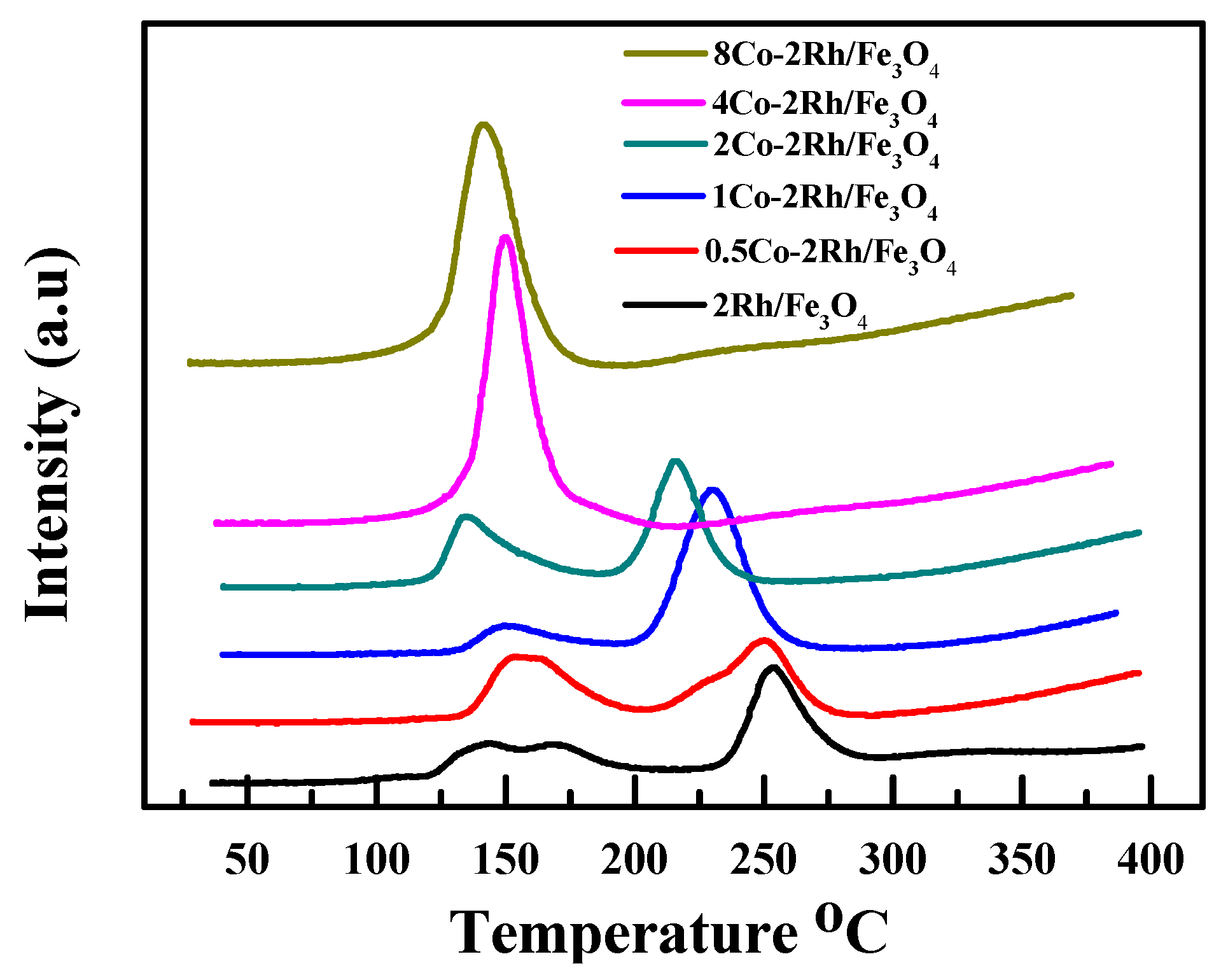
2.4. H2-TPR, H2-TPD, and TG-DTA
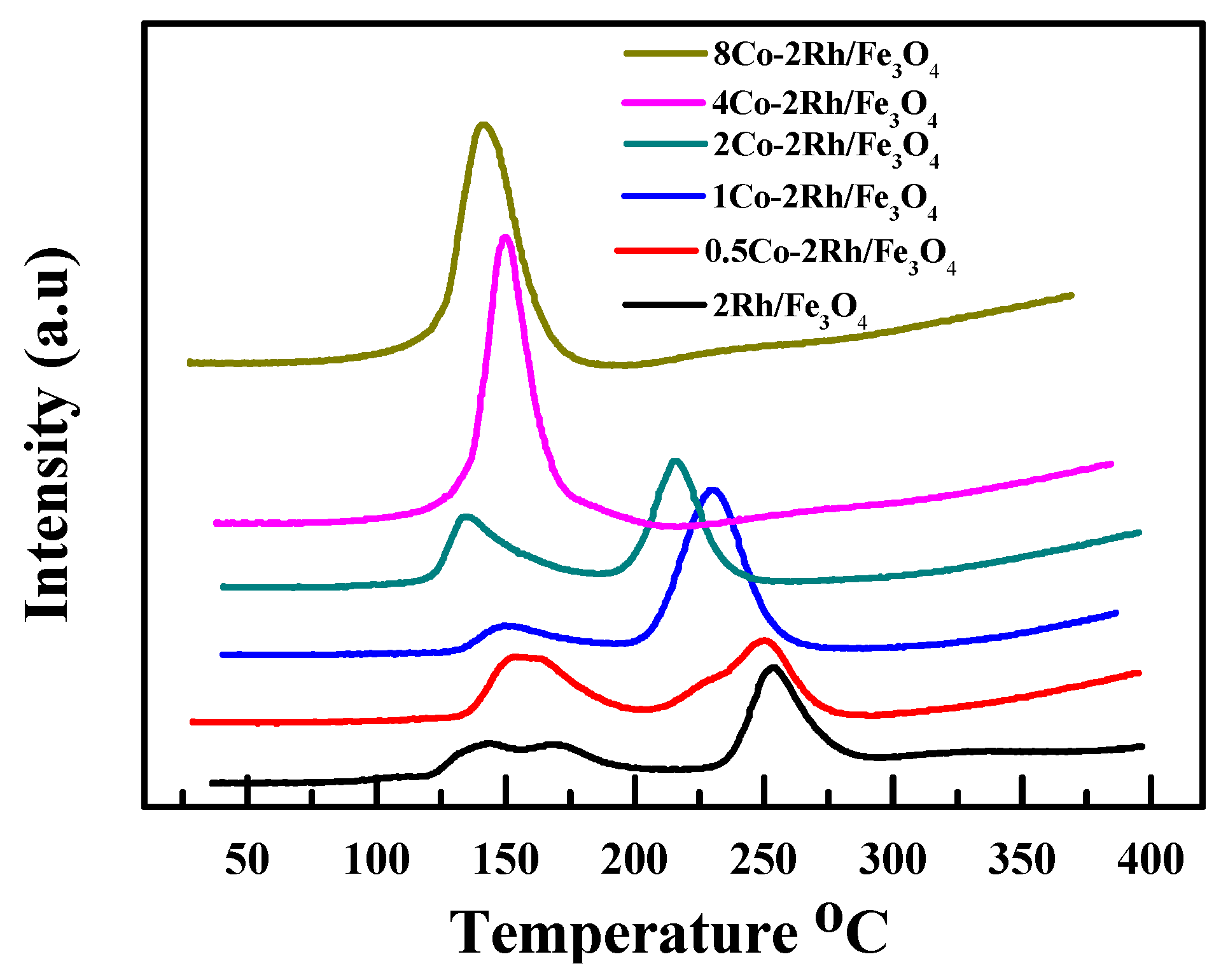
TPR was used to probe the Rh oxidation state in the cobalt-modified Rh/Fe
3O
4 catalyst precursors (before reduction, existing as oxides). The results are summarized in
Figure 3. Two obvious peaks were observed for the Rh/Fe
3O
4 catalyst. The RhO
x reduction peaks of the cobalt-modified materials were shifted to lower temperatures relative to those of the Rh/Fe
3O
4 catalyst. Moreover, only one peak was observed when the Co/Rh ratio was increased to 2:1. In addition, the area under the curve (AUC) also varied with the cobalt loading. When the Co/Rh ratio was increased from 0 to 1:2, the AUC increased. However, a further increase in the amount of cobalt added to the Rh/Fe
3O
4 catalyst led to a decrease in the AUC. These results indicated that the Rh surface atoms initially became more accessible and were then partially blocked by the added cobalt atoms. They also showed that the RhO
x reducibility was easily altered. Thus, the higher activities of the cobalt-modified Rh/Fe
3O
4 catalysts were attributed to the presence of a more reactive Rh surface species that was induced and stabilized by the cobalt atoms.
