
Solvent-Free Biginelli Reactions Catalyzed by Hierarchical Zeolite Utilizing a Ball Mill Technique: A Green Sustainable Process



 ,
, 
Abstract
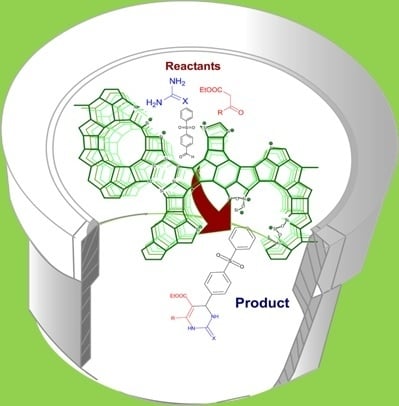
:
1. Introduction
2. Results and Discussions
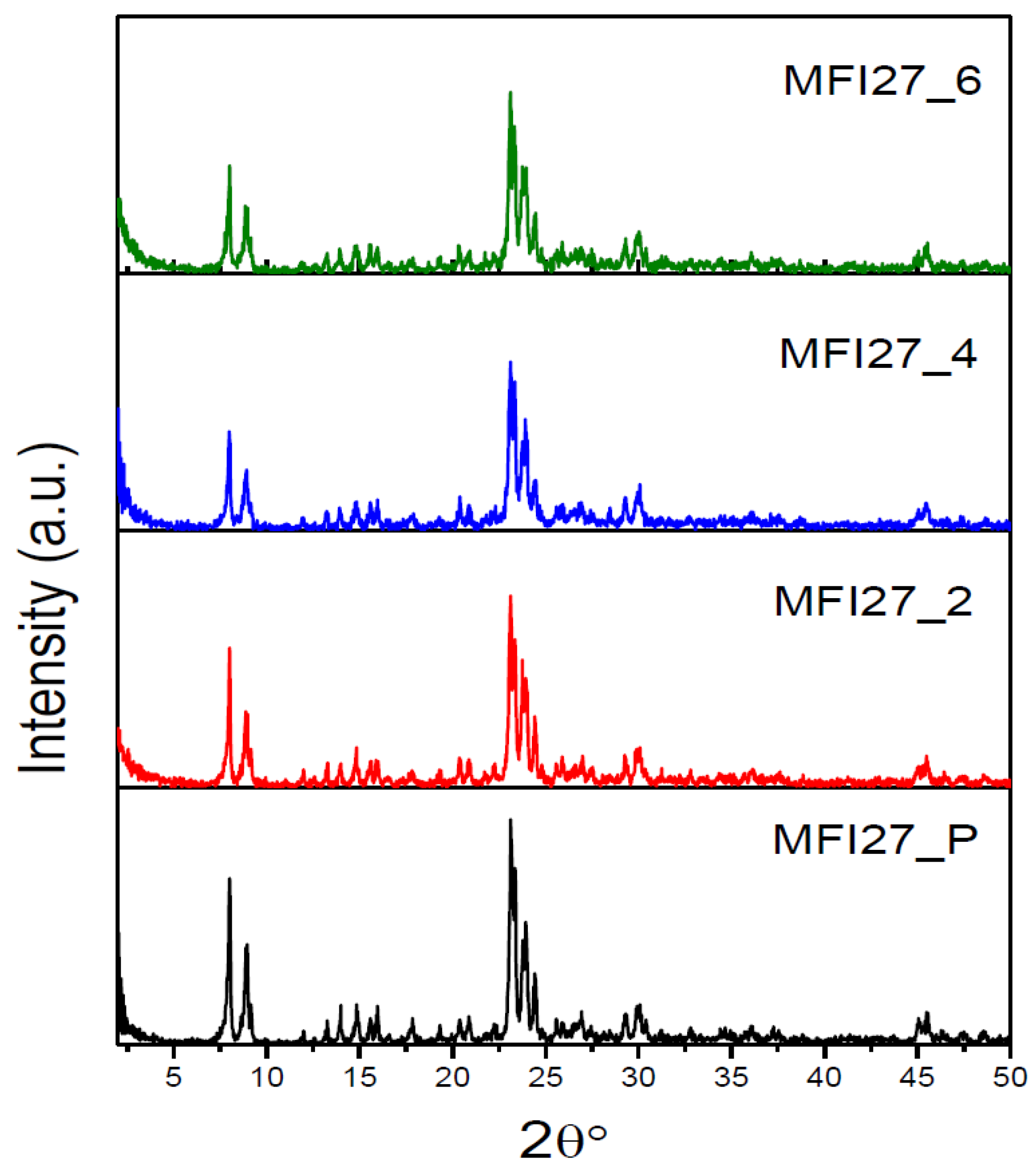
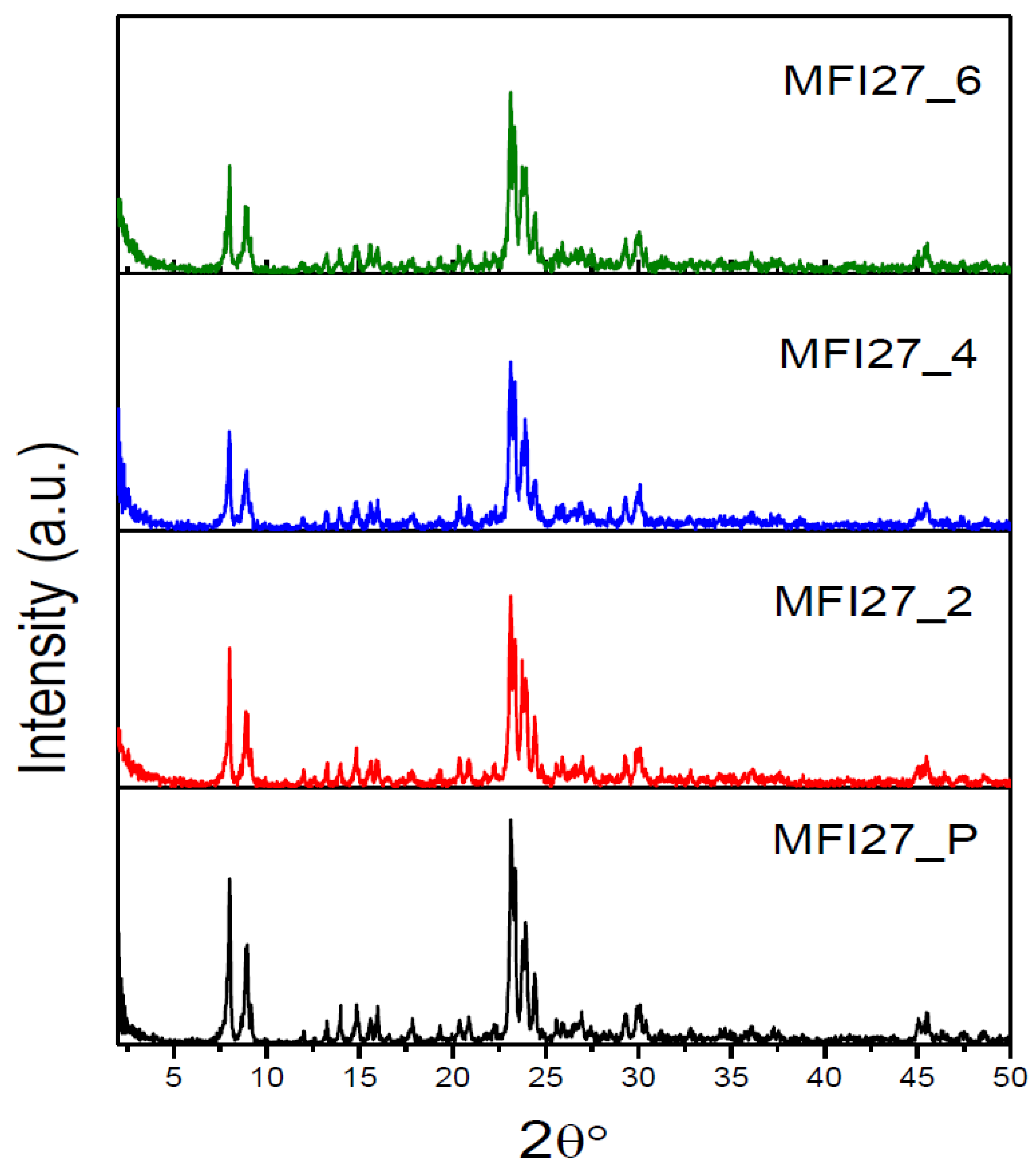
2.1. Powder X-ray Diffraction (PXRD)
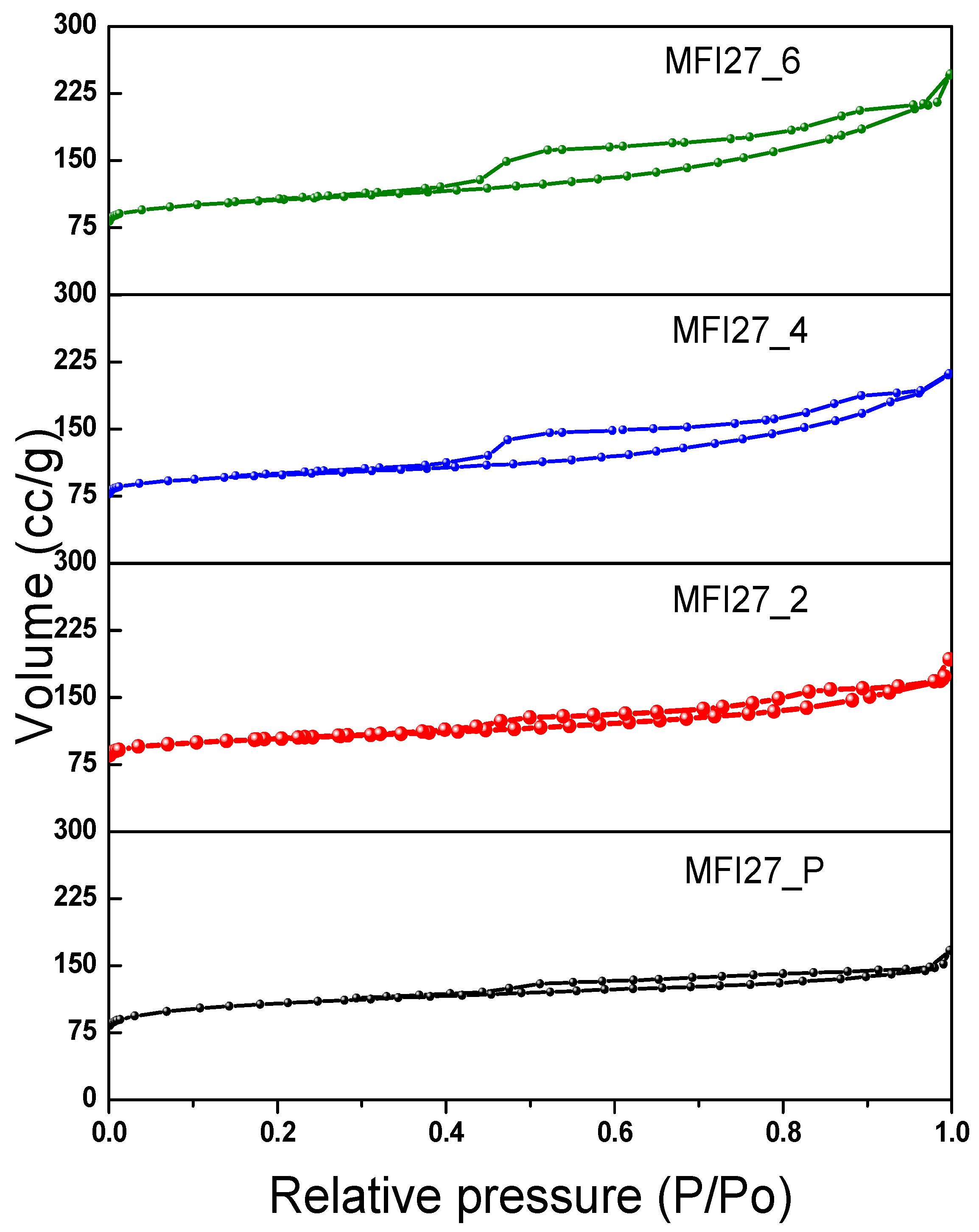
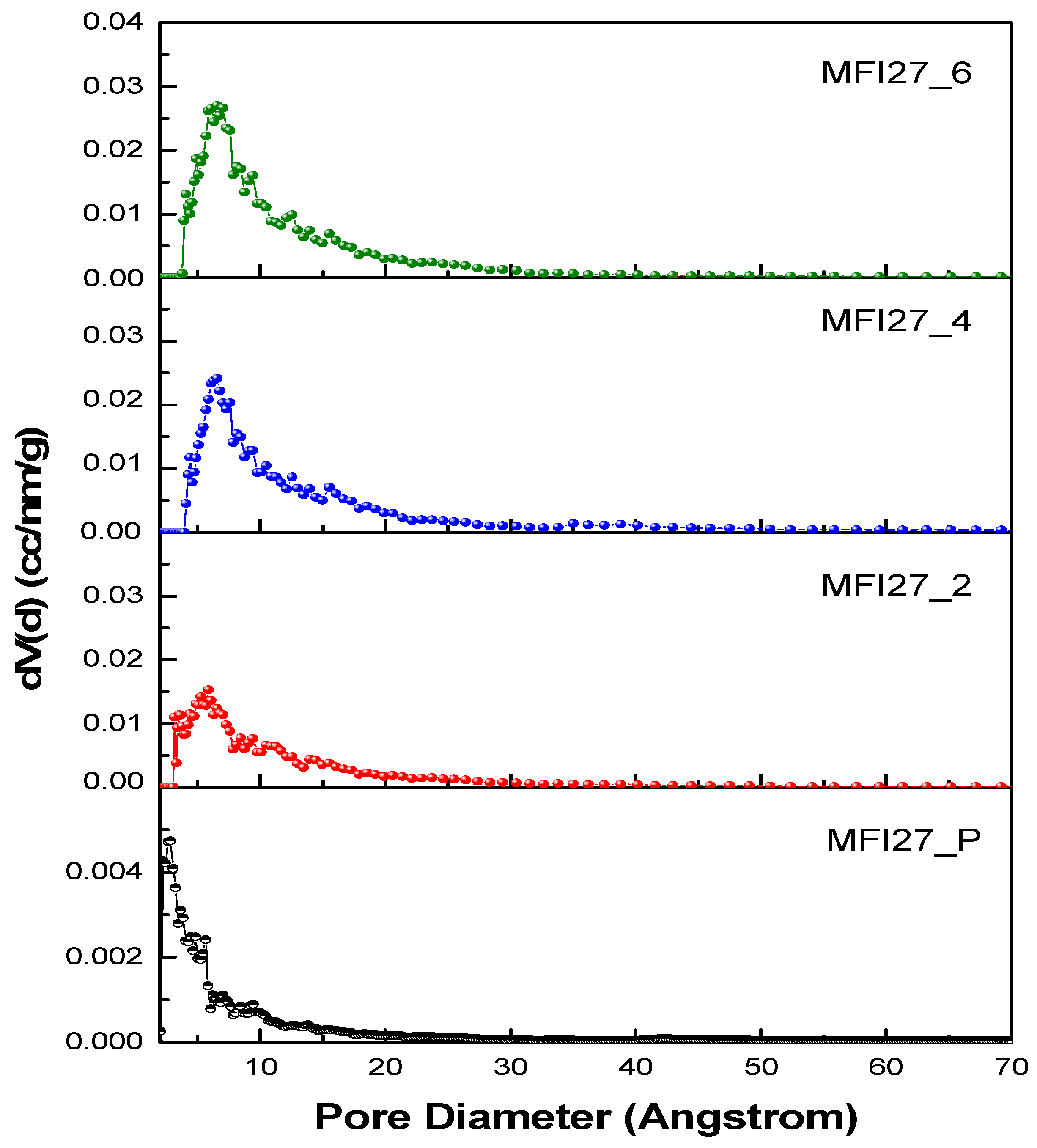
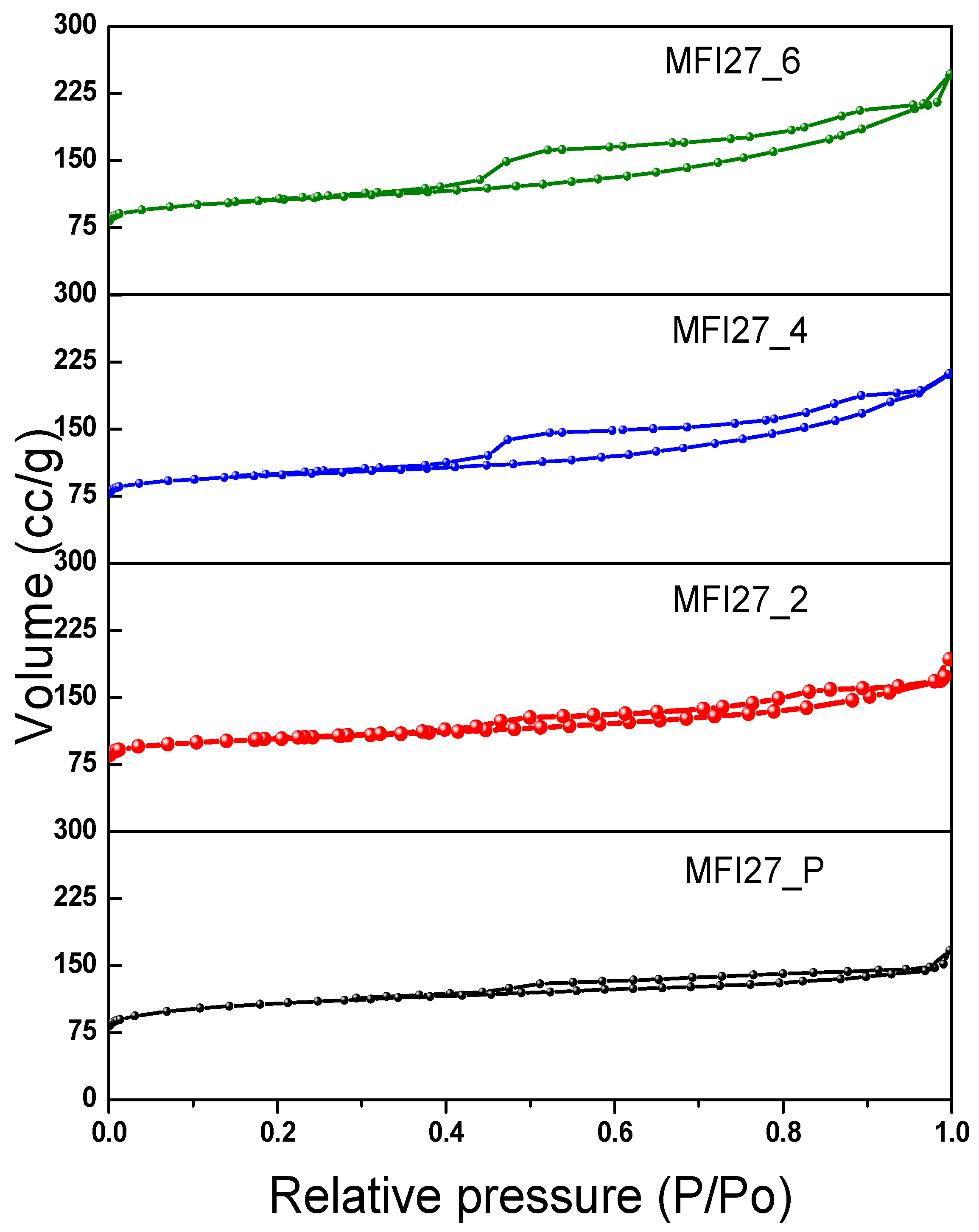
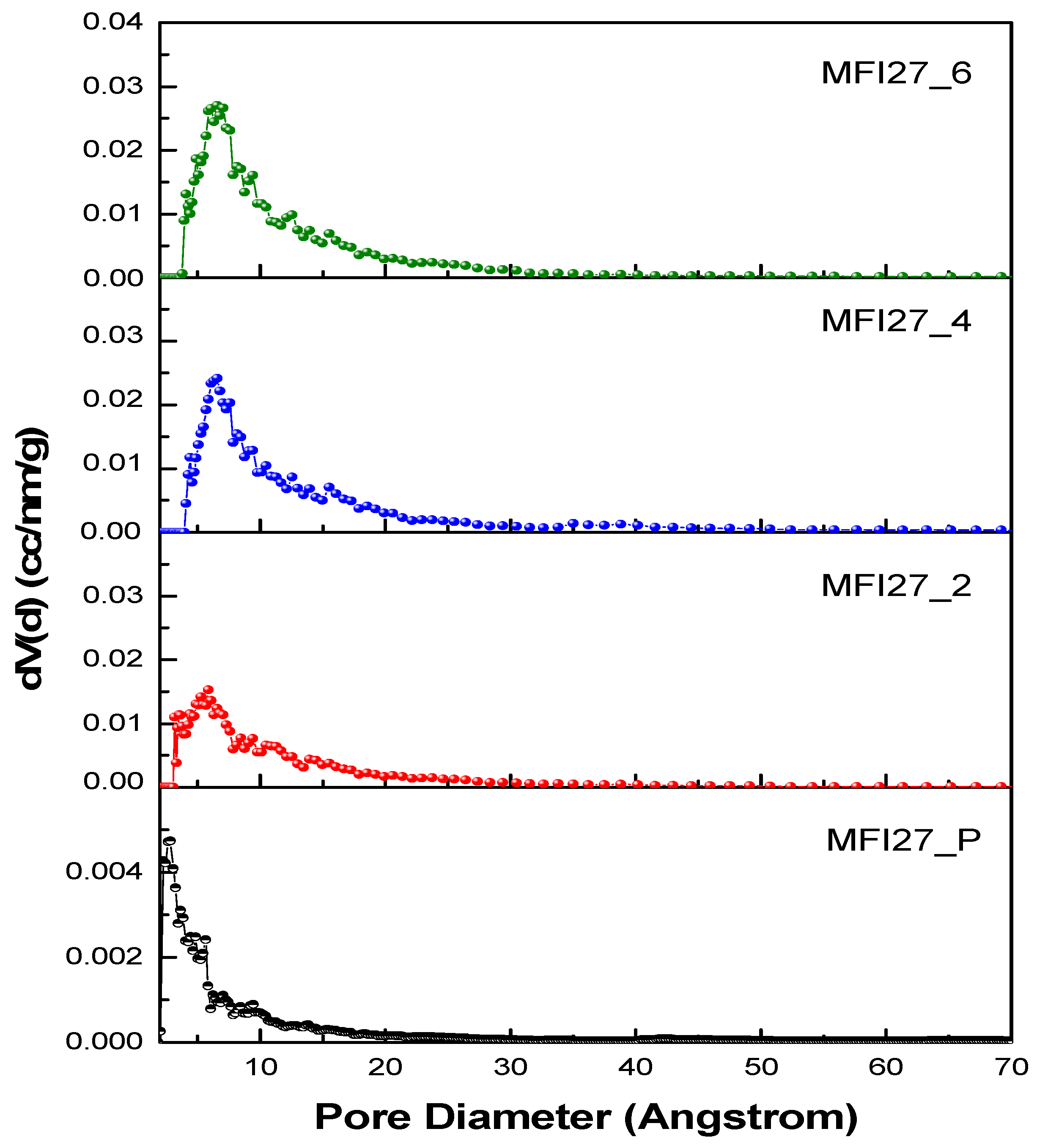
2.2. N2 Physisorption
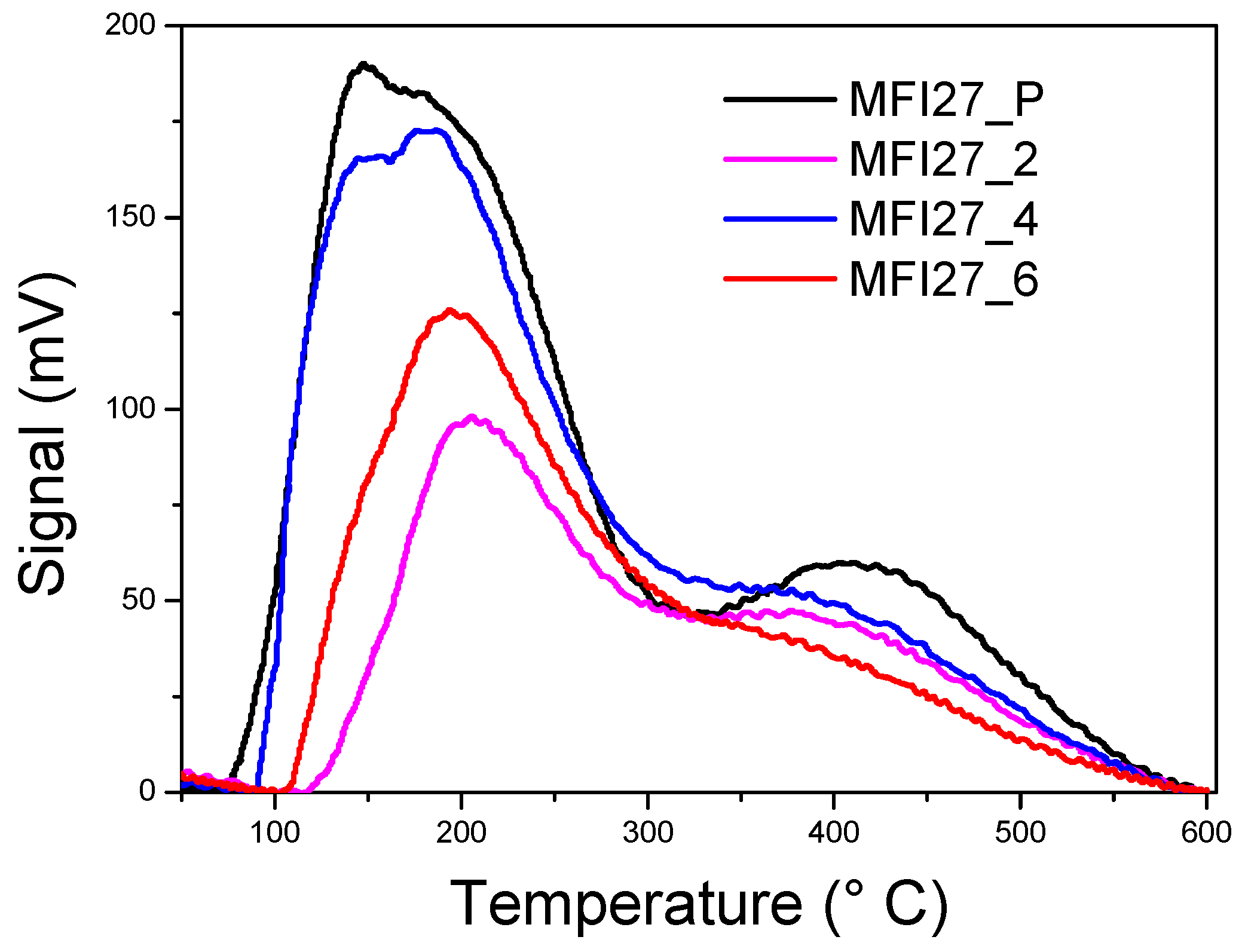
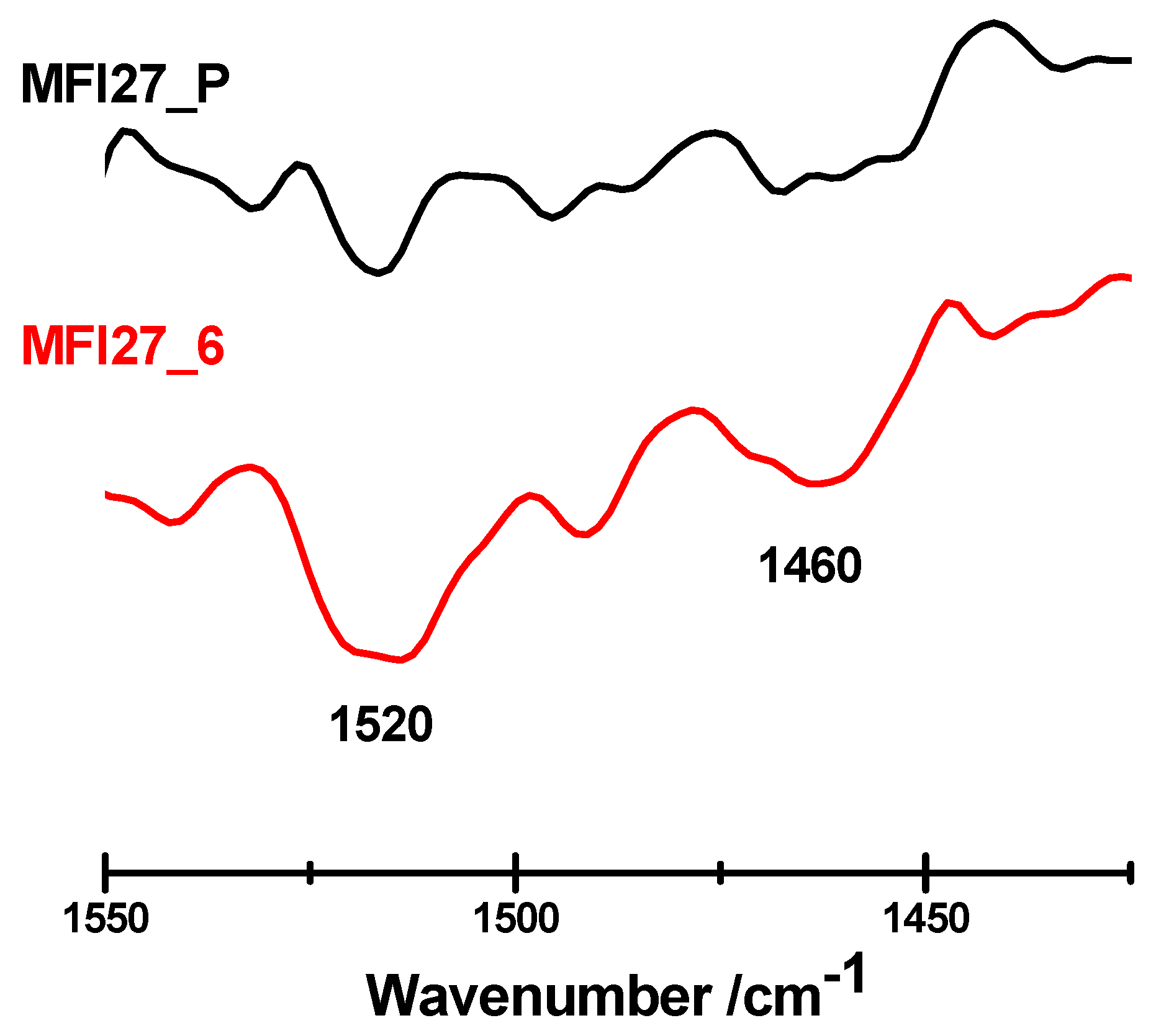
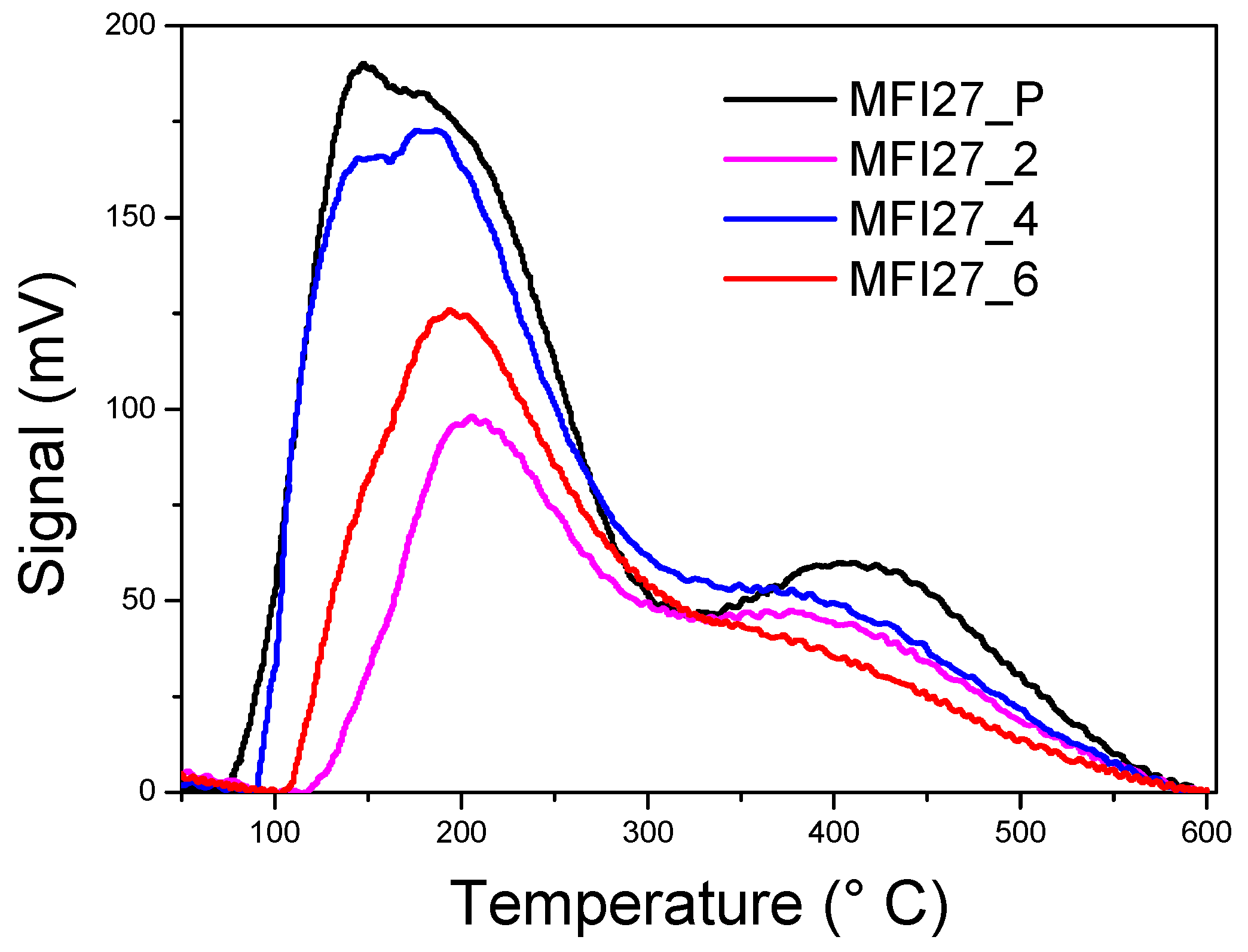
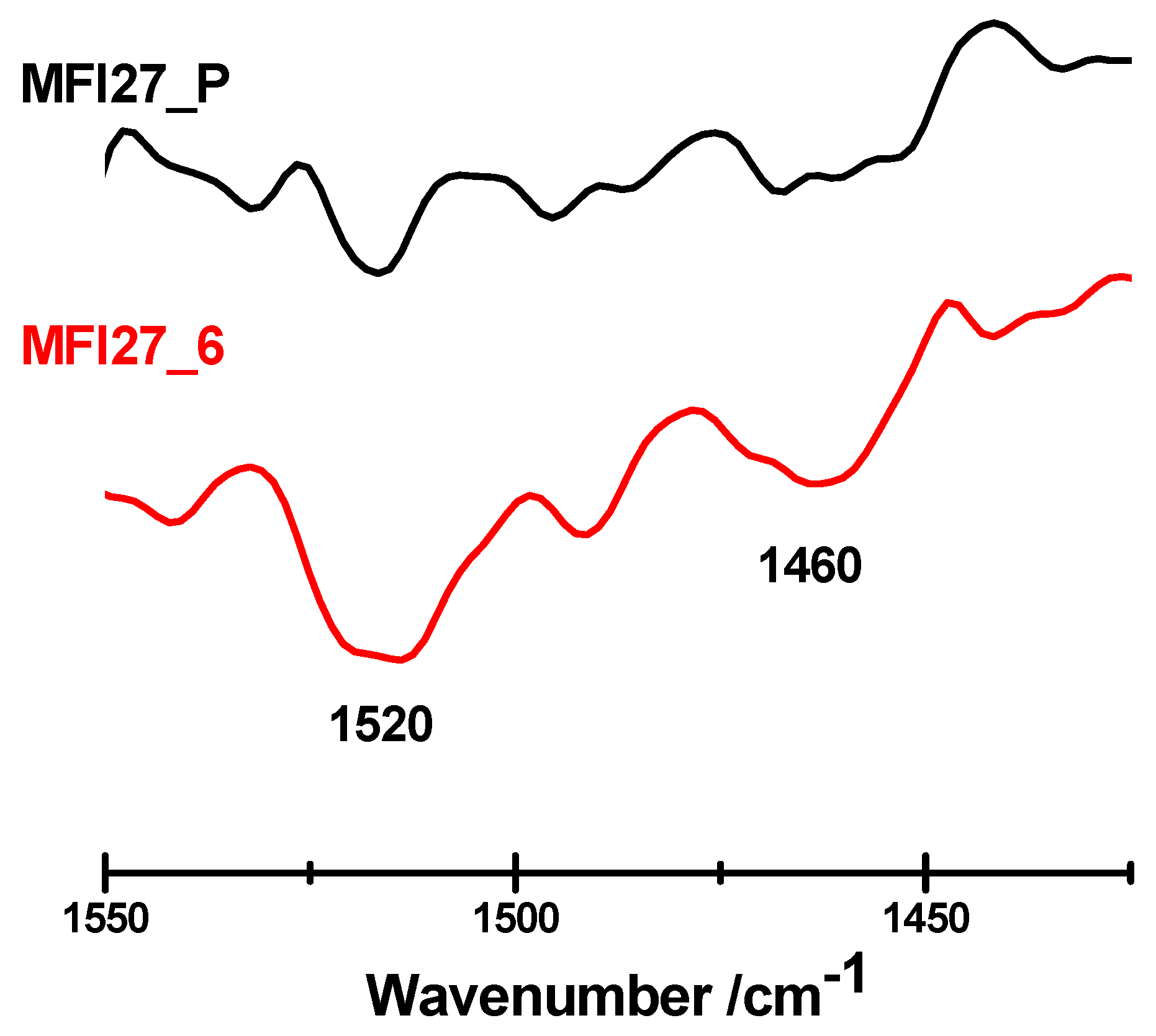
2.3. Evaluation of the Acid Sites
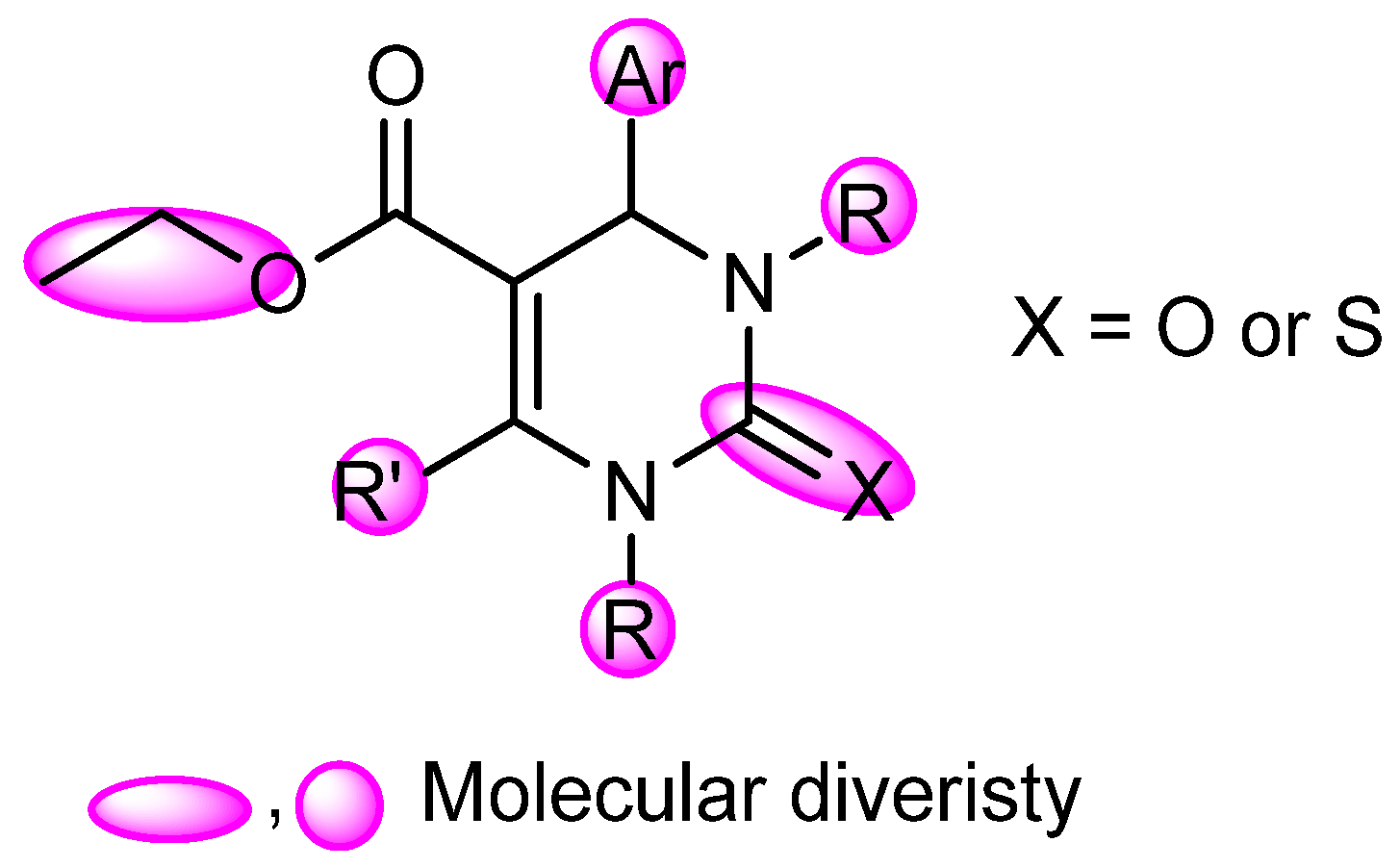
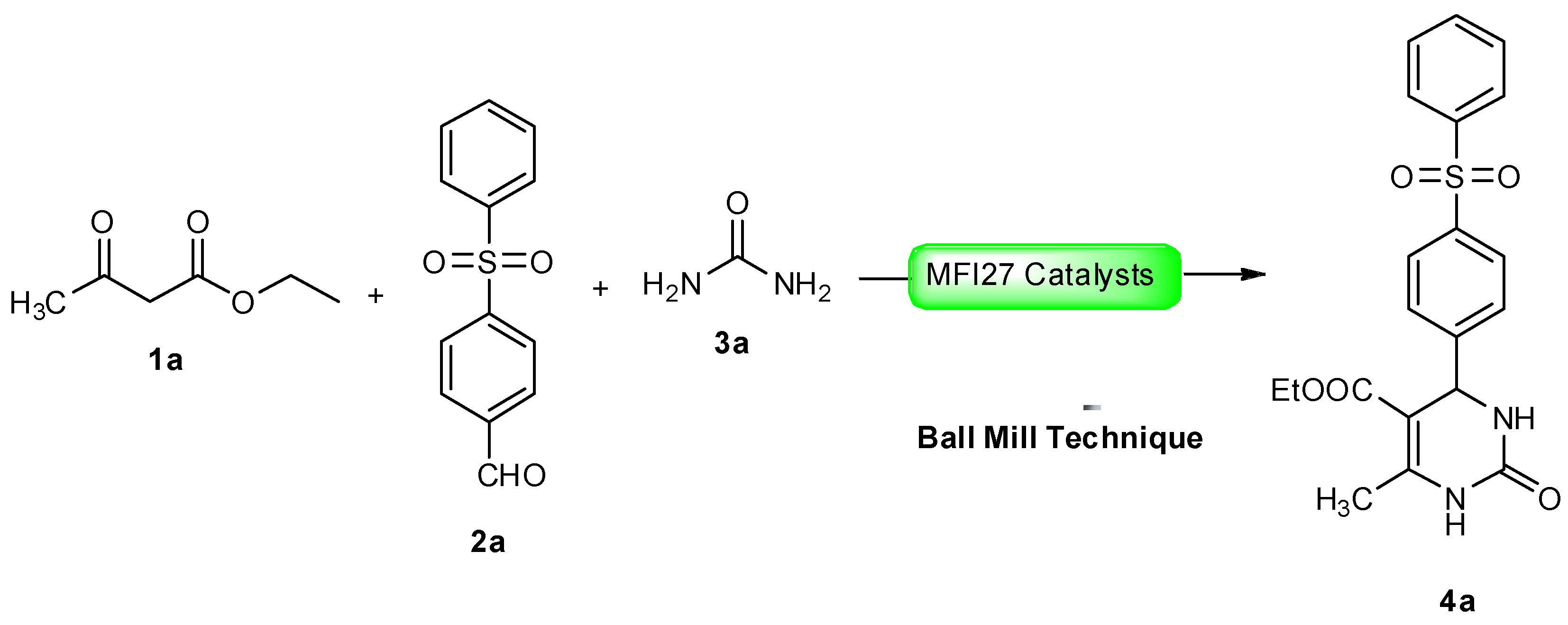
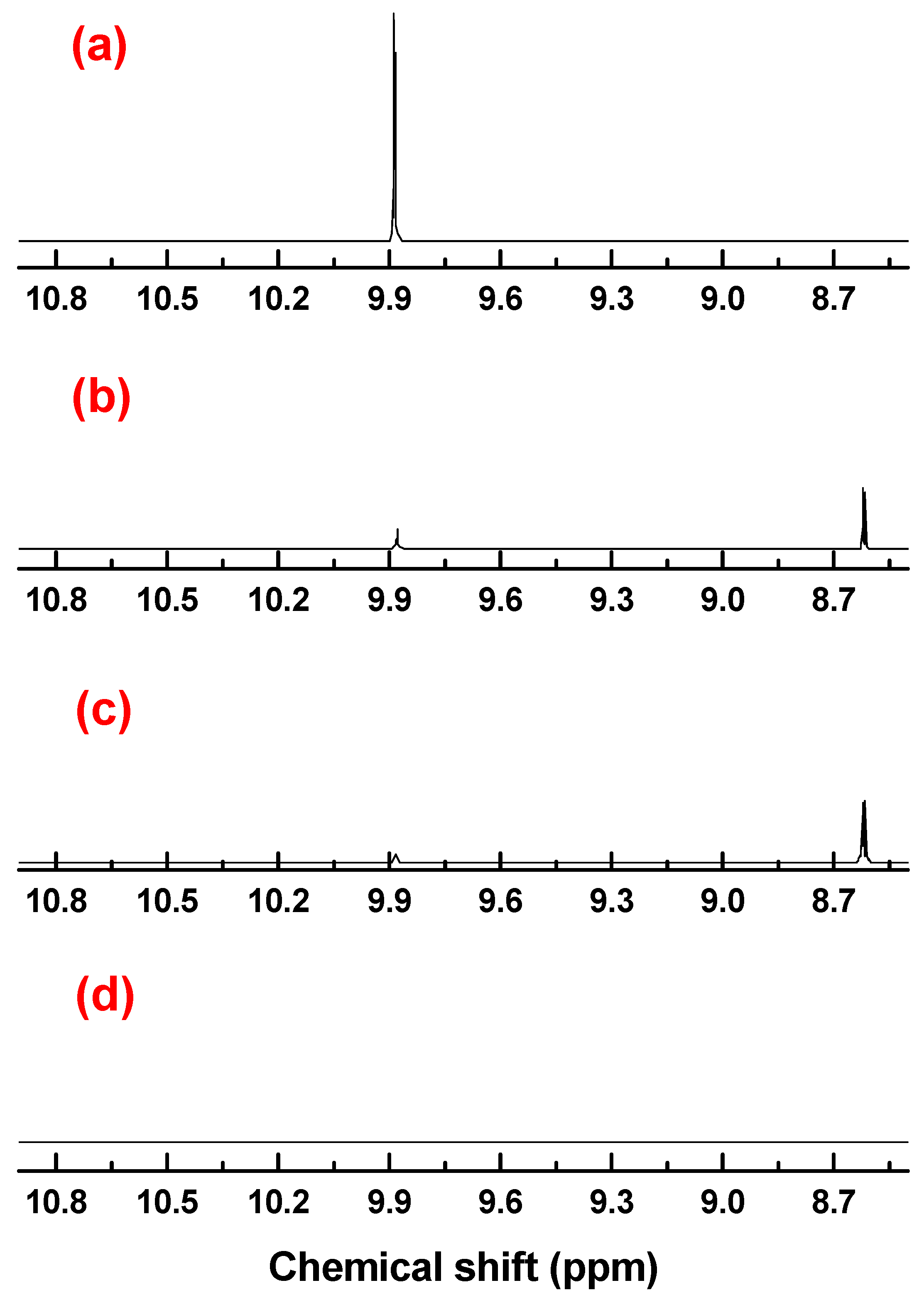
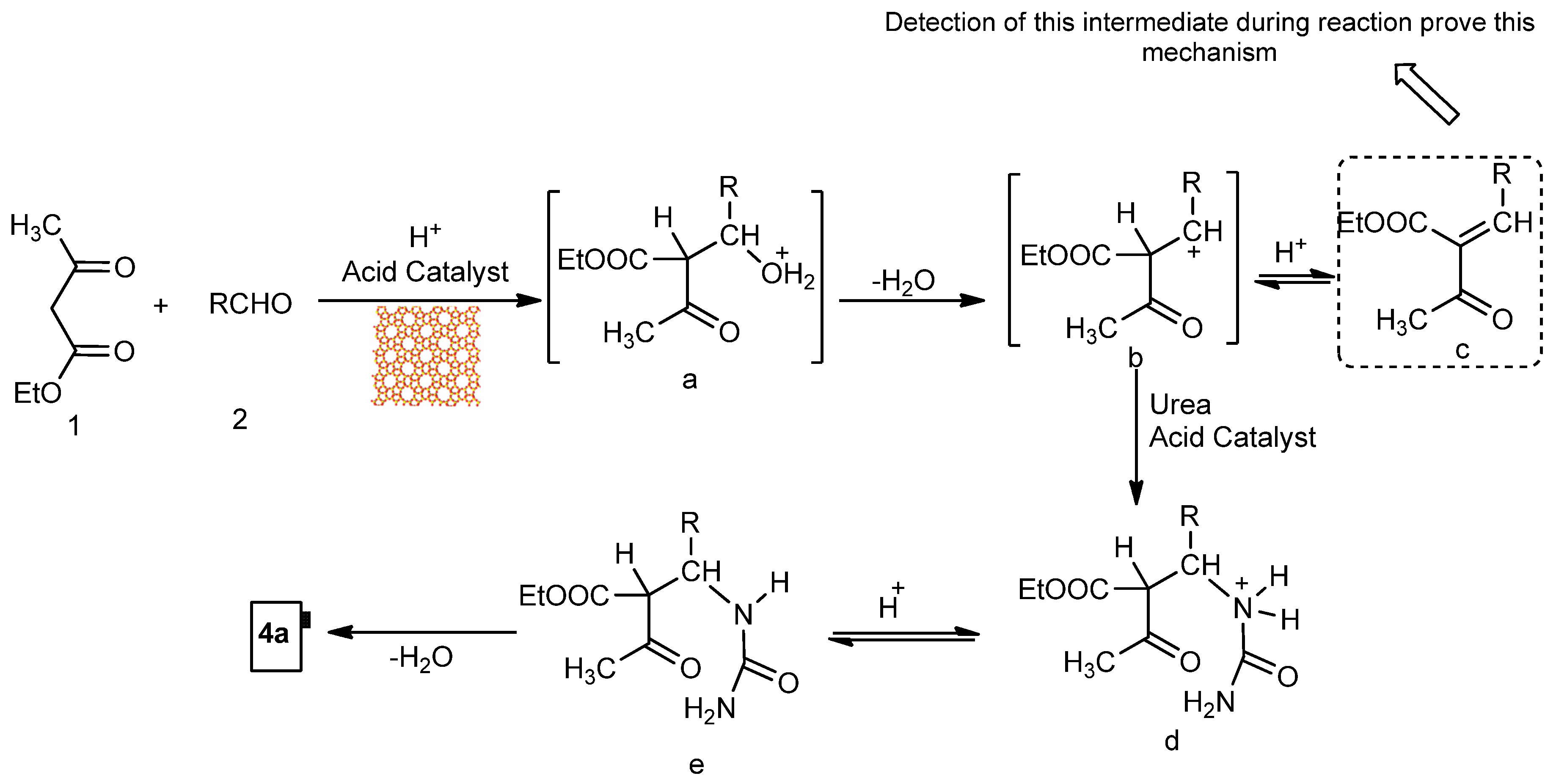
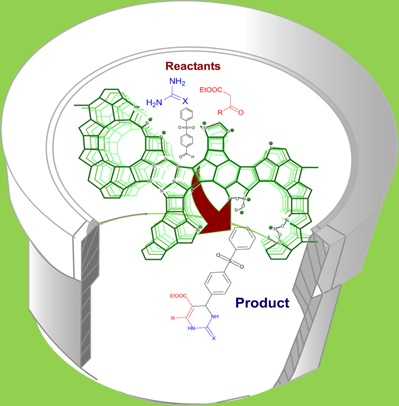
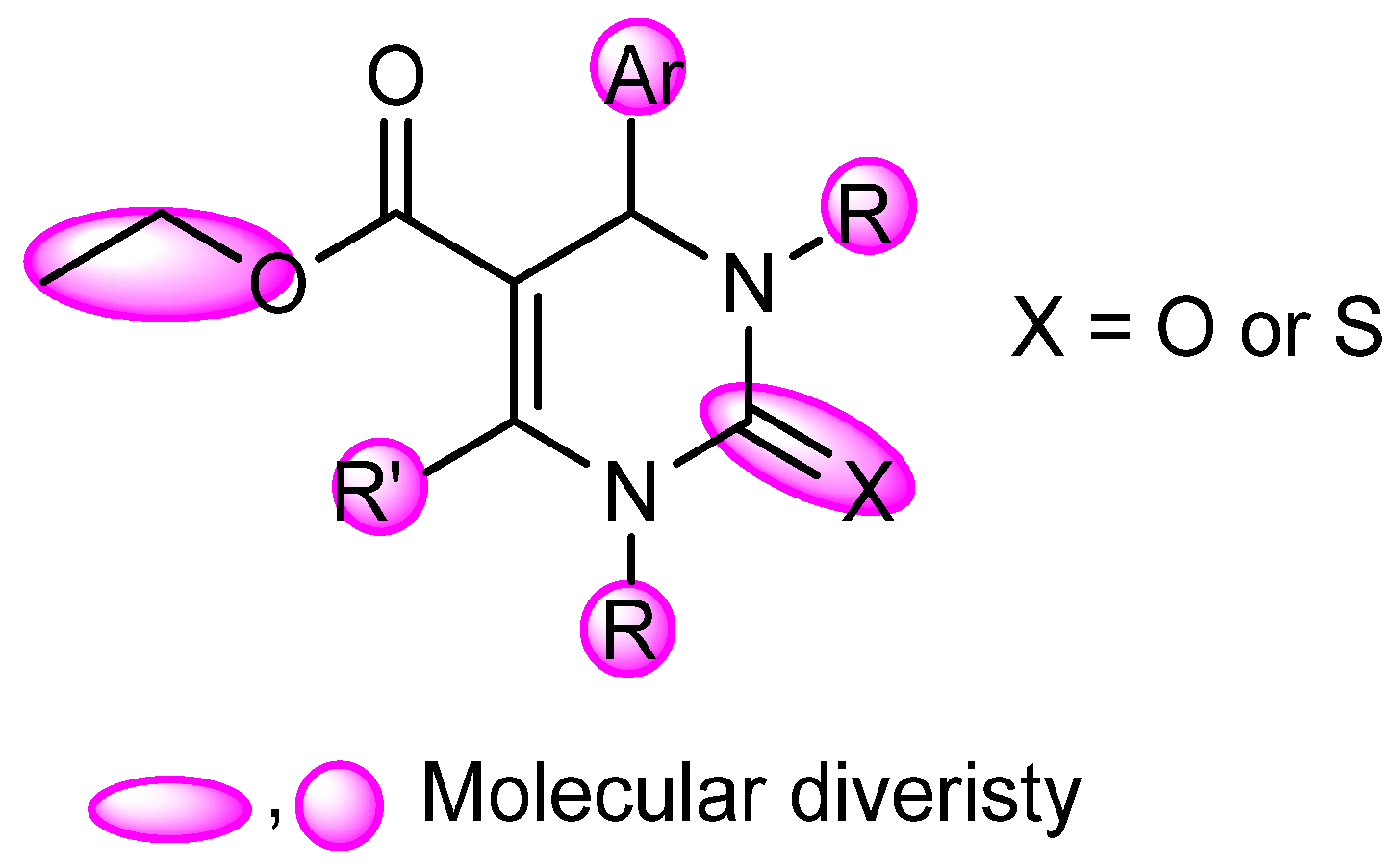
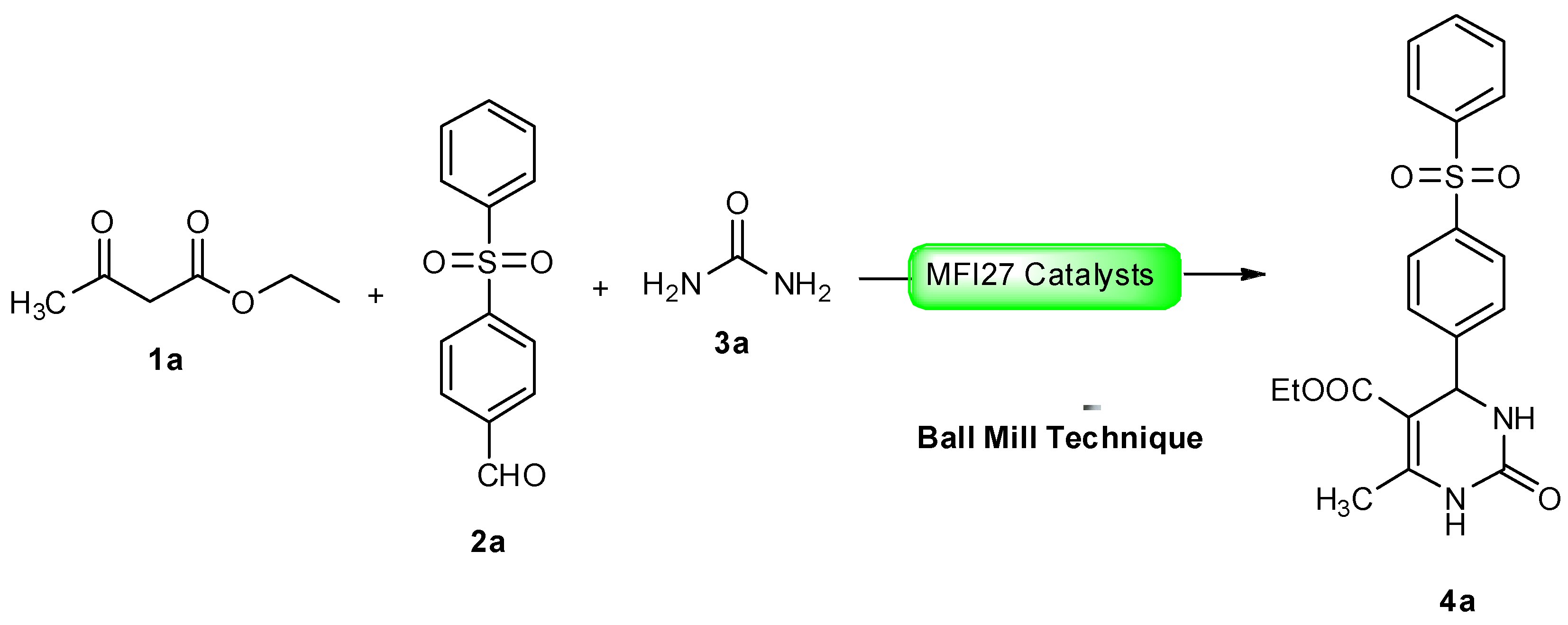
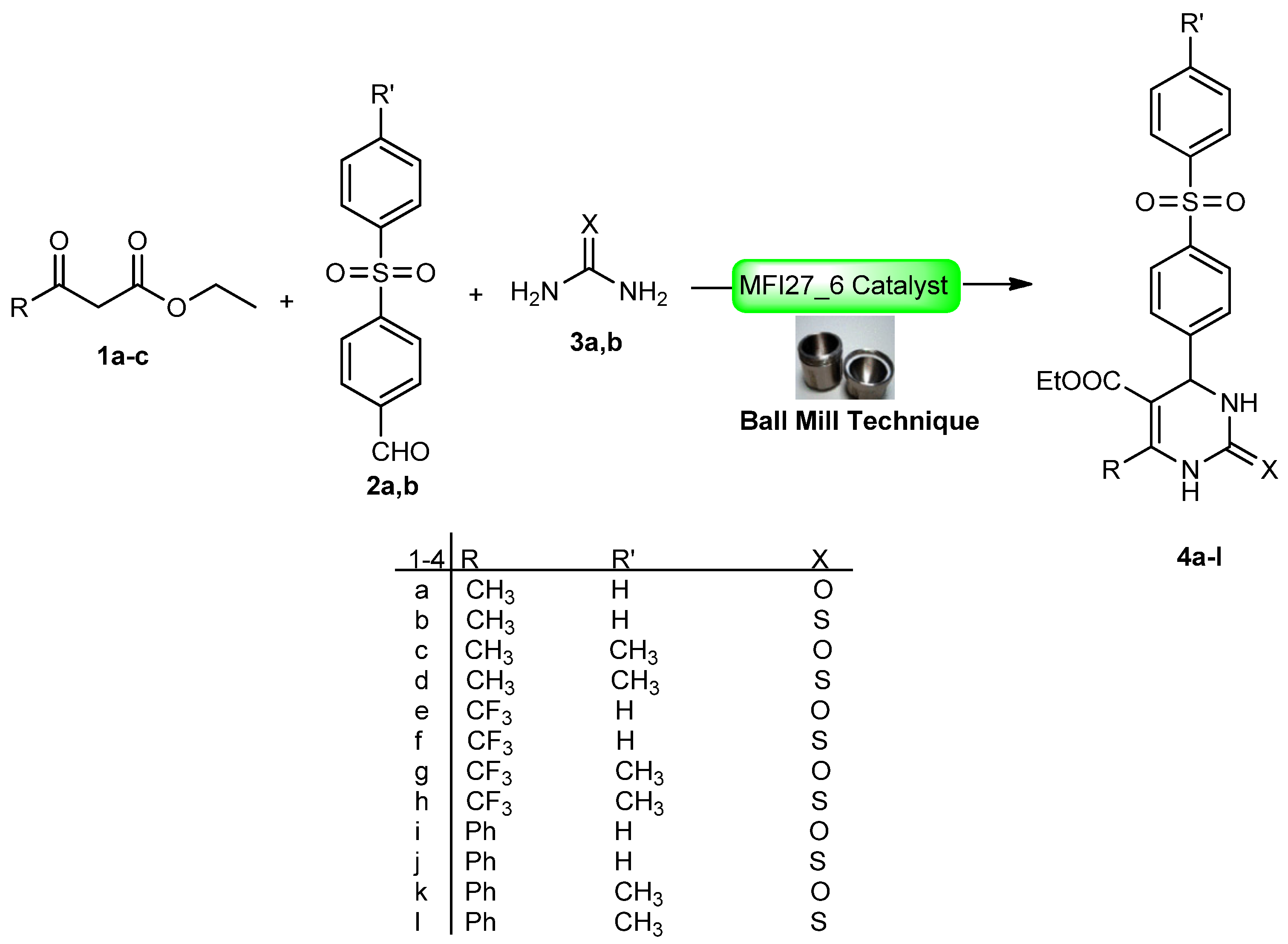
2.4. DHPM Synthesis
3. Experimental
3.1. Materials
3.2. Alkali-Treatment of ZSM-5
3.3. Characterization Techniques
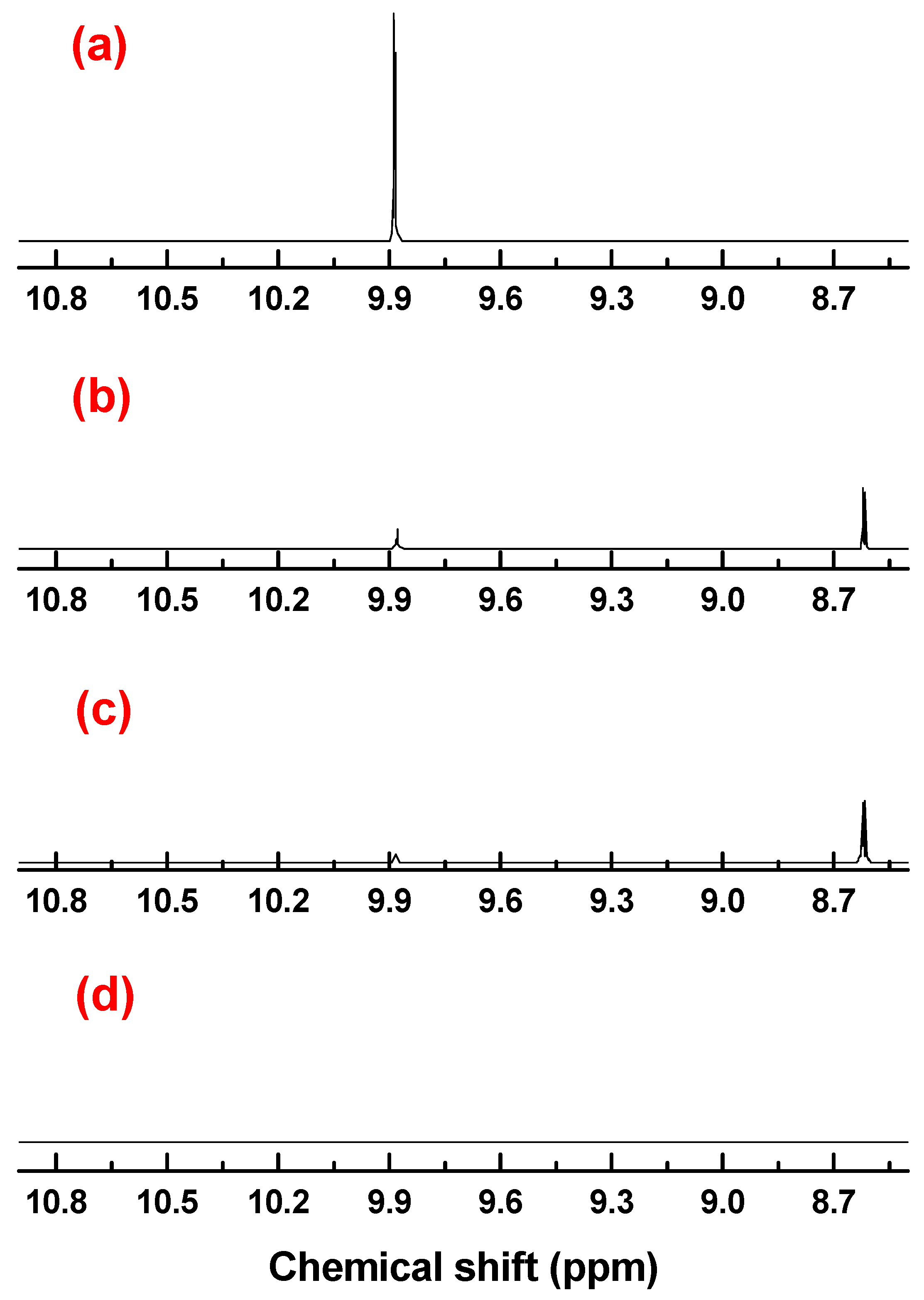
3.4. Characterizations of the Reaction Products
3.5. Typical Procedure for the Catalytic Test Reaction
Physical and spectral data of the title compounds 4a–l are listed below:
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Trost, B.M. The atom economy—A search for synthetic efficiency. Science 1991, 254, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. The E factor: Fifteen years on. Green Chem. 2007, 9, 1273–1283. [Google Scholar] [CrossRef]
- Kappe, C.O. Recent advances in the Biginelli dihydropyrimidine synthesis. New tricks from an old dog. Acc. Chem. Res. 2000, 33, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.O. The Biginelli reaction: Development and applications. Tetrahedron 1993, 49, 6937–7168. [Google Scholar]
- Kappe, C.O. Synthesis of octahydroquinazolinone derivatives using silica sulfuric acid as an efficient catalyst. Eur. J. Med. Chem. 2000, 35, 1043–1052. [Google Scholar] [CrossRef]
