Process Intensification of Alkynol Semihydrogenation in a Tube Reactor Coated with a Pd/ZnO Catalyst



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
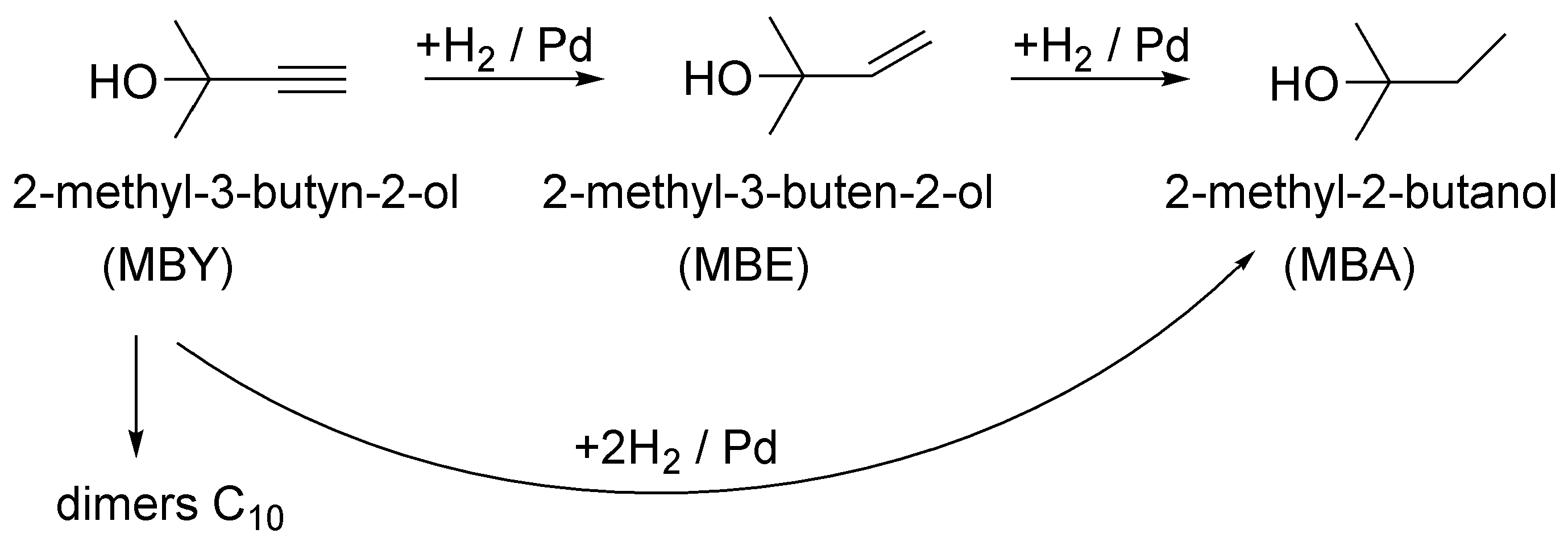
2. Results and Discussion
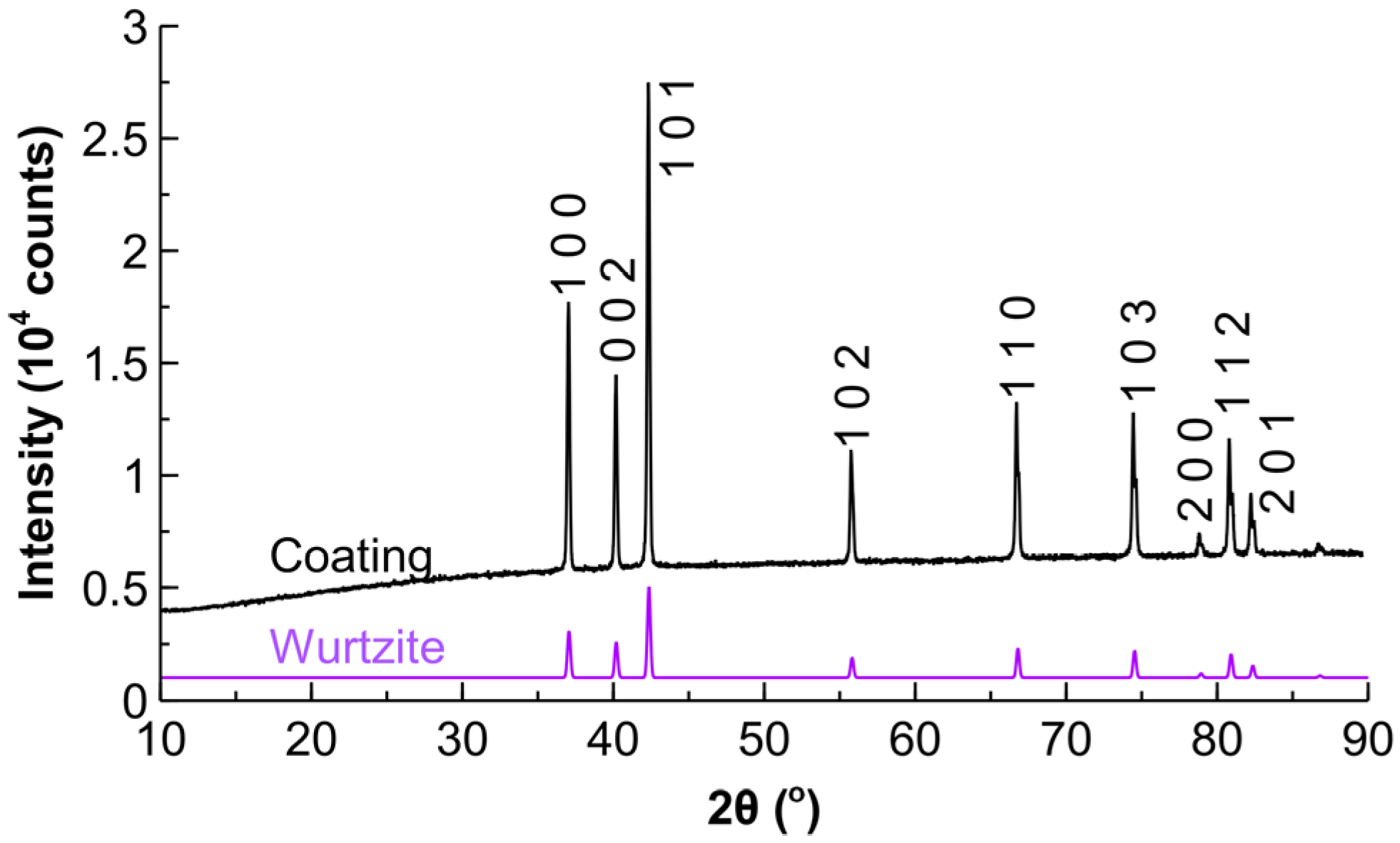
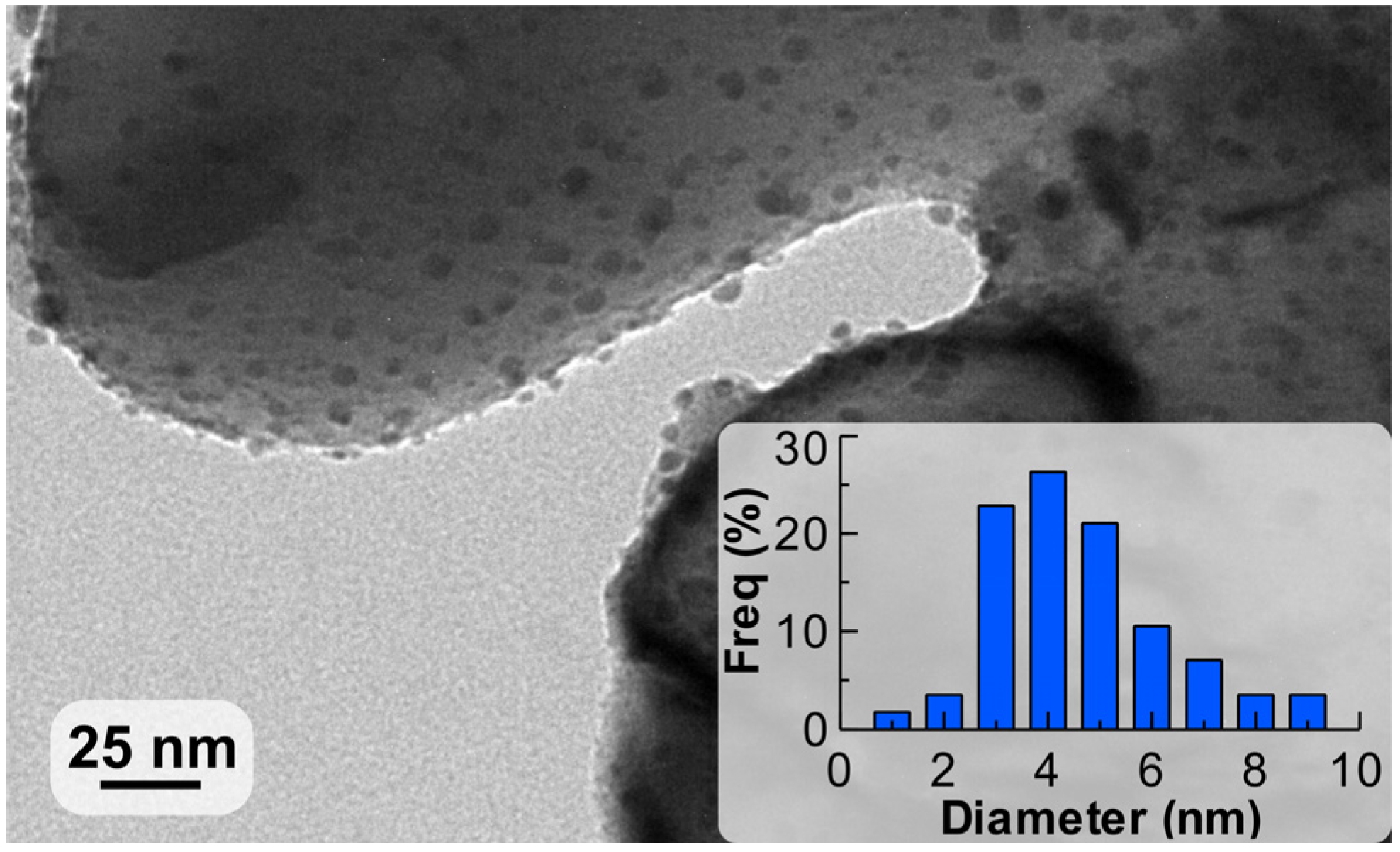
2.1. Characterisation
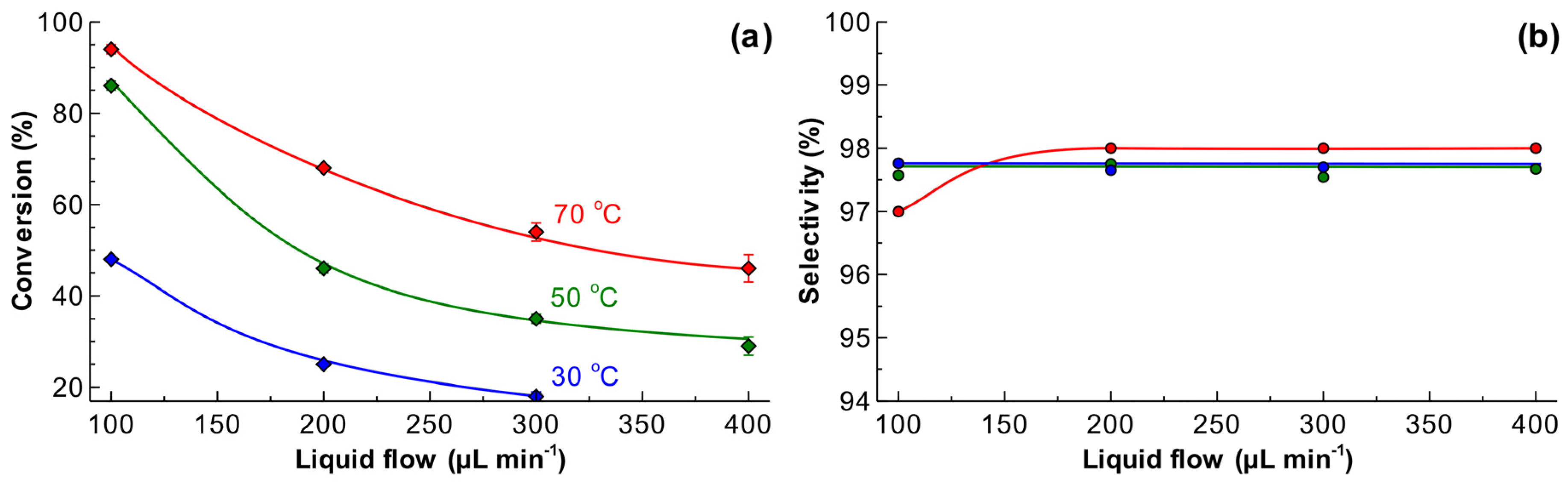
2.2. Effect of Liquid Flow Rates
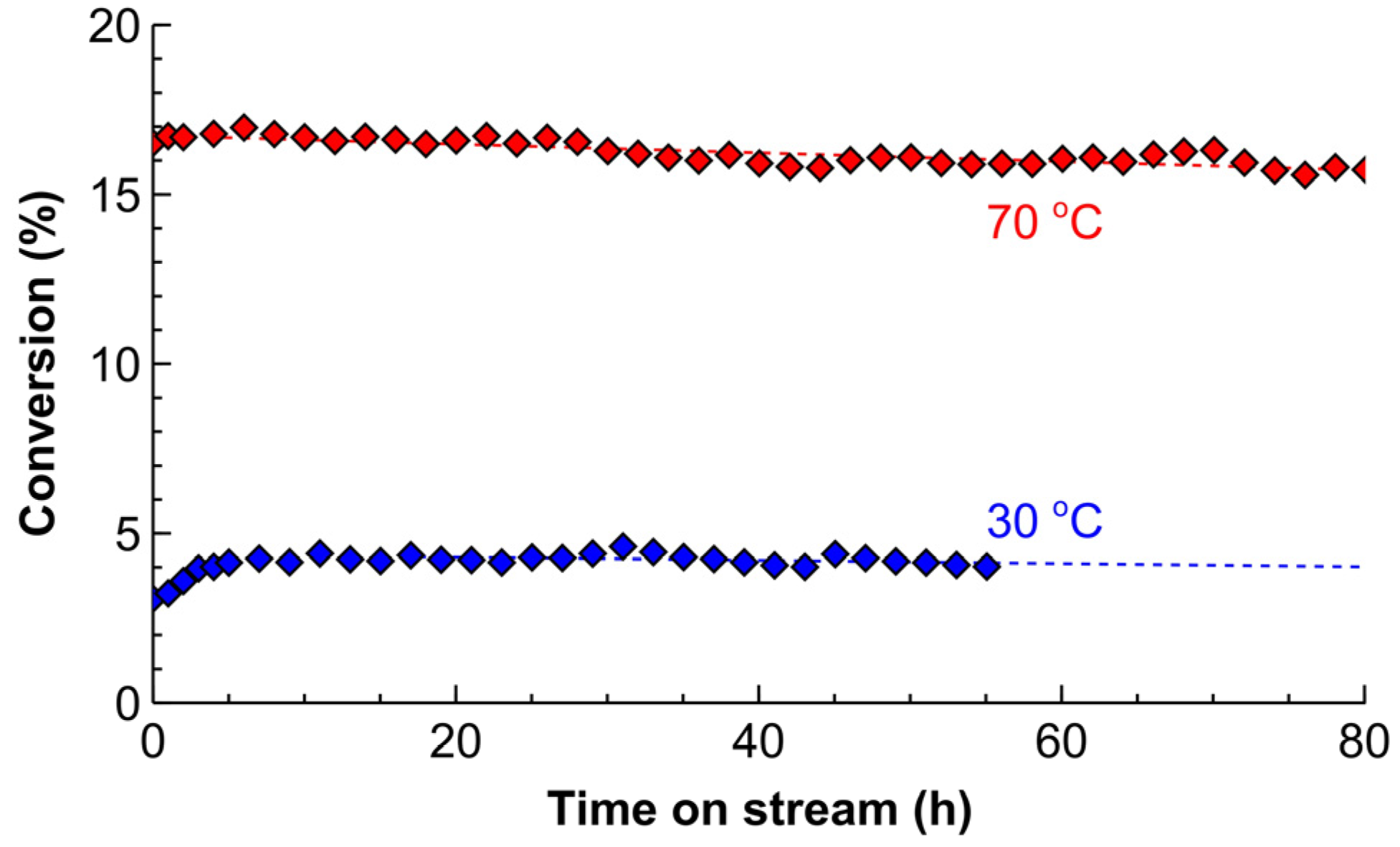