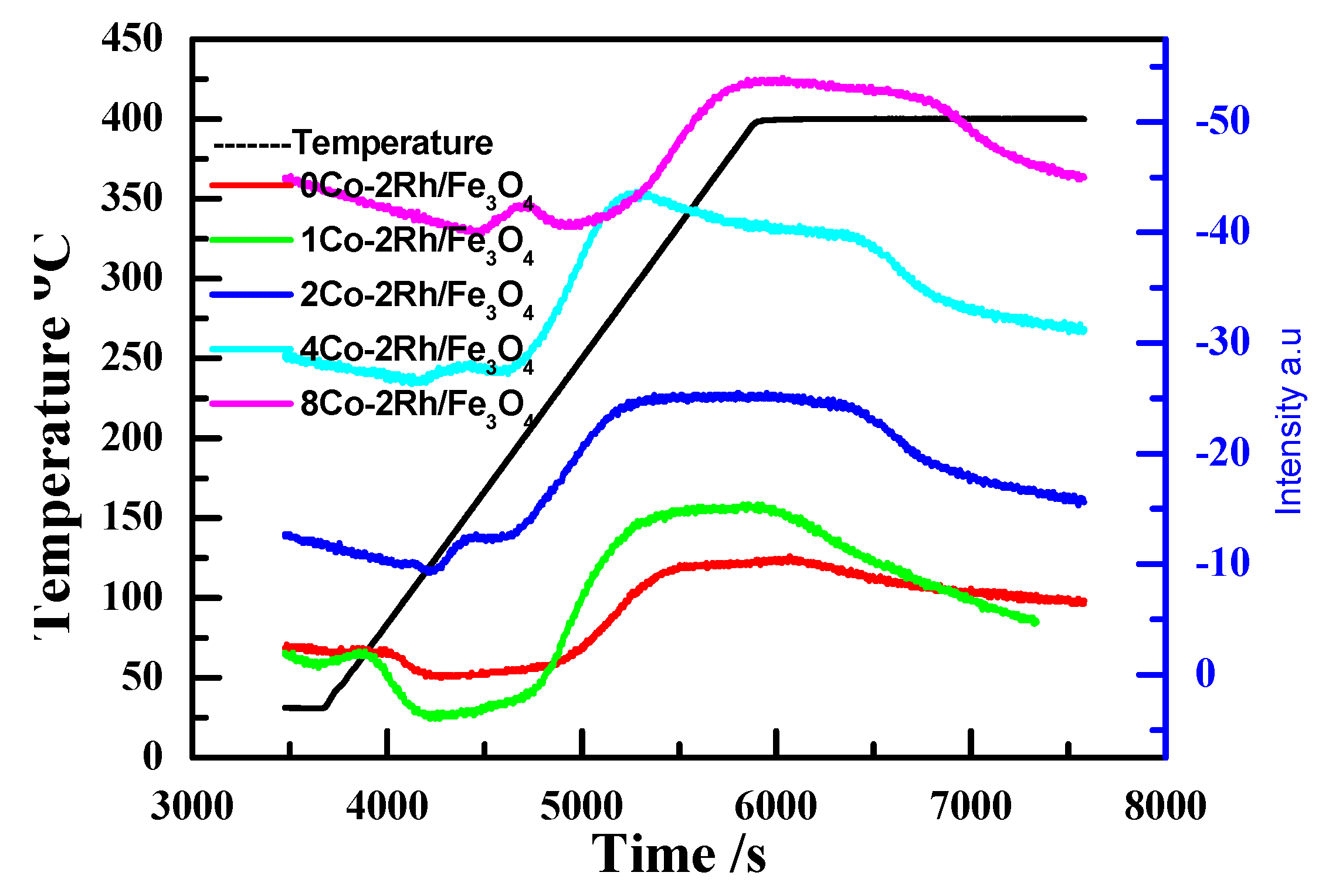
H
2-TPD was used to study the effect of the cobalt loading on Rh/Fe
3O
4. As shown in
Figure 4, all the catalysts except 8Co-2Rh/Fe
3O
4 had desorption peaks centered at similar temperatures, indicating that the introduction of cobalt into the system did not significantly affect the Rh dispersion. The peak area increased in the following order: 0.5Co-2Rh/Fe
3O
4 < 1Co-2Rh/Fe
3O
4 < 2Co-2Rh/Fe
3O
4 < 4Co-2Rh/Fe
3O
4. The accessibility of the Rh atoms on the Fe
3O
4 surface was expected to decrease with increasing cobalt loading; however, the Rh atoms were highly accessible after cobalt was introduced into the system, suggesting that the Rh dispersion was actually improved.
