
What Can We Learn in Electrocatalysis, from Nanoparticulated Precious and/or Non-Precious Catalytic Centers Interacting with Their Support?



Abstract
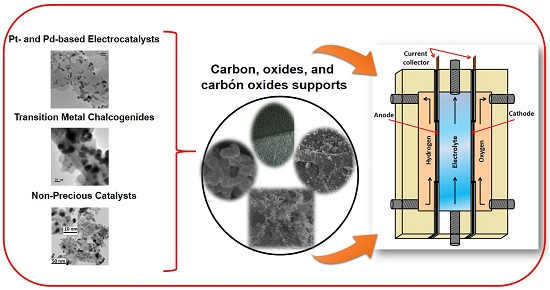
:
1. Introduction
2. Materials Design
2.1. Precious Catalysts
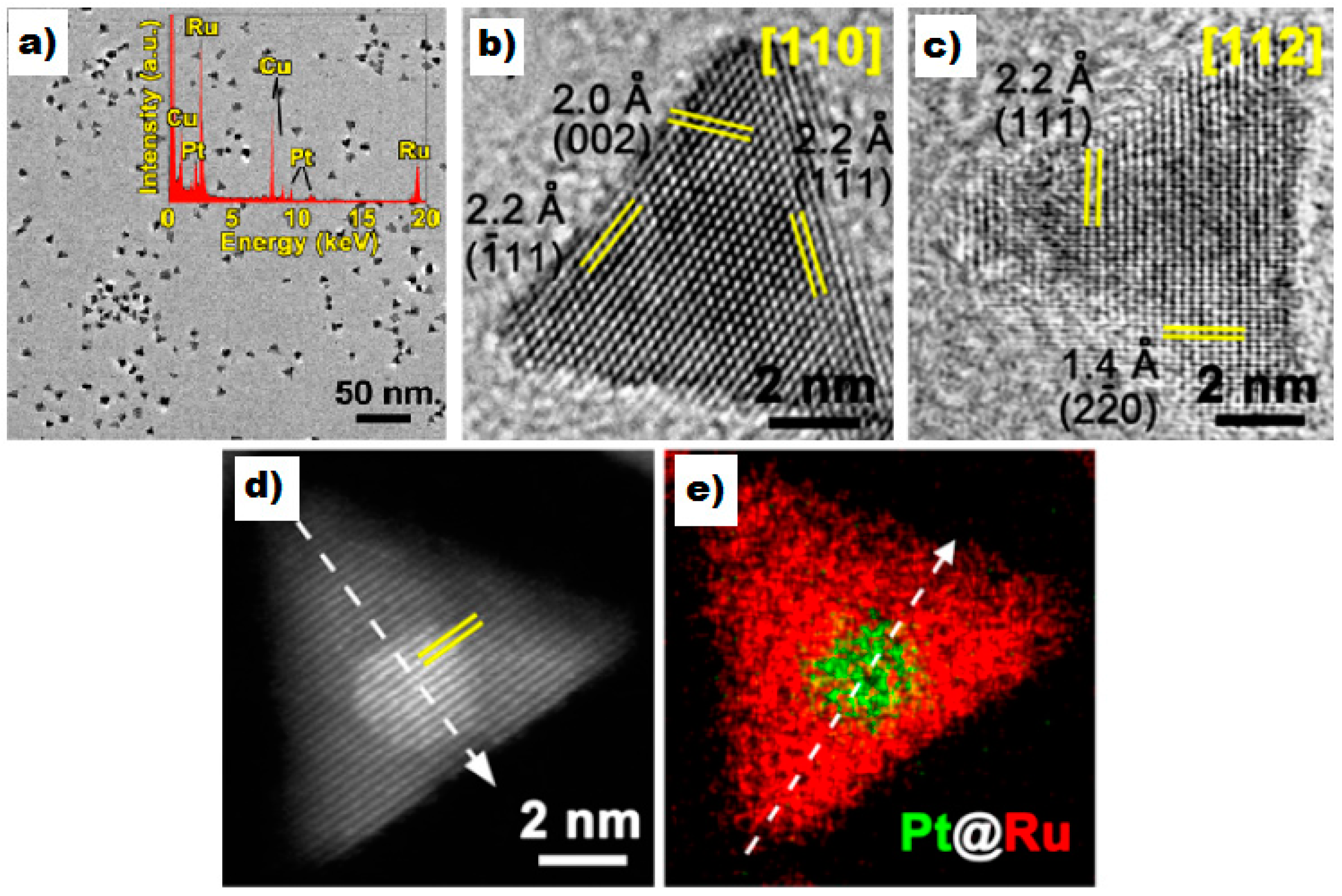
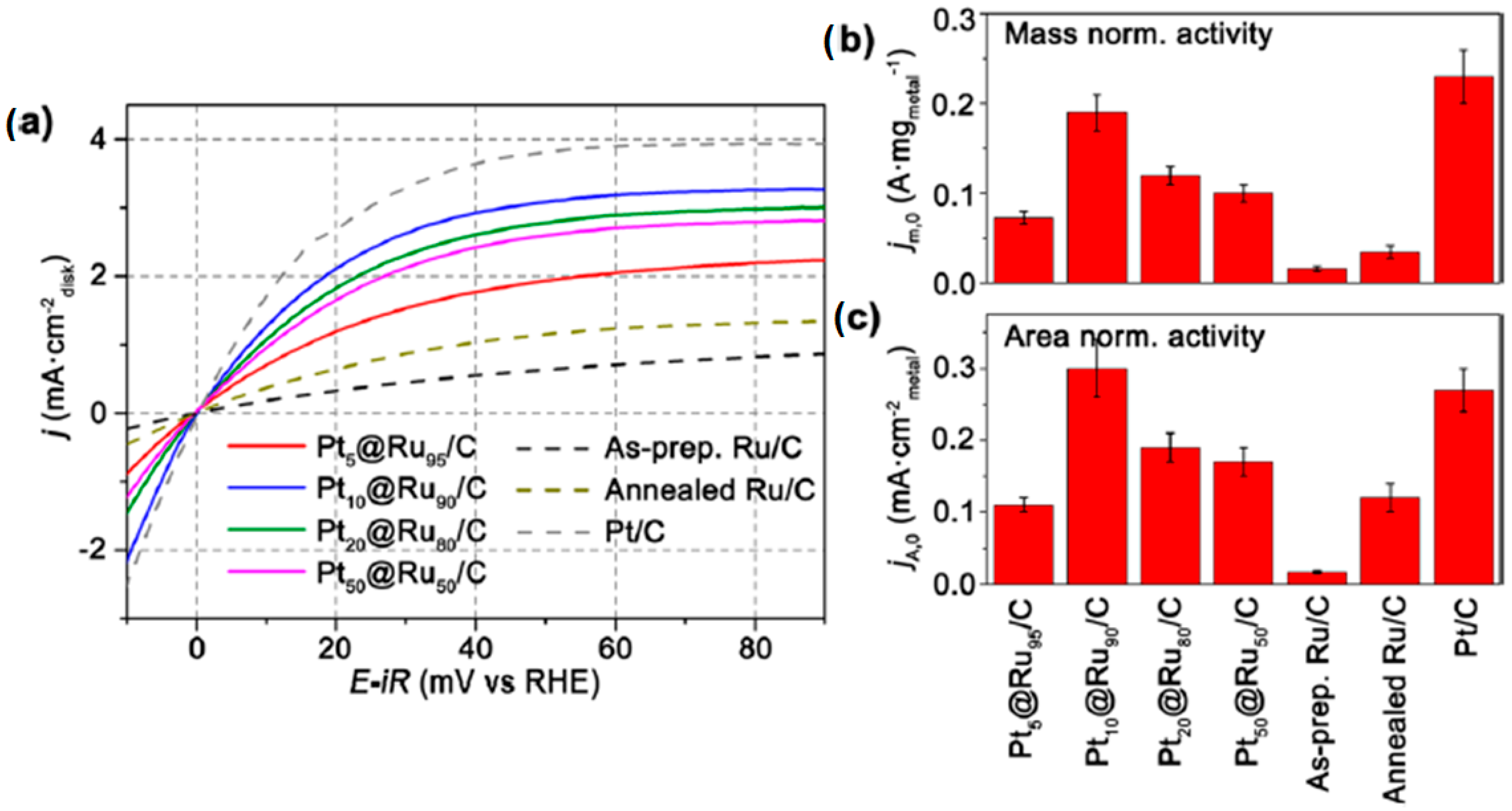
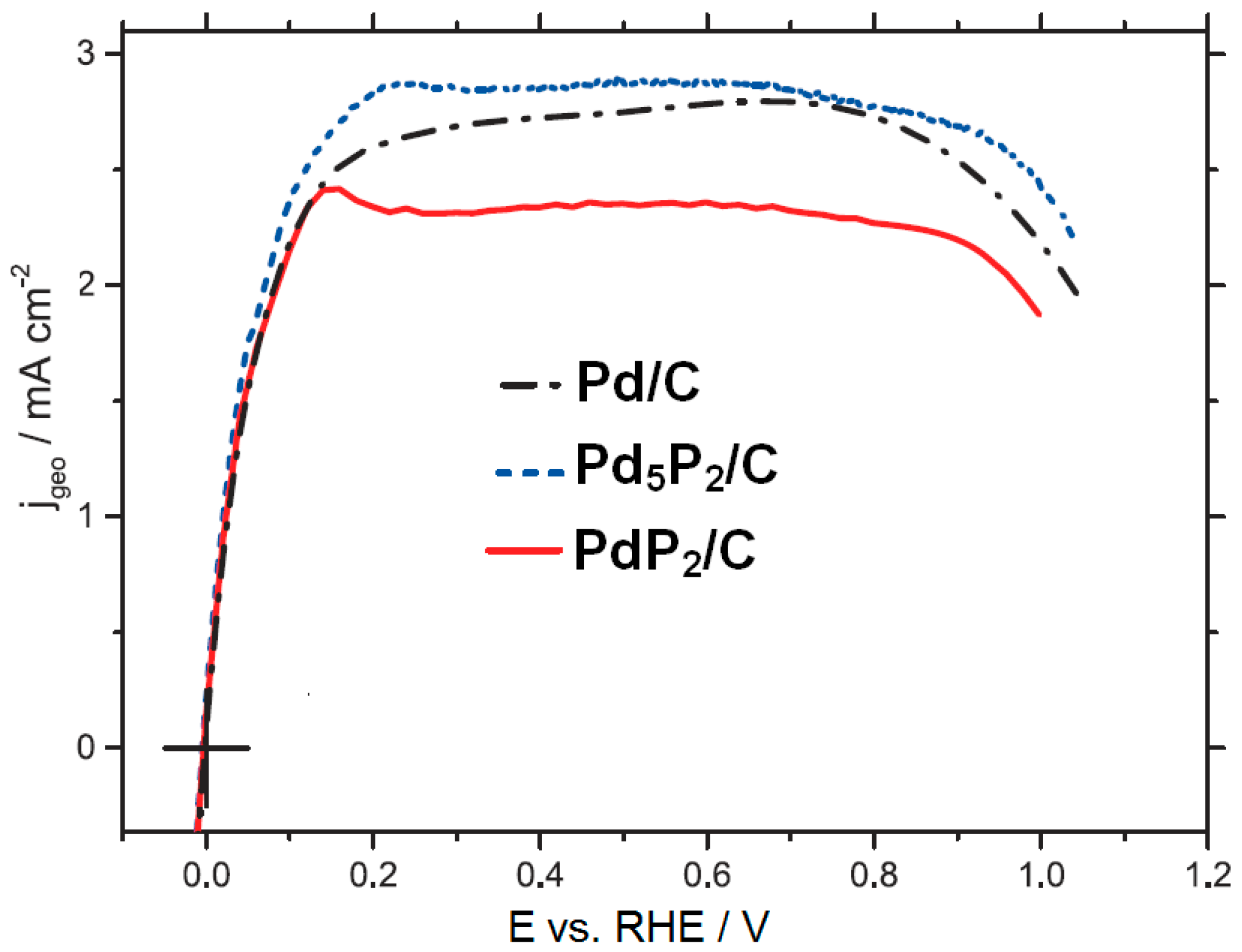
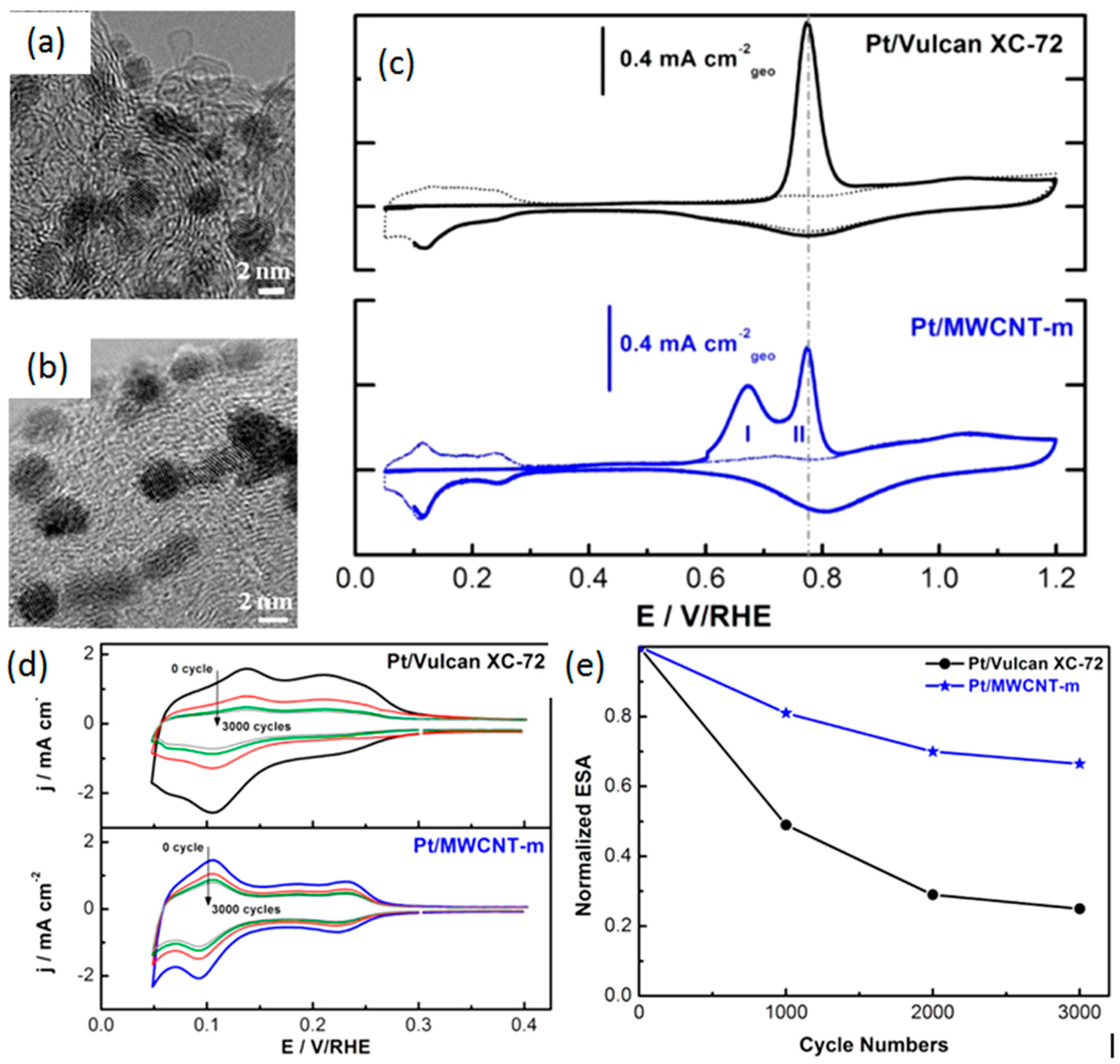
2.1.1. Pt- and Pd- Based Electrocatalysts
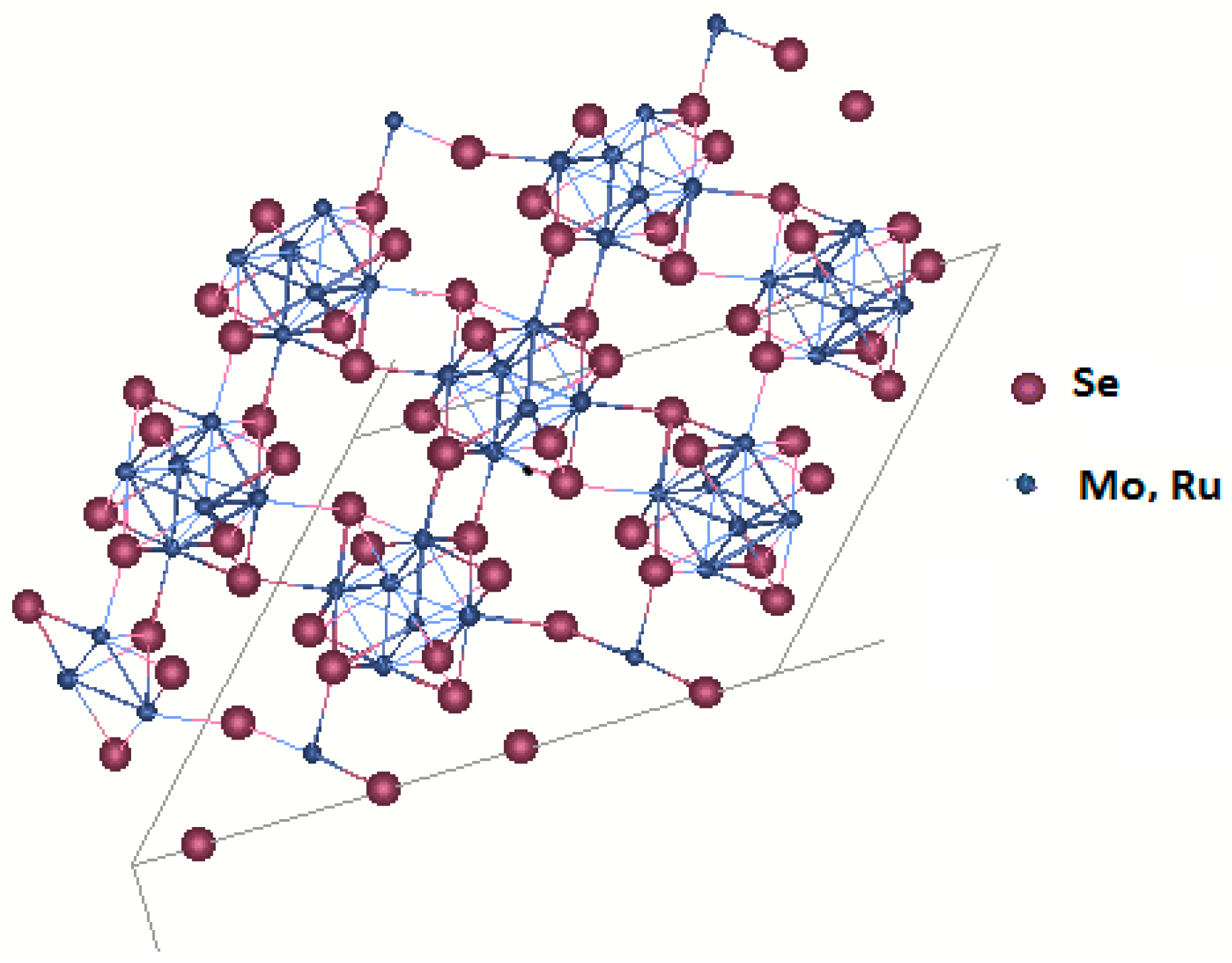
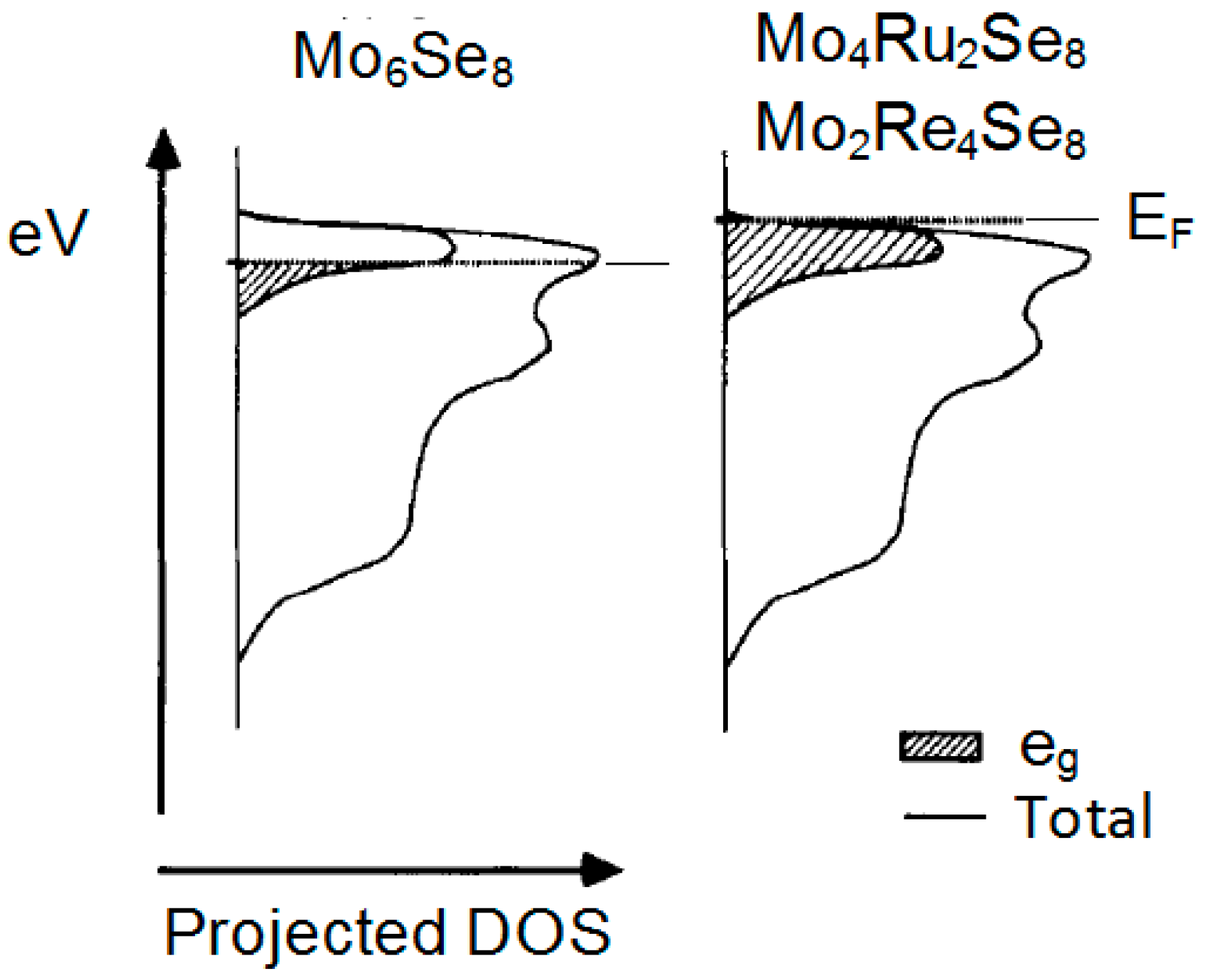
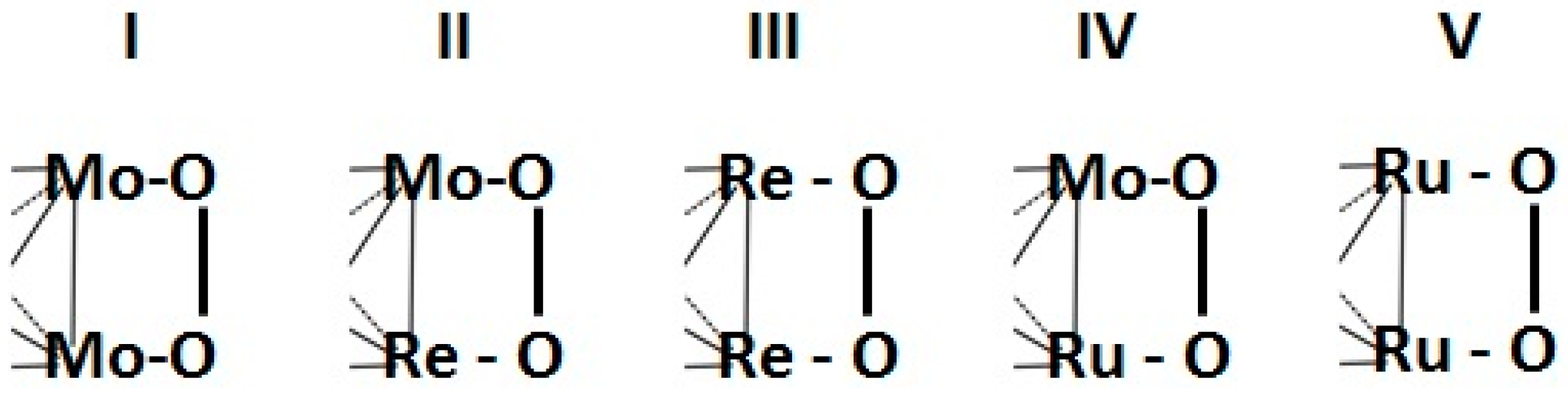
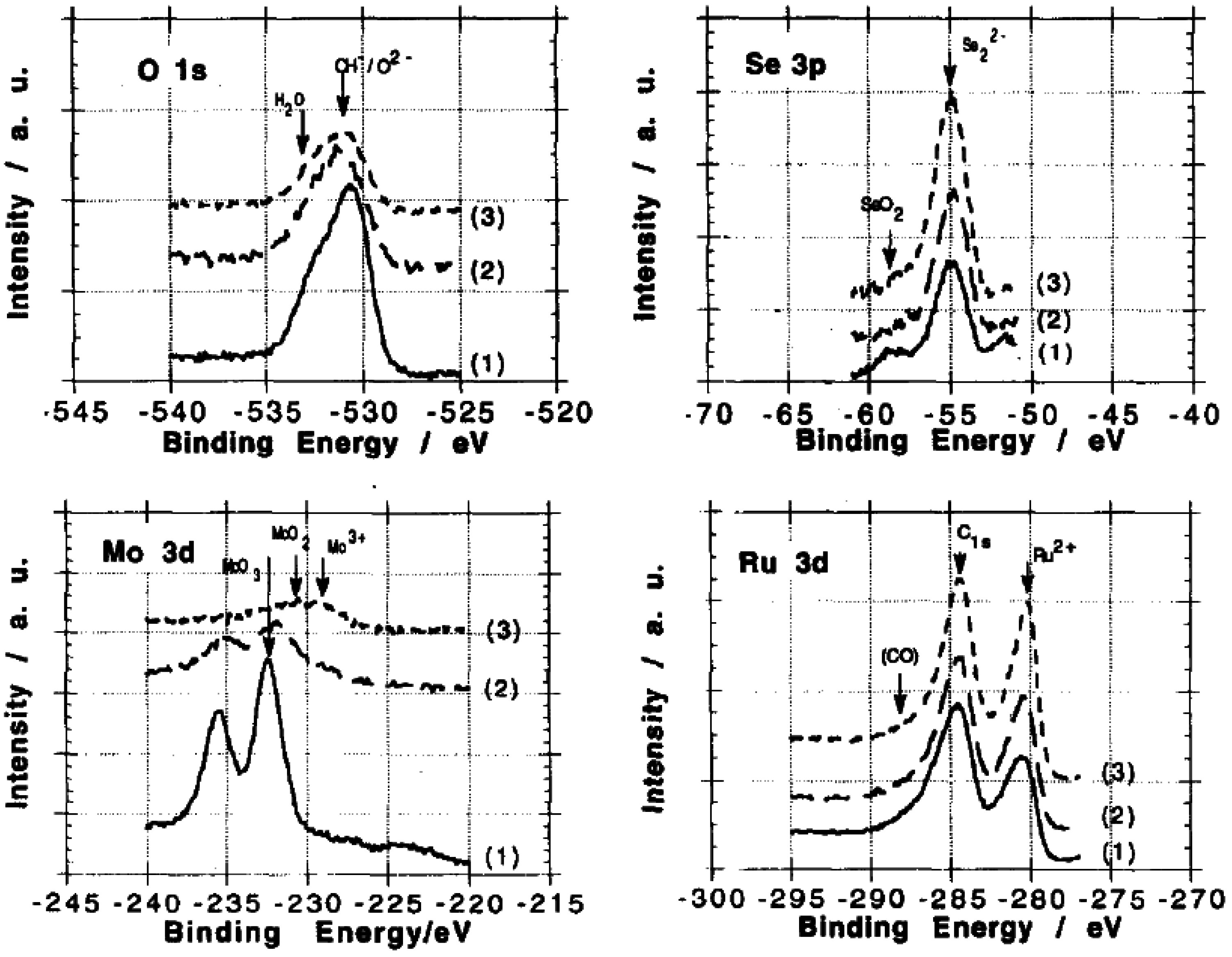
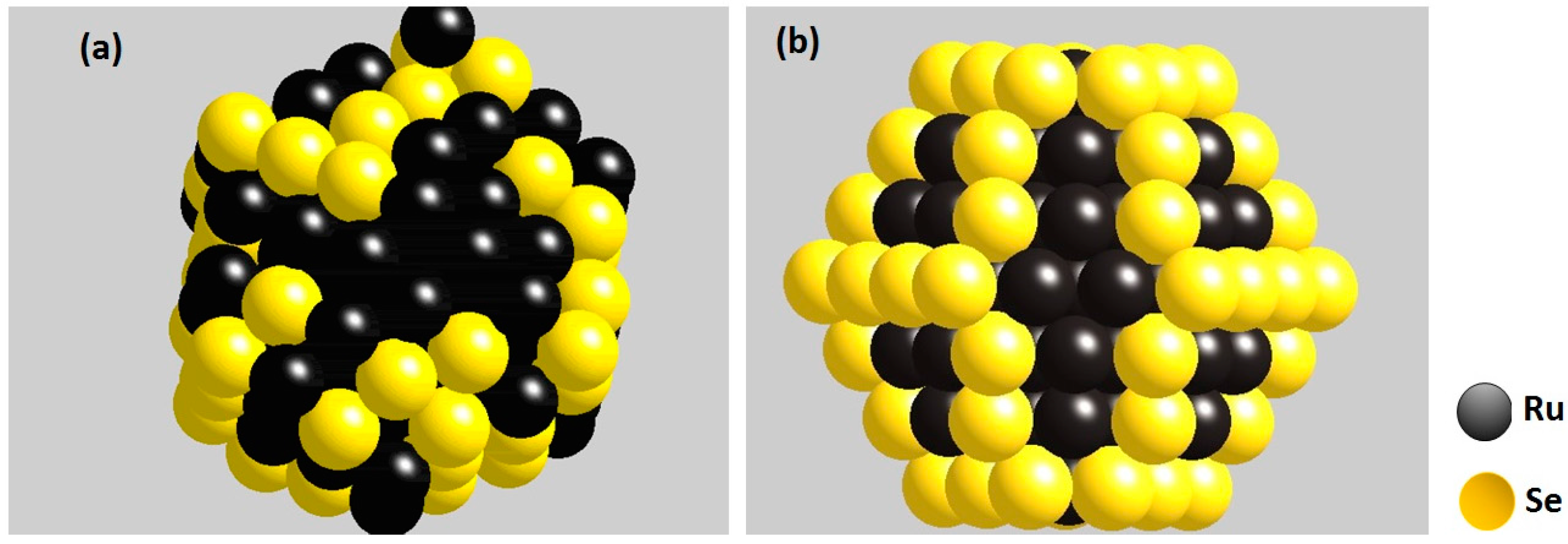
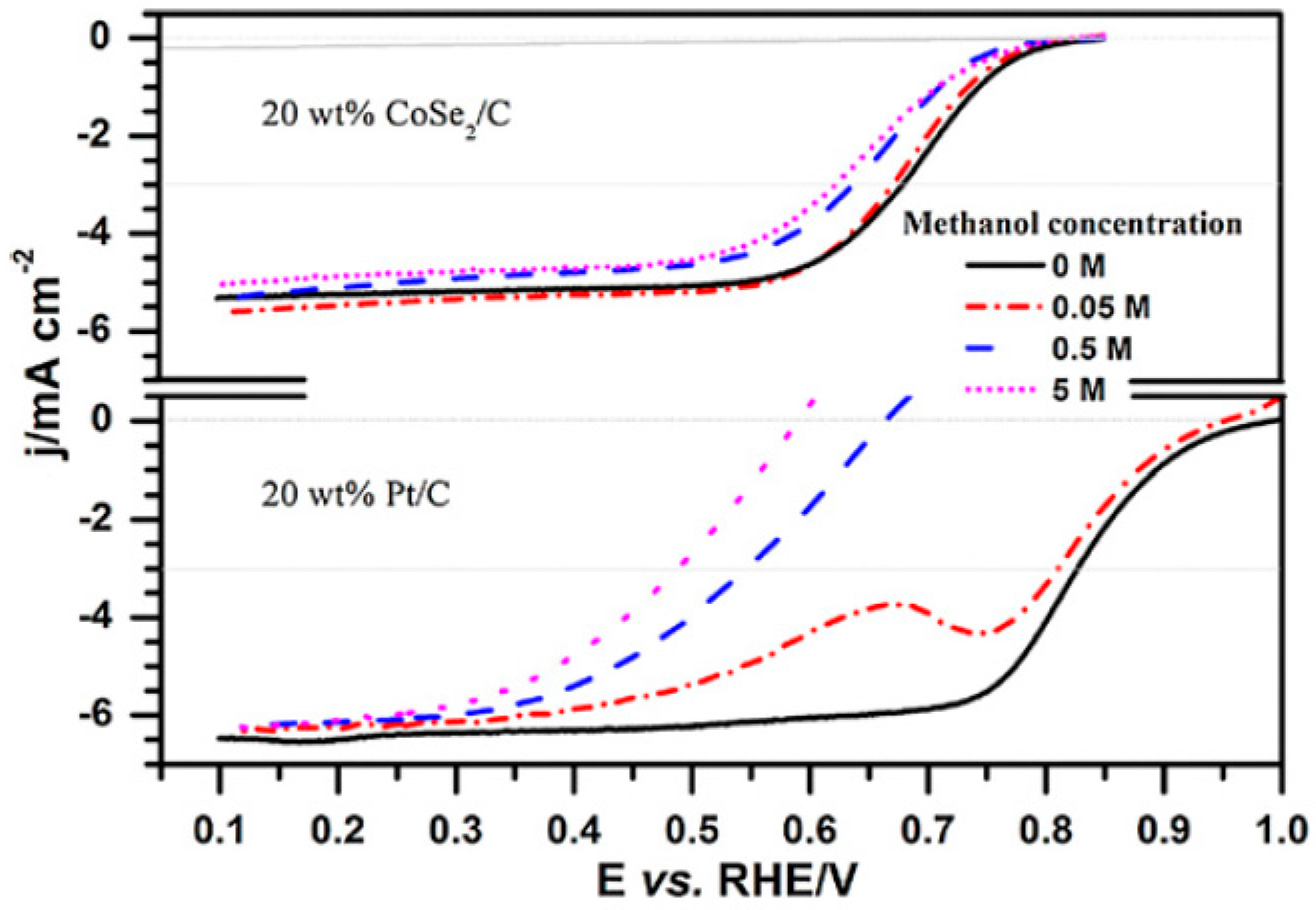
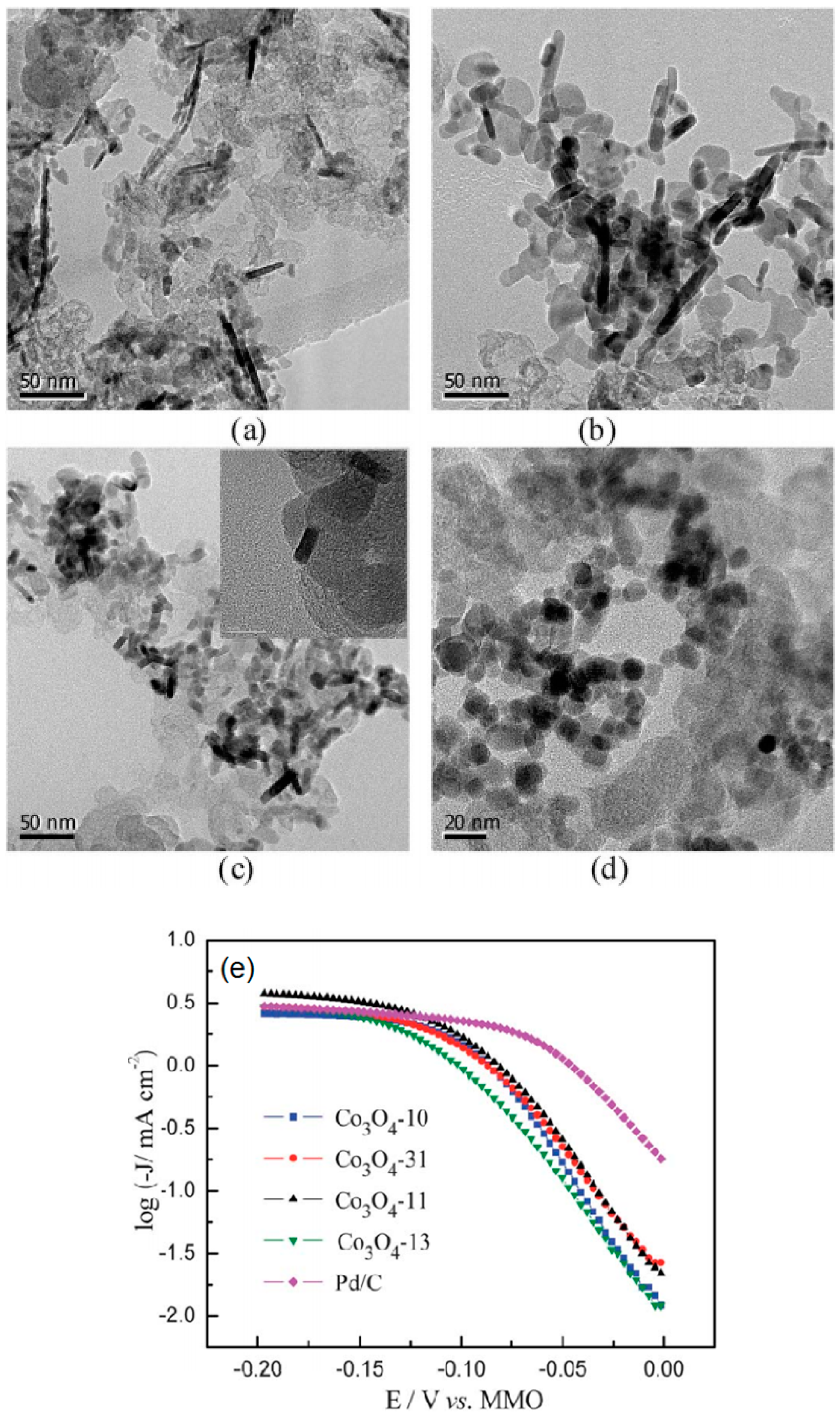
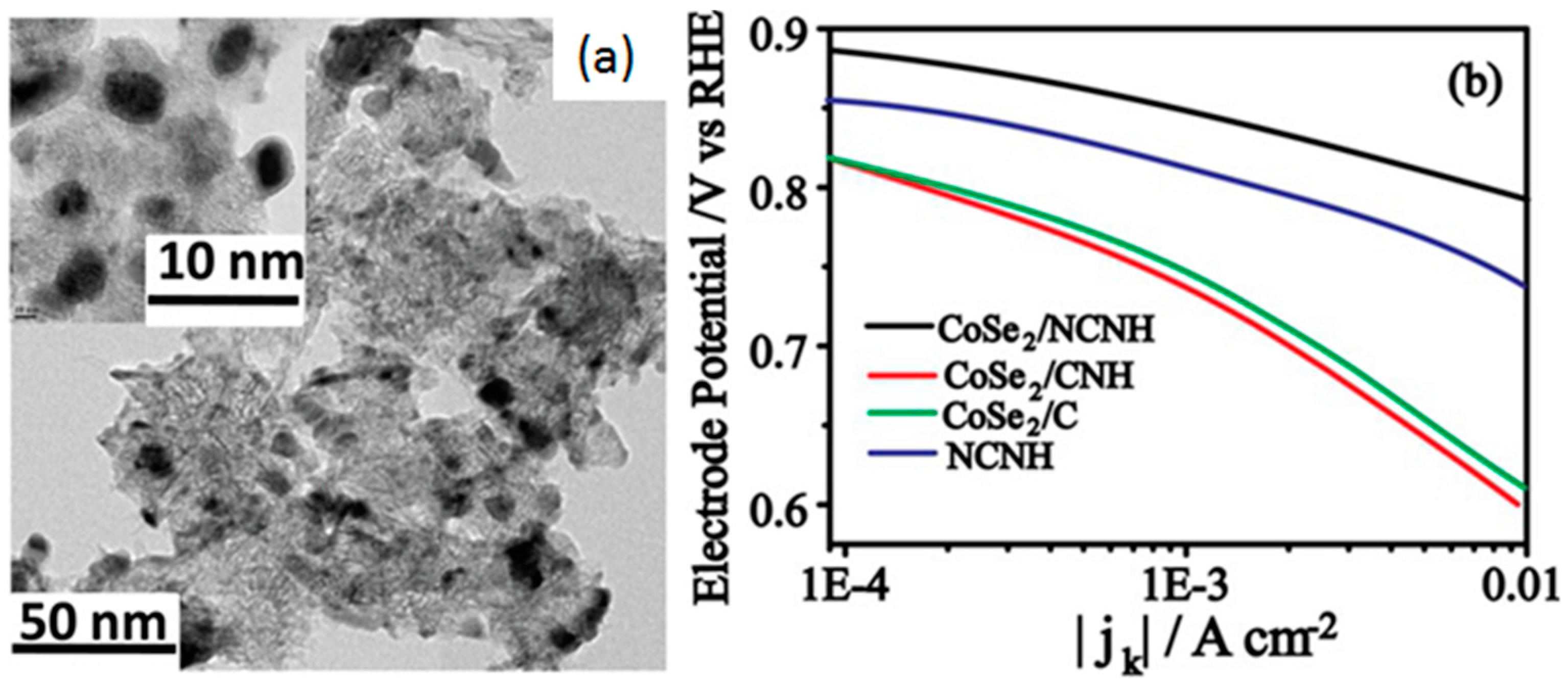
2.1.2. Transition Metal Chalcogenides
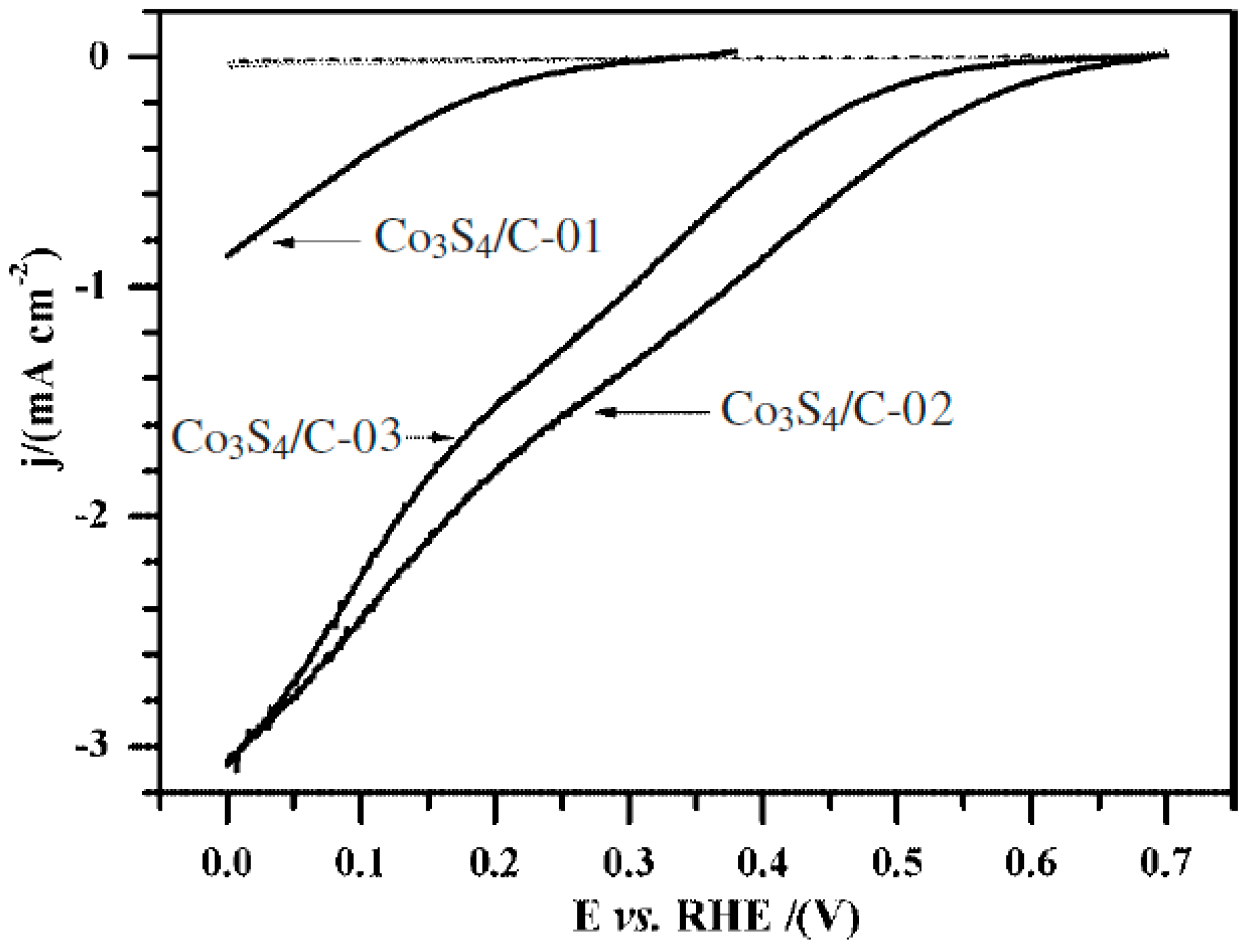
2.2. Non-Precious Catalysts
3. Fuel Cell Reactions
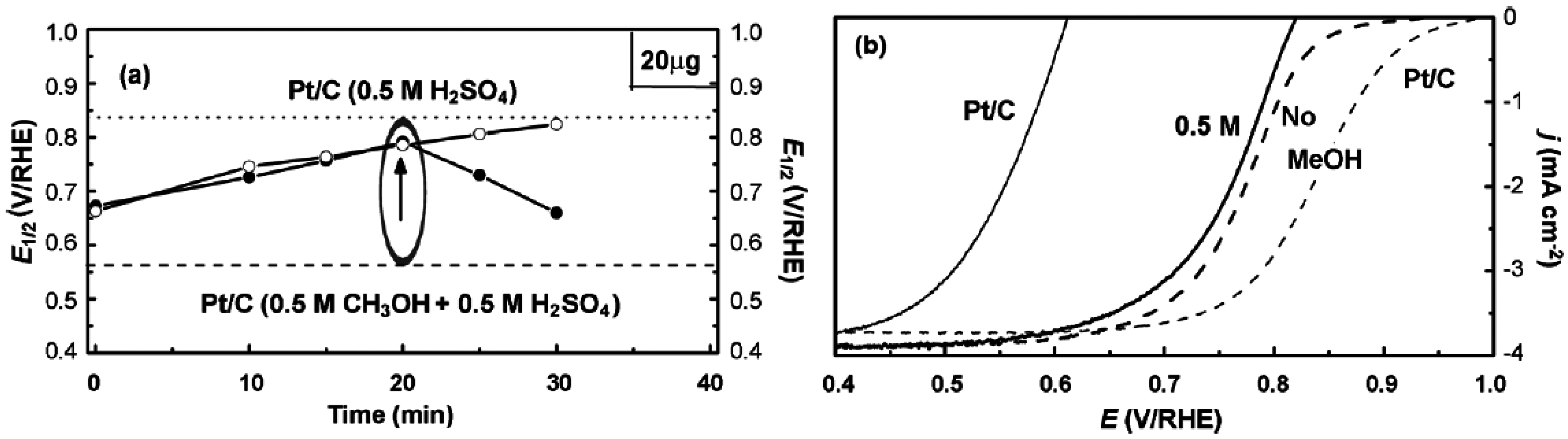
3.1. Hydrogen Oxidation Reaction—HOR
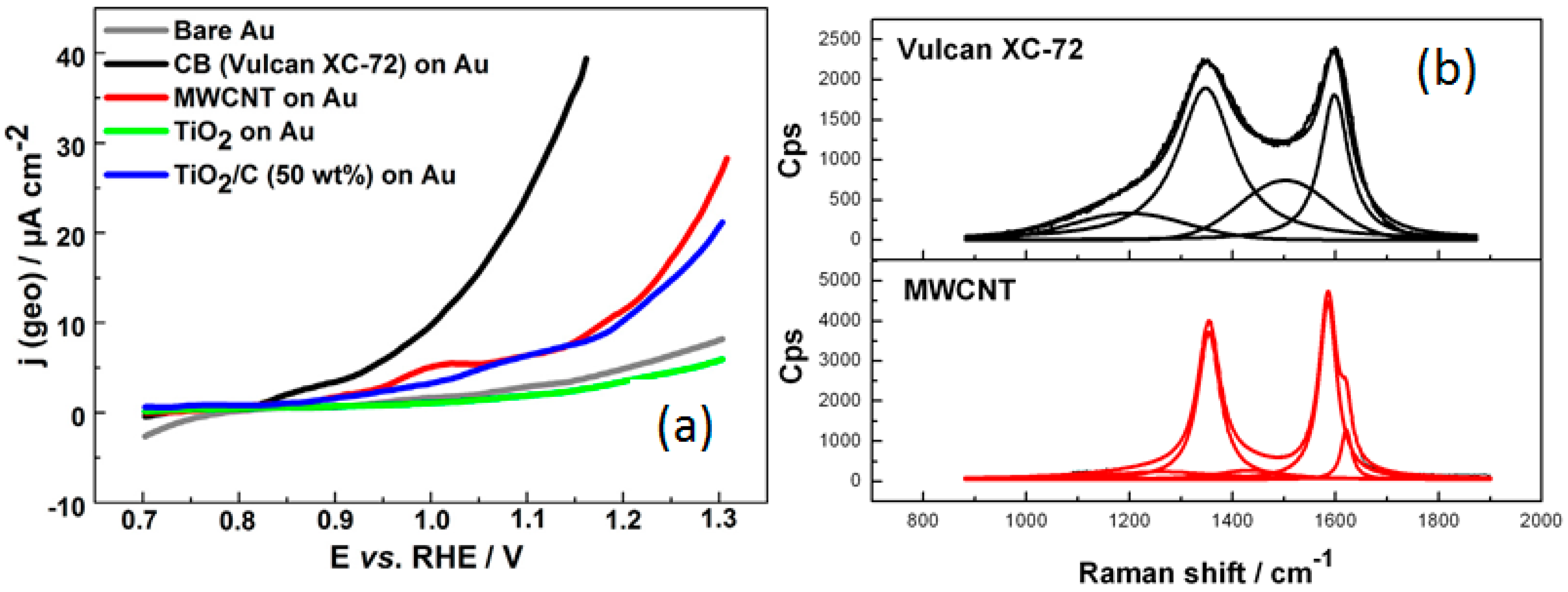
3.2. Oxygen Reduction Reaction—ORR, and Carbon Supports
3.2.1. Low Graphitic Carbon
3.2.2. High Graphitic Carbon
3.2.3. Oxide and Oxide-Carbon Composites
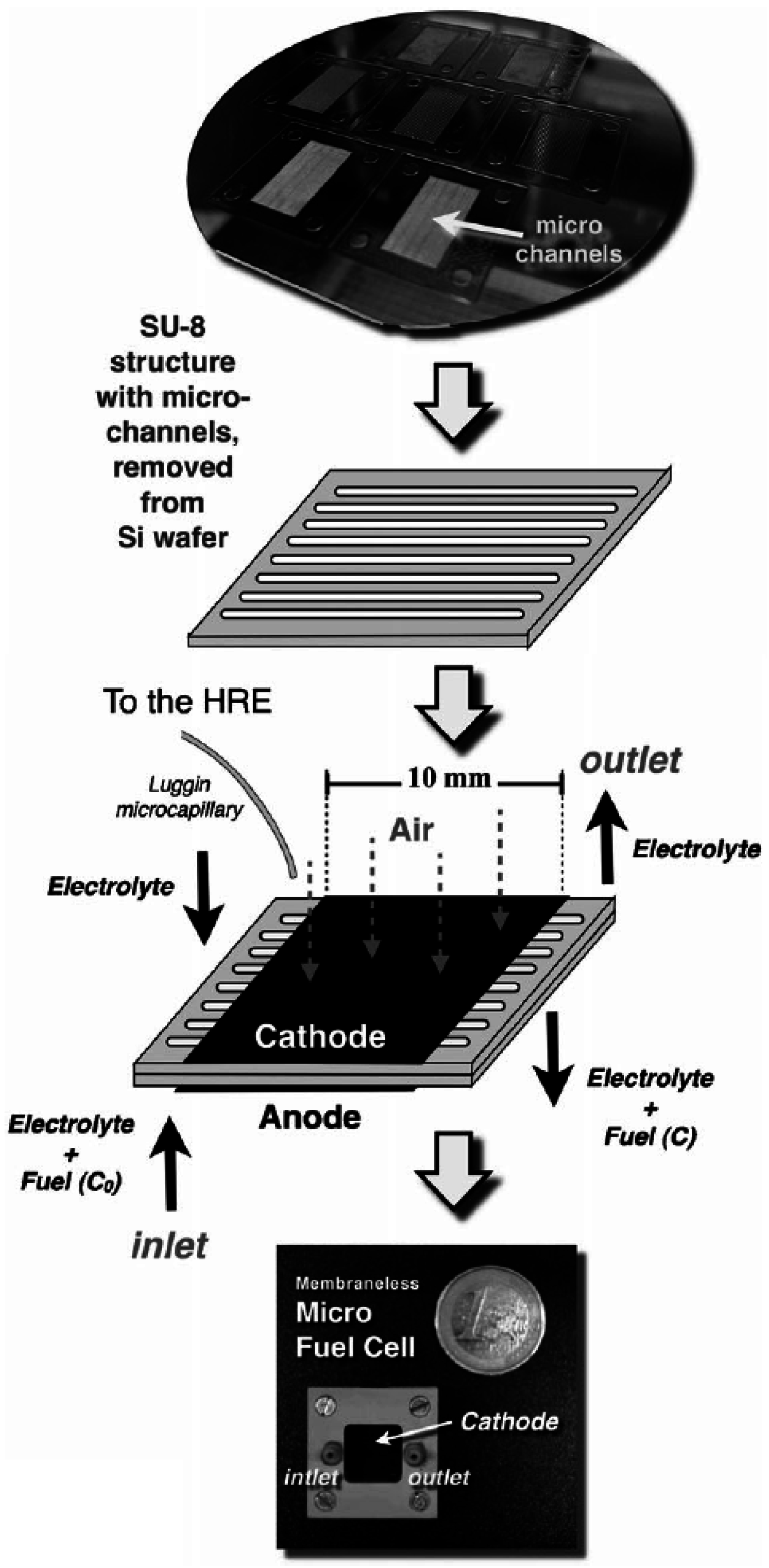
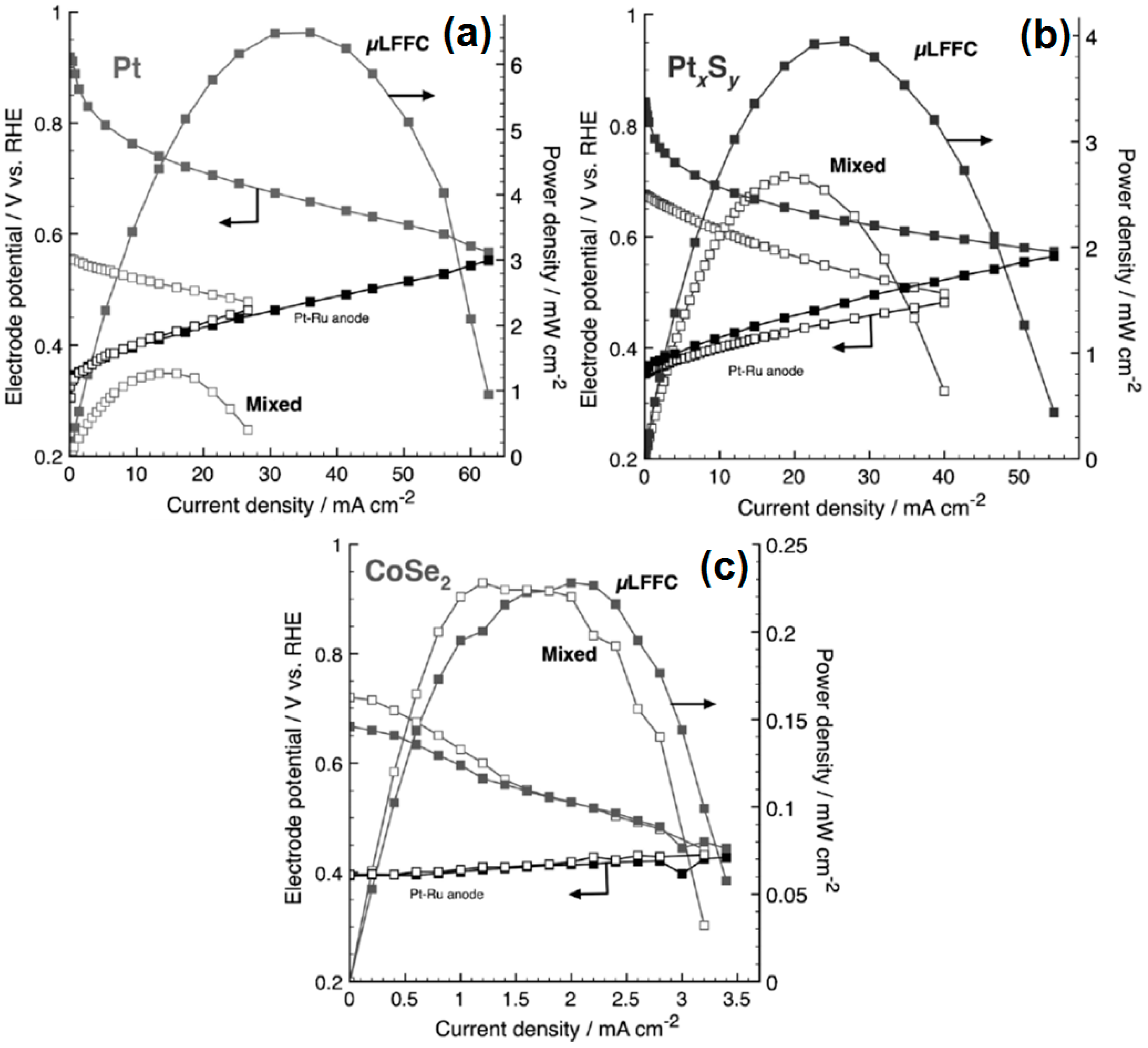
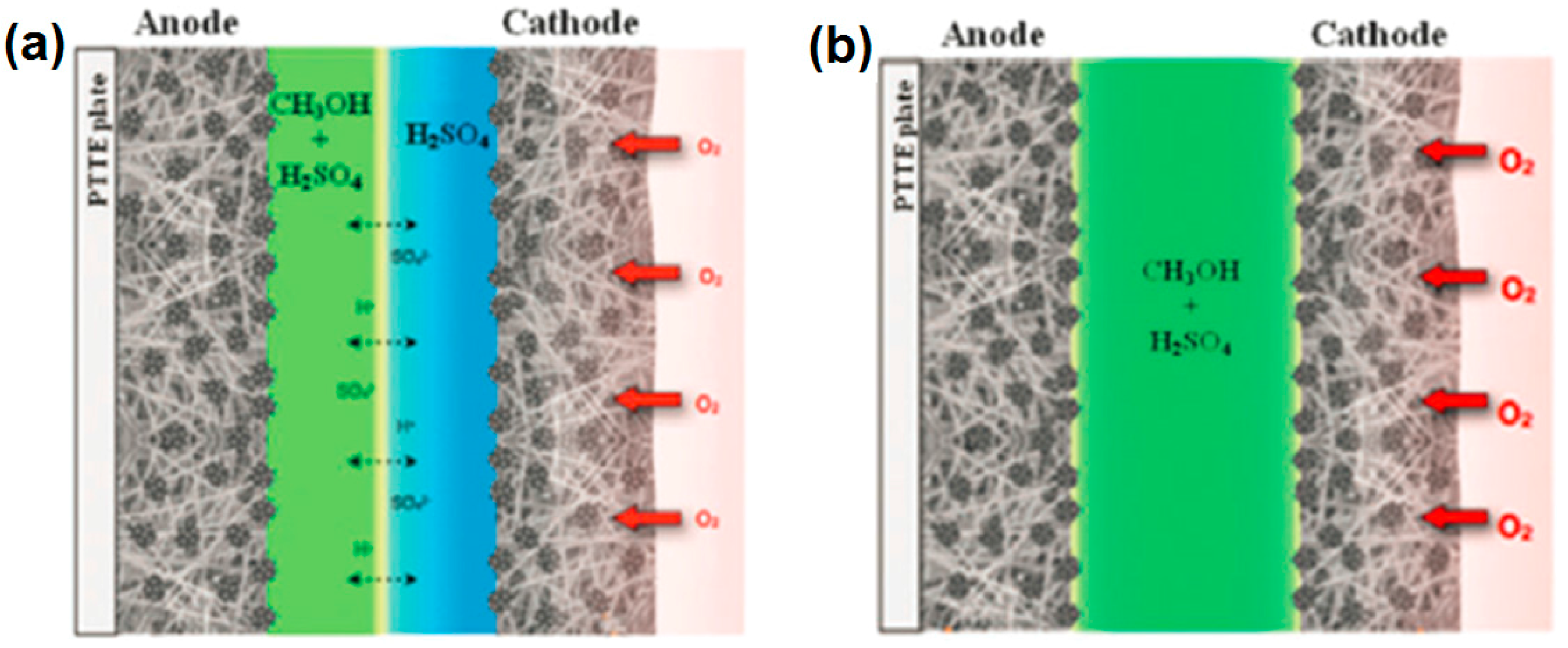
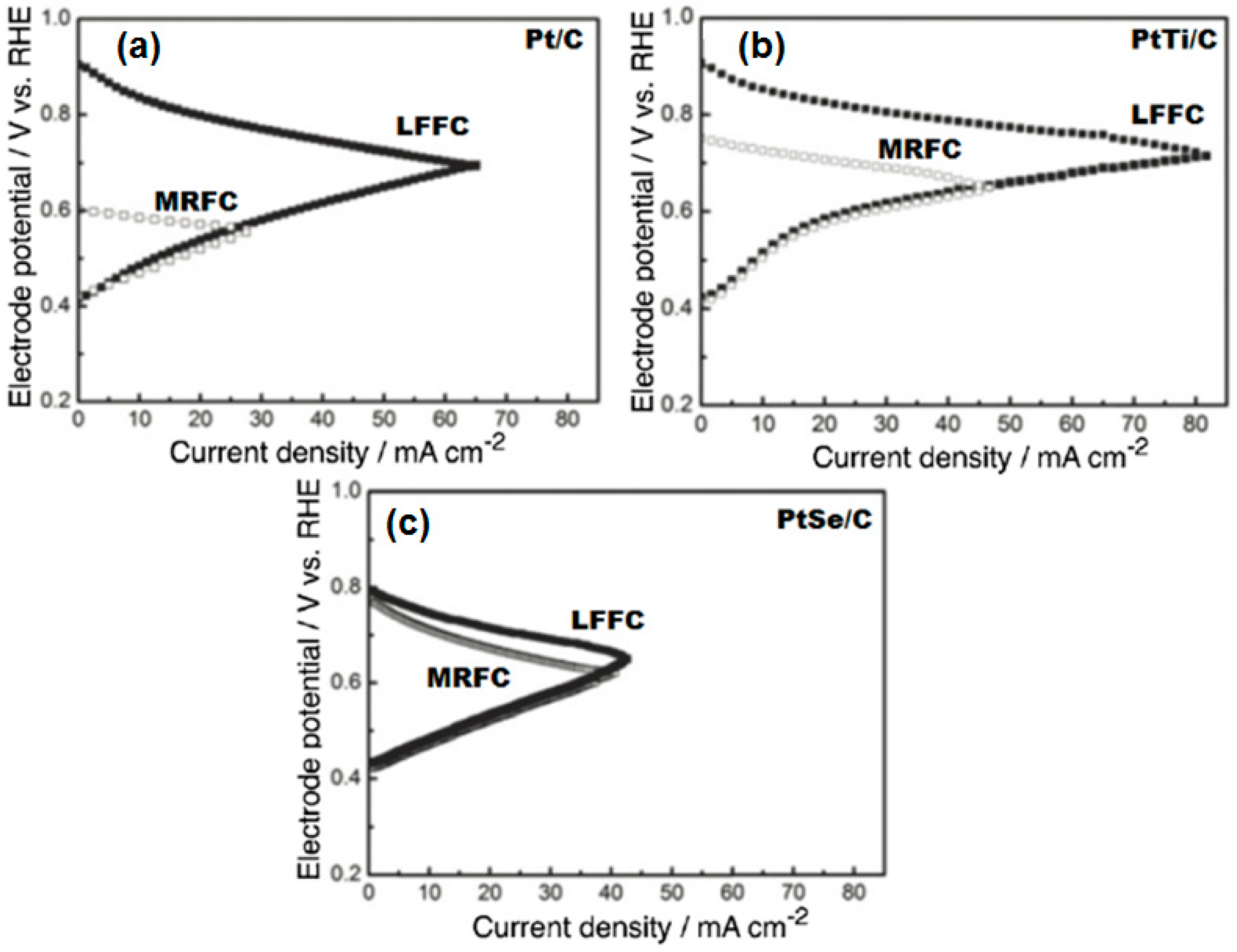
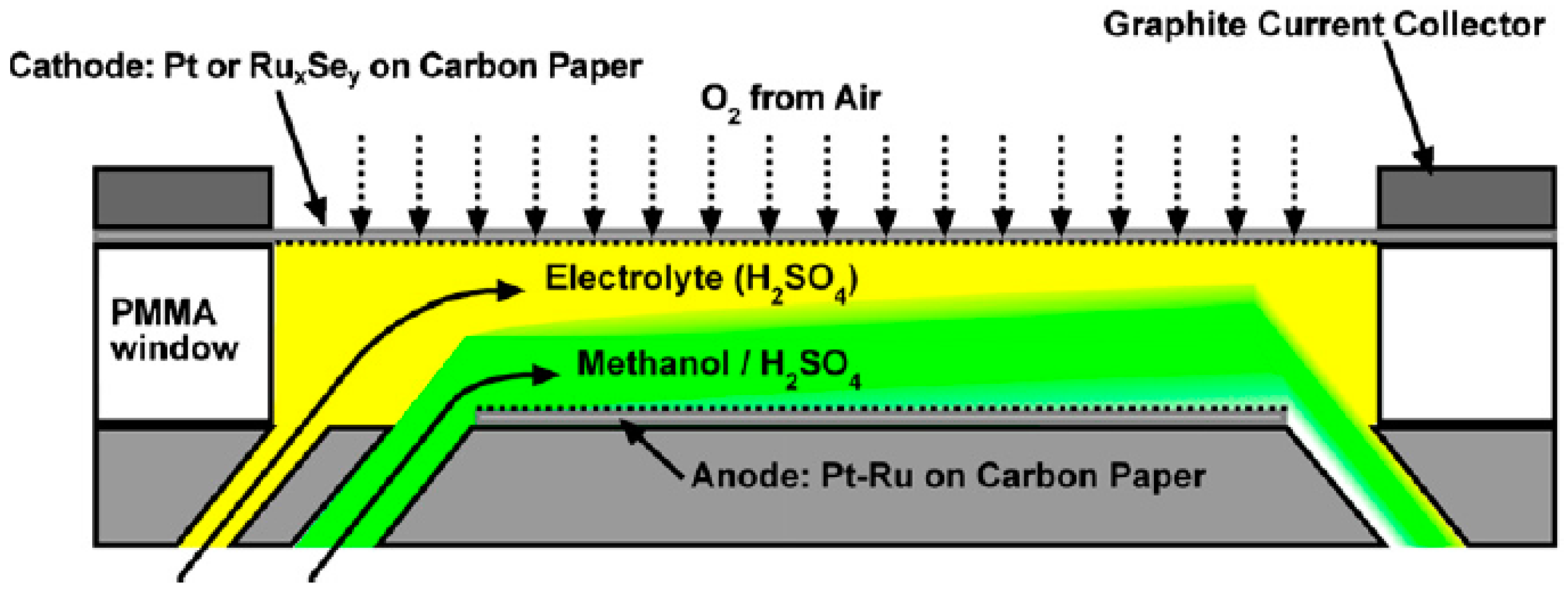
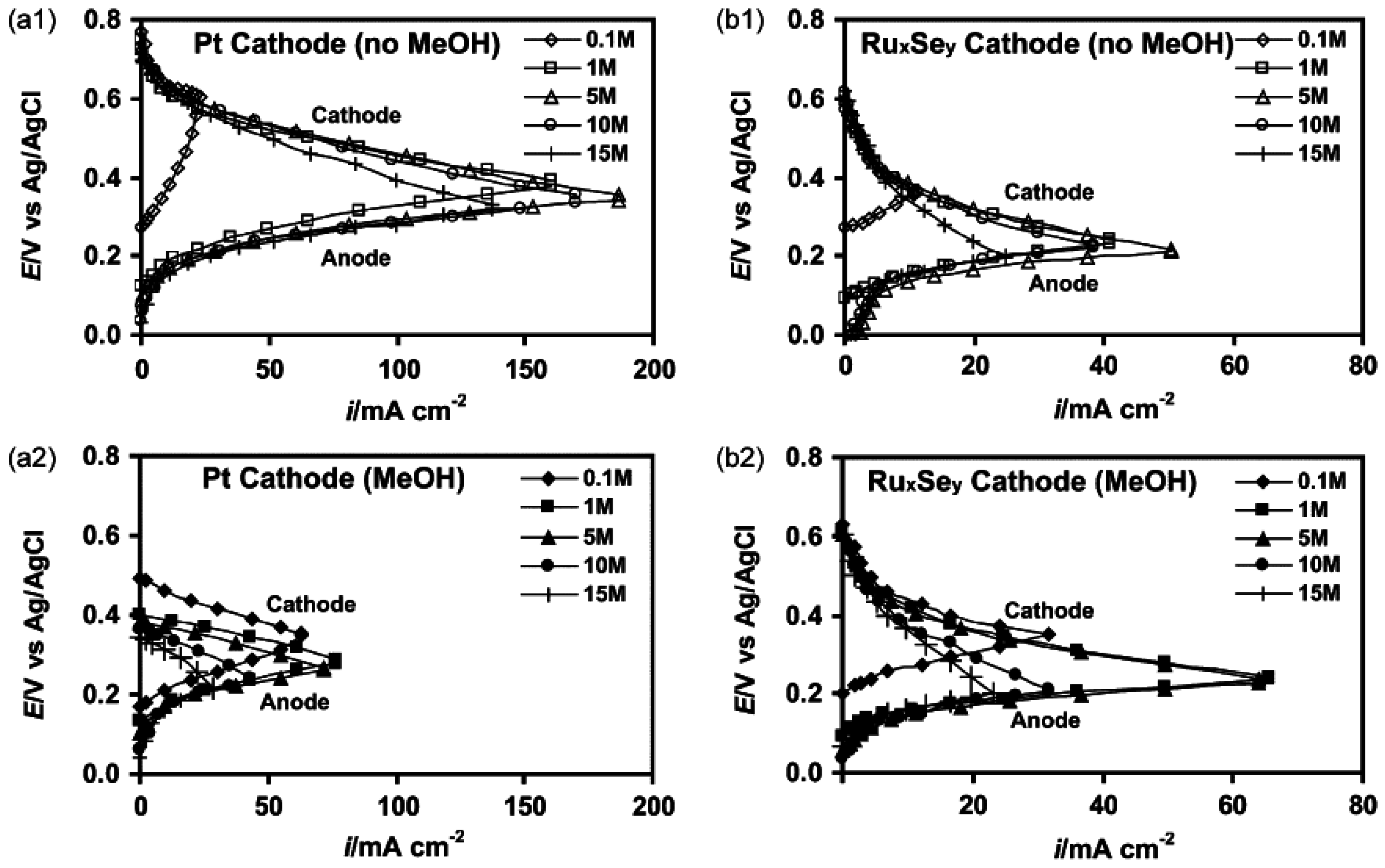
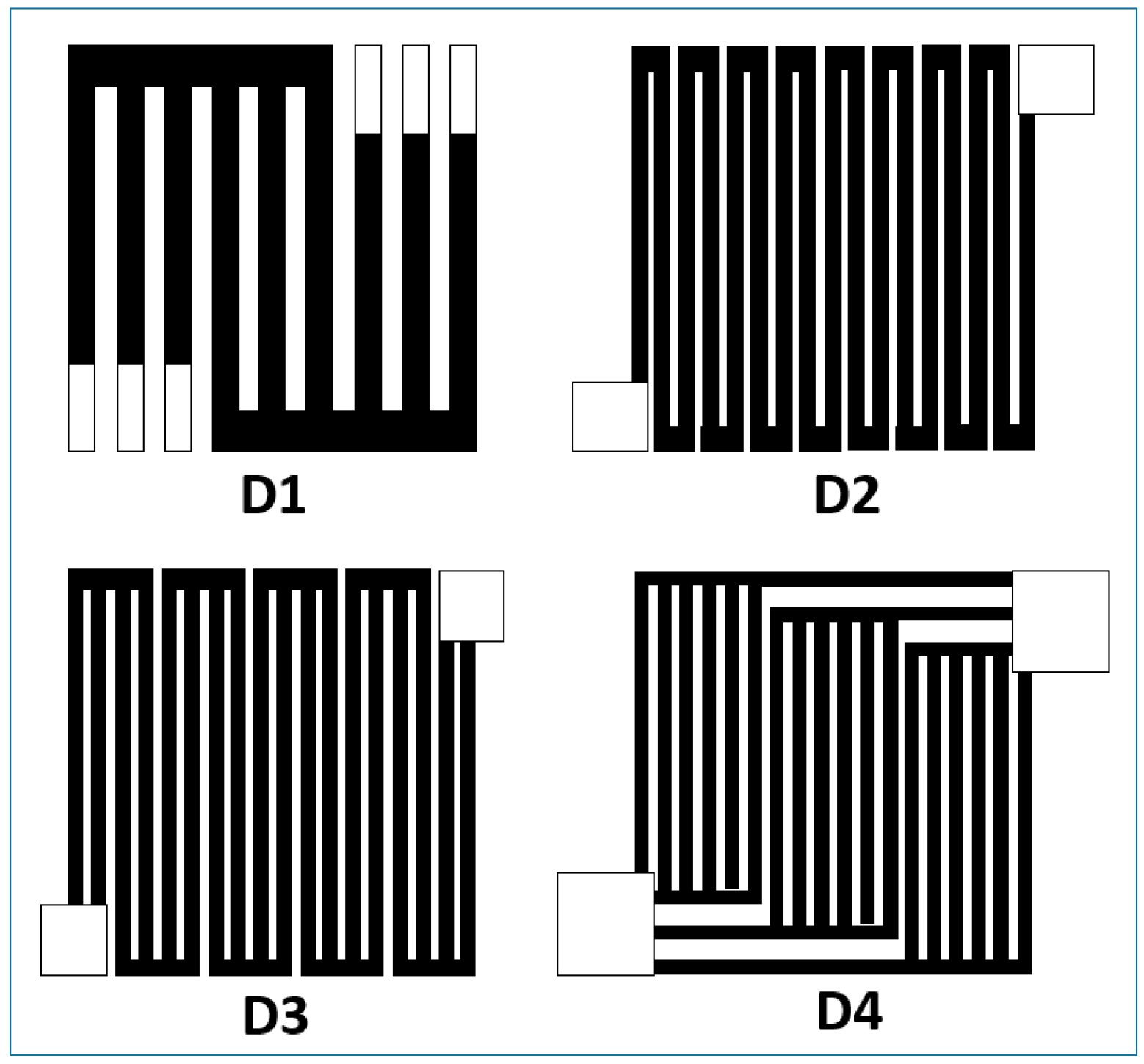
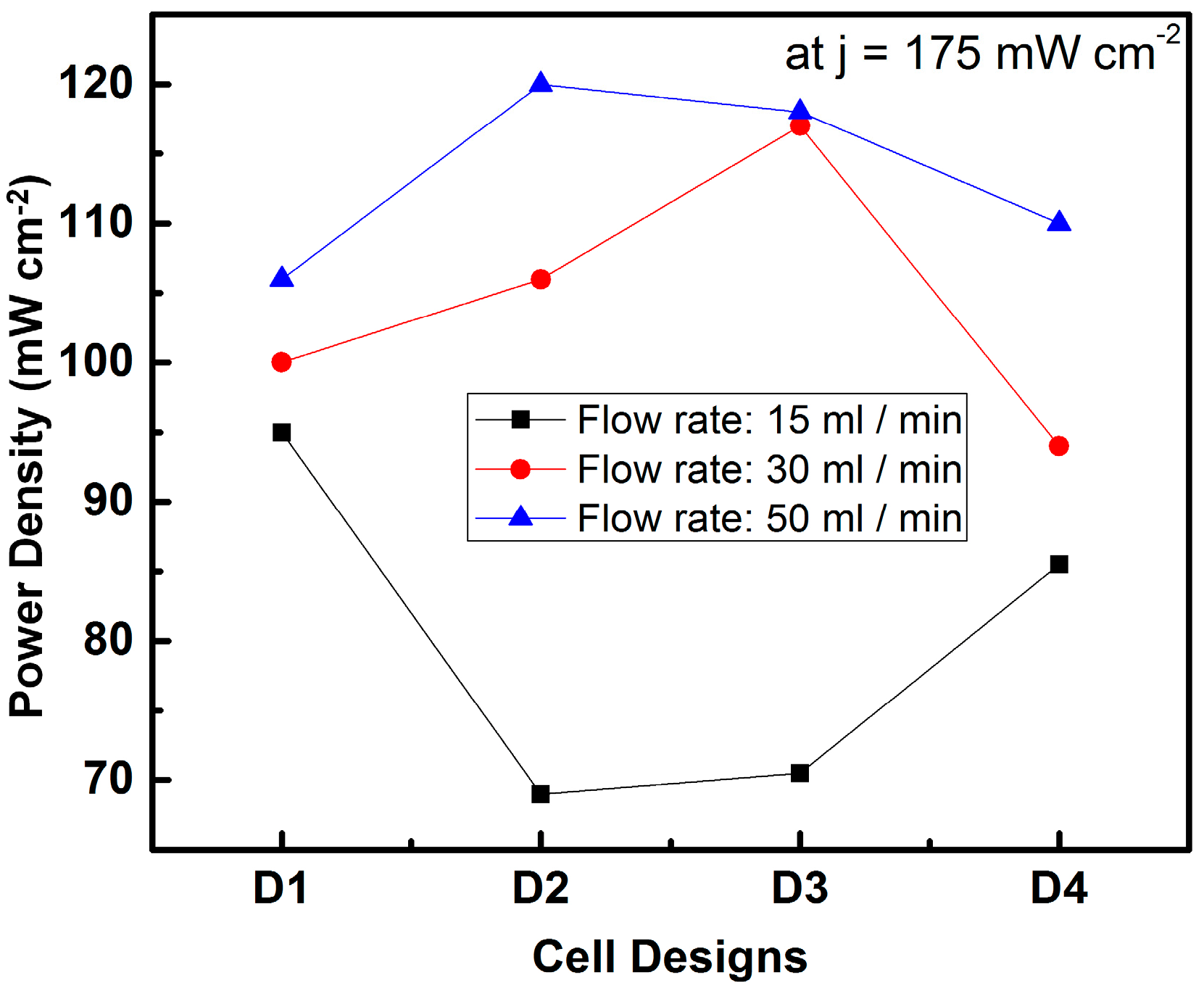
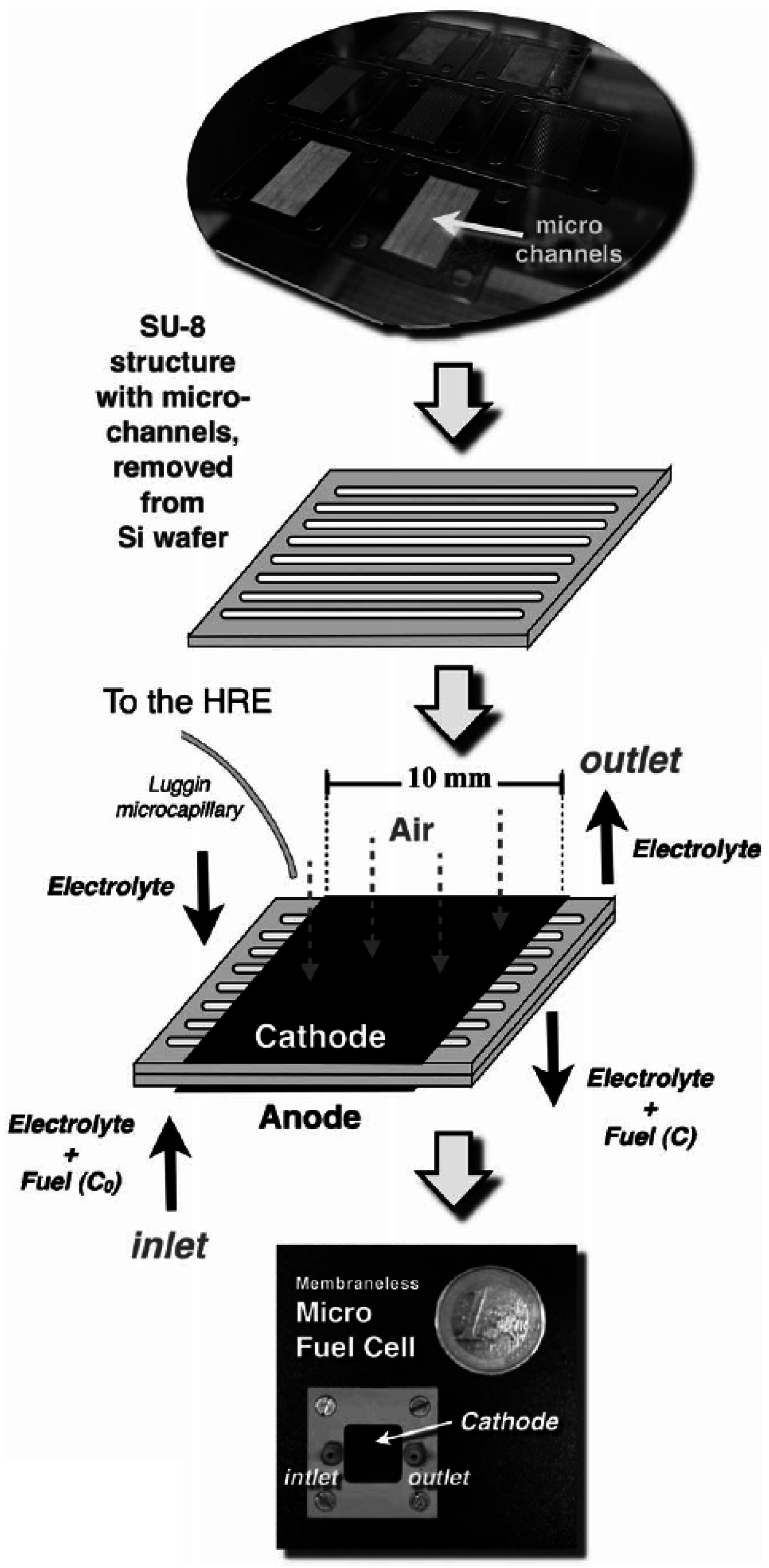
4. Micro-Fuel Cell Platform
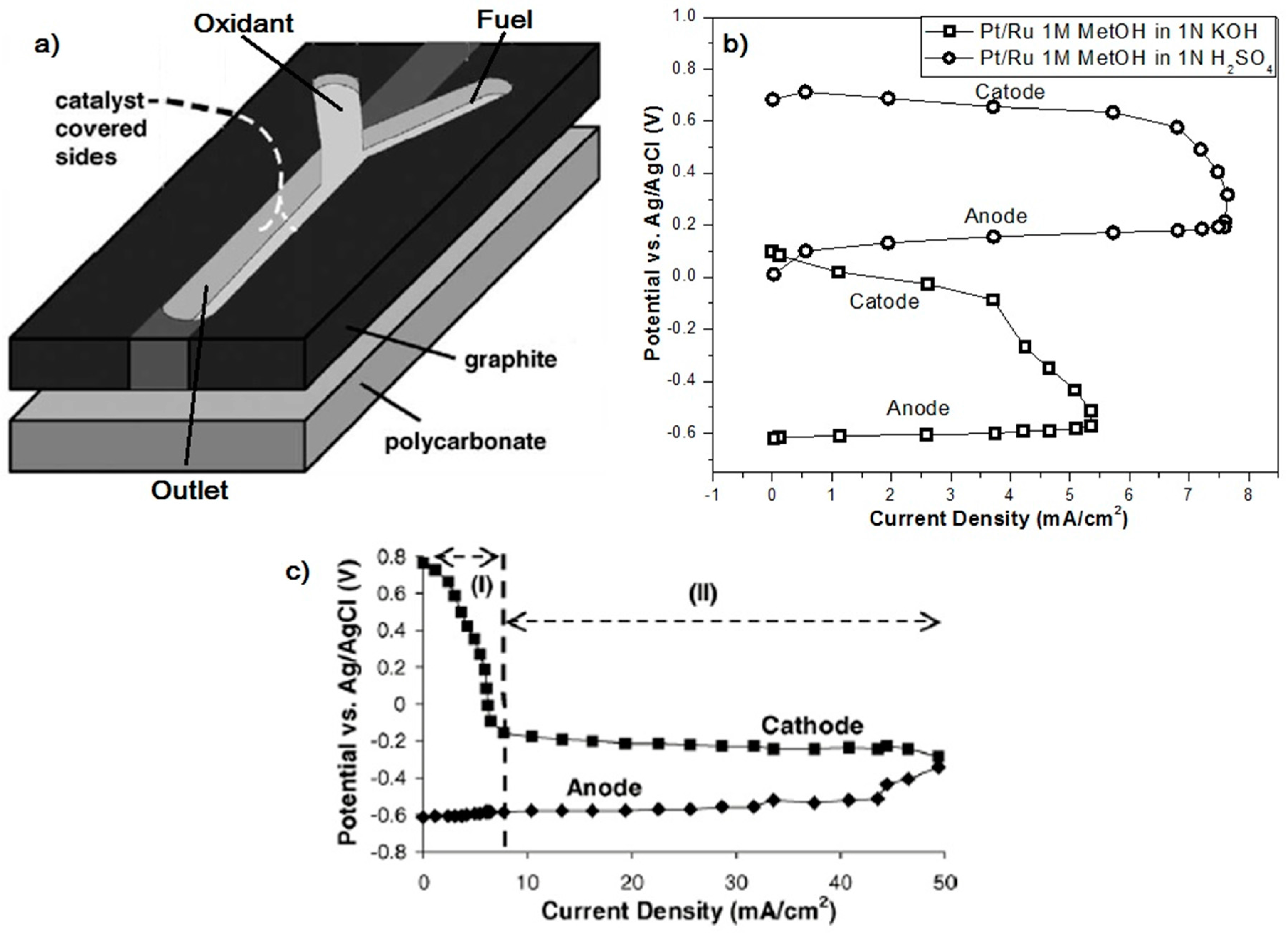
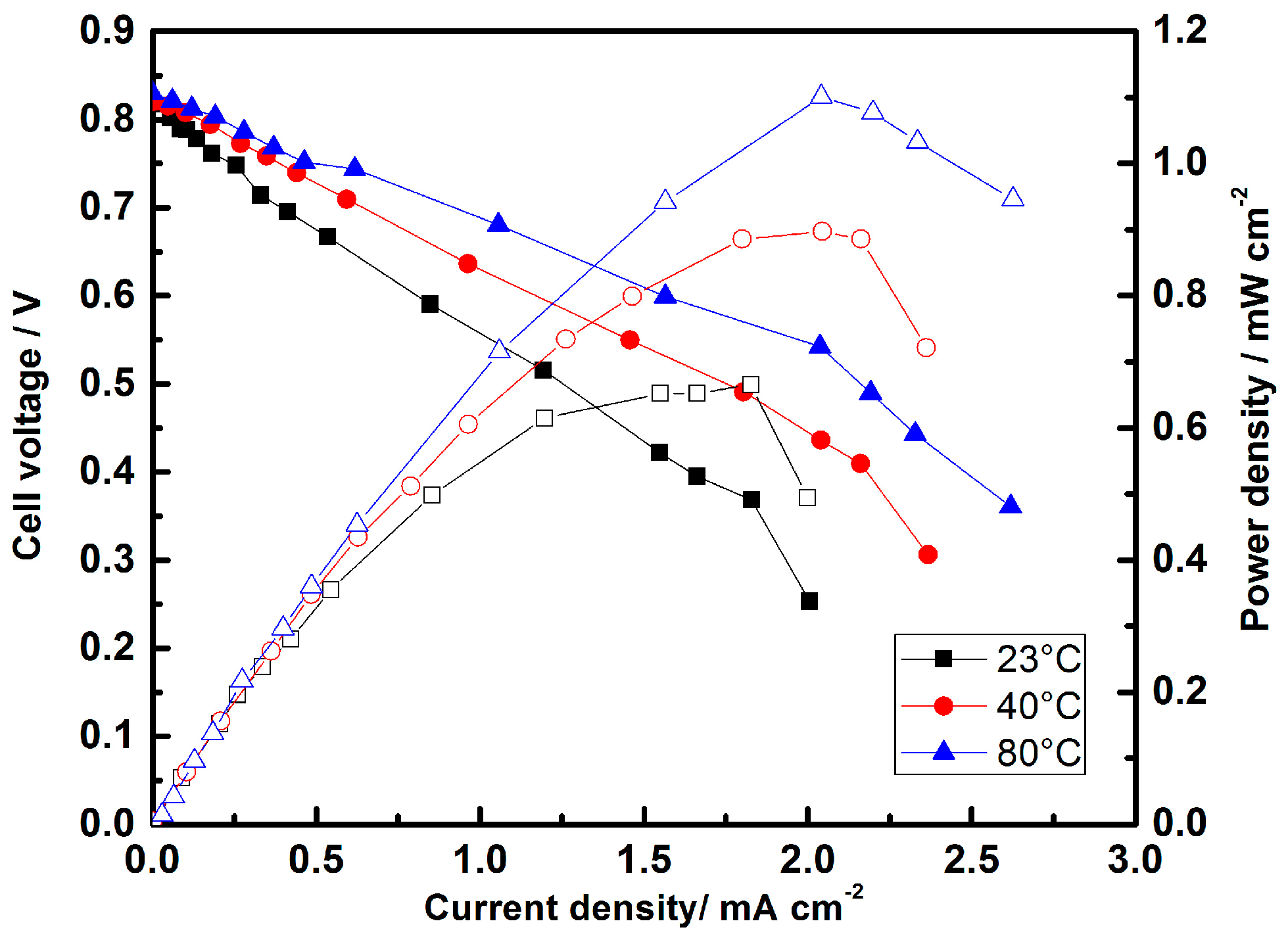
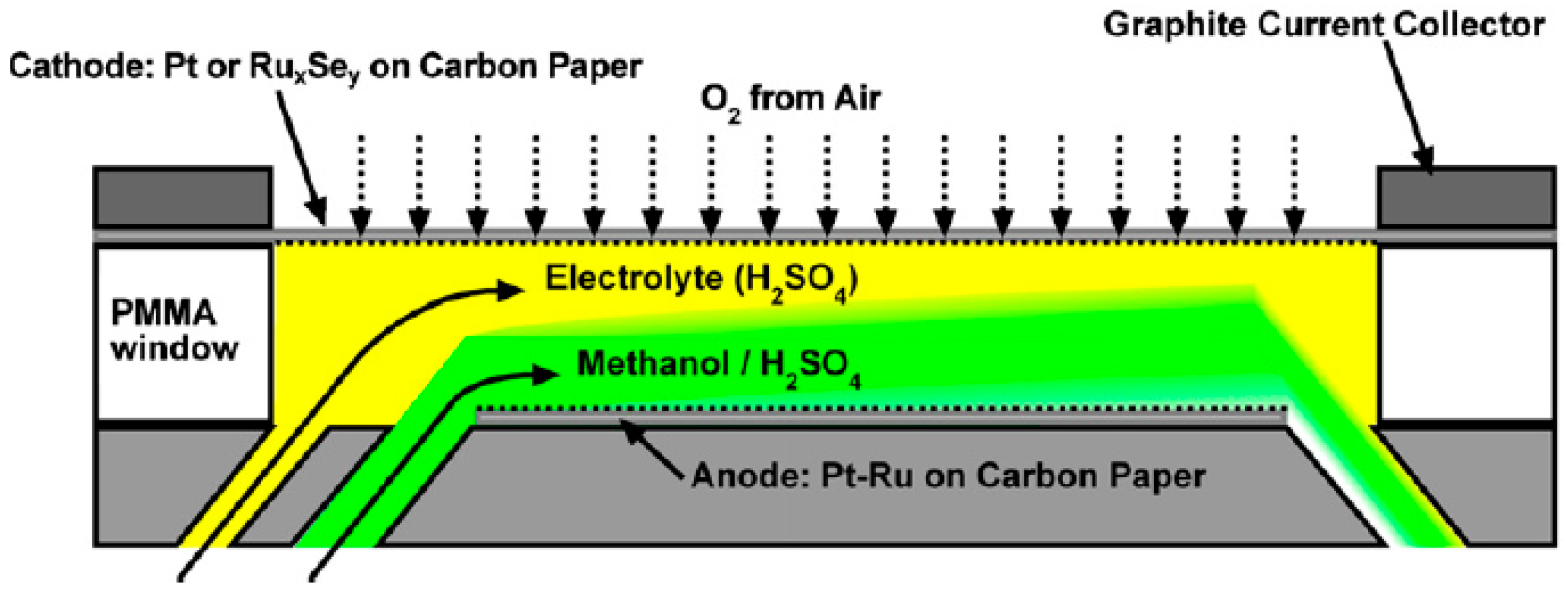
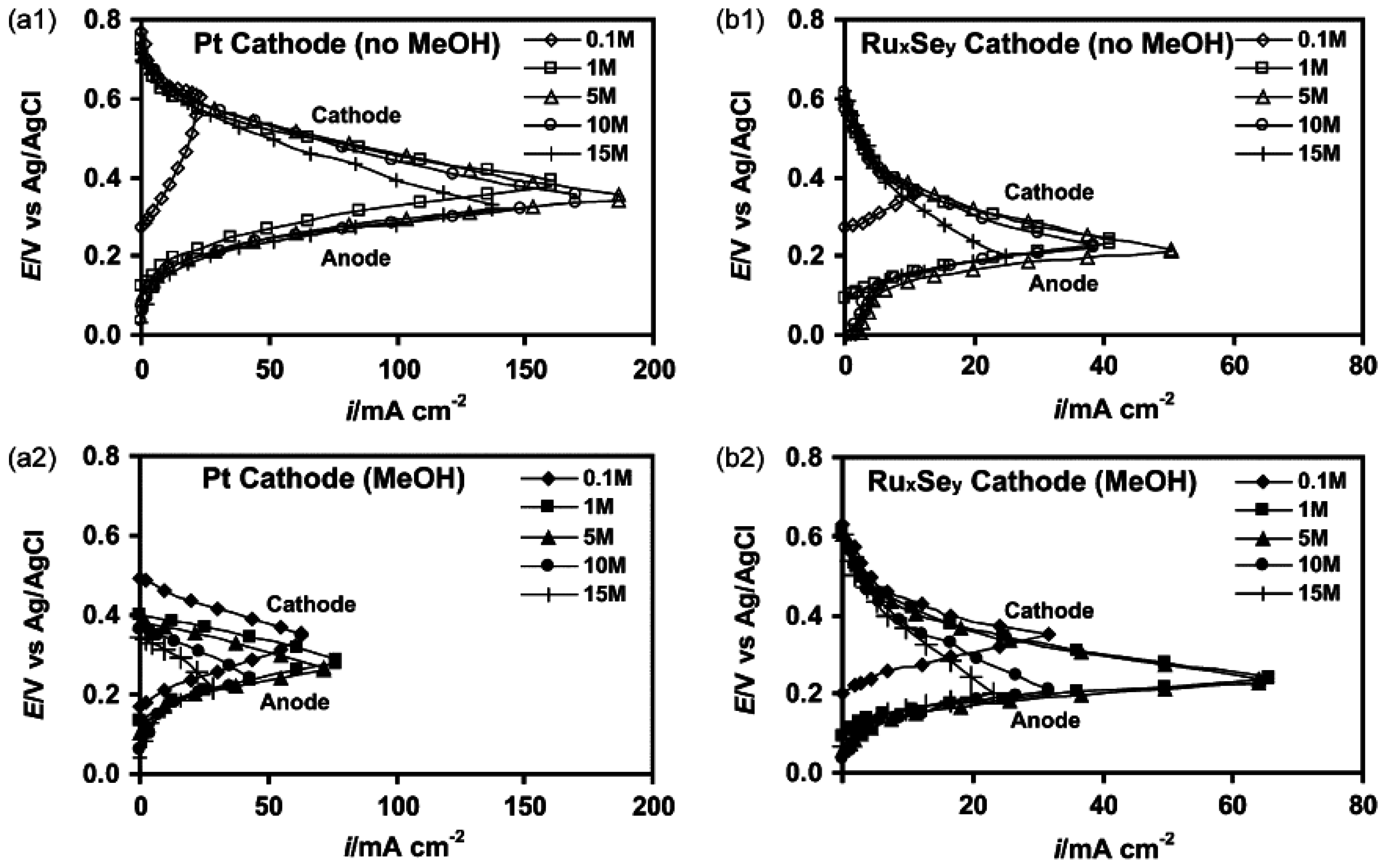
Micro-Laminar Flow Fuel Cells
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Perry, M.L.; Fuller, T.F. A historical perspective of fuel cell technology in the 20th century. J. Electrochem. Soc. 2002, 149, S59–S67. [Google Scholar] [CrossRef]
- Haiping, X.; Li, K.; Xuhui, W. Fuel cell power system and high power DC-DC converter. IEEE Trans. Power Electr. 2004, 19, 1250–1255. [Google Scholar]
- Jayasayee, K.; Veen, J.A.R.V.; Manivasagam, T.G.; Celebi, S.; Hensen, E.J.M.; de Bruijn, F.A. Oxygen reduction reaction (ORR) activity and durability of carbon supported PtM (Co, Ni, Cu) alloys: Influence of particle size and non-noble metals. Appl. Catal. B: Environ. 2012, 111–112, 515–526. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B: Environ. 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Ezeta, A.; Arce, E.M.; Solorza, O.; González, R.G.; Dorantes, H. Effect of the leaching of Ru-Se-Fe and Ru-Mo-Fe obtained by mechanical alloying on electrocatalytical behavior for the oxygen reduction reaction. J. Alloys Compd. 2009, 483, 429–431. [Google Scholar] [CrossRef]
- Suryanarayana, C. Mechanical alloying and milling. Prog. Mater. Sci. 2001, 46, 1–184. [Google Scholar] [CrossRef]
- Mukerjee, S.; Srinivasan, S.; Soriaga, M.P.; McBreen, J. Role of structural and electronic properties of Pt and Pt alloys on electrocatalysis of oxygen reduction: An in situ XANES and EXAFS investigation. J. Electrochem. Soc. 1995, 142, 1409–1422. [Google Scholar] [CrossRef]
- Chen, S.; Ferreira, P.J.; Sheng, W.; Yabuuchi, N.; Allard, L.F.; Shao-Horn, Y. Enhanced activity for oxygen reduction reaction on “Pt3Co” nanoparticles: Direct evidence of percolated and sandwich-segregation structures. J. Am. Chem. Soc. 2008, 130, 13818–13819. [Google Scholar] [CrossRef] [PubMed]
- Min, M.-K.; Cho, J.; Cho, K.; Kim, H. Particle size and alloying effects of Pt-based alloy catalysts for fuel cell applications. Electrochim. Acta 2000, 45, 4211–4217. [Google Scholar] [CrossRef]
- Guo, S.; Li, D.; Zhu, H.; Zhang, S.; Markovic, N.M.; Stamenkovic, V.R.; Sun, S. FePt and CoPt nanowires as efficient catalysts for the oxygen reduction reaction. Angew. Chem. Int. Ed. 2013, 52, 3465–3468. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Chang, Q.; Dodelet, J.-P.; Chenitz, R. Recent advances in electrocatalysts for oxygen reduction reaction. Chem. Rev. 2016, 116, 3594–3657. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Stephens, I.E.L.; Bondarenko, A.S.; Johansson, T.P.; Hansen, H.A.; Jaramillo, T.F.; Rossmeisl, J.; Chorkendorff, I.; Nørskov, J.K. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nat. Chem. 2009, 1, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.M.; Escudero-Escribano, M.; Velázquez-Palenzuela, A.; Christensen, L.H.; Chorkendorff, I.; Stephens, I.E.L. Benchmarking Pt-based electrocatalysts for low temperature fuel cell reactions with the rotating disk electrode: Oxygen reduction and hydrogen oxidation in the presence of CO. Electrochim. Acta 2015, 179, 647–657. [Google Scholar] [CrossRef]
- Alonso-Vante, N.; Jaegermann, W.; Tributsch, H.; Hoenle, W.; Yvon, K. Electrocatalysis of oxygen reduction by chalcogenides containing mixed transition metal clusters. J. Am. Chem. Soc. 1987, 109, 3251–3257. [Google Scholar] [CrossRef]
- Alonso-Vante, N. Chevrel phase and cluster-like chalcogenide materials. In Handbook of Fuel Cells; Vielstich, W., Lamm, A., Gasteiger, H., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2003; Volume 2, pp. 534–543. [Google Scholar]
- Friedrich, K.A.; Geyzers, K.P.; Linke, U.; Stimming, U.; Stumper, J. Co adsorption and oxidation on a Pt(111) electrode modified by ruthenium deposition: An ir spectroscopic study. J. Electroanal. Chem. 1996, 402, 123–128. [Google Scholar] [CrossRef]
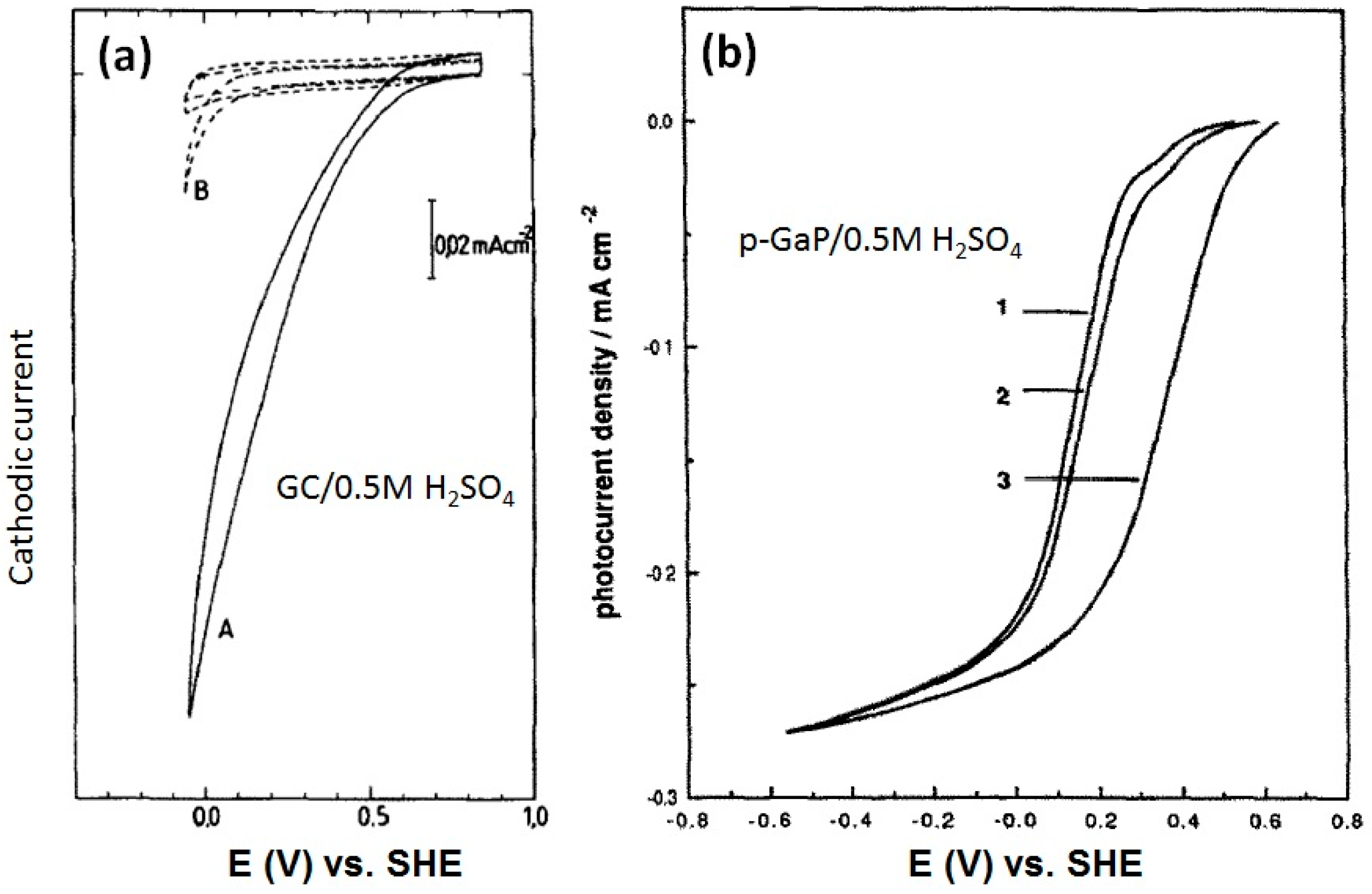
- Piazza, S.; Kühne, H.M.; Tributsch, H. Photoelectrochemical and electrocatalytic behaviour of p-type ruthenium disulphide electrodes. J. Electroanal. Chem. 1985, 196, 53–67. [Google Scholar] [CrossRef]
- Bewick, A.; Gutiérrez, C.; Larramona, G. An international journal devoted to all aspects of electrode kinetics, interfacial structure, properties of electrolytes, colloid and biological electrochemistryin-situ IR spectroscopy study of the ruthenium electrode in acid and alkaline solutions. J. Electroanal. Chem. 1992, 332, 155–167. [Google Scholar] [CrossRef]
- Alonso-Vante, N. Spectral sensitization of titanium dioxide electrodes via Ru-based chalcogenides thin layers. Sol. Energy Mater. Sol. C 1994, 31, 509–524. [Google Scholar] [CrossRef]
- Chang, C.C.; Wen, T.C. Kinetics of oxygen reduction at RuO2-coated titanium electrode in alkaline solution. J. Appl. Electrochem. 1997, 27, 355–363. [Google Scholar] [CrossRef]
- González-Cruz, R.; Solorza-Feria, O. Oxygen reduction in acid media by a RuxFeySez(CO)n cluster catalyst dispersed on a glassy carbon-supported nafion film. J. Sol. State Electrochem. 2003, 7, 289–295. [Google Scholar] [CrossRef]
- Cheng, H.; Yuan, W.; Scott, K. The influence of a new fabrication procedure on the catalytic activity of ruthenium–selenium catalysts. Electrochim. Acta 2006, 52, 466–473. [Google Scholar] [CrossRef]
- Nagabhushana, K.S.; Dinjus, E.; Bönnemann, H.; Zaikovskii, V.; Hartnig, C.; Zehl, G.; Dorbandt, I.; Fiechter, S.; Bogdanoff, P. Reductive annealing for generating Se doped 20 wt % Ru/C cathode catalysts for the oxygen reduction reaction. J. Appl. Electrochem. 2007, 37, 1515–1522. [Google Scholar] [CrossRef]
- Montiel, M.; García-Rodríguez, S.; Hernández-Fernández, P.; Díaz, R.; Rojas, S.; Fierro, J.L.G.; Fatás, E.; Ocón, P. Relevance of the synthesis route of Se-modified Ru/C as methanol tolerant electrocatalysts for the oxygen reduction reaction. J. Power Sources 2010, 195, 2478–2487. [Google Scholar] [CrossRef]
- Suárez-Alcántara, K.; Solorza-Feria, O. Kinetics and PEMFC performance of RuxMoySez nanoparticles as a cathode catalyst. Electrochim. Acta 2008, 53, 4981–4989. [Google Scholar] [CrossRef]
- Koper, M.T.M. Electrocatalysis on bimetallic and alloy surfaces. Surf. Sci. 2004, 548, 1–3. [Google Scholar] [CrossRef]
- He, T.; Kreidler, E.; Xiong, L.; Ding, E. Combinatorial screening and nano-synthesis of platinum binary alloys for oxygen electroreduction. J. Power Sources 2007, 165, 87–91. [Google Scholar] [CrossRef]
- Anastasijević, N.A.; Vesović, V.; Adžić, R.R. Determination of the kinetic parameters of the oxygen reduction reaction using the rotating ring-disk electrode. J. Electroanal. Chem. Interfacial Electrochem. 1987, 229, 305–316. [Google Scholar] [CrossRef]
- Kalevaru, V.N.; Benhmid, A.; Radnik, J.; Pohl, M.M.; Bentrup, U.; Martin, A. Marked influence of support on the catalytic performance of PdSb acetoxylation catalysts: Effects of Pd particle size, valence states, and acidity characteristics. J. Catal. 2007, 246, 399–412. [Google Scholar] [CrossRef]
- Bagotzky, V.S.; Shumilova, N.A.; Samoilov, G.P.; Khrushcheva, E.I. Electrochemical oxygen reduction on nickel electrodes in alkaline solutions—II. Electrochim. Acta 1972, 17, 1625–1635. [Google Scholar] [CrossRef]
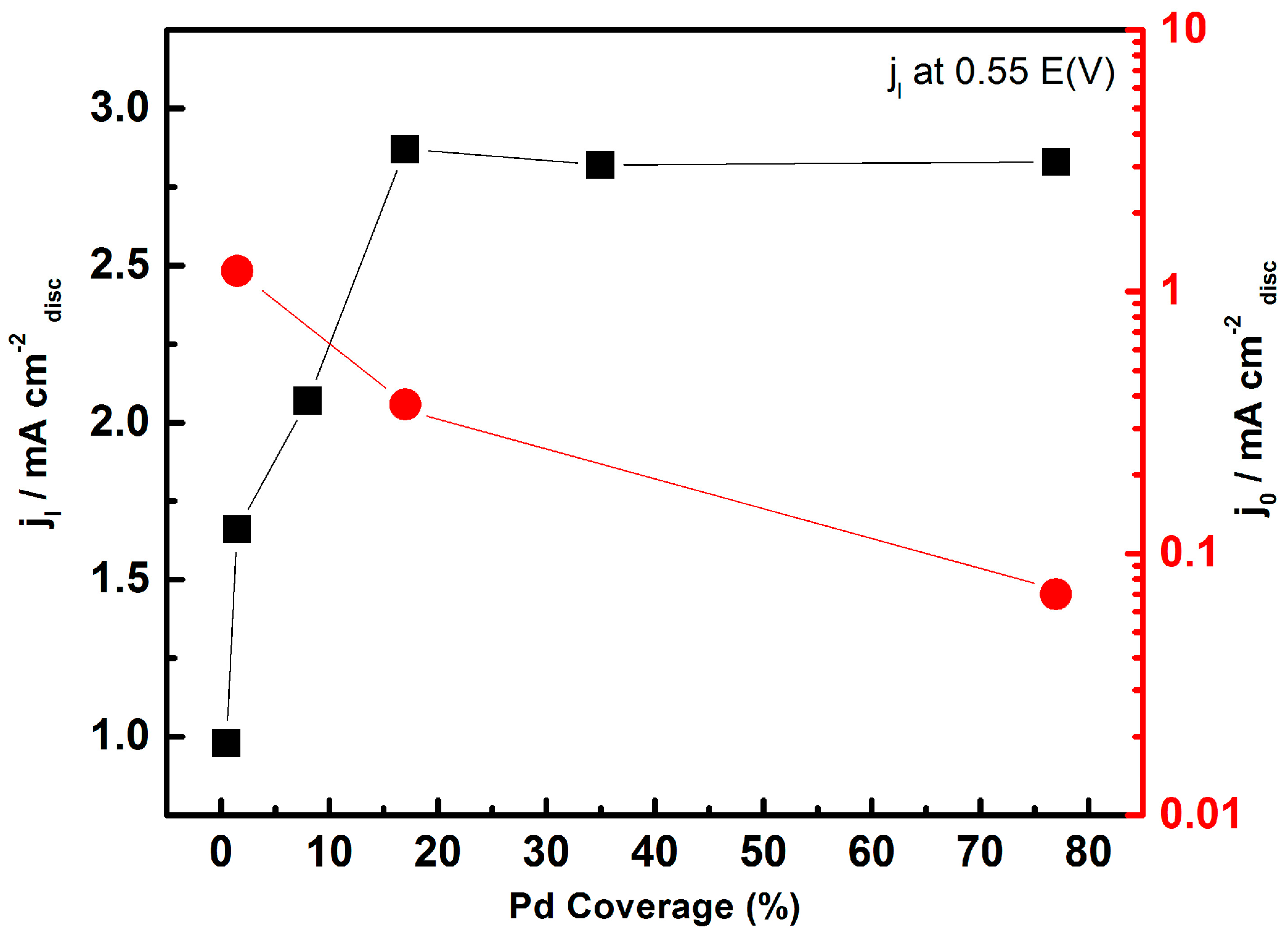
- Ju, W.; Favaro, M.; Durante, C.; Perini, L.; Agnoli, S.; Schneider, O.; Stimming, U.; Granozzi, G. Pd nanoparticles deposited on nitrogen-doped HOPG: New insights into the Pd-catalyzed oxygen reduction reaction. Electrochim. Acta 2014, 141, 89–101. [Google Scholar] [CrossRef]
- Chen, J.; Takanabe, K.; Ohnishi, R.; Lu, D.; Okada, S.; Hatasawa, H.; Morioka, H.; Antonietti, M.; Kubota, J.; Domen, K. Nano-sized tin on carbon black as an efficient electrocatalyst for the oxygen reduction reaction prepared using an MPG-C3N4 template. Chem. Commun. 2010, 46, 7492–7494. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Gross, A.; Yang, H. Shape and composition-controlled platinum alloy nanocrystals using carbon monoxide as reducing agent. Nano Lett. 2011, 11, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Vukmirovic, M.B.; Zhang, J.; Sasaki, K.; Nilekar, A.U.; Uribe, F.; Mavrikakis, M.; Adzic, R.R. Platinum monolayer electrocatalysts for oxygen reduction. Electrochim. Acta 2007, 52, 2257–2263. [Google Scholar] [CrossRef]