- Rovnyak, G.C.; Kimball, S.D.; Beyer, B.; Cucinotta, G.; DiMarco, J.D.; Gougoutas, J.; Hedberg, A.; Malley, M.; McCarthy, J.P.; Zhang, R. Calcium entry blockers and activators: Conformational and structural determinants of dihydropyrimidine calcium channel modulators. J. Med. Chem. 1995, 38, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Atwal, K.S.; Swanson, B.N.; Unger, S.E.; Floyd, D.M.; Moreland, S.; Hedberg, A.; O’Reilly, B.C. Dihydropyrimidine calcium channel blockers. 3. 3-Carbamoyl-4-aryl-1,2,3,4-tetrahydro-6-methyl-5-pyrimidinecarboxylic acid esters as orally effective antihypertensive agents. J. Med. Chem. 1991, 34, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Grover, G.J.; Dzwomczyk, S.; McMullen, D.M.; Normandin, D.E.; Parham, C.S.; Sleph, P.G.; Moreland, S. Pharmacologic profile of the dihydropyrimidine calcium channel blockers SQ 32,547 and SQ 32,946. J. Cardiovasc. Pharmacol. 1995, 26, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.O.; Fabian, W.M.F.; Semones, M.A. Conformational analysis of 4-aryl-dihydropyrimidine calcium channel modulators. A comparison of ab initio, semiempirical and X-ray crystallographic studies. Tetrahedron 1997, 53, 2803–2816. [Google Scholar] [CrossRef]
- Atwal, K.S.; Rovnyak, G.C.; Kimball, S.D.; Floyd, D.M.; Moreland, S.; Swanson, B.N.; Gougoutas, J.Z.; Schwartz, J.; Smillie, K.M.; Malley, M.F. Dihydropyrimidine calcium channel blockers. II. 3-Substituted-4-aryl-1,4-dihydro-6-methyl-5-pyrimidinecarboxylic acid esters as potent mimics of dihydropyridines. J. Med. Chem. 1990, 33, 2629–2635. [Google Scholar] [CrossRef] [PubMed]
- Atwal, K.S.; Rovnyak, G.C.; O’Reilly, B.C.; Schwartz, J. Substituted 1,4-dihydropyrimidines. 3. Synthesis of selectively functionalized 2-hetero-1,4-dihydropyrimidines. J. Org. Chem. 1989, 54, 5898–5907. [Google Scholar] [CrossRef]
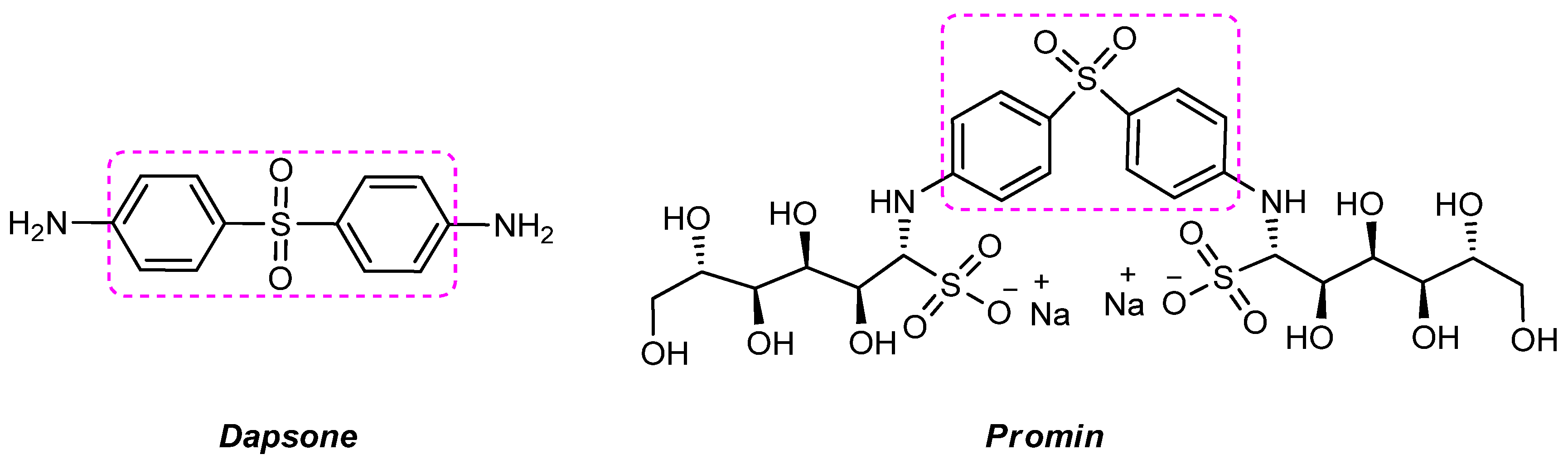
- El-Gaby, M.S.; Ali, G.A.E.H.; El-Maghraby, A.A.; El-Rahman, M.T.A.; Helal, M.H. Synthesis, characterization and in vitro antimicrobial activity of novel 2-thioxo-4-thiazolidinones and 4,4′-bis(2-thioxo-4-thiazolidinone-3-yl)diphenylsulfones. Eur. J. Med. Chem. 2009, 44, 4148–4152. [Google Scholar] [CrossRef] [PubMed]