2.3. Catalyst Deactivation
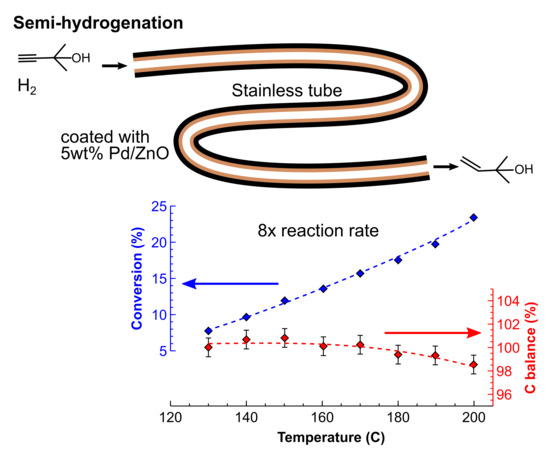
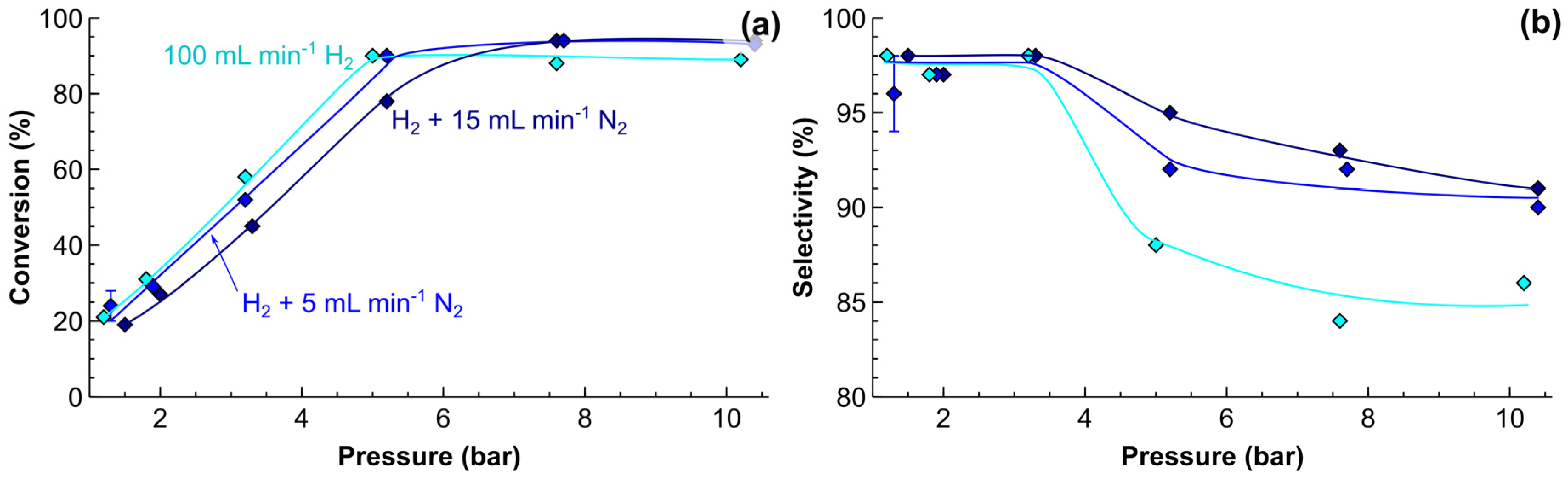
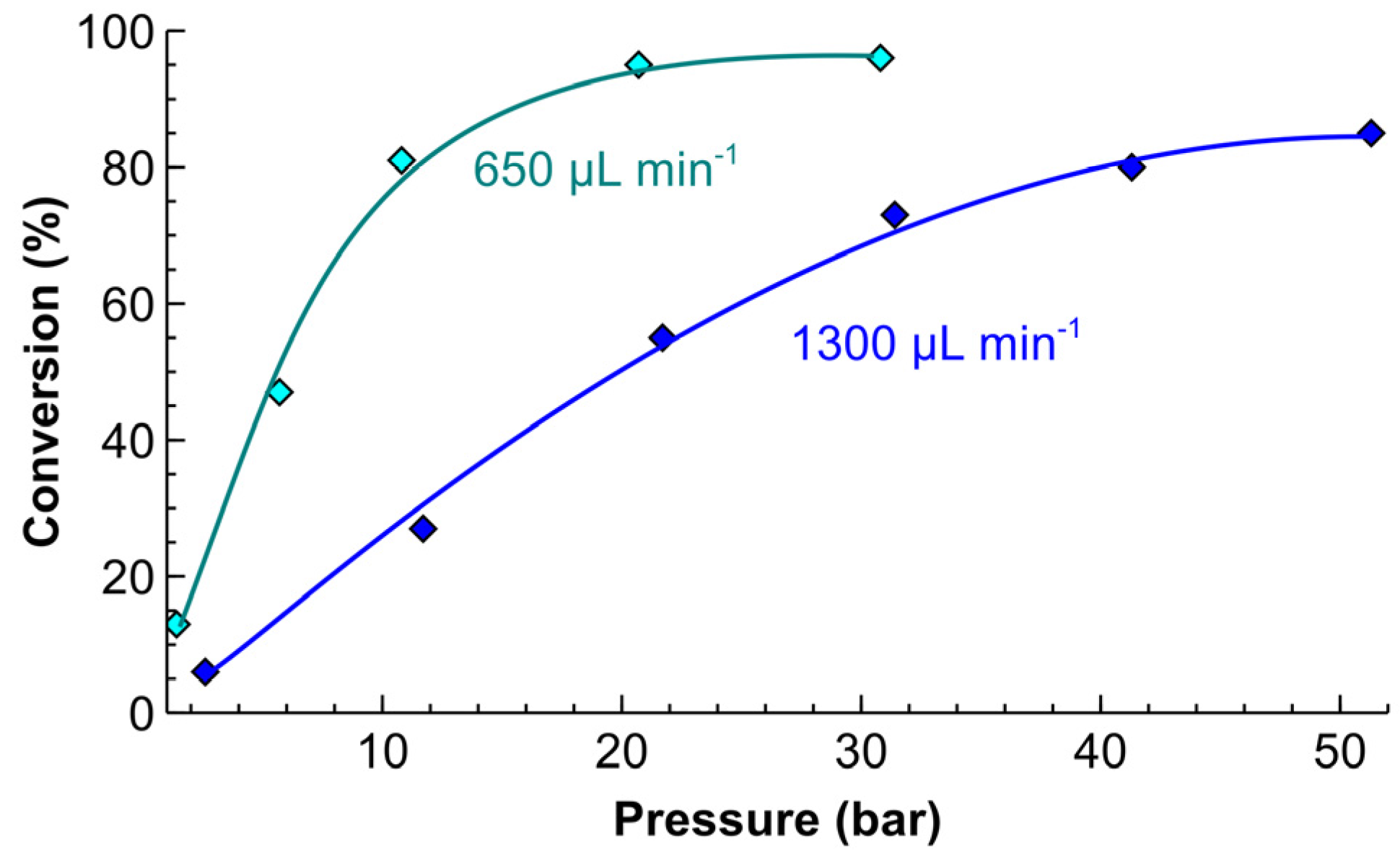
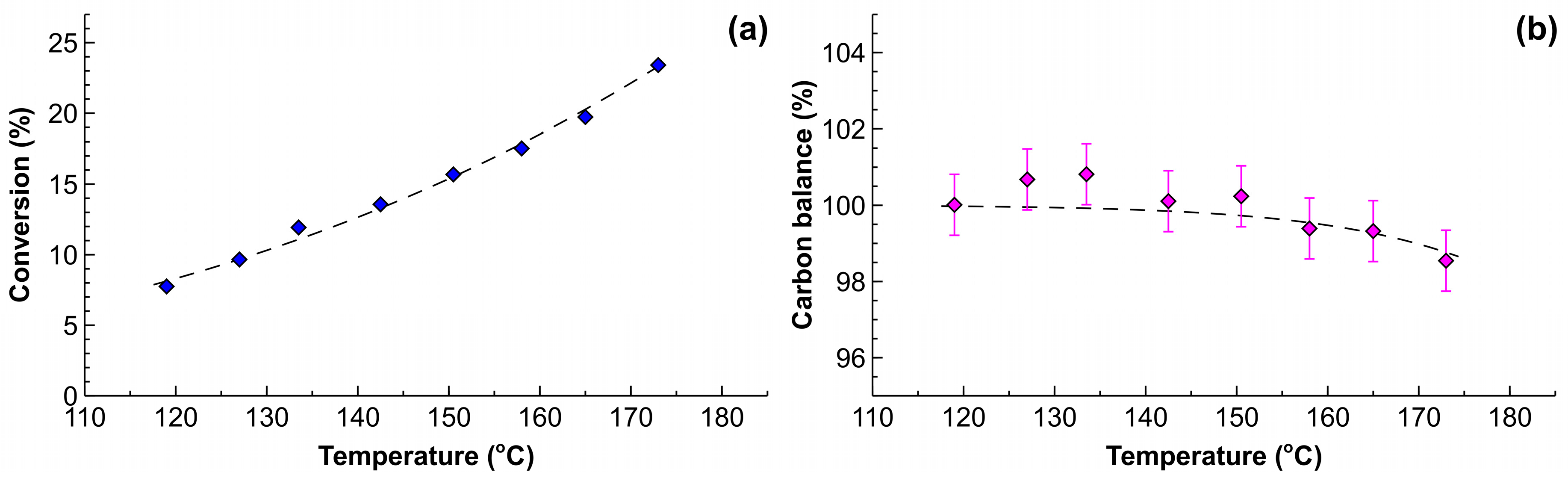
2.4. Process Intensification
3. Materials and Methods
3.1. Characterisation
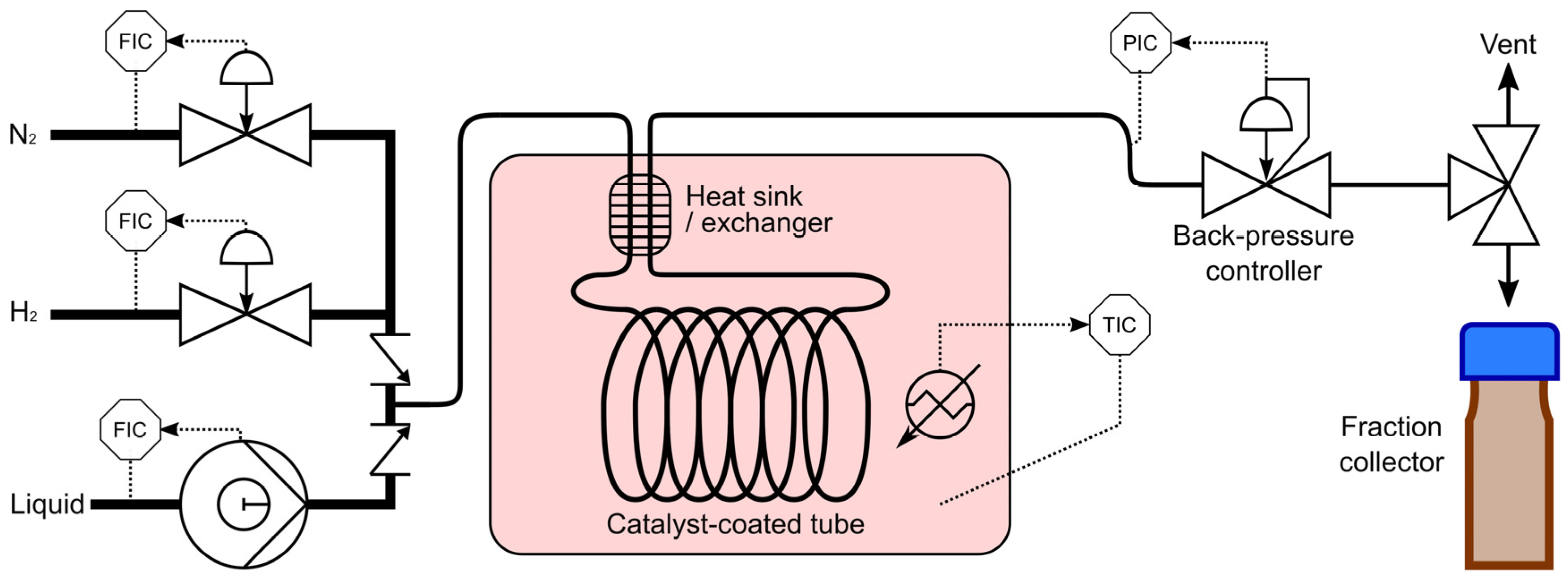
3.2. Catalyst Testing
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jähnisch, K.; Hessel, V.; Löwe, H.; Baerns, M. Chemistry