- Salvador-Pascual, J.J.; Citalán-Cigarroa, S.; Solorza-Feria, O. Kinetics of oxygen reduction reaction on nanosized Pd electrocatalyst in acid media. J. Power Sources 2007, 172, 229–234. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, C.M.; Bard, A.J. Hydrogen peroxide production in the oxygen reduction reaction at different electrocatalysts as quantified by scanning electrochemical microscopy. Anal. Chem. 2009, 81, 8094–8100. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.-H.; Sasaki, K.; Adzic, R.R. Pd–Fe nanoparticles as electrocatalysts for oxygen reduction. J. Am. Chem. Soc. 2006, 128, 3526–3527. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Liu, P.; Zhang, J.; Adzic, R. Origin of enhanced activity in palladium alloy electrocatalysts for oxygen reduction reaction. J. Phys. Chem. B 2007, 111, 6772–6775. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Manthiram, A. Tuning the electrocatalytic activity and durability of low cost Pd70Co30 nanoalloy for oxygen reduction reaction in fuel cells. Electrochem. Commun. 2008, 10, 740–744. [Google Scholar] [CrossRef]
- Kirubakaran, A.; Jain, S.; Nema, R.K. A review on fuel cell technologies and power electronic interface. Renew. Sust. Energy Rev. 2009, 13, 2430–2440. [Google Scholar] [CrossRef]
- Mora-Hernández, J.M.; Ezeta-Mejía, A.; Reza-San Germán, C.; Citalán-Cigarroa, S.; Arce-Estrada, E.M. Electrochemical activity towards ORR of mechanically alloyed PdCo supported on vulcan carbon and carbon nanospheres. J. Appl. Electrochem. 2014, 44, 1307–1315. [Google Scholar] [CrossRef]
- Lee, Y.-W.; Ko, A.R.; Kim, D.-Y.; Han, S.-B.; Park, K.-W. Octahedral Pt-Pd alloy catalysts with enhanced oxygen reduction activity and stability in proton exchange membrane fuel cells. RSC Adv. 2012, 2, 1119–1125. [Google Scholar] [CrossRef]
- Antolini, E. Palladium in fuel cell catalysis. Energy Environ. Sci. 2009, 2, 915–931. [Google Scholar] [CrossRef]
- Calvo, S.R.; Balbuena, P.B. Density functional theory analysis of reactivity of PtxPdy alloy clusters. Surf. Sci. 2007, 601, 165–171. [Google Scholar] [CrossRef]
- Shen, P.K.; Xu, C. Alcohol oxidation on nanocrystalline oxide Pd/C promoted electrocatalysts. Electrochem. Commun. 2006, 8, 184–188. [Google Scholar] [CrossRef]
- Zhang, S.; Shao, Y.; Yin, G.; Lin, Y. Electrostatic self-assembly of a Pt-around-Au nanocomposite with high activity towards formic acid oxidation. Angew. Chem. Int. Ed. 2010, 49, 2211–2214. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.; Larsen, R.; Masel, R.I. Performance characterization of Pd/C nanocatalyst for direct formic acid fuel cells. J. Power Sources 2005, 144, 28–34. [Google Scholar] [CrossRef]
- Zhu, Y.; Khan, Z.; Masel, R.I. The behavior of palladium catalysts in direct formic acid fuel cells. J. Power Sources 2005, 139, 15–20. [Google Scholar] [CrossRef]
- Alonso-Vante, N. Tailoring of metal cluster-like materials for the molecular oxygen reduction reaction. Pure Appl. Chem. 2008, 80, 2103–2114. [Google Scholar] [CrossRef]
- Gago Aldo, S.; Habrioux, A.; Alonso-Vante, N. Tailoring nanostructured catalysts for electrochemical energy conversion systems. Nanotechnol. Rev. 2012, 1, 427–453. [Google Scholar] [CrossRef]
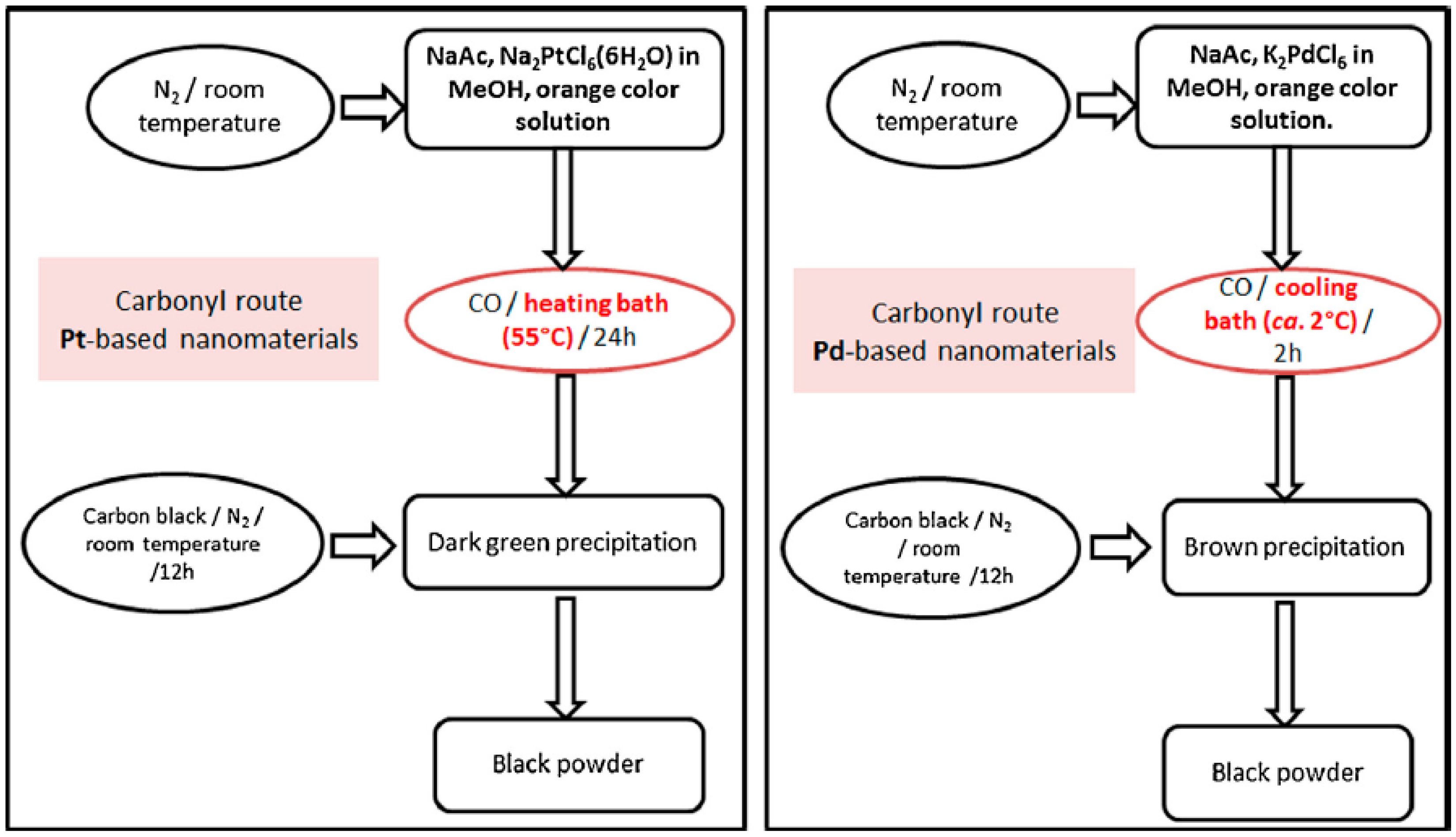
- Alonso-Vante, N. Carbonyl tailored electrocatalysts. Fuel Cells 2006, 6, 182–189. [Google Scholar] [CrossRef]
- Le Rhun, V.; Garnier, E.; Pronier, S.; Alonso-Vante, N. Electrocatalysis on nanoscale ruthenium-based material manufactured by carbonyl decomposition. Electrochem. Commun. 2000, 2, 475–479. [Google Scholar] [CrossRef]
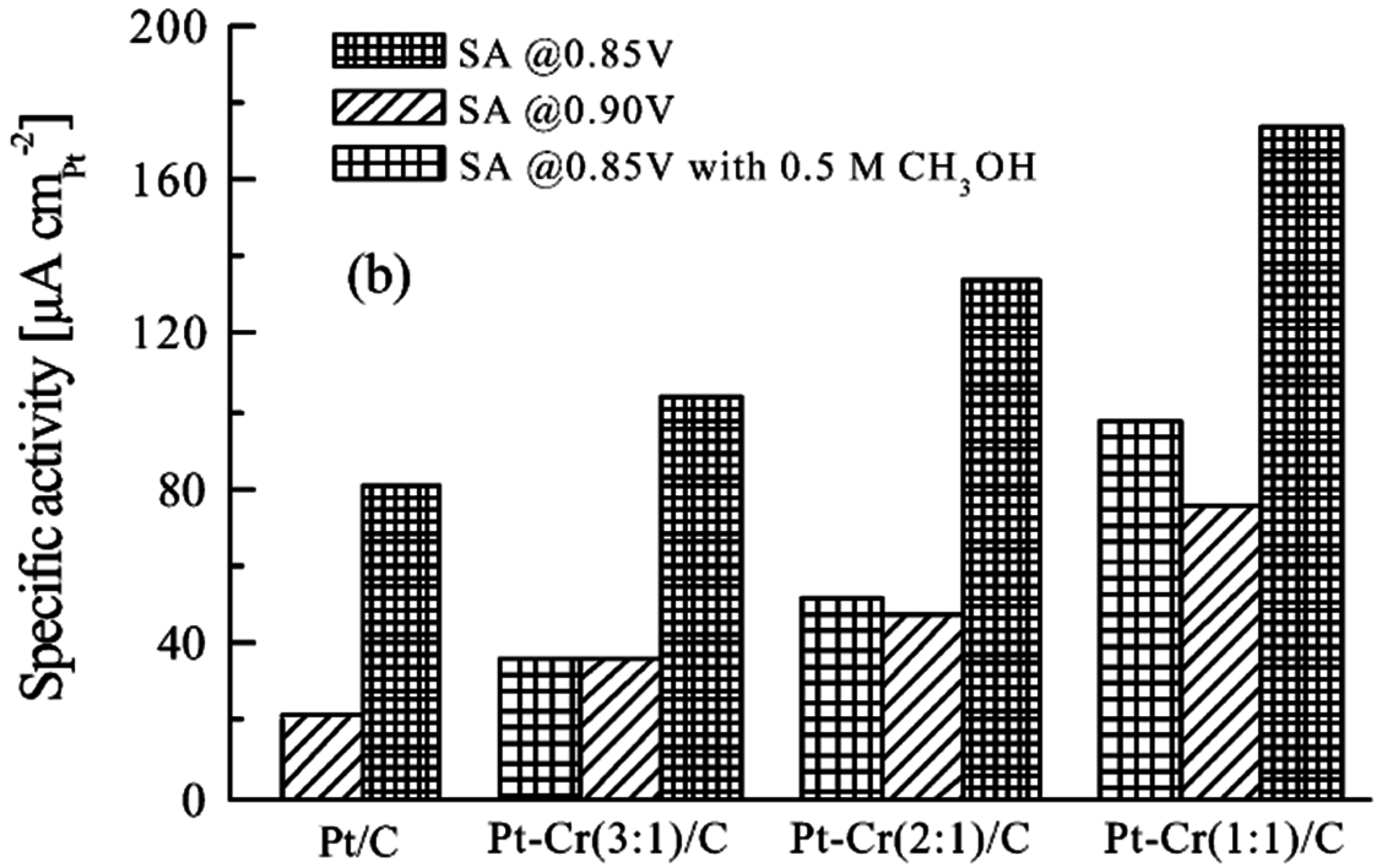
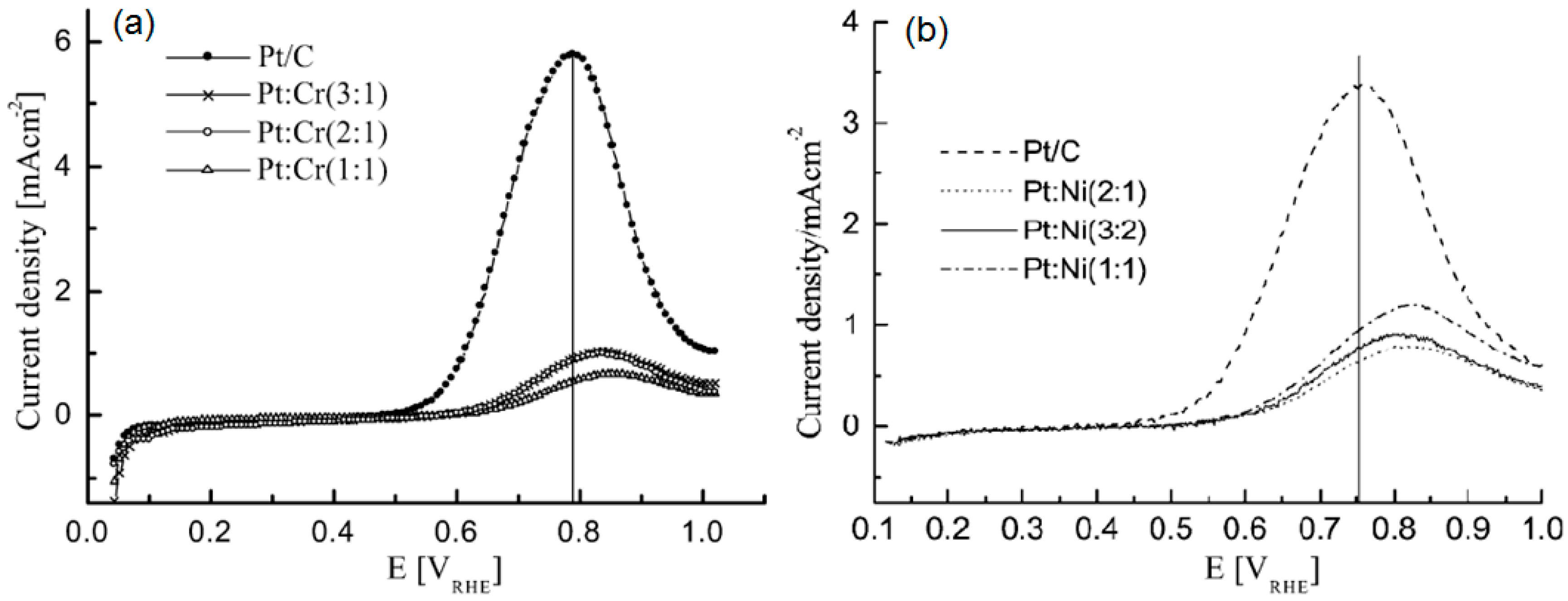
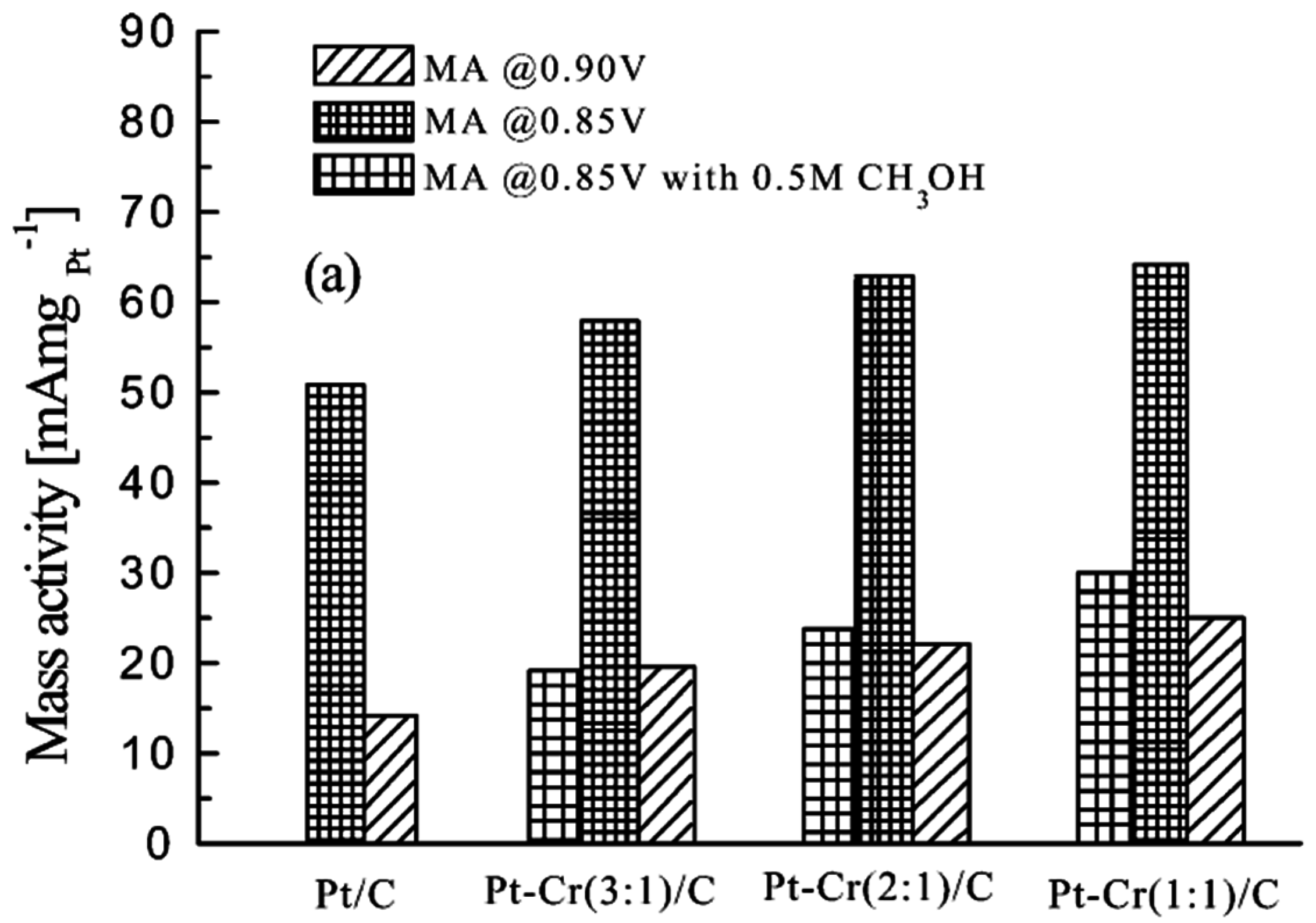
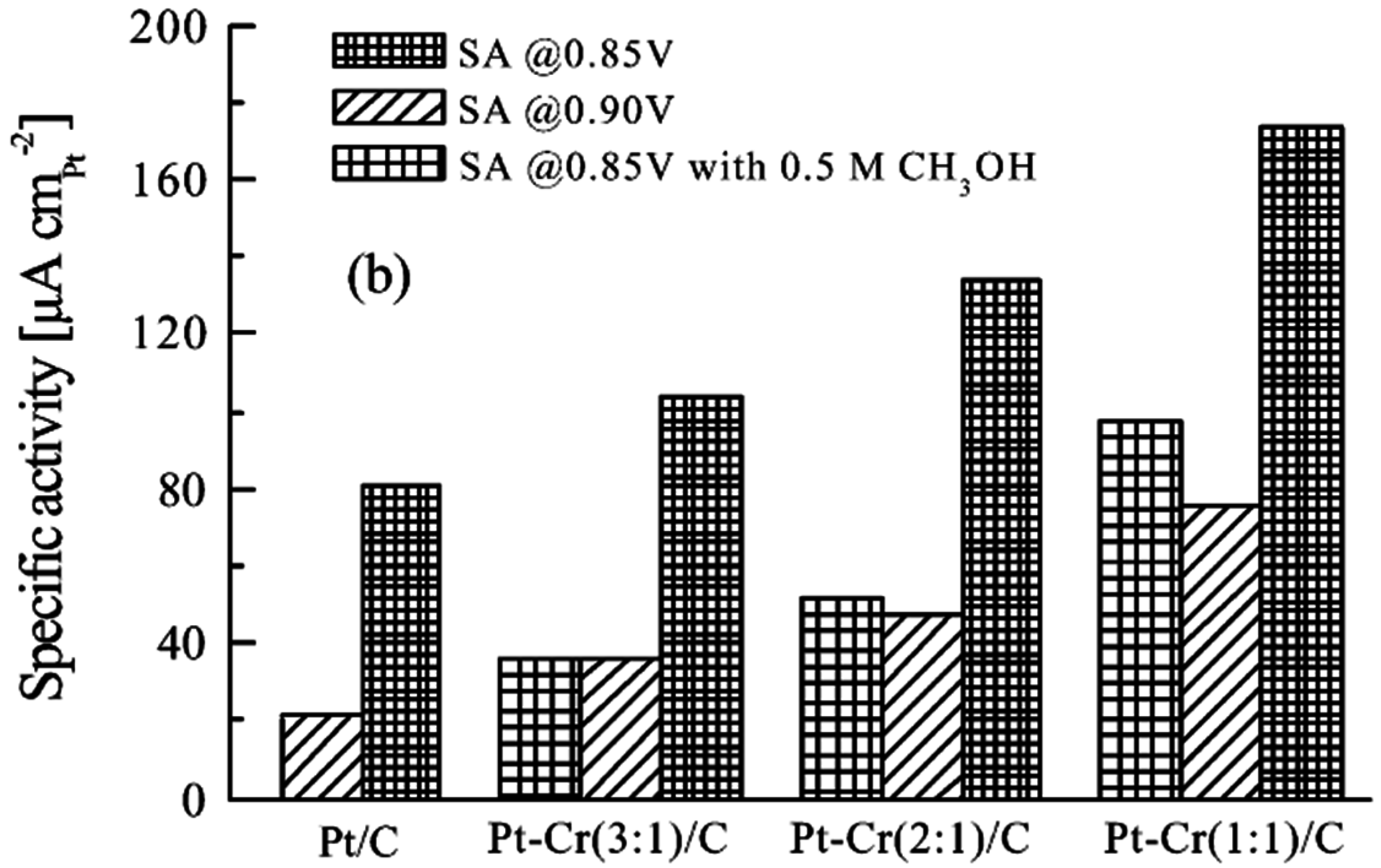
- Yang, H.; Alonso-Vante, N.; Lamy, C.; Akins, D.L. High methanol tolerance of carbon-supported Pt-Cr alloy nanoparticle electrocatalysts for oxygen reduction. J. Electrochem. Soc. 2005, 152, A704–A709. [Google Scholar] [CrossRef]
- Yang, H.; Alonso-Vante, N.; Léger, J.-M.; Lamy, C. Tailoring, structure, and activity of carbon-supported nanosized Pt–Cr alloy electrocatalysts for oxygen reduction in pure and methanol-containing electrolytes. J. Phys. Chem. B 2004, 108, 1938–1947. [Google Scholar] [CrossRef]
- Yang, H.; Vogel, W.; Lamy, C.; Alonso-Vante, N. Structure and electrocatalytic activity of carbon-supported Pt–Ni alloy nanoparticles toward the oxygen reduction reaction. J. Phys. Chem. B 2004, 108, 11024–11034. [Google Scholar] [CrossRef]
- Luo, Y.; Mora-Hernández, J.M.; Alonso-Vante, N. Morphological impact onto organic fuel oxidation of nanostructured palladium synthesized via carbonyl chemical route. J. Electroanal. Chem. 2016, 765, 79–86. [Google Scholar] [CrossRef]
- Luo, Y.; Mora-Hernández, J.M.; Estudillo-Wong, L.A.; Arce-Estrada, E.M.; Alonso-Vante, N. Nanostructured palladium tailored via carbonyl chemical route towards oxygen reduction reaction. Electrochim. Acta 2015, 173, 771–778. [Google Scholar] [CrossRef]
- Longoni, G.; Chini, P. Synthesis and chemical characterization of platinum carbonyl dianions [Pt3(CO)6]n2− (n = .apprx.10,6,5,4,3,2,1). A new series of inorganic oligomers. J. Am. Chem. Soc. 1976, 98, 7225–7231. [Google Scholar] [CrossRef]
- Manzo-Robledo, A.; Boucher, A.C.; Pastor, E.; Alonso-Vante, N. Electro-oxidation of carbon monoxide and methanol on carbon-supported Pt–Sn nanoparticles: A dems study. Fuel Cells 2002, 2, 109–116. [Google Scholar] [CrossRef]
- Yang, H.; Coutanceau, C.; Léger, J.-M.; Alonso-Vante, N.; Lamy, C. Methanol tolerant oxygen reduction on carbon-supported Pt–Ni alloy nanoparticles. J. Electroanal. Chem. 2005, 576, 305–313. [Google Scholar] [CrossRef]
- Ma, J.; Gago, A.S.; Alonso-Vante, N. Performance study of platinum nanoparticles supported onto MWCNT in a formic acid microfluidic fuel cell system. J. Electrochem. Soc. 2013, 160, F859–F866. [Google Scholar] [CrossRef]
- Kondo, T.; Iwasaki, Y.; Honma, Y.; Takagi, Y.; Okada, S.; Nakamura, J. Formation of nonbonding π electronic states of graphite due to Pt-C hybridization. Phys. Rev. B 2009, 80, 233408. [Google Scholar] [CrossRef]
- Ma, J.; Habrioux, A.; Miyao, T.; Kakinuma, K.; Inukai, J.; Watanabe, M.; Alonso-Vante, N. Correlation between surface chemical composition with catalytic activity and selectivity of organic-solvent synthesized Pt-Ti nanoparticles. J. Mater. Chem. A 2013, 1, 8798–8804. [Google Scholar] [CrossRef]
- Alonso-Vante, N.; Schubert, B.; Tributsch, H. Transition metal cluster materials for multi-electron transfer catalysis. Mater. Chem. Phys. 1989, 22, 281–307. [Google Scholar] [CrossRef]
- Chevrel, R.; Sergent, M.; Prigent, J. Sur de nouvelles phases sulfurées ternaires du molybdène. J. Solid State Chem. 1971, 3, 515–519. (In French) [Google Scholar] [CrossRef]
- Perrin, A.; Chevrel, R.; Sergent, M.; Fischer, Ø. Synthesis and electrical properties of new chalcogenide compounds containing mixed (Mo, Me)6 octahedral clusters (Me = Ru or Rh). J. Sol. State Chem. 1980, 33, 43–47. [Google Scholar] [CrossRef]
- Perrin, A.; Sergent, M.; Fischer, O. New compounds of the type Mo2Re4X8 (M = S, Se) containing octahedral Mo2Re4 clusters. Mater. Res. Bull. 1978, 13, 259–264. [Google Scholar] [CrossRef]
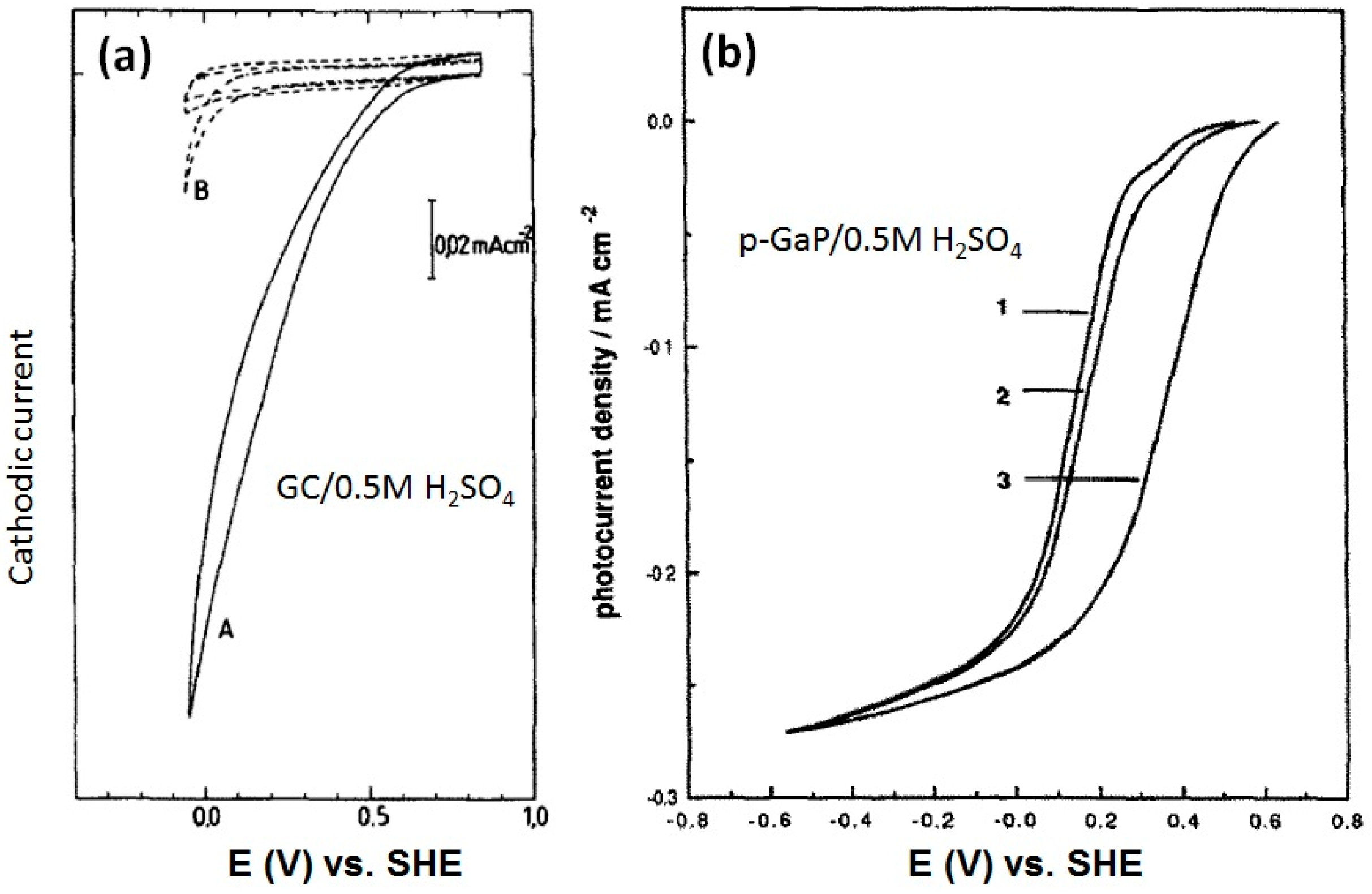
- Alonso-Vante, N.; Tributsch, H. Energy conversion catalysis using semiconducting transition metal cluster compounds. Nature 1986, 323, 431–432. [Google Scholar] [CrossRef]
- Alonso-Vante, N.; Tributsch, H. what. J. Electroanal. Chem. Interfacial Electrochem. 1987, 229, 223–237. [Google Scholar]
- Kühne, H.M.; Tributsch, H. Oxygen and chlorine evolution on ruthenium-iron-disulphide mediated by low energy photons. Ber. Bunsenges. Physikalische Chem. 1984, 88, 10–16. [Google Scholar] [CrossRef]
- Kühne, H.M.; Tributsch, H. Energetics and dynamics of the interface of RuS2 and implications for photoelectrolysis of water. J. Electroanal. Chem. Interfacial Electrochem. 1986, 201, 263–282. [Google Scholar] [CrossRef]
- Vante, N.A.; Schubert, B.; Tributsch, H.; Perrin, A. Influence of d-state density and chemistry of transition metal cluster selenides on electrocatalysis. J. Catal. 1988, 112, 384–391. [Google Scholar] [CrossRef]
- Hughbanks, T.; Hoffmann, R. Molybdenum chalcogenides: Clusters, chains, and extended solids. The approach to bonding in three dimensions. J. Am. Chem. Soc. 1983, 105, 1150–1162. [Google Scholar] [CrossRef]
- Jaegermann, W.; Pettenkofer, C.; Alonso Vante, N.; Schwarzlose, T.; Tributsch, H. Chevrel phase type compounds: Electronic, chemical and structural factors in oxygen reduction electrocatalysis. Ber. Bunsenges. Phys. Chem. 1990, 94, 513–520. [Google Scholar] [CrossRef]
- Solorza-Feria, O.; Ellmer, K.; Giersig, M.; Alonso-Vante, N. Novel low-temperature synthesis of semiconducting transition metal chalcogenide electrocatalyst for multielectron charge transfer: Molecular oxygen reduction. Electrochim. Acta 1994, 39, 1647–1653. [Google Scholar] [CrossRef]
- Kochubey, D.I.; Nikitenko, S.G.; Parmon, V.N.; Gruzdkov, Yu.A.; Tributsch, H.; Alonso-Vante, N. In situ EXAFS-electrochemical study of reduction of molecular oxygen on Mo□Ru□Se thin layers electrodes in acidic media. Phys. B Condens. Matter 1995, 208–209, 694–696. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Paulus, U.A.; Gasteiger, H.A.; Alonso-Vante, N.; Behm, R.J. Oxygen reduction on Ru1.92Mo0.08SeO4, Ru/carbon, and Pt/carbon in pure and methanol-containing electrolytes. J. Electrochem. Soc. 2000, 147, 2620–2624. [Google Scholar] [CrossRef]
- Dassenoy, F.; Vogel, W.; Alonso-Vante, N. Structural studies and stability of cluster-like RuxSey electrocatalysts. J. Phys. Chem. B 2002, 106, 12152–12157. [Google Scholar] [CrossRef]
- Alonso-Vante, N.; Borthen, P.; Fieber-Erdmann, M.; Strehblow, H.H.; Holub-Krappe, E. An in situ grazing incidence X-ray absorption study of ultra thin ruxsey cluster-like electrocatalyst layers. Electrochim. Acta 2000, 45, 4227–4236. [Google Scholar] [CrossRef]
- Malakhov, I.V.; Nikitenko, S.G.; Savinova, E.R.; Kochubey, D.I.; Alonso-Vante, N. In situ EXAFS study to probe active centers of Ru chalcogenide electrocatalysts during oxygen reduction reaction. J. Phys. Chem. B 2002, 106, 1670–1676. [Google Scholar] [CrossRef]
- Babu, P.K.; Lewera, A.; Jong, H.C.; Hunger, R.; Jaegermann, W.; Alonso-Vante, N.; Wieckowski, A.; Oldfield, E. Selenium becomes metallic in Ru-Se fuel cell catalysts: An EC-NMR and XPS investigation. J. Am. Chem. Soc. 2007, 129, 15140–15141. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Wieckowski, A.; Inukai, J.; Alonso-Vante, N. Oxygen reduction reaction on ruthenium and rhodium nanoparticles modified with selenium and sulfur. J. Electrochem. Soc. 2006, 153, A869–A874. [Google Scholar] [CrossRef]
