Abstract
Linear aliphatic polyesters are degradable thermoplastic polymers, which can be obtained by ring-opening polymerization (ROP) of cyclic esters through a coordination-insertion mechanism. Aluminum based organometallic complexes have a leading position as efficient catalysts for this polymerization process. Aluminumalkyl complexes bearing salicylaldiminato ligands, although less explored, have been shown to be efficient and versatile catalysts for the ROP of various cyclic esters. These species have the potential to function as active catalysts in the ROP because of their less coordinatively saturated nature with respect to analogous SALEN-type complexes. They have been used as efficient catalysts in the ROP of commercially available cyclic esters, such as ε-caprolactone, l-lactide, rac-lactide, and glycolide. Moreover, they resulted in efficient catalysts for the ROP of cyclic esters with large ring-size and for the ROP of functionalized lactide. Furthermore, they have been used in the co- and ter-polymerization of various cyclic esters affording well controlled polymerization and a plethora of microstructural architectures, ranging from random to block to multiblock.
1. Introduction
Aliphatic polyesters are a class of natural and synthetic polymers with good mechanical and thermal properties. Among them, linear aliphatic polyesters are degradable thermoplastic polymers. Their degradation process can occur through an enzymatic route or by hydrolytic cleavage of the ester bonds. Materials with different degradation rates can be obtained by varying the lengths of the aliphatic chains as well as by copolymerization processes [1,2]. To date, aliphatic polyesters with short aliphatic chains—such as poly(glycolide) (PGA), poly(lactide) (PLA), and poly(ε-caprolactone) (PCL)—have been extensively investigated as biocompatible and bioresorbable materials, since their hydrolysis generates metabolites which are excreted via the citric acid cycle. They have already found a broad range of applications in the biomedical field, such as bone screws, sutures, tissue engineering scaffolds, and drug delivery systems; or as ecological materials used for packaging or films in agriculture [3]. On the contrary, aliphatic polyesters with long aliphatic chains have been recently envisaged as “poly(ethylene)-like” materials, and they are suitable for long term applications [4].
The uniqueness of the aliphatic polyesters lies in their immense diversity and synthetic versatility. Indeed, they can be prepared by a variety of monomers via enzymatic route or synthetic approaches, i.e., ring-opening polymerization (ROP) of cyclic esters or polycondensation routes [5]. Although the enzymatic route can be regarded as an environmentally-friendly synthetic process, which can occur in mild conditions, it is too expensive for large applications. On the contrary, the step-growth polymerization or polycondensation of diols with diacids (or diesters), or of hydroxyacids is a low-cost process. However, it suffers from several drawbacks and it is scarcely controlled. In contrast to these limitations, the ROP of cyclic esters—despite the restriction on monomers—can provide high-molecular weight aliphatic polyesters under mild conditions. In the presence of proper catalyst and conditions, the ROP may proceed in a controlled manner, and in some cases it may also display the features of a “living” polymerization, enabling the preparation of materials with predictable molecular weights, narrow dispersity, as well as the synthesis of block copolymers by sequential addition of different monomers. Cyclic diesters and lactones—such as glycolide (GA), lactide (LA), ε-caprolactone (CL)—have been the most investigated monomers in the ROP. Recently, the ROP of large ring-size lactones, such as pentadecalactone (PDL), has also been developed.
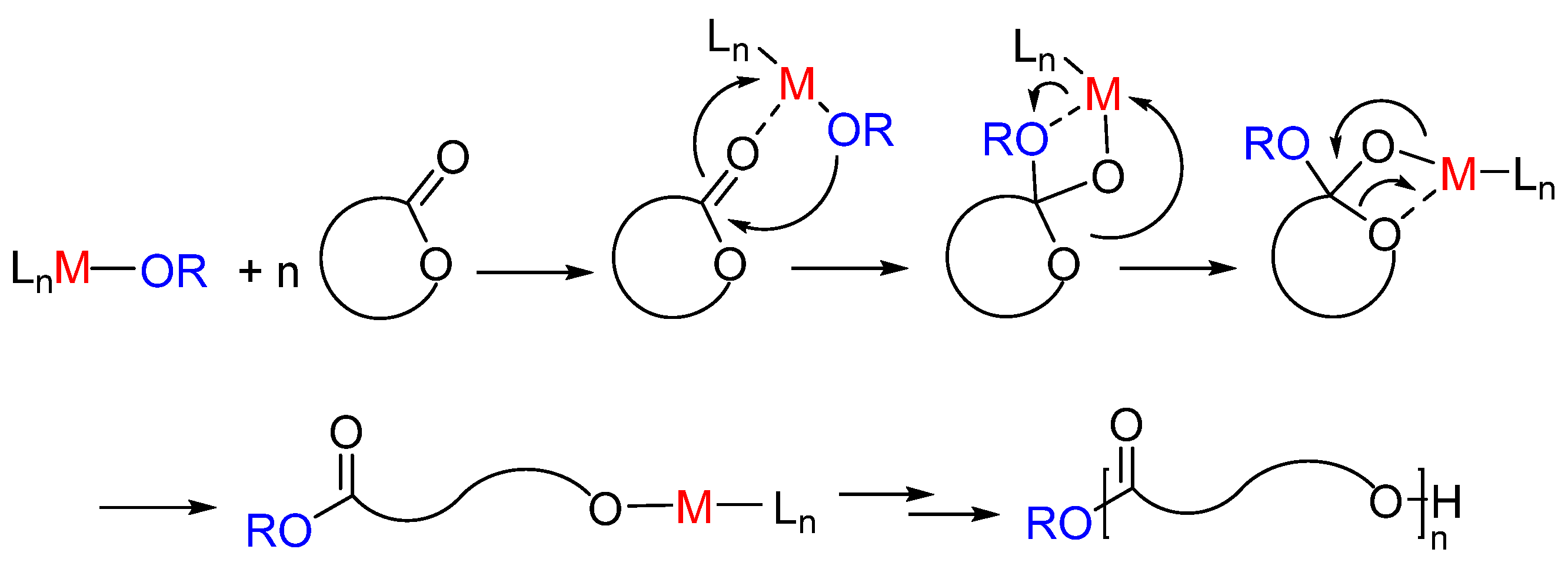
The ROP processes can be promoted by homoleptic metal alkoxide complexes, organometallic complexes, enzymes, or simple organic molecules [6,7,8,9,10]. Depending on monomers, catalytic system, nature of active species, the ROP can proceed as a coordinative, anionic, or cationic polymerization mechanism. Between these methods, the coordinative ROP by metal-based catalysts allows a better control on the polymer microstructure. Different metal-based catalysts are able to mediate the ROP of cyclic esters by coordination-insertion mechanism. In this mechanism, the first step is the coordination of the monomer to the metal center through the carbonyl oxygen, followed by the insertion of the monomer in the metal-initiator group bond, typically an alkoxide. The polymerization proceeds by propagation by a metal alkoxide species (Scheme 1).
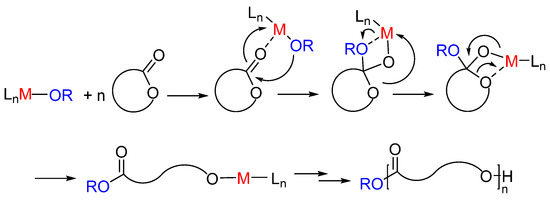
Scheme 1.
Ring-opening polymerization of cyclic ester by coordination-insertion mechanism.
The most frequently used and industrial catalyst is tin(II) octanoate (SnOct2). It is an efficient catalyst for the ROP of a wide range of cyclic esters. However, it also promotes transesterification reactions as side reactions, which leads to a decreased control in the polymerization, manifested by broad molecular weight dispersities in the obtained polyesters.
To achieve better control over chain growth, novel single-site metal-based catalysts have been developed, which can be described by the general formula LnM-OR. The enchainment of monomer occurs at a metal center, M, the active site, after coordination and then insertion into the metal-alkoxide bond (OR). The ancillary ligand (L) remains bound to the metal throughout the entire catalytic reaction, and its role is to tune the reactivity and selectivity of the metal, minimizing the side reactions. The ancillary ligand might also remove or minimize the mechanistic complexity resulting from the aggregation-disaggregation processes, in which multiple-site alkoxides are usually engaged.
A large variety of aluminum complexes active in the ROP of lactones and lactides have been described with different coordination environments [6,7,8,9,10]. Recent literature reviews described the use of various aluminum complexes as polymerization catalysts [11,12,13]. This review focus on aluminum-based salicylaldiminato complexes and their reactivity in the ring-opening homo and copolymerization of commercially available and conventional lactones and lactides. The ROP of macrolactones and novel-synthetized functionalized cyclic esters by the same class of catalyst is also described.
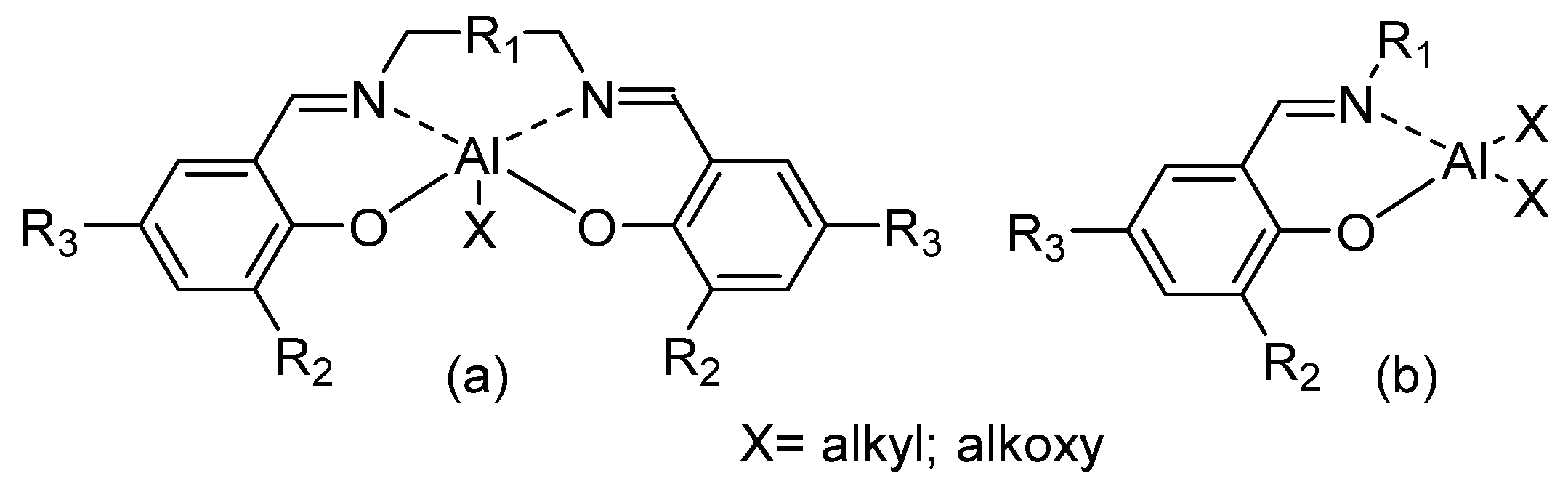
2. Dimethyl(salicilaldiminato)aluminum Complexes
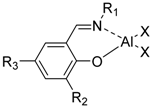
Aluminum complexes bearing tetradentate salicilaldiminato ligands (SALEN, Figure 1a) have been largely studied and used in many catalytic reactions [14,15,16,17,18,19,20]. After the discovery of the stereoselective polymerization of racemic lactides by chiral binaphthyl Schiff base aluminum complex [21], a large synthetic effort has been devoted to the preparation of five-coordinate SALEN aluminum complexes and their use in the ROP of lactones and lactides. The relationship between structure of the catalytic complexes and stereoselectivity of the polymerization has been investigated and elucidated [6,7,8,9,10,22]. Recent organometallic literature has featured reports of many alkylaluminum compounds bearing bidentate N,N-, N,S-, or N,O-donor ligands [23,24,25,26,27,28,29,30]. These species have the potential to function as active catalysts because of their less coordinatively saturated nature. Interestingly, “half-SALEN” salicylaldiminato aluminum complexes (Figure 1b) have been shown as efficient catalysts for ROP of cyclic esters.