in microstructured reactors. Angew. Chem. Int. Ed. 2004, 43, 406–446. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, G.S.; Pissavini, S. From Batch to Continuous Flow Processing in Chemicals Manufacturing. AIChE J. 2011, 57, 828–834. [Google Scholar] [CrossRef]
- Roberge, D.M.; Ducry, L.; Bieler, N.; Cretton, P.; Zimmermann, B. Microreactor Technology: A Revolution for the Fine Chemical and Pharmaceutical Industries? Chem. Eng. Technol. 2005, 28, 318–323. [Google Scholar] [CrossRef]
- Ford, M.E. (Ed.) Catalysis of Organic Reactions; Marcel Dekker: New York, NY, USA, 2001; ISBN 082470486X. [Google Scholar]
- Irfan, M.; Glasnov, T.N.; Kappe, C.O. Heterogeneous catalytic hydrogenation reactions in continuous-flow reactors. ChemSusChem 2011, 4, 300–316. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.F. Flow chemistry-Microreaction technology comes of age. AIChE J. 2017, 63, 858–869. [Google Scholar] [CrossRef]
- Chatterjee, S. FDA Perspective on Continuous Manufacturing. In Proceedings of the IFPAC Annual Meeting, Baltimore, MD, USA, 22–25 January 2012. [Google Scholar]
- Lawrence Yu Continuous Manufacturing Has a Strong Impact on Drug Quality. Available online: https://blogs.fda.gov/fdavoice/index.php/2016/04/continuous-manufacturing-has-a-strong-impact-on-drug-quality/ (accessed on 6 July 2017).
