Recent Advancements in Stereoselective Olefin Metathesis Using Ruthenium Catalysts

Abstract
:1. Introduction
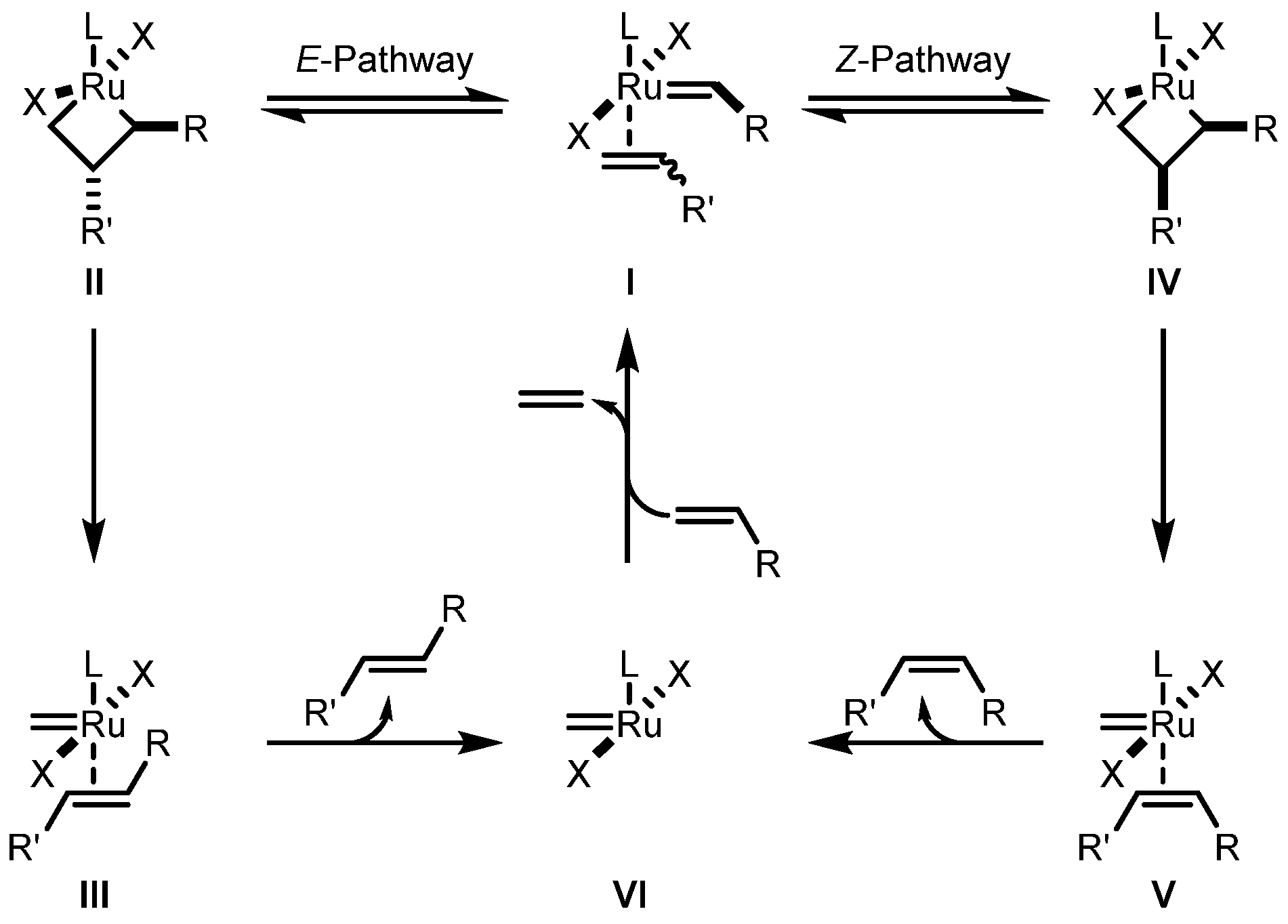
2. Z-Selective Olefin Metathesis
2.1. General Introduction
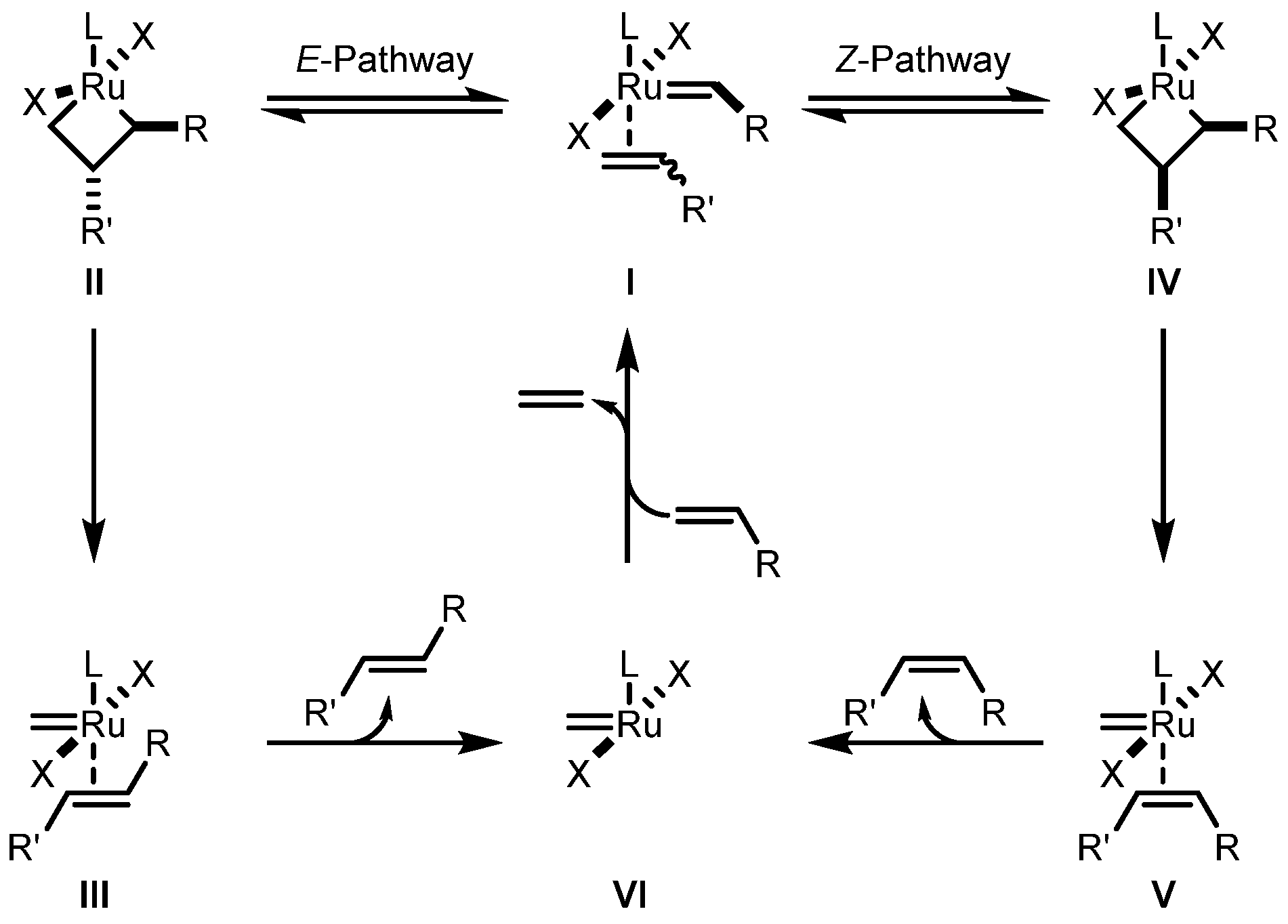
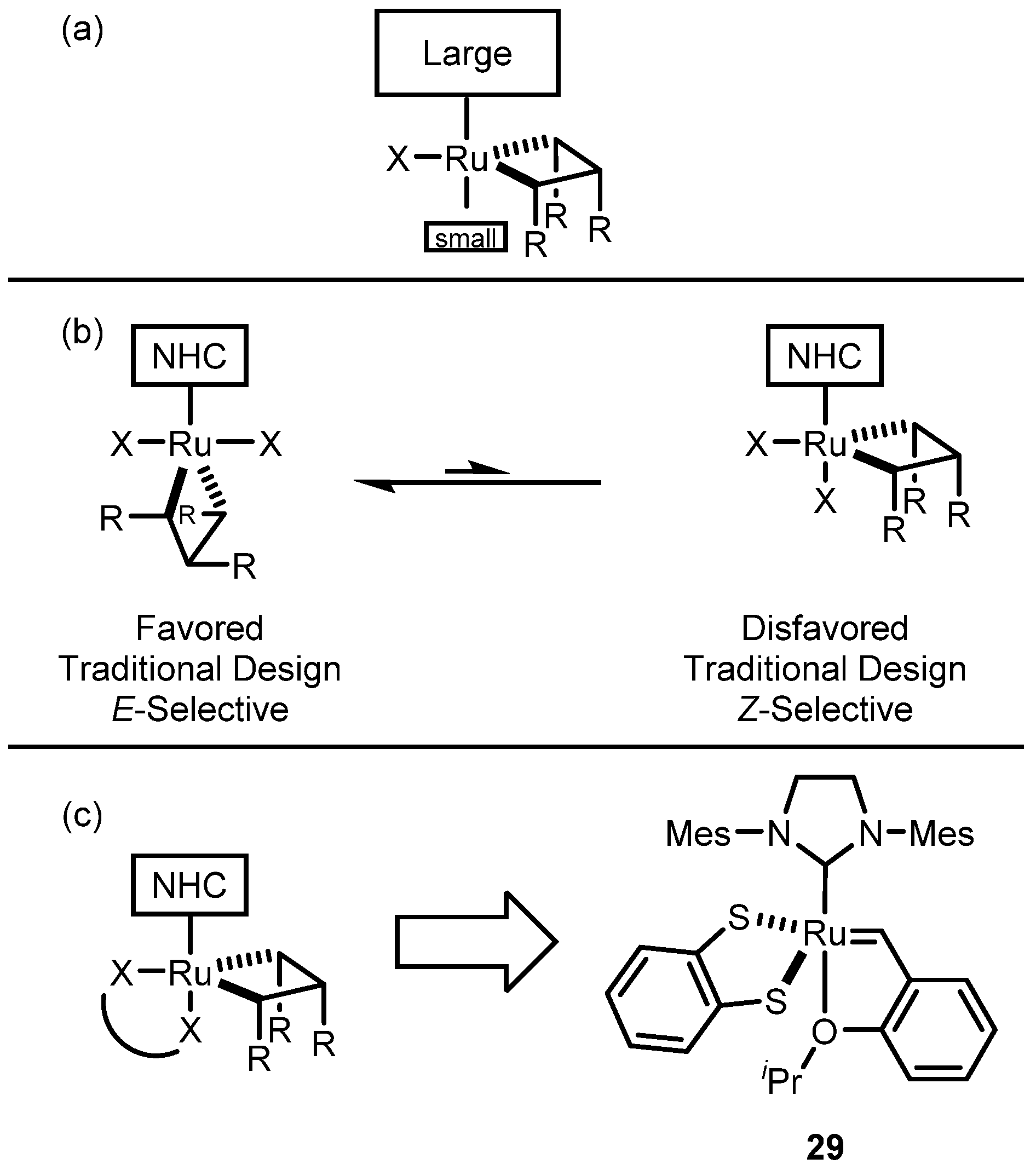
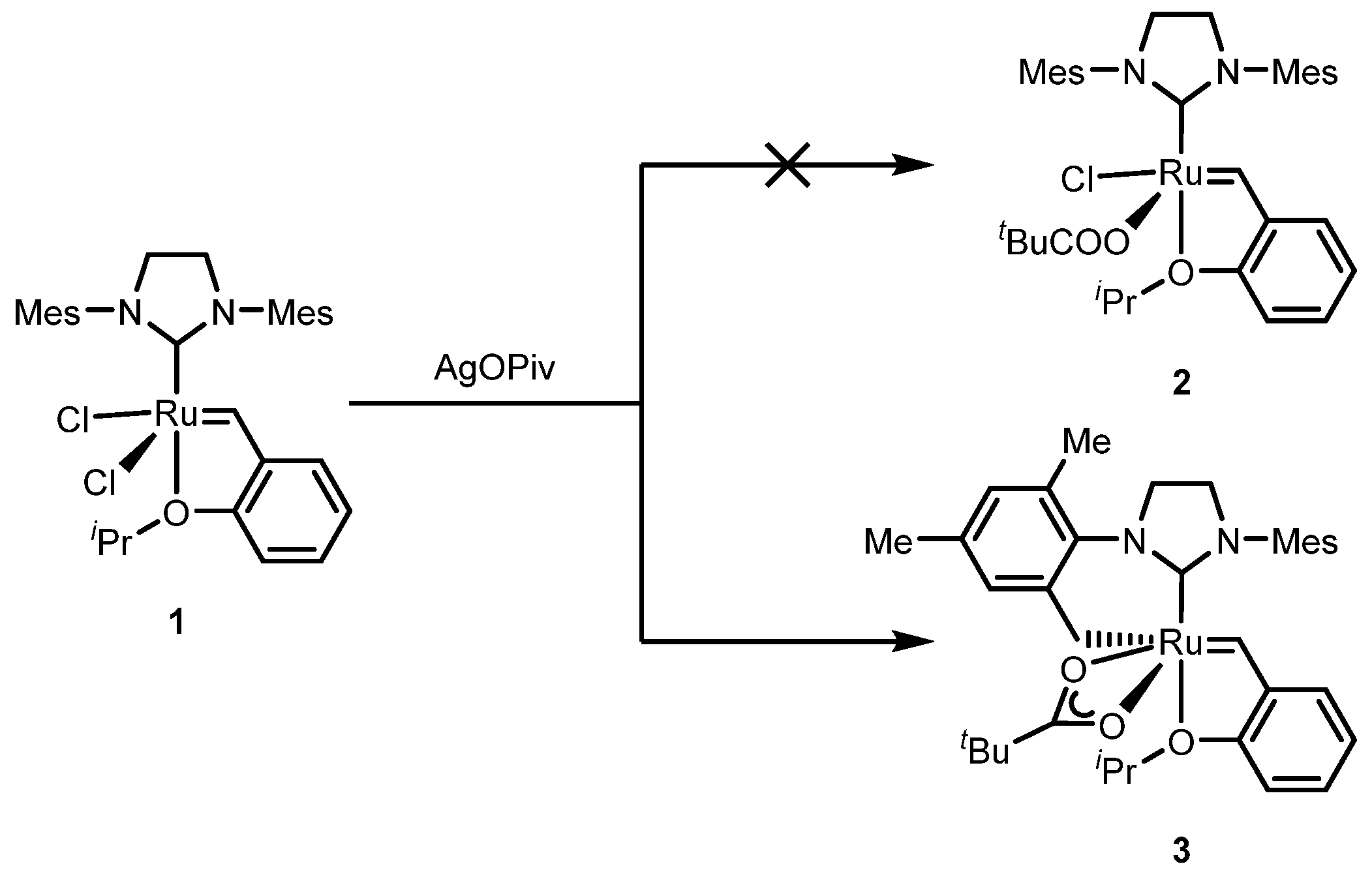
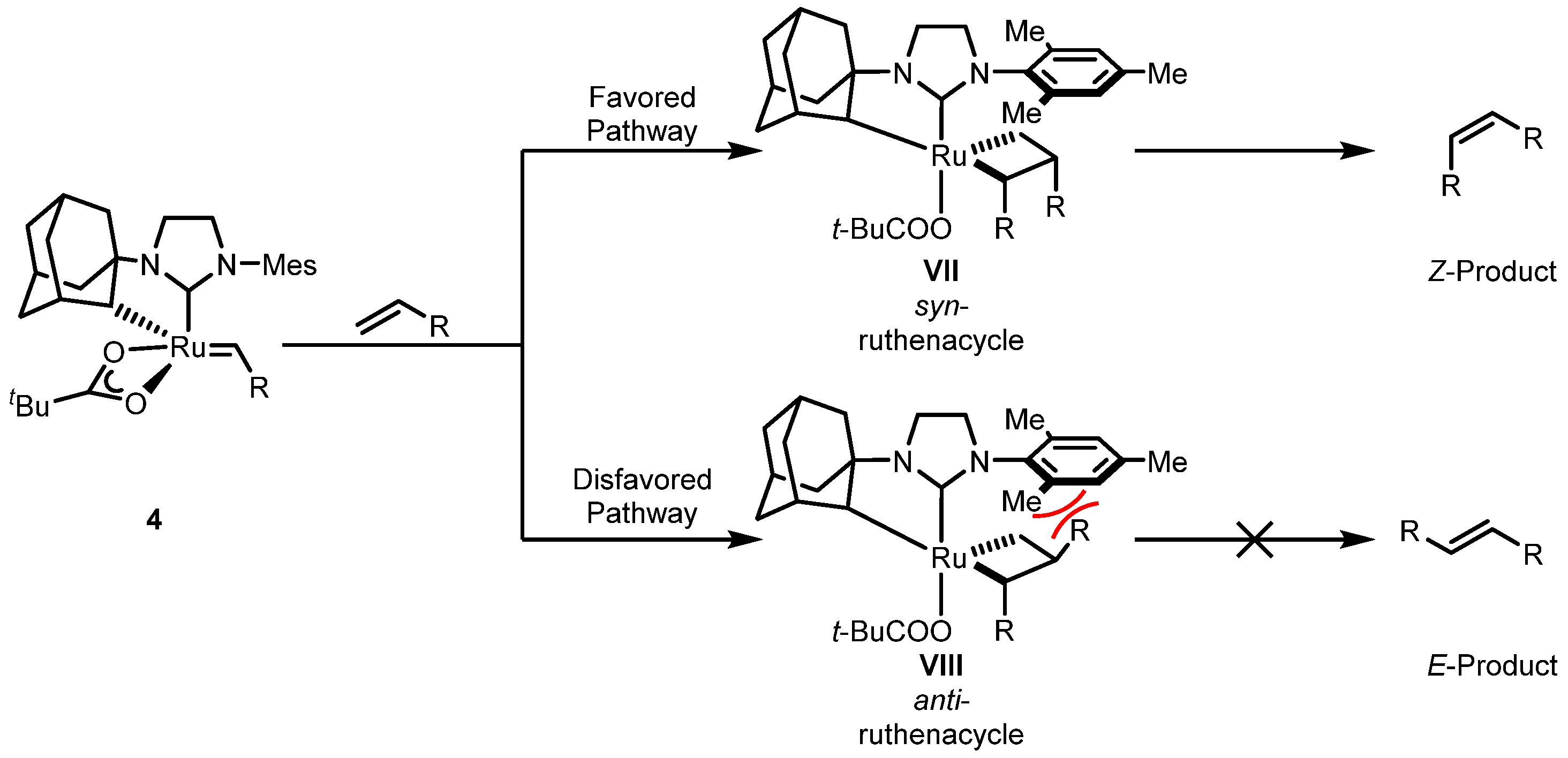
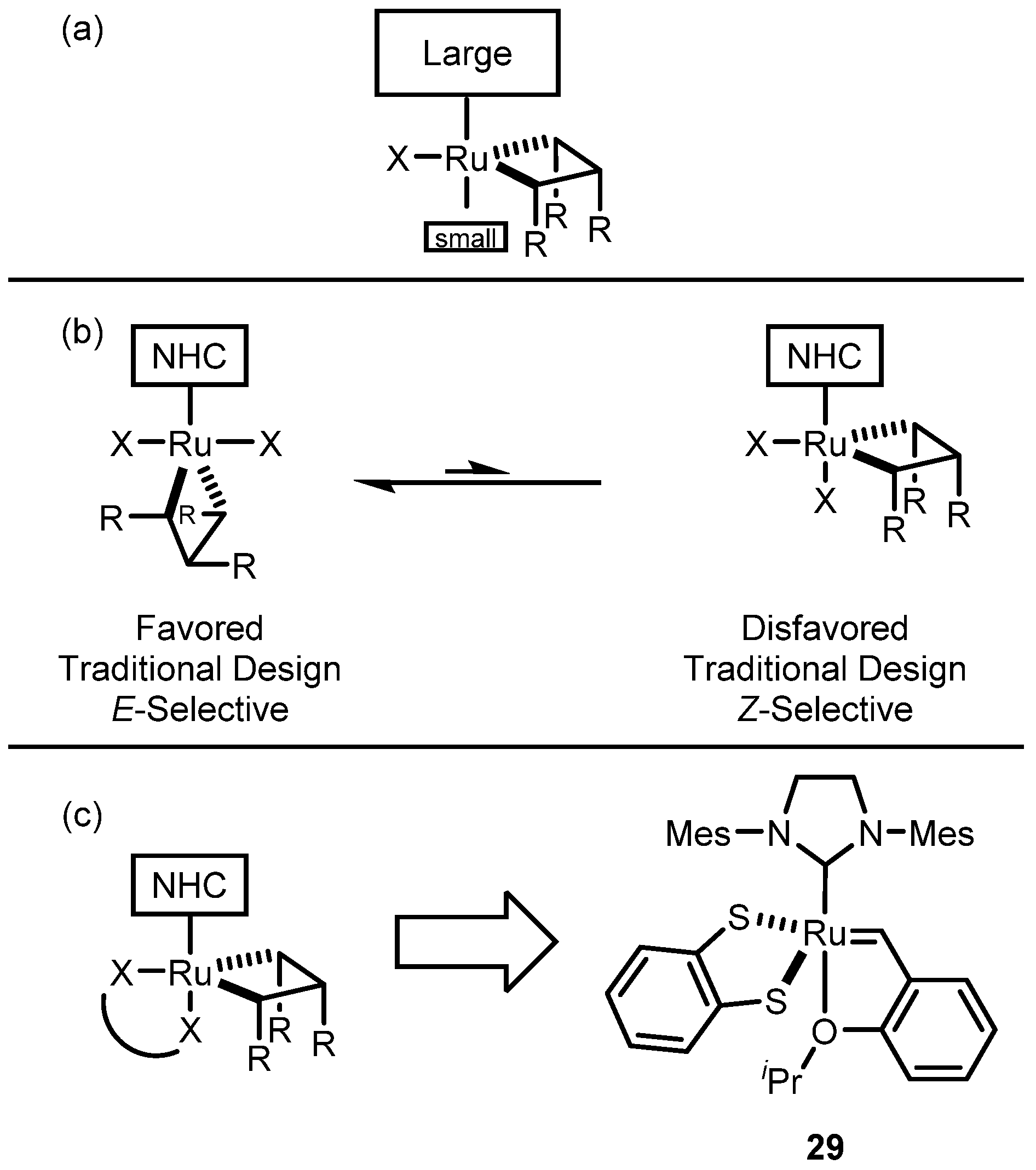
2.2. Cyclometalated Catalysts
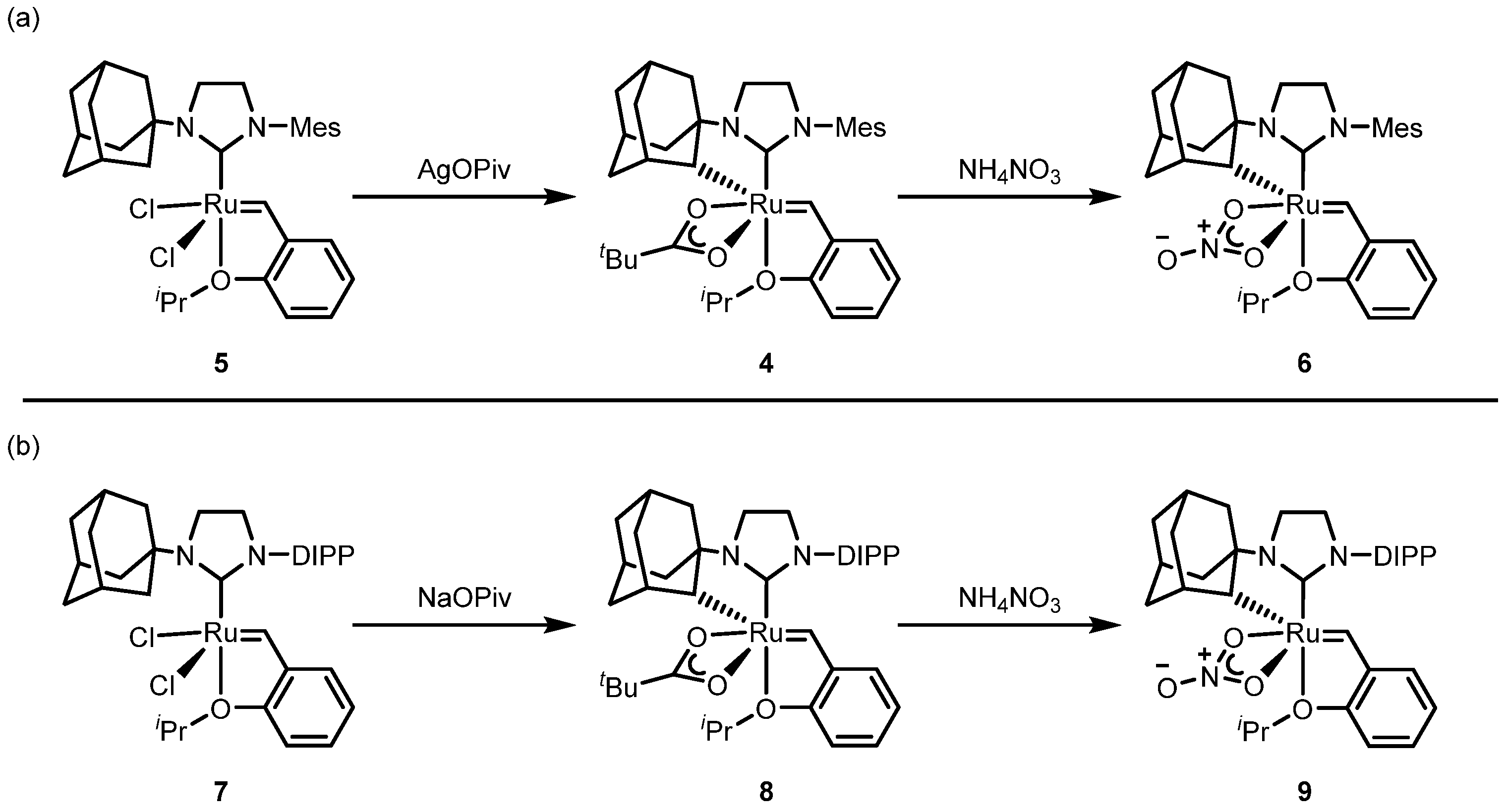
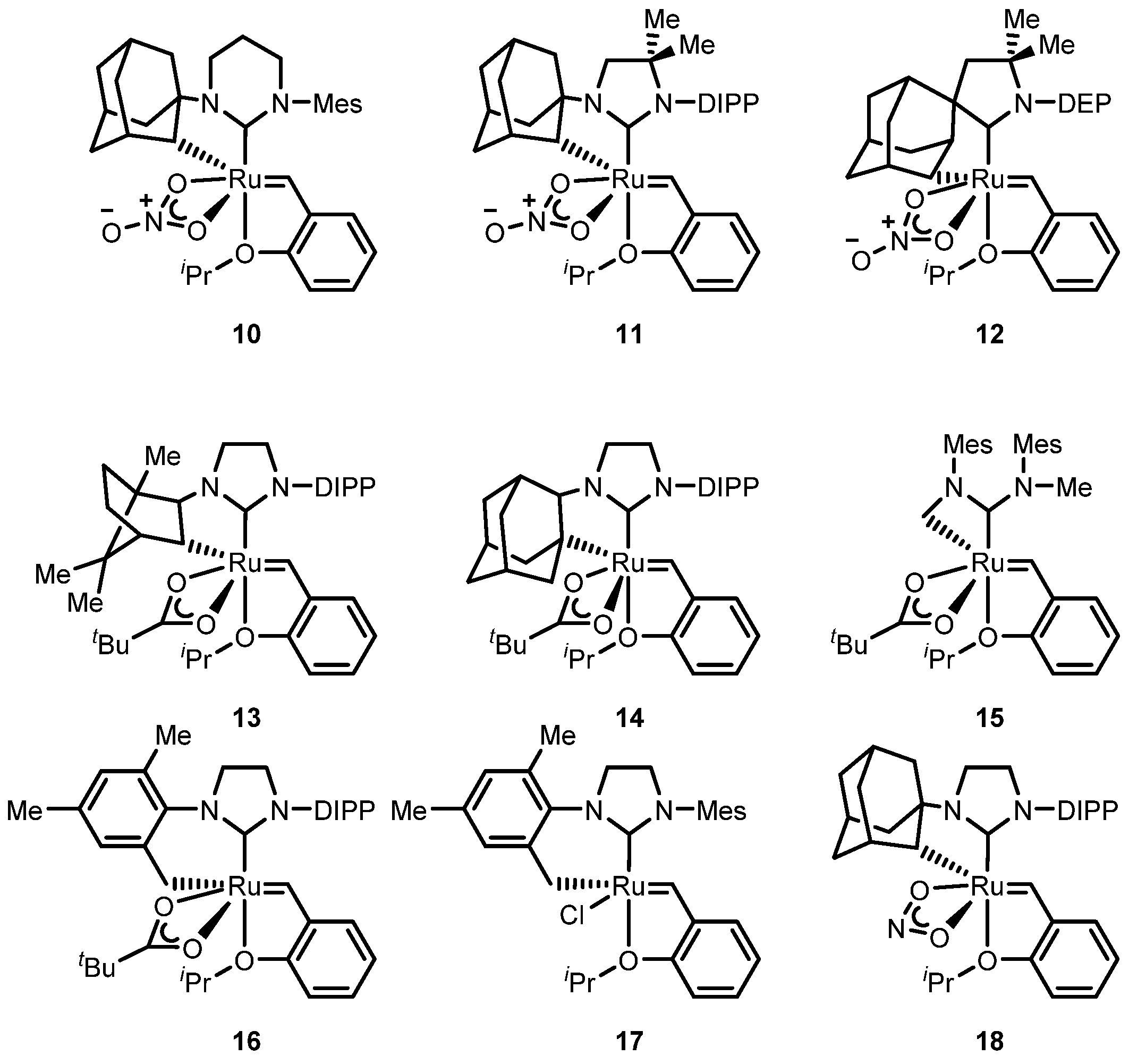
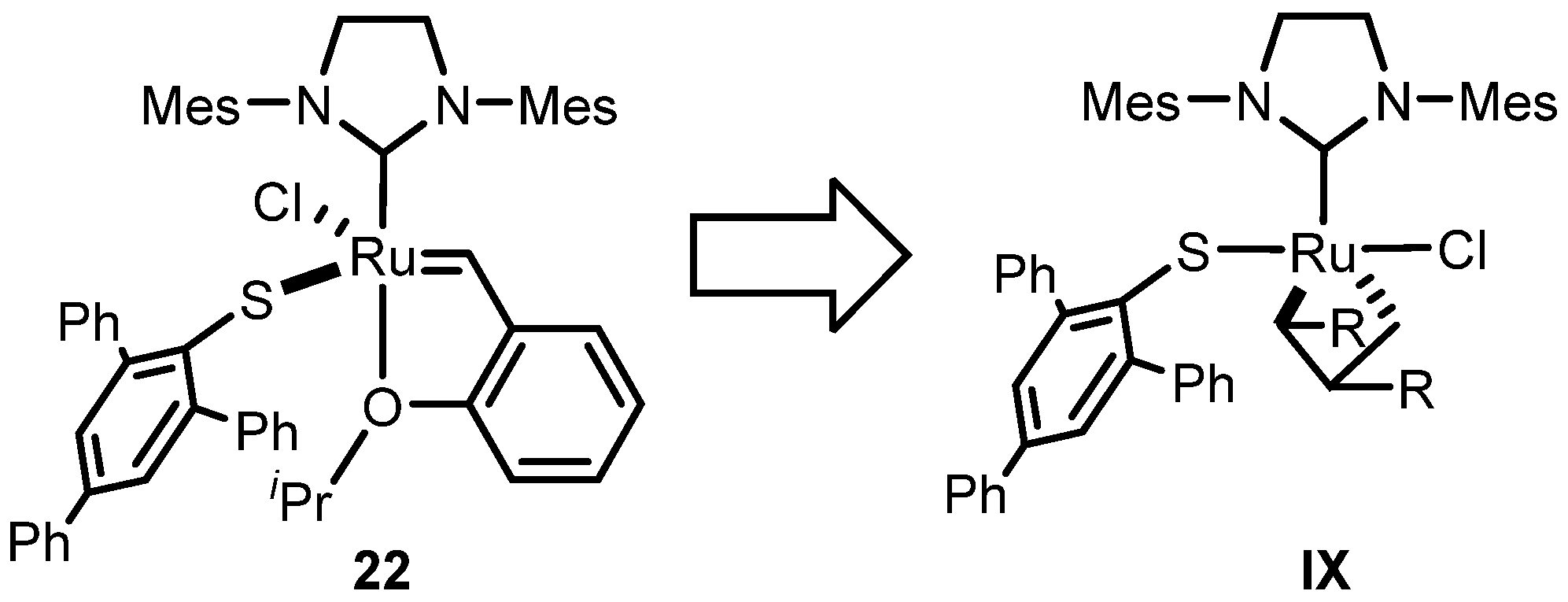
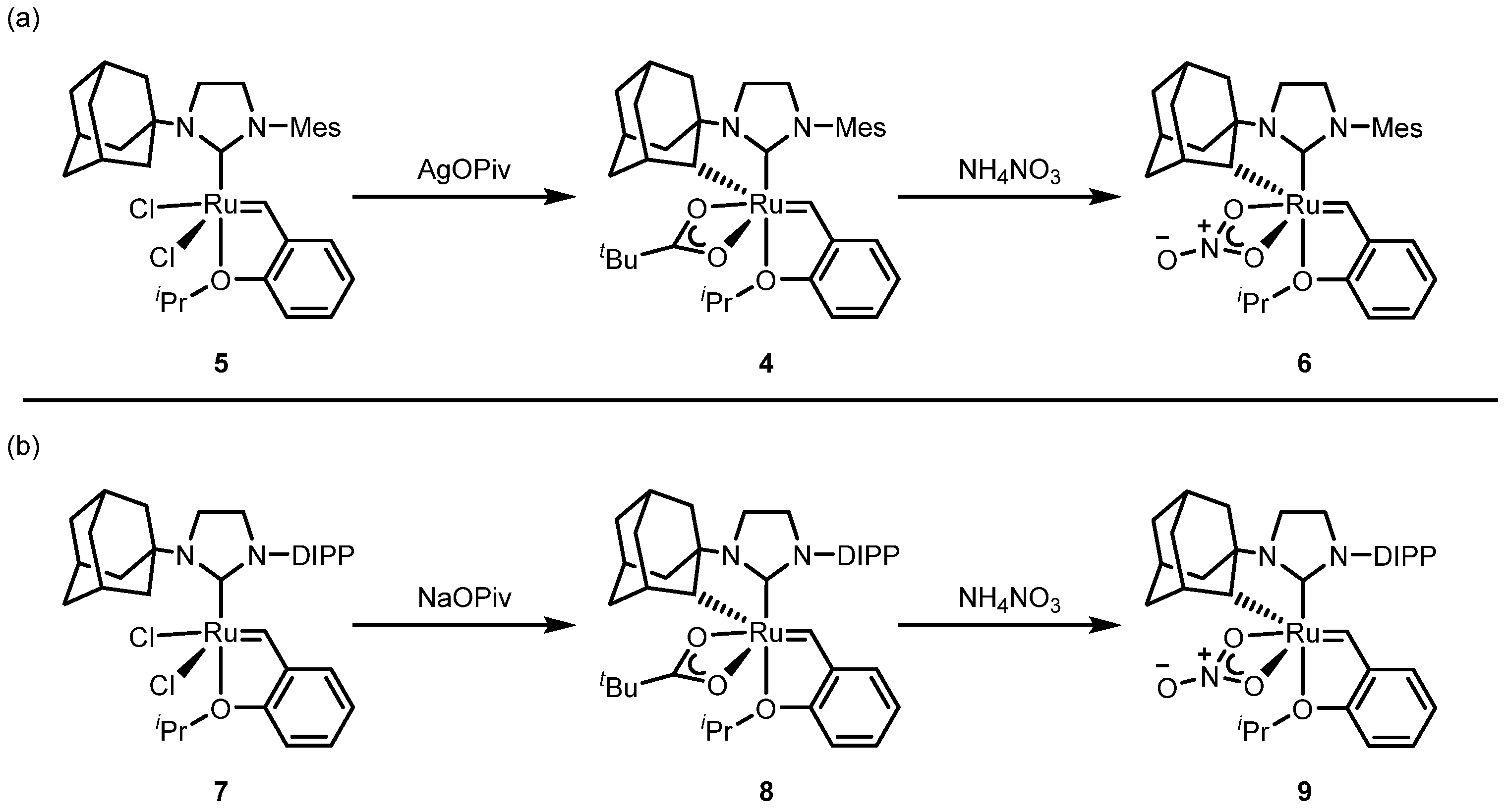
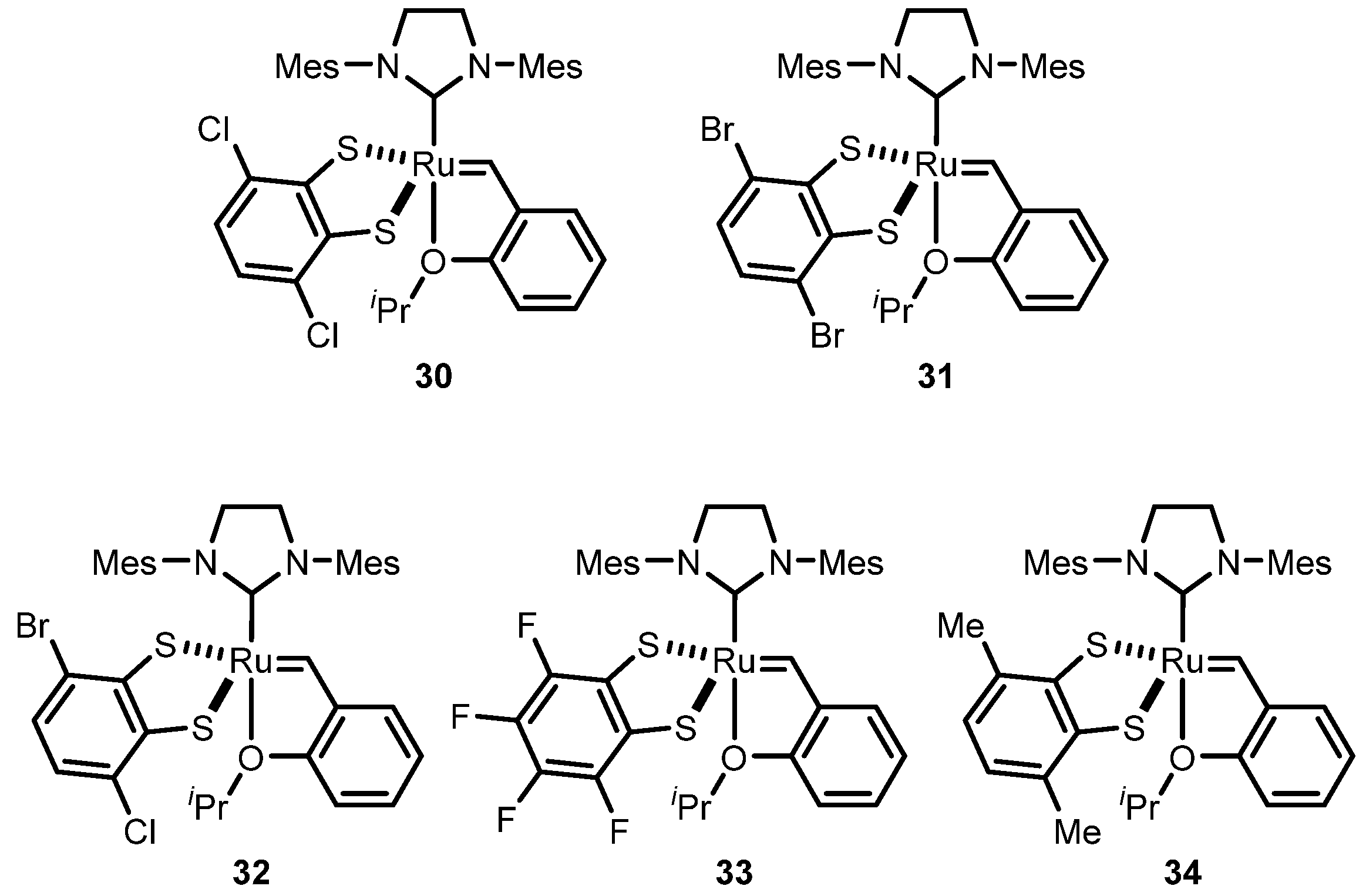
2.2.1. Cyclometalated Catalysts Development
2.2.2. Z-Selective Homodimerization
2.2.3. Z-Selective Cross Metathesis
2.2.4. Z-Selective Macrocyclic Ring-Closing Metathesis
2.2.5. Z-Selective Ethenolysis
2.2.6. Chemoselectivity in Z-Selective Olefin Metathesis
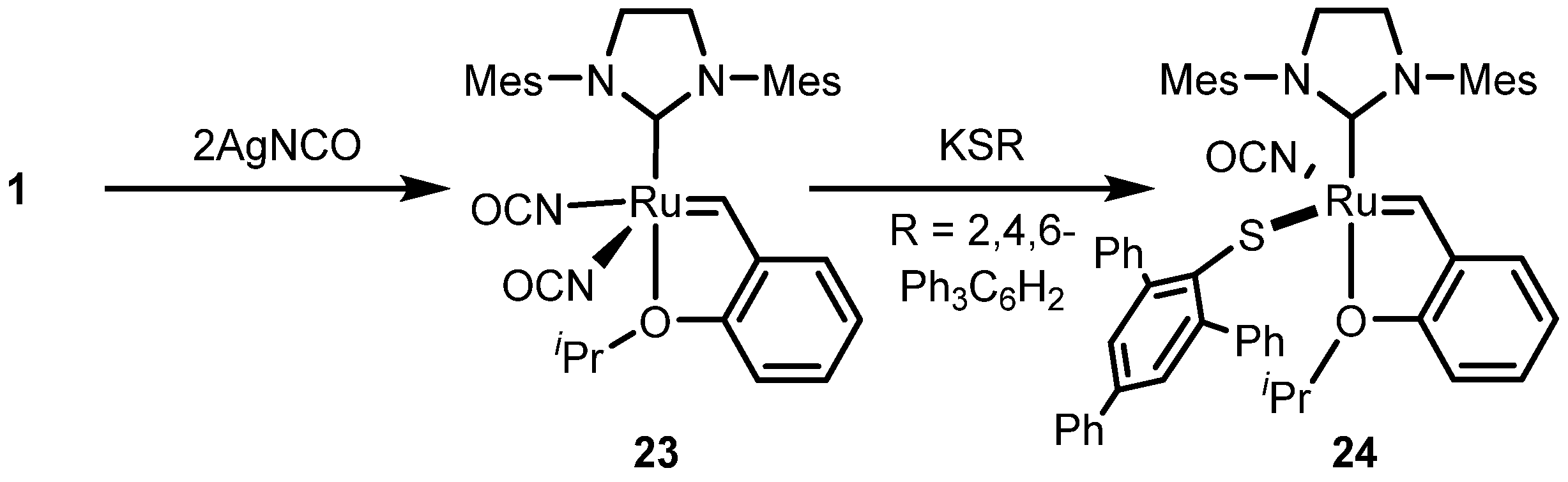
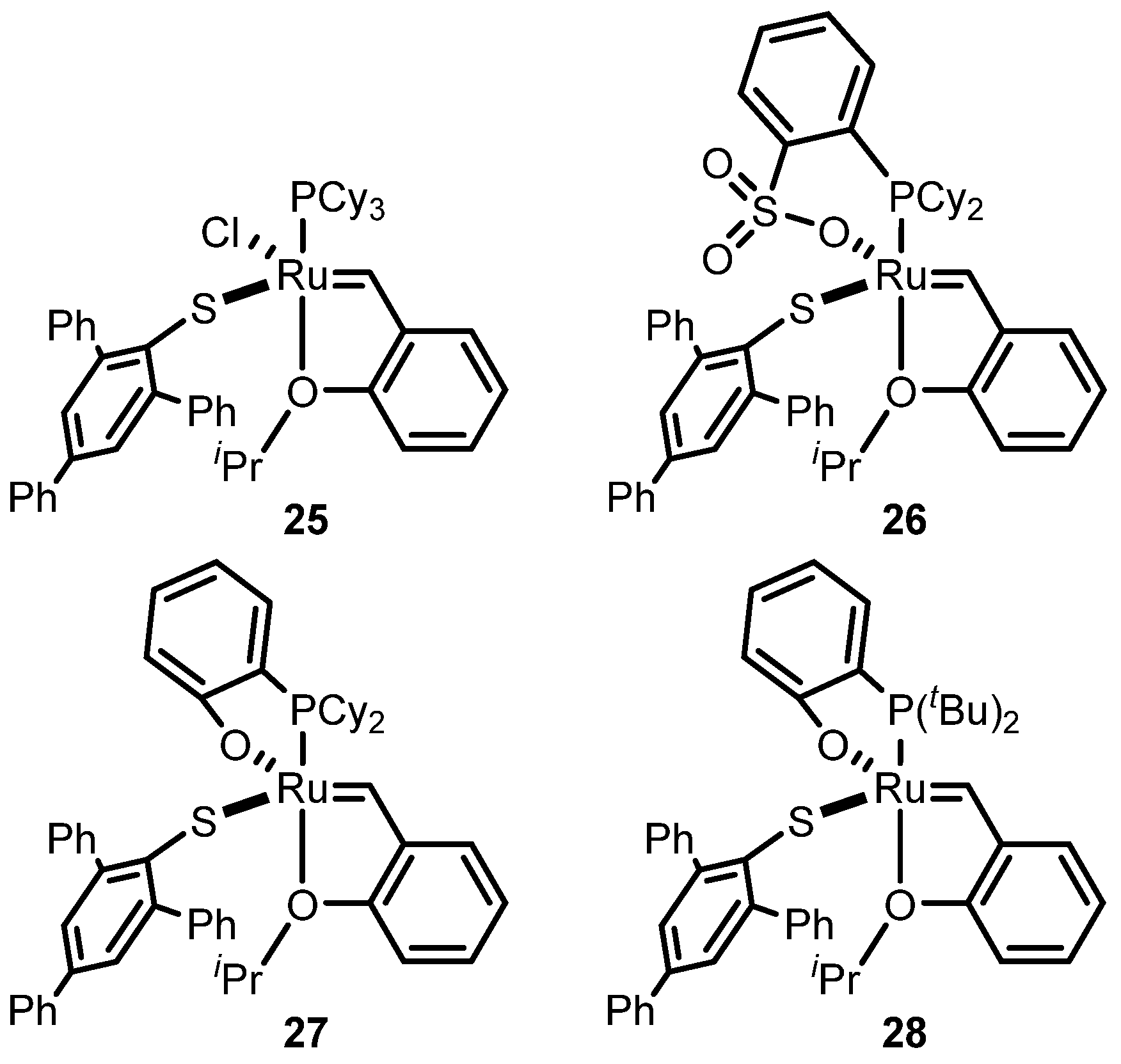
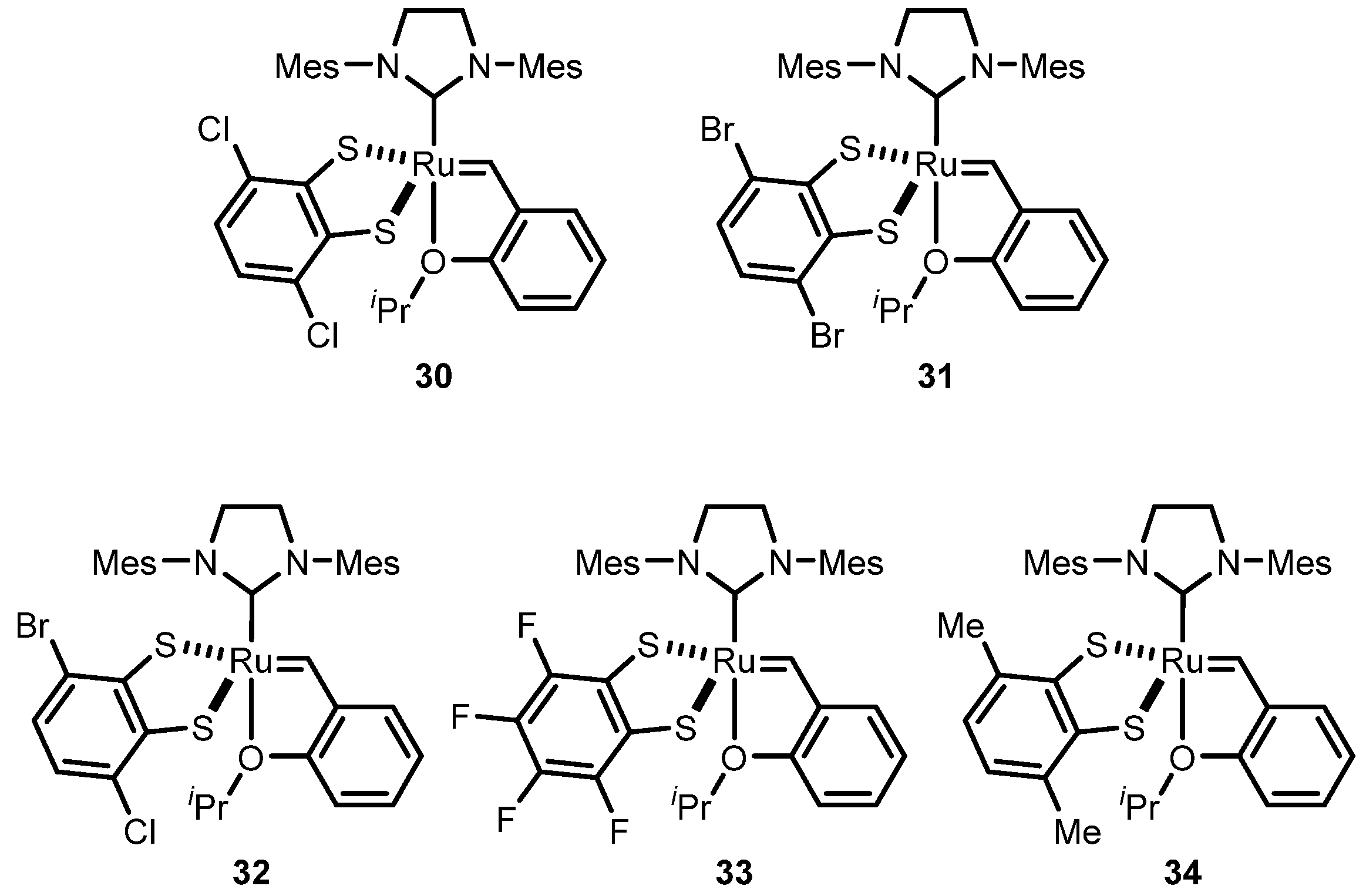
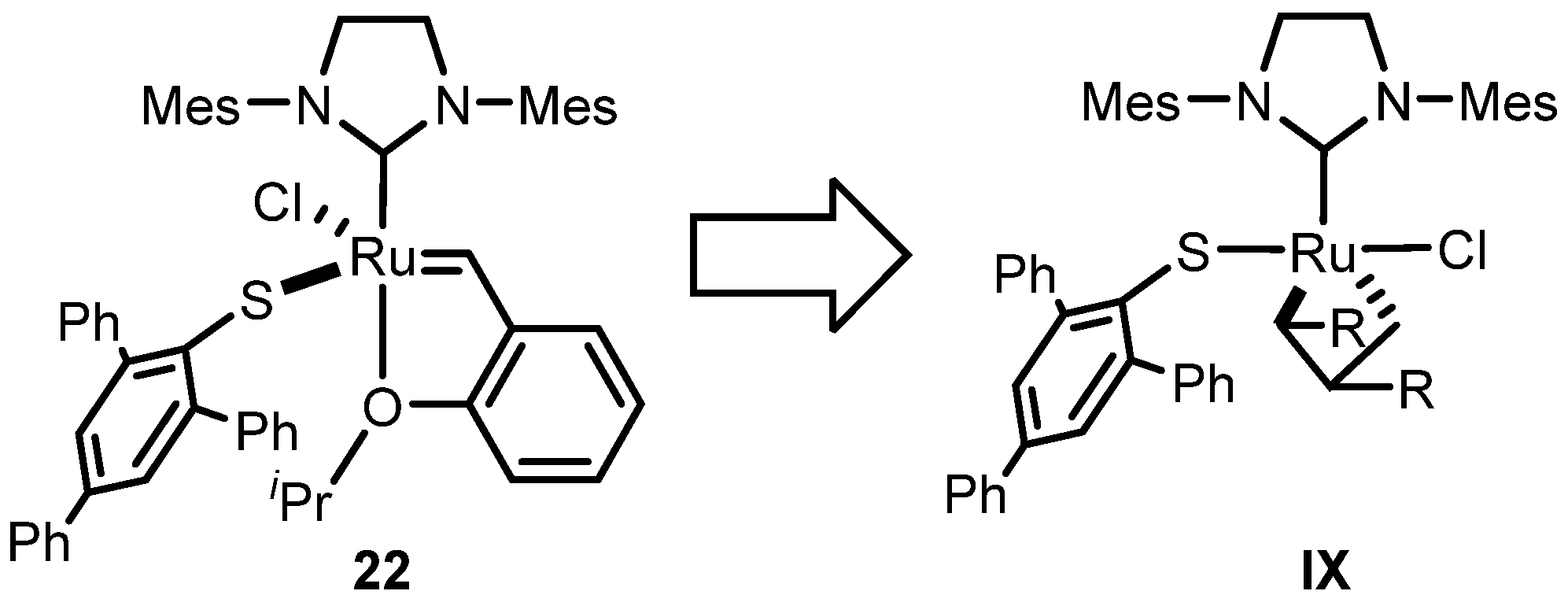
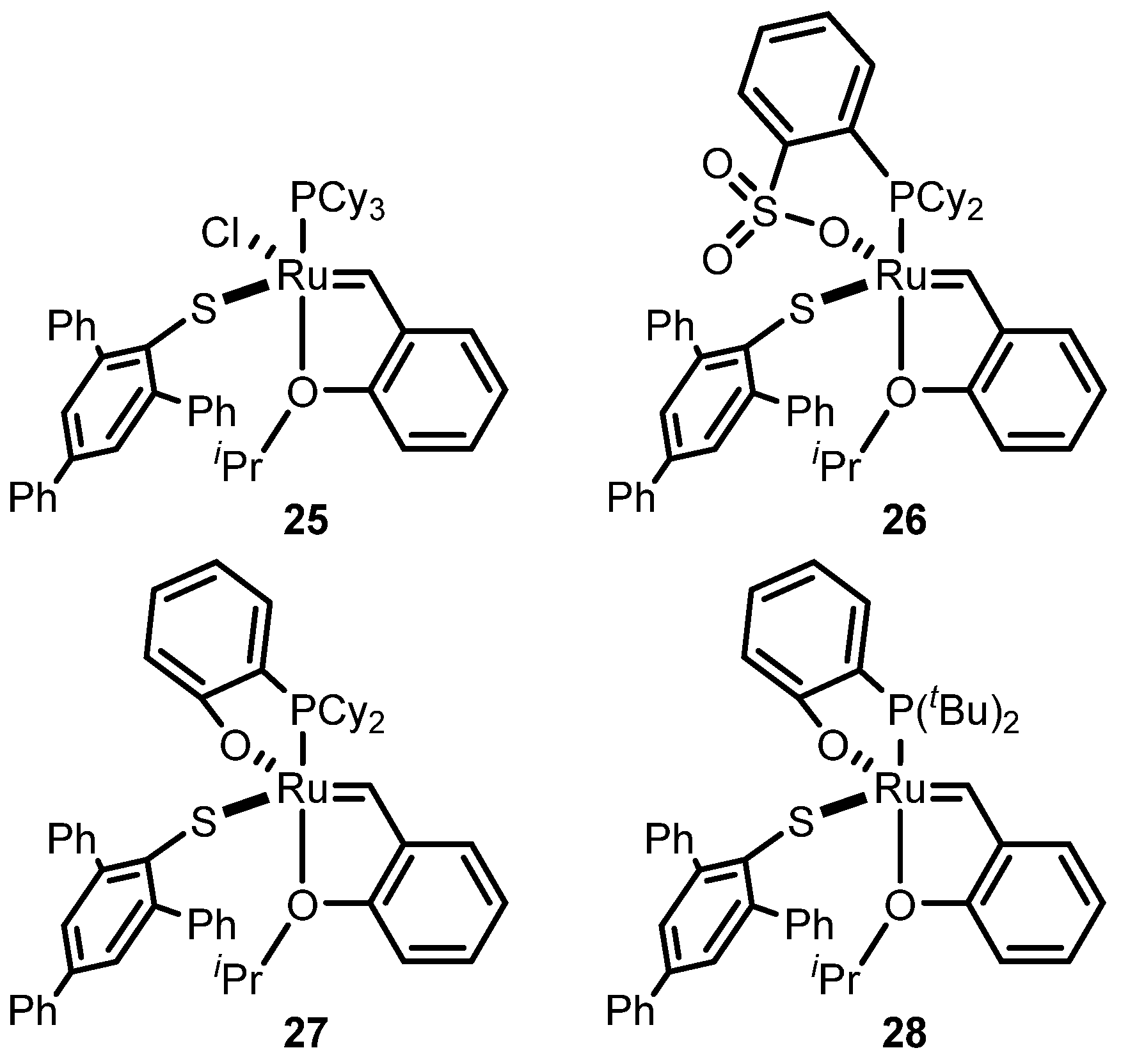
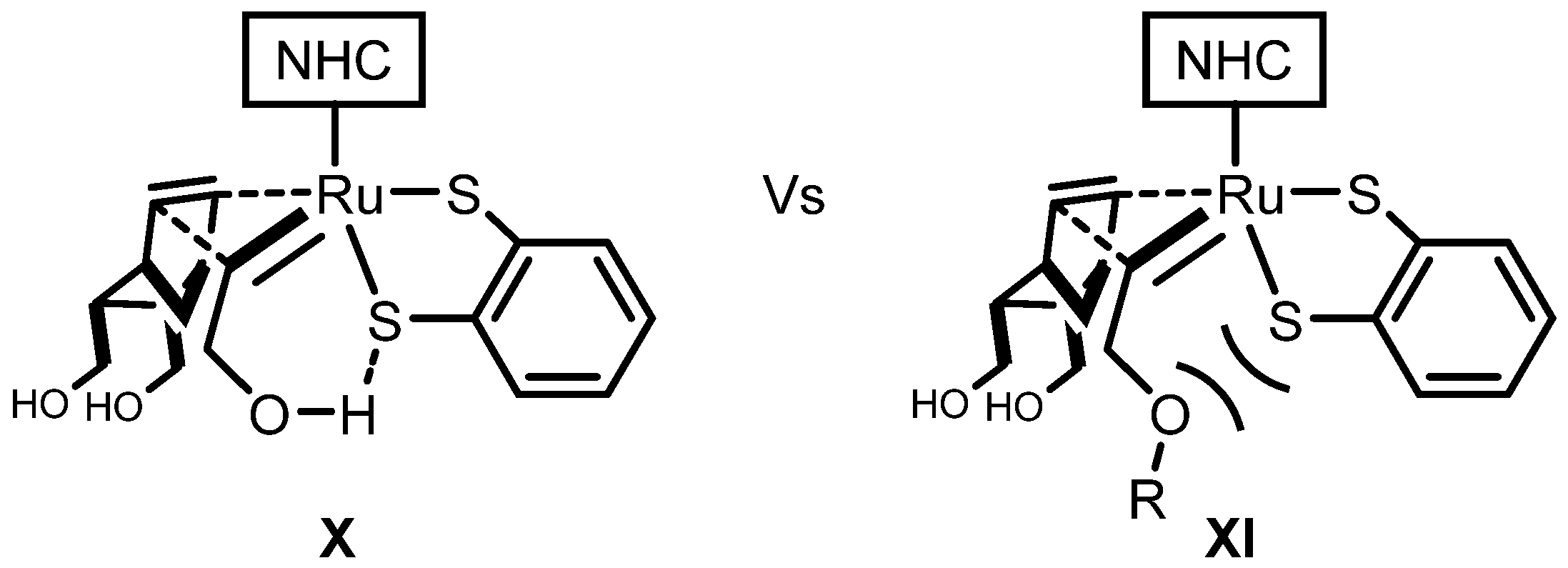
2.3. Monothiolate Catalyst
Monothiolate Catalyst Development and Applications
2.4. Dithiolate Catalysts
2.4.1. Catalyst Development
2.4.2. Z-Selective Ring-Opening Cross Metathesis
2.4.3. Z-Selective Cross Metathesis
3. Asymmetric Olefin Metathesis
3.1. General Introduction
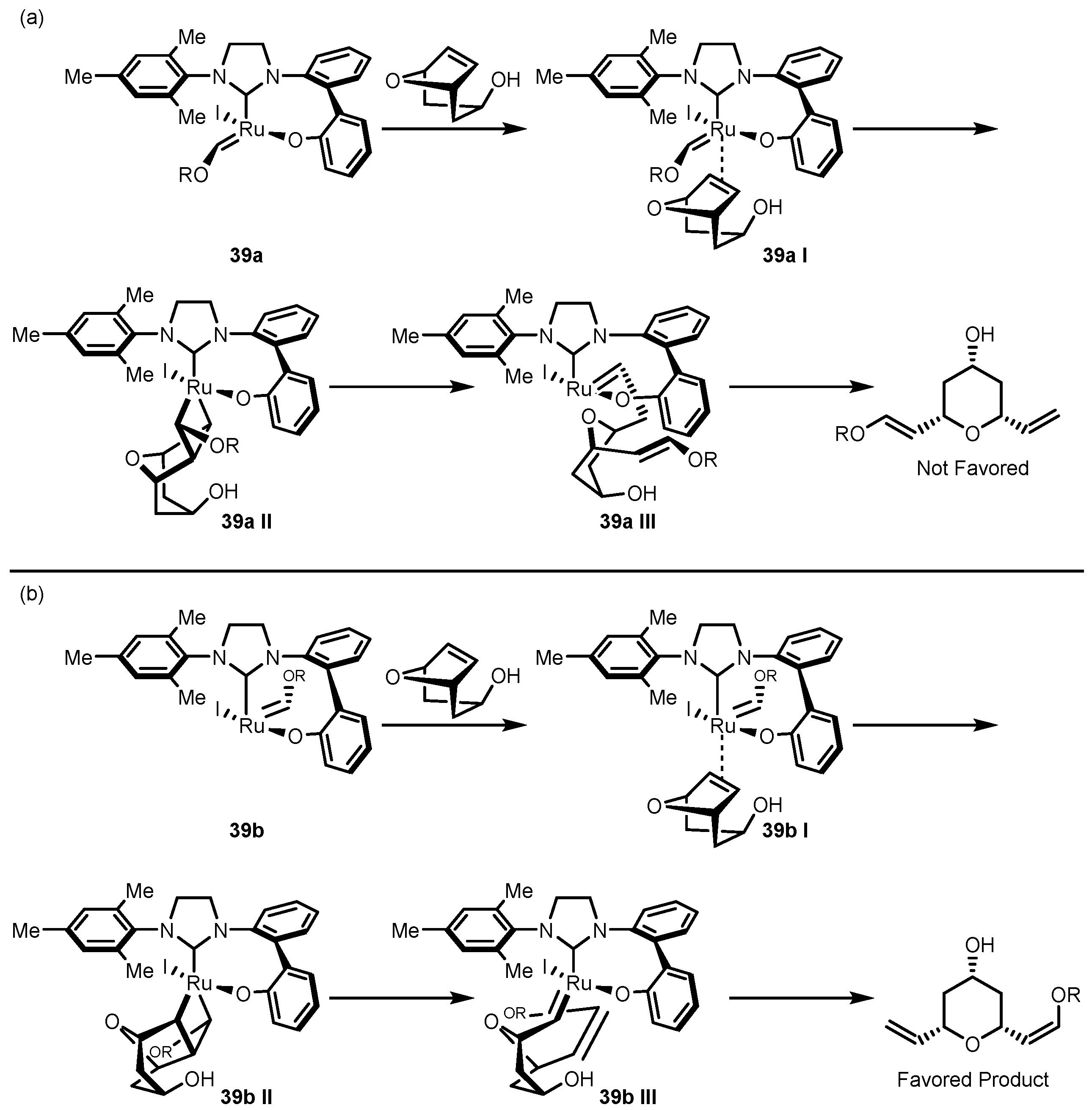
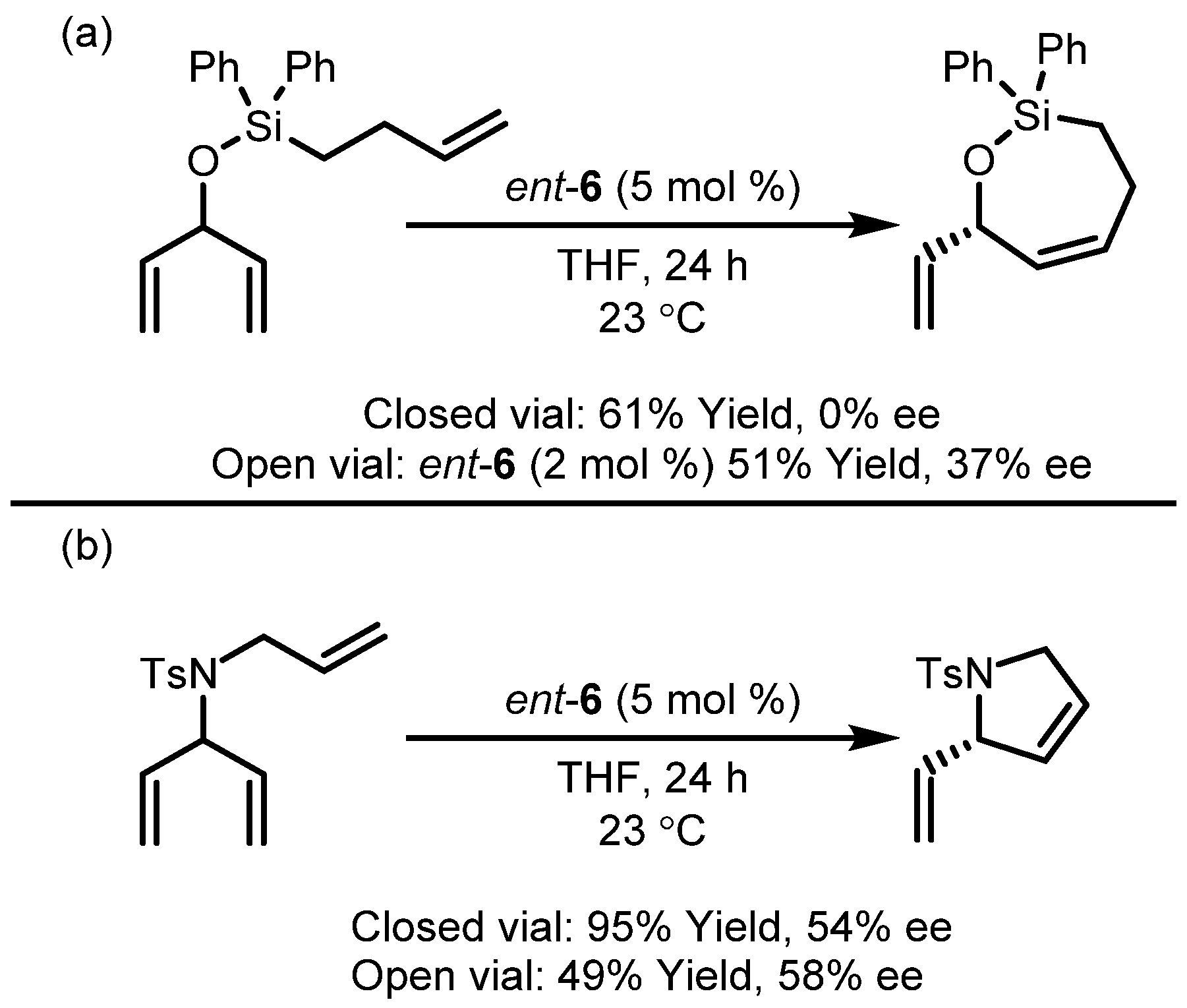
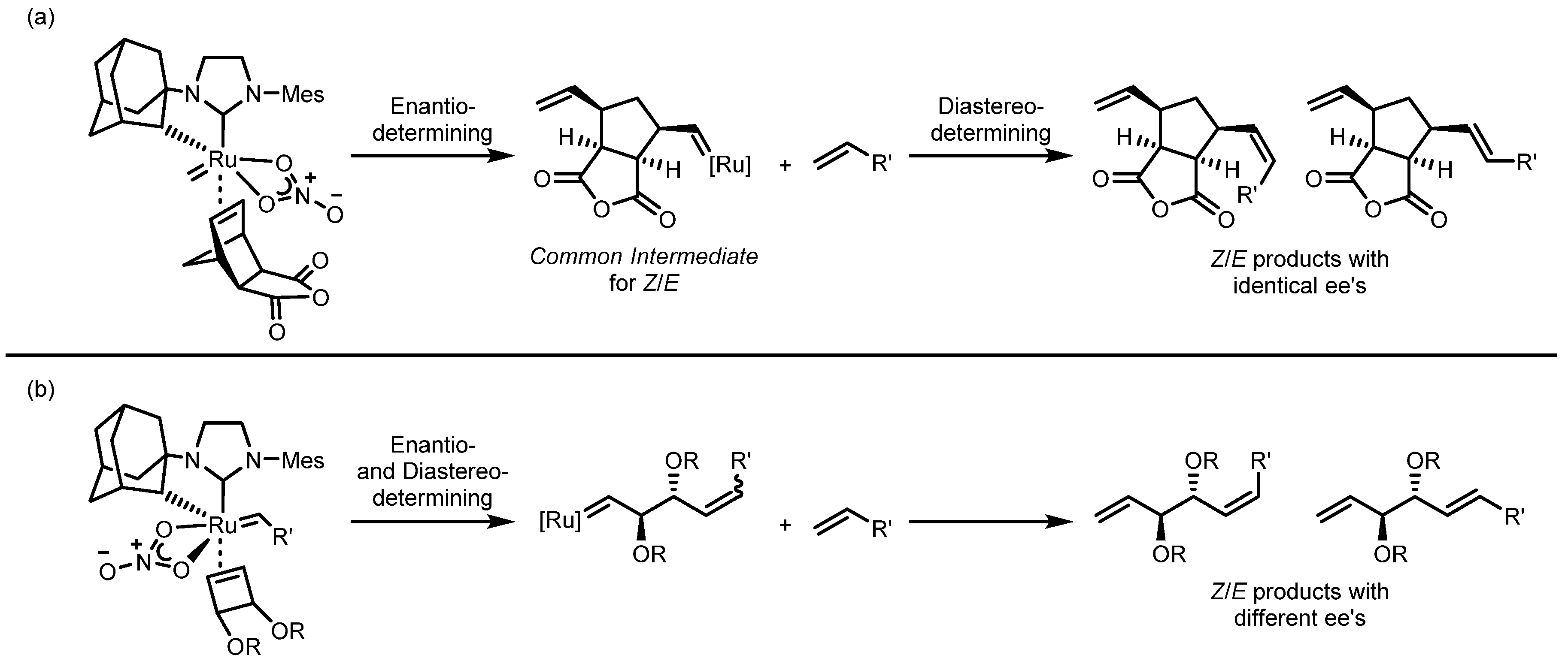
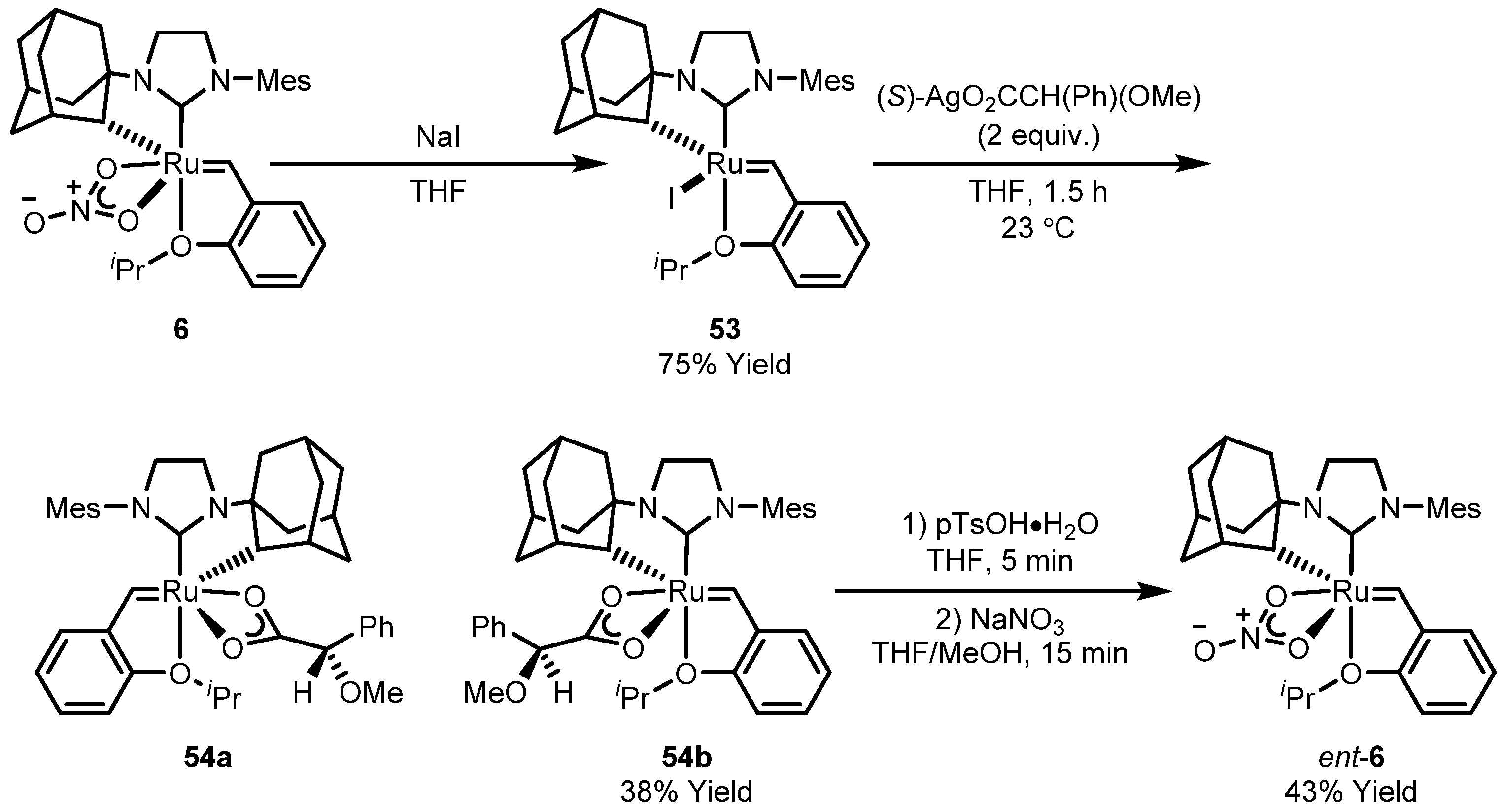
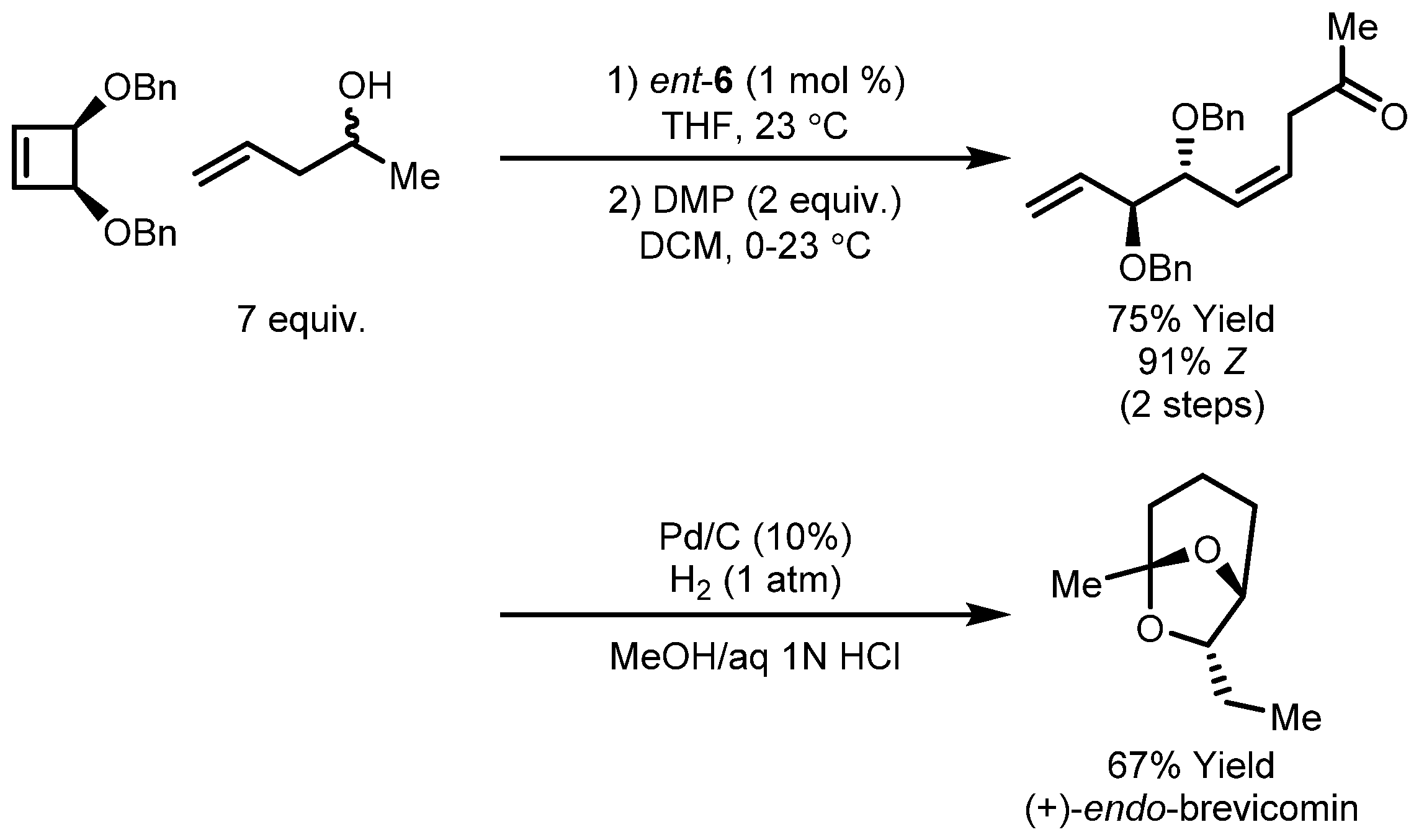
3.2. Asymmetric Ring-Closing Metathesis
3.3. Asymmetric Ring-Opening/Cross Metathesis
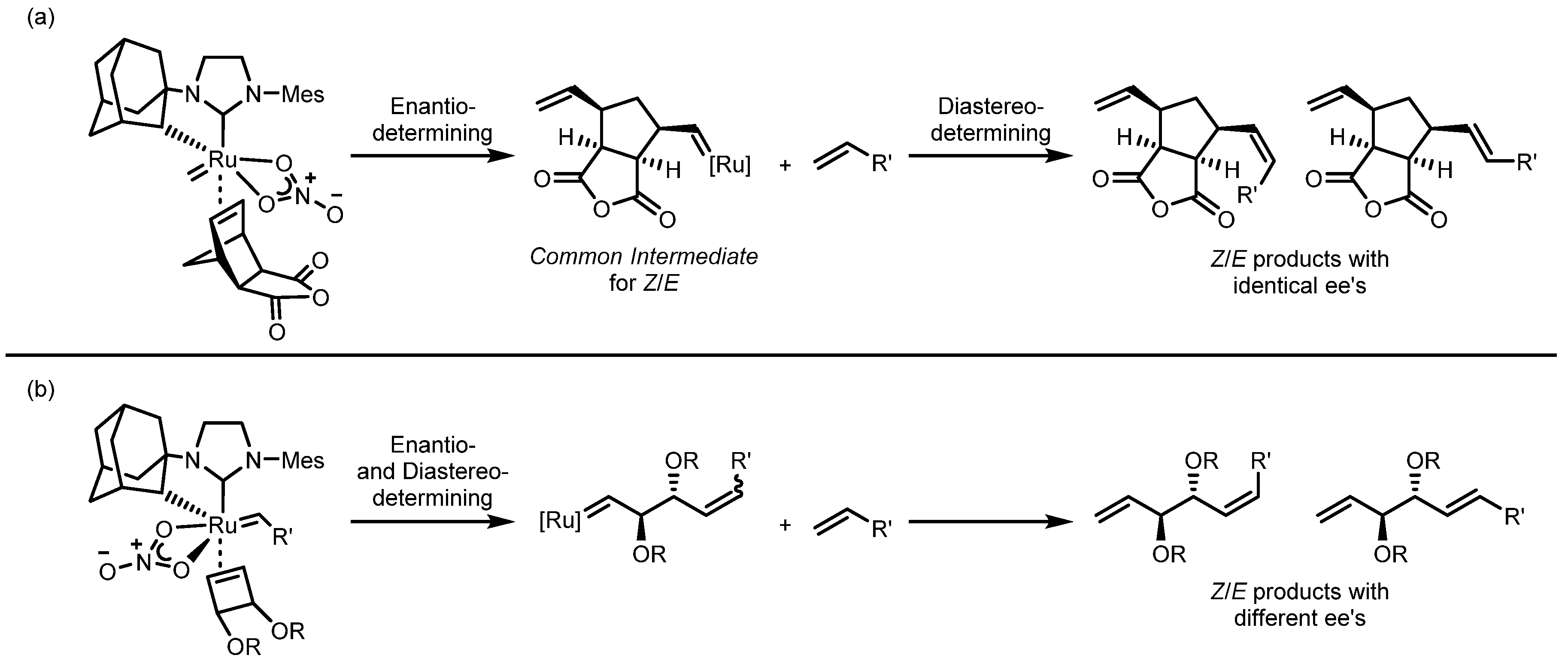
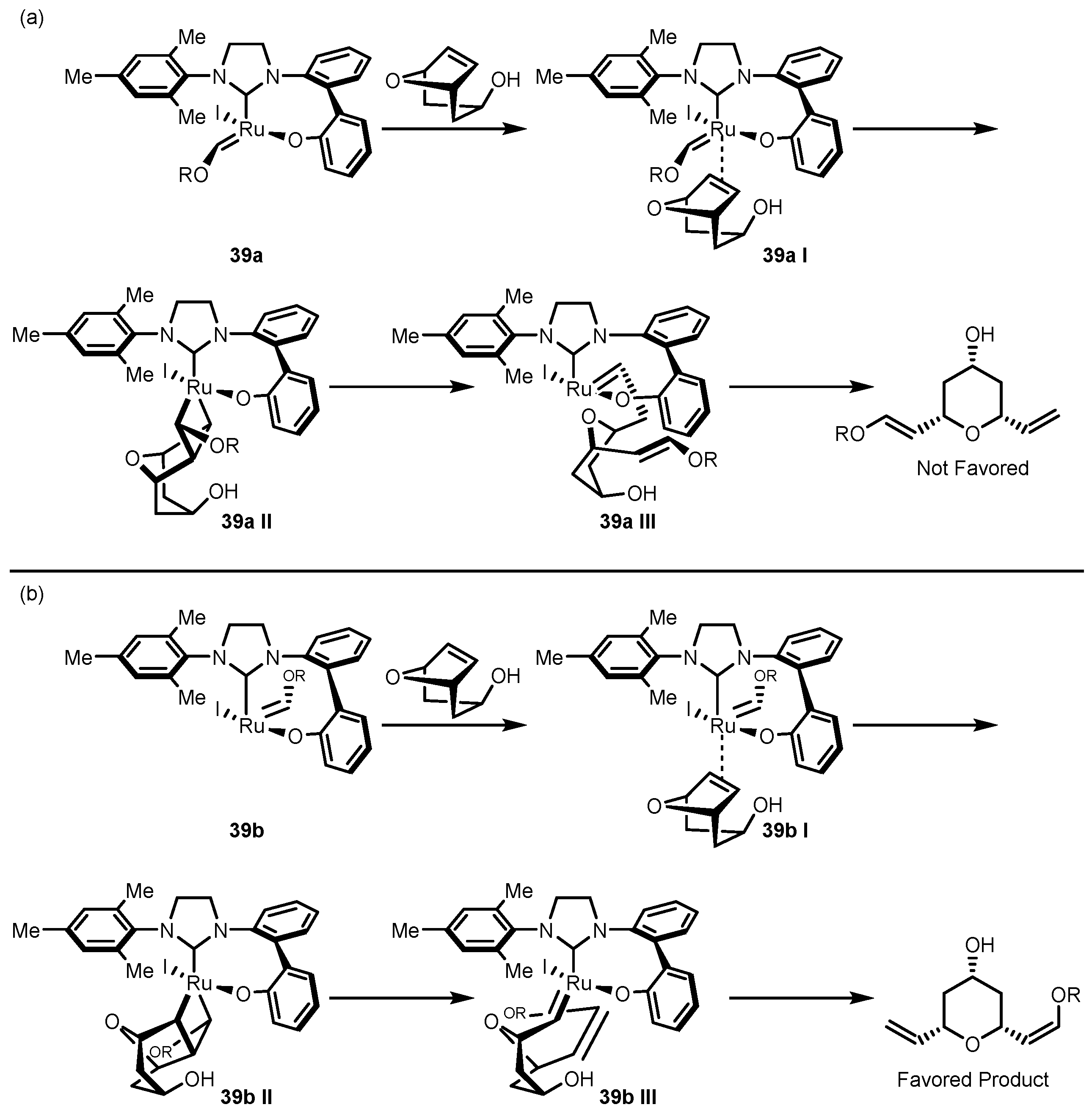
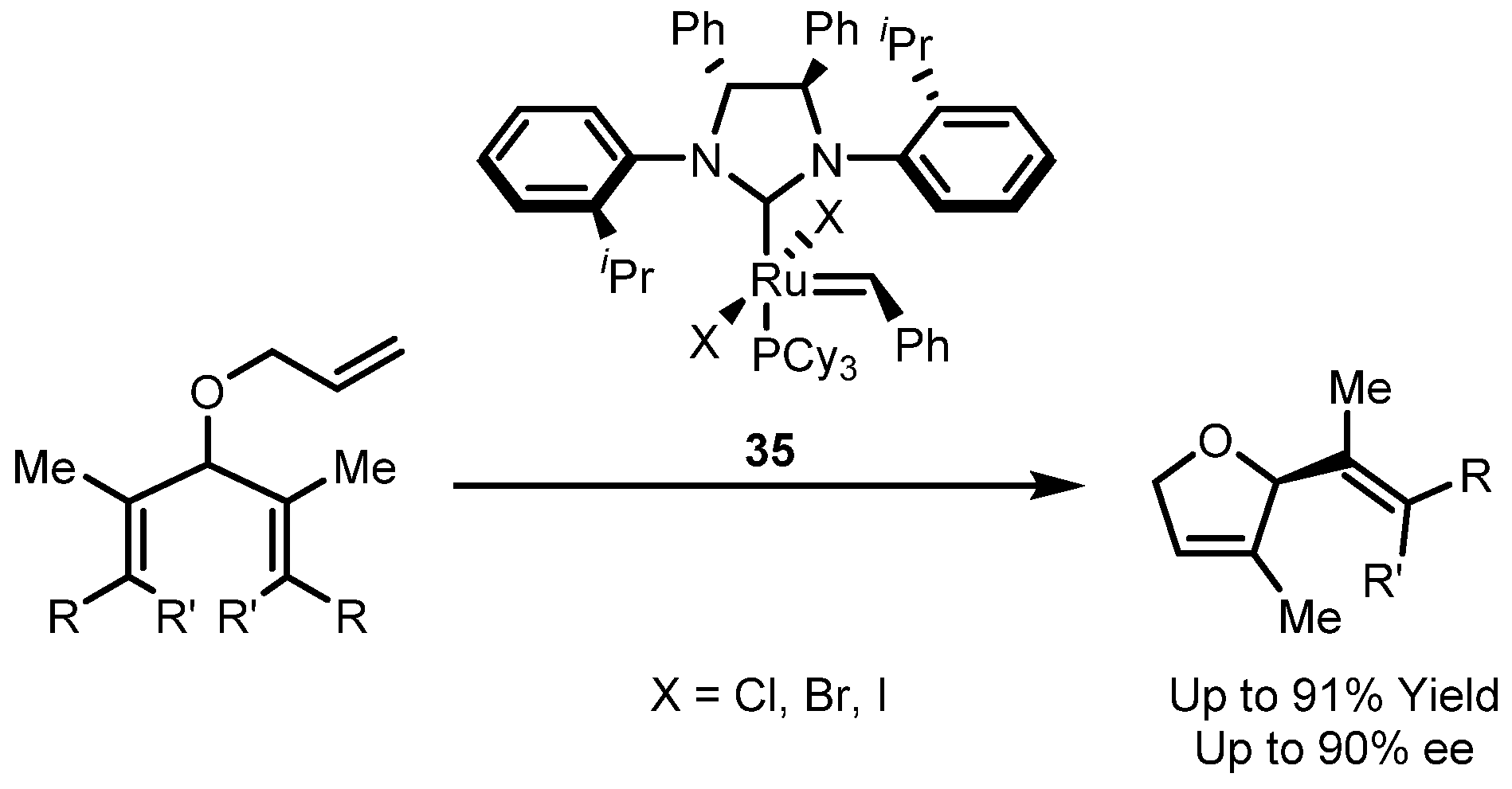
3.4. Asymmetric Cross Metathesis
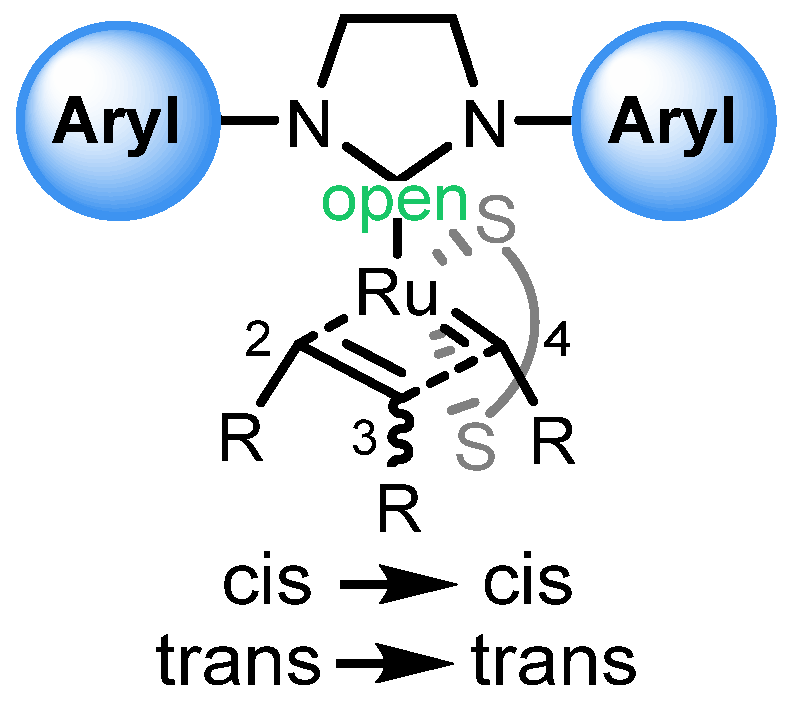
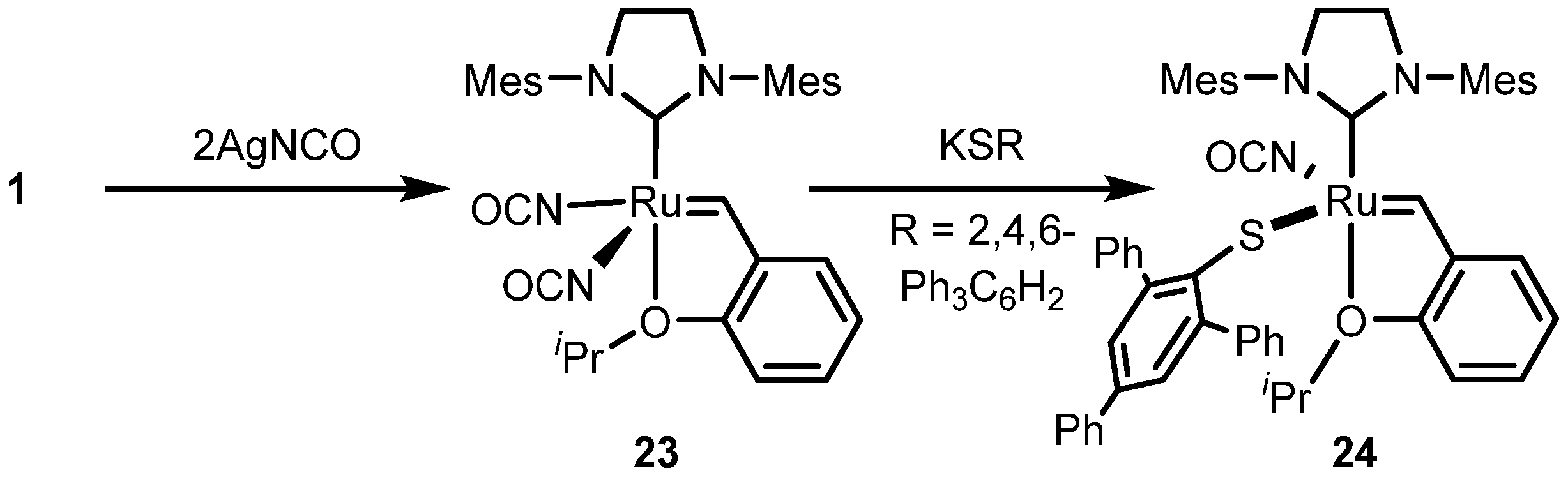
4. Stereoretentive Olefin Metathesis
5. Stereoselective Olefin Metathesis in Synthetic Applications
6. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fürstner, A. Olefin Metathesis and Beyond. Angew. Chem. 2000, 39, 3012–3043. [Google Scholar] [CrossRef]
- Trnka, T.M.; Grubbs, R.H. The Development of L2X2 RuCHR Olefin Metathesis Catalysts: An Organometallic Success Story. Acc. Chem. Res. 2001, 34, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Schrock, R.R. High Oxidation State Multiple Metal—Carbon Bonds. Chem. Rev. 2002, 102, 145–180. [Google Scholar] [CrossRef] [PubMed]
- Schrock, R.R.; Hoveyda, A.H. Molybdenum and Tungsten Imido Alkylidene Complexes as Efficient Olefin-Metathesis Catalysts. Angew. Chem. Int. Ed. 2003, 42, 4592–4633. [Google Scholar] [CrossRef] [PubMed]
- Handbook of Metathesis; Grubbs, R.H.; O'Leary, D.J. (Eds.) Wiley-VCH: Weinheim, Germany, 2003.
- Vougioukalakis, G.C.; Grubbs, R.H. Ruthenium-Based Heterocyclic Carbene-Coordinated Olefin Metathesis Catalysts. Chem. Rev. 2010, 110, 1746–1787. [Google Scholar] [CrossRef] [PubMed]
- Samojłowicz, C.; Bieniek, M.; Grela, K. Ruthenium-Based Olefin Metathesis Catalysts Bearing N-Heterocyclic Carbene Ligands. Chem. Rev. 2009, 109, 3708–3742. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Vila, A.M.; Monsaert, S.; Bajek, A.; Verpoort, F. Ruthenium-Based Olefin Metathesis Catalysts Derived from Alkynes. Chem. Rev. 2010, 110, 4865–4909. [Google Scholar] [CrossRef] [PubMed]
- William, A.A.; George, M.N. Polymeric Bicyclo-(2, 2, 1)-2-Heptene. U.S. Patent 2,721,189 A, 18 October 1955. [Google Scholar]
- Truett, W.L.; Johnson, D.R.; Robinson, I.M.; Montague, B.A. Polynorbornene by Coördination Polymerization 1. J. Am. Chem. Soc. 1960, 82, 2337–2340. [Google Scholar] [CrossRef]
- Ji, J.-X.; Chan, A.S.C.; Helmchen, G.; Kazmaier, U.; Frster, S.; Ojima, I.; Kaloko, J.J.; Chaterpaul, S.J.; Teng, Y.-H.G.; Lin, C.-F.; et al. Asymmetric Carbon–Carbon Bond-Forming Reactions. In Catalytic Asymmetric Synthesis; Ojima, I., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 437–770. [Google Scholar]
- Grubbs, R.H. Metathesis in Natural Product Synthesis: Strategies, Substrates and Catalysts; Cossy, J., Arseniyadis, S., Meyer, C., Eds.; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Ring Opening Metathesis Polymerisation and Related Chemistry: State of the Art and Visions for the New Century: State of the Art and Visions for the New Century, Proceedings of the NATO Advanced Study Institute on Ring Opening Metathesis Polymerisation and Related Chemistry: Polaica-Zdroy, Poland, 3–15 September 2000; Khosravi, E.; Szymańska-Buzar, T.; Advanced Study Institute on Ring Opening Metathesis Polymerisation and Related Chemistry (Eds.) NATO Science Series 2, Mathematics, Physics and Chemistry; Kluwer: Dordrecht, The Netherlands, 2002.
- Schrock, R.R. Synthesis of stereoregular ROMP polymers using molybdenum and tungsten imido alkylidene initiators. Dalton Trans. 2011, 40, 7484–7495. [Google Scholar] [CrossRef] [PubMed]
- Jean-Louis Hérisson, P.; Chauvin, Y. Catalyse de transformation des oléfines par les complexes du tungstène. II. Télomérisation des oléfines cycliques en présence d’oléfines acycliques. Makromol. Chem. 1971, 141, 161–176. (In French) [Google Scholar] [CrossRef]
- Hoveyda, A.H.; Schrock, R.R. Catalytic Asymmetric Olefin Metathesis. Chemistry 2001, 7, 945–950. [Google Scholar] [CrossRef]
- Kress, S.; Blechert, S. Asymmetric catalysts for stereocontrolled olefin metathesis reactions. Chem. Soc. Rev. 2012, 41, 4389–4408. [Google Scholar] [CrossRef] [PubMed]
- Gottumukkala, A.L.; Madduri, A.V.R.; Minnaard, A.J. Z-Selectivity: A Novel Facet of Metathesis. ChemCatChem 2012, 4, 462–467. [Google Scholar] [CrossRef]
- Siau, W.-Y.; Zhang, Y.; Zhao, Y. Stereoselective Synthesis of Z-Alkenes. In Stereoselective Alkene Synthesis; Wang, J., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 327, pp. 33–58. [Google Scholar]
- Shahane, S.; Bruneau, C.; Fischmeister, C. Z Selectivity: Recent Advances in one of the Current Major Challenges of Olefin Metathesis. ChemCatChem 2013, 5, 3436–3459. [Google Scholar] [CrossRef]
- Hoveyda, A.H. Evolution of Catalytic Stereoselective Olefin Metathesis: From Ancillary Transformation to Purveyor of Stereochemical Identity. J. Org. Chem. 2014, 79, 4763–4792. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Schowner, R.; Buchmeiser, M.R. Molybdenum imido alkylidene and tungsten oxo alkylidene N-heterocyclic carbene complexes for olefin metathesis. Monatshefte Für Chem. Chem. Mon. 2015, 146, 1037–1042. [Google Scholar] [CrossRef]
- Schrock, R.R. Metathesis by Molybdenum and Tungsten Catalysts. Chim. Int. J. Chem. 2015, 69, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Marx, V.M.; Rosebrugh, L.E.; Herbert, M.B.; Grubbs, R.H. Cyclometalated Ruthenium Alkylidene Complexes: A Powerful Family of Z-Selective Olefin Metathesis Catalysts. In Ruthenium in Catalysis; Dixneuf, P.H., Bruneau, C., Eds.; Springer: Cham, Germany, 2014; Volume 48, pp. 1–17. [Google Scholar]
- Fürstner, A.; Davies, P.W. Alkyne metathesis. Chem. Commun. 2005, 2307–2320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Moore, J.S. Alkyne Metathesis: Catalysts and Synthetic Applications. Adv. Synth. Catal. 2007, 349, 93–120. [Google Scholar] [CrossRef]
- Wang, Y.; Jimenez, M.; Hansen, A.S.; Raiber, E.-A.; Schreiber, S.L.; Young, D.W. Control of Olefin Geometry in Macrocyclic Ring-Closing Metathesis Using a Removable Silyl Group. J. Am. Chem. Soc. 2011, 133, 9196–9199. [Google Scholar] [CrossRef] [PubMed]
- Gallenkamp, D.; Fürstner, A. Stereoselective Synthesis of E,Z-Configured 1,3-Dienes by Ring-Closing Metathesis. Application to the Total Synthesis of Lactimidomycin. J. Am. Chem. Soc. 2011, 133, 9232–9235. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.J.; Khan, R.K.M.; Torker, S.; Hoveyda, A.H. Broadly Applicable Z- and Diastereoselective Ring-Opening/Cross-Metathesis Catalyzed by a Dithiolate Ru Complex. Angew. Chem. Int. Ed. 2014, 53, 1968–1972. [Google Scholar] [CrossRef] [PubMed]
- Coe, S.; Pereira, N.; Geden, J.V.; Clarkson, G.J.; Fox, D.J.; Napier, R.M.; Neve, P.; Shipman, M. Ring closing metathesis reactions of α-methylene-β-lactams: Application to the synthesis of a simplified phyllostictine analogue with herbicidal activity. Org. Biomol. Chem. 2015, 13, 7655–7663. [Google Scholar] [CrossRef] [PubMed]
- Teo, P.; Grubbs, R.H. Facile Synthesis of Effcient and Selective Ruthenium Olefin Metathesis Catalysts with Sulfonate and Phosphate Ligands. Organometallics 2010, 29, 6045–6050. [Google Scholar] [CrossRef]