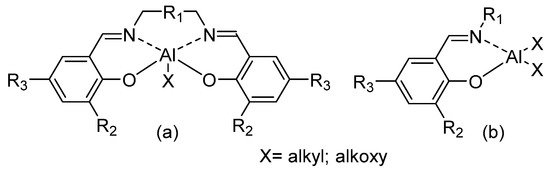
Figure 1.
SALEN-Al (a) vs. “half-SALEN” salicylaldiminato-Al (b) complexes.
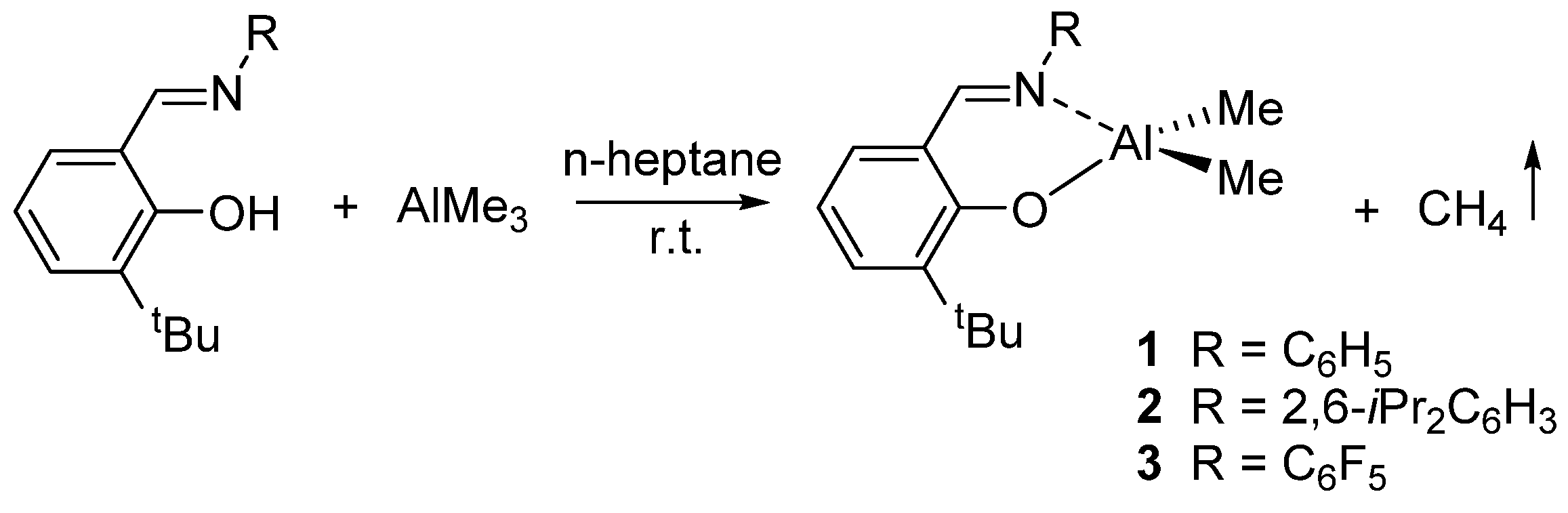
Our group recently investigated the reactivity of this class of catalysts in the ROP of lactones and lactides. Following the reports by Coates [31] and by Fujita [32] describing titanium complexes carrying salicylaldiminato ligands with electron-withdrawing substituents that showed increased activity in the polymerization of olefins, we synthesized the dimethyl(salicylaldiminato)aluminum compounds {3-tBu-2-(O)C6H3CH=N-R}AlMe2 [R = C6H5 (1); 2,6-iPr2C6H3 (2); C6F5 (3)] (Scheme 2) and tested their reactivity in olefin polymerization [33]. Toluene solutions of 1, 2, and 3, when activated with 1 equiv of the Lewis acid B(C6F5)3, polymerized ethylene at 1 atm to solid polyethylene with very low activity.
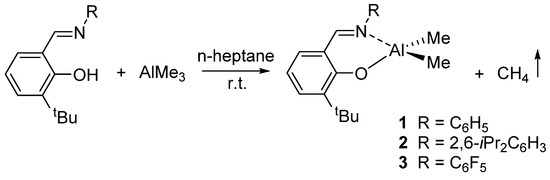
Scheme 2.
Synthesis of dimethyl(salicylaldiminato)aluminium compounds 1–3 [33].
Along these studies, bearing in mind the similarities between titanium and tin, these studies have been extended to the synthesis of bis(phenoxy-imine)tin(IV) alkyl complexes, and their reactivity against ionizing agents and ethylene [34].
In 2002, Baugh and Sissano reported the synthesis and characterization of dimethylaluminum complexes bearing simple salicylaldiminato N,O-ligands, without substituents on the aryl ring. These complexes were investigated either as neutral or as borane-activated cationic forms in the polymerization of different monomers, such as methyl methacrylate, ε-caprolactone, and propylene oxide. The neutral complexes usually did not promote polymerization, however the polymerization/oligomerization of all three monomers was achieved when the catalysts were activated with B(C6F5)3 or [Ph3C]+[B(C6F5)4]−. The cationic complexes indeed catalyzed the ring-opening cationic polymerization of tetrahydrofuran. The polymerization of CL by the cationic species produced just traces of polymers or low molecular weight, alcohol-soluble polymer [35].
We have tested the dimethyl(salicylaldiminato)aluminum compounds 1 and 2 and the perfluoro derivative 3 in the ring-opening polymerization of caprolactone and lactides, disclosing a well-controlled polymerization with absence of transesterification reaction, and a certain living behavior [36]. This interesting result prompted us to explore the reactivity of the same catalytic system also in the ROP not only of other conventional cyclic esters, i.e., glycolide [37], but also of large ring macrolactone [38] and of novel synthetized thiol-functionalized lactide [39].
A large variety of aluminum-based salicylaldiminato complexes have been described and used in the ROP of various cyclic esters, their structural features are summarized in Table 1 with reference to the proper literature.

Table 1.
Structural features of the (salicilaldiminato)aluminum complexes used in ring-opening polymerization (ROP) of cyclic esters.
3. Salicylaldiinato-Aluminum Complexes in the Ring-Opening Polymerization of ε-Caprolactone and Other Small Ring Size Lactones
The ring-opening polymerization of the ε-caprolactone (CL), firstly performed by Carothers in the early 1930s, yields the poly(ε-caprolactone) (PCL), a highly processable polymer, soluble in a wide range of organic solvents. It shows a melting point of about 65 °C and a glass-transition temperature of −60 °C, well below room temperature. The PCL is highly hydrophobic with a degradation time of the order of two years. It degrades by hydrolytic degradation as well as by enzymatic attack. Hydrolysis yields 6-hydroxy caproic acid, which enters the citric acid cycle and it is completely metabolized. Thus, PCL has been exploited in the biomedical field to develop long-term delivery system or scaffolds for bone tissue engineering and for degradable substituents of fossil-fuel derived common plastic.
The first report by Baugh and Sissano in 2002, relative to the synthesis and characterization of dimethylaluminum complexes bearing simple salicylaldiminato N,O-ligands without substituents on the aryl ring showed that these complexes, in their cationic form, catalyzed the ring-opening cationic polymerization of tetrahydrofuran [35]. However, polymerization of CL by the cationic species formed by activation with B(C6F5)3 produced just traces of polymers; when [Ph3C]+[B(C6F5)4]− was used, high yields of low molecular weight alcohol-soluble polymer were produced, probably due to the occurrence of transesterification processes during polymerization. Subsequently, the use of analogous N,O-ligand-based aluminum compounds for the polymerization of lactones was filed in a patent application [49].
Nomura et al. described the use of Al-salicylaldimine complexes prepared in situ by mixing two equiv of salicylaldimine ligand with AlEt3 and benzyl alcool for the polymerization of CL. Various salicylaldimines were used; the complexes were not isolated. The steric effects of the salicylidene moiety were analyzed. The activity was higher in the presence of a methyl or an isopropyl substituent at the 3-position of the salicylidene moiety; the most efficient catalysts, 4 and 5, were obtained by the ligands with the sterically demanding 2,4,6-tri-tertbutylphenylimine moiety and a methyl substituent at the 3-position of the salicylidene moiety (Scheme 3) [45].
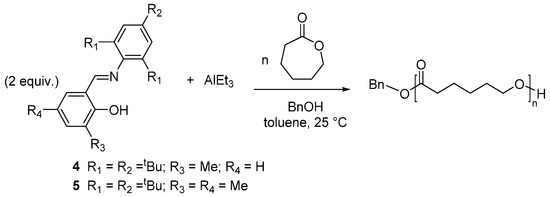
Scheme 3.
Ring-opening polymerization of ε-caprolactone (CL) by in situ prepared salicylaldimine-Al complexes [45].
Interestingly, the authors underlined the opposite behavior observed for analogous SALEN-type aluminum complexes, where the bulky substituents hampered the ROP of lactide [50]. This difference may probably lay in the coordination ability of two phenoxy-imine ligands versus the SALEN ligand toward the Al center. Indeed, Nuclear Magnetic Resonance (NMR) analysis of the in situ prepared salicylaldimine-Al complexes indicated that only one of the phenoxy-imine was bidentate, while the other resulted in monodentate [45].