- Deadman, B.J.; Collins, S.G.; Maguire, A.R. Taming Hazardous Chemistry in Flow: The Continuous Processing of Diazo and Diazonium Compounds. Chem. A Eur. J. 2015, 21, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Hessel, V.; Kralisch, D.; Kockmann, N.; Noël, T.; Wang, Q. Novel process windows for enabling, accelerating, and uplifting flow chemistry. ChemSusChem 2013, 6, 746–789. [Google Scholar] [CrossRef] [PubMed]
- Ehrfeld, W.; Hessel, V.; Löwe, H. Microreactors; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000; ISBN 3527295909. [Google Scholar]
- Pastre, J.C.; Browne, D.L.; Ley, S.V. Flow chemistry syntheses of natural products. Chem. Soc. Rev. 2013, 42, 8849–8869. [Google Scholar] [CrossRef] [PubMed]
- Frost, C.G.; Mutton, L. Heterogeneous catalytic synthesis using microreactor technology. Green Chem. 2010, 12, 1687–1703. [Google Scholar] [CrossRef]
- Mason, B.P.; Price, K.E.; Steinbacher, J.L.; Bogdan, A.R.; McQuade, T.D.; McQuade, D.T. Greener approaches to organic synthesis using microreactor technology. Chem. Rev. 2007, 107, 2300–2318. [Google Scholar] [CrossRef] [PubMed]
- Wakami, H.; Yoshida, J. Grignard Exchange Reaction Using a Microflow System: From Bench to Pilot Plant. Org. Process Res. Dev. 2005, 9, 787–791. [Google Scholar] [CrossRef]
- Zhang, X.; Stefanick, S.; Villani, F.J. Application of Microreactor Technology in Process Development. Org. Process Res. Dev. 2004, 8, 455–460. [Google Scholar] [CrossRef]
- Müller, S.T.R.; Wirth, T. Diazo Compounds in Continuous-Flow Technology. ChemSusChem 2015, 8, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Pieber, B.; Kappe, C.O. Generation and Synthetic Application of Trifluoromethyl Diazomethane Utilizing Continuous Flow Technologies. Org. Lett. 2016, 18, 1076–1079. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.S.; Laffan, D.; Thomson, C.; Williams, M.T. Analysis of the reactions used for the preparation of drug candidate molecules. Org. Biomol. Chem. 2006, 4, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Bonrath, W.; Medlock, J.; Schutz, J.; Wüstenberg, B.; Netscher, T. Hydrogenation in the Vitamins and Fine Chemicals Industry—An Overview. In Hydrogenation; InTech: Rijeka, Yugoslavia, 2012; pp. 69–90. ISBN 978-953-51-0785-9. [Google Scholar]
- Bonrath, W.; Eggersdorfer, M.; Netscher, T. Catalysis in the industrial preparation of vitamins and nutraceuticals. Catal. Today 2007, 121, 45–57. [Google Scholar] [CrossRef]
- Eggersdorfer, M.; Laudert, D.; Létinois, U.; McClymont, T.; Medlock, J.; Netscher, T.; Bonrath, W. One hundred years of vitamins-a success story of the natural sciences. Angew. Chem. Int. Ed. 2012, 51, 12960–12990. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.A.; Mellor, J.; Wells, R.K.P.K. Pd catalysed hexyne hydrogenation modified by Bi and by Pb. J. Catal. 2009, 261, 208–216. [Google Scholar] [CrossRef]
- Cherkasov, N.; Ibhadon, A.O.; McCue, A.; Anderson, J.A.; Johnston, S.K. Palladium–bismuth intermetallic and surface-poisoned catalysts for the semi-hydrogenation of 2-methyl-3-butyn-2-ol. Appl. Catal. A Gen. 2015, 497, 22–30. [Google Scholar] [CrossRef]
- Coq, B.; Figueras, F. Bimetallic palladium catalysts: influence of the co-metal on the catalyst performance. J. Mol. Catal. A Chem. 2001, 173, 117–134. [Google Scholar] [CrossRef]
- López, N.; Vargas-Fuentes, C. Promoters in the hydrogenation of alkynes in mixtures: insights from density functional theory. Chem. Commun. 2012, 48, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Cherkasov, N.; Cravotto, G.; Borretto, E.; Ibhadon, A.O.; Medlock, J.; Bonrath, W. Ultrasound- and Microwave-Assisted Preparation of Lead-Free Palladium Catalysts: Effects on the Kinetics of Diphenylacetylene Semi-Hydrogenation. ChemCatChem 2015, 7, 952–959. [Google Scholar] [CrossRef]
- Cherkasov, N.; Ibhadon, A.O.; Rebrov, E.V. Solvent-free Semihydrogenation of Acetylene Alcohols in a Capillary Reactor coated with a Pd-Bi/TiO2 Catalyst. Appl. Catal. A Gen. 2016, 515, 108–115. [Google Scholar] [CrossRef]
- Nijhuis, T.A.; van Koten, G.; Moulijn, J.A. Optimized palladium catalyst systems for the selective liquid-phase hydrogenation of functionalyzed alkynes. Appl. Catal. A Gen. 2003, 238, 259–271. [Google Scholar] [CrossRef]
- Kundu, A.; Saroha, A.K.; Nigam, K.D.P. Liquid distribution studies in trickle-bed reactors. Chem. Eng. Sci. 2001, 56, 5963–5967. [Google Scholar] [CrossRef]
- Van Herk, D.; Castaño, P.; Makkee, M.; Moulijn, J.A.; Kreutzer, M.T. Catalyst testing in a multiple-parallel, gas–liquid, powder-packed bed microreactor. Appl. Catal. A Gen. 2009, 365, 199–206. [Google Scholar] [CrossRef]
- Elias, Y.; Rudolf von Rohr, P.; Bonrath, W.; Medlock, J.; Buss, A. A porous structured reactor for hydrogenation reactions. Chem. Eng. Process. Process Intensif. 2015, 95, 175–185. [Google Scholar] [CrossRef]
- Kapteijn, F.; Nijhuis, T.A.; Heiszwolf, J.J.; Moulijn, J.A. New non-traditional multiphase catalytic reactors based on monolithic structures. Catal. Today 2001, 66, 133–144. [Google Scholar] [CrossRef]
- Tsoligkas, A.N.; Simmons, M.J.H.; Wood, J. Influence of orientation upon the hydrodynamics of gas-liquid flow for square channels in monolith supports. Chem. Eng. Sci. 2007, 62, 4365–4378. [Google Scholar] [CrossRef]