- Trnka, T.M.; Morgan, J.P.; Sanford, M.S.; Wilhelm, T.E.; Scholl, M.; Choi, T.-L.; Ding, S.; Day, M.W.; Grubbs, R.H. Synthesis and Activity of Ruthenium Alkylidene Complexes Coordinated with Phosphine and N-Heterocyclic Carbene Ligands. J. Am. Chem. Soc. 2003, 125, 2546–2558. [Google Scholar] [CrossRef] [PubMed]
- Leitao, E.M.; Dubberley, S.R.; Piers, W.E.; Wu, Q.; McDonald, R. Thermal Decomposition Modes for Four-Coordinate Ruthenium Phosphonium Alkylidene Olefin Metathesis Catalysts. Chem. Eur. J. 2008, 14, 11565–11572. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Grubbs, R.H. Chelated Ruthenium Catalysts for Z-Selective Olefin Metathesis. J. Am. Chem. Soc. 2011, 133, 8525–8527. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Xu, X.; Dong, X.; Keitz, B.K.; Herbert, M.B.; Grubbs, R.H.; Houk, K.N. Z-Selectivity in Olefin Metathesis with Chelated Ru Catalysts: Computational Studies of Mechanism and Selectivity. J. Am. Chem. Soc. 2012, 134, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Wang, Z.-X.; Wang, X. A Thorough DFT Study of the Mechanism of Homodimerization of Terminal Olefins through Metathesis with a Chelated Ruthenium Catalyst: From Initiation to Z Selectivity to Regeneration. Organometallics 2012, 31, 7222–7234. [Google Scholar] [CrossRef]
- Keitz, B.K.; Endo, K.; Patel, P.R.; Herbert, M.B.; Grubbs, R.H. Improved Ruthenium Catalysts for Z-Selective Olefin Metathesis. J. Am. Chem. Soc. 2012, 134, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Rosebrugh, L.E.; Herbert, M.B.; Marx, V.M.; Keitz, B.K.; Grubbs, R.H. Highly Active Ruthenium Metathesis Catalysts Exhibiting Unprecedented Activity and Z -Selectivity. J. Am. Chem. Soc. 2013, 135, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Herbert, M.B.; Grubbs, R.H. Investigations into Ruthenium Metathesis Catalysts with Six-Membered Chelating NHC Ligands: Relationship between Catalyst Structure and Stereoselectivity. Organometallics 2013, 32, 5128–5135. [Google Scholar] [CrossRef] [PubMed]
- Bronner, S.M.; Herbert, M.B.; Patel, P.R.; Marx, V.M.; Grubbs, R.H. Ru-based Z-Selective metathesis catalysts with modified cyclometalated carbene ligands. Chem. Sci. 2014, 5, 4091–4098. [Google Scholar] [CrossRef] [PubMed]
- Herbert, M.B.; Suslick, B.A.; Liu, P.; Zou, L.; Dornan, P.K.; Houk, K.N.; Grubbs, R.H. Cyclometalated Z-Selective Ruthenium Metathesis Catalysts with Modified N-Chelating Groups. Organometallics 2015, 34, 2858–2869. [Google Scholar] [CrossRef]
- Pribisko, M.A.; Ahmed, T.S.; Grubbs, R.H. Z-Selective ruthenium metathesis catalysts: Comparison of nitrate and nitrite X-type ligands. Polyhedron 2014, 84, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Herbert, M.B.; Grubbs, R.H. Z-Selective Cross Metathesis with Ruthenium Catalysts: Synthetic Applications and Mechanistic Implications. Angew. Chem. Int. Ed. 2015, 54, 5018–5024. [Google Scholar] [CrossRef] [PubMed]
- Keitz, B.K.; Endo, K.; Herbert, M.B.; Grubbs, R.H. Z-Selective Homodimerization of Terminal Olefins with a Ruthenium Metathesis Catalyst. J. Am. Chem. Soc. 2011, 133, 9686–9688. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.K.; Choi, T.-L.; Sanders, D.P.; Grubbs, R.H. A General Model for Selectivity in Olefin Cross Metathesis. J. Am. Chem. Soc. 2003, 125, 11360–11370. [Google Scholar] [CrossRef] [PubMed]
- Quigley, B.L.; Grubbs, R.H. Ruthenium-catalysed Z-Selective cross metathesis of allylic-substituted olefins. Chem. Sci. 2014, 5, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, M.; Fujita, M. Modern Supramolecular Chemistry: Strategies for Macrocycle Synthesis; Diederich, F., Stang, P.J., Tykwinski, R.R., Eds.; WILEY-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Ohloff, G.; Pickenhagen, W.; Kraft, P. Scent and Chemistry: The Molecular World of Odors; Verlag Helvetica Chimica Acta: Zürich, Switzerland; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Marx, V.M.; Herbert, M.B.; Keitz, B.K.; Grubbs, R.H. Stereoselective Access to Z and E Macrocycles by Ruthenium-Catalyzed Z-Selective Ring-Closing Metathesis and Ethenolysis. J. Am. Chem. Soc. 2013, 135, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Burdett, K.A.; Harris, L.D.; Margl, P.; Maughon, B.R.; Mokhtar-Zadeh, T.; Saucier, P.C.; Wasserman, E.P. Renewable Monomer Feedstocks via Olefin Metathesis: Fundamental Mechanistic Studies of Methyl Oleate Ethenolysis with the First-Generation Grubbs Catalyst. Organometallics 2004, 23, 2027–2047. [Google Scholar] [CrossRef]
- Lysenko, Z.; Maughon, B.R.; Bicerano, J.; Burdett, K.A.; Christenson, C.P.; Cummins, C.H.; Dettloff, M.L.; Maher, J.M.; Schrock, A.K.; Thomas, P.J.; et al. Intergrated Chemical Processes for Industrial Utilization of Seed Oils. U.S. Patent 7,576,227, 18 August 2009. [Google Scholar]
- Olson, E.S. Chain-Selective Synthesis of Fuel Components and Chemical Feedstocks. U.S. Patent 8,420,840, 16 April 2013. [Google Scholar]
- Dubois, J.-L. Carburant Aviation Contenant une Proportion de Composes Organiques Ex-Biomasse. EP 2,406,354 B1, 27 April 2016. [Google Scholar]
- Bidange, J.; Fischmeister, C.; Bruneau, C. Ethenolysis: A Green Catalytic Tool to Cleave Carbon-Carbon Double Bonds. Chem. Eur. J. 2016, 22, 12226–12244. [Google Scholar] [CrossRef] [PubMed]
- Spekreijse, J.; Sanders, J.P.M.; Bitter, J.H.; Scott, E.L. The Future of Ethenolysis in Biobased Chemistry. ChemSusChem 2017, 10, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Koh, M.J.; Shen, X.; Romiti, F.; Schrock, R.R.; Hoveyda, A.H. Kinetically controlled E-selective catalytic olefin metathesis. Science 2016, 352, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, H.; Herbert, M.B.; Liu, P.; Dong, X.; Xu, X.; Keitz, B.K.; Ung, T.; Mkrtumyan, G.; Houk, K.N.; Grubbs, R.H. Z-Selective Ethenolysis with a Ruthenium Metathesis Catalyst: Experiment and Theory. J. Am. Chem. Soc. 2013, 135, 5848–5858. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.S.; Grubbs, R.H. Alkene Chemoselectivity in Ruthenium-Catalyzed Z-Selective Olefin Metathesis. Angew. Chem. Int. Ed. 2013, 52, 9001–9004. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.-X.; Cannon, J.S.; Taylor, B.L.H.; Engle, K.M.; Houk, K.N.; Grubbs, R.H. Z-Selective Cross-Metathesis and Homodimerization of 3 E-1,3-Dienes: Reaction Optimization, Computational Analysis, and Synthetic Applications. J. Am. Chem. Soc. 2016, 138, 14039–14046. [Google Scholar] [CrossRef] [PubMed]
- Ibrahem, I.; Yu, M.; Schrock, R.R.; Hoveyda, A.H. Highly Z- and Enantioselective Ring-Opening/Cross-Metathesis Reactions Catalyzed by Stereogenic-at-Mo Adamantylimido Complexes. J. Am. Chem. Soc. 2009, 131, 3844–3845. [Google Scholar] [CrossRef] [PubMed]
- Flook, M.M.; Jiang, A.J.; Schrock, R.R.; Hoveyda, A.H. Z-Selective Olefin Metathesis Processes Catalyzed by a Molybdenum Hexaisopropylterphenoxide Monopyrrolide Complex. J. Am. Chem. Soc. 2009, 131, 7962–7963. [Google Scholar] [CrossRef] [PubMed]
- Jiang, A.J.; Zhao, Y.; Schrock, R.R.; Hoveyda, A.H. Highly Z-Selective Metathesis Homocoupling of Terminal Olefins. J. Am. Chem. Soc. 2009, 131, 16630–16631. [Google Scholar] [CrossRef] [PubMed]
- Peryshkov, D.V.; Schrock, R.R.; Takase, M.K.; Müller, P.; Hoveyda, A.H. Z-Selective Olefin Metathesis Reactions Promoted by Tungsten Oxo Alkylidene Complexes. J. Am. Chem. Soc. 2011, 133, 20754–20757. [Google Scholar] [CrossRef] [PubMed]
- Occhipinti, G.; Hansen, F.R.; Törnroos, K.W.; Jensen, V.R. Simple and Highly Z-Selective Ruthenium-Based Olefin Metathesis Catalyst. J. Am. Chem. Soc. 2013, 135, 3331–3334. [Google Scholar] [CrossRef] [PubMed]
- Occhipinti, G.; Koudriavtsev, V.; Törnroos, K.W.; Jensen, V.R. Theory-assisted development of a robust and Z-selective olefin metathesis catalyst. Dalton Trans. 2014, 43, 11106–11117. [Google Scholar] [CrossRef] [PubMed]
- Smit, W.; Koudriavtsev, V.; Occhipinti, G.; Törnroos, K.W.; Jensen, V.R. Phosphine-Based Z-Selective Ruthenium Olefin Metathesis Catalysts. Organometallics 2016, 35, 1825–1837. [Google Scholar] [CrossRef]
- Khan, R.K.M.; Torker, S.; Hoveyda, A.H. Readily Accessible and Easily Modifiable Ru-Based Catalysts for Efficient and Z -Selective Ring-Opening Metathesis Polymerization and Ring-Opening/Cross-Metathesis. J. Am. Chem. Soc. 2013, 135, 10258–10261. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.K.M.; Torker, S.; Hoveyda, A.H. Reactivity and Selectivity Differences between Catecholate and Catechothiolate Ru Complexes. Implications Regarding Design of Stereoselective Olefin Metathesis Catalysts. J. Am. Chem. Soc. 2014, 136, 14337–14340. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.J.; Khan, R.K.M.; Torker, S.; Yu, M.; Mikus, M.S.; Hoveyda, A.H. High-value alcohols and higher-oxidation-state compounds by catalytic Z-selective cross-metathesis. Nature 2015, 517, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.K.M.; O’Brien, R.V.; Torker, S.; Li, B.; Hoveyda, A.H. Z- and Enantioselective Ring-Opening/Cross-Metathesis with Enol Ethers Catalyzed by Stereogenic-at-Ru Carbenes: Reactivity, Selectivity, and Curtin–Hammett Kinetics. J. Am. Chem. Soc. 2012, 134, 12774–12779. [Google Scholar] [CrossRef] [PubMed]