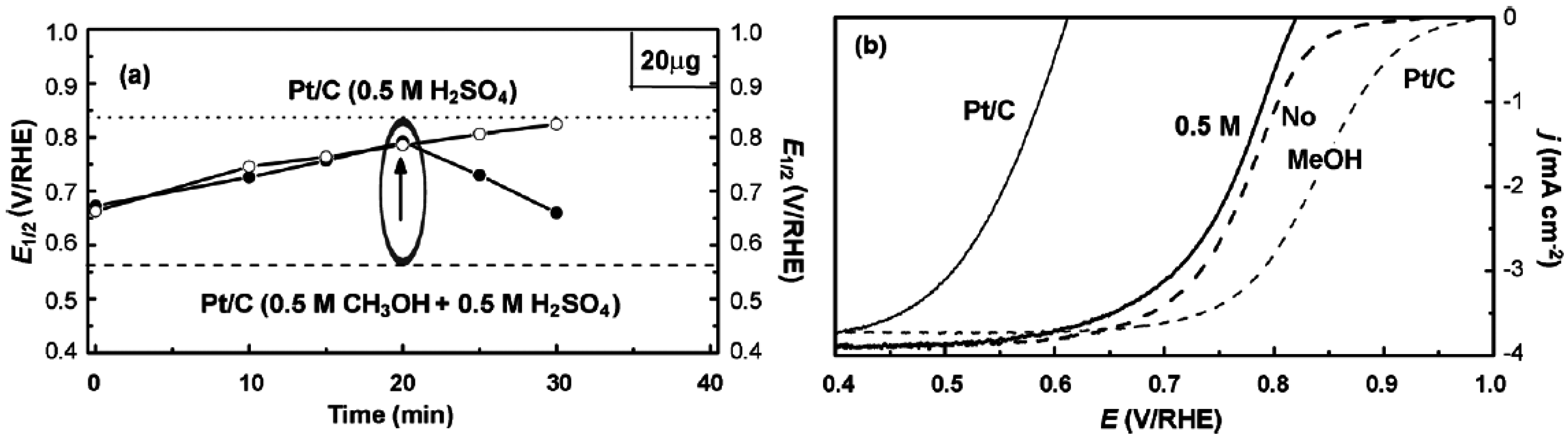
- Ma, J.; Gago, A.S.; Vogel, W.; Alonso-Vante, N. Tailoring and tuning the tolerance of a Pt chalcogenide cathode electrocatalyst to methanol. ChemCatChem 2013, 5, 701–705. [Google Scholar] [CrossRef]
- Nie, Y.; Li, L.; Wei, Z. Recent advancements in Pt and Pt-free catalysts for oxygen reduction reaction. Chem. Soc. Rev. 2015, 44, 2168–2201. [Google Scholar] [CrossRef] [PubMed]
- Borup, R.; Meyers, J.; Pivovar, B.; Kim, Y.S.; Mukundan, R.; Garland, N.; Myers, D.; Wilson, M.; Garzon, F.; Wood, D.; et al. Scientific aspects of polymer electrolyte fuel cell durability and degradation. Chem. Rev. 2007, 107, 3904–3951. [Google Scholar] [CrossRef] [PubMed]
- Su, D.S.; Sun, G. Nonprecious-metal catalysts for low-cost fuel cells. Angew. Chem. Int. Ed. 2011, 50, 11570–11572. [Google Scholar] [CrossRef] [PubMed]
- Behret, H.; Binder, H.; Sandstede, G. Electrocatalytic oxygen reduction with thiospinels and other sulphides of transition metals. Electrochim. Acta 1975, 20, 111–117. [Google Scholar] [CrossRef]
- Feng, Y.; Alonso-Vante, N. Nonprecious metal catalysts for the molecular oxygen-reduction reaction. Phys. Status Solidi 2008, 245, 1792–1806. [Google Scholar] [CrossRef]
- Puntes, V.F.; Krishnan, K.M.; Alivisatos, A.P. Colloidal nanocrystal shape and size control: The case of cobalt. Science 2001, 291, 2115–2117. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.T.; Shen, C.M.; Wang, Y.G.; Su, Y.K.; Yang, T.Z.; Gao, H.J. Stable cobalt nanoparticles passivated with oleic acid and triphenylphosphine. Nanotechnology 2004, 15, 70. [Google Scholar] [CrossRef]
- Yin, Y.; Rioux, R.M.; Erdonmez, C.K.; Hughes, S.; Somorjai, G.A.; Alivisatos, A.P. Formation of hollow nanocrystals through the nanoscale kirkendall effect. Science 2004, 304, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Sidik, R.A.; Anderson, A.B. Co9S8 as a catalyst for electroreduction of O2: Quantum chemistry predictions. J. Phys. Chem. B 2006, 110, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Vayner, E.; Sidik, R.A.; Anderson, A.B.; Popov, B.N. Experimental and theoretical study of cobalt selenide as a catalyst for O2 electroreduction. J. Phys. Chem. C 2007, 111, 10508–10513. [Google Scholar] [CrossRef]
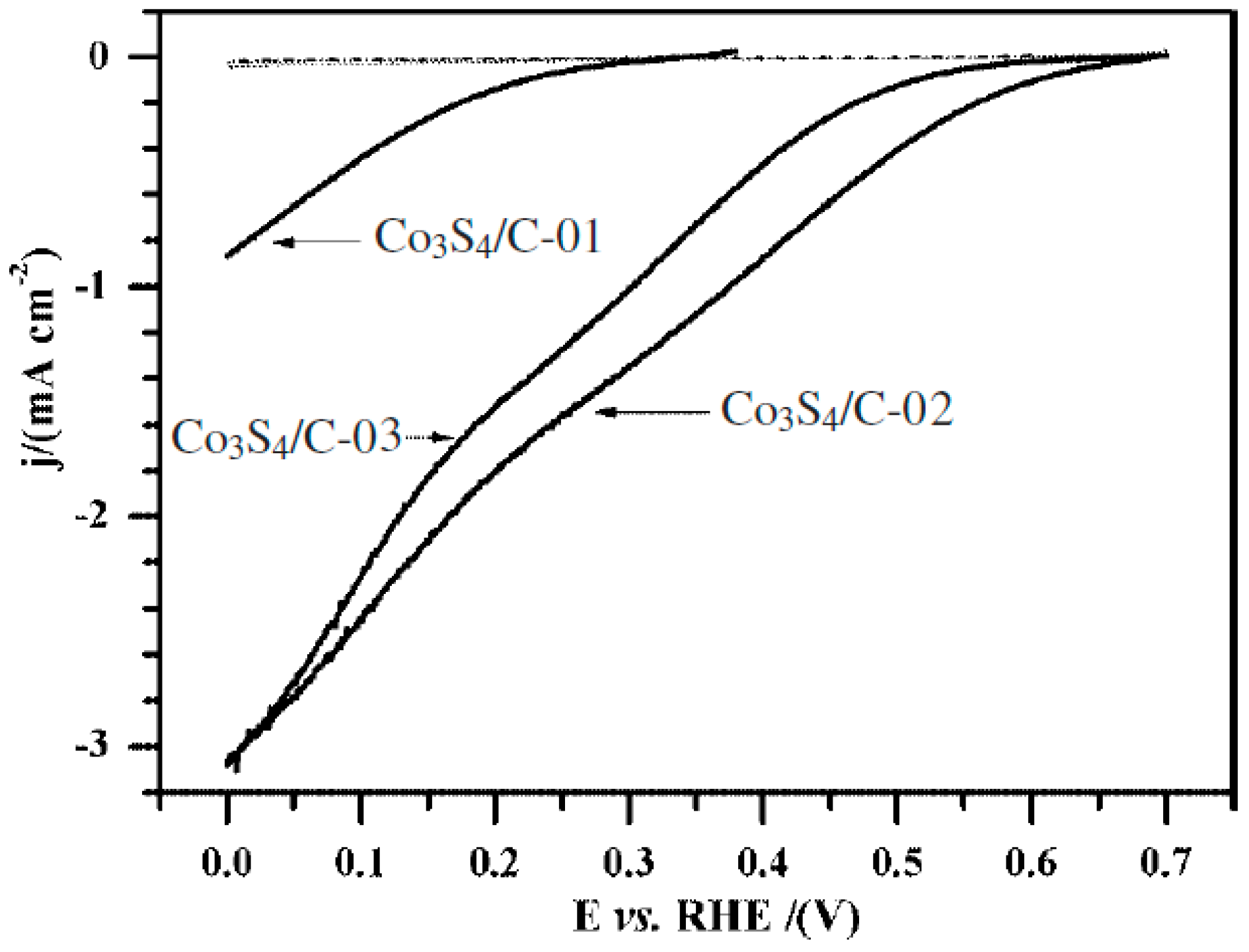
- Feng, Y.; He, T.; Alonso-Vante, N. In situ free-surfactant synthesis and ORR- electrochemistry of carbon-supported Co3S4 and CoSe2 nanoparticles. Chem. Mater. 2008, 20, 26–28. [Google Scholar] [CrossRef]
- Feng, Y.; He, T.; Alonso-Vante, N. Oxygen reduction reaction on carbon-supported CoSe2 nanoparticles in an acidic medium. Electrochim. Acta 2009, 54, 5252–5256. [Google Scholar] [CrossRef]
- Susac, D.; Sode, A.; Zhu, L.; Wong, P.C.; Teo, M.; Bizzotto, D.; Mitchell, K.A.R.; Parsons, R.R.; Campbell, S.A. A methodology for investigating new nonprecious metal catalysts for PEM fuel cells. J. Phys. Chem. B 2006, 110, 10762–10770. [Google Scholar] [CrossRef] [PubMed]
- Susac, D.; Zhu, L.; Teo, M.; Sode, A.; Wong, K.C.; Wong, P.C.; Parsons, R.R.; Bizzotto, D.; Mitchell, K.A.R.; Campbell, S.A. Characterization of FeS2-based thin films as model catalysts for the oxygen reduction reaction. J. Phys. Chem. C 2007, 111, 18715–18723. [Google Scholar] [CrossRef]
- Zhu, L.; Susac, D.; Teo, M.; Wong, K.C.; Wong, P.C.; Parsons, R.R.; Bizzotto, D.; Mitchell, K.A.R.; Campbell, S.A. Investigation of CoS2-based thin films as model catalysts for the oxygen reduction reaction. J. Catal. 2008, 258, 235–242. [Google Scholar] [CrossRef]
- Ahlberg, E.; Elfström Broo, A. Oxygen reduction at sulphide minerals. 1. A rotating ring disc electrode (RRDE) study at galena and pyrite. Int. J. Miner. Process. 1996, 46, 73–89. [Google Scholar] [CrossRef]
- Bonakdarpour, A.; Delacote, C.; Yang, R.; Wieckowski, A.; Dahn, J.R. Loading of Se/Ru/C electrocatalyst on a rotating ring-disk electrode and the loading impact on a H2O2 release during oxygen reduction reaction. Electrochem. Commun. 2008, 10, 611–615. [Google Scholar] [CrossRef]
- Alonso-Vante, N. Platinum and non-platinum nanomaterials for the molecular oxygen reduction reaction. ChemPhysChem 2010, 11, 2732–2744. [Google Scholar] [CrossRef] [PubMed]
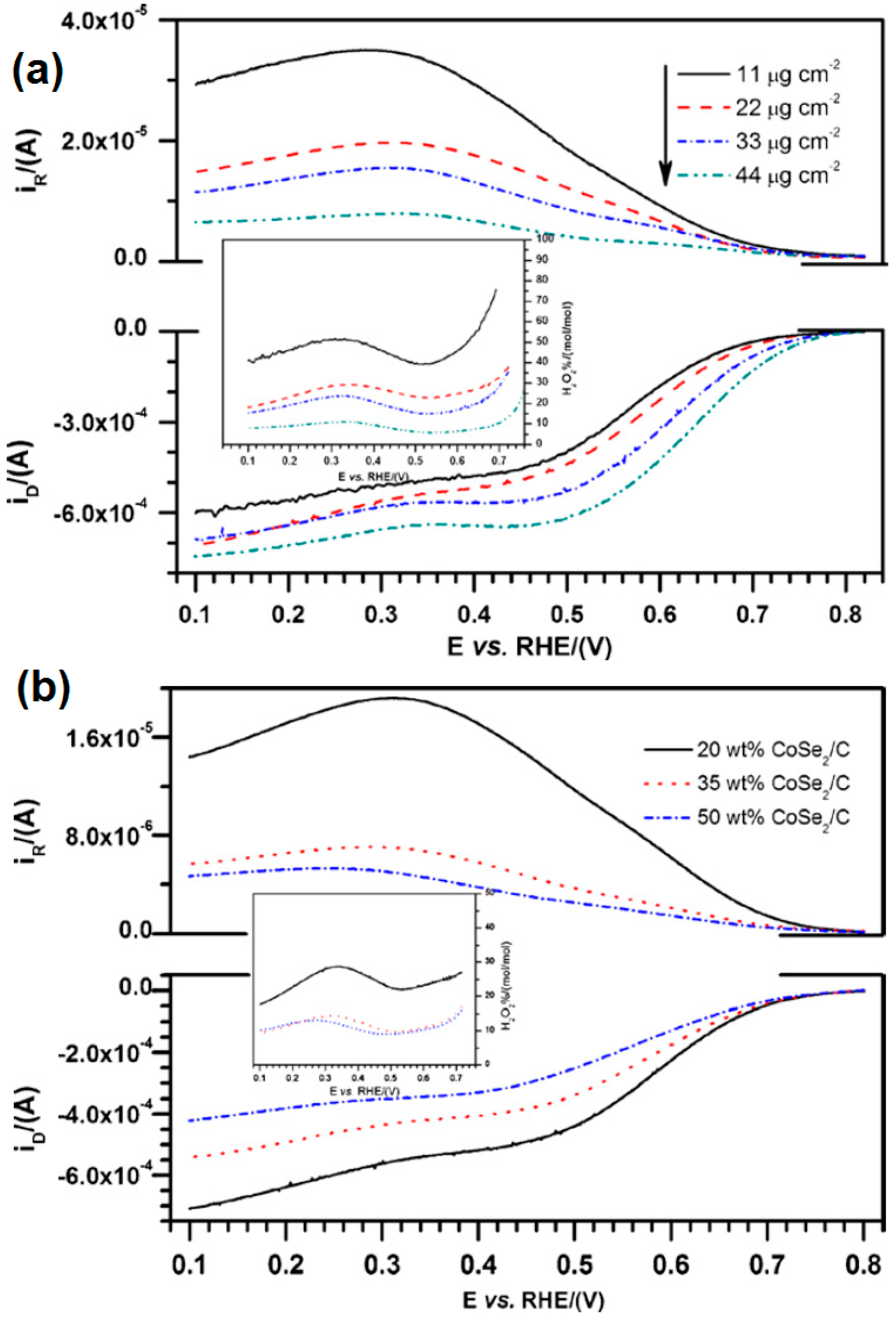
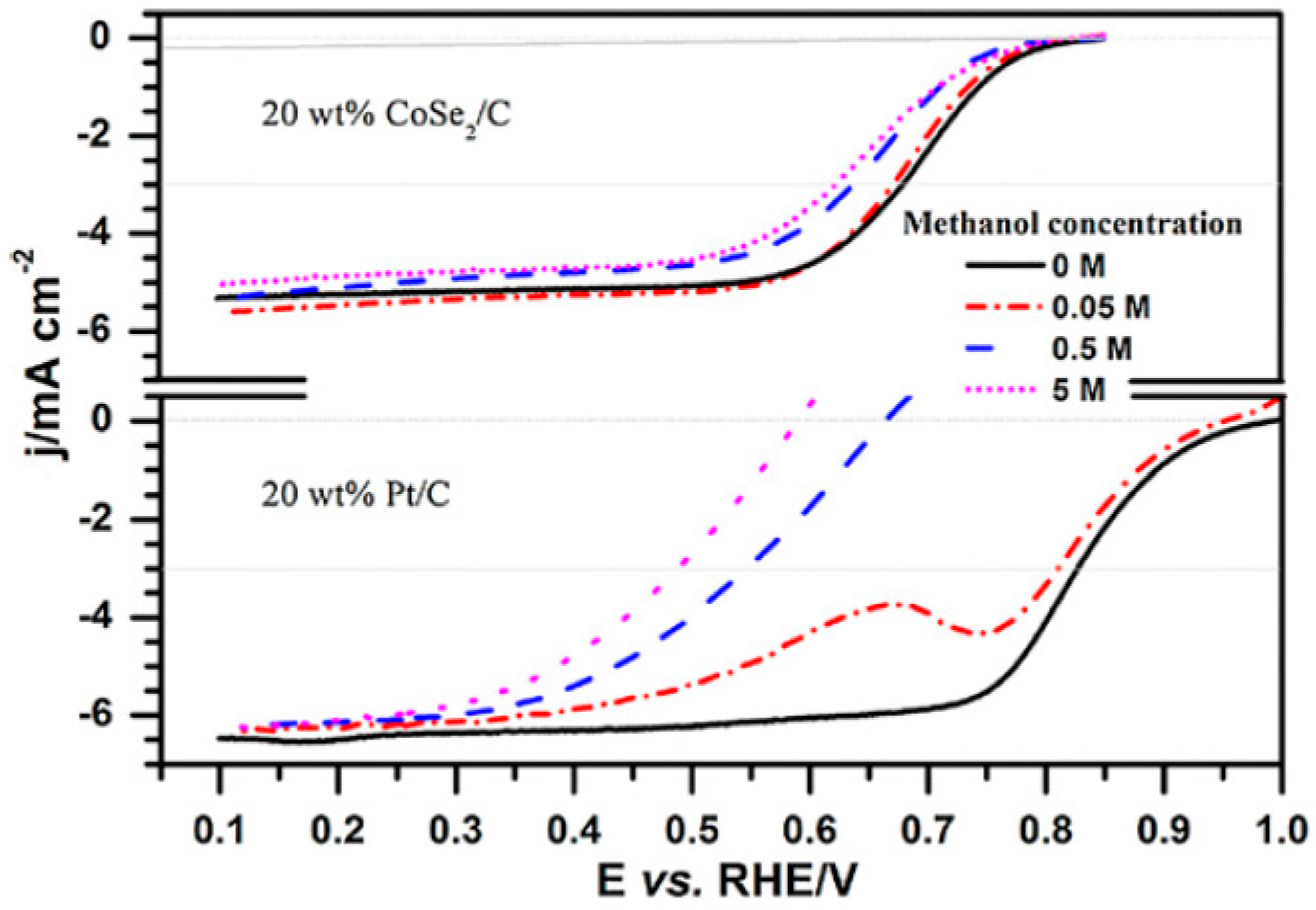
- Feng, Y.; Alonso-Vante, N. Carbon-supported cubic CoSe2 catalysts for oxygen reduction reaction in alkaline medium. Electrochim. Acta 2012, 72, 129–133. [Google Scholar] [CrossRef]
- Feng, Y.J.; He, T.; Alonso-Vante, N. Carbon-supported CoSe2 nanoparticles for oxygen reduction reaction in acid medium. Fuel Cells 2010, 10, 77–83. [Google Scholar]
- Liang, Y.; Li, Y.; Wang, H.; Zhou, J.; Wang, J.; Regier, T.; Dai, H. Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction. Nat. Mater. 2011, 10, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, P. Hydrogénations et déshydrogénations par catalyse. Ber. Dtsch. Chem. Ges. 1911, 44, 1984–2001. [Google Scholar] [CrossRef]
- Yang, B.; Burch, R.; Hardacre, C.; Headdock, G.; Hu, P. Understanding the optimal adsorption energies for catalyst screening in heterogeneous catalysis. ACS Catal. 2014, 4, 182–186. (In French) [Google Scholar] [CrossRef]
- Maruyama, J.; Abe, I. Formation of platinum-free fuel cell cathode catalyst with highly developed nanospace by carbonizing catalase. Chem. Mater. 2005, 17, 4660–4667. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Wilkinson, D.P.; Wang, H. Progress in preparation of non-noble electrocatalysts for PEM fuel cell reactions. J. Power Sources 2006, 156, 171–182. [Google Scholar] [CrossRef]
- Wood, T.E.; Tan, Z.; Schmoeckel, A.K.; O’Neill, D.; Atanasoski, R. Non-precious metal oxygen reduction catalyst for PEM fuel cells based on nitroaniline precursor. J. Power Sources 2008, 178, 510–516. [Google Scholar] [CrossRef]
- Rand, D.A.J.; Woods, R. Cyclic voltammetric studies on iridium electrodes in sulphuric acid solutions. J. Electroanal. Chem. Interfacial Electrochem. 1974, 55, 375–381. [Google Scholar] [CrossRef]
- Croissant, M.J.; Napporn, T.; Léger, J.M.; Lamy, C. Electrocatalytic oxidation of hydrogen at platinum-modified polyaniline electrodes. Electrochim. Acta 1998, 43, 2447–2457. [Google Scholar] [CrossRef]
- Cao, L.; Sun, G.; Li, H.; Xin, Q. Carbon-supported IrSn catalysts for direct ethanol fuel cell. Fuel Cells Bull. 2007, 2007, 12–16. [Google Scholar] [CrossRef]
- Lee, K.; Zhang, L.; Zhang, J. IrxCO1−x (x = 0.3–1.0) alloy electrocatalysts, catalytic activities, and methanol tolerance in oxygen reduction reaction. J. Power Sources 2007, 170, 291–296. [Google Scholar] [CrossRef]
- Santos, E.; Schmickler, W. Electrocatalysis of hydrogen oxidation—Theoretical foundations. Angew. Chem. Int. Ed. 2007, 46, 8262–8265. [Google Scholar] [CrossRef] [PubMed]
- Mokrane, S.; Makhloufi, L.; Alonso-Vante, N. Electrochemical behaviour of platinum nanoparticles supported on polypyrrole (PPY)/C composite. ECS Trans. 2008, 6, 93–103. [Google Scholar]
- Shao, M. Palladium-based electrocatalysts for hydrogen oxidation and oxygen reduction reactions. J. Power Sources 2011, 196, 2433–2444. [Google Scholar] [CrossRef]
- Kwon, K.; Jin, S.-A.; Lee, K.H.; You, D.J.; Pak, C. Performance enhancement of Pd-based hydrogen oxidation catalysts using tungsten oxide. Catal. Today 2014, 232, 175–178. [Google Scholar] [CrossRef]
- Zhang, H.; Shen, P.K. Recent development of polymer electrolyte membranes for fuel cells. Chem. Rev. 2012, 112, 2780–2832. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Bivens, A.P.; Myint, M.; Zhuang, Z.; Forest, R.V.; Fang, Q.; Chen, J.G.; Yan, Y. Non-precious metal electrocatalysts with high activity for hydrogen oxidation reaction in alkaline electrolytes. Energy Environ. Sci. 2014, 7, 1719–1724. [Google Scholar] [CrossRef]
- Spendelow, J.S.; Wieckowski, A. Electrocatalysis of oxygen reduction and small alcohol oxidation in alkaline media. PhysChemChemPhys 2007, 9, 2654–2675. [Google Scholar] [CrossRef] [PubMed]
- Neyerlin, K.C.; Gu, W.; Jorne, J.; Gasteiger, H.A. Study of the exchange current density for the hydrogen oxidation and evolution reactions. J. Electrochem. Soc. 2007, 154, B631–B635. [Google Scholar] [CrossRef]
- Durst, J.; Siebel, A.; Simon, C.; Hasche, F.; Herranz, J.; Gasteiger, H.A. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ. Sci. 2014, 7, 2255–2260. [Google Scholar] [CrossRef]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.; Norskov, J.K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Guo, Y.; Jiang, Y.-Y.; Zhu, W.; Xu, Y.-S.; Zhao, Z.-Q.; Liu, J.-X.; Li, W.-X.; Jin, C.-H.; Yan, C.-H.; et al. Robust phase control through hetero-seeded epitaxial growth for face-centered cubic Pt@Ru nanotetrahedrons with superior hydrogen electro-oxidation activity. J. Phys. Chem. C 2015, 119, 17697–17706. [Google Scholar] [CrossRef]
- Kucernak, A.R.J.; Fahy, K.F.; Sundaram, V.N.N. Facile synthesis of palladium phosphide electrocatalysts and their activity for the hydrogen oxidation, hydrogen evolutions, oxygen reduction and formic acid oxidation reactions. Catal. Today 2016, 262, 48–56. [Google Scholar] [CrossRef]
- Carenco, S.; Portehault, D.; Boissière, C.; Mézailles, N.; Sanchez, C. Nanoscaled metal borides and phosphides: Recent developments and perspectives. Chem. Rev. 2013, 113, 7981–8065. [Google Scholar] [CrossRef] [PubMed]
- Henkes, A.E.; Vasquez, Y.; Schaak, R.E. Converting metals into phosphides: A general strategy for the synthesis of metal phosphide nanocrystals. J. Am. Chem. Soc. 2007, 129, 1896–1897. [Google Scholar] [CrossRef] [PubMed]
- Barry, B.M.; Gillan, E.G. Low-temperature solvothermal synthesis of phosphorus-rich transition-metal phosphides. Chem. Mater. 2008, 20, 2618–2620. [Google Scholar] [CrossRef]
- Alexander, A.-M.; Hargreaves, J.S.J. Alternative catalytic materials: Carbides, nitrides, phosphides and amorphous boron alloys. Chem. Soc. Rev. 2010, 39, 4388–4401. [Google Scholar] [CrossRef] [PubMed]
- Shervedani, R.K.; Lasia, A. Studies of the hydrogen evolution reaction on Ni-P electrodes. J. Electrochem. Soc. 1997, 144, 511–519. [Google Scholar] [CrossRef]