- Wolf, R.; Matz, H.; Orion, E.; Tuzun, B.; Tuzun, Y. Dapsone: Unapproved uses or indications. Clin. Dermatol. 2000, 18, 37–53. [Google Scholar] [CrossRef]
- GFaget, H.; Pogge, R.C.; Johansen, F.A.; Dinan, J.F.; Prejean, B.M.; Eccles, C.G. The promin treatment of leprosy. A progress report. Int. J. Lepr. Other Mycobact. Dis. 1966, 34, 298–310. [Google Scholar]
- Barbuceanu, S.F.; Saramet, I.; Almajan, G.L.; Draghici, C.; Barbuceanu, F.; Bancescu, G. New heterocyclic compounds from 1,2,4-triazole and 1,3,4-thiadiazole class bearing diphenylsulfone moieties. Synthesis, characterization and antimicrobial activity evaluation. Eur. J. Med. Chem. 2012, 49, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Barbuceanu, S.F.; Almajan, G.L.; Saramet, I.; Draghici, C.; Socoteanu, R.; Barbuceanu, F.; Bancescu, G. New S-alkylated 1,2,4-triazoles incorporating diphenyl sulfone moieties with potential antibacterial activity. J. Serbian Chem. Soc. 2009, 74, 1041–1049. [Google Scholar] [CrossRef]
- Barbuceanu, S.F.; Almajan, G.L.; Saramet, I.; Draghici, C.; Tarcomnicu, A.I.; Bancescu, G. Synthesis, characterization and evaluation of antibacterial activity of some thiazolo[3,2-b][1,2,4]triazole incorporating diphenylsulfone moieties. Eur. J. Med. Chem. 2009, 44, 4752–4757. [Google Scholar] [CrossRef] [PubMed]
- Almajan, G.L.; Barbuceanu, S.F.; Almajan, E.R.; Draghici, C.; Saramet, I. Synthesis, characterization and antibacterial activity of some triazole Mannich bases carrying diphenylsulfone moieties. Eur. J. Med. Chem. 2009, 44, 3083–3089. [Google Scholar] [CrossRef] [PubMed]
- Almajan, G.L.; Barbuceanu, S.F.; Innocenti, A.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Inhibition of the cytosolic and tumor-associated carbonic anhydrase isozymes I, II and IX with some 1,3,4-oxadiazole- and 1,2,4-triazole-thiols. J. Enzyme Inhib. Med. Chem. 2008, 23, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Brindaban, A.; Jana, J.U. Indium(III) chloride-catalyzed one-pot synthesis of dihydropyrimidinones by a three-component coupling of 1,3-dicarbonyl compounds, aldehydes, and urea: An improved procedure for the Biginelli reaction. J. Org. Chem. 2000, 65, 6270–6272. [Google Scholar]