Some of these complexes were also effective in the polymerization of δ-valerolactone (VL). The polymerization of (BL) was also attempted and resulted much slower even at 70 °C; however, poly(β-butyrolactone) with a narrow polydispersity was obtained until the monomer conversion reached 60% [45].
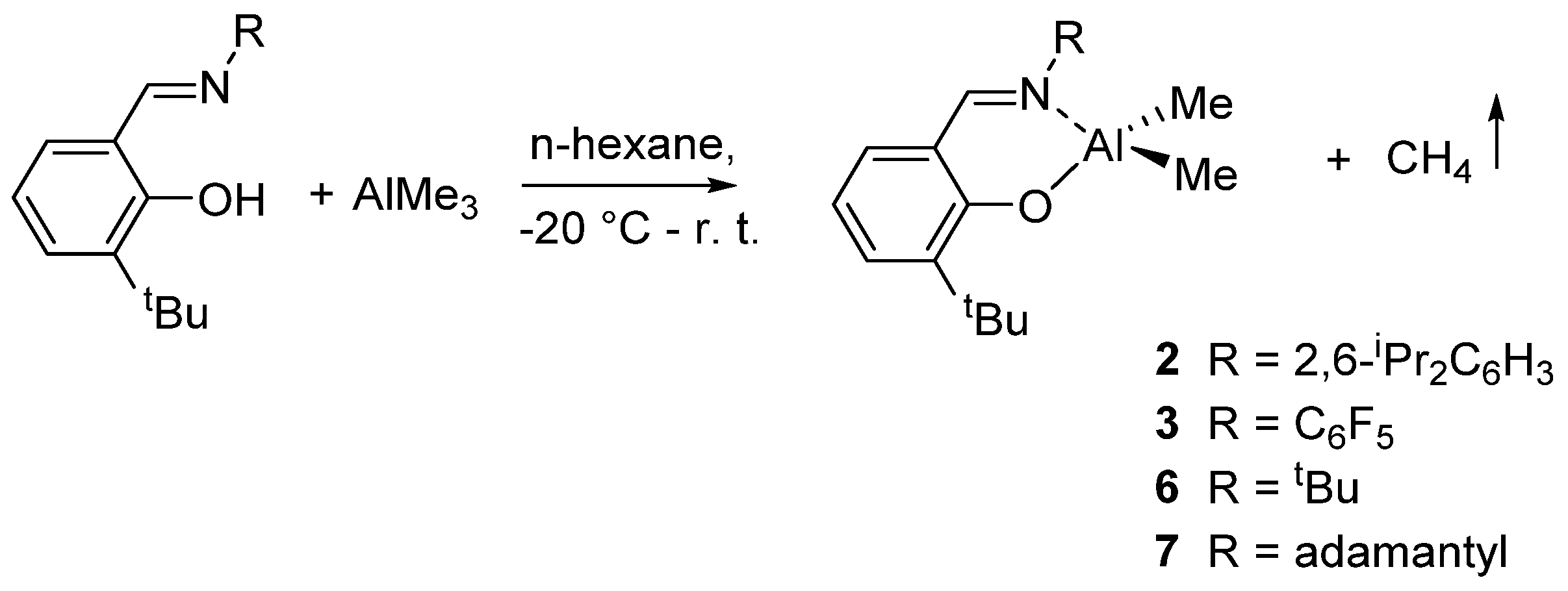
Other Al complexes bearing different phenoxy-imine ligands of type {3-tBu-2-(O)C6H3CH=N-R}AlMe2 [R = 2,6-iPr2C6H3 (2); C6F5 (3); tBu (6); adamantly (7)] were described by Nomura et al.. The complexes were prepared by reaction of 1 equiv of ligand with AlMe3 (Scheme 4), isolated, and characterized, some of these also by X-ray crystallography [43].
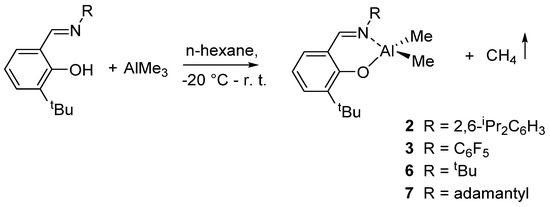
Scheme 4.
Synthesis of dimethyl(salicylaldiminato)aluminum compounds 2, 3, 6, and 7 [43].
The catalytic activity in the ring-opening polymerization (ROP) of ε-caprolactone initiated by 2, 3, 6, or 7 in combination with nBuOH increased in the order: 3 > 2 > 6 > 7, indicating that the imino substituent (R) plays an important role for the efficient ROP [43]. The role of the imino substituents for efficient and controlled ROP was further remarked in an extension of this study, showing that in the presence of C6F5 as imino substituent, the ROP of ε-caprolactone took place in a living manner [40]. Notably, the ROP reactions were accompanied by a certain degree of transesterification as side reaction except for the polymerization carried out in the presence of complexes bearing the C6F5 substituent. Thus, a positive effect on the control of the polymerization was observed in the presence of fluoro substituents, and it could be related to electronic effect [41]. The polymerization of β-butyrolactone and δ-valerolactone afforded negligible amount of polymers, while with the rac-lactide, atactic polymers were obtained [42].
The dimethylaluminum compounds {3-tBu-2-(O)C6H3CH=N-R}AlMe2 [R = C6H5 (1); 2,6-iPr2C6H3 (2); C6F5 (3)] were used as initiators in the ring-opening polymerization (ROP) of ε-caprolactone (and also l-lactide and rac-lactide, see below) in combination with MeOH [36]. In the absence of MeOH, the ROP did not take place. The analysis of the end-groups of the polymer showed the presence of methyl ester and hydroxyl end groups, supporting a coordination-insertion mechanism operating in these systems. Analogously to the results obtained by Nomura (see above), polymerization tests in the presence of complex 3 at 70 °C displayed a linear correlation between the polymer molecular weight and the conversion, evidencing the “living” character of the polymerization.
4. Salicylaldiminato-Aluminum Complexes in the Ring-Opening Polymerization of Lactide and in the Copolymerization of ε-Caprolactone with Lactide
4.1. Polymerization of Lactide
The dimethyl(salicylaldiminato)aluminum compounds 1, 2, and 3 were used in the ring-opening polymerization of l-lactide (l-LA) and rac-lactide (rac-LA) in toluene solution at 70 °C, in the presence of 1 equiv of MeOH [36]. Monomodal curves and narrow molecular weight distributions were observed. For compound 3, the living behavior in the polymerization of l-LA and rac-LA was also assessed. In the case of the ROP of rac-LA, slightly isotactic enriched polylactides were obtained with compounds 1 and 3, while an atactic polymer was obtained with 2 [36].
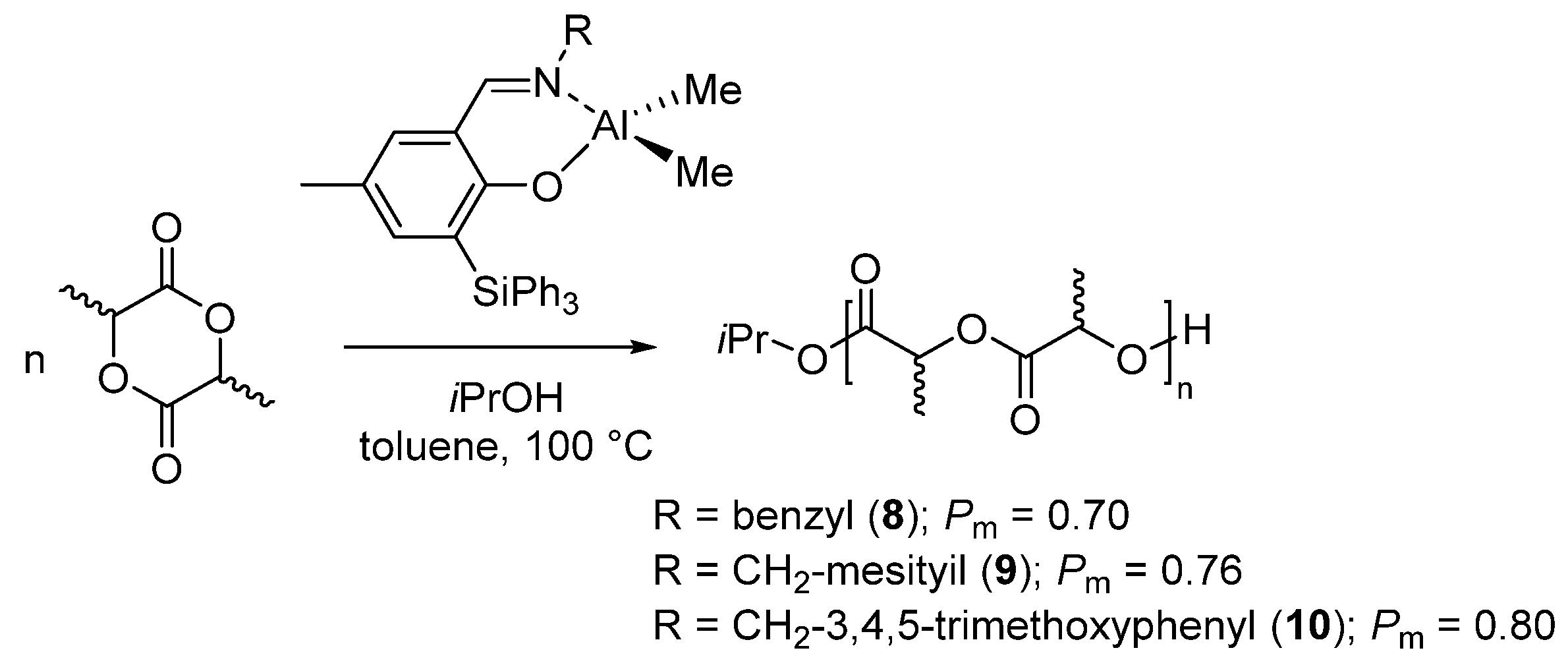
A series of dialkylaluminum {ONR}AlMe2 bearing a phenoxy-imine {ONR} ligand, were prepared and structurally characterized in solution and in the solid state by Carpentier et al. [46,47]. Ligands with with different R-imino substituents and functionalized by a bulky o-SiPh3 in the phenoxy moiety were synthetized. The complexes prepared thereof, in combination with an alcohol, promoted the living (immortal) ring-opening polymerization of rac-LA with various microstructures. Isotactic poly(lactides), with Pm up to 0.80 were obtained in the presence of complexes having benzyl-type imino substituents, 8–10 (Scheme 5) [46]. The isoselectivity increased by increasing the bulkiness of the aryl-imino group. However, the stereoselectivity changed drastically in the presence of a trityl group as imino substituent (complex 46 in Table 1) and a slighly heterotactic polymer was obtained. Moreover, when the imino substituent was derived by aniline, mainly atactic polymers were obtained (complexes 40–42, Table 1) [46]. Notably, as observed in the case of complex 2 [36], when the imino substituent was the 2,6-diisopropyl, a truly atactic polymer was obtained (complex 40, Table 1; Pm = 0.50). This behavior, although hardly rationalized, suggested again a pivotal role of the imino substituent for both reactivity and stereoselectivity.