- Tsoligkas, A.N.; Simmons, M.J.H.; Wood, J.; Frost, C.G. Kinetic and selectivity studies of gas-liquid reaction under Taylor flow in a circular capillary. Catal. Today 2007, 128, 36–46. [Google Scholar] [CrossRef]
- Cherkasov, N.; Ibhadon, A.O.; Rebrov, E.V. Novel synthesis of thick wall coatings of titania supported Bi poisoned Pd catalysts and application in selective hydrogenation of acetylene alcohols in capillary microreactors. Lab Chip 2015, 15, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Rebrov, E.V.; Berenguer-Murcia, A.; Wheatley, A.E.H.; Johnson, B.F.G.; Schouten, J.C. Thin catalytic coatings on microreactor walls A way to make industrial processes more efficient. Chim. Oggi 2009, 27, 4–7. [Google Scholar]
- Rebrov, E.V.; Klinger, E.A.; Berenguer-murcia, A.; Sulman, E.M.; Schouten, J.C. Selective Hydrogenation of 2-Methyl-3-butyne-2-ol in a Wall-Coated Capillary Microreactor with a Pd2.5Zn7.5/TiO2 Catalyst. Org. Process Res. Dev. 2009, 13, 991–998. [Google Scholar] [CrossRef]
- Protasova, L.N.; Rebrov, E.V.; Skelton, H.E.; Wheatley, A.E.H.; Schouten, J.C. A kinetic study of the liquid-phase hydrogenation of citral on Au/TiO2 and Pt–Sn/TiO2 thin films in capillary microreactors. Appl. Catal. A Gen. 2011, 399, 12–21. [Google Scholar] [CrossRef]
- Bakker, J.J.W.; Zieverink, M.M.P.; Reintjens, R.W.E.G.; Kapteijn, F.; Moulijn, J.A.; Kreutzer, M.T. Heterogeneously Catalyzed Continuous-Flow Hydrogenation Using Segmented Flow in Capillary Columns. ChemCatChem 2011, 3, 1155–1157. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, N.; Al-Rawashdeh, M.; Ibhadon, A.O.; Rebrov, E.V. Scale up study of capillary microreactors in solvent-free semihydrogenation of 2-methyl-3-butyn-2-ol. Catal. Today 2016, 273, 205–212. [Google Scholar] [CrossRef]
- Li, D.; Haneda, H. Morphologies of zinc oxide particles and their effects on photocatalysis. Chemosphere 2003, 51, 129–137. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, L.; Bao, Y.; Zhang, Y.; Wang, J.; Fu, M.; Wu, J.; Ye, D. The Applications of Morphology Controlled ZnO in Catalysis. Catalysts 2016, 6, 188. [Google Scholar] [CrossRef]
- Johnston, S.K.; Cherkasov, N.; Pérez-Barrado, E.; Aho, A.; Murzin, D.Y.; Ibhadon, A.O.; Francesconi, M.G. Pd3Sn nanoparticles on TiO2 and ZnO supports as catalysts for semi-hydrogenation: Synthesis and catalytic performance. Appl. Catal. A Gen. 2017, 544, 40–45. [Google Scholar] [CrossRef]
- Okhlopkova, L.B.; Cherepanova, S.V.; Prosvirin, I.P.; Kerzhentsev, M.A.; Ismagilov, Z.R. Semi-hydrogenation of 2-methyl-3-butyn-2-ol on Pd-Zn nanoalloys prepared by polyol method: Effect of composition and heterogenizationNo Title. Appl. Catal. A 2018, 549, 245–253. [Google Scholar] [CrossRef]
- Semagina, N.; Grasemann, M.; Xanthopoulos, N.; Renken, A.; Kiwi-Minsker, L. Structured catalyst of Pd/ZnO on sintered metal fibers for 2-methyl-3-butyn-2-ol selective hydrogenation. J. Catal. 2007, 251, 213–222. [Google Scholar] [CrossRef]
- Borodziński, A.; Bonarowska, M. Relation between Crystallite Size and Dispersion on Supported Metal Catalysts. Langmuir 1997, 13, 5613–5620. [Google Scholar] [CrossRef]
- Crespo-Quesada, M.; Grasemann, M.; Semagina, N.; Renken, A.; Kiwi-Minsker, L. Kinetics of the solvent-free hydrogenation of 2-methyl-3-butyn-2-ol over a structured Pd-based catalyst. Catal. Today 2009, 147, 247–254. [Google Scholar] [CrossRef]
- Rebrov, E.V.; Berenguer-Murcia, A.; Skelton, H.E.; Johnson, B.F.G.; Wheatley, A.E.H.; Schouten, J.C. Capillary microreactors wall-coated with mesoporous titania thin film catalyst supports. Lab Chip 2009, 9, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Okhlopkova, L.B.; Matus, E.V.; Prosvirin, I.P.; Kerzhentsev, M.A.; Ismagilov, Z.R. Selective hydrogenation of 2-methyl-3-butyn-2-ol catalyzed by embedded polymer-protected PdZn nanoparticles. J. Nanoparticle Res. 2015, 17, 1–15. [Google Scholar] [CrossRef]
- Shen, L.; Mao, S.; Li, J.; Li, M.; Chen, P.; Li, H.; Chen, Z.; Wang, Y. PdZn intermetallic on a CN@ZnO hybrid as an efficient catalyst for the semihydrogenation of alkynols. J. Catal. 2017, 350, 13–20. [Google Scholar] [CrossRef]
- Vernuccio, S.; Goy, R.; Meier, A.; Rudolf von Rohr, P.; Medlock, J. Kinetics and mass transfer of the hydrogenation of 2-methyl-3-butyn-2-ol in a structured Pd/ZnO/Al2O3 reactor. Chem. Eng. J. 2017, 316, 121–130. [Google Scholar] [CrossRef]
- Silvestre-Albero, J.; Rupprechter, G.; Freund, H. Atmospheric pressure studies of selective 1,3-butadiene hydrogenation on Pd single crystals: effect of CO addition. J. Catal. 2005, 235, 52–59. [Google Scholar] [CrossRef]
- Bos, A.N.R.; Westerterp, K.R. Mechanism and kinetics of the selective hydrogenation of ethyne and ethene. Chem. Eng. Process. Process Intensif. 1993, 32, 1–7. [Google Scholar] [CrossRef]
- Kennedy, D.R.; Webb, G.; Jackson, S.D.; Lennon, D. Propyne hydrogenation over alumina-supported palladium and platinum catalysts. Appl. Catal. A Gen. 2004, 259, 109–120. [Google Scholar] [CrossRef]
- Tew, M.W.; Emerich, H.; van Bokhoven, J.A. Formation and Characterization of PdZn Alloy: A Very Selective Catalyst for Alkyne Semihydrogenation. J. Phys. Chem. C 2011, 115, 8457–8465. [Google Scholar] [CrossRef]
- Duca, D.; Liotta, L.F.; Deganello, G. Selective hydrogenation of phenylacetylene on pumice-supported palladium catalysts. J. Catal. 1995, 154, 69–79. [Google Scholar] [CrossRef]