- Seiders, T.J.; Ward, D.W.; Grubbs, R.H. Enantioselective Ruthenium-Catalyzed Ring-Closing Metathesis. Org. Lett. 2001, 3, 3225–3228. [Google Scholar] [CrossRef] [PubMed]
- Funk, T.W.; Berlin, J.M.; Grubbs, R.H. Highly Active Chiral Ruthenium Catalysts for Asymmetric Ring-Closing Olefin Metathesis. J. Am. Chem. Soc. 2006, 128, 1840–1846. [Google Scholar] [CrossRef] [PubMed]
- Berlin, J.M.; Goldberg, S.D.; Grubbs, R.H. Highly Active Chiral Ruthenium Catalysts for Asymmetric Cross- and Ring-Opening Cross-Metathesis. Angew. Chem. Int. Ed. 2006, 45, 7591–7595. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhuizen, J.J.; Garber, S.B.; Kingsbury, J.S.; Hoveyda, A.H. A Recyclable Chiral Ru Catalyst for Enantioselective Olefin Metathesis. Efficient Catalytic Asymmetric Ring-Opening/Cross Metathesis in Air. J. Am. Chem. Soc. 2002, 124, 4954–4955. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhuizen, J.J.; Gillingham, D.G.; Garber, S.B.; Kataoka, O.; Hoveyda, A.H. Chiral Ru-Based Complexes for Asymmetric Olefin Metathesis: Enhancement of Catalyst Activity through Steric and Electronic Modifications. J. Am. Chem. Soc. 2003, 125, 12502–12508. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhuizen, J.J.; Campbell, J.E.; Giudici, R.E.; Hoveyda, A.H. A Readily Available Chiral Ag-Based N-Heterocyclic Carbene Complex for Use in Efficient and Highly Enantioselective Ru-Catalyzed Olefin Metathesis and Cu-Catalyzed Allylic Alkylation Reactions. J. Am. Chem. Soc. 2005, 127, 6877–6882. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.-A.; Collins, S.K. A Highly Active Chiral Ruthenium-Based Catalyst for Enantioselective Olefin Metathesis. Organometallics 2007, 26, 2945–2949. [Google Scholar] [CrossRef]
- Grisi, F.; Costabile, C.; Gallo, E.; Mariconda, A.; Tedesco, C.; Longo, P. Ruthenium-Based Complexes Bearing Saturated Chiral N-Heterocyclic Carbene Ligands: Dynamic Behavior and Catalysis. Organometallics 2008, 27, 4649–4656. [Google Scholar] [CrossRef]
- Stenne, B.; Timperio, J.; Savoie, J.; Dudding, T.; Collins, S.K. Desymmetrizations Forming Tetrasubstituted Olefins Using Enantioselective Olefin Metathesis. Org. Lett. 2010, 12, 2032–2035. [Google Scholar] [CrossRef] [PubMed]
- Tiede, S.; Berger, A.; Schlesiger, D.; Rost, D.; Lühl, A.; Blechert, S. Highly Active Chiral Ruthenium-Based Metathesis Catalysts through a Monosubstitution in the N-Heterocyclic Carbene. Angew. Chem. Int. Ed. 2010, 49, 3972–3975. [Google Scholar] [CrossRef] [PubMed]
- Gawin, R.; Pieczykolan, M.; Malińska, M.; Woźniak, K.; Grela, K. Testing New Ruthenium Complexes bearing Chiral 1,2,4-Triazol-5-ylideneLigands as Catalysts for Asymmetric Olefin Metathesis. Synlett 2013, 24, 1250–1254. [Google Scholar]
- Paradiso, V.; Bertolasi, V.; Grisi, F. Novel Olefin Metathesis Ruthenium Catalysts Bearing Backbone-Substituted Unsymmetrical NHC Ligands. Organometallics 2014, 33, 5932–5935. [Google Scholar] [CrossRef]
- Paradiso, V.; Bertolasi, V.; Costabile, C.; Grisi, F. Ruthenium olefin metathesis catalysts featuring unsymmetrical N-heterocyclic carbenes. Dalton Trans 2016, 45, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Hartung, J.; Dornan, P.K.; Grubbs, R.H. Enantioselective Olefin Metathesis with Cyclometalated Ruthenium Complexes. J. Am. Chem. Soc. 2014, 136, 13029–13037. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.C.; Ayyangar, N.R.; Zweifel, G. Hydroboration. XVIII. The Reaction of Diisopinocampheylborane with Representative cis-Acyclic, Cyclic, and Bicyclic Olefins. A Convenient Systhesis of Optically Active Alcohols and Olefins of High Optical Purity and Established Configuration. J. Am. Chem. Soc. 1964, 86, 397–403. [Google Scholar] [CrossRef]
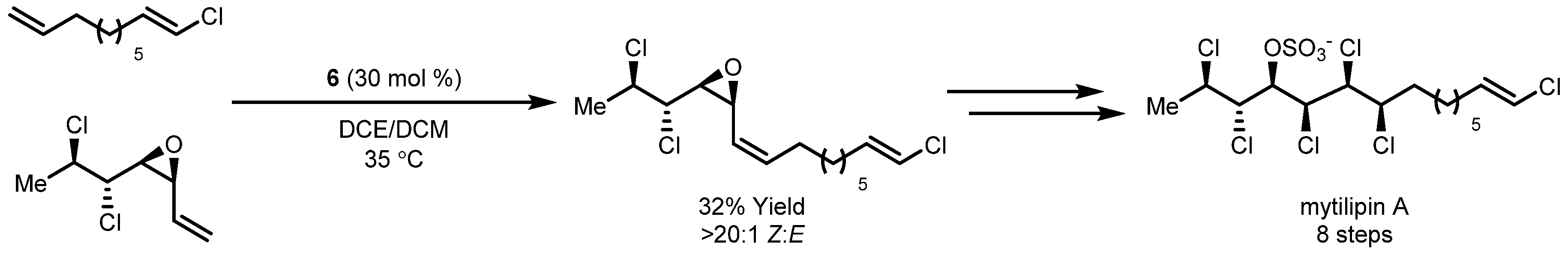
- Werrel, S.; Walker, J.C.L.; Donohoe, T.J. Application of catalytic Z-Selective olefin metathesis in natural product synthesis. Tetrahedron Lett. 2015, 56, 5261–5268. [Google Scholar] [CrossRef]
- Kannenberg, A.; Rost, D.; Eibauer, S.; Tiede, S.; Blechert, S. A Novel Ligand for the Enantioselective Ruthenium-Catalyzed Olefin Metathesis. Angew. Chem. Int. Ed. 2011, 50, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.K.M.; Zhugralin, A.R.; Torker, S.; O’Brien, R.V.; Lombardi, P.J.; Hoveyda, A.H. Synthesis, Isolation, Characterization, and Reactivity of High-Energy Stereogenic-at-Ru Carbenes: Stereochemical Inversion through Olefin Metathesis and Other Pathways. J. Am. Chem. Soc. 2012, 134, 12438–12441. [Google Scholar] [CrossRef] [PubMed]
- Gillingham, D.G.; Kataoka, O.; Garber, S.B.; Hoveyda, A.H. Efficient Enantioselective Synthesis of Functionalized Tetrahydropyrans by Ru-Catalyzed Asymmetric Ring-Opening Metathesis/Cross-Metathesis (AROM/CM). J. Am. Chem. Soc. 2004, 126, 12288–12290. [Google Scholar] [CrossRef] [PubMed]
- Hartung, J.; Grubbs, R.H. Highly Z-Selective and Enantioselective Ring-Opening/Cross-Metathesis Catalyzed by a Resolved Stereogenic-at-Ru Complex. J. Am. Chem. Soc. 2013, 135, 10183–10185. [Google Scholar] [CrossRef] [PubMed]
- Hartung, J.; Grubbs, R.H. Catalytic, Enantioselective Synthesis of 1,2-anti-Diols by Asymmetric Ring-Opening/Cross-Metathesis. Angew. Chem. Int. Ed. 2014, 53, 3885–3888. [Google Scholar] [CrossRef] [PubMed]
- Bilhou, J.L.; Basset, J. Metathese des olefines avec les precurseurs zerovalents du tungstene (W(CO)5L, L = CO, PPh3, P(OPh)3, P(n-C4H9)3) associes a C2H5AlCl2 et O2. Une etude infrarouge. J. Organomet. Chem. 1977, 132, 395–407. [Google Scholar] [CrossRef]
- Katz, T.J.; Hersh, W.H. The stereochemistry of the olefin metathesis reaction. Tetrahedron Lett. 1977, 18, 585–588. [Google Scholar] [CrossRef]
- Couturier, J.-L.; Paillet, C.; Leconte, M.; Basset, J.-M.; Weiss, K. A Cyclometalated Aryloxy(chloro)neopentylidenetungsten Complex: A Highly Active and Stereoselective Catalyst for the Metathesis of cis- and trans-2-Pentene, Norbornene, 1-Methyl-norbornene, and Ethyl Oleate. Angew. Chem. Int. Ed. Engl. 1992, 31, 628–631. [Google Scholar] [CrossRef]
- Johns, A.M.; Ahmed, T.S.; Jackson, B.W.; Grubbs, R.H.; Pederson, R.L. High Trans Kinetic Selectivity in Ruthenium-Based Olefin Cross-Metathesis through Stereoretention. Org. Lett. 2016, 18, 772–775. [Google Scholar] [CrossRef] [PubMed]
- Olefin Metathesis: Theory and Practice; Grela, K. (Ed.) Wiley: Hoboken, NJ, USA, 2014.
- Pederson, R.L.; Fellows, I.M.; Ung, T.A.; Ishihara, H.; Hajela, S.P. Applications of Olefin Cross Metathesis to Commercial Products. Adv. Synth. Catal. 2002, 344, 728–735. [Google Scholar] [CrossRef]
- Henrick, C.A. The synthesis of insect sex phermones. Tetrahedron 1977, 33, 1845–1889. [Google Scholar] [CrossRef]
- Ortiz, A.; Quesada, A.; Sanchez, A. Potential for Use of Synthetic Sex Pheromone for Mating Disruption of the Olive Pyralid Moth, Euzophera pinguis. J. Chem. Ecol. 2004, 30, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Howse, P.E.; Stevens, I.D.R.; Jones, O.T. Insect Pheromones and their Use in Pest Management; Springer: Dordrecht, Netherlands, 1998. [Google Scholar]
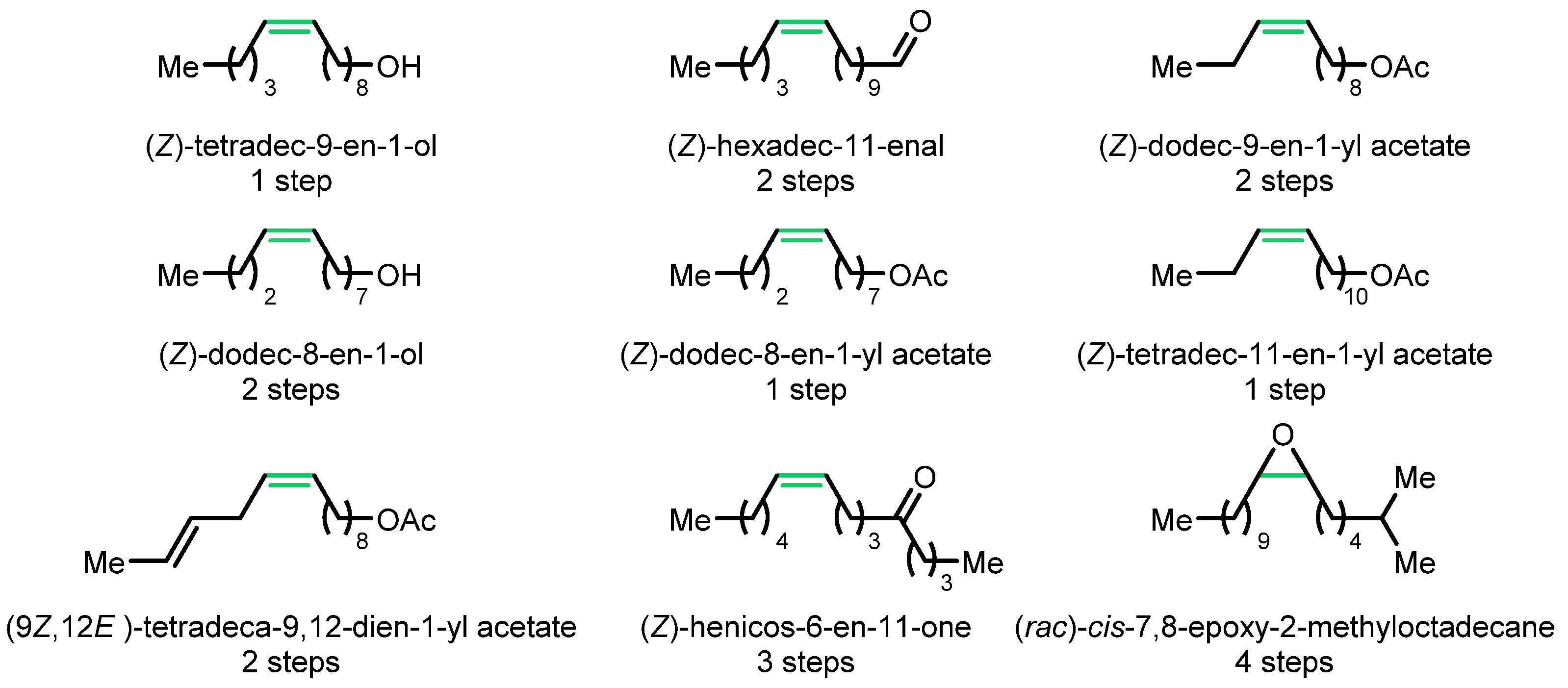
- Herbert, M.B.; Marx, V.M.; Pederson, R.L.; Grubbs, R.H. Concise Syntheses of Insect Pheromones Using Z-Selective Cross Metathesis. Angew. Chem. Int. Ed. 2013, 52, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Inomata, S.; Yamamoto, M. Lepidopteran Sex Pheromones. In The Chemistry of Pheromones and Other Semiochemicals I; Schulz, S., Ed.; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2004; pp. 51–96. [Google Scholar]
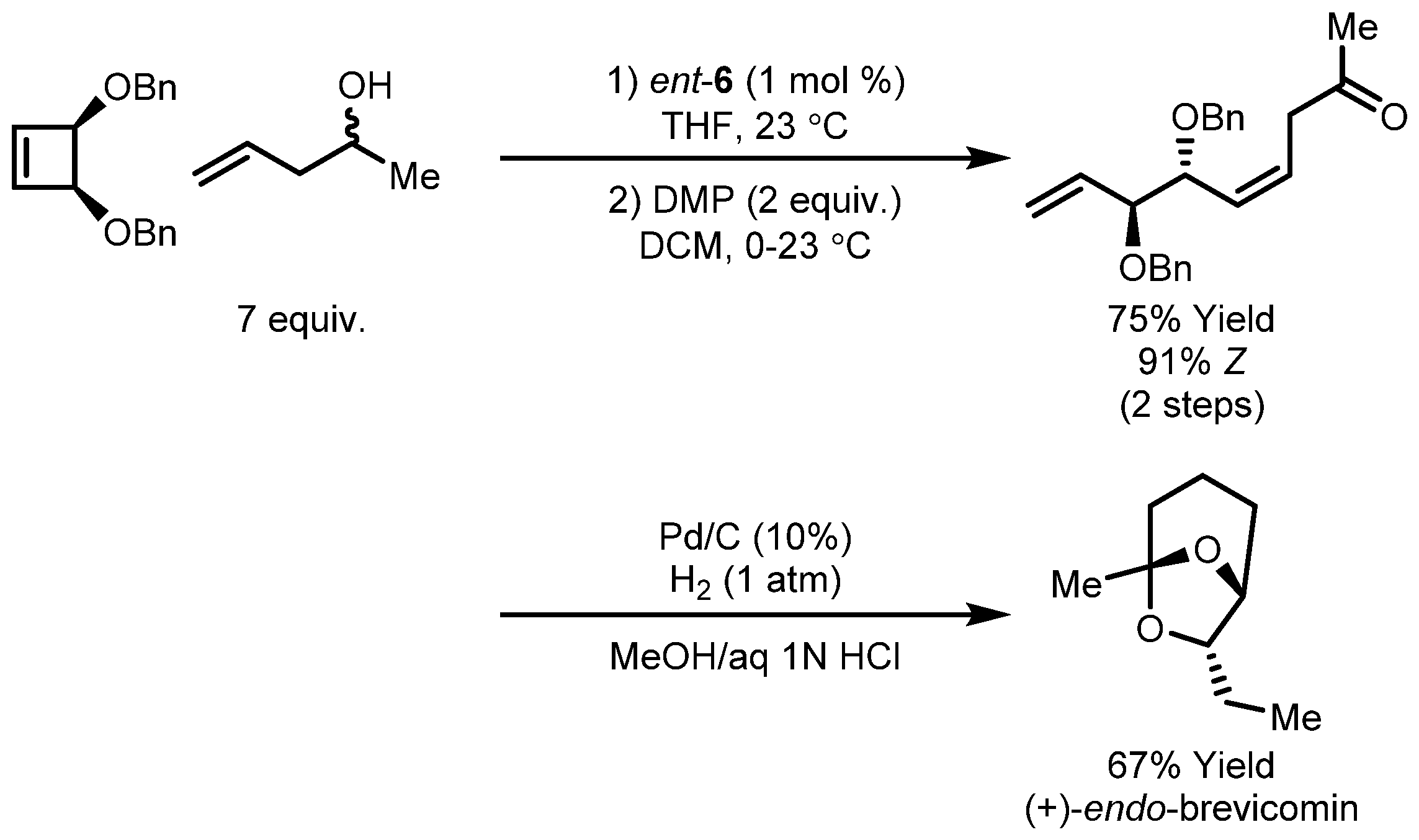
- Bernardi, R.; Fuganti, C.; Grasselli, P. On the steric course of addition of Grignard reagents onto α,β-dialkoxy and chiral aldehydes. Synthesis of (+) and (−)-exo and endo-brevicomin. Tetrahedron Lett. 1981, 22, 4021–4024. [Google Scholar] [CrossRef]
- Mori, K.; Seu, Y.-B. Synthesis of both the enantiomers of endo-brevicomin, the aggregation pheromone of Dryocoetes autographus. Tetrahedron 1985, 41, 3429–3431. [Google Scholar] [CrossRef]
- Sato, F.; Takahashi, O.; Kato, T.; Kobayashi, Y. Stereocontrolled synthesis of four possible stereoisomers of vicinal diol derivatives via relative 1,2-asymmetric induction. Preparation of optically active exo- and endo-brevicomin. J. Chem. Soc. Chem. Commun. 1985, 22, 1638–1641. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Sakurai, K.; Takano, S. Preparation of (2R,3S)-1,2-epoxypent-4-en-3-ol, a new chiral building block for the synthesis of (+)-endo- and (–)-exo-brevicomin. J. Chem. Soc. Chem. Commun. 1985, 1759–1761. [Google Scholar] [CrossRef]