- Lu, J.; Bai, Y.; Wang, Z.; Yang, B.; Ma, H. One-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones using lanthanum chloride as a catalyst. Tetrahedron Lett. 2000, 41, 9075–9078. [Google Scholar] [CrossRef]
- Hu, E.H.; Sidler, D.R.; Dolling, U.H. Unprecedented catalytic three component one-pot condensation reaction: An efficient synthesis of 5-alkoxycarbonyl-4-aryl-3,4-dihydropyrimidin-2(1H)-ones. J. Org. Chem. 1998, 63, 3454–3457. [Google Scholar] [CrossRef]
- Liu, C.; Wang, J.; Li, Y. One-pot synthesis of 3,4-dihydropyrimidin-2(1H)-(thio)ones using strontium(II) nitrate as a catalyst. J. Mol. Catal. A 2006, 258, 367–370. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, B.V.S.; Reddy, K.B.; Raj, K.S.; Prasad, A.R. Ultrasound-accelerated synthesis of 3,4-dihydropyrimidin-2(1H)-ones with ceric ammonium nitrate. J. Chem. Soc. Perkin Trans. 2001, 1, 1939–1941. [Google Scholar] [CrossRef]
- Al-Mutairi, E.; Narasimharao, K.; Mokhtar, M. Heteropolyacid generated on the surface of iron phosphate nanotubes: Structure and catalytic activity studies. RSC Adv. 2015, 5, 63917–63929. [Google Scholar] [CrossRef]
- Maradur, S.P.; Gokavi, G.S. Heteropoly acid catalyzed synthesis of 3,4-dihydropyrimidin-2(1H)-ones. Catal. Commun. 2007, 8, 279–284. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V. Catalysis by heteropoly acids and multicomponent polyoxometalates in liquid-phase reactions. Chem. Rev. 1998, 98, 171–198. [Google Scholar] [CrossRef] [PubMed]
- Miras, H.N.; Yan, J.; Long, D.L.; Cronin, L. Engineering polyoxometalates with emergent properties. Chem. Soc. Rev. 2012, 41, 7403–7430. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Hu, C. Heterogeneous photocatalysis by solid polyoxometalates. J. Mol. Catal. A 2007, 262, 136–148. [Google Scholar] [CrossRef]
- Watson, B.A.; Barteau, M.A.; Haggerty, L.; Lenhoff, A.M.; Weber, R.S. Scanning tunneling microscopy and tunneling spectroscopy of ordered hetero- and isopolyanion arrays on graphite. Langmuir 1992, 8, 1145–1148. [Google Scholar] [CrossRef]
- Sun, C.Y.; Liu, S.X.; Liang, D.D.; Shao, K.Z.; Ren, Y.H.; Su, Z.M. Highly stable crystalline catalysts based on a microporous metal–organic framework and polyoxometalates. J. Am. Chem. Soc. 2009, 131, 1883–1888. [Google Scholar] [CrossRef] [PubMed]