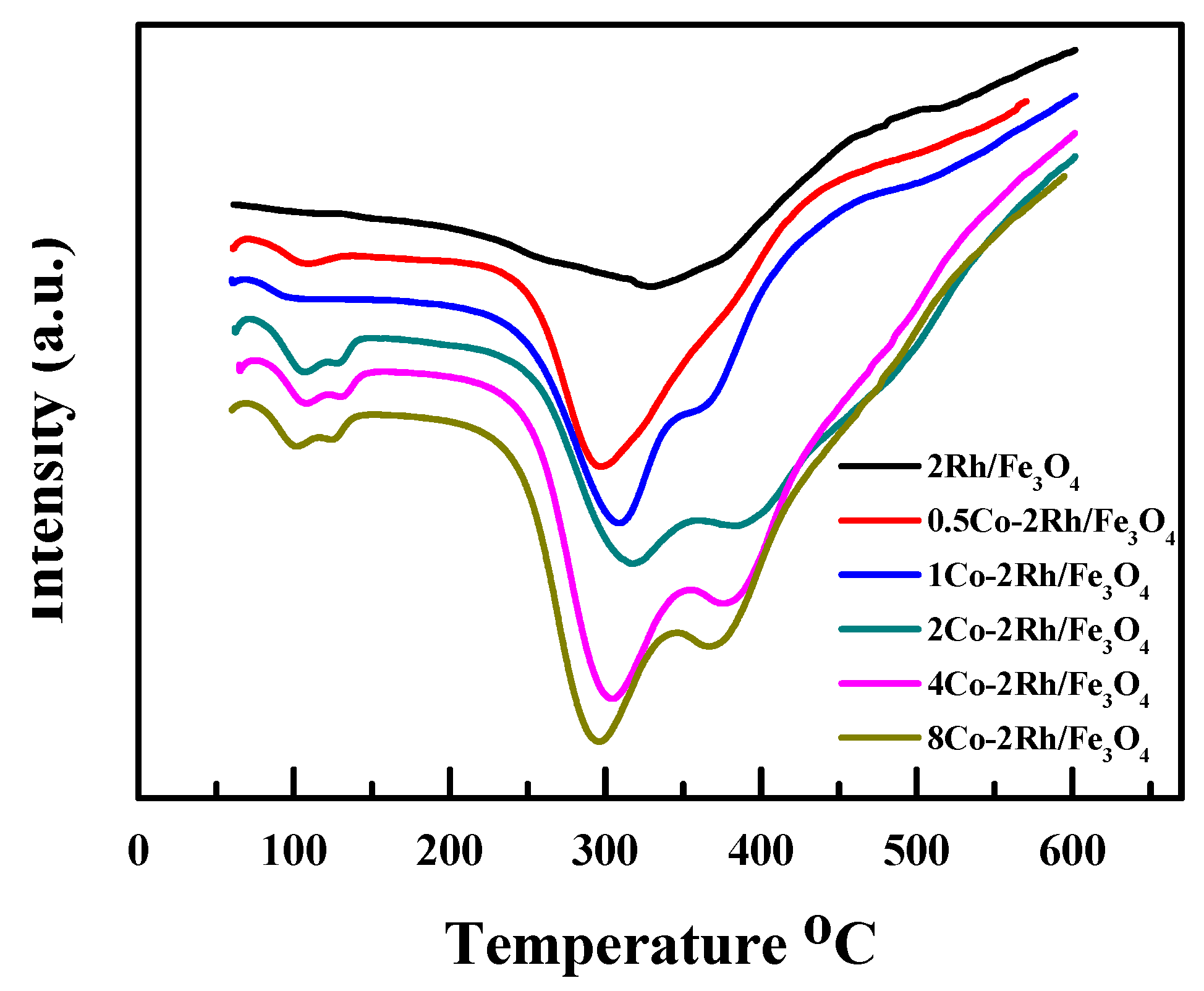
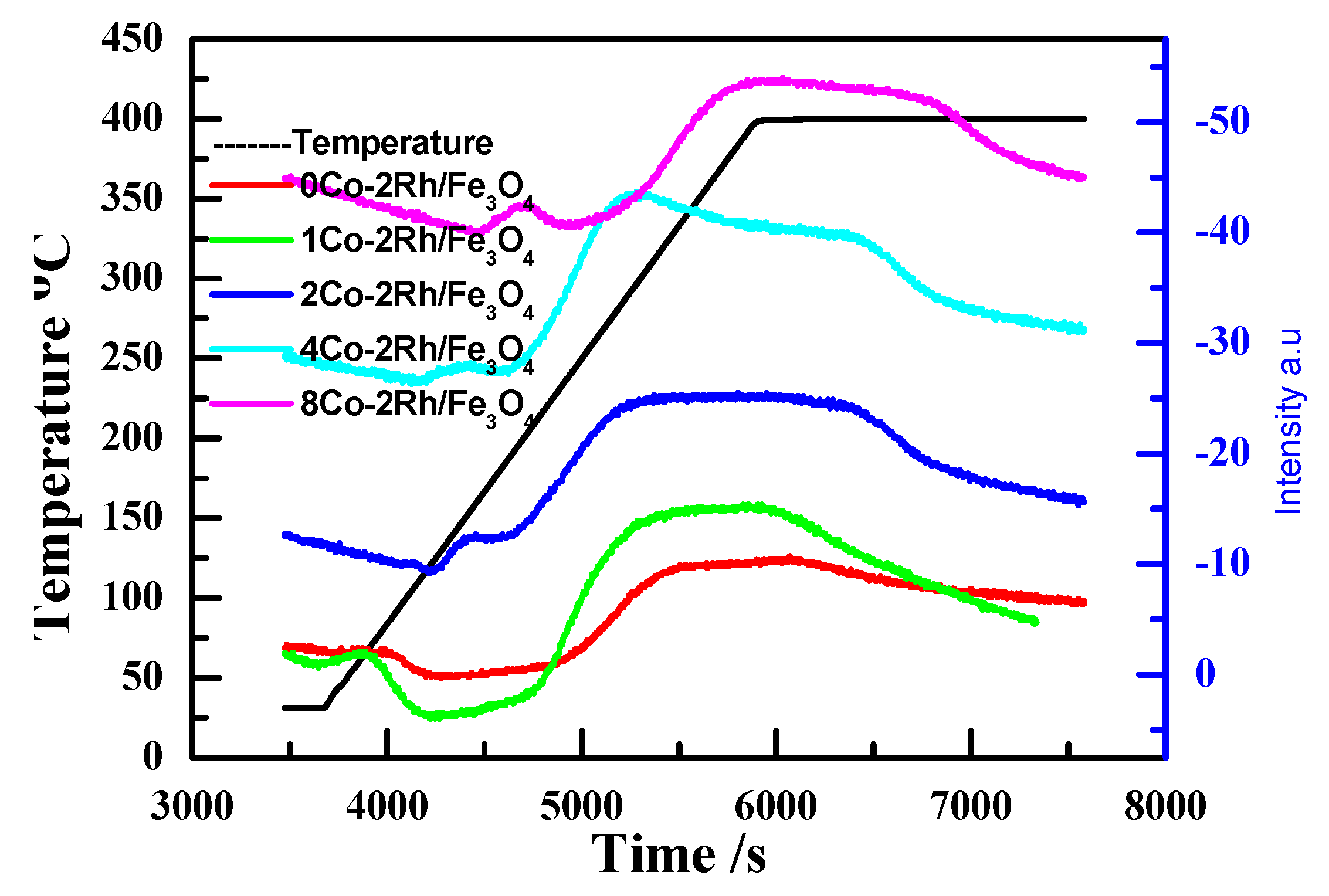
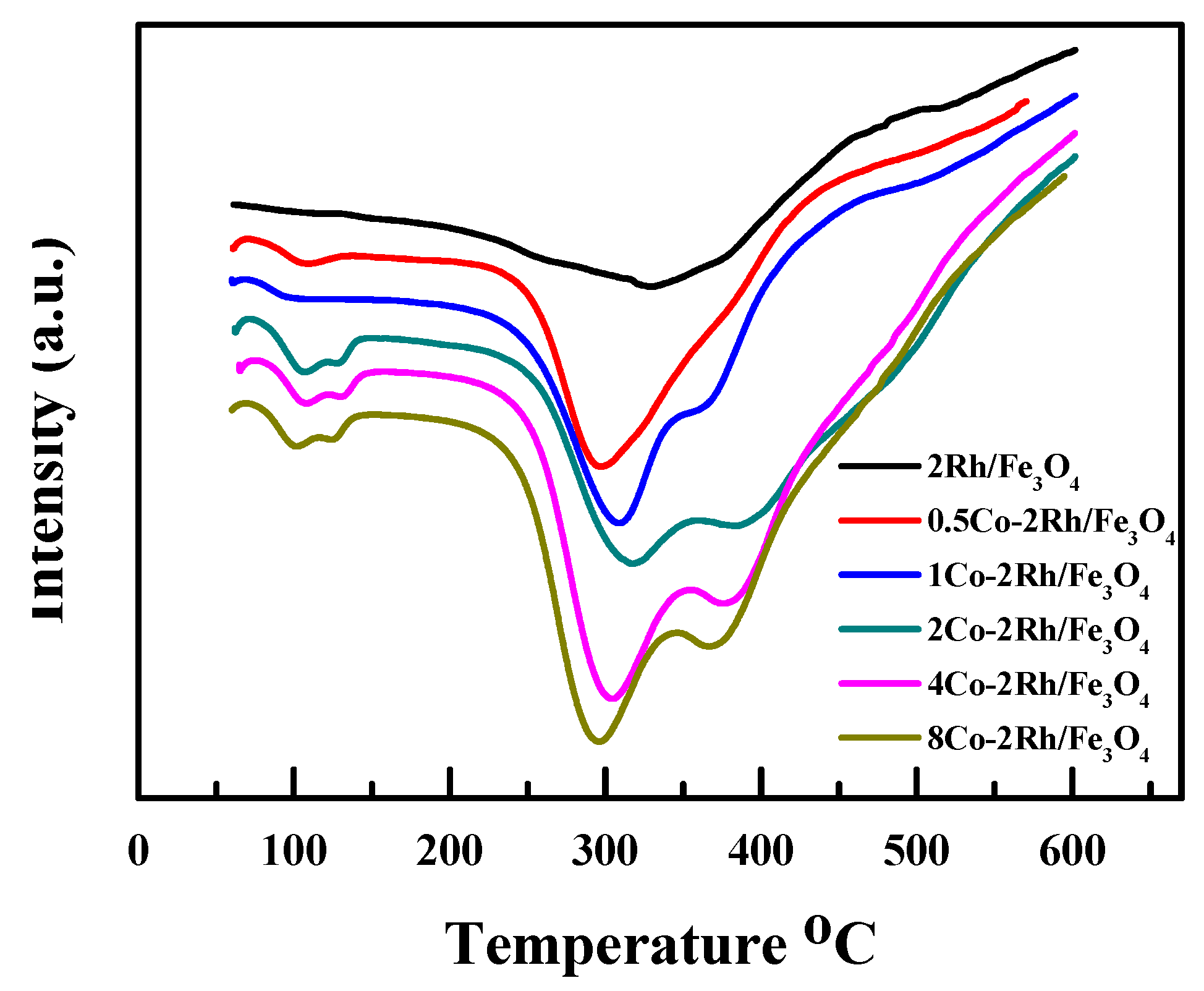
Figure 5 shows the differential thermal analyses (DTA) profiles of Rh/Fe
3O
4, 0.5Co-2Rh/Fe
3O
4, 1Co-2Rh/Fe
3O
4, 2Co-2Rh/Fe
3O
4, 4Co-2Rh/Fe
3O
4, and 8Co-2Rh/Fe
3O
4. Only one peak was observed for Rh/Fe
3O
4 and 0.5Co-2Rh/Fe
3O
4, and the 0.5Co-2Rh/Fe
3O
4 peak was shifted to a lower temperature relative to the Rh/Fe
3O
4 peak. However, two peaks were observed for 1Co-2Rh/Fe
3O
4, 2Co-2Rh/Fe
3O
4, 4Co-2Rh/Fe
3O
4, and 8Co-2Rh/Fe
3O
4. The low-temperature peak shifted to lower temperatures, and the high-temperature peak shifted to higher temperatures as the Co/Rh ratio increased from 1:2 to 2:1. The high-temperature peak of 8Co-2Rh/Fe