- Yusufoglu, A.; Antons, S.; Scharf, H.D. Enantioselective synthesis and absolute configuration of both enantiomers of endo-brevicomin. J. Org. Chem. 1986, 51, 3485–3487. [Google Scholar] [CrossRef]
- Mulzer, J.; Angermann, A.; Münch, W. Enantio- and Diastereocontrolled Synthesis of Chiral 1,2-Diol Derivatives from (R)-2,3-Di-O-isopropylideneglyceraldehyde:endo- and exo-brevicomin. Liebigs Ann. Chem. 1986, 1986, 825–838. [Google Scholar] [CrossRef]
- Oehlschlager, A.C.; Johnston, B.D. Synthesis of the enantiomers of endo-brevicomin. J. Org. Chem. 1987, 52, 940–943. [Google Scholar] [CrossRef]
- Redlich, H.; Bruns, W.; Francke, W.; Schurig, V.; Payne, T.L.; Vité, J.P. Chiral building units from carbohydrates-XIII. Tetrahedron 1987, 43, 2029–2034. [Google Scholar] [CrossRef]
- Chong, J.M.; Mar, E.K. Enantioselective syntheses of endo- and exo-brevicomin via α-alkoxystannanes. Tetrahedron 1989, 45, 7709–7716. [Google Scholar] [CrossRef]
- Noda, Y.; Kikuchi, M. A Short Synthesis of (+)-endo-Brevicomin. Chem. Lett. 1989, 18, 1755–1756. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Oehlschlager, A.C. An example of kinetic resolution by bakers’ yeast: Synthesis of enantiomers of endo-brevicomin from the same precursor. J. Org. Chem. 1989, 54, 255–257. [Google Scholar] [CrossRef]
- Matsumoto, K.; Suzuki, N.; Ohta, H. Synthesis of (+)-endo- and (+)-exo-brevicomin viaEnzyme-Mediated Hydrolysis of an Enol Ester. Tetrahedron Lett. 1990, 31, 7163–7166. [Google Scholar] [CrossRef]
- Pedrocchi-Fantoni, G.; Servi, S. Hydrolytic and reductive action of fermenting yeast on a keto acetate: Synthesis of (+)-endo-brevicomin. J. Chem. Soc. [Perkin 1] 1991, 1764–1765. [Google Scholar] [CrossRef]
- Cere, V.; Mazzini, C.; Paolucci, C.; Pollicino, S.; Fava, A. Dihyro- and tetrahydrofuran building blocks from 1,4:3,6-dianhydromannitol. 1. Synthesis of (1S,5R,7R)-endo-(-)- and (1S,5R,7S)-(-)-exo-brevicomin and (R)-(+)-dodecanolide. J. Org. Chem. 1993, 58, 4567–4571. [Google Scholar] [CrossRef]
- Soderquist, J.A.; Ranel, A.M. (+)-exo-brevicomin via an organometallic boulevard. Tetrahedron Lett. 1993, 34, 5031–5034. [Google Scholar] [CrossRef]
- Gypser, A.; Flasche, M.; Scharf, H.-D. D-Erythronolactone and 2,3-O-Isopropylidene-l-erythrose as C4 Building Units: An Efficient Synthesis of both Enantiomers ofendo-Brevicomin and its 7-Vinyl Analogues. Liebigs Ann. Chem. 1994, 1994, 775–780. [Google Scholar] [CrossRef]
- Kim, M.-J.; Choi, G.-B.; Kim, J.-Y.; Kim, H.-J. Lipase-catalyzed transesterification as a practical route to homochiral acyclic anti-1,2-diols. A new synthesis of (+)- and (−)-endo-brevicomin. Tetrahedron Lett. 1995, 36, 6253–6256. [Google Scholar] [CrossRef]
- Vettel, S.; Lutz, C.; Knochel, P. Enantioselective Synthesis of Protected α-Hydroxy Aldehydes and Related 1,2-Amino Alcohols. Applications to the Synthesis of (-)-exo- and (-)-endo-Brevicomin. Synlett 1996, 1996, 731–733. [Google Scholar] [CrossRef]
- Burke, S.D.; Müller, N.; Beaudry, C.M. Desymmetrization by Ring-Closing Metathesis Leading to 6,8-Dioxabicyclo[3.2.1]octanes: A New Route for the Synthesis of (+)-exo- and endo-Brevicomin. Org. Lett. 1999, 1, 1827–1829. [Google Scholar] [CrossRef] [PubMed]
- Gallos, J.K.; Kyradjoglou, L.C.; Koftis, T.V. A Concise Synthesis of (-)-endo-Brevicomin. Heterocycles 2001, 55, 781–784. [Google Scholar] [CrossRef]
- Kim, S.-G.; Park, T.-H.; Kim, B.J. Efficient total synthesis of (+)-exo-, (−)-endo-brevicomin and their derivatives via asymmetric organocatalysis and olefin cross-metathesis. Tetrahedron Lett. 2006, 47, 6369–6372. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Jung, Y.; Moon, H. A Facile Total Synthesis of (-)-Frontalin, (-)-endo-Brevicomin and (-)-exo-Brevicomin through PtCl4 Catalyzed Hydroalkoxylation Reaction. Bull. Korean Chem. Soc. 2009, 30, 771–772. [Google Scholar]
- Singh, S.; Guiry, P.J. Microwave-Assisted Synthesis of Substituted Tetrahydropyrans Catalyzed by ZrCl4 and Its Application in the Asymmetric Synthesis of exo- and endo-brevicomin. J. Org. Chem. 2009, 74, 5758–5761. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, R.M.; Brownlee, R.G.; Bellas, T.E.; Wood, D.L.; Browne, L.E. Brevicomin: Principal Sex Attractant in the Frass of the Female Western Pine Beetle. Science 1968, 159, 889–891. [Google Scholar] [CrossRef] [PubMed]
- Kukovinets, O.S.; Kasradze, V.G.; Salimova, E.V.; Odinokov, V.N.; Galin, F.Z.; Fedorov, P.I. Insect pheromones and their analogs LIX. A new method for the synthesis of components of the sex pheromones of insects of the genusMalacosoma. Chem. Nat. Compd. 1999, 35, 358–360. [Google Scholar] [CrossRef]
- Bjoerkling, F.; Norin, T.; Unelius, C.R.; Miller, R.B. A stereospecific synthesis of all four isomers of 9,11-tetradecadienyl acetate using a general method applicable to 1,3-dienes. J. Org. Chem. 1987, 52, 292–294. [Google Scholar] [CrossRef]
- Williams, D.E.; Lassota, P.; Andersen, R.J. Motuporamines A−C, Cytotoxic Alkaloids Isolated from the Marine Sponge Xestospongia exigua (Kirkpatrick). J. Org. Chem. 1998, 63, 4838–4841. [Google Scholar] [CrossRef]
- Goldring, W.P.D.; Weiler, L. Cytotoxic Alkaloids Motuporamines A−C: Synthesis and Structural Verification. Org. Lett. 1999, 1, 1471–1473. [Google Scholar] [CrossRef] [PubMed]
- Ciminiello, P.; Fattorusso, E.; Forino, M.; Di Rosa, M.; Ianaro, A.; Poletti, R. Structural Elucidation of a New Cytotoxin Isolated from Mussels of the Adriatic Sea †. J. Org. Chem. 2001, 66, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Ciminiello, P.; Dell’Aversano, C.; Fattorusso, E.; Forino, M.; Magno, S.; Di Rosa, M.; Ianaro, A.; Poletti, R. Structure and Stereochemistry of a New Cytotoxic Polychlorinated Sulfolipid from Adriatic Shellfish. J. Am. Chem. Soc. 2002, 124, 13114–13120. [Google Scholar] [CrossRef] [PubMed]
- Ciminiello, P.; Dell’Aversano, C.; Fattorusso, E.; Forino, M.; Magno, S.; Di Meglio, P.; Ianaro, A.; Poletti, R. A new cytotoxic polychlorinated sulfolipid from contaminated Adriatic mussels. Tetrahedron 2004, 60, 7093–7098. [Google Scholar] [CrossRef]
- Nilewski, C.; Geisser, R.W.; Carreira, E.M. Total synthesis of a chlorosulpholipid cytotoxin associated with seafood poisoning. Nature 2009, 457, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Bedke, D.K.; Shibuya, G.M.; Pereira, A.; Gerwick, W.H.; Haines, T.H.; Vanderwal, C.D. Relative Stereochemistry Determination and Synthesis of the Major Chlorosulfolipid from Ochromonas danica. J. Am. Chem. Soc. 2009, 131, 7570–7572. [Google Scholar] [CrossRef] [PubMed]
- Bedke, D.K.; Shibuya, G.M.; Pereira, A.R.; Gerwick, W.H.; Vanderwal, C.D. A Concise Enantioselective Synthesis of the Chlorosulfolipid Malhamensilipin A. J. Am. Chem. Soc. 2010, 132, 2542–2543. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, G.M.; Kanady, J.S.; Vanderwal, C.D. Stereoselective Dichlorination of Allylic Alcohol Derivatives to Access Key Stereochemical Arrays of the Chlorosulfolipids. J. Am. Chem. Soc. 2008, 130, 12514–12518. [Google Scholar] [CrossRef] [PubMed]
- Yoshimitsu, T.; Fukumoto, N.; Tanaka, T. Enantiocontrolled Synthesis of Polychlorinated Hydrocarbon Motifs: A Nucleophilic Multiple Chlorination Process Revisited. J. Org. Chem. 2009, 74, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Yoshimitsu, T.; Fukumoto, N.; Nakatani, R.; Kojima, N.; Tanaka, T. Asymmetric Total Synthesis of (+)-Hexachlorosulfolipid, a Cytotoxin Isolated from Adriatic Mussels. J. Org. Chem. 2010, 75, 5425–5437. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, T.; Shibata, M.; Kaneko, K.; Okino, T.; Matsuda, F. Asymmetric Total Synthesis of Danicalipin A and Evaluation of Biological Activity. Org. Lett. 2011, 13, 904–907. [Google Scholar] [CrossRef] [PubMed]
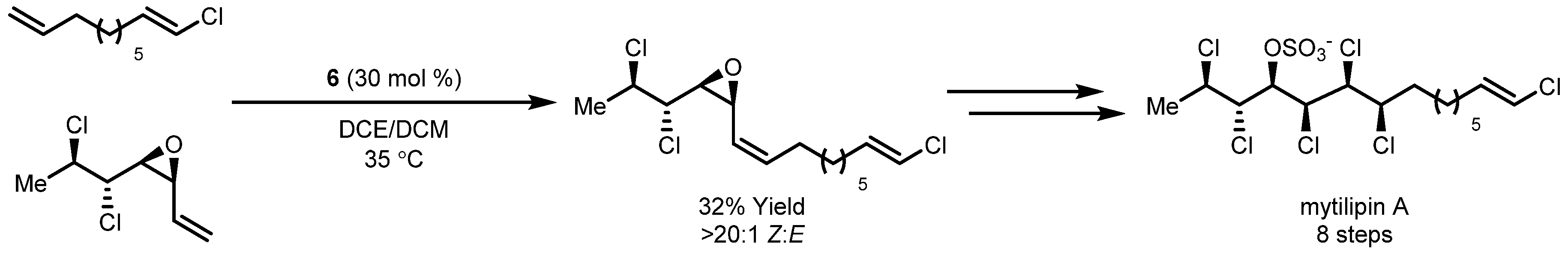
- Chung, W.; Carlson, J.S.; Bedke, D.K.; Vanderwal, C.D. A Synthesis of the Chlorosulfolipid Mytilipin A via a Longest Linear Sequence of Seven Steps. Angew. Chem. Int. Ed. 2013, 52, 10052–10055. [Google Scholar] [CrossRef] [PubMed]
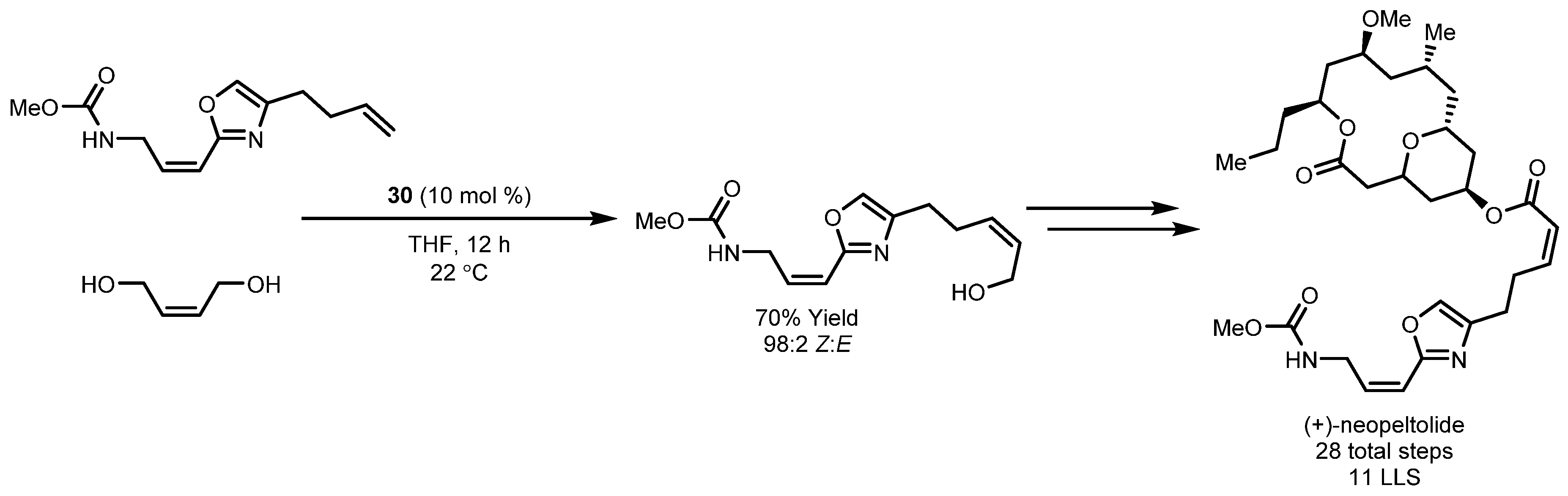
- Wright, A.E.; Botelho, J.C.; Guzmán, E.; Harmody, D.; Linley, P.; McCarthy, P.J.; Pitts, T.P.; Pomponi, S.A.; Reed, J.K. Neopeltolide, a Macrolide from a Lithistid Sponge of the Family Neopeltidae⊥. J. Nat. Prod. 2007, 70, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Youngsaye, W.; Lowe, J.T.; Pohlki, F.; Ralifo, P.; Panek, J.S. Total Synthesis and Stereochemical Reassignment of (+)-Neopeltolide. Angew. Chem. Int. Ed. 2007, 46, 9211–9214. [Google Scholar] [CrossRef] [PubMed]