- Hu, C.C.; Bai, A. Optimization of hydrogen evolving activity on nickel–phosphorus deposits using experimental strategies. J. Appl. Electrochem. 2001, 31, 565–572. [Google Scholar] [CrossRef]
- Popczun, E.J.; McKone, J.R.; Read, C.G.; Biacchi, A.J.; Wiltrout, A.M.; Lewis, N.S.; Schaak, R.E. Nanostructured nickel phosphide as an electrocatalyst for the hydrogen evolution reaction. J. Am. Chem. Soc. 2013, 135, 9267–9270. [Google Scholar] [CrossRef] [PubMed]
- Kucernak, A.R.J.; Naranammalpuram Sundaram, V.N. Nickel phosphide: The effect of phosphorus content on hydrogen evolution activity and corrosion resistance in acidic medium. J. Mater. Chem. A 2014, 2, 17435–17445. [Google Scholar] [CrossRef]
- Popczun, E.J.; Read, C.G.; Roske, C.W.; Lewis, N.S.; Schaak, R.E. Highly active electrocatalysis of the hydrogen evolution reaction by cobalt phosphide nanoparticles. Angew. Chem. Int. Ed. 2014, 53, 5427–5430. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Sk, M.A.; Thia, L.; Ge, X.; Lim, R.J.; Wang, J.-Y.; Lim, K.H.; Wang, X. Molybdenum phosphide as an efficient electrocatalyst for the hydrogen evolution reaction. Energy Environ. Sci. 2014, 7, 2624–2629. [Google Scholar] [CrossRef]
- McEnaney, J.M.; Chance Crompton, J.; Callejas, J.F.; Popczun, E.J.; Read, C.G.; Lewis, N.S.; Schaak, R.E. Electrocatalytic hydrogen evolution using amorphous tungsten phosphide nanoparticles. Chem. Commun. 2014, 50, 11026–11028. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Pan, J.; Huang, A.; Zhuang, L.; Lu, J. Alkaline polymer electrolyte fuel cells completely free from noble metal catalysts. Proc. Natl. Acad. Sci. USA 2008, 105, 20611–20614. [Google Scholar] [CrossRef]
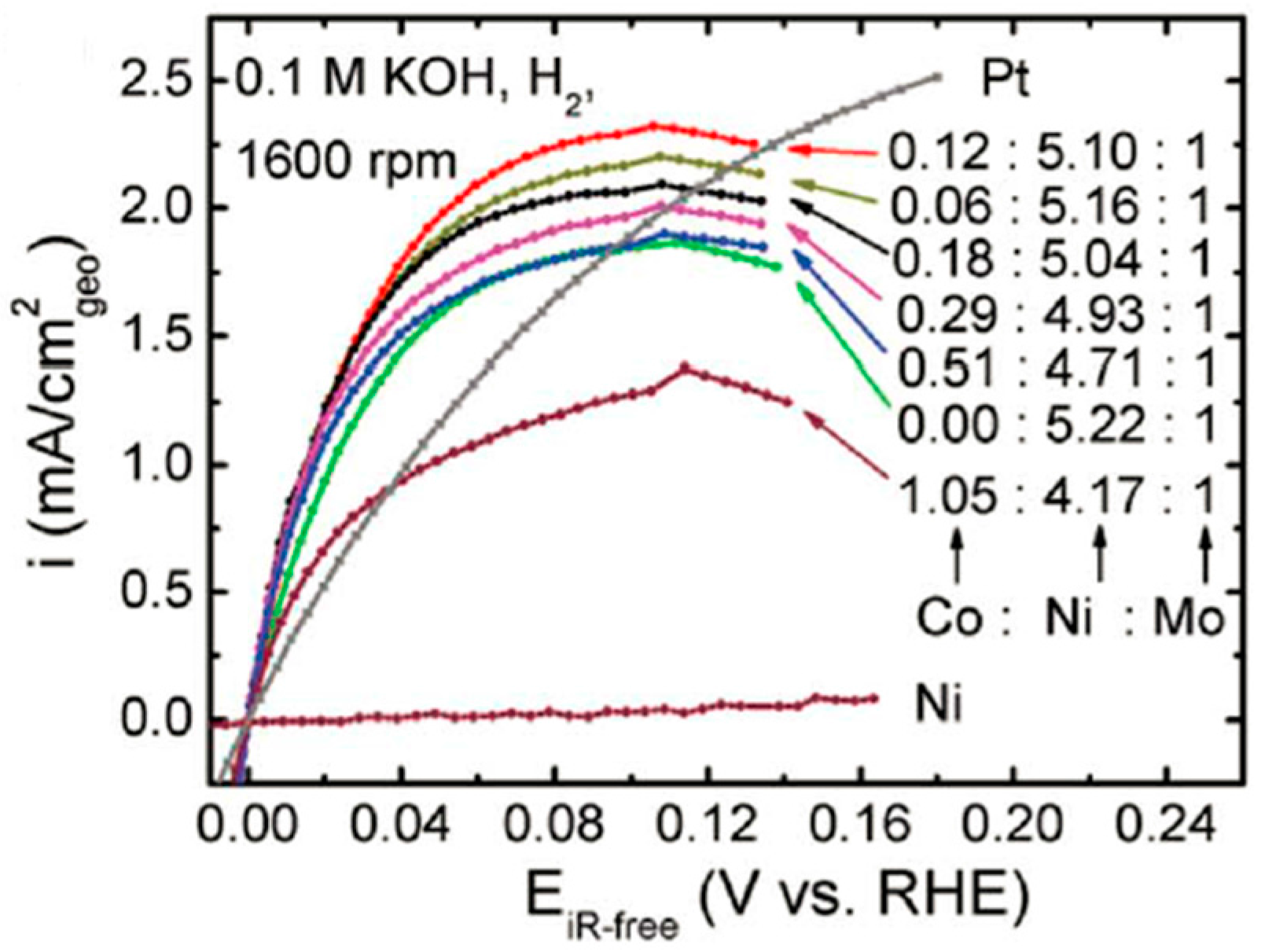
- Sakamoto, T.; Asazawa, K.; Sanabria-Chinchilla, J.; Martinez, U.; Halevi, B.; Atanassov, P.; Strasser, P.; Tanaka, H. Combinatorial discovery of Ni-based binary and ternary catalysts for hydrazine electrooxidation for use in anion exchange membrane fuel cells. J. Power Sources 2014, 247, 605–611. [Google Scholar] [CrossRef]
- Brown, D.E.; Mahmood, M.N.; Man, M.C.M.; Turner, A.K. Preparation and characterization of low overvoltage transition metal alloy electrocatalysts for hydrogen evolution in alkaline solutions. Electrochim. Acta 1984, 29, 1551–1556. [Google Scholar] [CrossRef]
- Bakos, I.; Paszternák, A.; Zitoun, D. Pd/Ni synergestic activity for hydrogen oxidation reaction in alkaline conditions. Electrochim. Acta 2015, 176, 1074–1082. [Google Scholar] [CrossRef]
- Subbaraman, R.; Danilovic, N.; Lopes, P.P.; Tripkovic, D.; Strmcnik, D.; Stamenkovic, V.R.; Markovic, N.M. Origin of anomalous activities for electrocatalysts in alkaline electrolytes. J. Phys. Chem. C 2012, 116, 22231–22237. [Google Scholar] [CrossRef]
- Henning, S.; Herranz, J.; Gasteiger, H.A. Bulk-palladium and palladium-on-gold electrocatalysts for the oxidation of hydrogen in alkaline electrolyte. J. Electrochem. Soc. 2015, 162, F178–F189. [Google Scholar] [CrossRef]
- Subbaraman, R.; Tripkovic, D.; Strmcnik, D.; Chang, K.-C.; Uchimura, M.; Paulikas, A.P.; Stamenkovic, V.; Markovic, N.M. Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)2-Pt interfaces. Science 2011, 334, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
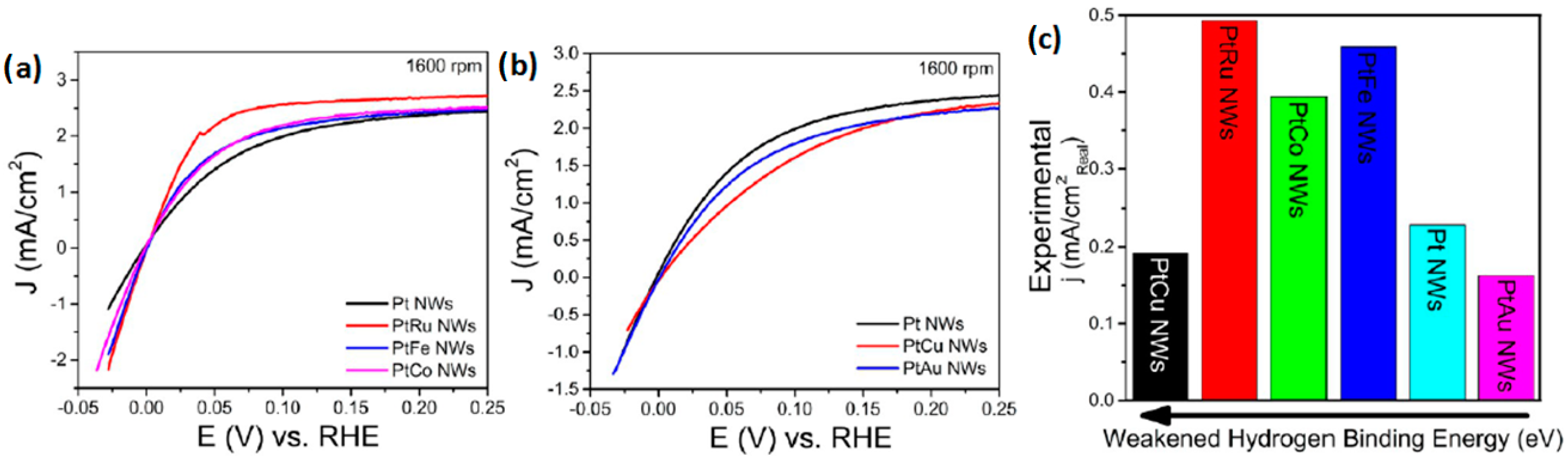
- Scofield, M.E.; Zhou, Y.; Yue, S.; Wang, L.; Su, D.; Tong, X.; Vukmirovic, M.B.; Adzic, R.R.; Wong, S.S. Role of chemical composition in the enhanced catalytic activity of Pt-based alloyed ultrathin nanowires for the hydrogen oxidation reaction under alkaline conditions. ACS Catal. 2016, 6, 3895–3908. [Google Scholar] [CrossRef]
- Scofield, M.E.; Koenigsmann, C.; Wang, L.; Liu, H.; Wong, S.S. Tailoring the composition of ultrathin, ternary alloy ptrufe nanowires for the methanol oxidation reaction and formic acid oxidation reaction. Energy Environ. Sci. 2015, 8, 350–363. [Google Scholar] [CrossRef]
- Alia, S.M.; Larsen, B.A.; Pylypenko, S.; Cullen, D.A.; Diercks, D.R.; Neyerlin, K.C.; Kocha, S.S.; Pivovar, B.S. Platinum-coated nickel nanowires as oxygen-reducing electrocatalysts. ACS Catal. 2014, 4, 1114–1119. [Google Scholar] [CrossRef]
- Kandoi, S.; Ferrin, P.A.; Mavrikakis, M. Hydrogen on and in selected overlayer near-surface alloys and the effect of subsurface hydrogen on the reactivity of alloy surfaces. Top. Catal. 2010, 53, 384–392. [Google Scholar] [CrossRef]
- Rheinländer, P.J.; Herranz, J.; Durst, J.; Gasteiger, H.A. Kinetics of the hydrogen oxidation/evolution reaction on polycrystalline platinum in alkaline electrolyte reaction order with respect to hydrogen pressure. J. Electrochem. Soc. 2014, 161, F1448–F1457. [Google Scholar] [CrossRef]
- Montero, M.A.; Gennero de Chialvo, M.R.; Chialvo, A.C. Kinetics of the hydrogen oxidation reaction on nanostructured rhodium electrodes in alkaline solution. J. Power Sources 2015, 283, 181–186. [Google Scholar] [CrossRef]
- Montero, M.A.; Fernández, J.L.; Gennero de Chialvo, M.R.; Chialvo, A.C. Characterization and kinetic study of a nanostructured rhodium electrode for the hydrogen oxidation reaction. J. Power Sources 2014, 254, 218–223. [Google Scholar] [CrossRef]
- Montero, M.A.; Fernández, J.L.; Gennero de Chialvo, M.R.; Chialvo, A.C. Kinetic study of the hydrogen oxidation reaction on nanostructured iridium electrodes in acid solutions. J. Phys. Chem. C 2013, 117, 25269–25275. [Google Scholar] [CrossRef]
- Pasti, I.A.; Gavrilov, N.M.; Mentus, S.V. Potentiodynamic investigation of oxygen reduction reaction on polycrystalline platinum surface in acidic solutions: The effect of the polarization rate on the kinetic parameters. Int. J. Electrochem. Sci. 2012, 7, 11076–11090. [Google Scholar]
- Lopes, P.P.; Strmcnik, D.; Jirkovsky, J.S.; Connell, J.G.; Stamenkovic, V.; Markovic, N. Double layer effects in electrocatalysis: The oxygen reduction reaction and ethanol oxidation reaction on Au(111), Pt(111) and Ir(111) in alkaline media containing Na and Li cations. Catal. Today 2016, 262, 41–47. [Google Scholar] [CrossRef]
- Wang, J.X.; Markovic, N.M.; Adzic, R.R. Kinetic analysis of oxygen reduction on Pt(111) in acid solutions: Intrinsic kinetic parameters and anion adsorption effects. J. Phys. Chem. B 2004, 108, 4127–4133. [Google Scholar] [CrossRef]
- Markovic, N.; Gasteiger, H.; Ross, P.N. Kinetics of oxygen reduction on Pt(hkl) electrodes: Implications for the crystallite size effect with supported Pt electrocatalysts. J. Electrochem. Soc. 1997, 144, 1591–1597. [Google Scholar] [CrossRef]
- Paulus, U.A.; Schmidt, T.J.; Gasteiger, H.A.; Behm, R.J. Oxygen reduction on a high-surface area Pt/vulcan carbon catalyst: A thin-film rotating ring-disk electrode study. J. Electroanal. Chem. 2001, 495, 134–145. [Google Scholar] [CrossRef]
- Strmcnik, D.; Escudero-Escribano, M.; Kodama, K.; StamenkovicVojislav, R.; Cuesta, A.; Marković, N.M. Enhanced electrocatalysis of the oxygen reduction reaction based on patterning of platinum surfaces with cyanide. Nat. Chem. 2010, 2, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Antolini, E.; Gonzalez, E.R. Alkaline direct alcohol fuel cells. J. Power Sources 2010, 195, 3431–3450. [Google Scholar] [CrossRef]
- Blizanac, B.B.; Ross, P.N.; Markovic, N.M. Oxygen electroreduction on Ag(111): The Ph effect. Electrochim. Acta 2007, 52, 2264–2271. [Google Scholar] [CrossRef]
- Cifrain, M.; Kordesch, K.V. Advances, aging mechanism and lifetime in AFCs with circulating electrolytes. J. Power Sources 2004, 127, 234–242. [Google Scholar] [CrossRef]
- Koscher, G.A.; Kordesch, K. Alkaline methanol–air system. J. Sol. State Electrochem. 2003, 7, 632–636. [Google Scholar] [CrossRef]
- Lin, B.Y.S.; Kirk, D.W.; Thorpe, S.J. Performance of alkaline fuel cells: A possible future energy system? J. Power Sources 2006, 161, 474–483. [Google Scholar] [CrossRef]
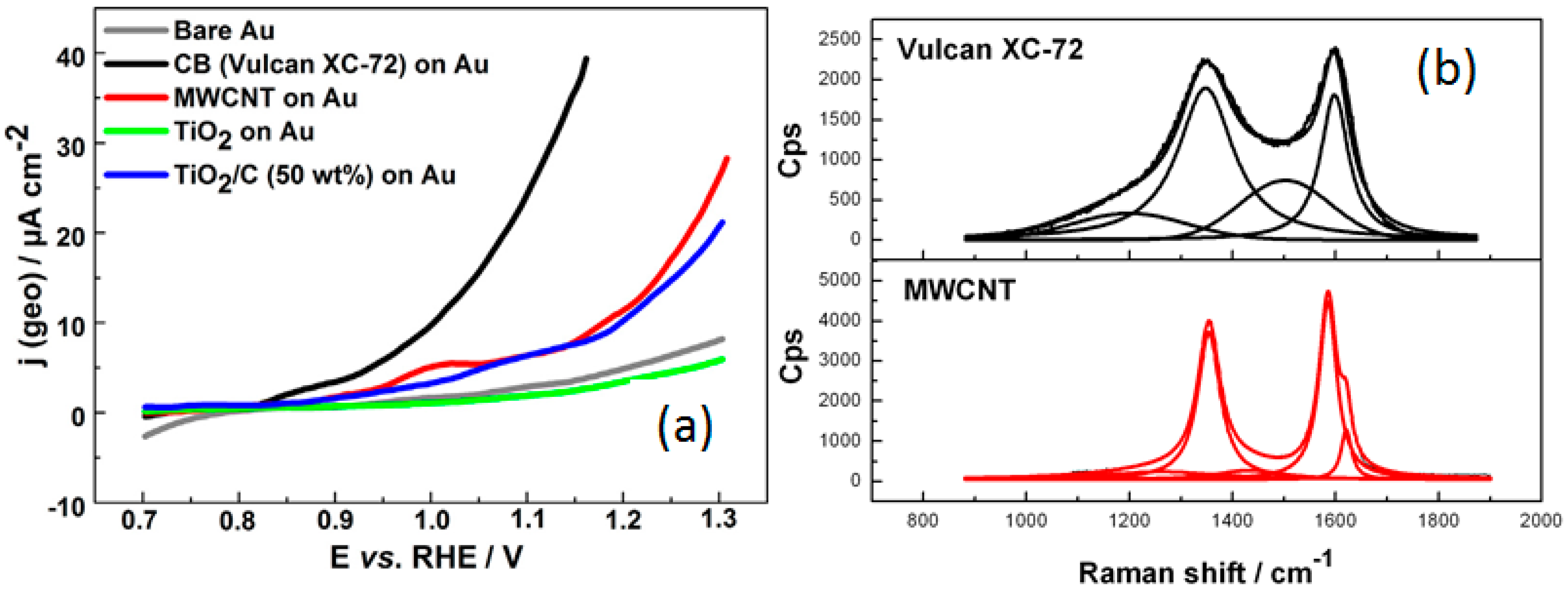
- Lewera, A.; Timperman, L.; Roguska, A.; Alonso-Vante, N. Metal–support interactions between nanosized Pt and metal oxides (WO3 and TiO2) studied using X-ray photoelectron spectroscopy. J. Phys. Chem. C 2011, 115, 20153–20159. [Google Scholar] [CrossRef]
- Luo, Y.; Alonso-Vante, N. The effect of support on advanced Pt-based cathodes towards the oxygen reduction reaction. State of the art. Electrochim. Acta 2015, 179, 108–118. [Google Scholar] [CrossRef]
- Yang, W.; Wang, X.; Yang, F.; Yang, C.; Yang, X. Carbon nanotubes decorated with Pt nanocubes by a noncovalent functionalization method and their role in oxygen reduction. Adv. Mater. 2008, 20, 2579–2587. [Google Scholar] [CrossRef]
- Orfanidi, A.; Daletou, M.K.; Neophytides, S.G. Preparation and characterization of Pt on modified multi-wall carbon nanotubes to be used as electrocatalysts for high temperature fuel cell applications. Appl. Catal. B: Environ. 2011, 106, 379–389. [Google Scholar] [CrossRef]
- Li, W.; Liang, C.; Zhou, W.; Qiu, J.; Zhou, Z.; Sun, G.; Xin, Q. Preparation and characterization of multiwalled carbon nanotube-supported platinum for cathode catalysts of direct methanol fuel cells. J. Phys. Chem. B 2003, 107, 6292–6299. [Google Scholar] [CrossRef]
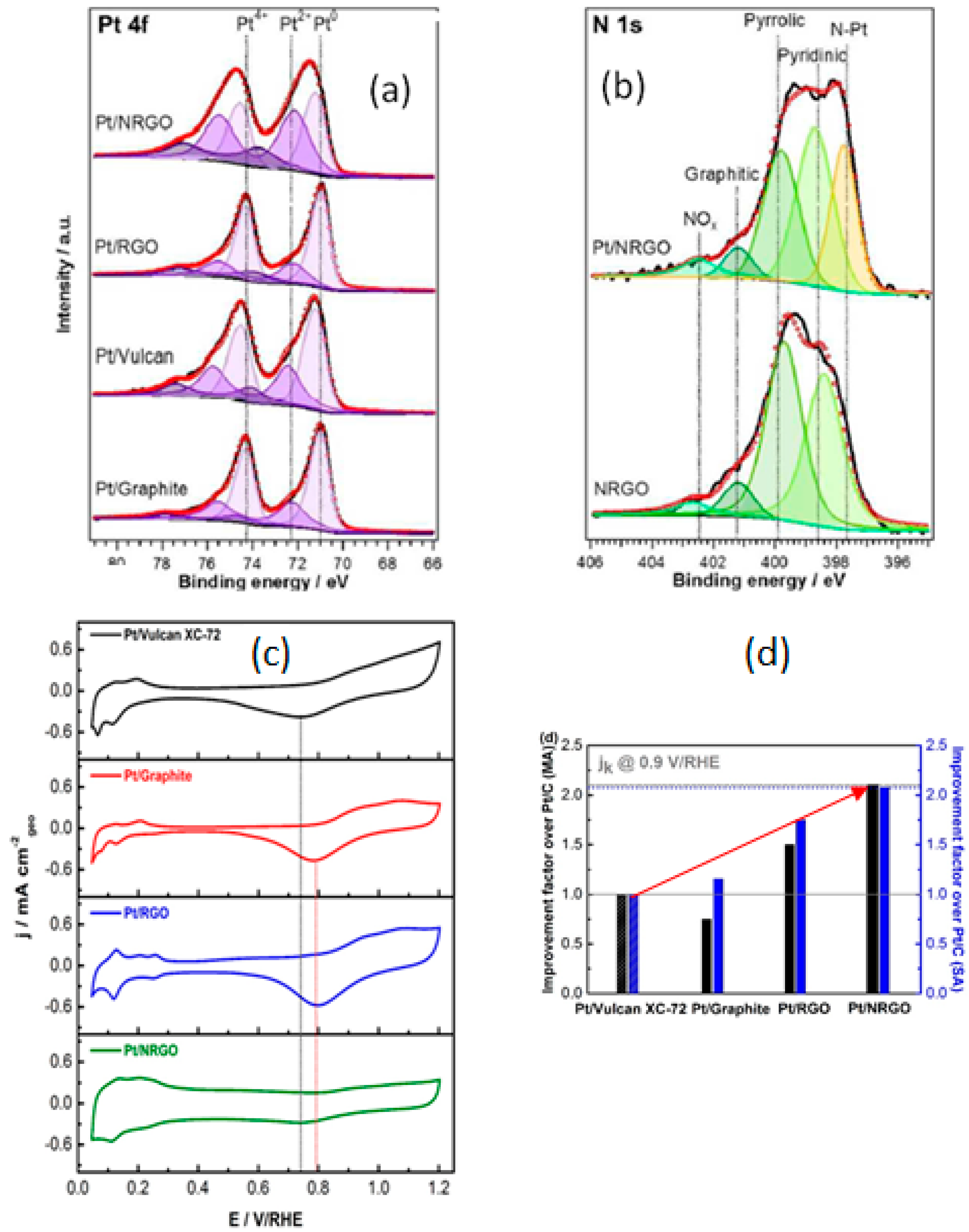
- Chen, Y.; Wang, J.; Liu, H.; Banis, M.N.; Li, R.; Sun, X.; Sham, T.-K.; Ye, S.; Knights, S. Nitrogen doping effects on carbon nanotubes and the origin of the enhanced electrocatalytic activity of supported Pt for proton-exchange membrane fuel cells. J. Phys. Chem. C 2011, 115, 3769–3776. [Google Scholar] [CrossRef]
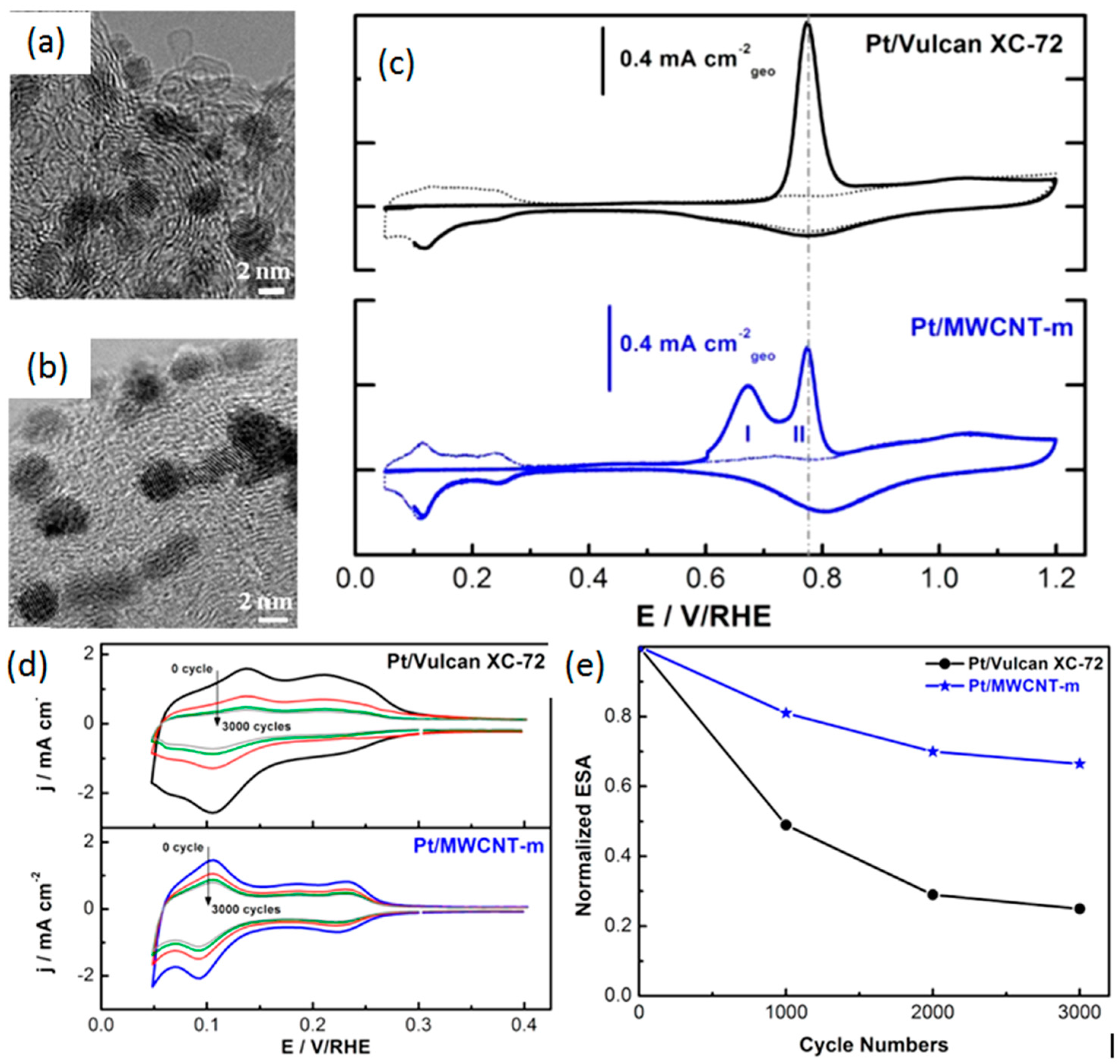
- Wang, S.; Jiang, S.P.; White, T.J.; Guo, J.; Wang, X. Electrocatalytic activity and interconnectivity of Pt nanoparticles on multiwalled carbon nanotubes for fuel cells. J. Phys. Chem. C 2009, 113, 18935–18945. [Google Scholar] [CrossRef]
- Hijazi, I.; Bourgeteau, T.; Cornut, R.; Morozan, A.; Filoramo, A.; Leroy, J.; Derycke, V.; Jousselme, B.; Campidelli, S. Carbon nanotube-templated synthesis of covalent porphyrin network for oxygen reduction reaction. J. Am. Chem. Soc. 2014, 136, 6348–6354. [Google Scholar] [CrossRef] [PubMed]
- Hasche, F.; Oezaslan, M.; Strasser, P. Activity, stability and degradation of multi walled carbon nanotube (MXCNT) supported Pt fuel cell electrocatalysts. PCCP 2010, 12, 15251–15258. [Google Scholar] [CrossRef] [PubMed]