3O
4 was observed at a lower temperature than those of 1Co-2Rh/Fe
3O
4, 2Co-2Rh/Fe
3O
4, and 4Co-2Rh/Fe
3O
4. These results indicated that the Rh–P interaction strength was altered by the added cobalt. Our previous work suggested that a weak Rh–phosphine interaction was desirable for MFTD production with fast reaction kinetics, whereas a strong Rh–phosphine complex was required to synthesize DFTD [
10], which might explain the observed increase in the reaction rate and DFTD selectivity with increasing Co/Rh ratio up to a ratio of 2:1 and decrease in these variables at higher Co/Rh ratios (i.e., Co/Rh = 4:1).
3. Experimental Section
3.1. Materials
All chemicals used in the experiments were analytical grade and used without further purification unless otherwise noted.
3.2. Catalyst Preparation
The 2Rh/Fe3O4 catalyst was prepared by co-precipitation. Briefly, 104 mmol (42 g) Fe(NO3)3·9H2O (Alfa Aesar, Tianjin, China) and 2.08 mmol (0.5 g) RhCl3 (Alfa Aesar, Tianjin, China) were dissolved in 150 mL of distilled water. This solution was then added dropwise to 600 mL of a 0.47 M Na2CO3 (Sinopharm, Shanghai, China) solution under vigorous stirring for approximately 1 h. The reaction mixture was stirred continuously for an additional 2 h. After filtration, the precipitate was washed with 150–200 mL of distilled water, dried at 120 °C for 16 h, and calcined at 400 °C in air for 4 h. The obtained solid was then reduced under an H2 flow at 400 °C for 2 h to yield the 2Rh/Fe3O4 catalyst. The 0.5Co-2Rh/Fe3O4, 1Co-2Rh/Fe3O4, 2Co-2Rh/Fe3O4, 4Co-2Rh/Fe3O4, and 8Co-2Rh/Fe3O4 catalysts, which had different cobalt loadings, were prepared in the same way.
3.3. Catalyst Characterization
3.3.1. Temperature-Programmed Reduction
Temperature-programmed reduction (TPR) experiments were performed on a FINESORB-3010 analyzer equipped with a TCD detector. The catalyst (approximately 100 mg) was pretreated under dry air at 383 K for 1 h. The TPR profile was obtained by heating the sample from room temperature to 673 K at a rate of 10 K/min under an H2/Ar (10% v/v) flow.