- Custar, D.W.; Zabawa, T.P.; Scheidt, K.A. Total Synthesis and Structural Revision of the Marine Macrolide Neopeltolide. J. Am. Chem. Soc. 2008, 130, 804–805. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.K.; Kwon, M.S.; Lee, E. Total Synthesis of (+)-Neopeltolide by a Prins Macrocyclization. Angew. Chem. Int. Ed. 2008, 47, 3242–3244. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H.; Naito, S.; Goto, T.; Sasaki, M. Total Synthesis of (+)-Neopeltolide. Angew. Chem. Int. Ed. 2008, 47, 4737–4739. [Google Scholar] [CrossRef] [PubMed]
- Paterson, I.; Miller, N.A. Total synthesis of the marine macrolide (+)-neopeltolide. Chem. Commun. 2008, 4708–4710. [Google Scholar] [CrossRef] [PubMed]
- Guinchard, X.; Roulland, E. Total Synthesis of the Antiproliferative Macrolide (+)-Neopeltolide. Org. Lett. 2009, 11, 4700–4703. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H.; Saito, A.; Sasaki, M. A Concise Total Synthesis of (+)-Neopeltolide. Angew. Chem. Int. Ed. 2010, 49, 3041–3044. [Google Scholar] [CrossRef] [PubMed]
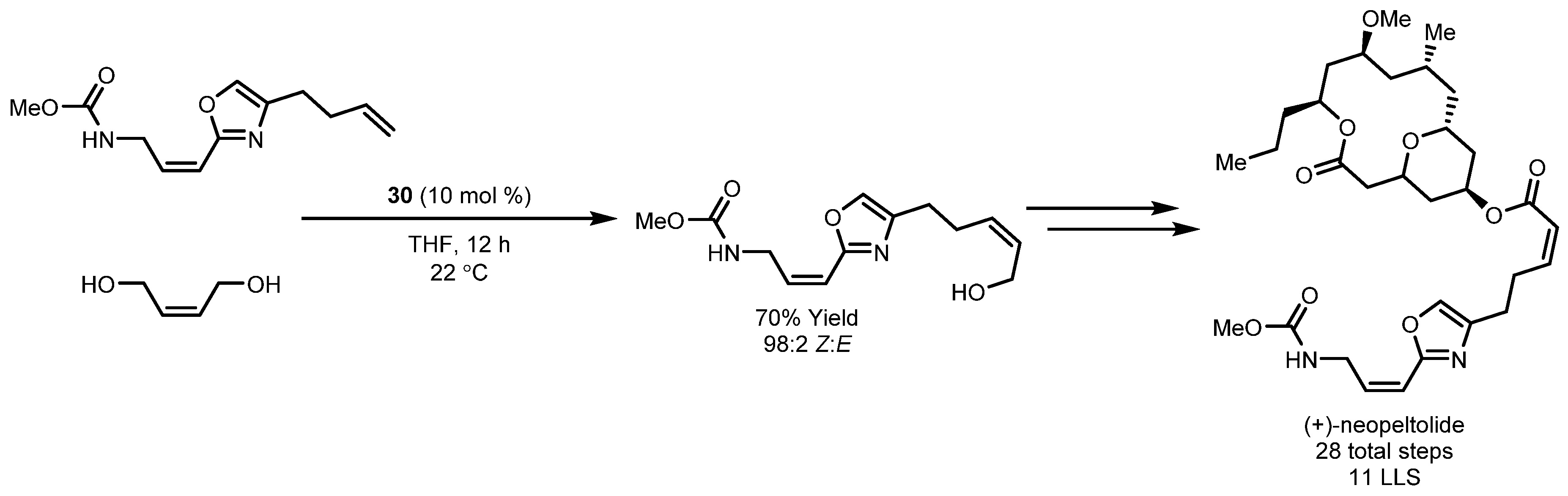
- Yu, M.; Schrock, R.R.; Hoveyda, A.H. Catalyst-Controlled Stereoselective Olefin Metathesis as a Principal Strategy in Multistep Synthesis Design: A Concise Route to (+)-Neopeltolide. Angew. Chem. Int. Ed. 2015, 54, 215–220. [Google Scholar] [CrossRef] [PubMed]
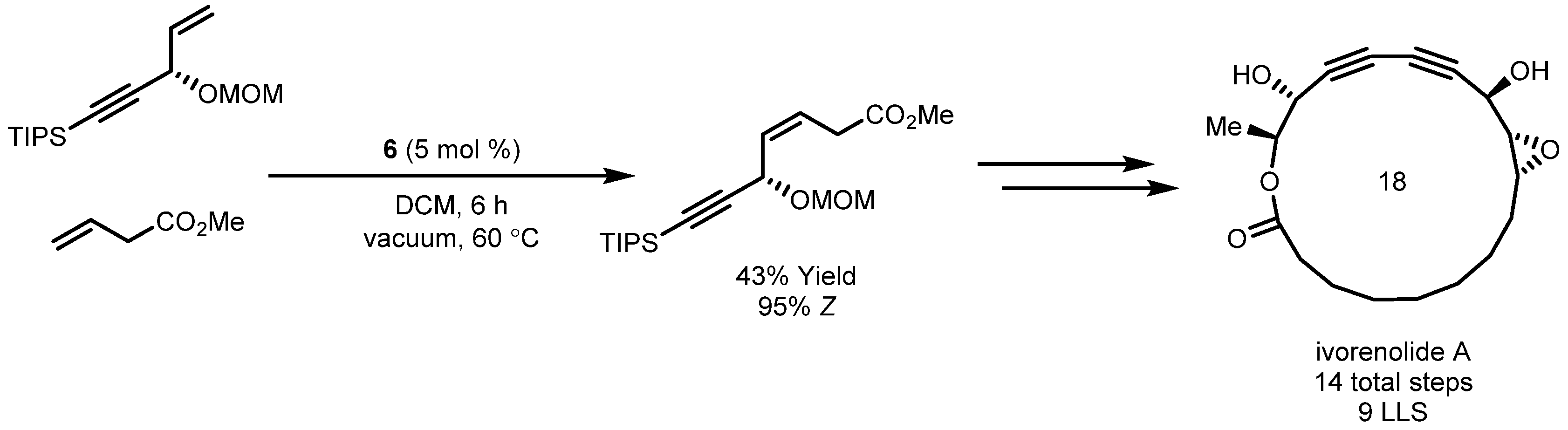
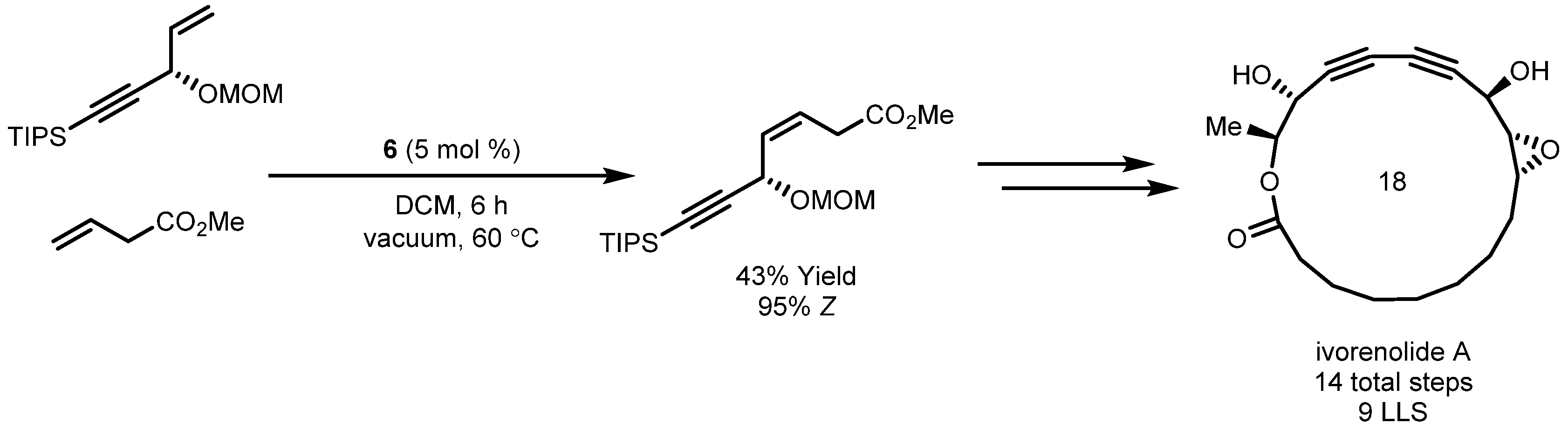
- De Léséleuc, M.; Godin, É.; Parisien-Collette, S.; Lévesque, A.; Collins, S.K. Catalytic Macrocyclization Strategies Using Continuous Flow: Formal Total Synthesis of Ivorenolide A. J. Org. Chem. 2016, 81, 6750–6756. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, Y.; Yang, S.-P.; Zhou, Y.; Wu, W.-B.; Tang, W.; Zuo, J.-P.; Li, Y.; Yue, J.-M. Ivorenolide A, an Unprecedented Immunosuppressive Macrolide from Khaya ivorensis: Structural Elucidation and Bioinspired Total Synthesis. J. Am. Chem. Soc. 2012, 134, 20605–20608. [Google Scholar] [CrossRef]
- Glaser, C. Beiträge zur Kenntniss des Acetenylbenzols. Berichte Dtsch. Chem. Ges. 1869, 2, 422–424. [Google Scholar] [CrossRef]
- Hay, A.S. Oxidative Coupling of Acetylenes. II 1. J. Org. Chem. 1962, 27, 3320–3321. [Google Scholar] [CrossRef]
- Song, W.; Wang, Y.; Qu, J.; Lin, Q. Selective Functionalization of a Genetically Encoded Alkene-Containing Protein via “Photoclick Chemistry” in Bacterial Cells. J. Am. Chem. Soc. 2008, 130, 9654–9655. [Google Scholar] [CrossRef] [PubMed]
- Van Hest, J.C.M.; Kiick, K.L.; Tirrell, D.A. Efficient Incorporation of Unsaturated Methionine Analogues into Proteins in Vivo. J. Am. Chem. Soc. 2000, 122, 1282–1288. [Google Scholar] [CrossRef]
- Bernardes, G.J.L.; Chalker, J.M.; Errey, J.C.; Davis, B.G. Facile Conversion of Cysteine and Alkyl Cysteines to Dehydroalanine on Protein Surfaces: Versatile and Switchable Access to Functionalized Proteins. J. Am. Chem. Soc. 2008, 130, 5052–5053. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.A.; Boutureira, O.; Lercher, L.; Bhushan, B.; Paton, R.S.; Davis, B.G. Rapid Cross-Metathesis for Reversible Protein Modifications via Chemical Access to Se -Allyl-selenocysteine in Proteins. J. Am. Chem. Soc. 2013, 135, 12156–12159. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; van der Donk, W.A. Convergent Synthesis of Peptide Conjugates Using Dehydroalanines for Chemoselective Ligations. Org. Lett. 2001, 3, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
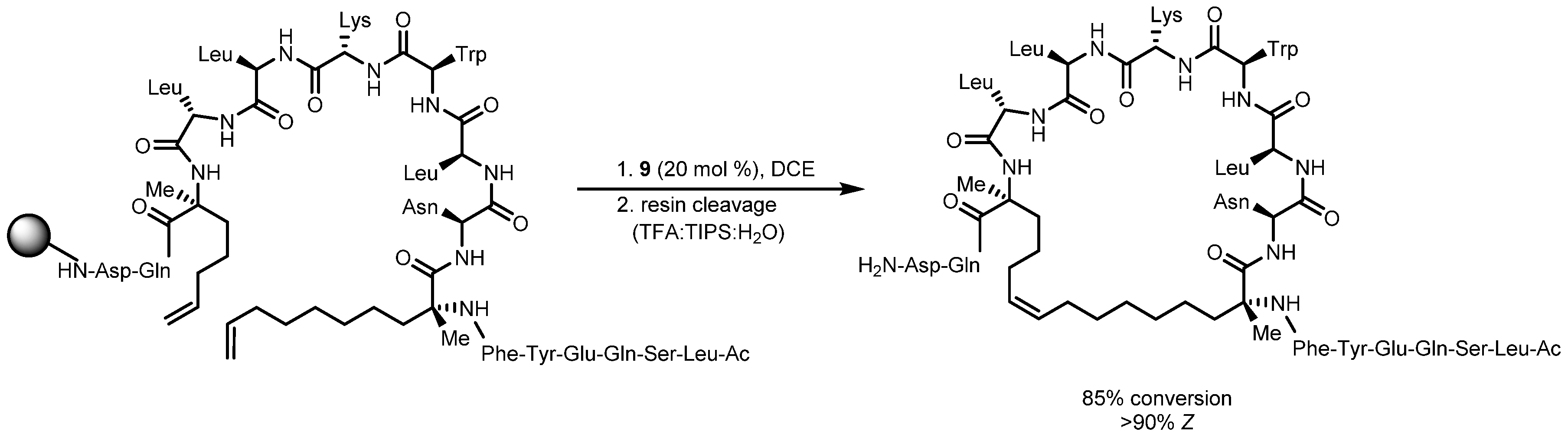
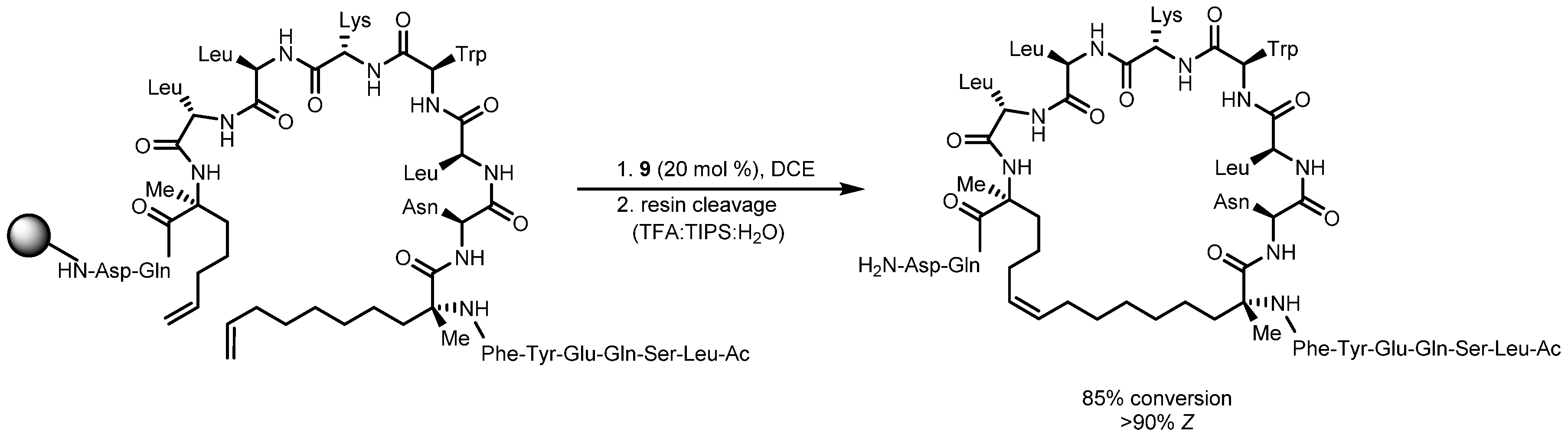
- Mangold, S.L.; O’Leary, D.J.; Grubbs, R.H. Z-Selective Olefin Metathesis on Peptides: Investigation of Side-Chain Influence, Preorganization, and Guidelines in Substrate Selection. J. Am. Chem. Soc. 2014, 136, 12469–12478. [Google Scholar] [CrossRef] [PubMed]
- Mangold, S.L.; Grubbs, R.H. Stereoselective synthesis of macrocyclic peptides via a dual olefin metathesis and ethenolysis approach. Chem Sci 2015, 6, 4561–4569. [Google Scholar] [CrossRef] [PubMed]
- Louie, J.; Bielawski, C.W.; Grubbs, R.H. Tandem Catalysis: The Sequential Mediation of Olefin Metathesis, Hydrogenation, and Hydrogen Transfer with Single-Component Ru Complexes. J. Am. Chem. Soc. 2001, 123, 11312–11313. [Google Scholar] [CrossRef] [PubMed]
- Camm, K.D.; Martinez Castro, N.; Liu, Y.; Czechura, P.; Snelgrove, J.L.; Fogg, D.E. Tandem ROMP–Hydrogenation with a Third-Generation Grubbs Catalyst. J. Am. Chem. Soc. 2007, 129, 4168–4169. [Google Scholar] [CrossRef]
- Sutton, A.E.; Seigal, B.A.; Finnegan, D.F.; Snapper, M.L. New Tandem Catalysis: Preparation of Cyclic Enol Ethers through a Ruthenium-Catalyzed Ring-Closing Metathesis−Olefin Isomerization Sequence. J. Am. Chem. Soc. 2002, 124, 13390–13391. [Google Scholar] [CrossRef] [PubMed]
- Seigal, B.A.; Fajardo, C.; Snapper, M.L. Tandem Catalysis: Generating Multiple Contiguous Carbon−Carbon Bonds through a Ruthenium-Catalyzed Ring-Closing Metathesis/Kharasch Addition. J. Am. Chem. Soc. 2005, 127, 16329–16332. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.G.; Snapper, M.L. Preparation of Alkenyl Cyclopropanes through a Ruthenium-Catalyzed Tandem Enyne Metathesis−Cyclopropanation Sequence. J. Am. Chem. Soc. 2006, 128, 52–53. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.; Krehl, S. A single precatalyst tandem RCM–allylic oxidation sequence. Chem. Commun. 2011, 47, 5879–5881. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.; Krehl, S.; Hauke, S. Assisted Tandem Catalytic Cross Metathesis–Oxidation: In One Flask from Styrenes to 1,2-Diketones and Further to Quinoxalines. J. Org. Chem. 2013, 78, 5427–5435. [Google Scholar] [CrossRef] [PubMed]
- Beligny, S.; Eibauer, S.; Maechling, S.; Blechert, S. Sequential Catalysis: A Metathesis/Dihydroxylation Sequence. Angew. Chem. Int. Ed. 2006, 45, 1900–1903. [Google Scholar] [CrossRef] [PubMed]
- Scholte, A.A.; An, M.H.; Snapper, M.L. Ruthenium-Catalyzed Tandem Olefin Metathesis−Oxidations. Org. Lett. 2006, 8, 4759–4762. [Google Scholar] [CrossRef] [PubMed]
- Neisius, N.M.; Plietker, B. Diastereoselective Ru-Catalyzed Cross-Metathesis−Dihydroxylation Sequence. An Efficient Approach toward Enantiomerically Enriched syn-Diols. J. Org. Chem. 2008, 73, 3218–3227. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Ishigame, T.; Oshima, N.; Hoshiya, N.; Shimawaki, K.; Arisawa, M.; Shuto, S. One-Pot Ring-Closing Metathesis (RCM)/Oxidation by an Assisted Tandem Ruthenium Catalysis for the Synthesis of 2-Quinolones. Adv. Synth. Catal. 2011, 353, 2676–2680. [Google Scholar] [CrossRef]
- Dornan, P.K.; Wickens, Z.K.; Grubbs, R.H. Tandem Z-Selective Cross-Metathesis/Dihydroxylation: Synthesis of anti-1,2-Diols. Angew. Chem. Int. Ed. 2015, 54, 7134–7138. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, J.L.; Shi, G.; Zeng, L.; Gu, Z.; M. MacDougal, J. Absolute Stereochemistries of Sylvaticin and 12,15-cis-Sylvaticin, Bioactive C-20,23-cis Non-adjacent Bistetrahydrofuran Annonaceous Acetogenins, from Rollinia mucosa. Heterocycles 1995, 41, 1785–1796. [Google Scholar] [CrossRef]
- Abdel Ghani, S.B.; Chapman, J.M.; Figadère, B.; Herniman, J.M.; Langley, G.J.; Niemann, S.; Brown, R.C.D. Total Synthesis and Stereochemical Assignment of cis-Uvariamicin I and cis-Reticulatacin. J. Org. Chem. 2009, 74, 6924–6928. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Cragg, G.M.; Polonsky, J.; Herald, D.L.; Goswami, A.; Smith, C.R.; Moretti, C.; Schmidt, J.M.; Weisleder, D. Isolation and structure of rolliniastatin 1 from the South American tree Rollinia mucosa. Can. J. Chem. 1987, 65, 1433–1435. [Google Scholar] [CrossRef]
- Řezanka, T.; Hanuš, L.; Dembitsky, V.M. Chagosensine, a New Chlorinated Macrolide from the Red Sea SpongeLeucetta chagosensis. Eur. J. Org. Chem. 2003, 2003, 4073–4079. [Google Scholar] [CrossRef]
- Klein, E.; Rojahn, W. Die permanganatoxydation von 1,5-dienverbindungen. Tetrahedron 1965, 21, 2353–2358. [Google Scholar] [CrossRef]
- Carlsen, P.H.J.; Katsuki, T.; Martin, V.S.; Sharpless, K.B. A greatly improved procedure for ruthenium tetroxide catalyzed oxidations of organic compounds. J. Org. Chem. 1981, 46, 3936–3938. [Google Scholar] [CrossRef]
- De Champdoré, M.; Lasalvia, M.; Piccialli, V. OsO4-Catalyzed oxidative cyclization of geranyl and neryl acetate to cis-2,5-bis(hydroxymethyl)tetrahydrofurans. Tetrahedron Lett. 1998, 39, 9781–9784. [Google Scholar] [CrossRef]
- Piccialli, V. Oxidative Cyclization of Dienes and Polyenes Mediated by Transition-Metal-Oxo Species. Synthesis 2007, 2007, 2585–2607. [Google Scholar] [CrossRef]
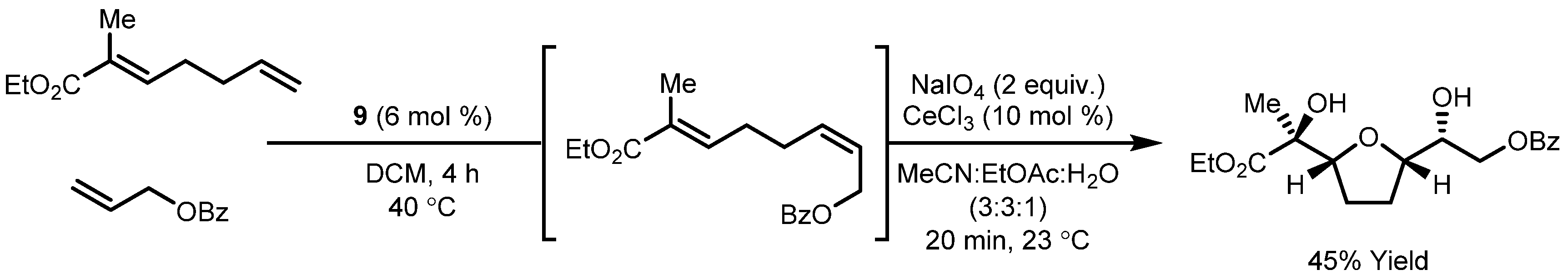
- Dornan, P.K.; Lee, D.; Grubbs, R.H. Tandem Olefin Metathesis/Oxidative Cyclization: Synthesis of Tetrahydrofuran Diols from Simple Olefins. J. Am. Chem. Soc. 2016, 138, 6372–6375. [Google Scholar] [CrossRef] [PubMed]