- Kongkanand, A.; Vinodgopal, K.; Kuwabata, S.; Kamat, P.V. Highly dispersed Pt catalysts on single-walled carbon nanotubes and their role in methanol oxidation. J. Phys. Chem. B 2006, 110, 16185–16188. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Yin, G.P.; Zhang, J.; Wang, Z.B.; Gao, Y.Z. High utilization platinum deposition on single-walled carbon nanotubes as catalysts for direct methanol fuel cell. Electrochim. Acta 2007, 52, 7042–7050. [Google Scholar] [CrossRef]
- Su, L.; Jia, W.; Zhang, L.; Beacham, C.; Zhang, H.; Lei, Y. Facile synthesis of a platinum nanoflower monolayer on a single-walled carbon nanotube membrane and its application in glucose detection. J. Phys. Chem. C 2010, 114, 18121–18125. [Google Scholar] [CrossRef]
- Seo, M.H.; Choi, S.M.; Seo, J.K.; Noh, S.H.; Kim, W.B.; Han, B. The graphene-supported palladium and palladium–yttrium nanoparticles for the oxygen reduction and ethanol oxidation reactions: Experimental measurement and computational validation. Appl. Catal. B: Environ. 2013, 129, 163–171. [Google Scholar] [CrossRef]
- Rao, C.V.; Reddy, A.L.M.; Ishikawa, Y.; Ajayan, P.M. Synthesis and electrocatalytic oxygen reduction activity of graphene-supported Pt3Co and Pt3Cr alloy nanoparticles. Carbon 2011, 49, 931–936. [Google Scholar] [CrossRef]
- Kou, R.; Shao, Y.; Wang, D.; Engelhard, M.H.; Kwak, J.H.; Wang, J.; Viswanathan, V.V.; Wang, C.; Lin, Y.; Wang, Y.; et al. Enhanced activity and stability of Pt catalysts on functionalized graphene sheets for electrocatalytic oxygen reduction. Electrochem. Commun. 2009, 11, 954–957. [Google Scholar] [CrossRef]
- Li, Y.; Tang, L.; Li, J. Preparation and electrochemical performance for methanol oxidation of Pt/graphene nanocomposites. Electrochem. Commun. 2009, 11, 846–849. [Google Scholar] [CrossRef]
- Moldovan, M.S.; Bulou, H.; Dappe, Y.J.; Janowska, I.; Bégin, D.; Pham-Huu, C.; Ersen, O. On the evolution of Pt nanoparticles on few-layer graphene supports in the high-temperature range. J. Phys. Chem. C 2012, 116, 9274–9282. [Google Scholar] [CrossRef]
- Shao, Y.; Zhang, S.; Wang, C.; Nie, Z.; Liu, J.; Wang, Y.; Lin, Y. Highly durable graphene nanoplatelets supported Pt nanocatalysts for oxygen reduction. J. Power Sources 2010, 195, 4600–4605. [Google Scholar] [CrossRef]
- Jahan, M.; Bao, Q.; Loh, K.P. Electrocatalytically active graphene–porphyrin MOF composite for oxygen reduction reaction. J. Am. Chem. Soc. 2012, 134, 6707–6713. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Guo, S.; Zuo, J.-L.; Sun, S. Synthesis and assembly of Pd nanoparticles on graphene for enhanced electrooxidation of formic acid. Nanoscale 2013, 5, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Di Noto, V.; Negro, E.; Lavina, S.; Gross, S.; Pace, G. Pd-Co carbon-nitride electrocatalysts for polymer electrolyte fuel cells. Electrochim. Acta 2007, 53, 1604–1617. [Google Scholar] [CrossRef]
- Di Noto, V.; Negro, E.; Polizzi, S.; Riello, P.; Atanassov, P. Preparation, characterization and single-cell performance of a new class of Pd-carbon nitride electrocatalysts for oxygen reduction reaction in PEMFCs. Appl. Catal. B: Environ. 2012, 111–112, 185–199. [Google Scholar] [CrossRef]
- Di Noto, V.; Negro, E.; Polizzi, S.; Vezzù, K.; Toniolo, L.; Cavinato, G. Synthesis, studies and fuel cell performance of “core–shell” electrocatalysts for oxygen reduction reaction based on a PtNix carbon nitride “shell” and a pyrolyzed polyketone nanoball “core”. Int. J. Hydrogen Energy 2014, 39, 2812–2827. [Google Scholar] [CrossRef]
- Di Noto, V.; Negro, E.; Polizzi, S.; Agresti, F.; Giffin, G.A. Synthesis–structure–morphology interplay of bimetallic “core–shell” carbon nitride nano-electrocatalysts. ChemSusChem 2012, 5, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Datye, A.K.; Kalakkad, D.S.; Yao, M.H.; Smith, D.J. Comparison of metal-support interactions in Pt/TiO2 and Pt/CeO2. J. Catal. 1995, 155, 148–153. [Google Scholar] [CrossRef]
- Koudelka, M.; Monnier, A.; Sanchez, J.; Augustynski, J. Correlation between the surface composition of Pt/TiO2 catalysts and their adsorption behaviour in aqueous solutions. J. Mol. Catal. 1984, 25, 295–305. [Google Scholar] [CrossRef]
- Jiang, D.-e.; Overbury, S.H.; Dai, S. Structures and energetics of Pt clusters on TiO2: Interplay between metal–metal bonds and metal–oxygen bonds. J. Phys. Chem. C 2012, 116, 21880–21885. [Google Scholar] [CrossRef]
- Bauer, A.; Lee, K.; Song, C.; Xie, Y.; Zhang, J.; Hui, R. Pt nanoparticles deposited on TiO2 based nanofibers: Electrochemical stability and oxygen reduction activity. J. Power Sources 2010, 195, 3105–3110. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Ganesan, P.; Popov, B.N. Electrocatalytic activity and stability of niobium-doped titanium oxide supported platinum catalyst for polymer electrolyte membrane fuel cells. Appl. Catal. B: Environ. 2010, 96, 224–231. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Ganesan, P.; Popov, B.N. Titania supported platinum catalyst with high electrocatalytic activity and stability for polymer electrolyte membrane fuel cell. Appl. Catal. B: Environ. 2011, 102, 71–77. [Google Scholar] [CrossRef]
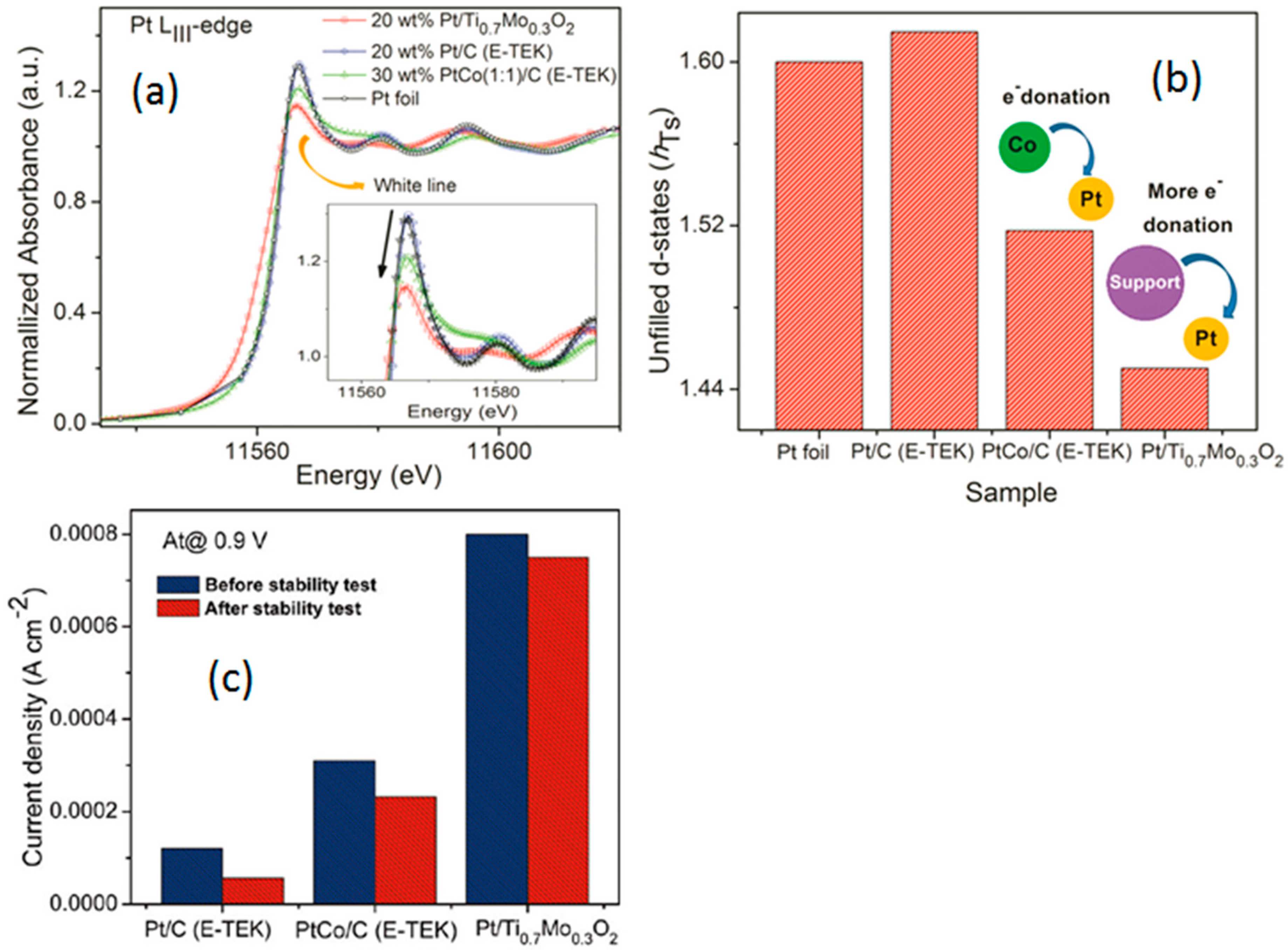
- Ho, V.T.T.; Pan, C.-J.; Rick, J.; Su, W.-N.; Hwang, B.-J. Nanostructured Ti0.7Mo0.3O2 support enhances electron transfer to Pt: High-performance catalyst for oxygen reduction reaction. J. Am. Chem. Soc. 2011, 133, 11716–11724. [Google Scholar] [CrossRef] [PubMed]
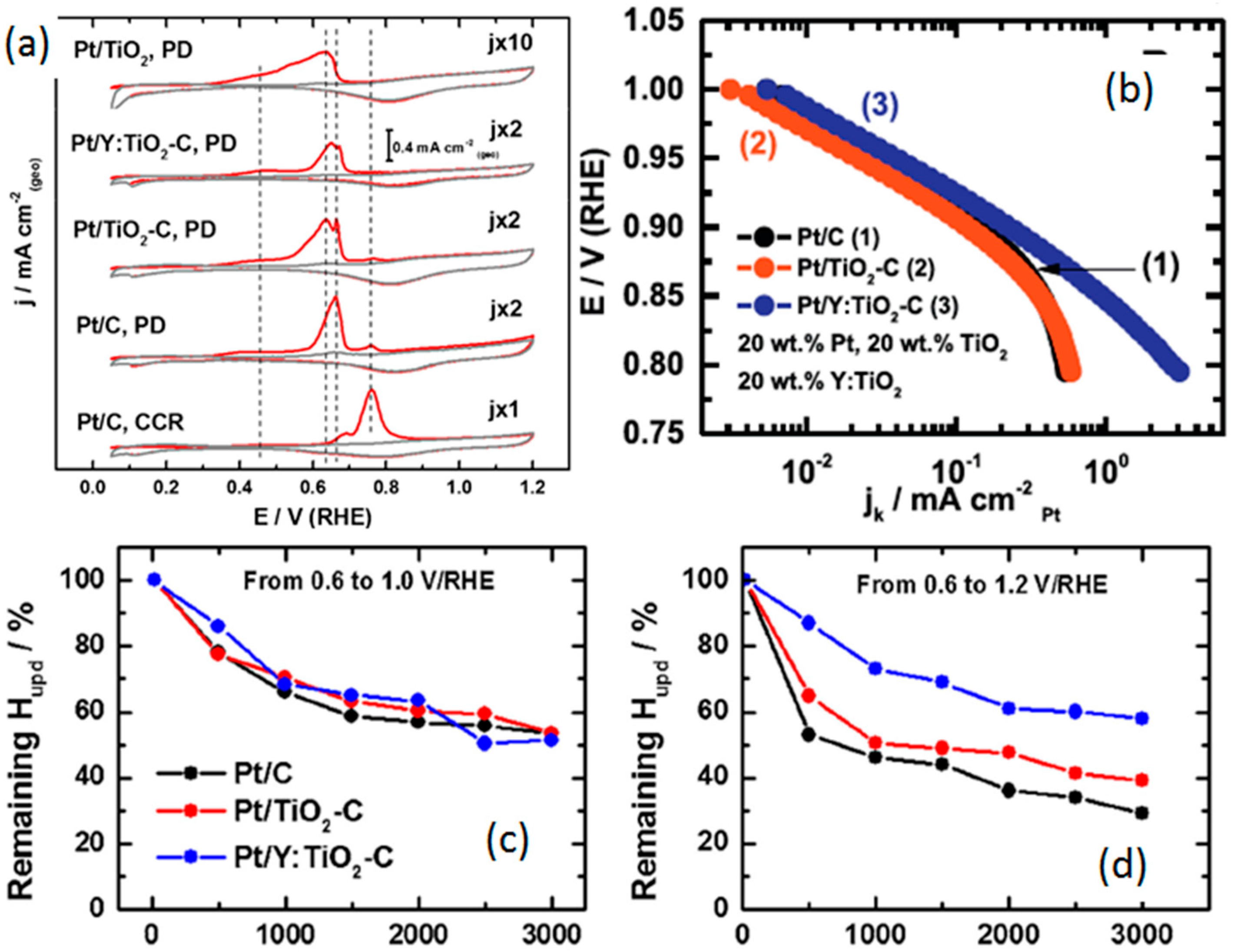
- Estudillo-Wong, L.A.; Luo, Y.; Diaz-Real, J.A.; Alonso-Vante, N. Enhanced oxygen reduction reaction stability on platinum nanoparticles photo-deposited onto oxide-carbon composites. Appl. Catal. B: Environ. 2015, 187, 291–300. [Google Scholar] [CrossRef]
- Luo, Y.; Calvillo, L.; Daiguebonne, C.; Daletou, M.K.; Granozzi, G.; Alonso-Vante, N. A highly efficient and stable oxygen reduction reaction on Pt/CeOx/C electrocatalyst obtained via a sacrificial precursor based on a metal-organic framework. Appl. Catal. B: Environ. 2016, 189, 39–50. [Google Scholar] [CrossRef]
- Ruiz Camacho, B.; Morais, C.; Valenzuela, M.A.; Alonso-Vante, N. Enhancing oxygen reduction reaction activity and stability of platinum via oxide-carbon composites. Catal. Today 2013, 202, 36–43. [Google Scholar] [CrossRef]
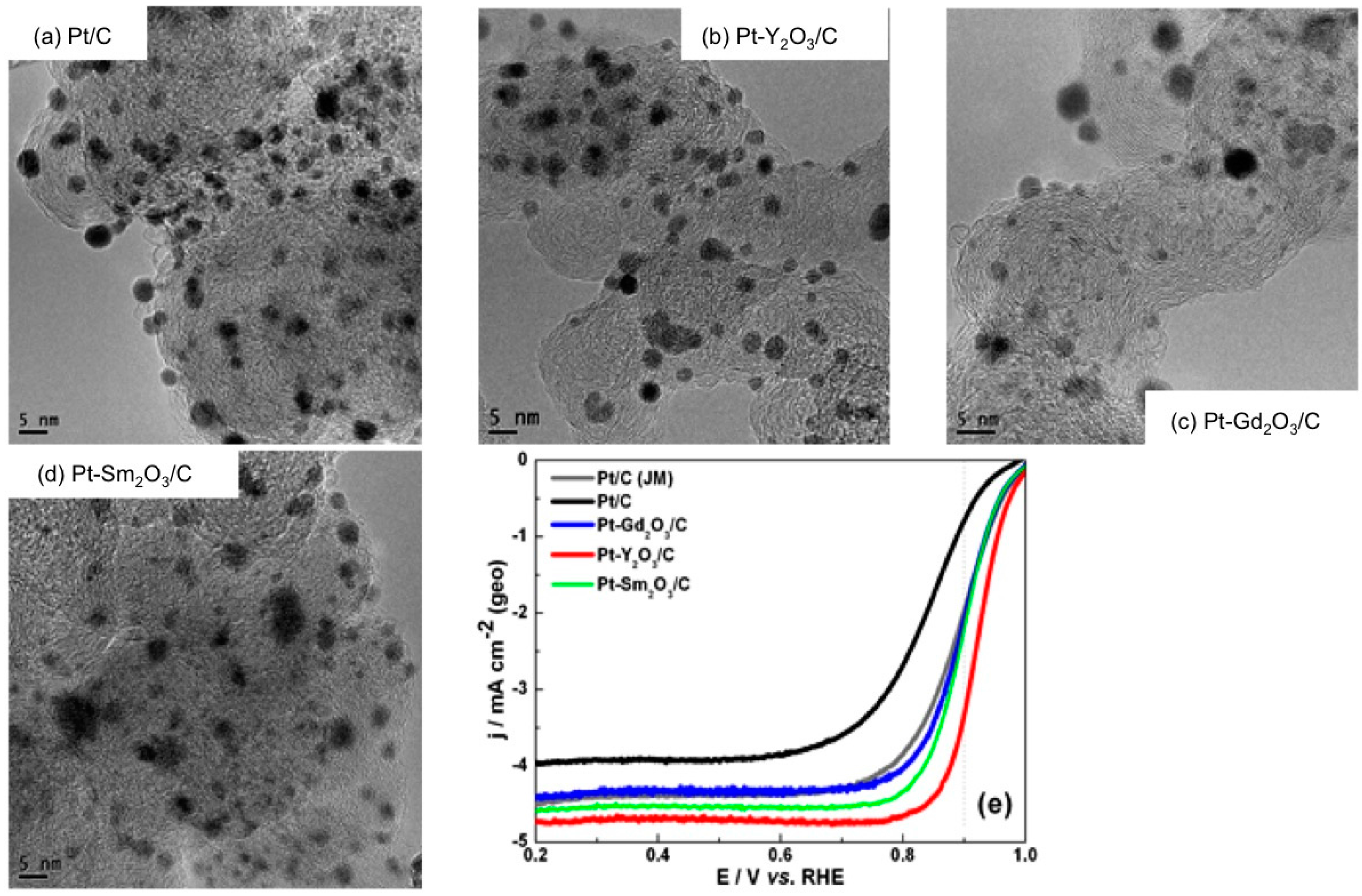
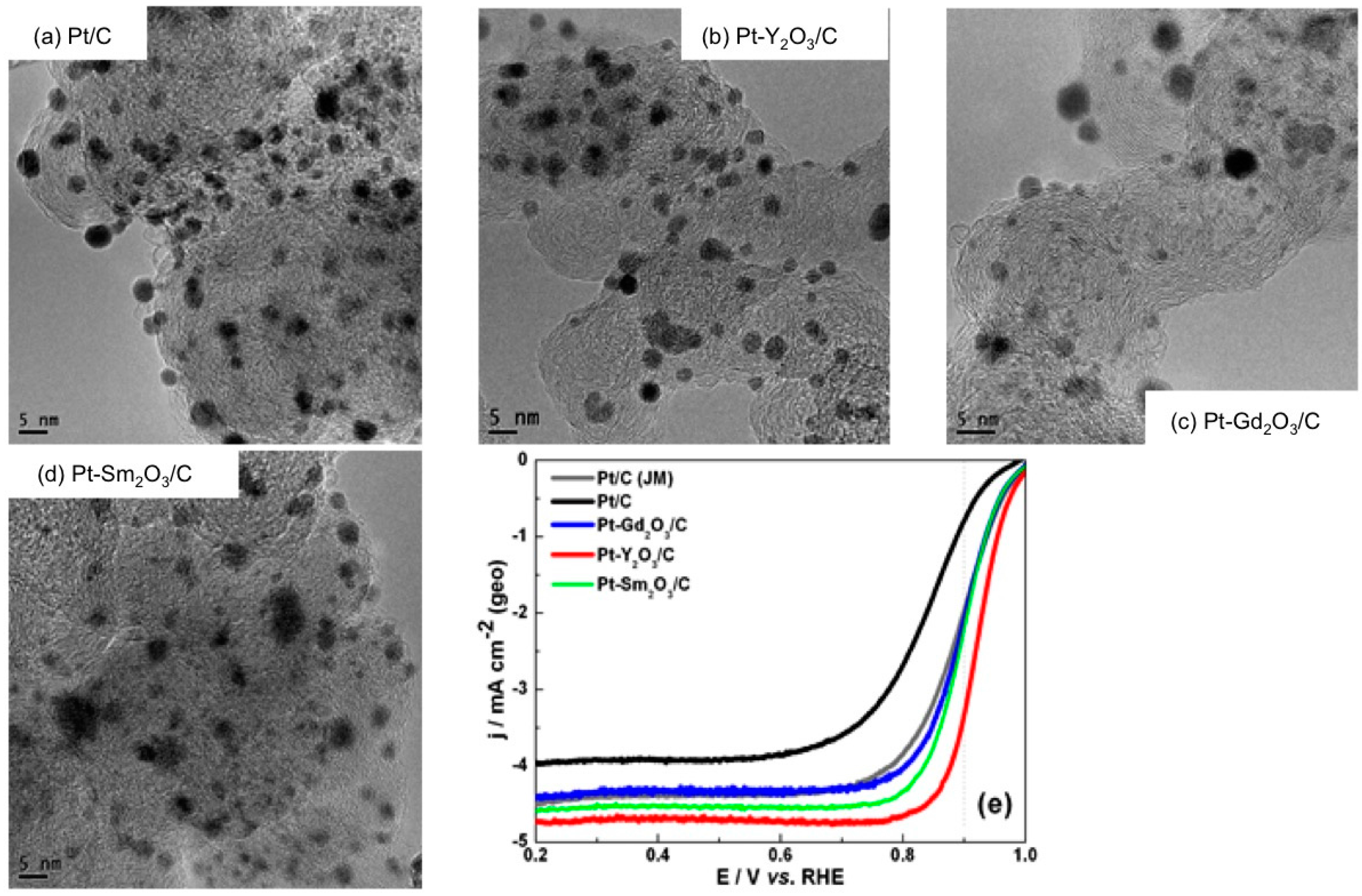
- Luo, Y.; Habrioux, A.; Calvillo, L.; Granozzi, G.; Alonso-Vante, N. Thermally induced strains on the catalytic activity and stability of Pt–M2O3/C (M = Y or Gd) catalysts towards oxygen reduction reaction. ChemCatChem 2015, 7, 1573–1582. [Google Scholar] [CrossRef]
- Luo, Y.; Habrioux, A.; Calvillo, L.; Granozzi, G.; Alonso-Vante, N. Yttrium oxide/gadolinium oxide-modified platinum nanoparticles as cathodes for the oxygen reduction reaction. ChemPhysChem 2014, 15, 2136–2144. [Google Scholar] [CrossRef] [PubMed]
- Tauster, S.J. Strong metal-support interactions. Acc. Chem. Res. 1987, 20, 389–394. [Google Scholar] [CrossRef]
- Tauster, S.J.; Fung, S.C.; Garten, R.L. Strong metal-support interactions. Group 8 noble metals supported on titanium dioxide. J. Am. Chem. Soc. 1978, 100, 170–175. [Google Scholar] [CrossRef]
- Ma, J.; Habrioux, A.; Morais, C.; Lewera, A.; Vogel, W.; Verde-Gómez, Y.; Ramos-Sanchez, G.; Balbuena, P.B.; Alonso-Vante, N. Spectroelectrochemical probing of the strong interaction between platinum nanoparticles and graphitic domains of carbon. ACS Catal. 2013, 3, 1940–1950. [Google Scholar] [CrossRef]
- Ma, J.; Habrioux, A.; Pisarek, M.; Lewera, A.; Alonso-Vante, N. Induced electronic modification of Pt nanoparticles deposited onto graphitic domains of carbon materials by uv irradiation. Electrochem. Commun. 2013, 29, 12–16. [Google Scholar] [CrossRef]
- Fleisch, T.H.; Hicks, R.F.; Bell, A.T. An XPS study of metal-support interactions on PdSiO2 and PdLa2O3. J. Catal. 1984, 87, 398–413. [Google Scholar] [CrossRef]
- Dall’Agnol, C.; Gervasini, A.; Morazzoni, F.; Pinna, F.; Strukul, G.; Zanderighi, L. Hydrogenation of carbon monoxide: Evidence of a strong metal-support interaction in RhZrO2 catalysts. J. Catal. 1985, 96, 106–114. [Google Scholar] [CrossRef]
- Wu, G.; Zelenay, P. Nanostructured nonprecious metal catalysts for oxygen reduction reaction. Acc. Chem. Res. 2013, 46, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Mack, N.H.; Gao, W.; Ma, S.; Zhong, R.; Han, J.; Baldwin, J.K.; Zelenay, P. Nitrogen-doped graphene-rich catalysts derived from heteroatom polymers for oxygen reduction in nonaqueous lithium–O2 battery cathodes. ACS Nano 2012, 6, 9764–9776. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; More, K.L.; Johnston, C.M.; Zelenay, P. High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron, and cobalt. Science 2011, 332, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Wang, M.; Cai, W.; Cui, Y.; Gao, F.; Peng, L.; Chen, W.; Ding, W. Acid-resistant catalysis without use of noble metals: Carbon nitride with underlying nickel. ACS Catal. 2014, 4, 2536–2543. [Google Scholar] [CrossRef]
- Jiang, T.; Zhang, Q.; Wang, T.-J.; Zhang, Q.; Ma, L.-L. High yield of pentane production by aqueous-phase reforming of xylitol over Ni/HZSM-5 and Ni/MCM22 catalysts. Energy Convers. Manag. 2012, 59, 58–65. [Google Scholar] [CrossRef]
- Luo, Y.; Estudillo-Wong, L.A.; Alonso-Vante, N. Carbon supported Pt-Y2O3 and Pt-Gd2O3 nanoparticles prepared via carbonyl chemical route towards oxygen reduction reaction: Kinetics and stability. Int. J. Hydrogen Energy 2016, in press. [Google Scholar] [CrossRef]
- Vogel, W.; Timperman, L.; Alonso-Vante, N. Probing metal substrate interaction of Pt nanoparticles: Structural XRD analysis and oxygen reduction reaction. Appl. Catal. A 2010, 377, 167–173. [Google Scholar] [CrossRef]
- Ma, J.; Habrioux, A.; Gago, A.; Alonso-Vante, N. Towards understanding the essential role played by the platinum-support interaction on electrocatalytic activity. ECS Trans. 2013, 45, 25–33. [Google Scholar] [CrossRef]
- Ma, J.; Habrioux, A.; Guignard, N.; Alonso-Vante, N. Functionalizing effect of increasingly graphitic carbon supports on carbon-supported and TiO2–carbon composite-supported Pt nanoparticles. J. Phys. Chem. C 2012, 116, 21788–21794. [Google Scholar] [CrossRef]