- Bigi, F.; Carloni, S.; Frullanti, B.; Maggi, R.; Sartori, G. A revision of the Biginelli reaction under solid acid catalysis. Solvent-free synthesis of dihydropyrimidines over montmorillonite KSF. Tetrahedron Lett. 1999, 40, 3465–3468. [Google Scholar] [CrossRef]
- Rani, V.R.; Srinivas, N.; Kishan, M.R.; Kulkarni, S.J.; Raghavan, K.V. Zeolite-catalyzed cyclocondensation reaction for the selective synthesis of 3,4-dihydropyrimidin-2(1H)-ones. Green Chem. 2001, 3, 305–306. [Google Scholar] [CrossRef]
- Tajbakhsh, M.; Mohajerani, B.; Heravi, M.M.; Ahmadi, A.N. Natural HEU type zeolite catalyzed Biginelli reaction for the synthesis of 3,4-dihydropyrimidin-2(1H) one derivatives. J. Mol. Catal. A 2005, 235, 216–219. [Google Scholar] [CrossRef]
- Zendehdel, M.; Mohbinikhaledi, A.; Asgari, A. Zeolite an efficient catalyst for the Biginelli condensation reaction. J. Incl. Phenom. Macrocycl. Chem. 2008, 60, 353–357. [Google Scholar] [CrossRef]
- Ogura, M.; Shinomiya, S.Y.; Tateno, J.; Nara, Y.; Nomura, M.; Kikuchi, E.; Matsukata, M. Alkali-treatment technique-new method for modification of structural and acid-catalytic properties of ZSM-5 zeolites. Appl. Catal. A 2001, 219, 33–43. [Google Scholar] [CrossRef]
- Groen, J.C.; Zhu, W.; Brouwer, S.; Huynink, S.J.; Kapteijn, F.; Moulijn, J.A.; Pérez-Ramírez, J. Direct demonstration of enhanced diffusion in mesoporous ZSM-5 zeolite obtained via controlled desilication. J. Am. Chem. Soc. 2007, 129, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Meunier, F.C.; Verboekend, D.; Gilson, J.P.; Groen, J.C.; Pérez-Ramírez, J. Influence of crystal size and probe molecule on diffusion in hierarchical ZSM-5 zeolites prepared by desilication. Microporous Mesoporous Mater. 2012, 148, 115–121. [Google Scholar] [CrossRef]
- Christensen, C.H.; Johannsen, K.; Törnqvist, E.; Schmidt, I.; Topsøe, H.; Christensen, C.H. Mesoporous zeolite single crystal catalysts: Diffusion and catalysis in hierarchical zeolites. Catal. Today 2007, 128, 117–122. [Google Scholar] [CrossRef]