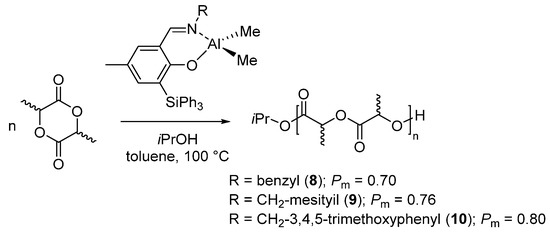
Scheme 5.
Ring-opening polymerization of rac-lactide by phenoxy-imino aluminum complex bearing o-triphenylsilyl mojety [46].
Recently, a series of Al complexes bearing N,O- and N,S-Schiff base ligands were prepared, and compared in the CL and LA polymerization. The Al complexes with N,S-Schiff base ligands showed higher polymerization rate respect to the others. Mechanism studies by Density Functional Theory (DFT) calculation predicted lower free energies of activation for the complexes bearing N,S Schiff base ligand due to a catalyst-substrate destabilization effect [48].
4.2. Copolymerization of ε-Caprolactone and Lactide
Copolymerization of LA with CL can allow the production of a range of biodegradable materials with improved properties in comparison to those of the close relative homopolyesters. Block and random copolymers have been obtained with this class of catalyst. By sequential addition of the two monomers, first the l-LA (or the rac-LA) and then the CL, di-block copolymers poly-(l-lactide-block-ε-caprolactone) and poly(rac-lactide-block-ε-caprolactone) were obtained in the presence of compound 3. Formation of di-block copolymers was established by NMR and Gel Permeation Chromatography (GPC) characterization [36]. Complex 3 was able to copolymerize CL with l-LA and CL with rac-LA in a random fashion; the obtained copolymers had random sequences, with the percentage of heterodiads higher than 50%. The chain microstructure of the copolymers was analyzed by NMR: according to the general case of a binary copolymerization, eight different triads were observed. Interestingly, the signal related to presence of the triad having one single “lactic” ester unit between two CL units was never detected. That triad is indicative of the occurrence of transesterification reactions, since it cannot result from the insertion of the lactide monomer into the chain. The absence of transesterification reaction was further confirmed by the GPC analysis, disclosing in all cases narrow molecular weight distributions. Copolymerization reactions were better controlled than those carried out in the presence of other catalysts—i.e., SnOct2—where the transesterification was responsible for the randomized structure. Thermal analysis of the copolymers of CL with l-LA, performed by DSC, showed that the copolymers were amorphous; the Tg values of random copolymer of CL with l-LA were in agreement with the theoretical one, calculated by the Fox equation; this observation was a further support to the random structure [36].
5. Salicylaldiminato-Aluminum Complexes in the Ring-Opening Polymerization of Glycolide
Poly(glycolide) (PGA) was the first synthetic biodegradable polymer used commercially as biomedical material. Due to its excellent fiber forming ability, PGA was initially investigated for developing resorbable sutures. It is a highly crystalline polymer (45%–55%) and therefore it shows excellent mechanical properties, it exhibits a high tensile modulus, E, approximately 12.5 GPa. The glass transition temperature of the polymer ranges from 35 to 40 °C and the melting point is greater than 200 °C [51].
The best strategy to synthesize PGA is by ring-opening polymerization of the glycolide (GA) [52]. We have reported the ROP of GA catalyzed by the dimethyl(salicilaldiminato)aluminum compounds 11 and 12, having different steric hindrance at the ortho position of the phenolato ring (Scheme 6) [37].
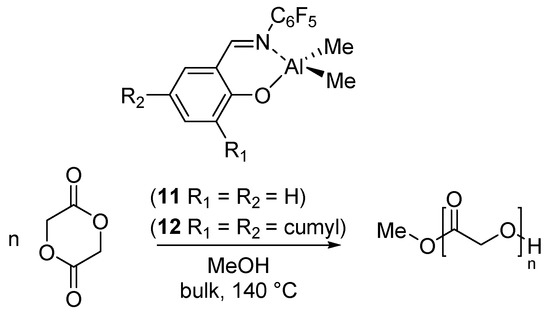
Scheme 6.
Ring-opening polymerization of glycolide (GA) by dimethyl(salicilaldiminato)aluminum compounds 11 and 12 [37].
The polymerizations were performed in bulk, at 140 °C, using 1 equiv of MeOH as initiator. Due to the scarce solubility of the polymer in common solvents, the molecular weight estimation by Size Exclusion Chromatography (SEC) was hampered. A detailed analysis of the end groups by 1H NMR indicated that the first step was the insertion of the glycolide in the Al-OCH3 bond and confirmed that a mechanism of coordination-insertion polymerization was operative also for the ROP of GA.
The thermogram of the PGA prepared with catalyst 12 showed a melting peak at 226 °C with an endotherm of fusion of 83.8 J·g−1, the glass transition temperature was not observed.
6. Co-Polymerizations of Glycolide with Other Cyclic Esters
6.1. Copolymerization of Lactide and Glycolide
The poly(lactide-co-glycolide) (PLGA) copolymer is by far one of the most used biodegradable aliphatic polyesters in biomedical application. PLGA can be obtained by ring-opening copolymerization of GA and LA [53]. The rate of degradation as well as the physical properties of PLGA depend on several parameters—including the LA/GA ratio, molecular weight, microstructure, and tacticity—which should be properly modulated during the copolymerization step [54,55]. Therefore, although the polymer has been known a long time, the search for an efficient ROP initiator for the PLGA production is still a field of increasing interest.
Dimethyl(salicylaldiminato) aluminum compounds, 11 and 12 were reported by us as precatalysts for the copolymerization of GA with rac-LA [37]. The versatility of this class of catalyst was assessed: random and block copolymers were obtained in different experimental conditions. The copolymerization of GA and rac-LA carried out in bulk produced random copolymer with short average block lengths were obtained. The average length of lactidyl blocks (LLL) and glycolidyl block (LGG) were assessed by NMR and linearly depended on the copolymer composition. Notably, a perfect random copolymer, with block length values close to 2, was obtained for catalyst 12 and monomers feed ratio of 50:50. The random microstructure of the copolymers was confirmed by thermal analysis. The copolymers were amorphous and displayed a unique glass transition temperature, Tg, except for the sample prepared with 80 mol % of GA. Moreover, the Tg values linearly increased by decreasing the GA content in the copolymer, which in turn reflected the feed composition. It should be underlined that GA shows usually higher reactivity respect to the LA [6]. The final random microstructure was not only the result of the monomers reactivity but it also depended on the occurrence of transesterification side processes, which took place together with the main copolymerization reaction. Indeed, signals due to sequences relative to transesterification processes of the second mode were revealed by NMR. These side reactions can occasionally occur between the growing chain and preformed segments, while they cannot be generated by a coordination-insertion polymerization of the monomers. The quantification of these signals showed that the behavior of the catalysts 11 and 12 was in contrast with that of the classical Sn(Oct)2 catalyst [56]. In particular, in the copolymerization of GA and rac-LA catalyzed by dimethyl(salicylaldiminato) aluminum compounds the transesterification due to the attack of active lactidyl chain end on already formed glycolidyl segments was favored. Indeed, as described above for the CL-LA copolymerization, the tendency of these complexes to break the lactidyl unit into two lactyl fragments is low [36]. Moreover, it resulted that transesterifications of the second mode were slightly higher for catalyst 12, bearing bulky cumyl groups as ortho-phenoxy substituents. This steric bulkiness could probably slow down the chain propagation rate, therefore it could have an influence on the relative rate of chain propagation versus transesterification reaction.
The analysis of the end-groups of the copolymers confirmed a coordination-insertion mechanism, proceeding through acyl-oxygen cleavage of both the monomers, with the insertion of either GA or rac-LA into the Al-OCH3 bond as first step. However, the GA was preferably first inserted into the Al-OCH3 bond, which was in agreement with the higher reactivity of this monomer with respect to that of LA. This preference was more significant with catalyst 12. The molecular weight values were determined by NMR and resulted in agreement with the theoretical ones. Molecular weights up to 154,000 g/mol were achieved with a monomer to catalyst ratio of 100:1.
When the copolymerization of GA and rac-LA with the same catalysts 11 and 12 were performed in solution (monomers feed of 50:50 mol %) at 90 °C the GA was almost exclusively incorporated. On the contrary, by increasing the temperature up to 130 °C the GA content in the copolymer chains ranged between 49% and 66% showing that, in these experimental conditions, comparable incorporation of both monomers was obtained. A detailed analysis of the microstructure for copolymer produced at higher temperature revealed that with catalyst 11 the average block lengths were higher than 2, indicating a blockier copolymer than the one obtained in bulk. Accordingly, the thermograms of these samples showed two Tg values and in one case a melting endotherm of glycolide blocks crystalline phase. On the contrary, for copolymer prepared with catalyst 12 at higher temperatures, the calculated average block lengths were shorter and the amount of transesterification reactions were higher; consequently, due to the more random microstructure, a unique glass transition was observed.
Moreover, by sequentially addition of the two monomers (i.e., first the rac-LA was polymerized, and then, after rac-LA was consumed the GA was added) the synthesis of block copolymer poly(rac-lactide)-block-poly(glycolide) was achieved by using catalyst 11 in xylenes at 130 °C. The NMR signals, both in the 13C and 1H spectra, were compatible with the desired microstructure, which was definitely proven by a Diffusion-Ordered Spectroscopy (DOSY) NMR experiment. This result was a further evidence of the tendentially living behavior of the ring-opening polymerization promoted by dimethyl(salicylaldiminato) aluminum compounds [37].