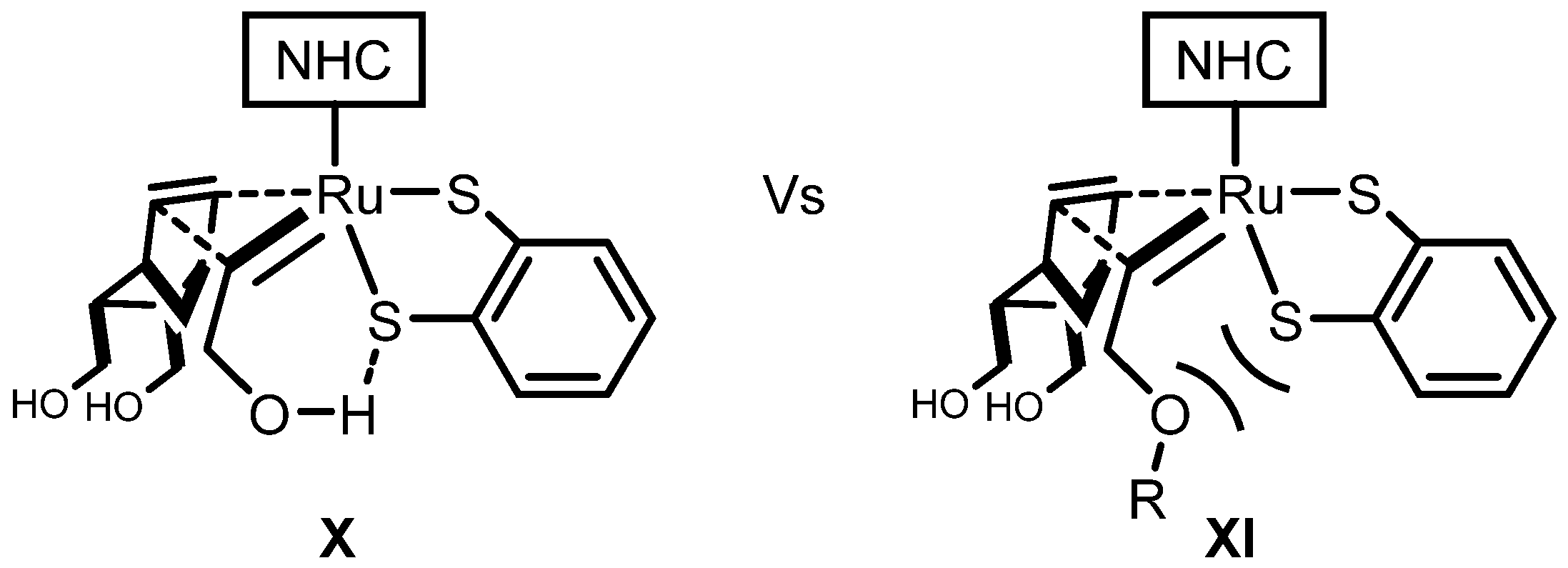
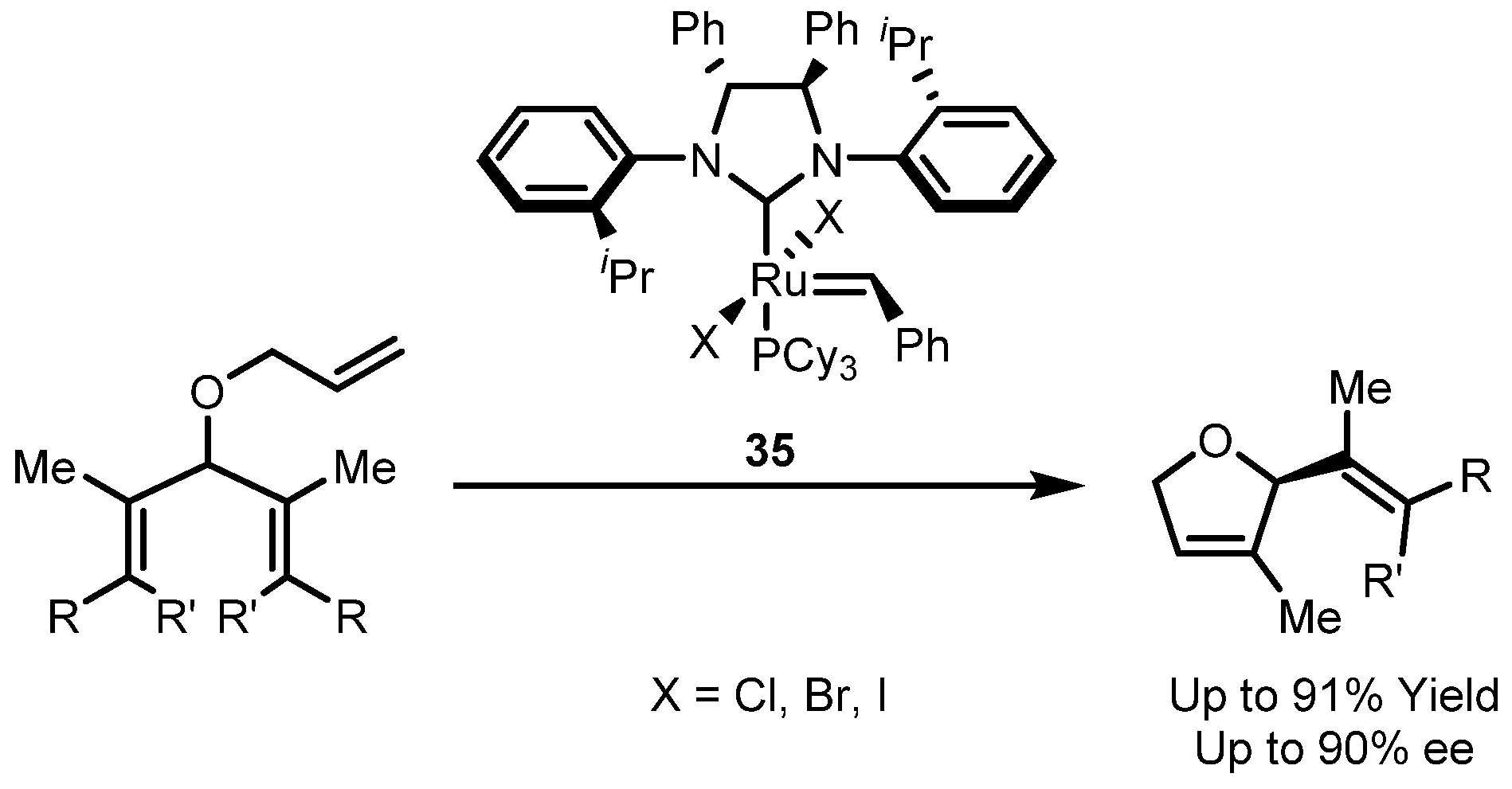
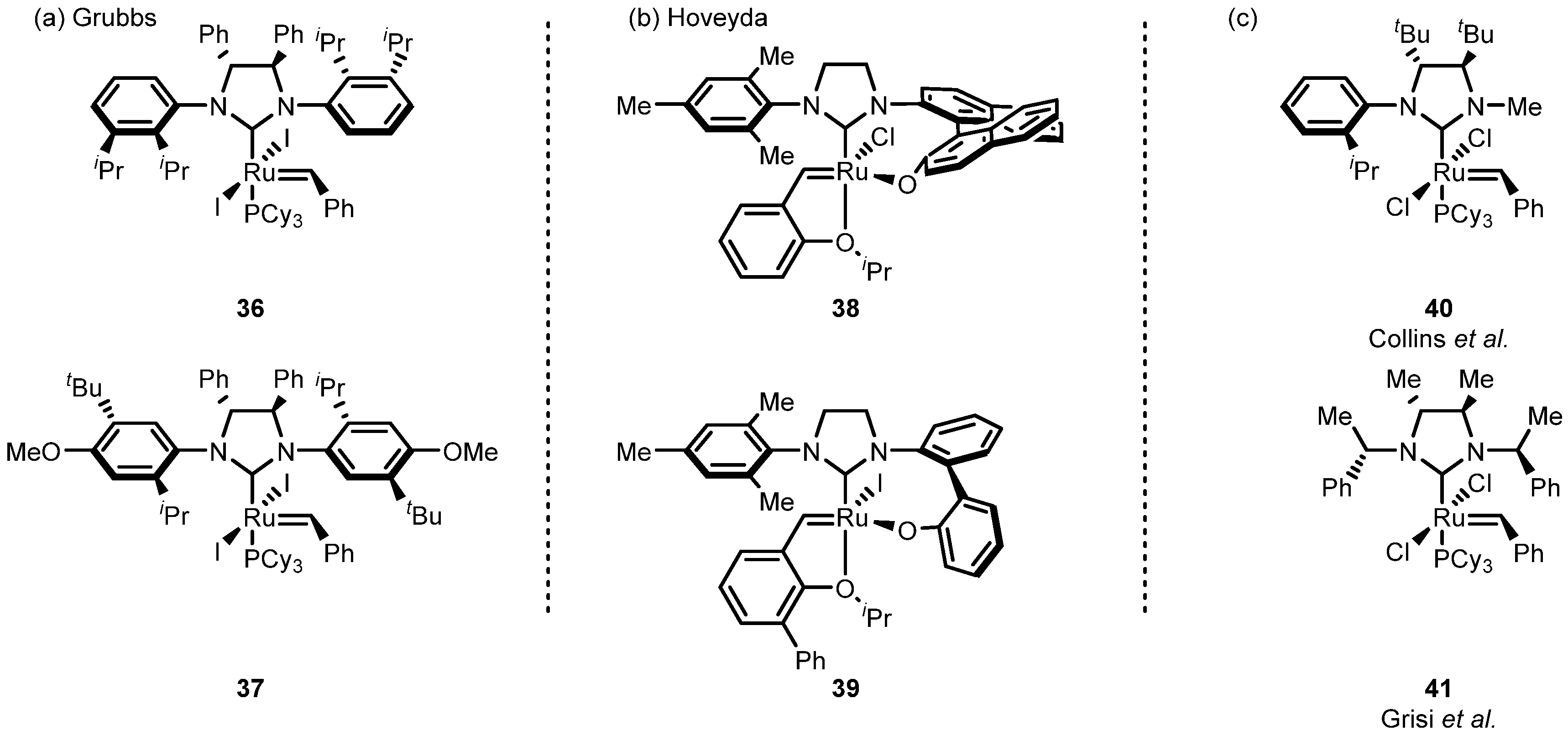
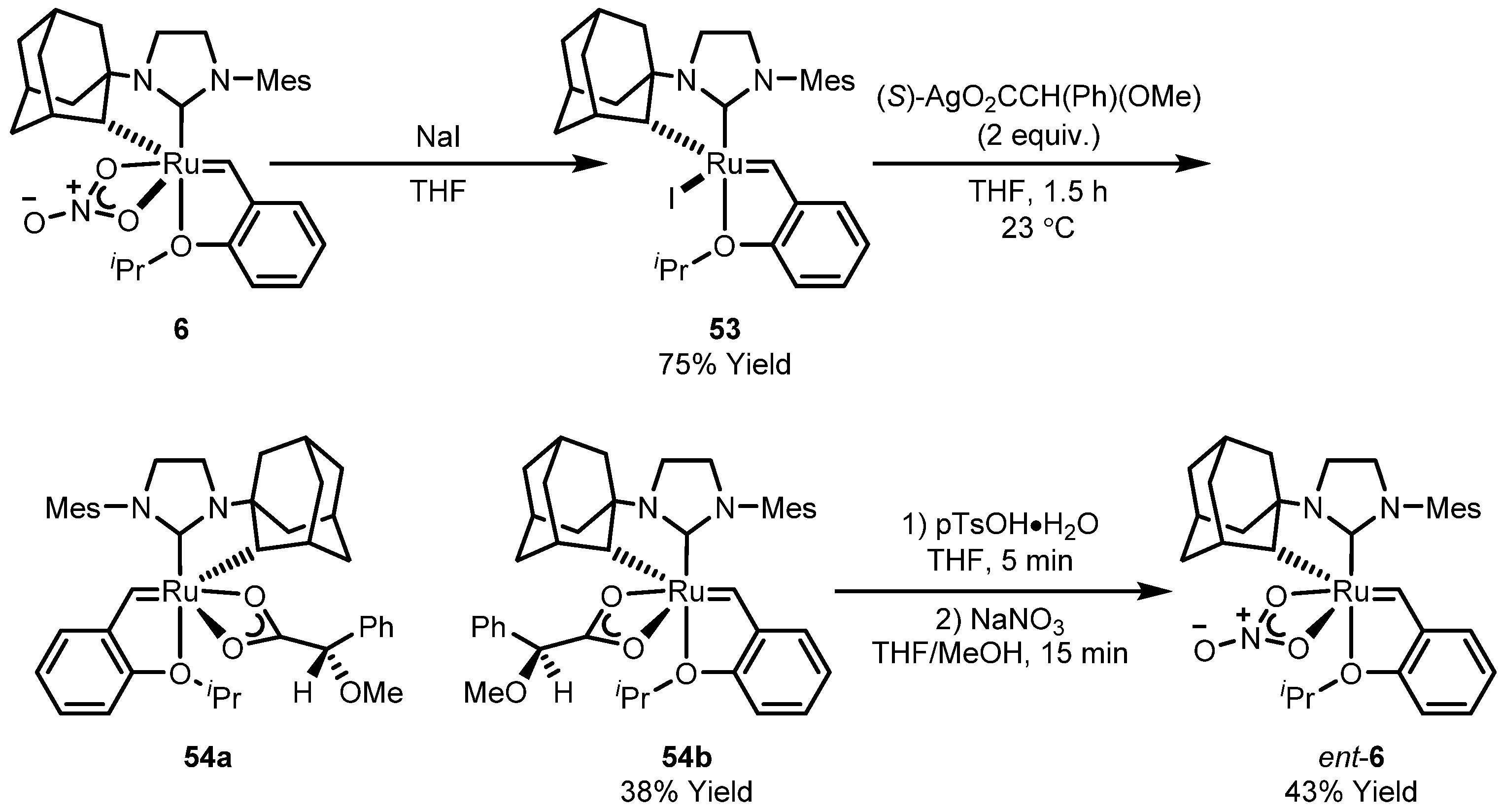
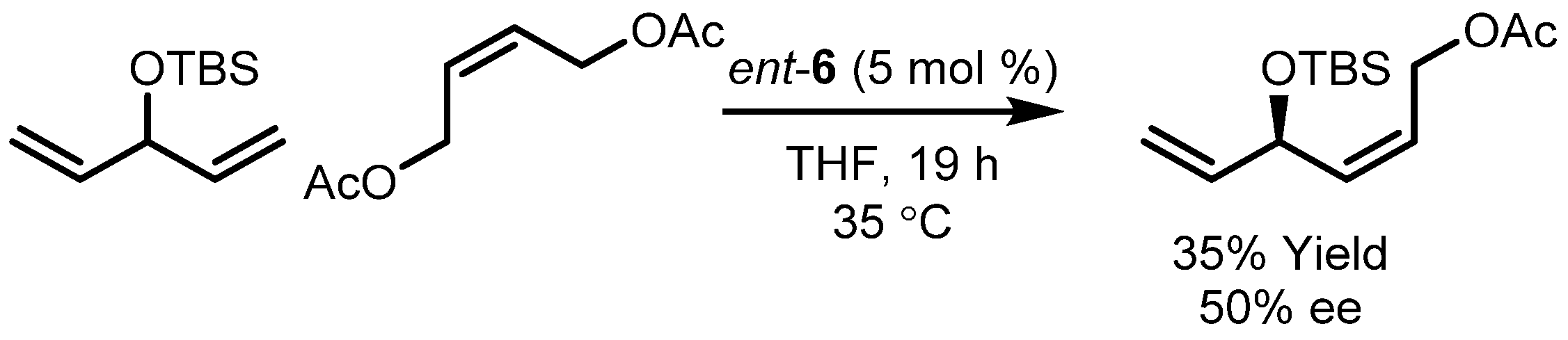
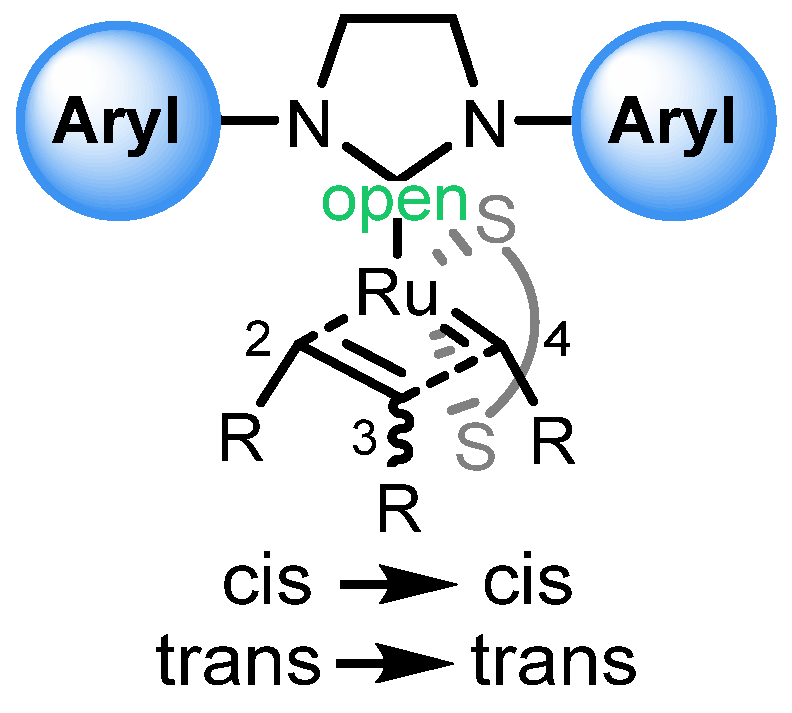