- Tanaka, K. Solvent-free Organic Synthesis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Kaupp, G. Waste-free synthesis and production all across chemistry with the benefit of self-assembled crystal packings. J. Phys. Org. Chem. 2008, 21, 630–643. [Google Scholar] [CrossRef]
- Sahoo, P.K.; Bose, A.; Mal, P. Solvent-free ball-milling Biginelli reaction by subcomopnent synthesis. Eur. J. Org. Chem. 2015, 2015, 6994–6998. [Google Scholar] [CrossRef]
- Fernandez, C.; Stan, I.; Gilson, J.P.; Thomas, K.; Vicente, A.; Bonila, A.; Pérez-Ramírez, J. Hierarchical ZSM-5 zeolites in shape-selective xylene isomerization: Role of mesoporosity and acid site speciation. Chem. Eur. J. 2010, 16, 6224–6233. [Google Scholar] [CrossRef] [PubMed]
- Van Donk, S.; Janssen, A.H.; Bitter, J.H.; de Jong, K.P. Generation, characterization, and impact of mesopores in zeolite catalysts. Catal. Rev. 2003, 45, 297–319. [Google Scholar] [CrossRef]
- Cizmek, A.; Subotic, B.; Aiello, R.; Crea, F.; Nastro, A.; Tuoto, C. Dissolution of high-silica zeolites in alkaline solutions I. Dissolution of silicalite-1 and ZSM-5 with different aluminum content. Microporous Mater. 1995, 4, 159–168. [Google Scholar] [CrossRef]
- JAhn, H.; Kolvenbach, R.; Neudeck, C.; Al-Khattaf, S.S.; Jentys, A.; Lercher, J.A. Tailoring mesoscopically structured H-ZSM5 zeolites for toluene methylation. J. Catal. 2014, 311, 271–280. [Google Scholar]
- Wang, R.; Liu, Z.-Q. Solvent-free and catalyst-free Biginelli reaction to synthesize ferrocenoyl dihydropyrimidine and kinetic method to express radical-scavenging ability. Org. Chem. 2012, 77, 3952–3958. [Google Scholar] [CrossRef] [PubMed]
- Oliverio, M.; Costanzo, P.; Nardi, M.; Rivalta, I.; Procopio, A. Facile ecofriendly synthesis of monastrol and its structural isomers via Biginelli reaction. ACS Sustain. Chem. Eng. 2014, 2, 1228–1233. [Google Scholar] [CrossRef]
- Mokhtar, M.; Saleh, T.S.; Ahmed, N.S.; Al-Thabaiti, S.A.; Al-Shareef, R.A. An eco-friendly N-sulfonylation of amines using stable and reusable Zn–Al–hydrotalcite solid base catalyst under ultrasound irradiation. Ultrason. Sonochem. 2011, 18, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.S.; Narasimharao, K.; Ahmed, N.S.; Basahel, S.N.; Al-Thabaiti, S.A.; Mokhtar, M. Mg–Al hydrotalcite as an efficient catalyst for microwave assisted regioselective 1,3-dipolar cycloaddition of nitrilimines with the enaminone derivatives: A green protocol. J. Mol. Catal. A 2013, 367, 12–22. [Google Scholar] [CrossRef]