6.2. Glycolide, ε-Caprolactone, and rac-Lactide co- and ter-Polymerization
The dimethyl(salicylaldiminato) aluminum compounds were shown to be also efficient initiator for the random copolymerization of GA and CL. The aluminum compounds 11, 12, and 3 were used in the ring-opening copolymerization of GA and CL in the presence of one equivalent of MeOH [44]. The copolymerizations were performed in bulk, at 140 °C varying systematically the monomers feed ratio. A detailed analysis of 1H NMR spectra and thermal characterization by DSC of copolymers having different composition and/or prepared with the different catalysts elucidated how the reactivity of the two monomers, the transesterification reaction, and the steric hindrance at the ortho position of the phenolato ring of the catalyst played a role on determining the final microstructure of the copolymer chains. Due to its higher reactivity, GA was polymerized first and faster than CL, thus, long glycolidyl blocks were formed during the copolymerization, while the CL was gradually inserted by transesterification reaction. Indeed, while the glycolidyl block lengths, LGG, linearly increased by increasing the amount of GA, the caproyl block lengths, LCap, did not vary significantly with the composition. The value of coefficient of the transesterification of the second mode—i.e., the one involving the attack of the active caproyl chain growing on preformed glycolidyl segment—increased by increasing the amount of CL in the feed. LGG values lower than 1 were obtained for all the catalyst, when the content of CL was 70 mol %, indicating that glycolidyl blocks, GG, have been almost all cleaved into glycolyl units, G.
Although GA is usually polymerized faster than CL, the net difference in the reactivity of the CL and GA comonomers could be reduced by increasing the bulkiness of the substituents in the ortho positions of the phenolato rings. Indeed, the insertion rate of the GA monomer seems to be decreased by the bulkiest complex 12, which led to a comparable insertion rate of the two monomers, and showed the highest propensity to furnish random copolymers. However, for the least encumbered catalyst, 11, the transesterification reactions were more frequent, thus allowing the randomization of the copolymer chains.
The thermal behavior of these copolymers reflected the copolymer microstructure. Indeed, for copolymers having a content of GA higher than 50 mol %, the DSC thermograms displayed neat melting point due to the long glycolidyl blocks. Increasing the amount of CL, only a single glass transition was observed for the copolymers. Interestingly, for the copolymers obtained with catalyst 12, the experimental Tg values were in agreement with the theoretical ones calculated with the Fox’s equation, thus indicating the achievement of a random copolymerization.
The terpolymerization of GA, CL, and rac-LA was also investigated in the presence of dimethyl(salicylaldiminato) aluminum compounds [44]. Poly(glycolide-co-lactide-co-caprolactone) terpolymer (PGLC) are interesting materials in drug delivery and tissue engineering. However, besides dimethyl(salicylaldiminato) aluminum compounds, only three metal initiators were reported for the ring-opening terpolymerization of GA, LA, and CL [44].
The behavior of the three different catalyst 11, 12, and 3, was studied for equimolar amount of the three monomers, in bulk at 140 °C using one equivalent of MeOH as initiator. Glycolide was generally more incorporated than rac-LA and CL for all the catalysts. However, a difference in the preferred incorporation of rac-LA respect to CL was observed. While for the least encumbered catalyst, 11, the copolymer chains were richer in rac-LA than CL, an opposite behavior was observed for catalyst 12 and 3, having bulkier substituents. This was explained on the basis of the higher coordination ability of the rac-LA, which led a major incorporation of this monomer when catalyst 11 was used. On the contrary, the more flexible and less bulky CL was preferentially incorporated with catalysts 12 and 3, due to the more hindered ligands.
Transesterification reactions involving the attack of glycolidyl or caproyl active chain ends on lactidyl segments were absent or negligible, once more confirming the low propensity of these catalytic systems in cleaving of lactidyl blocks.
Short average glycolidyl block lengths, below 2, were obtained for catalyst 11 and 3, indicating a truly random microstructure. On the contrary, for the most hindered catalyst 12 longer glycolidyl blocks were assessed, evidencing a blockier microstructure. Accodingly, the DSC thermograms revealed that for catalysts 11 and 3, a unique glass transition temperature was observed. Thus, samples retained a single phase and the Tg values were in agreement with the theoretical ones determined by Fox’s equation. Two glass transitions were observed for the sample prepared with catalyst 12, as a consequence of the longer GG blocks.
7. Ring-Opening Polymerization of Macrolactones
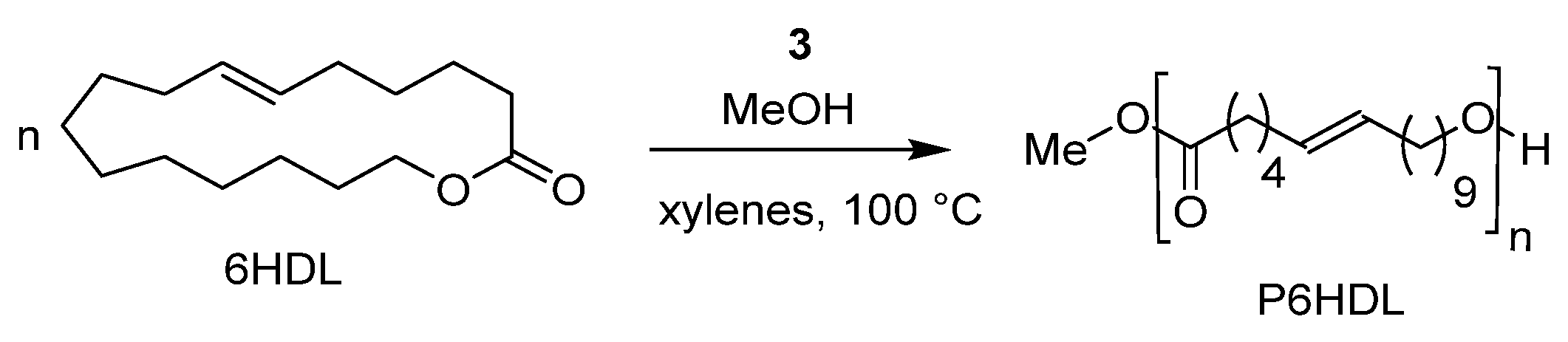
The ROP of an unsaturated large lactone, the ω-6-hexadecenlactone (6HDL) was explored for the first time by using this class of catalysts. It is well known that large lactones have low polymerizability. Indeed, only few metal initiators able to catalyze their ROP have been reported up to now. Interestingly, the dimethyl(salicylaldiminato) aluminum compound 3 resulted in active catalysts for the ROP of 6HDL to high molecular weight polymer, P6HDL, in a controlled fashion (Scheme 7). Such a monomer could represent a useful platform for the synthesis of semicrystalline and functional polyethylene-like polyesters [38].
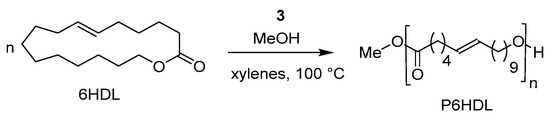
Scheme 7.
Ring-opening polymerization of ω-6-hexadecenlactone (6HDL) by dimethyl(salicilaldiminato)aluminum componds 3 [38].
Polymerization runs were performed in xylenes solution at 100 °C, in the presence of complex 3 and one equivalent of methanol. The molecular weight linearly increased with time and conversion while the dispersity values of 1.6 remained constant during the polymerization, thus displaying a pseudo-living character of the polymerization. However, the monomer conversion levelled off around 60% after 27 h, which was explained either on the basis of the hampered diffusion of the monomer in the high viscous polymerization mixture or on the possible equilibrium between cyclic oligomers and linear polymers, which is typical for entropy-driven polymerization [57,58,59]. In addition, the copolymerization of the 6HDL with CL and rac-LA was reported in conditions analogous to those used for the homopolymerization [38]. The simultaneous copolymerization of a 50/50 mixture of 6HDL and CL produced a truly random semicrystalline copolymer, with average sequence block lengths around 2. On the other hand, sequential copolymerizations allowed the synthesis of linear block copolymers of 6HDL with CL and/or rac-LA thanks to the pseudo-living behavior of the catalytic system and the absence of transesterification reactions.
8. Ring-Opening Polymerization of Functionalized Monomers
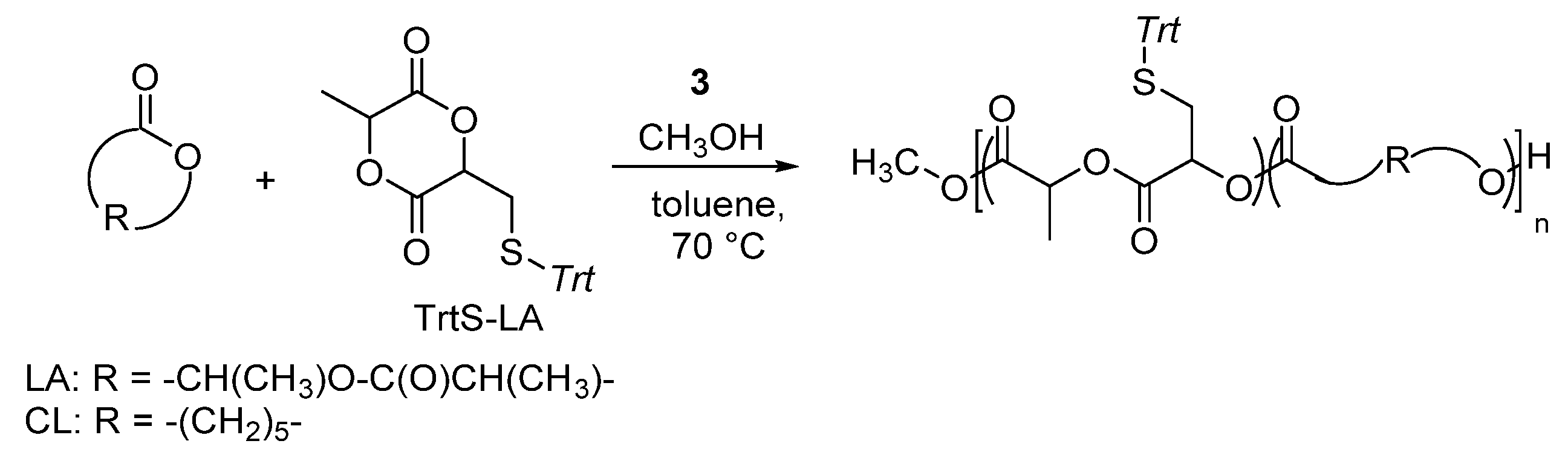
Dimethyl(salicylaldiminato)aluminum complex 3 was also described for the ROP of a functionalized cyclic diester. A lactide-type monomer bearing a thiol-protected group as trityl thioether, the 3-methyl-6-(tritylthiomethyl)-1,4-dioxane-2,5-dione (TrtS-LA) was indeed efficiently copolymerizated via ROP with LA and CL in the presence of complex 3 and 1 equivalent of MeOH as initiator, in toluene solution at 70 °C using a monomer-to-catalyst feed ratio of 100 to 1 (Scheme 8) [39].