3.3.2. H2 Temperature-Programmed Desorption
The metal dispersion was determined by H2 temperature-programmed desorption (TPD) using a FINESORB-3010 analyzer equipped with a mass detector (DM 300, AMETEK, San Diago, CA, USA). In a typical experiment, 100 mg of the catalyst (particle size of 160–200 µm, dried at 383 K for 5 h before each measurement) was placed in a U-shaped quartz tube. The sample was pretreated by passing hydrogen over the catalyst at a flow rate of 50 mL/min at 673 K for 1 h. The sample was then cooled to room temperature and kept in an H2 flow (50 mL/min) for 1 h. The system was subsequently purged with pure Ar (flow rate of 50 mL/min) at room temperature for an additional 1 h. In the TPD experiments, the samples were heated from 20 °C to 400 °C at a rate of 10 °C/min under a 50 mL/min Ar flow. The desorbed H2 was continuously monitored by mass spectrometry (m/z = 2).
3.3.3. Thermogravimetric and Differential Thermal Analyses
Thermogravimetric analysis was used to determine the ligand degradation temperature (TA Instruments, model DTG-60, Shimadzu, Kyoto, Japan). The 2Rh/Fe3O4 and (0.5%–8%) Co-2Rh/Fe3O4 catalysts were modified with PPh3 and then pretreated with syngas under the reaction conditions. The samples were subsequently heated to 600 °C at 10 °C/min under a 60 mL/min nitrogen flow.
3.4. DCPD Hydroformylation
3.4.1. MFTD Synthesis
DCPD hydroformylation to monoformyltricyclodecenes (MFTD) was conducted in a 200 mL stainless steel autoclave reactor with a glass liner insert. The reaction temperature was controlled by an oil bath on a hot plate connected to a digital thermocouple. In a typical experiment, the catalyst (200 mg), solvent (20 mL), phosphine ligand (0.76 mmol), and DCPD (20.0 g) were placed in the autoclave. Then, the reactor was purged twice with syngas (1:1 CO/H2). Afterwards, the initial pressure was adjusted to 4 MPa at room temperature. The reaction medium was then stirred, and the system was heated to approximately 95 °C and maintained at that temperature for 1.5 h.
3.4.2. DFTD Synthesis
The catalyst (200 mg), acetone solvent (20 mL), phosphine ligand (0.76 mmol), and DCPD (5.0 g) were mixed in the autoclave reactor. The reaction was performed in two stages. In the first step, the reactor was charged at an initial pressure of 4 MPa and room temperature, heated to 95 °C, and then maintained at that temperature for 1.5 h to selectively produce MFTD. In the second stage, the pressure was increased to 7 MPa, and the temperature was further increased to 140 °C. The reaction was performed under these conditions for 4 h to convert the MFTD to DFTD.
3.5. Reaction Product Analysis
The reaction effluents were analyzed using a GC2014 (Shimadzu, Kyoto, Japan) instrument equipped with an FID detector. The following definitions are used in the Results and Discussion section: the DCPD conversion is defined as the number of moles of DCPD consumed divided by the initial number of moles of DCPD. The selectivity to MFTD (DFTD) is defined as the number of moles of MFTD (DFTD) produced divided by the number of moles of DCPD consumed. Finally, the MFTD (DFTD) yield is defined as the DCPD conversion multiplied by the MFTD (DFTD) selectivity.
4. Conclusions
Six different Rh-based catalysts were prepared on Fe3O4 supports by co-precipitation, and their catalytic performances in DCPD hydroformylation were evaluated. When the DCPD conversion over the monometallic Rh/Fe3O4 catalyst was nearly complete, the selectivity to the desired DFTD product was only 21.3%, but the DFTD selectivity increased to 90.6% when cobalt was introduced into the system at a Co/Rh ratio of 2:1.
The catalysts were extensively characterized to provide insight into the effect of the cobalt loading on the hydroformylation selectivity and kinetics. The TPR results indicated that the added cobalt likely enhanced the catalytic performance by giving rise to a more reactive Rh surface species. H2-TPD analysis showed that the Rh dispersion was improved with increasing cobalt loading. TG-DTA analysis showed that the cobalt loading affected the Rh–P interaction strength, which resulted in different reaction rates and product distributions.
In summary, modifying Rh/Fe3O4 catalysts with cobalt can significantly enhance the reaction rate and DFTD selectivity with nearly complete DCPD conversion. The improved catalytic performance was likely due to the formation of a more reactive Rh species with a different Rh–phosphine interaction strength on the catalyst surface.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}