- Suresh, J.S.; Sandhu, J.S. Past, present and future of the Biginelli reaction: A critical perspective. ARKIVOC Online J. Org. Chem. 2012. [Google Scholar] [CrossRef]
- McNaught, A.D.; Wilkinson, A. (Eds.) IUPAC Compendium of Chemical Technology (the ‘‘Gold Book’’), 2nd ed.; Blackwell Scientific Publications: Oxford, UK, 1997.
- Kaupp, G. Mechanochemistry: The varied applications of mechanical bond-breaking. CrystEngComm 2009, 11, 388–403. [Google Scholar] [CrossRef]
- Ulman, A.; Urankar, E. A novel synthesis of 4-[alkyl(aryl)sulfonyl]benzaldehydes: Alkyl(aryl)sulfinate anion as a nucleophile in aromatic substitutions. J. Org. Chem. 1989, 54, 4691–4692. [Google Scholar] [CrossRef]
- Yang, H.; Li, Y.; Jiang, M.; Wang, J.; Fu, H. General copper-catalyzed transformations of functional groups from arylboronic acids in water. Chem. Eur. J. 2011, 17, 5652–5660. [Google Scholar] [CrossRef] [PubMed]
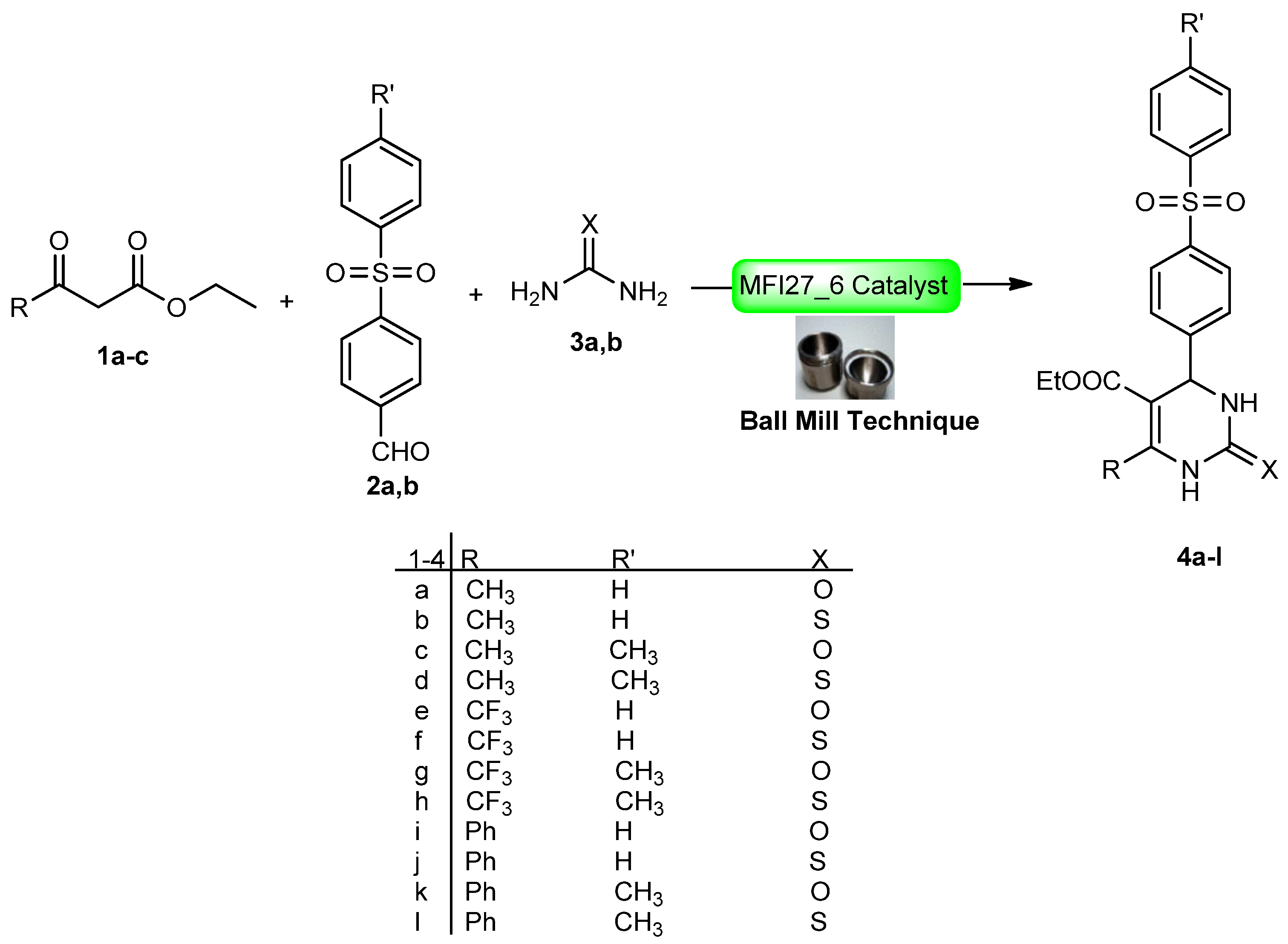
| Compound | Time (min) | Yield (%) | Compound | Time (min) | Yield (%) |
|---|---|---|---|---|---|
| 4a | 10 | 96 | 4g | 30 | 86 |
| 4b | 10 | 96 | 4h | 30 | 89 |
| 4c | 20 | 89 | 4i | 20 | 90 |
| 4d | 20 | 90 | 4j | 20 | 92 |
| 4e | 30 | 88 | 4k | 30 | 86 |
| 4f | 30 | 91 | 4l | 30 | 88 |
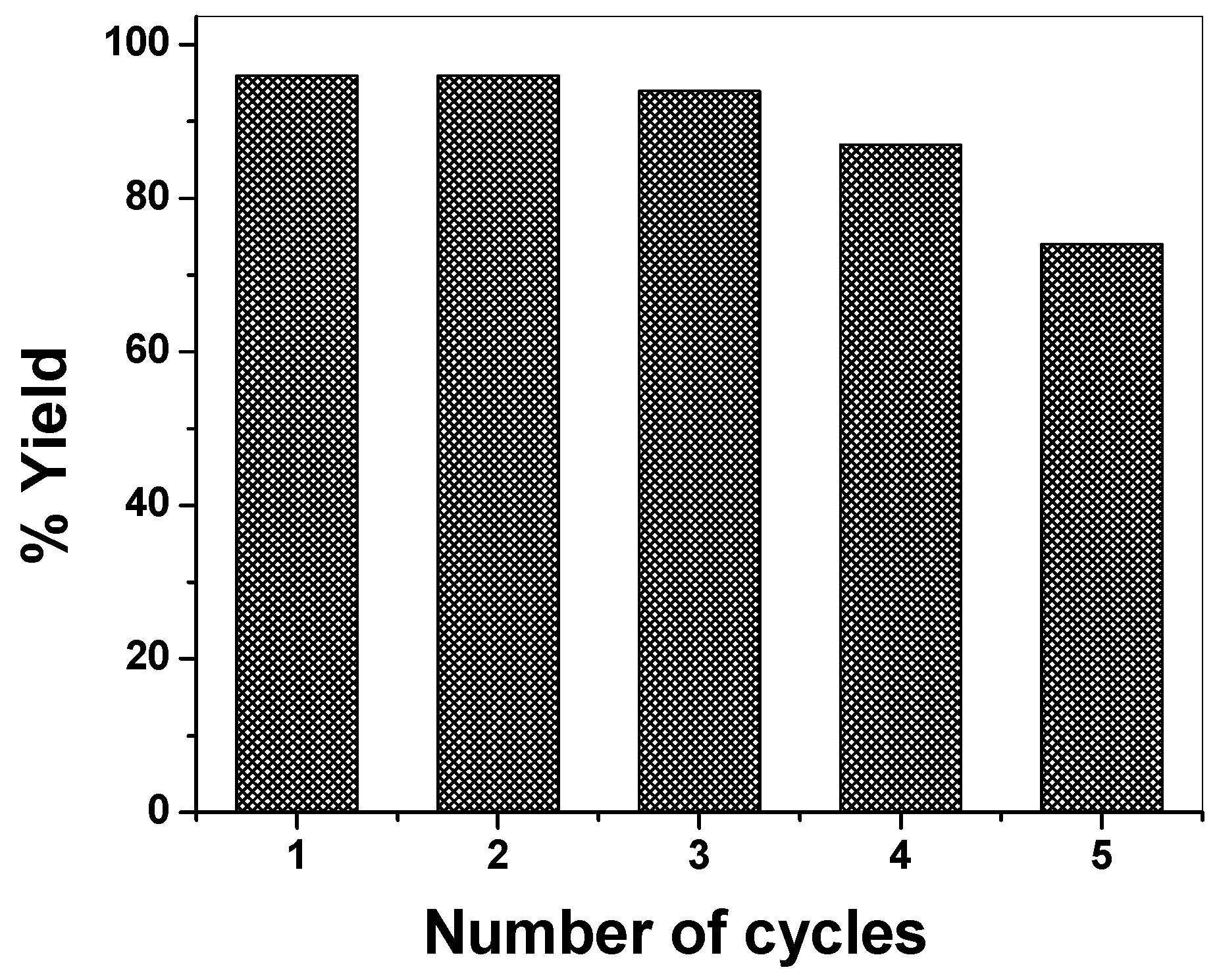

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
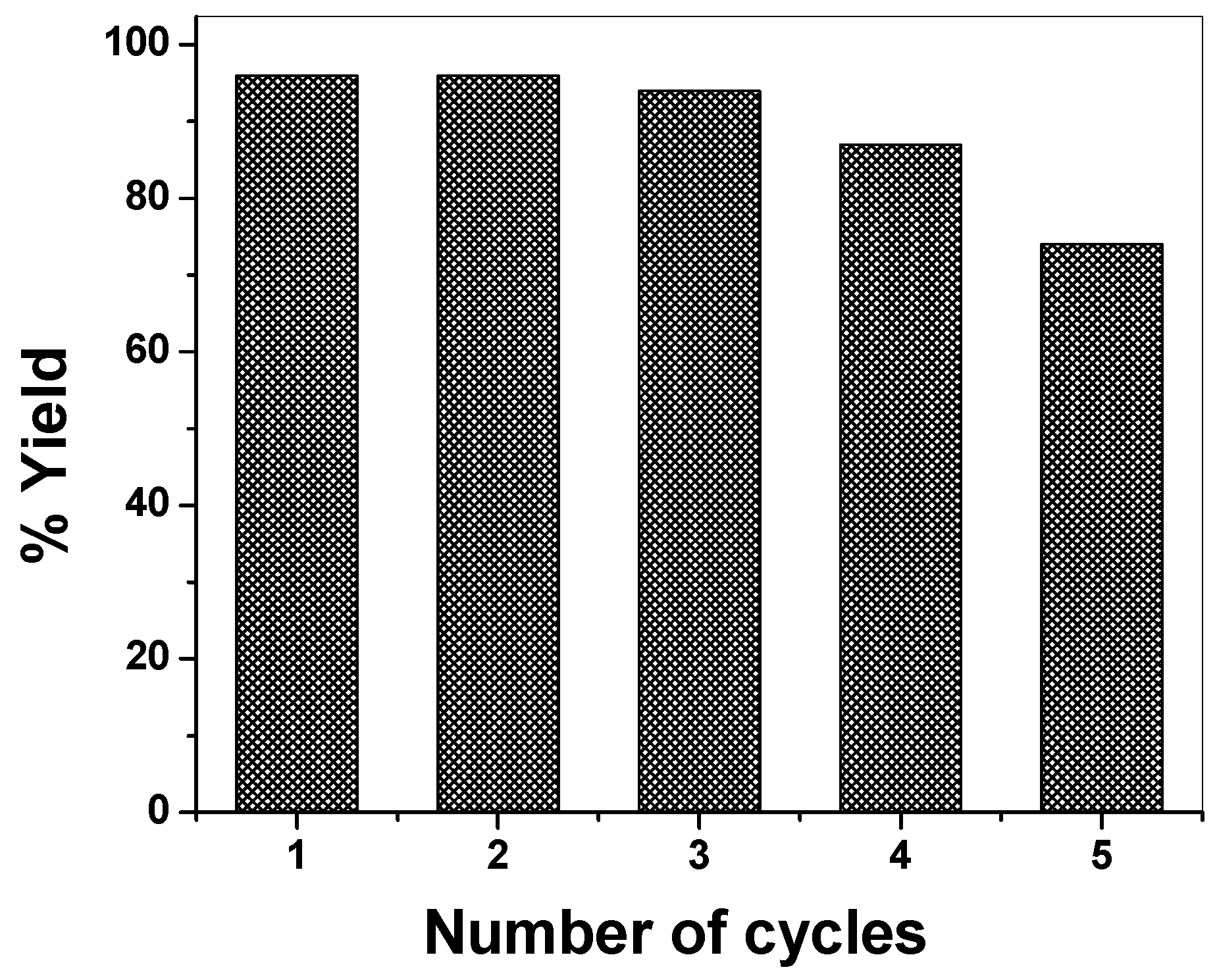{kind=link}
{kind=link}
{kind=link}
{kind=link}
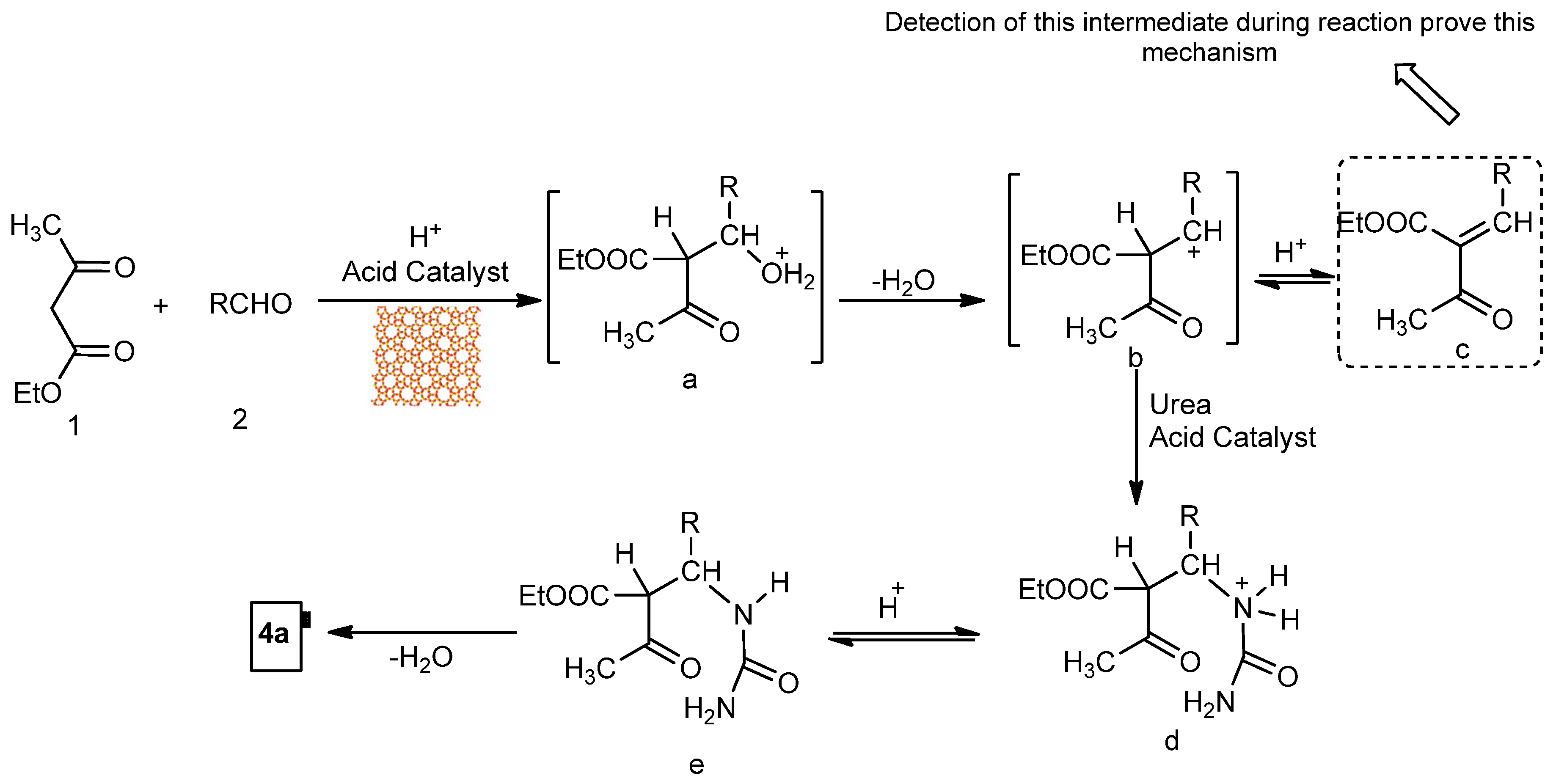{kind=link}
| General Biginelli Reaction: | ||||||
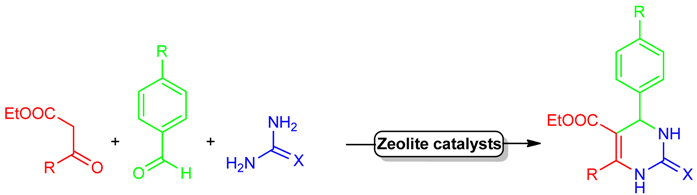 | ||||||
| Entry | Type of Zeolite Used | Method | Time (h) | Yield | Reference | Temperature (°C) |
| 1 | Commercial HZSM-5 | Reflux in toluene | 24 | 21% | [33] a | 100 |
| 2 | Commercial HY | Reflux in toluene | 24 | 80% | [33] a | 100 |
| 3 | MCM-41 | Reflux in toluene | 24 | 38% | [33] a | 100 |
| 4 | Natural zeolite Heulandite | Reflux in acetic acid | 5 | 75% | [34] a | 100 |