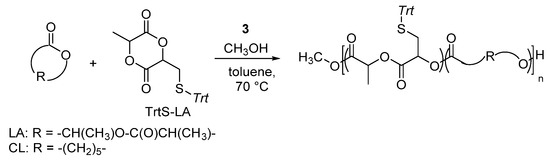
Scheme 8.
Ring-opening copolymerization of 3-methyl-6-(tritylthiomethyl)-1,4-dioxane-2,5-dione (TrtS-LA) with LA and/or CL by dimethyl(salicilaldiminato)aluminum compounds 3 [39].
By varying the co-monomers’ mixture and/or their molar ratio, different aliphatic polyesters with pendant tritylthiomethyl groups were synthesized in high yield (up to 92%). The composition nicely reflected the feed and the resulting TrtS-LA was always totally incorporated. Moreover, the analysis of the molecular weight by NMR and GPC revealed a good agreement between the experimental and the Mn. GPC traces displayed for all samples monomodal molecular weight distribution with narrow dispersities in the range 1.1–1.4. Complex 3 allowed good control over the chain growth during the copolymerization of TrtS-LA with LA and/or CL, without affecting the functional groups, under relatively mild conditions.
It is worth noting that the homopolymerization of the TrtS-LA in the same experimental conditions was not achieved. The analysis of the copolymer microstructure by 13C NMR revealed that consecutive TrtS-LA units were not incorporated in the copolymers, even when the functional monomers were fully converted. Average block lengths close to 1 were calculated for a copolymer prepared by copolymerizing a 50/50 CL/TrtS-LA mixture. Such a result indicated that isolated TrtS-LA monomeric units were incorporated during the copolymerization and, therefore, the resulting functional groups were well-spread along the polymeric chain.
Thus, the dimethyl(salicylaldiminato)aluminum complexes were able to promote the ROP of a thiol-protected functionalized lactide, without affecting the functionalities and with good incorporation rate [39].
9. Conclusions
In the last few decades, the degradable and biocompatible aliphatic poly(ester)s have appeared to be a more and more efficiently renewable alternative to petrol based plastics for a wide range of applications in various fields—from agriculture to highly sophisticated biomedical uses. Several methods and initiators have been developed for the production of these materials by ROP of cyclic esters. Although organocatalysis have been exploited in this research field and showed high potential to be developed further in force of the possibilities to avoid dangerous metals [60], at the moment some optimal features still remain a prerogative of metallorganic catalysis. Up to now, the ROP of cyclic esters in the presence of metal-based initiators still represent the most convenient method for the preparation of these materials with controlled and predictable molecular weights, stereochemistry, and end groups.
In this regard, a large variety of aluminum-based organometallic compounds have been described in the literature as initiators for the ROP of cyclic esters. In this field, salicylaldiminato aluminum complexes played a special role. Due to their less coordinatively-saturated nature with respect to the parent SALEN-type aluminum-based complexes, they have the potential to function as active catalysts. In addition to this, the synthesis of the salicylaldimine proligand is straightforward and allows an easy introduction of different steric and electronic features, thus opening the way to the tuning of catalyst performance by modification of the ligand framework. This class of catalysts appeared to be effective in the ring-opening homo and copolymerization of commercially available conventional lactones and lactides, such as GL, CL, and LA. Not only were the ROP of macrolactones and novel-synthetized functionalized cyclic esters by the same class of catalyst also successfully achieved in the presence of this class of catalysts. They also effectively became active catalysts in the preparation of copolymers of CL and LA, GL and CL, and GL and LA, affording various copolymeric structures from diblocks, to multiblock, to random. Moreover, the ter-polymerization of CL, LA, and GL was also achieved. In this regard, it is worth mentioning the ability of this class of catalysts to produce a large variety of copolymers microstructures, from random to blocks to multiblocks either in the case of commercial and traditional cyclic esters, or in the case of ‘non-conventional’ monomers, such as macrolactones and functionalized monomers. The versatility of this class of catalysts was ascertained in several homo- and copolymerizations, and will open the way to the synthesis of novel materials having different and peculiar features by further copolymerization of other different monomers. The ability of such complexes to readily tune the polymer features, producing polymers with narrow molecular weight dispersities, end group fidelity, and almost complete absence of transesterification side reactions, provides attractive options in the synthesis of advanced polymer architecture. Moreover, the simple formulation, straightforward synthesis, and easy activation are advantageous features with respect to other aluminum catalysts bearing more complex polydentate ligand systems. Metallorganic chemistry offers incredible possibilities to readily tune the ability of the catalysts by modifying the sterical and electronic peculiarities of complexes. Modification of the ligand framework will surely open the way to more different features. Just to mention few examples: tridentate NOO Schiff base ligand aluminum complexes have been synthetized, which displayed a bimetallic character and the ability to induce the stereoselective polymerization of rac-lactide to isotactic PLA (Pm = 0.76) [61]. The capability of this class of complexes to catalyze the polymerization of monomers other than cyclic esters—i.e., trimethylene carbonate—has been demonstrated [62]. In addition to that, Schiff-base macrocyclic systems that can bind in N,O fashion at the Al centre, have been also prepared and employed in the ROP of CL and LA, [63]. Very recently ther phenoxy-imine complexes bearing, inter-alia, N-alkyl and N-benzyl substituents were synthetized and employed in the ROP of cyclic esters [64].
The use of different metal with the same ligand framework can open new possibilities and enlarge the catalysts performances [65]. For instance, the possible presence of toxic metal in the materials which are made to be used in biomedical applications still remains a big concern, and methods to purify the polymers, and/or reduce the residual metal are required. Recent research efforts have been directed toward the search of less toxic—or more biocompatible—metal-based initiators [66]. Li and Na complexes bearing imino-phenoxide ligands, for example, have also been synthetized and used for the l-lactide polymerization [67]. Although the research area is flourishing, further studies need to be carried out in the near future before the materials could be commercialized and used in biomedical applications.
There are therefore still plenty of possibilities for the research, and we believe that spectacular results can be further achieved in this field.
Acknowledgments
The research was supported by the Italian Ministry of University and Research (Fondi di Ateneo per la Ricerca, FAR 2015, Università del Sannio).
Author Contributions
Tiziana Fuoco and Daniela Pappalardo equally contributed to the review of the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Scott, G.; Gilead, D. Degradable polymers. Principles and Applications; Chapman & Hall: London, UK, 1995. [Google Scholar]
- Jarrett, P.; Benedict, C.; Bell, J.P.; Cameron, J.A.; Huang, S.J. Mechanism of the biodegradation of polycaprolactone. In Polymers as Biomaterials; Shalaby, S.W., Hoffman, A.S., Ratner, B.D., Horbett, T.A., Eds.; Plenum Press: New York, NY, USA, 1984. [Google Scholar]