| Entry | R | Cat (xx mol %) | Yield (%) | Z (%) |
|---|---|---|---|---|
| 1 | CH2Ph | 4 (2) | 81 | 92 |
| 2 | CH2Ph | 6 (0.1) | 91 | 92 |
| 3 | CH2Ph | 9 (0.1) | >95 | >95 |
| 4 | CH2Ph | 9 (0.01) | 74 | >95 |
| 5 | (CH2)8CO2Me | 4 (2) | >95 | 73 |
| 6 | (CH2)8CO2Me | 6 (0.1) | 85 | 91 |
| 7 | (CH2)8CO2Me | 9 (0.1) | >95 | >95 |
| 8 | (CH2)3OH | 6 (0.1) | 67 | 81 |
| 9 | (CH2)3OH | 9 (0.1) | 77 | 95 |

| Entry | R | R’ | R” | Cat (xx mol %) | Yield (%) | Z (%) |
|---|---|---|---|---|---|---|
| 1* | CH2Ph | CH2OAc | CH2OAc | 4 (5) | 60 | 84 |
| 2 | CH2Ph | CH2OAc | CH2OAc | 6 (1) | 58 | 91 |
| 3 | (CH2)3Me | (CH2)7OAc | H | 6 (0.5) | 70 | 91 |
| 4 | (CH2)3Me | (CH2)7OAc | H | 9 (0.5) | 71 | >95 |