| 5 | Metal/Y zeolite | Reflux in ethanol | 12 | 20%–38% b | [35] a | 70 |
| Samples | Si/Al | SBET a (m2/g) | Smicro (m2/g) | Smeso (m2/g) | VTotal b (cm3/g) | Vmicro c (cm3/g) | Vmeso d (cm3/g) | Hierarchy Factor e |
|---|---|---|---|---|---|---|---|---|
| MFI27_P | 13 | 399 | 360 | 39 | 0.25 | 0.16 | 0.09 | 0.063 |
| MFI27_2 | 10.9 | 356 | 268 | 88 | 0.29 | 0.12 | 0.17 | 0.102 |
| MFI27_4 | 9.8 | 334 | 196 | 138 | 0.32 | 0.08 | 0.24 | 0.103 |
| MFI27_6 | 8.6 | 363 | 200 | 163 | 0.38 | 0.08 | 0.30 | 0.095 |
| Entry | Catalyst | Catalyst Weight (g) | Time (min) | Yield (%) | Ball-Mill Frequency (Hz) |
|---|---|---|---|---|---|
| 1 | MFI27_P | 0.25 | 60 | 21 | 30 |
| 2 | MFI27_2 | 0.25 | 40 | 83 | 30 |
| 3 | MFI27_4 | 0.25 | 35 | 86 | 30 |
| 4 | MFI27_6 | 0.25 | 20 | 91 | 30 |
| 5 | MFI27_6 | 0.35 | 10 | 96 | 30 |
| 6 | MFI27_6 | 0.35 | 10 | 90 | 25 |
| 7 | MFI27_6 | 0.35 | 10 | 81 | 15 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahid, A.; Ahmed, N.S.; Saleh, T.S.; Al-Thabaiti, S.A.; Basahel, S.N.; Schwieger, W.; Mokhtar, M. Solvent-Free Biginelli Reactions Catalyzed by Hierarchical Zeolite Utilizing a Ball Mill Technique: A Green Sustainable Process. Catalysts 2017, 7, 84. https://doi.org/10.3390/catal7030084
Shahid A, Ahmed NS, Saleh TS, Al-Thabaiti SA, Basahel SN, Schwieger W, Mokhtar M. Solvent-Free Biginelli Reactions Catalyzed by Hierarchical Zeolite Utilizing a Ball Mill Technique: A Green Sustainable Process. Catalysts. 2017; 7(3):84. https://doi.org/10.3390/catal7030084
Chicago/Turabian StyleShahid, Ameen, Nesreen S. Ahmed, Tamer S. Saleh, Shaeel Ahmed Al-Thabaiti, Sulaiman N. Basahel, Wilhelm Schwieger, and Mohamed Mokhtar. 2017. "Solvent-Free Biginelli Reactions Catalyzed by Hierarchical Zeolite Utilizing a Ball Mill Technique: A Green Sustainable Process" Catalysts 7, no. 3: 84. https://doi.org/10.3390/catal7030084
APA StyleShahid, A., Ahmed, N. S., Saleh, T. S., Al-Thabaiti, S. A., Basahel, S. N., Schwieger, W., & Mokhtar, M. (2017). Solvent-Free Biginelli Reactions Catalyzed by Hierarchical Zeolite Utilizing a Ball Mill Technique: A Green Sustainable Process. Catalysts, 7(3), 84. https://doi.org/10.3390/catal7030084