- Matter, P.H.; Zhang, L.; Ozkan, U.S. The role of nanostructure in nitrogen-containing carbon catalysts for the oxygen reduction reaction. J. Catal. 2006, 239, 83–96. [Google Scholar] [CrossRef]
- Wang, D.; Xin, H.L.; Hovden, R.; Wang, H.; Yu, Y.; Muller, D.A.; DiSalvo, F.J.; Abruña, H.D. Structurally ordered intermetallic platinum–cobalt core–shell nanoparticles with enhanced activity and stability as oxygen reduction electrocatalysts. Nat. Mater. 2013, 12, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xin, H.L.; Wang, H.; Yu, Y.; Rus, E.; Muller, D.A.; DiSalvo, F.J.; Abruña, H.D. Facile synthesis of carbon-supported Pd–Co core–shell nanoparticles as oxygen reduction electrocatalysts and their enhanced activity and stability with monolayer pt decoration. Chem. Mater. 2012, 24, 2274–2281. [Google Scholar] [CrossRef]
- Wang, D.; Xin, H.L.; Yu, Y.; Wang, H.; Rus, E.; Muller, D.A.; Abruña, H.D. Pt-decorated PdCo@Pd/C core–shell nanoparticles with enhanced stability and electrocatalytic activity for the oxygen reduction reaction. J. Am. Chem. Soc. 2010, 132, 17664–17666. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Park, G.S.; Lee, H.I.; Kim, S.T.; Cao, R.; Liu, M.; Cho, J. Ketjenblack carbon supported amorphous manganese oxides nanowires as highly efficient electrocatalyst for oxygen reduction reaction in alkaline solutions. Nano Lett. 2011, 11, 5362–5366. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Gan, L.; Heggen, M.; Rudi, S.; Strasser, P. Compositional segregation in shaped Pt alloy nanoparticles and their structural behaviour during electrocatalysis. Nat. Mater. 2013, 12, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Chhina, H.; Campbell, S.; Kesler, O. An oxidation-resistant indium tin oxide catalyst support for proton exchange membrane fuel cells. J. Power Sources 2006, 161, 893–900. [Google Scholar] [CrossRef]
- Millington, B.; Whipple, V.; Pollet, B.G. A novel method for preparing proton exchange membrane fuel cell electrodes by the ultrasonic-spray technique. J. Power Sources 2011, 196, 8500–8508. [Google Scholar] [CrossRef]
- Malacrida, P.; Escudero-Escribano, M.; Verdaguer-Casadevall, A.; Stephens, I.E.L.; Chorkendorff, I. Enhanced activity and stability of Pt-La and Pt-Ce alloys for oxygen electroreduction: The elucidation of the active surface phase. J. Mater. Chem. A 2014, 2, 4234–4243. [Google Scholar] [CrossRef]
- Escudero-Escribano, M.; Verdaguer-Casadevall, A.; Malacrida, P.; Grønbjerg, U.; Knudsen, B.P.; Jepsen, A.K.; Rossmeisl, J.; Stephens, I.E.L.; Chorkendorff, I. Pt5Gd as a highly active and stable catalyst for oxygen electroreduction. J. Am. Chem. Soc. 2012, 134, 16476–16479. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Fernandez, P.; Masini, F.; McCarthy, D.N.; Strebel, C.E.; Friebel, D.; Deiana, D.; Malacrida, P.; Nierhoff, A.; Bodin, A.; Wise, A.M.; et al. Mass-selected nanoparticles of PtxY as model catalysts for oxygen electroreduction. Nat. Chem. 2014, 6, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.K.; McGinn, P.J. Carbon supported Pt–Y electrocatalysts for the oxygen reduction reaction. J. Power Sources 2011, 196, 1127–1131. [Google Scholar] [CrossRef]
- Nishanth, K.G.; Sridhar, P.; Pitchumani, S. Enhanced oxygen reduction reaction activity through spillover effect by Pt–Y(OH)3/C catalyst in direct methanol fuel cells. Electrochem. Commun. 2011, 13, 1465–1468. [Google Scholar] [CrossRef]
- Xu, J.; Gao, P.; Zhao, T.S. Non-precious Co3O4 nano-rod electrocatalyst for oxygen reduction reaction in anion-exchange membrane fuel cells. Energy Environ. Sci. 2012, 5, 5333–5339. [Google Scholar] [CrossRef]
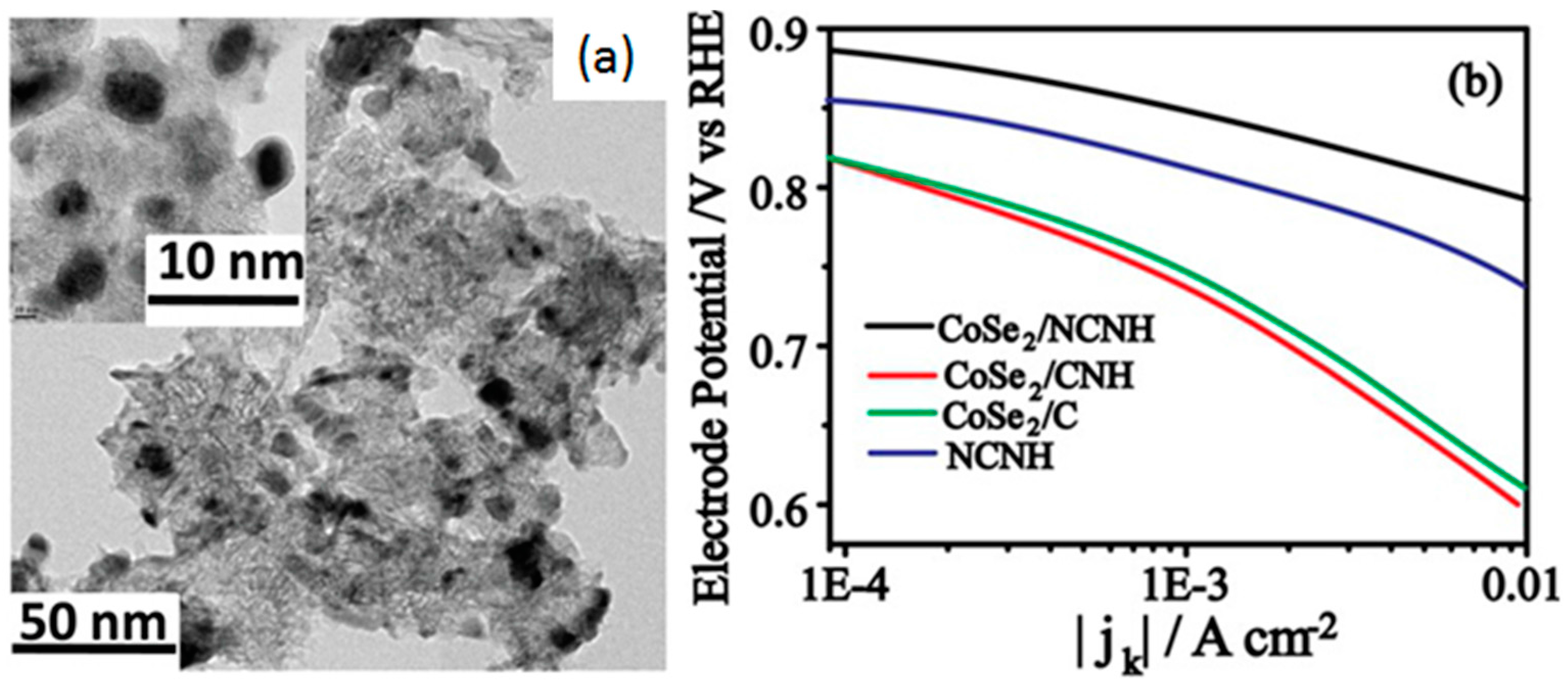
- Unni, S.M.; Mora-Hernandez, J.M.; Kurungot, S.; Alonso-Vante, N. CoSe2 supported on nitrogen-doped carbon nanohorns as a methanol-tolerant cathode for air-breathing microlaminar flow fuel cells. ChemElectroChem 2015, 2, 1339–1345. [Google Scholar] [CrossRef]
- Zhu, S.; Xu, G. Single-walled carbon nanohorns and their applications. Nanoscale 2010, 2, 2538–2549. [Google Scholar] [CrossRef] [PubMed]
- Kasuya, D.; Yudasaka, M.; Takahashi, K.; Kokai, F.; Iijima, S. Selective production of single-wall carbon nanohorn aggregates and their formation mechanism. J. Phys. Chem. B 2002, 106, 4947–4951. [Google Scholar] [CrossRef]
- Guo, D.-J.; Jing, Z.-H. A novel co-precipitation method for preparation of Pt-CeO2 composites on multi-walled carbon nanotubes for direct methanol fuel cells. J. Power Sources 2010, 195, 3802–3805. [Google Scholar] [CrossRef]
- Wang, X.; Li, W.; Chen, Z.; Waje, M.; Yan, Y. Durability investigation of carbon nanotube as catalyst support for proton exchange membrane fuel cell. J. Power Sources 2006, 158, 154–159. [Google Scholar] [CrossRef]
- Wu, G.; More, K.L.; Xu, P.; Wang, H.-L.; Ferrandon, M.; Kropf, A.J.; Myers, D.J.; Ma, S.; Johnston, C.M.; Zelenay, P. A carbon-nanotube-supported graphene-rich non-precious metal oxygen reduction catalyst with enhanced performance durability. Chem. Commun. 2013, 49, 3291–3293. [Google Scholar] [CrossRef] [PubMed]
- Jaouen, F.; Proietti, E.; Lefevre, M.; Chenitz, R.; Dodelet, J.-P.; Wu, G.; Chung, H.T.; Johnston, C.M.; Zelenay, P. Recent advances in non-precious metal catalysis for oxygen-reduction reaction in polymer electrolyte fuel cells. Energy Environ. Sci. 2011, 4, 114–130. [Google Scholar] [CrossRef]
- Li, Q.; Xu, P.; Gao, W.; Ma, S.; Zhang, G.; Cao, R.; Cho, J.; Wang, H.-L.; Wu, G. Graphene/graphene-tube nanocomposites templated from cage-containing metal-organic frameworks for oxygen reduction in Li–O2 batteries. Adv. Mater. 2014, 26, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Estudillo-Wong, L.A.; Vargas-Gómez, A.M.; Arce-Estrada, E.M.; Manzo-Robledo, A. TiO2/C composite as a support for Pd-nanoparticles toward the electrocatalytic oxidation of methanol in alkaline media. Electrochim. Acta 2013, 112, 164–170. [Google Scholar] [CrossRef]
- Xiong, L.; Manthiram, A. Synthesis and characterization of methanol tolerant Pt/TiOx/C nanocomposites for oxygen reduction in direct methanol fuel cells. Electrochim. Acta 2004, 49, 4163–4170. [Google Scholar] [CrossRef]
- Andrew Lin, K.-Y.; Hsu, F.-K.; Lee, W.-D. Magnetic cobalt-graphene nanocomposite derived from self-assembly of MOFs with graphene oxide as an activator for peroxymonosulfate. J. Mater. Chem. A 2015, 3, 9480–9490. [Google Scholar] [CrossRef]
- Guo, D.-J.; Cui, S.-K.; Sun, H. Preparation of Pt–CeO2/MWNT nano-composites by reverse micellar method for methanol oxidation. J. Nanoparticle Res. 2009, 11, 707–712. [Google Scholar] [CrossRef]
- Von Kraemer, S.; Wikander, K.; Lindbergh, G.; Lundblad, A.; Palmqvist, A.E.C. Evaluation of TiO2 as catalyst support in Pt-TiO2/C composite cathodes for the proton exchange membrane fuel cell. J. Power Sources 2008, 180, 185–190. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, X.; Liu, Z.; Zhang, H.; Yu, P.; Mao, L. Pt–Ru/CeO2/carbon nanotube nanocomposites: An efficient electrocatalyst for direct methanol fuel cells. Langmuir 2010, 26, 12383–12389. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Habrioux, A.; Luo, Y.; Ramos-Sanchez, G.; Calvillo, L.; Granozzi, G.; Balbuena, P.B.; Alonso-Vante, N. Electronic interaction between platinum nanoparticles and nitrogen-doped reduced graphene oxide: Effect on the oxygen reduction reaction. J. Mater. Chem. A 2015, 3, 11891–11904. [Google Scholar] [CrossRef]
- Sreekuttan, M.U.; Campos-Roldan, C.A.; Mora-Hernandez, J.M.; Luo, Y.; Estudillo-Wong, L.A.; Alonso-Vante, N. The effect of carbon-based substrates onto non-precious and precious electrocatalytic centers. ECS Trans. 2015, 69, 35–42. [Google Scholar] [CrossRef]
- Lee, C.-L.; Huang, C.-H.; Huang, K.-L.; Tsai, Y.-L.; Yang, C.-C. A comparison of three carbon nanoforms as catalyst supports for the oxygen reduction reaction. Carbon 2013, 60, 392–400. [Google Scholar] [CrossRef]
- Liu, Z.-T.; Huang, K.-L.; Wu, Y.-S.; Lyu, Y.-P.; Lee, C.-L. A comparison of physically and chemically defective graphene nanosheets as catalyst supports for cubic Pd nanoparticles in an alkaline oxygen reduction reaction. Electrochim. Acta 2015, 186, 552–561. [Google Scholar] [CrossRef]
- Thanh Ho, V.T.; Pillai, K.C.; Chou, H.-L.; Pan, C.-J.; Rick, J.; Su, W.-N.; Hwang, B.-J.; Lee, J.-F.; Sheu, H.-S.; Chuang, W.-T. Robust non-carbon Ti0.7Ru0.3O2 support with co-catalytic functionality for Pt: Enhances catalytic activity and durability for fuel cells. Energy Environ. Sci. 2011, 4, 4194–4200. [Google Scholar] [CrossRef]
- Timperman, L.; Alonso-Vante, N. Oxide substrate effect toward electrocatalytic enhancement of platinum and ruthenium–selenium catalysts. Electrocatalysis 2011, 2, 181–191. [Google Scholar] [CrossRef]
- Scotti, G.; Kanninen, P.; Matilainen, V.-P.; Salminen, A.; Kallio, T. Stainless steel micro fuel cells with enclosed channels by laser additive manufacturing. Energy 2016, 106, 475–481. [Google Scholar] [CrossRef]
- Wang, R.; Xu, Z. Recycling of non-metallic fractions from waste electrical and electronic equipment (WEEE): A review. Waste Manag. 2014, 34, 1455–1469. [Google Scholar] [CrossRef] [PubMed]
- Cadena, L.E.S.; Arroyo, Z.G.; Lara, M.A.G.; Quiroz, Q.D. Cell-phone recycling by solvolysis for recovery of metals. J. Mater. Sci. Chem. Eng. 2015, 3, 52–57. [Google Scholar] [CrossRef]
- Xiao, Z.; Feng, C.; Chan, P.C.H.; Hsing, I.M. Monolithically integrated planar microfuel cell arrays. Sensors Actuators B: Chem. 2008, 132, 576–586. [Google Scholar] [CrossRef]
- Cha, S.-W.; O’Hayre, R.; Lee, S.J.; Saito, Y.; Prinz, F.B. Geometric scale effect of flow channels on performance of fuel cells. J. Electrochem. Soc. 2004, 151, A1856–A1864. [Google Scholar] [CrossRef]
- Lu, Y.; Reddy, R.G. Performance of micro-PEM fuel cells with different flow fields. J. Power Sources 2010, 195, 503–508. [Google Scholar] [CrossRef]
- Hockaday, R.; Navas, C. Micro-fuel cells TM for portable electronics. Fuel Cells Bull. 1999, 2, 9–12. [Google Scholar] [CrossRef]
- Mex, L.; Ponath, N.; Müller, J. Miniaturized fuel cells based on microsystem technologies. Fuel Cells Bull. 2001, 4, 9–12. [Google Scholar] [CrossRef]
- Breakthrough in micro fuel cell design, architecture. Fuel Cells Bull. 2002, 1. Available online: http://thirdworld.nl/breakthrough-in-micro-fuel-cell-design-architecture (accessed on 15 September 2016).