- Raquez, J.-M.; Mincheva, R.; Coulembier, O.; Dubois, P. Ring-opening polymerization of cyclic esters: Industrial synthesis, properties, applications, and perspectives. In Polymer Science: A Comprehensive Reference; Elsevier: Amsterdam, The Netherlands, 2012; pp. 761–777. [Google Scholar]
- Ikada, Y.; Tsuji, H. Biodegradable polyesters for medical and ecological applications. Macromol. Rapid Commun. 2000, 21, 117–132. [Google Scholar] [CrossRef]
- Albertsson, A.-C.; Varma, I.K. Recent developments in ring opening polymerization of lactones for biomedical applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef] [PubMed]
- Dechy-Cabaret, O.; Martin-Vaca, B.; Bourissou, D. Controlled ring-opening polymerization of lactide and glycolide. Chem. Rev. 2004, 104, 6147–6176. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.; Marshall, E.L. Metal complexes as catalysts for polymerization reactions. In Comprehensive Coordination Chemistry II; Ward, M.D., Ed.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 9, pp. 1–74. [Google Scholar]
- Wu, J.; Yu, T.-L.; Chen, C.-T.; Lin, C.-C. Recent developments in main group metal complexes catalyzed/initiated polymerization of lactides and related cyclic esters. Coord. Chem. Rev. 2006, 250, 602–626. [Google Scholar] [CrossRef]
- Thomas, C.M. Stereocontrolled ring-opening polymerization of cyclic esters: Synthesis of new polyester microstructures. Chem. Soc. Rev. 2010, 39, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, P.J.; Du, H.; Feijen, J. Single site catalysts for stereoselective ring-opening polymerization of lactides. Polym. Chem. 2011, 2, 520–527. [Google Scholar] [CrossRef]
- MacDonald, J.P.; Shaver, M.P. Aluminum salen and salan polymerization catalysts: from monomer scope to macrostructure control. In Green Polymer Chemistry: Biobased Materials and Biocatalysis; American Chemical Society: Washington, DC, USA, 2015; Volume 1192, pp. 147–167. [Google Scholar]
- Jianming, R.; Anguo, X.; Hongwei, W.; Hailin, Y. Review—Recent development of ring-opening polymerization of cyclic esters using aluminum complexes. Des. Monomers Polym. 2014, 1, 345–355. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, S.; Zhou, S. Aluminum alkyl complexes: Synthesis, structure, and application in ROP of cyclic esters. Dalton Trans. 2016, 45, 4471–4485. [Google Scholar] [CrossRef] [PubMed]
- Atwood, D.A.; Harvey, M.J. Group 13 compounds incorporating salen ligands. Chem. Rev. 2001, 101, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.A.; Gibson, V.C.; Irvine, D.J. Nickel-catalyzed generation of Schiff base aluminum enolate initiators for controlled methacrylate polymerization. Angew. Chem. Int. Ed. 2000, 39, 2141–2144. [Google Scholar] [CrossRef]
- Cameron, P.A.; Gibson, V.C.; Irvine, D.J. A Living Polymerisation Process. World Pat. Appl. WO 00/00525, 6 January 2000. [Google Scholar]
- Cameron, P.A.; Jhurry, D.; Gibson, V.C.; White, A.J.P.; Williams, D.J.; Williams, S. Controlled polymerization of lactides at ambient temperature using [5-Cl-salen]AlOMe. Macromol. Rapid Commun. 1999, 20, 616–618. [Google Scholar] [CrossRef]
- Irvine, D.J.; Gibson, V.C.; Cameron, P.A. Aluminium Compounds for Producing Vinylic Polymers. World Pat. Appl. WO 00/00496, 6 January 2000. [Google Scholar]
- Le Borgne, A.; Vincens, V.; Jouglard, M.; Spassky, N. Ring-opening oligomerization reactions using aluminium complexes of Schiff’s bases as initiators. Macromol. Symp. 1993, 73, 37–46. [Google Scholar] [CrossRef]
- Ovitt, T.M.; Coates, G.W. Stereoselective ring-opening polymerization of rac-lactide with a single-site, racemic aluminum alkoxide catalyst: Synthesis of stereoblock poly(lactic acid). J. Polym. Sci. Part A 2000, 38, 4686–4692. [Google Scholar] [CrossRef]
- Spassky, N.; Wisniewski, M.; Pluta, C.; Le Borgne, A. Highly stereoelective polymerization of rac-(d,l)-lactide with a chiral Schiff’s base/aluminium alkoxide initiator. Macromol. Chem. Phys. 1996, 197, 2627–2637. [Google Scholar] [CrossRef]
- Nomura, N.; Ishii, R.; Yamamoto, Y.; Kondo, T. Stereoselective ring-opening polymerization of a racemic lactide by using achiral salen- and homosalen-aluminum complexes. Chem. Eur. J. 2007, 13, 4433–4451. [Google Scholar] [CrossRef] [PubMed]
- Aeilts, S.L.; Coles, M.P.; Swenson, D.C.; Jordan, R.F.; Young, V.G. Aluminum alkyl complexes containing guanidinate ligands. Organometallics 1998, 17, 3265–3270. [Google Scholar] [CrossRef]
- Coles, M.P.; Swenson, D.C.; Jordan, R.F.; Young, V.G. Aluminum complexes incorporating bulky nitrogen and sulfur donor ligands. Organometallics 1998, 17, 4042–4048. [Google Scholar] [CrossRef]
- Ong, C.M.; Stephan, D.W. 1,2-Cyclopentadienyl diimine—Group 13 complexes. Inorg. Chem. 1999, 38, 5189–5191. [Google Scholar] [CrossRef] [PubMed]
- Gibson, V.C.; Redshaw, C.; White, A.J.P.; Williams, D.J. Synthesis and structural characterization of aluminium imino-amide and pyridyl-amide complexes: Bulky monoanionic N,N chelate ligands via methyl group transfer. J. Organomet. Chem. 1998, 550, 453–456. [Google Scholar] [CrossRef]
- Wissing, E.; Jastrzebski, J.T.B.H.; Boersma, J.; van Koten, G. An unexpected 1,2-alkyl shift within a chelate bonded organoaluminium-enamine. J. Organomet. Chem. 1993, 459, 11–16. [Google Scholar] [CrossRef]
- Radzewich, C.E.; Guzei, I.A.; Jordan, R.F. Three-coordinate cationic aluminum alkyl complexes incorporating β-diketiminate ligands. J. Am. Chem. Soc. 1999, 121, 8673–8674. [Google Scholar] [CrossRef]
- Cameron, P.A.; Gibson, V.C.; Redshaw, C.; Segal, J.A.; Solan, G.A.; White, A.J.P.; Williams, D.J. Synthesis and characterisation of neutral dialkylaluminium complexes stabilised by salicylaldiminato ligands, and their conversion to monoalkylaluminium cations. J. Chem. Soc. Dalton Trans. 2001. [Google Scholar] [CrossRef]
- Hogerheide, M.P.; Wesseling, M.; Jastrzebski, J.T.B.H.; Boersma, J.; Kooijman, H.; Spek, A.L.; van Koten, G. Versatility in phenolate bonding in organoaluminum complexes containing mono- and bis-ortho-chelating phenolate ligands. X-ray structures of Al{OC6H2(CH2NMe2)2-2,6-Me-4}3, Al(Me)2{OC6H2(CH2NMe2)2-2,6-Me-4}·N-AlMe3, and Al(Me)2{OC6H2(CH2NMe2)2-2,6-Me-4}·N-AlMe3·O-AlMe3. Organometallics 1995, 14, 4483–4492. [Google Scholar]
- Tian, J.; Hustad, P.D.; Coates, G.W. A new catalyst for highly syndiospecific living olefin polymerization: Homopolymers and block copolymers from ethylene and propylene. J. Am. Chem. Soc. 2001, 123, 5134–5135. [Google Scholar] [CrossRef] [PubMed]
- Saito, J.; Mitani, M.; Mohri, J.-i.; Ishii, S.-i.; Yoshida, Y.; Matsugi, T.; Kojoh, S.-i.; Kashiwa, N.; Fujita, T. Highly syndiospecific living polymerization of propylene using a titanium complex having two phenoxy-imine chelate ligands. Chem. Lett. 2001, 30, 576–577. [Google Scholar] [CrossRef]
- Pappalardo, D.; Tedesco, C.; Pellecchia, C. New neutral and cationic dialkylaluminium complexes bearing imino-amide or imino-phenoxide ligands: Synthesis, characterization and reactivity with olefins. Eur. J. Inorg. Chem. 2002, 2002, 621–628. [Google Scholar] [CrossRef]
- Annunziata, L.; Pappalardo, D.; Tedesco, C.; Pellecchia, C. Octahedral bis(phenoxy-imine)tin(IV) alkyl complexes: Synthesis, characterization, and reactivity toward ionizing species and ethylene. Organometallics 2005, 24, 1947–1952. [Google Scholar] [CrossRef]
- Baugh, L.S.; Sissano, J.A. Polymerization of methyl methacrylate and other polar monomers with alkylaluminum initiators bearing bidentate and tridentate N- and O-donor ligands. J. Polym. Sci. Part A 2002, 40, 1633–1651. [Google Scholar] [CrossRef]
- Pappalardo, D.; Annunziata, L.; Pellecchia, C. Living ring-opening homo- and copolymerization of ε-caprolactone and l- and d,l-lactides by dimethyl(salicylaldiminato)aluminum compounds. Macromolecules 2009, 42, 6056–6062. [Google Scholar] [CrossRef]