- Singh, U.K.; Vannice, M.A. Kinetics of liquid-phase hydrogenation reactions over supported metal catalysts—A review. Appl. Catal. A Gen. 2001, 213, 1–24. [Google Scholar] [CrossRef]
- Günther, A.; Khan, S.A.; Thalmann, M.; Trachel, F.; Jensen, K.F. Transport and reaction in microscale segmented gas—Liquid flow. Lab Chip 2004, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Warnier, M.J.F. Taylor Flow Hydrodynamics in Gas-Liquid-Solid Micro Reactors. Ph.D. Thesis, Eindhoven University of Technology, Eindhoven, The Netherlands, 2009. [Google Scholar]
- Albers, P.; Pietsch, J.; Parker, S.F. Poisoning and deactivation of palladium catalysts. J. Mol. Catal. A Chem. 2001, 173, 275–286. [Google Scholar] [CrossRef]
- Flahive, E.J.; Ewanicki, B.L.; Sach, N.W.; O’Neill-Slawecki, S.A.; Stankovic, N.S.; Yu, S.; Guinness, S.M.; Dunn, J. Development of an effective palladium removal process for VEGF oncology candidate AG13736 and a simple, efficient screening technique for scavenger reagent identification. Org. Process Res. Dev. 2008, 12, 637–645. [Google Scholar] [CrossRef]
- Panpranot, J.; Phandinthong, K.; Praserthdam, P.; Hasegawa, M.; Fujita, S.; Arai, M. A comparative study of liquid-phase hydrogenation on Pd/SiO2 in organic solvents and under pressurized carbon dioxide: Activity change and metal leaching/sintering. J. Mol. Catal. A Chem. 2006, 253, 20–24. [Google Scholar] [CrossRef]
- Nikoshvili, L.; Shimanskaya, E.; Bykov, A.; Yuranov, I.; Kiwi-Minsker, L.; Sulman, E. Selective hydrogenation of 2-methyl-3-butyn-2-ol over Pd-nanoparticles stabilized in hypercrosslinked polystyrene: Solvent effect. Catal. Today 2015, 241, 179–188. [Google Scholar] [CrossRef]
- Al-Rawashdeh, M.; Fluitsma, L.J.M.; Nijhuis, T.A.; Rebrov, E.V.; Hessel, V.; Schouten, J.C. Design criteria for a barrier-based gas-liquid flow distributor for parallel microchannels. Chem. Eng. J. 2012, 181–182, 549–556. [Google Scholar] [CrossRef]
- Al-Rawashdeh, M.; Yu, F.; Nijhuis, T.A.; Rebrov, E.V.; Hessel, V.; Schouten, J.C. Numbered-up gas–liquid micro/milli channels reactor with modular flow distributor. Chem. Eng. J. 2012, 207–208, 645–655. [Google Scholar] [CrossRef]
- Teschner, D.; Vass, E.; Havecker, M.; Zafeiratos, S.; Schnorch, P.; Sauer, H.; Knopgericke, A.; Schlogl, R.; Chamam, M.; Wootsch, A. Alkyne hydrogenation over Pd catalysts: A new paradigm. J. Catal. 2006, 242, 26–37. [Google Scholar] [CrossRef]
- Teschner, D.; Borsodi, J.; Kis, Z.; Szentmiklósi, L.; Révay, Z.; Knop-Gericke, A.; Schlögl, R.; Torres, D.; Sautet, P. Role of Hydrogen Species in Palladium-Catalyzed Alkyne Hydrogenation. J. Phys. Chem. C 2010, 114, 2293–2299. [Google Scholar] [CrossRef]
- Teschner, D.; Révay, Z.; Borsodi, J.; Hävecker, M.; Knop-Gericke, A.; Schlögl, R.; Milroy, D.; Jackson, S.D.; Torres, D.; Sautet, P. Understanding palladium hydrogenation catalysts: When the nature of the reactive molecule controls the nature of the catalyst active phase. Angew. Chem. 2008, 47, 9274–9278. [Google Scholar] [CrossRef] [PubMed]
- Teschner, D.; Borsodi, J.; Wootsch, A.; Révay, Z.; Hävecker, M.; Knop-Gericke, A.; Jackson, S.D.; Schlögl, R. The roles of subsurface carbon and hydrogen in palladium-catalyzed alkyne hydrogenation. Science 2008, 320, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Shokouhimehr, M. Magnetically Separable and Sustainable Nanostructured Catalysts for Heterogeneous Reduction of Nitroaromatics. Catalysts 2015, 5, 534–560. [Google Scholar] [CrossRef]
- Cherkasov, N.; Jadvani, V.; Mann, J.; Losovyj, Y.B.; Shifrina, Z.B.; Bronstein, L.M.; Rebrov, E.V. Hydrogenation of bio-oil into higher alcohols over Ru/Fe3O4-SiO2 catalysts. Fuel Process. Technol. 2017, 167, 738–746. [Google Scholar] [CrossRef]
- Argyle, M.; Bartholomew, C. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Mitsudome, T.; Takahashi, Y.; Ichikawa, S.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Metal-ligand core-shell nanocomposite catalysts for the selective semihydrogenation of alkynes. Angew. Chem. Int. Ed. 2013, 52, 1481–1485. [Google Scholar] [CrossRef] [PubMed]
- Molnár, Á.; Sárkány, A.; Varga, M. Hydrogenation of carbon–carbon multiple bonds: Chemo-, regio-and stereo-selectivity. J. Mol. Catal. A Chem. 2001, 173, 185–221. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]











© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cherkasov, N.; Bai, Y.; Rebrov, E. Process Intensification of Alkynol Semihydrogenation in a Tube Reactor Coated with a Pd/ZnO Catalyst. Catalysts 2017, 7, 358. https://doi.org/10.3390/catal7120358
Cherkasov N, Bai Y, Rebrov E. Process Intensification of Alkynol Semihydrogenation in a Tube Reactor Coated with a Pd/ZnO Catalyst. Catalysts. 2017; 7(12):358. https://doi.org/10.3390/catal7120358
Chicago/Turabian StyleCherkasov, Nikolay, Yang Bai, and Evgeny Rebrov. 2017. "Process Intensification of Alkynol Semihydrogenation in a Tube Reactor Coated with a Pd/ZnO Catalyst" Catalysts 7, no. 12: 358. https://doi.org/10.3390/catal7120358
APA StyleCherkasov, N., Bai, Y., & Rebrov, E. (2017). Process Intensification of Alkynol Semihydrogenation in a Tube Reactor Coated with a Pd/ZnO Catalyst. Catalysts, 7(12), 358. https://doi.org/10.3390/catal7120358