- Micro fuel cell developed at llnl. Fuel Cells Bull. 2002, 4. Available online: http://thirdworld.nl/breakthrough-in-micro-fuel-cell-design-architecture (accessed on 15 September 2016).
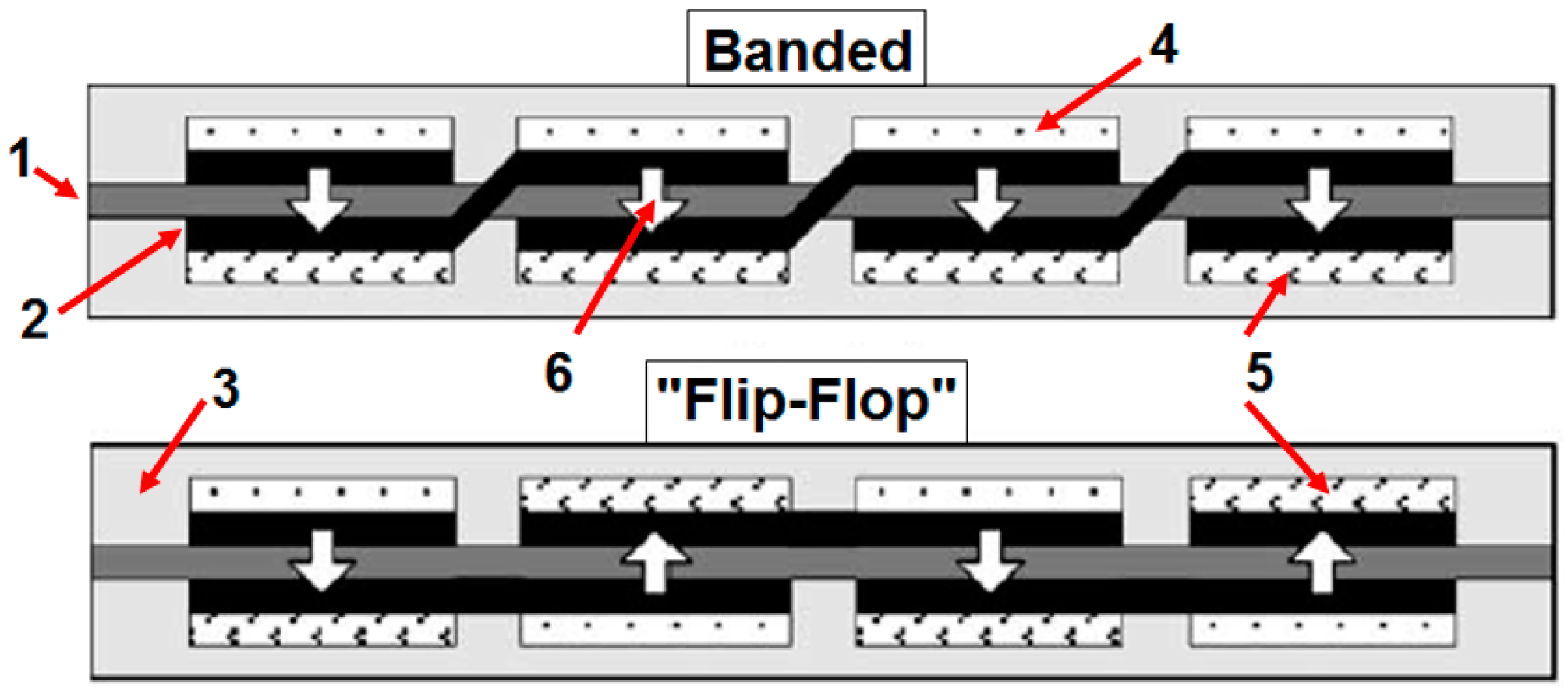
- Lee, S.J.; Chang-Chien, A.; Cha, S.W.; O’Hayre, R.; Park, Y.I.; Saito, Y.; Prinz, F.B. Design and fabrication of a micro fuel cell array with “flip-flop” interconnection. J. Power Sources 2002, 112, 410–418. [Google Scholar] [CrossRef]
- Blum, A.; Duvdevani, T.; Philosoph, M.; Rudoy, N.; Peled, E. Water-neutral micro direct-methanol fuel cell (DMFC) for portable applications. J. Power Sources 2003, 117, 22–25. [Google Scholar] [CrossRef]
- Yu, J.; Cheng, P.; Ma, Z.; Yi, B. Fabrication of miniature silicon wafer fuel cells with improved performance. J. Power Sources 2003, 124, 40–46. [Google Scholar] [CrossRef]
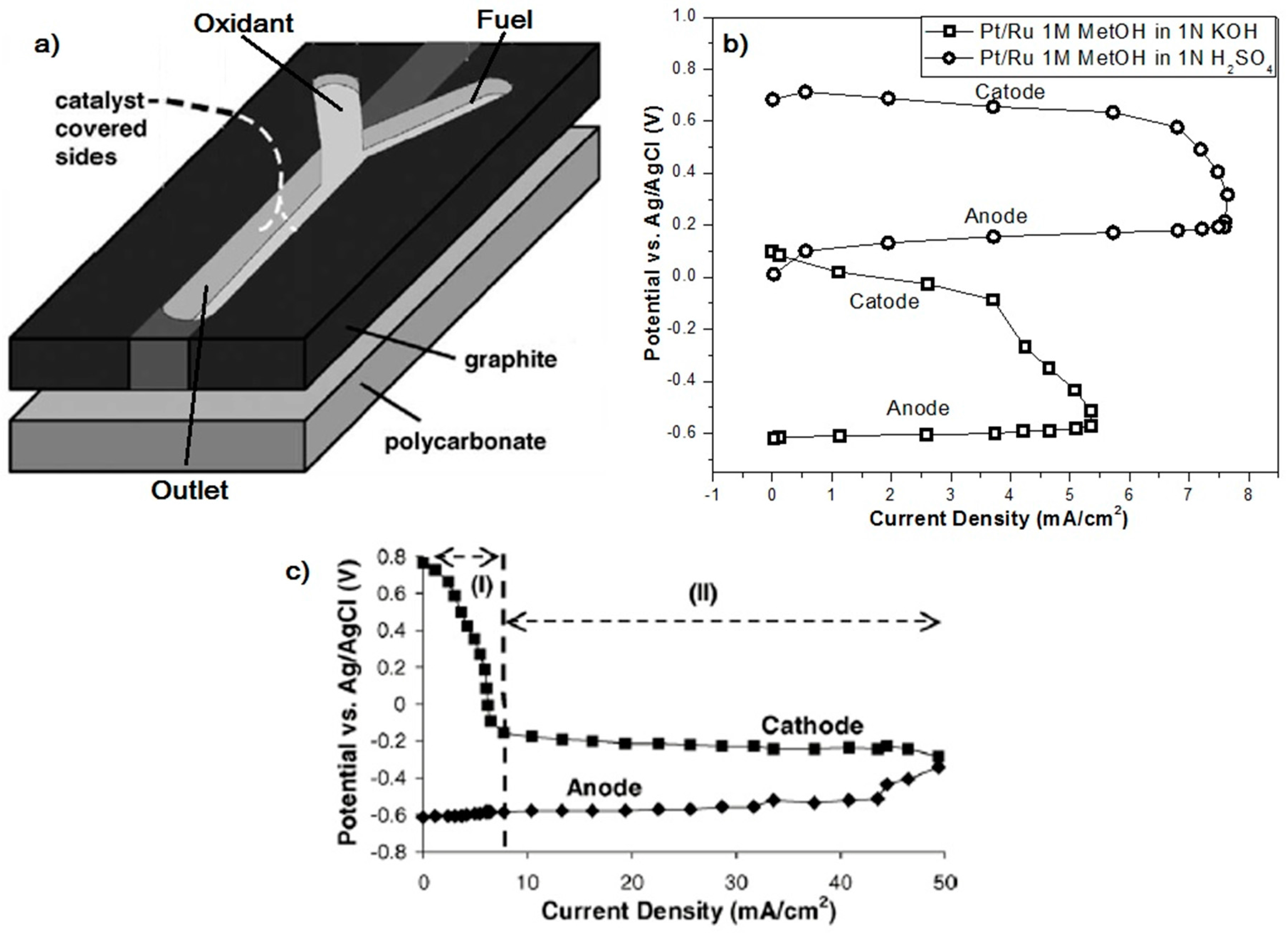
- Choban, E.R.; Markoski, L.J.; Wieckowski, A.; Kenis, P.J.A. Microfluidic fuel cell based on laminar flow. J. Power Sources 2004, 128, 54–60. [Google Scholar] [CrossRef]
- Choban, E.R.; Spendelow, J.S.; Gancs, L.; Wieckowski, A.; Kenis, P.J.A. Membraneless laminar flow-based micro fuel cells operating in alkaline, acidic, and acidic/alkaline media. Electrochim. Acta 2005, 50, 5390–5398. [Google Scholar] [CrossRef]
- McLean, G.F.; Niet, T.; Prince-Richard, S.; Djilali, N. An assessment of alkaline fuel cell technology. Int. J. Hydrogen Energy 2002, 27, 507–526. [Google Scholar] [CrossRef]
- Tripković, A.V.; Popović, K.D.; Lović, J.D.; Jovanović, V.M.; Kowal, A. Methanol oxidation at platinum electrodes in alkaline solution: Comparison between supported catalysts and model systems. J. Electroanal. Chem. 2004, 572, 119–128. [Google Scholar] [CrossRef]
- Yu, E.H.; Scott, K. Development of direct methanol alkaline fuel cells using anion exchange membranes. J. Power Sources 2004, 137, 248–256. [Google Scholar] [CrossRef]
- Hsieh, S.-S.; Yang, S.-H.; Kuo, J.-K.; Huang, C.-F.; Tsai, H.-H. Study of operational parameters on the performance of micro PEMFCs with different flow fields. Energy Convers. Manag. 2006, 47, 1868–1878. [Google Scholar] [CrossRef]
- Nakajima, H.; Konomi, T.; Kitahara, T. Direct water balance analysis on a polymer electrolyte fuel cell (PEFC): Effects of hydrophobic treatment and micro-porous layer addition to the gas diffusion layer of a PEFC on its performance during a simulated start-up operation. J. Power Sources 2007, 171, 457–463. [Google Scholar] [CrossRef]
- Hsieh, S.-S.; Huang, Y.-J. Measurements of current and water distribution for a micro-pem fuel cell with different flow fields. J. Power Sources 2008, 183, 193–204. [Google Scholar] [CrossRef]
- Park, B.Y.; Madou, M.J. Design, fabrication, and initial testing of a miniature pem fuel cell with micro-scale pyrolyzed carbon fluidic plates. J. Power Sources 2006, 162, 369–379. [Google Scholar] [CrossRef]
- Lin, P.-C.; Park, B.Y.; Madou, M.J. Development and characterization of a miniature PEM fuel cell stack with carbon bipolar plates. J. Power Sources 2008, 176, 207–214. [Google Scholar] [CrossRef]
- Whipple, D.T.; Jayashree, R.S.; Egas, D.; Alonso-Vante, N.; Kenis, P.J.A. Ruthenium cluster-like chalcogenide as a methanol tolerant cathode catalyst in air-breathing laminar flow fuel cells. Electrochim. Acta 2009, 54, 4384–4388. [Google Scholar] [CrossRef]
- Cheng, H.; Yuan, W.; Scott, K. Influence of thermal treatment on RuSe cathode materials for direct methanol fuel cells. Fuel Cells 2007, 7, 16–20. [Google Scholar] [CrossRef]