- Meduri, A.; Fuoco, T.; Lamberti, M.; Pellecchia, C.; Pappalardo, D. Versatile copolymerization of glycolide and rac-lactide by dimethyl(salicylaldiminato)aluminum compounds. Macromolecules 2014, 47, 534–543. [Google Scholar] [CrossRef]
- Fuoco, T.; Meduri, A.; Lamberti, M.; Venditto, V.; Pellecchia, C.; Pappalardo, D. Ring-opening polymerization of ω-6-hexadecenlactone by a salicylaldiminato aluminum complex: A route to semicrystalline and functional poly(ester)s. Polym. Chem. 2015, 6, 1727–1740. [Google Scholar] [CrossRef]
- Fuoco, T.; Finne-Wistrand, A.; Pappalardo, D. A route to aliphatic poly(ester)s with thiol pendant groups: From monomer design to editable porous scaffolds. Biomacromolecules 2016, 17, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Iwasa, N.; Nomura, K. Synthesis of Al complexes containing phenoxy-imine ligands and their use as the catalyst precursors for efficient living ring-opening polymerisation of ε-caprolactone. Dalton Trans. 2008. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, N.; Katao, S.; Liu, J.; Fujiki, M.; Furukawa, Y.; Nomura, K. Notable effect of fluoro substituents in the imino group in ring-opening polymerization of ε-caprolactone by Al complexes containing phenoxyimine ligands. Organometallics 2009, 28, 2179–2187. [Google Scholar] [CrossRef]
- Iwasa, N.; Fujiki, M.; Nomura, K. Ring-opening polymerization of various cyclic esters by Al complex catalysts containing a series of phenoxy-imine ligands: Effect of the imino substituents for the catalytic activity. J. Mol. Catal. A 2008, 292, 67–75. [Google Scholar] [CrossRef]
- Iwasa, N.; Liu, J.; Nomura, K. Notable effect of imino substituent for the efficient ring-opening polymerization of ε-caprolactone initiated by Al complexes containing phenoxy-imine ligand of type, Me2Al(L) [L: O-2-tBu-6-(RNCH)C6H3; R: 2,6-iPr2C6H3, tBu, adamantyl, C6F5]. Catal. Commun. 2008, 9, 1148–1152. [Google Scholar] [CrossRef]
- Fuoco, T.; Meduri, A.; Lamberti, M.; Pellecchia, C.; Pappalardo, D. Copolymerization and terpolymerization of glycolide with lactones by dimethyl(salicylaldiminato)aluminum compounds. J. Appl. Polym. Sci. 2015, 132, 42567. [Google Scholar] [CrossRef]
- Nomura, N.; Aoyama, T.; Ishii, R.; Kondo, T. Salicylaldimine−aluminum complexes for the facile and efficient ring-opening polymerization of ε-caprolactone. Macromolecules 2005, 38, 5363–5366. [Google Scholar] [CrossRef]
- Normand, M.; Dorcet, V.; Kirillov, E.; Carpentier, J.-F. {Phenoxy-imine}aluminum versus -indium complexes for the immortal ROP of lactide: Different stereocontrol, different mechanisms. Organometallics 2013, 32, 1694–1709. [Google Scholar] [CrossRef]
- Normand, M.; Roisnel, T.; Carpentier, J.-F.; Kirillov, E. Dinuclear vs. mononuclear complexes: Accelerated, metal-dependent ring-opening polymerization of lactide. Chem. Commun. 2013, 49, 11692–11694. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.-C.; Lu, W.-Y.; Chang, H.-Y.; Lai, Y.-C.; Chiang, M.Y.; Chen, H.-Y.; Chen, H.-Y. Comparative study of aluminum complexes bearing N,O- and N,S-Schiff base in ring-opening polymerization of ε-caprolactone and l-lactide. Inorg. Chem. 2015, 54, 11292–11298. [Google Scholar] [CrossRef] [PubMed]
- Partridge, M.; Davidson, M.; Eade, G. Polymerisation Reaction and Catalyst Therefor. World Pat. Appl. WO 2004052980, 24 June 2004. [Google Scholar]
- Nomura, N.; Ishii, R.; Akakura, M.; Aoi, K. Stereoselective ring-opening polymerization of racemic lactide using aluminum-achiral ligand complexes: Exploration of a chain-end control mechanism. J. Am. Chem. Soc. 2002, 124, 5938–5939. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca, H.; Ward, I.M. Structure and mechanical properties of PGA crystals and fibres. Polymer 2006, 47, 7070–7077. [Google Scholar] [CrossRef]
- Chujo, K.; Kobayashi, H.; Suzuki, J.; Tokuhara, S.; Tanabe, M. Ring-opening polymerization of glycolide. Makromol. Chem. 1967, 100, 262–266. [Google Scholar] [CrossRef]
- Pan, Z.; Ding, J. Poly(lactide-co-glycolide) porous scaffolds for tissue engineering and regenerative medicine. Inter. Focus 2012, 2, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Stayshich, R.M.; Meyer, T.Y. New insights into poly(lactic-co-glycolic acid) microstructure: Using repeating sequence copolymers to decipher complex NMR and thermal behavior. J. Am. Chem. Soc. 2010, 132, 10920–10934. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Stayshich, R.M.; Meyer, T.Y. Exploiting sequence to control the hydrolysis behavior of biodegradable PLGA copolymers. J. Am. Chem. Soc. 2011, 133, 6910–6913. [Google Scholar] [CrossRef] [PubMed]
- Dobrzynski, P.; Kasperczyk, J.; Janeczek, H.; Bero, M. Synthesis of biodegradable copolymers with the use of low toxic zirconium compounds. 1. Copolymerization of glycolide with l-lactide initiated by Zr(Acac)4. Macromolecules 2001, 34, 5090–5098. [Google Scholar] [CrossRef]
- Hodge, P.; Colquhoun, H.M. Recent work on entropically-driven ring-opening polymerizations: Some potential applications. Polym. Adv. Technol. 2005, 16, 84–94. [Google Scholar] [CrossRef]
- Strandman, S.; Gautrot, J.E.; Zhu, X.X. Recent advances in entropy-driven ring-opening polymerizations. Polym. Chem. 2011, 2, 791–799. [Google Scholar] [CrossRef]
- Hodge, P. Entropically driven ring-opening polymerization of strainless organic macrocycles. Chem. Rev. 2014, 114, 2278–2312. [Google Scholar] [CrossRef] [PubMed]
- Dove, A.P. Organic catalysis for ring-opening polymerization. ACS Macro Lett. 2012, 1, 1409–1412. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Karroonnirun, O. Stereoselective ring-opening polymerization of rac-lactides catalyzed by chiral and achiral aluminum half-salen complexes. Organometallics 2010, 29, 5627–5634. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Karroonnirun, O.; Wilson, S.J. Ring-opening polymerization of cyclic esters and trimethylene carbonate catalyzed by aluminum half-salen complexes. Inorg. Chem. 2011, 50, 6775–6787. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhao, K.-Q.; Prior, T.J.; Hughes, D.L.; Arbaoui, A.; Elsegood, M.R.J.; Redshaw, C. Structural studies of Schiff-base [2 + 2] macrocycles derived from 2,2′-oxydianiline and the ROP capability of their organoaluminium complexes. Dalton Trans. 2016, 45, 11990–12005. [Google Scholar] [CrossRef] [PubMed]
- Redshaw, C.; Wang, X.; Zhao, K.-Q.; Al-Khafaji, Y.; Mo, S.; Prior, T.J.; Elsegood, M.R.J. Organoaluminium complexes derived from Anilines or Schiff bases for ring opening polymerization of epsilon-caprolactone, delta-valerolactone and rac-lactide. Eur. J. Inorg. Chem. 2017. [Google Scholar] [CrossRef]
- Pradeepa, C.P.; Dasb, S.K. Coordination and supramolecular aspects of the metal complexes of chiral N-salicyl-β-amino alcohol Schiff base ligands: Towards understanding the roles of weak interactions in their catalytic reactions. Coordin. Chem. Rev. 2013, 257, 1699–1715. [Google Scholar] [CrossRef]
- Gorrasi, G.; Meduri, A.; Rizzarelli, P.; Carroccio, S.; Curcuruto, G.; Pellecchia, C.; Pappalardo, D. Preparation of poly(glycolide-co-lactide)s through a green process: Analysis of structural, thermal, and barrier properties. React. Funct. Polym. 2016, 109, 70–78. [Google Scholar] [CrossRef]
- Lu, W.-Y.; Hsiao, M.-W.; Hsu, S.C.N.; Peng, W.-T.; Chang, Y.-J.; Tsou, Y.-C.; Wu, T.-Y.; Lai, Y.-C.; Chen, Y.; Chen, H.-Y. Synthesis, characterization and catalytic activity of lithium and sodium iminophenoxide complexes towards ring-opening polymerization of l-lactide. Dalton Trans. 2012, 41, 3659–3667. [Google Scholar] [CrossRef] [PubMed]
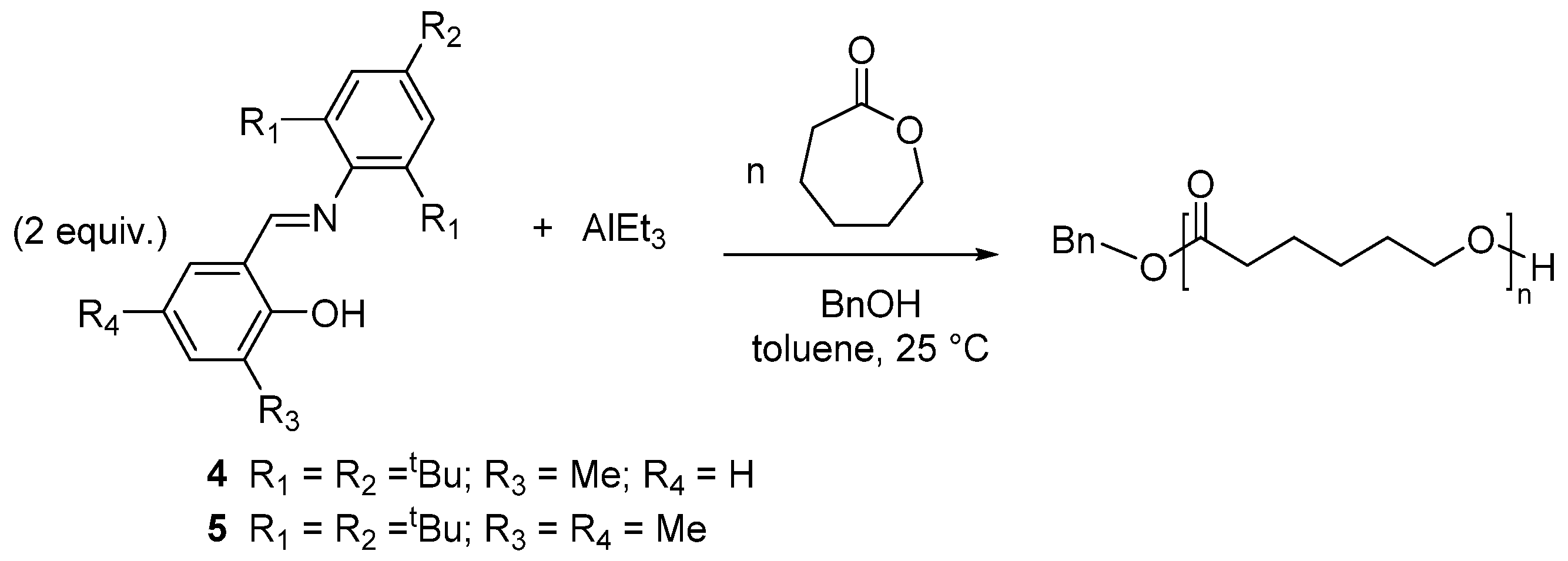
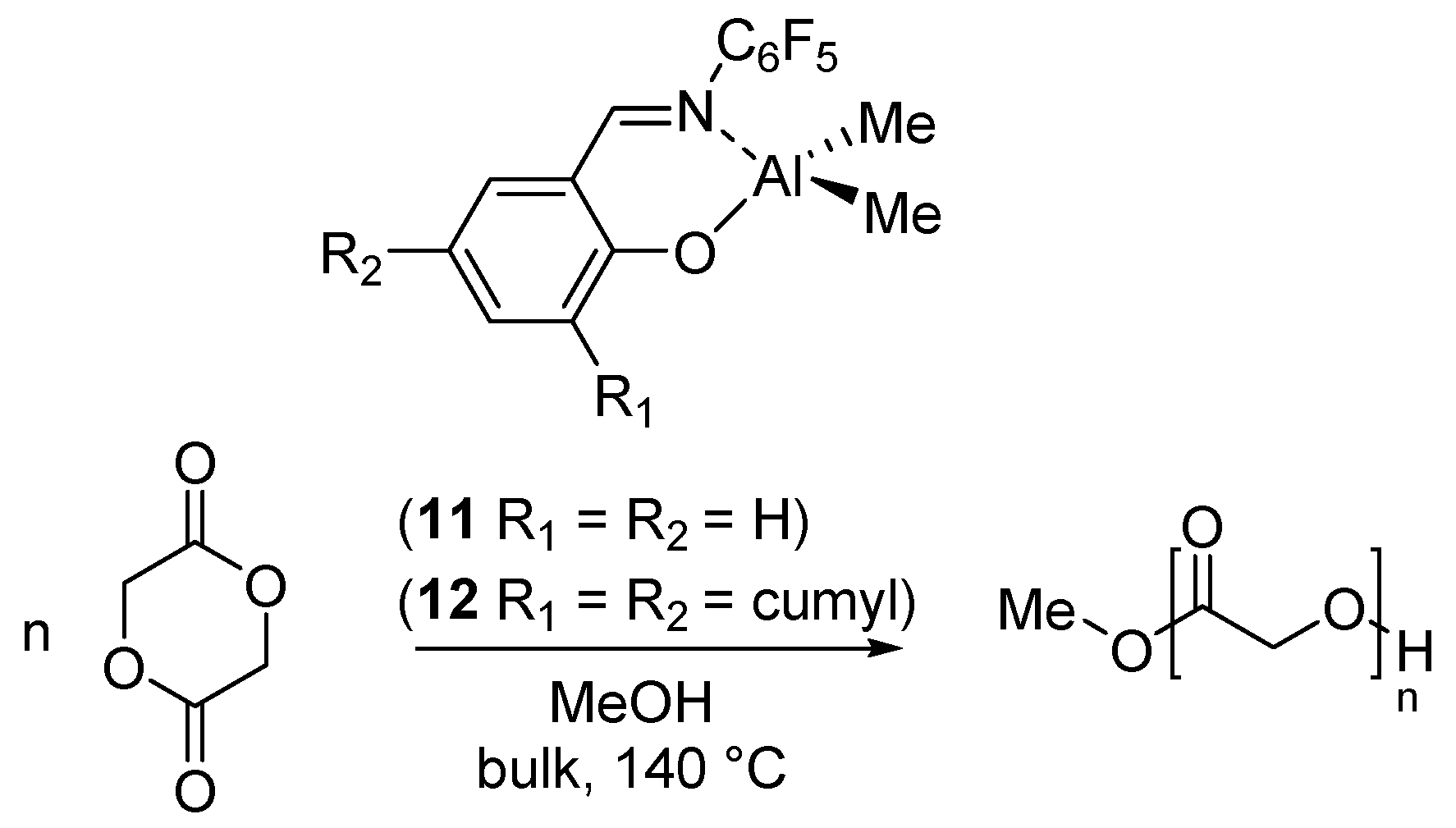
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).