| Entry | R | Yield (%) | Z (%) |
|---|---|---|---|
| 1 |  | 82 | >95 |
| 2 |  | 70 | >95 |
| 3 |  | 44 | 94 |
| 4 | 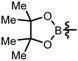 | 81 | 92 |
| 5 |  | 40 | >95 |

| Entry | Product | Cat | Yield (%) | Z (%) |
|---|---|---|---|---|
| 1 | 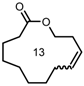 | 6 | 40 | 86 |
| 2 |  | 6 | 58 | 85 |
| 3 |  | 6 | 72 | 84 |
| 4 | 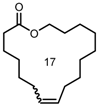 | 6 9 | 71 64 | 89 >95 |
| 5 |  | 6 9 | 50 36 | 68 >95 |
| 6 | 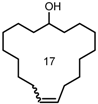 | 6 9 | 56 45 | 65 >95 |

| Entry | Substrate | Initial E (%) | Final E (%) |
|---|---|---|---|
| 1 | 19 | 55 | >95 |
| 2 | 20 | 80 | >95 |
| 3 | 21 | 80 | >95 |

| Entry | R | n | Initial E (%) | Final E (%) |
|---|---|---|---|---|
| 1 | Me | 3 | 52 | 90 |
| 2 | OAc | 7 | 78 | >95 |
| 3 | OH | 4 | 68 | 90 |
| 4 | CO2Me | 6 | 80 | >95 |
| 5 | NHPh | 3 | 80 | >95 |
| 6 | C(O)Me | 2 | 72 | >95 |

| Entry | R | Yield (%) | Z (%) |
|---|---|---|---|
| 1 | CH2Ph | 63 | >95 |
| 2 | (CH2)8CHO | 70 | >95 |
| 3 | (CH2)2COMe | 49 | >95 |
| 4 | (CH2)7CO2Me | 82 | >95 |
| 5 | CH2NHPh | 68 | >95 |
| 6 | CH2NHBoc | 54 | >95 |
| 7 | CH2OCO2Me | 79 | >95 |
| 8 | CH2BPin | 65 | >95 |
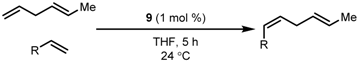
| Entry | R | R’ | 9 (xx mol %) | Yield (%) |
|---|---|---|---|---|
| 1 | (CH2)7OH | (CH2)9Me | 2 | 84 |
| 2 | CH2OCH2CCPh | (CH2)9Me | 3 | 82 |
| 3 | CH2NHBoc | (CH2)9Me | 4 | 60 |
| 4 | (CH2)8CHO | (CH2)9Me | 4 | 59 |
| 5 | CH2BPin | (CH2)9Me | 4 | 60 |
| 6 | (CH2)4OAc | (CH2)9Me | 4 | 69 |
| 7 | CH2CH2COMe | (CH2)9Me | 4 | 41 |
| 8 | CH2OCO2Me | (CH2)9Me | 4 | 42 |
| 9 | (CH2)6Br | (CH2)9Me | 4 | 34 |
| 10 | (CH2)2CN | (CH2)9Me | 4 | 12 |
| 11 | (CH2)7OH | (CH2)4OAc | 4 | 75 |
| 12 | (CH2)7OH | Ph | 4 | 15 |
| 13 | (CH2)7OH | 4-MeOPh | 4 | 25 |
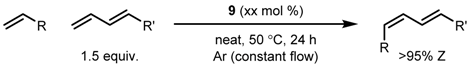
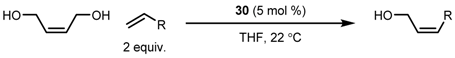
| Entry | R | Cat (xx mol %) | Solvent | T (°C) | Time (h) | Yield (%) | Z (%) |
|---|---|---|---|---|---|---|---|
| 1 | CH2Ph | 22 (0.25) | THF | 40 | 0.5 | 12 | 80 |
| 2 | 14 | 39 | |||||
| 2 | CH2Ph | 24 (0.25) | THF | 40 | 0.5 | 6 | 88 |
| 2 | 10 | 56 | |||||
| 3 | CH2SiMe3 | 22 (0.25) | THF | 60 | 18 | 12 | 95 |
| 4 | CH2SiMe3 | 24 (0.25) | THF | 60 | 16 | 9 | 96 |
| 5 | (CH2)5Me | 22 (0.01) | neat | 60 | 2 | 20 | 86 |
| 6 | (CH2)5Me | 24 (0.01) | neat | 60 | 1.5 | 13 | 88 |
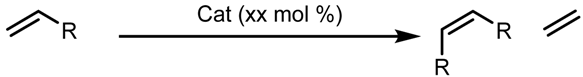
| Entry | R | 29 (xx mol %) | Time (h) | Yield (%) | Z (%) |
|---|---|---|---|---|---|
| 1 | Ph | 1 | 1 | 92 | 97 |
| 2 | m-FC6H4 | 1 | 1 | 93 | 96 |
| 3 | (CH2)2OTBS | 5 | 8 | 68 | >98 |
| 4 | (CH2)2C(O)NHPh | 5 | 8 | 65 | >98 |
| 5 | (CH2)7Me | 5 | 8 | 58 | >98 |
| 6 | 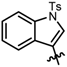 | 5 | 2 | 93 | 93 |
| 7 | 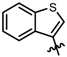 | 5 | 2 | 97 | >98 |
| 8 |  | 2 | 2 | 84 | 91 |
| 9 | 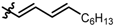 | 5 | 2 | 80 | >98 |
| 10 | OnBu | 2 | 2 | 95 | >98 |
| 11 | SEt | 5 | 12 | 80 | 92 |

| Entry | R | Time (h) | Yield (%) | Z (%) |
|---|---|---|---|---|
| 1 | (CH2)9Me | 4 | 72 | 96 |
| 2 | (CH2)2OTBS | 4 | 65 | 93 |
| 3 | (CH2)2OPNP | 4 | 74 | 96 |
| 4 | CH2OnBu | 12 | 57 | 91 |
| 5 | (CH2)2CO2Bn | 4 | 80 | 98 |
| 6 | (CH2)4Phth | 4 | 64 | 98 |
| 7 | (CH2)8CHO | 4 | 80 | 94 |
| 8 | (CH2)3CO2H | 4 | 70 | 96 |
| 9* | Ph | 4 | 53 | 94 |
| 10 | Cy | 4 | 59 | 98 |
| 11 | 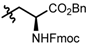 | 4 | 73 | 98 |
| 12 |  | 8 | 66 | 95 |
| 13 | 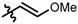 | 8 | 63 | 92 |
| 14 |  | 8 | 56 | 96 |
| 15 | 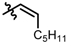 | 8 | 54 | 87 |
| Entry | Cat | Conversion (%) | ee (%) |
|---|---|---|---|
| 1 | 42 | 95 | 8 |
| 2 | 43 | 95 | 38 |
| 3 | 44 | 90 | 52 |
| 4 | 45 | 86 | 50 |
| 5 | 45 anti | 46 | 59 |

| Entry | Cat | Conversion (%) | ee (%) |
|---|---|---|---|
| 1 | 46 | >98 | 59 |
| 2 | 47 | 58 | 50 |
| 3 | 48 | 87 | 66 |

| Entry | Cat (xx mol %) | Solvent | Additive | Conversion (%) | ee (%) |
|---|---|---|---|---|---|
| 1 | 49 (2.5) | DCM | - | >98 | 18 |
| 2 | 49 (4.0) | THF | NaI | >95 | 53 |
| 3 | 50 (2.5) | DCM | - | >98 | 33 |
| 4 | 50 (4.0) | THF | NaI | >98 | 50 |
| 5 | 51 (2.5) | DCM | - | >98 | 19 |
| 6 | 51 (4.0) | THF | NaI | >95 | 52 |
| 7 | 52 (2.5) | DCM | - | >98 | 33 |
| 8 | 52 (4.0) | THF | NaI | >98 | 47 |

| Entry | Product | Yield (%) | ee (%) |
|---|---|---|---|
| 1 |  | 65 | 69 |
| 2 | 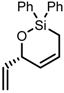 | 29 | 67 |
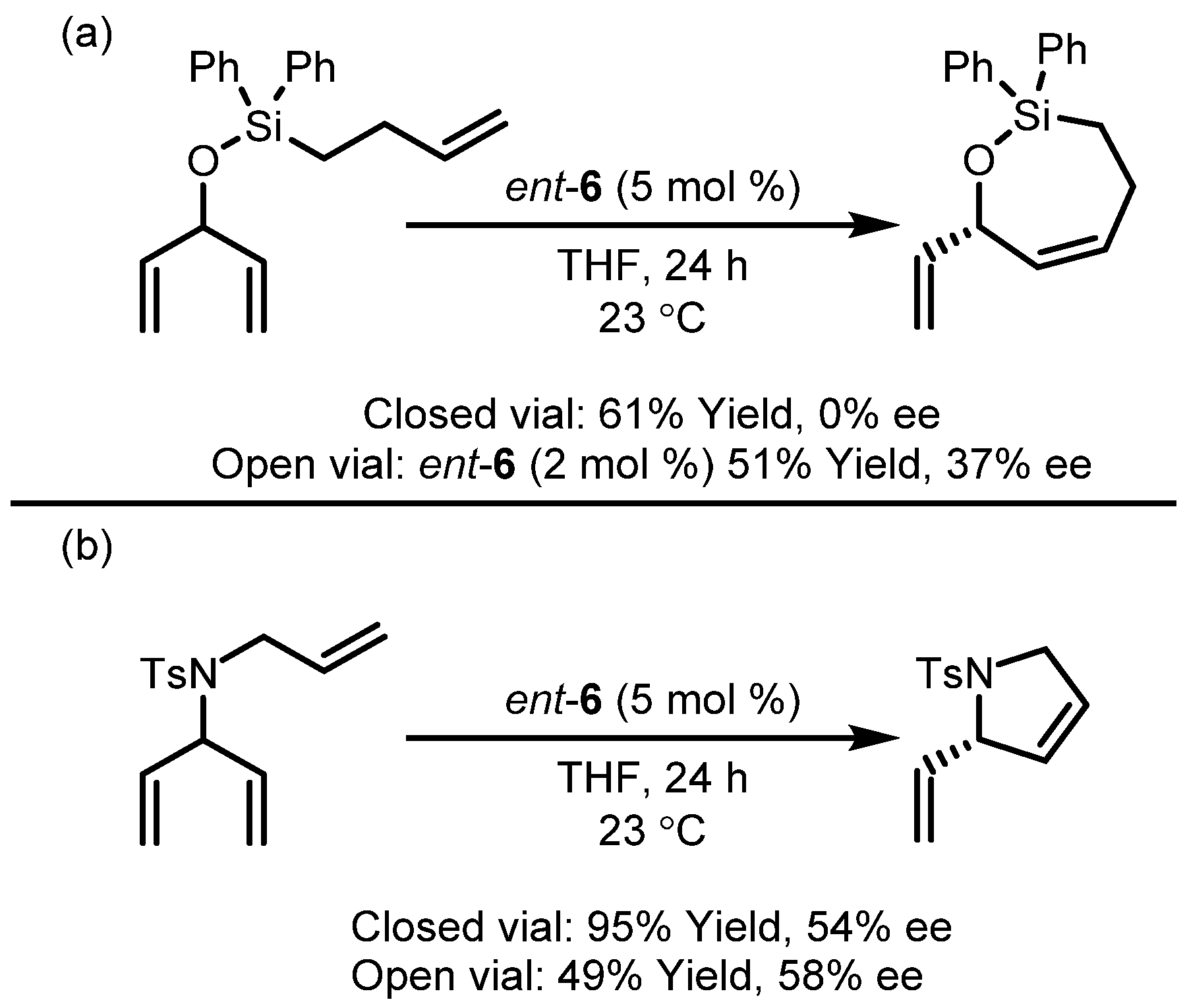
| 3 |  | 95 | 54 |
| 4 |  | 72 | 47 |
| 5 | 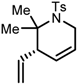 | 90 | 57 |
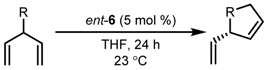
| Entry | Substrate | T (°C) | E:Z | Conversion (%) | ee (%) |
|---|---|---|---|---|---|
| 1 |  | 25 −10 | 24:1 13:1 | >98 >98 | 82 92 |
| 2 |  | 25 −10 | 21:1 23:1 | >98 >98 | 82 90 |
| 3 |  | 25 | 19:1 | 61 | 86 |
| 4 |  | 25 | >30:1 | >98 | 76 |
| 5 |  | −10 | 21:1 | >98 | 70 |

| Entry | R | Yield (%) | Z:E | ee (%) |
|---|---|---|---|---|
| 1 | OnBu | 80 | 95:5 | 96 |
| 2 | OCy | 64 | 98:2 | 96 |
| 3 | OPMP | 67 | 95:5 | 94 |
| 4 | OCH2CF3 | 65 | 94:6 | 92 |
| 5 | O(CH2)2Cl | 63 | 95:5 | 96 |
| 6 | SPh | 67 | 91:9 | 92 |
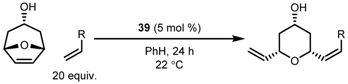
| Entry | Substrate | Yield (%) | Z:E | ee (%) |
|---|---|---|---|---|
| 1 |  | 64 | 95:5 | 93 |
| 2 |  | 58 | 98:2 | 75 |
| 3 | 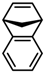 | 55 | 76:24 | >98 |
| 4 | 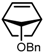 | 56 | 15:85 | 94 |
| 5 |  | 40 | 70:30 | 95 |
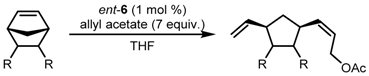
| Entry | R | R’ | Yield (%) | Z:E | ee Z (ee E) (%) |
|---|---|---|---|---|---|
| 1 | H | Bz | 67 | 75:25 | 91 (67) |
| 2 | Bz | H | 69 | 75:25 | 96 (82) |
| 3 | Bn | Ac | 79 | 85:15 | 95 |
| 4 | Bn | CH2C(O)Me | 65 | 90:10 | 92 (84) |

| Entry | Cat | Time (h) | Yield (%) | Z:E |
|---|---|---|---|---|
| 1 | 30 | 1 | 0 | <1:99 |
| 2 | 30 | 2 | 2 | <1:99 |
| 3 | 30 | 4 | 4 | <1:99 |
| 4 | 30 | 72 | 13 | <1:99 |
| 5 | 55 | 1 | 2 | <1:99 |
| 6 | 55 | 2 | 5 | <1:99 |
| 7 | 55 | 4 | 11 | <1:99 |
| 8 | 55 | 72 | 24 | <1:99 |
| 9 | 56 | 1 | 4 | <1:99 |
| 10 | 56 | 2 | 7 | <1:99 |
| 11 | 56 | 4 | 14 | <1:99 |
| 12 | 56 | 72 | 28 | <1:99 |
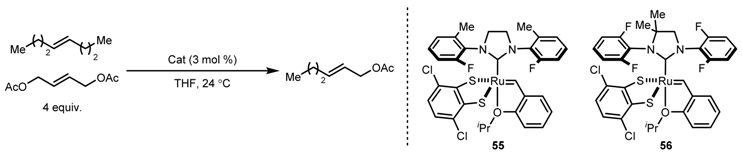
| Entry | m | n | Initial E:Z | Yield (%) | Final E:Z |
|---|---|---|---|---|---|
| 1 | CH2 | CH2 | 90:10 | 77 | >99:1 |
| 2 | CH2OCH2 | CH2OCH2 | 81:19 | 64 | 98:2 |
| 3 | CH2SCH2 | CH2SCH2 | 90:10 | 77 | 99:1 |
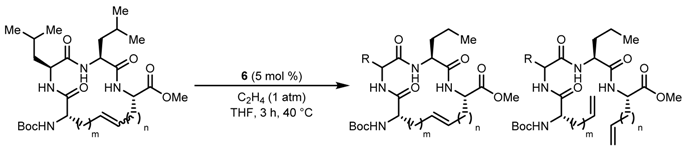
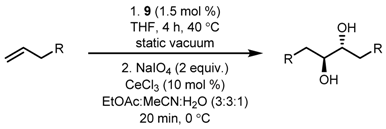
| Entry | R | Yield (%) |
|---|---|---|
| 1 | OCOnPr | 72 |
| 2 | OAc | 59 |
| 3 | OBz | 71 |
| 4 | OCO2Ph | 61 |
| 5 | NHTs | 70 |
| 6 | NHCBz | 53 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
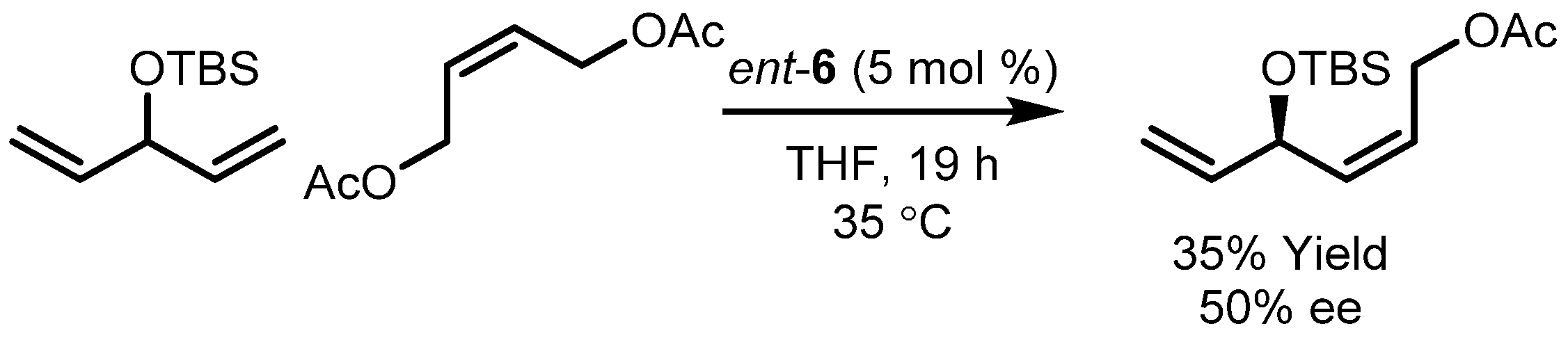{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montgomery, T.P.; Johns, A.M.; Grubbs, R.H. Recent Advancements in Stereoselective Olefin Metathesis Using Ruthenium Catalysts. Catalysts 2017, 7, 87. https://doi.org/10.3390/catal7030087
Montgomery TP, Johns AM, Grubbs RH. Recent Advancements in Stereoselective Olefin Metathesis Using Ruthenium Catalysts. Catalysts. 2017; 7(3):87. https://doi.org/10.3390/catal7030087
Chicago/Turabian StyleMontgomery, T. Patrick, Adam M. Johns, and Robert H. Grubbs. 2017. "Recent Advancements in Stereoselective Olefin Metathesis Using Ruthenium Catalysts" Catalysts 7, no. 3: 87. https://doi.org/10.3390/catal7030087
APA StyleMontgomery, T. P., Johns, A. M., & Grubbs, R. H. (2017). Recent Advancements in Stereoselective Olefin Metathesis Using Ruthenium Catalysts. Catalysts, 7(3), 87. https://doi.org/10.3390/catal7030087