- Colmenares, L.; Jusys, Z.; Behm, R.J. Activity, selectivity, and methanol tolerance of Se-modified Ru/C cathode catalysts. J. Phys. Chem. C 2007, 111, 1273–1283. [Google Scholar] [CrossRef]
- Cremers, C.; Scholz, M.; Seliger, W.; Racz, A.; Knechtel, W.; Rittmayr, J.; Grafwallner, F.; Peller, H.; Stimming, U. Developments for improved direct methanol fuel cell stacks for portable power. Fuel Cells 2007, 7, 21–31. [Google Scholar] [CrossRef]
- Itoe, R.N.; Wesson, G.D.; Kalu, E.E. Evaluation of oxygen transport parameters in H2SO4-CH3OH mixtures using electrochemical methods. J. Electrochem. Soc. 2000, 147, 2445–2450. [Google Scholar] [CrossRef]
- Gago, A.S.; Morales-Acosta, D.; Arriaga, L.G.; Alonso-Vante, N. Carbon supported ruthenium chalcogenide as cathode catalyst in a microfluidic formic acid fuel cell. J. Power Sources 2011, 196, 1324–1328. [Google Scholar] [CrossRef]
- Brushett, F.R.; Naughton, M.S.; Ng, J.W.D.; Yin, L.; Kenis, P.J.A. Analysis of Pt/C electrode performance in a flowing-electrolyte alkaline fuel cell. Int. J. Hydrogen Energy 2012, 37, 2559–2570. [Google Scholar] [CrossRef]
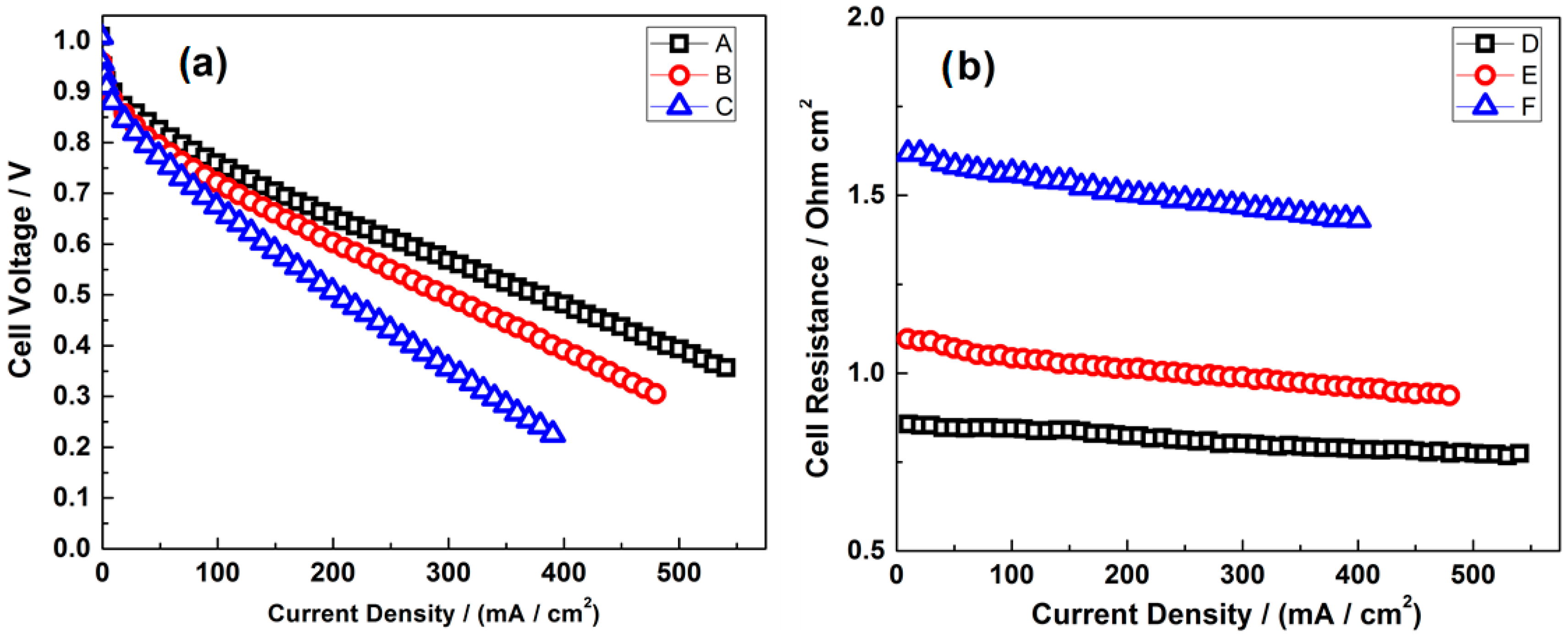
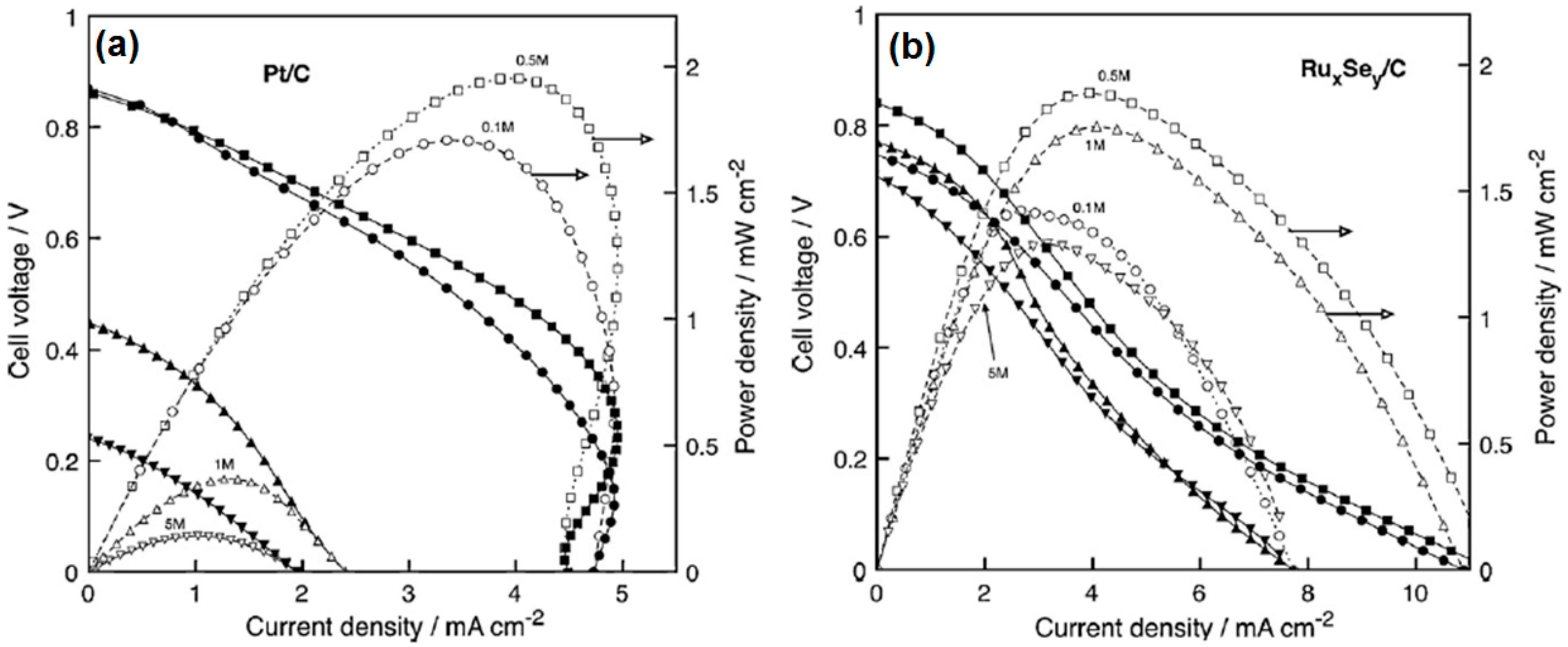
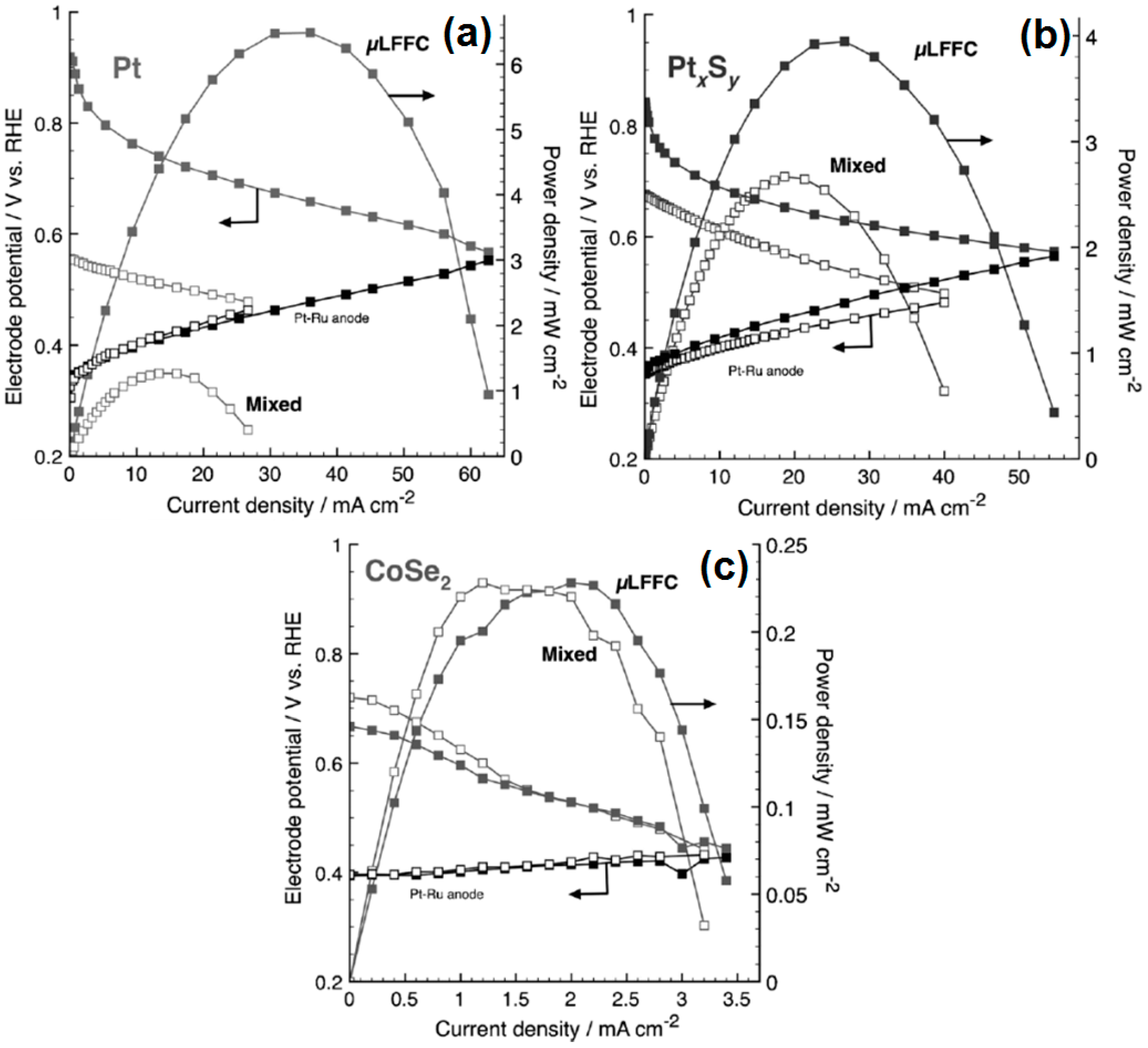
- Gago, A.S.; Gochi-Ponce, Y.; Feng, Y.-J.; Esquivel, J.P.; Sabaté, N.; Santander, J.; Alonso-Vante, N. Tolerant chalcogenide cathodes of membraneless micro fuel cells. ChemSusChem 2012, 5, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Gago, A.S.; Esquivel, J.-P.; Sabaté, N.; Santander, J.; Alonso-Vante, N. Comprehensive characterization and understanding of micro-fuel cells operating at high methanol concentrations. Beilstein J. Nanotechnol. 2015, 6, 2000–2006. [Google Scholar] [CrossRef] [PubMed]
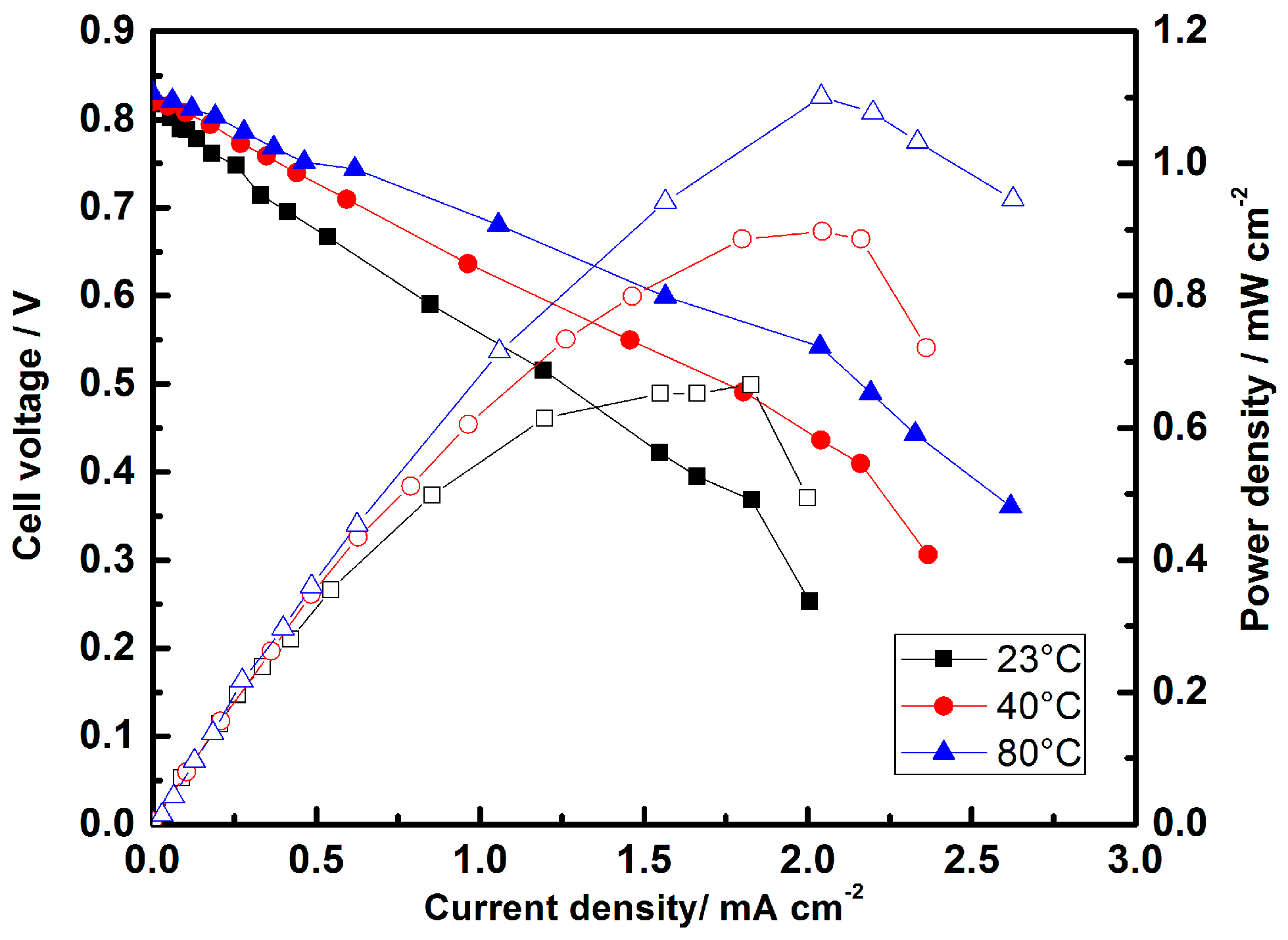
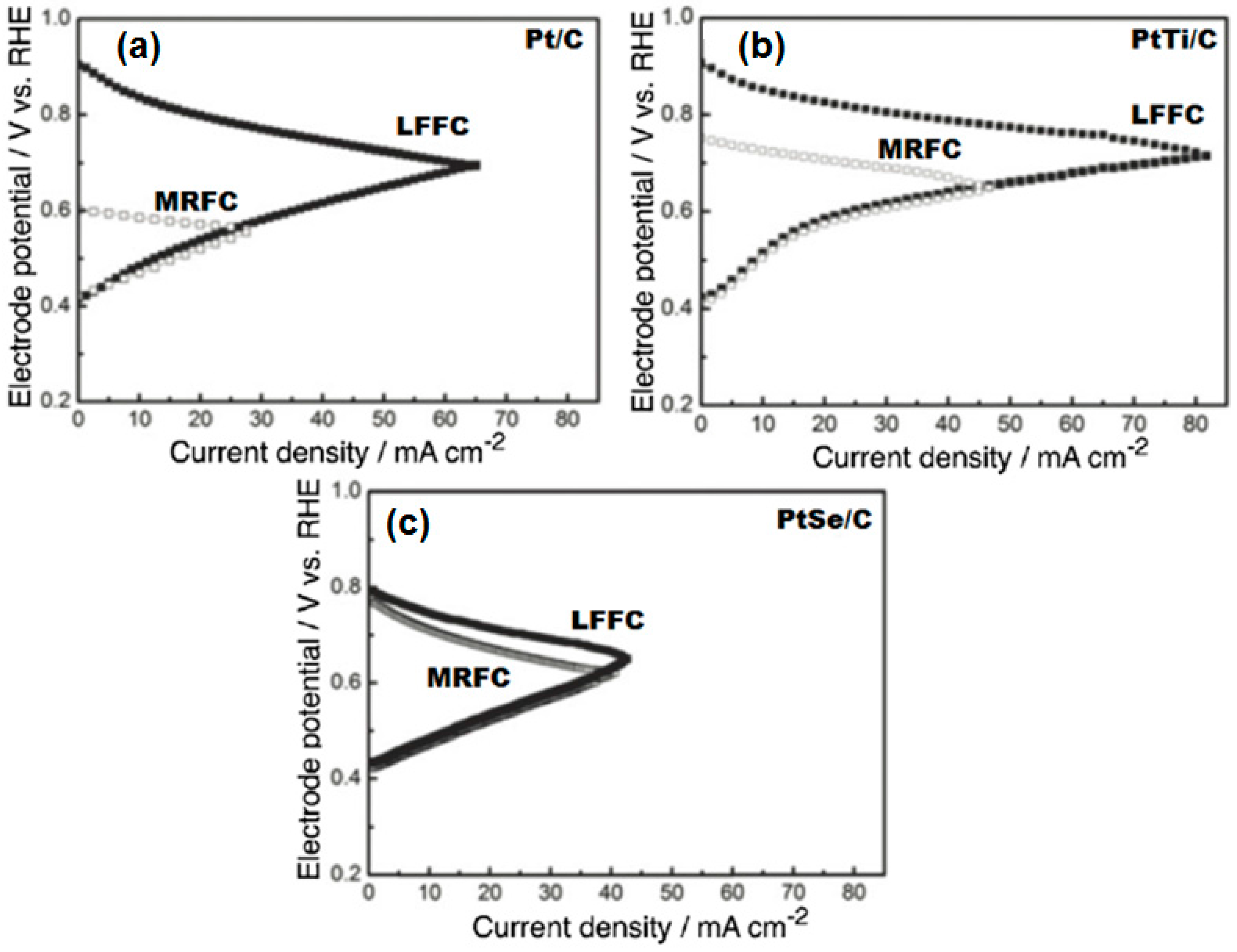
- Ma, J.; Habrioux, A.; Morais, C.; Alonso-Vante, N. Electronic modification of Pt via Ti and Se as tolerant cathodes in air-breathing methanol microfluidic fuel cells. PhysChemChemPhys 2014, 16, 13820–13826. [Google Scholar] [CrossRef] [PubMed]
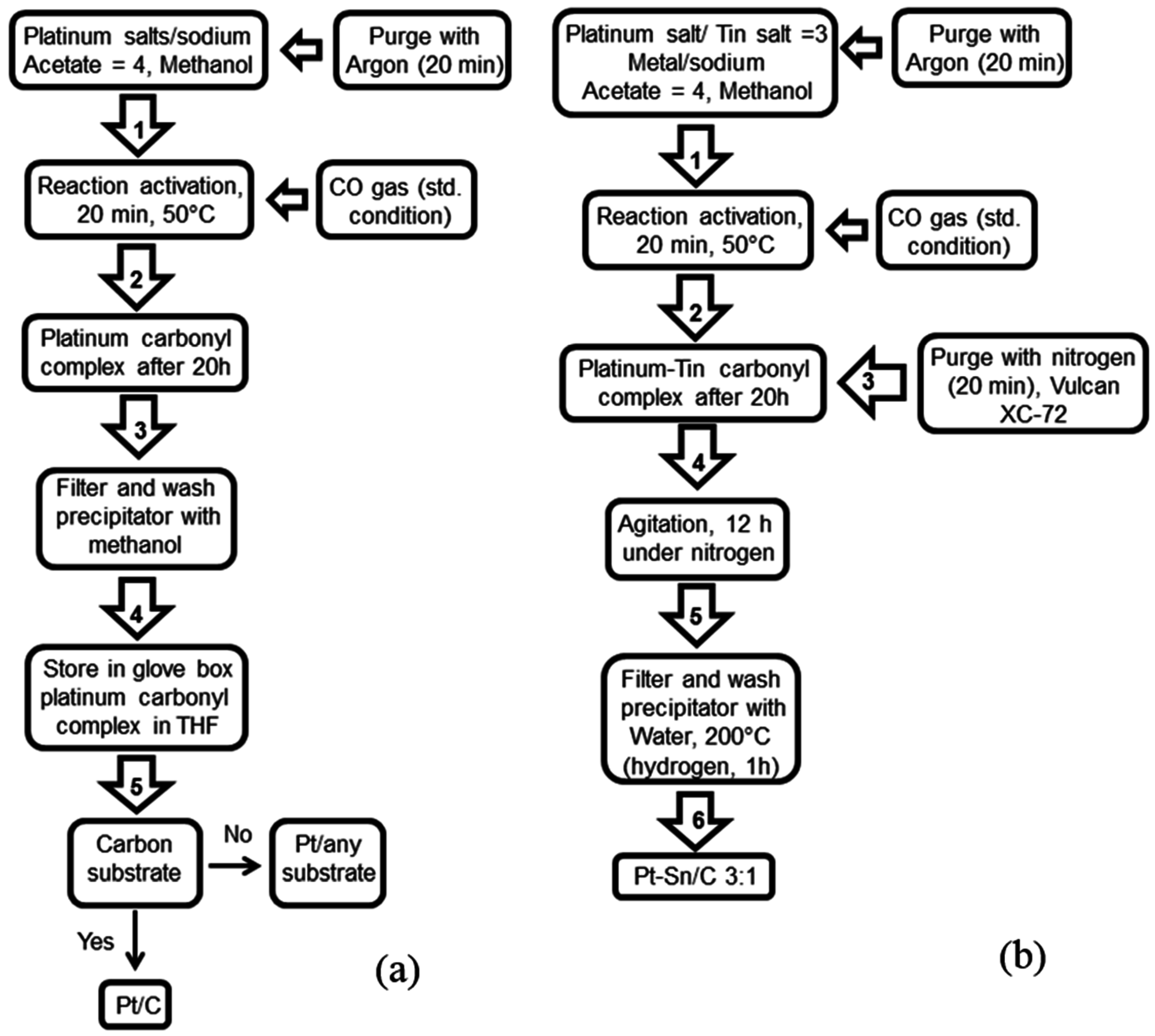
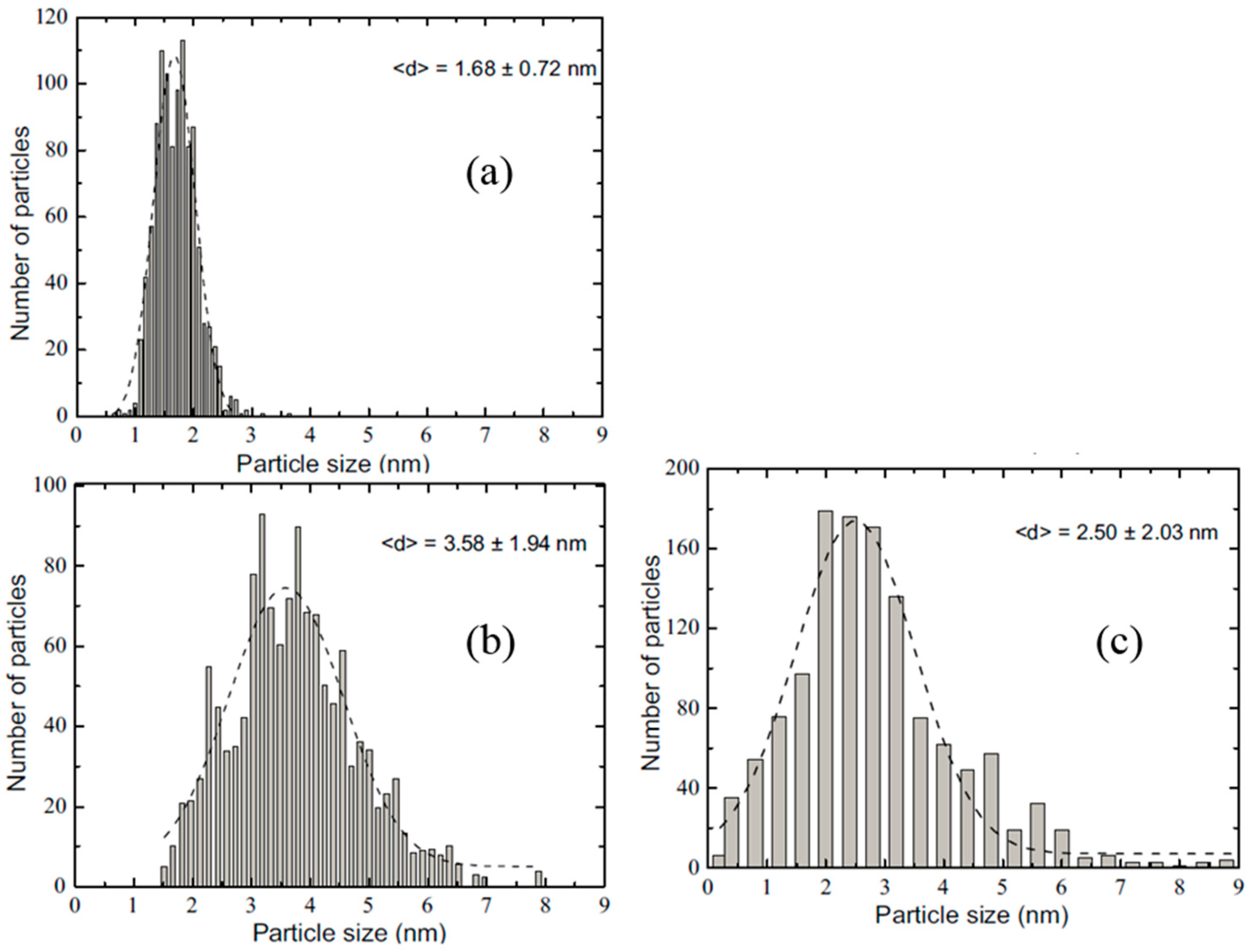
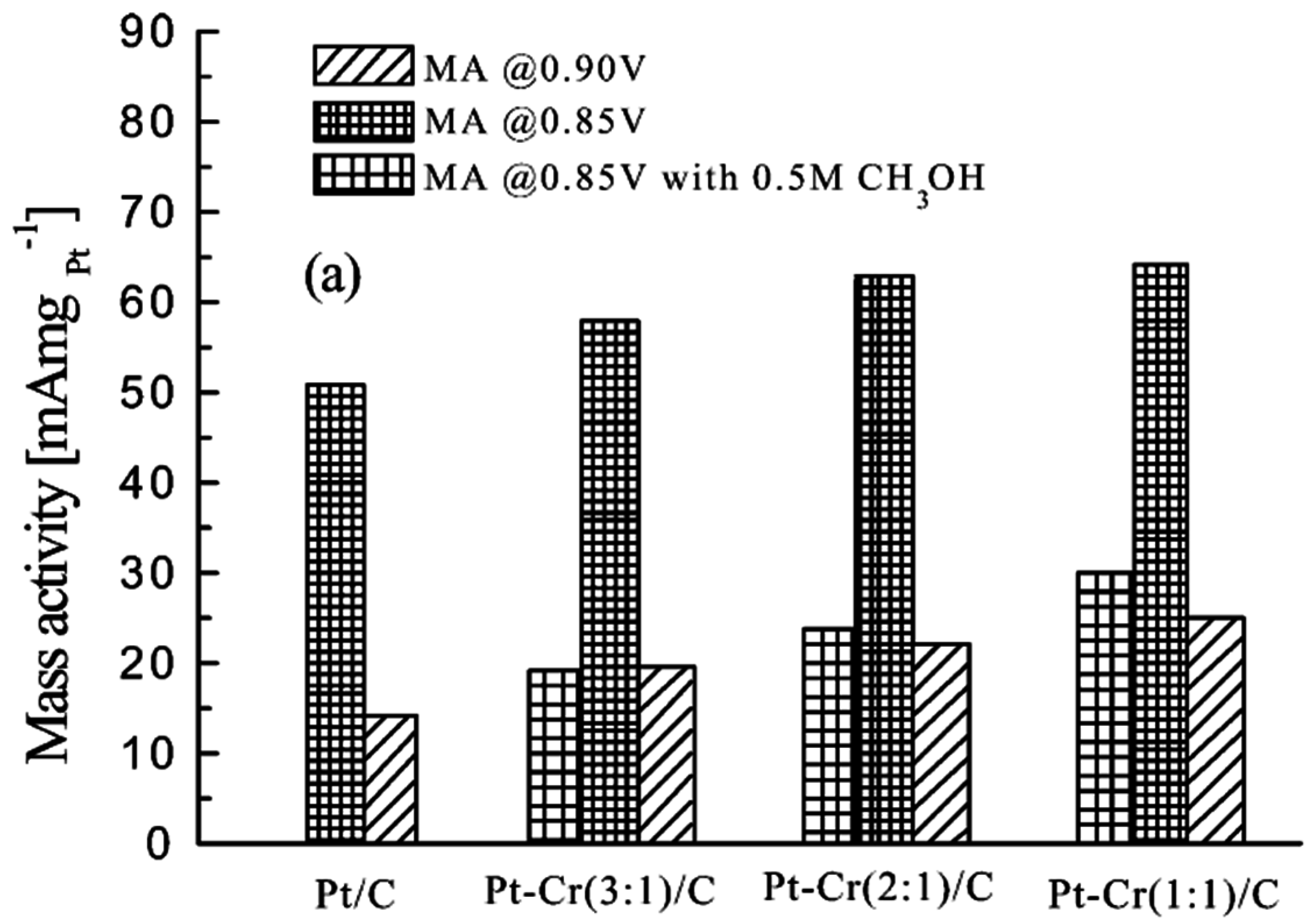


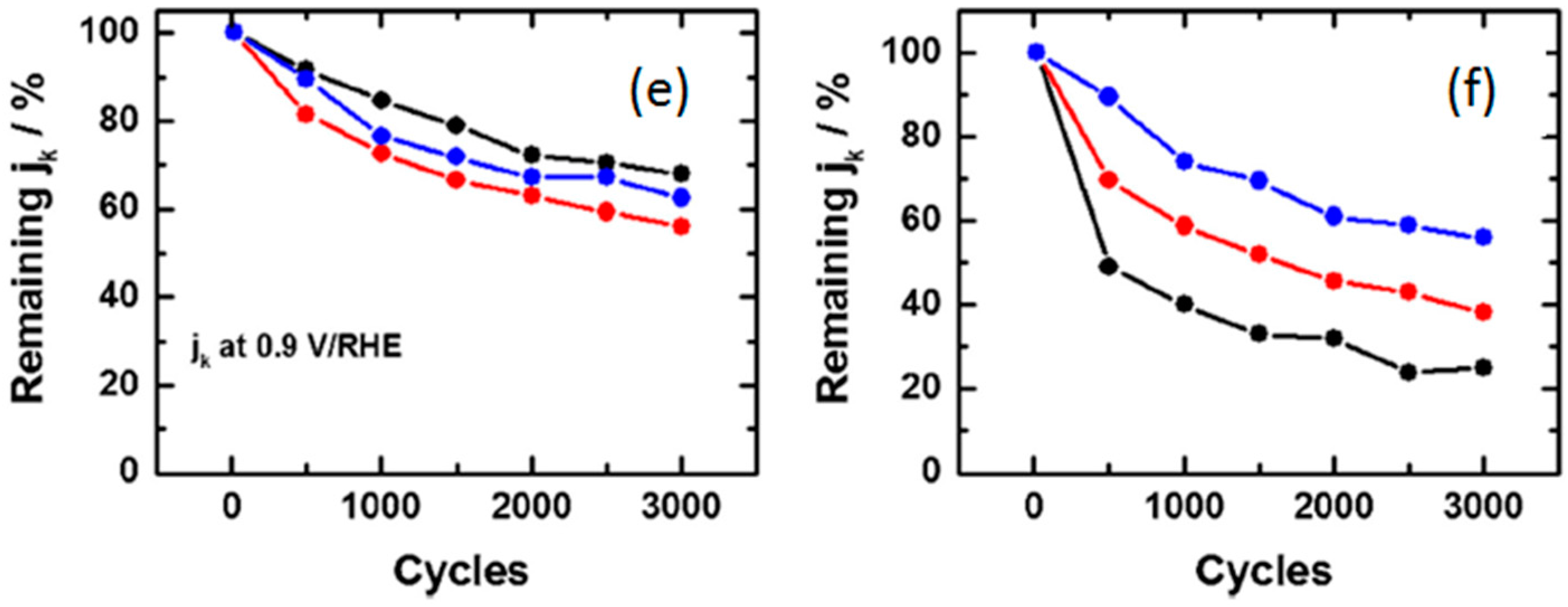
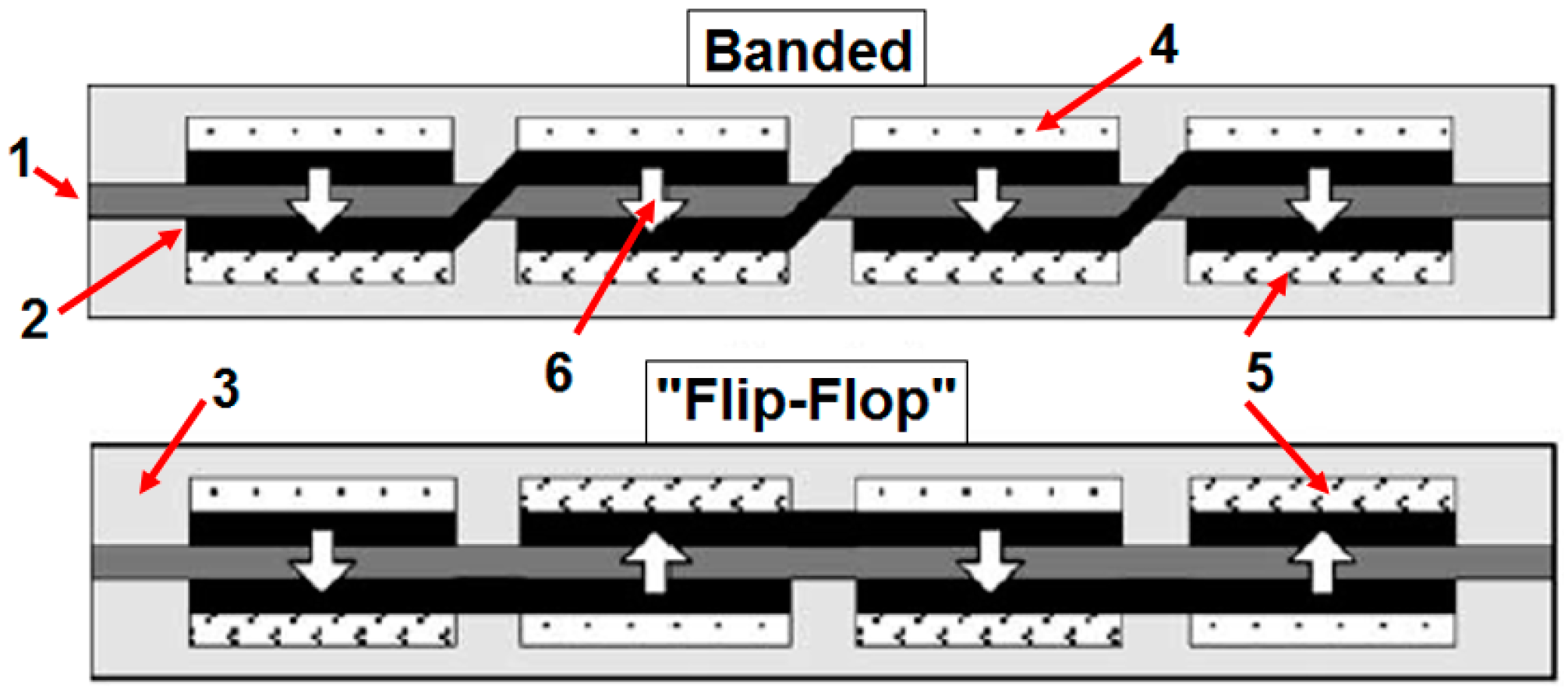
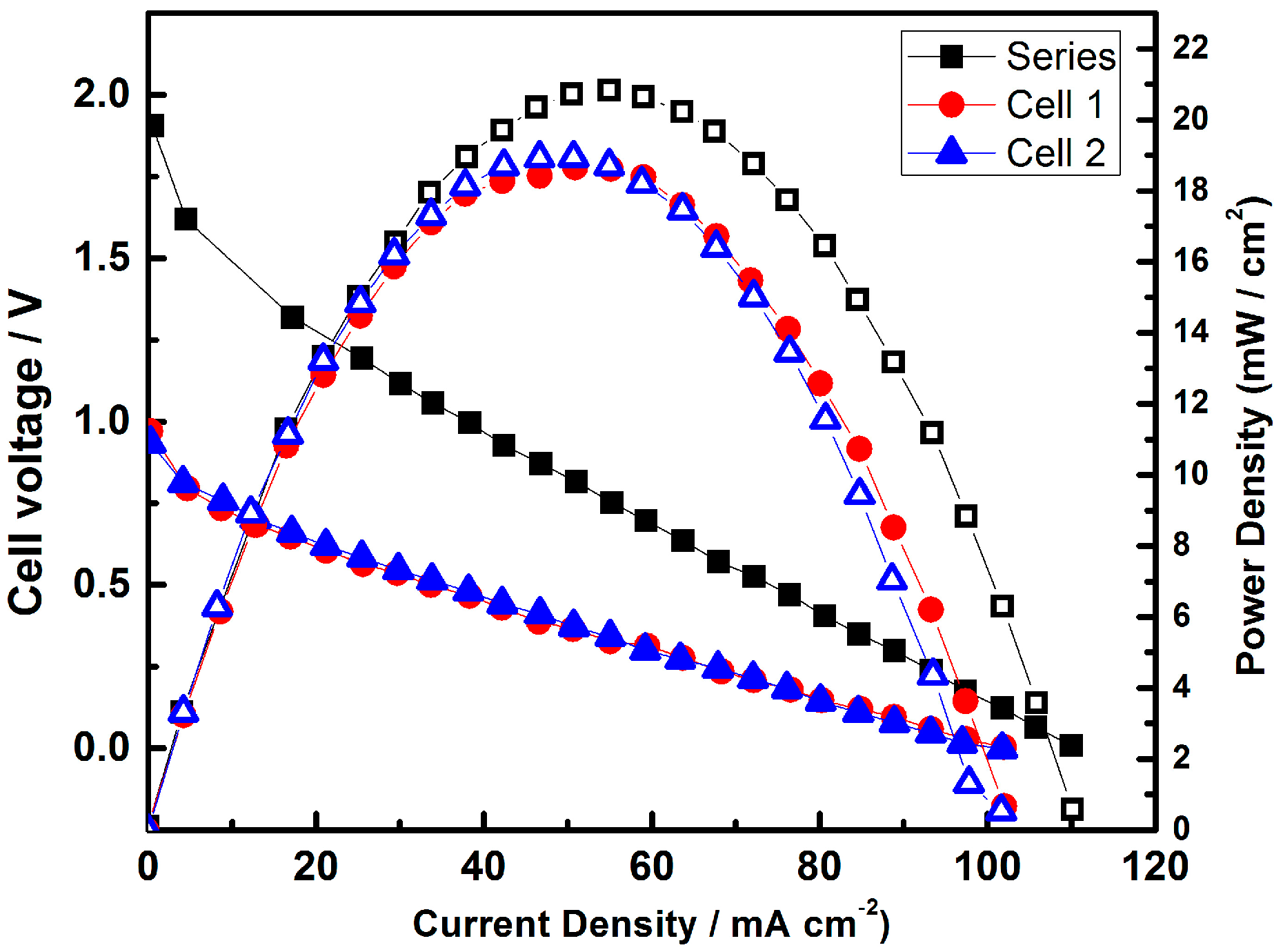
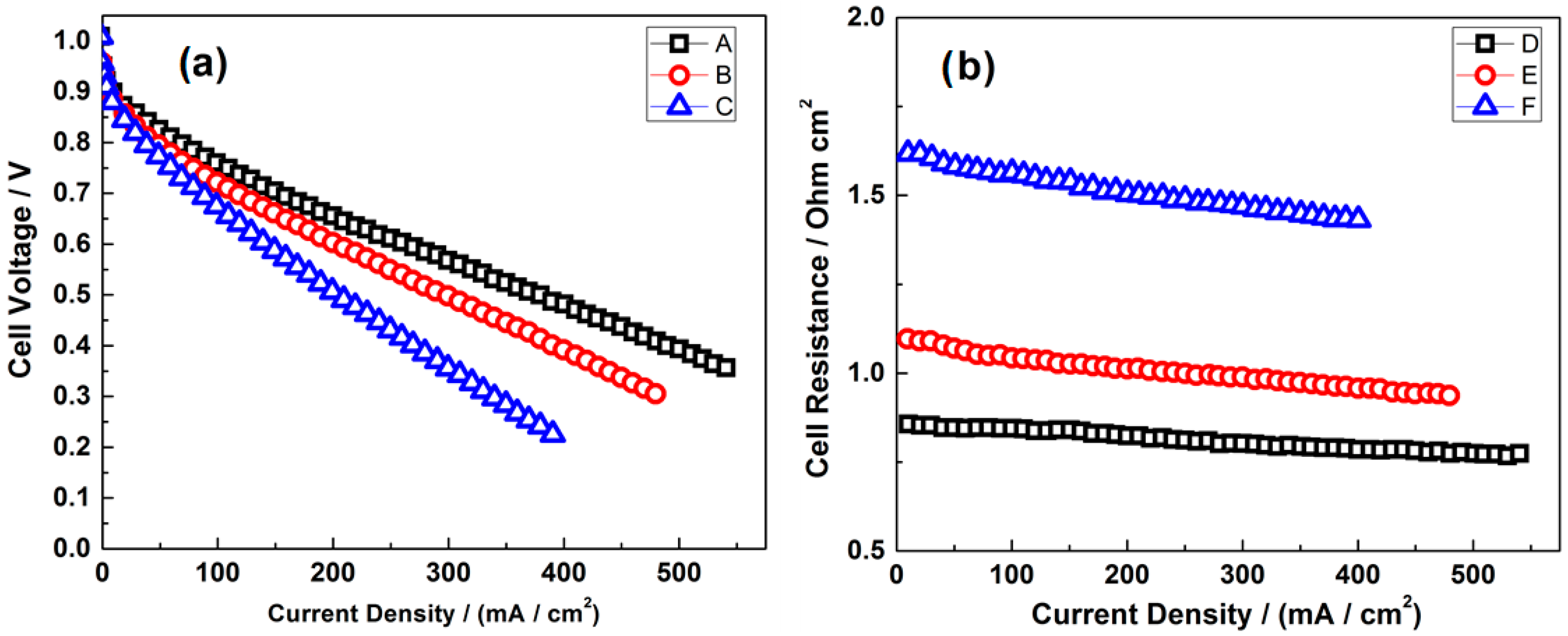

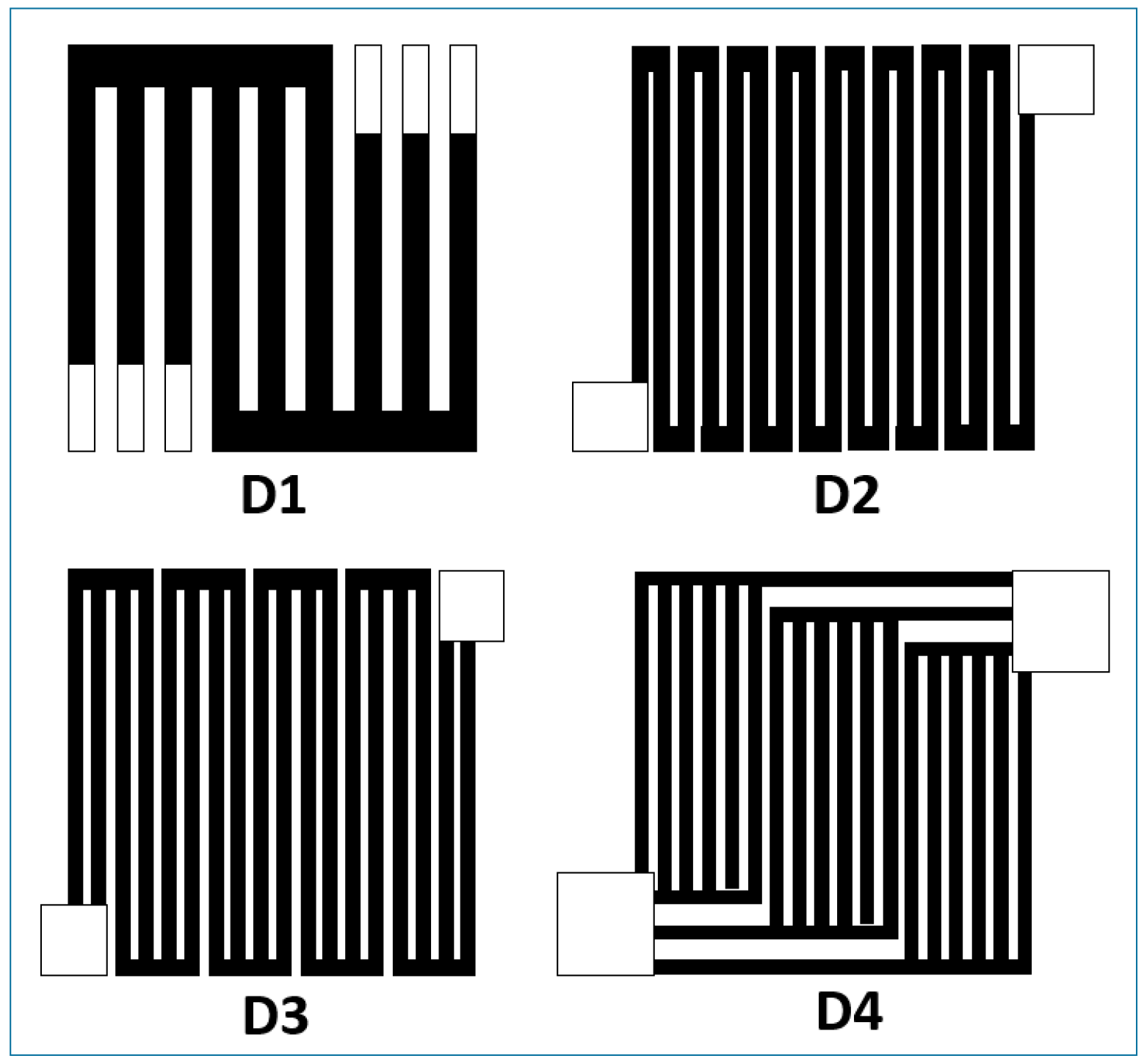
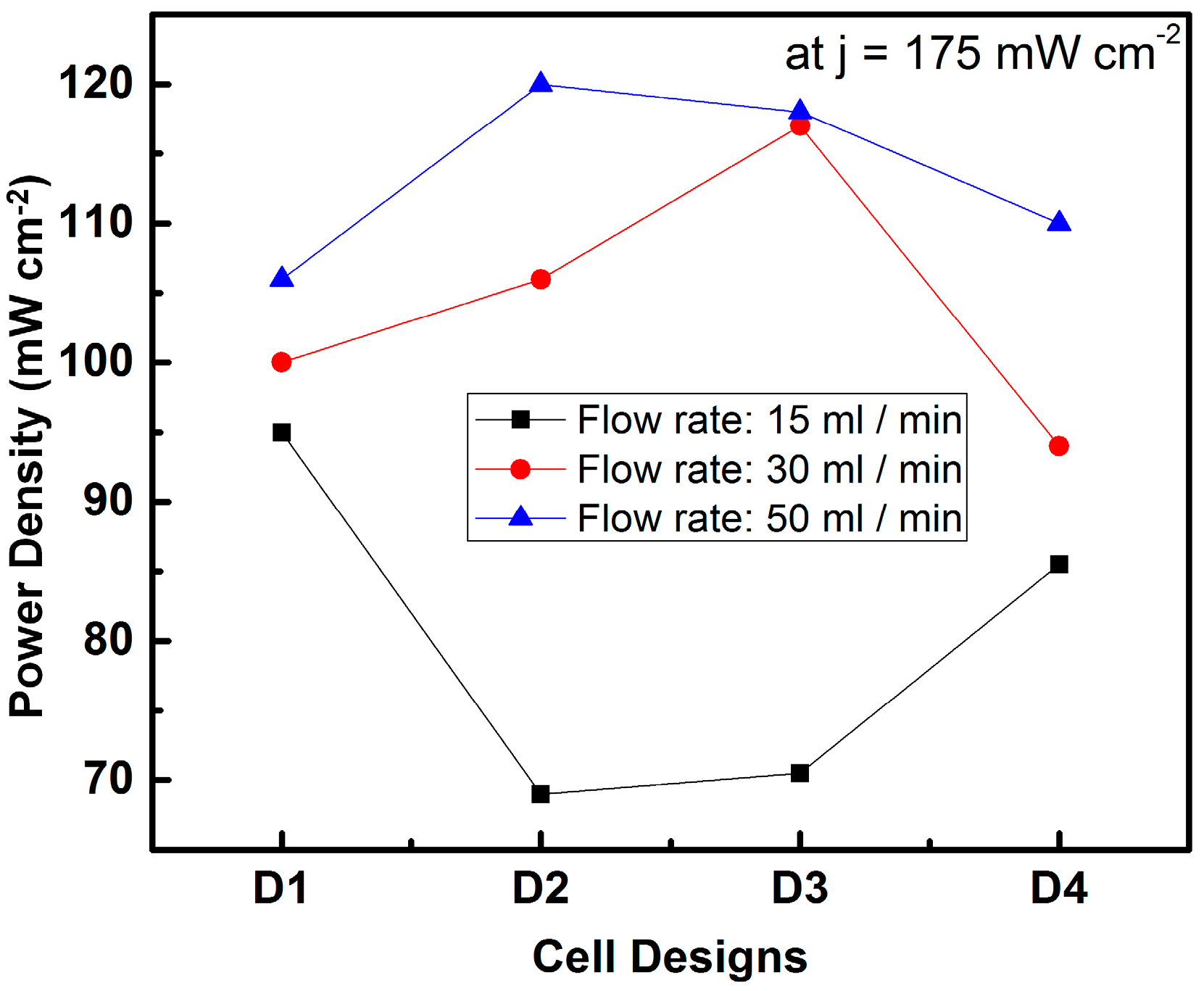
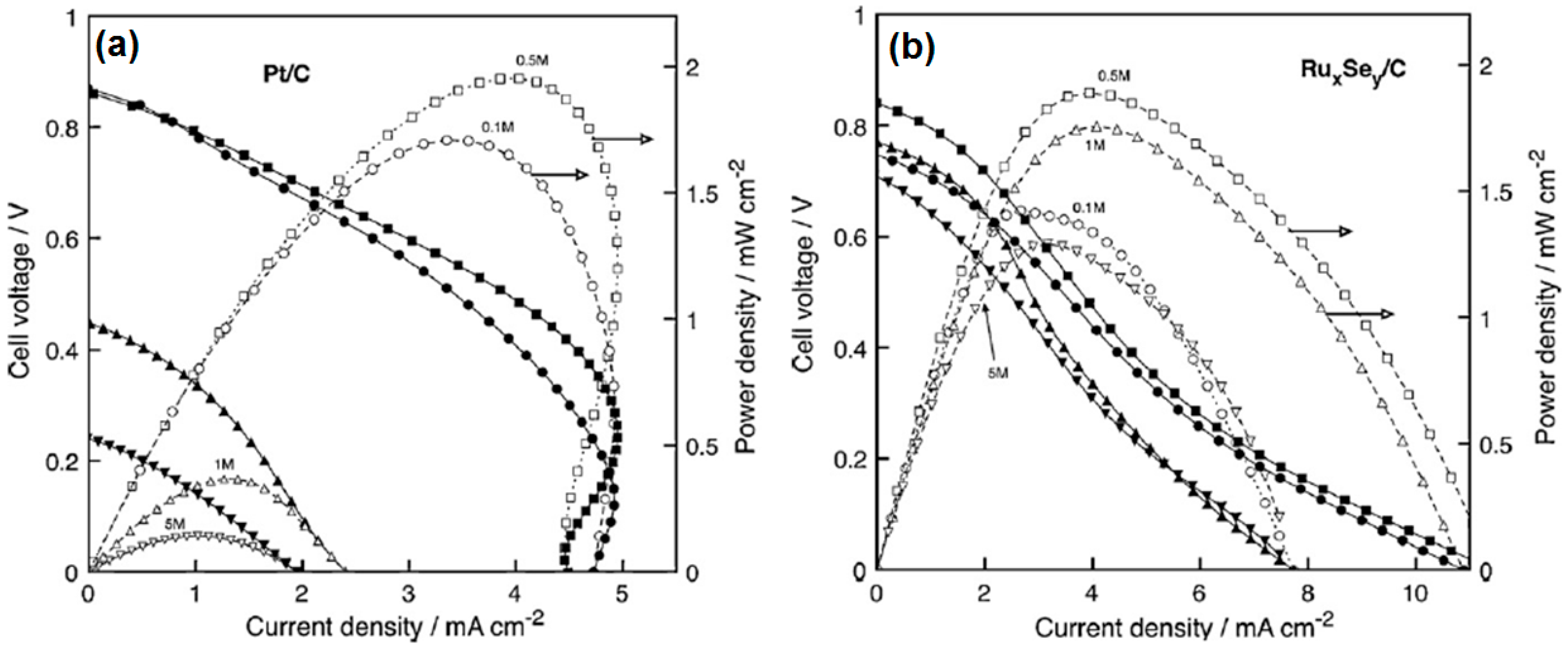
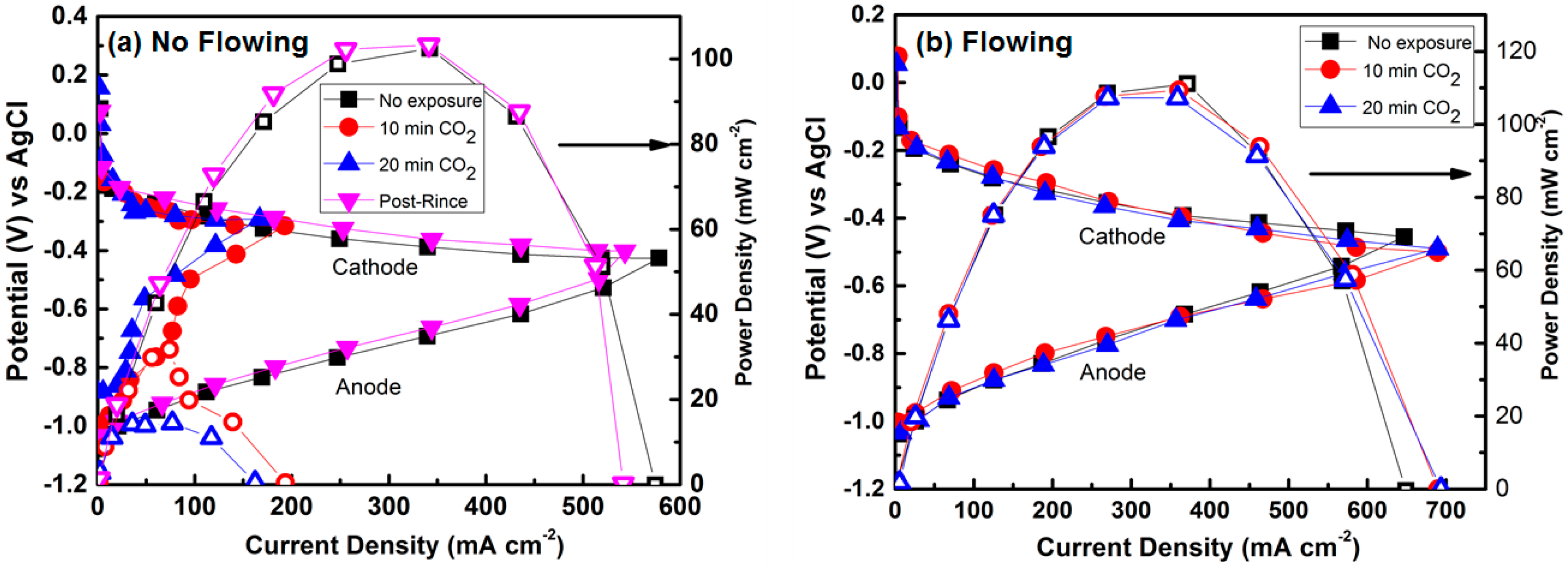

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mora-Hernández, J.M.; Luo, Y.; Alonso-Vante, N. What Can We Learn in Electrocatalysis, from Nanoparticulated Precious and/or Non-Precious Catalytic Centers Interacting with Their Support? Catalysts 2016, 6, 145. https://doi.org/10.3390/catal6090145
Mora-Hernández JM, Luo Y, Alonso-Vante N. What Can We Learn in Electrocatalysis, from Nanoparticulated Precious and/or Non-Precious Catalytic Centers Interacting with Their Support? Catalysts. 2016; 6(9):145. https://doi.org/10.3390/catal6090145
Chicago/Turabian StyleMora-Hernández, Juan Manuel, Yun Luo, and Nicolas Alonso-Vante. 2016. "What Can We Learn in Electrocatalysis, from Nanoparticulated Precious and/or Non-Precious Catalytic Centers Interacting with Their Support?" Catalysts 6, no. 9: 145. https://doi.org/10.3390/catal6090145
APA StyleMora-Hernández, J. M., Luo, Y., & Alonso-Vante, N. (2016). What Can We Learn in Electrocatalysis, from Nanoparticulated Precious and/or Non-Precious Catalytic Centers Interacting with Their Support? Catalysts, 6(9), 145. https://doi.org/10.3390/catal6090145