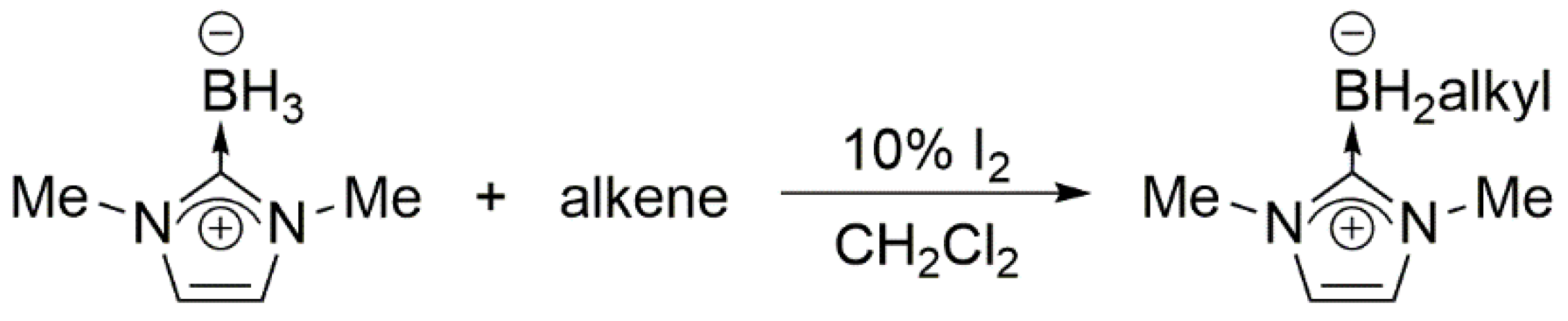
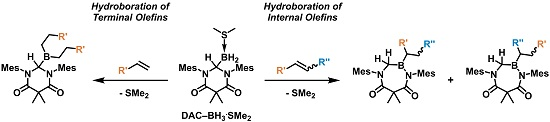
2. Results and Discussion
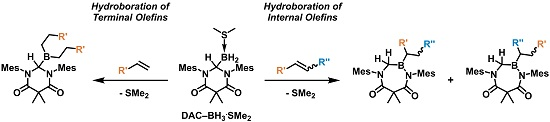
As summarized in
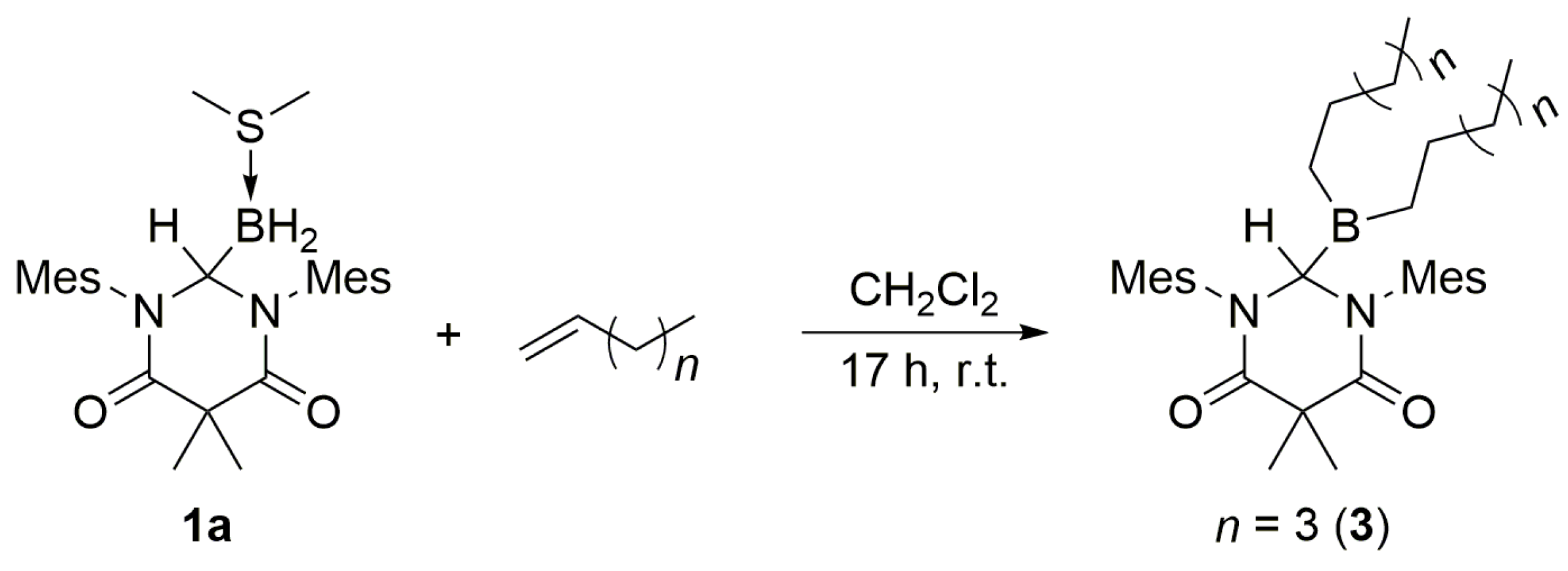
Scheme 3, the addition of an excess of a terminal olefin, such as 1-hexene, to a solution of
1a in CH
2Cl
2 resulted in the formation of
3, as determined by
1H,
13C, and
11B NMR spectroscopy as well as high resolution mass spectrometry. Inspection of the
1H NMR data revealed a diagnostic singlet at δ 6.09 ppm (C
6D
6), which was assigned to the methine group of
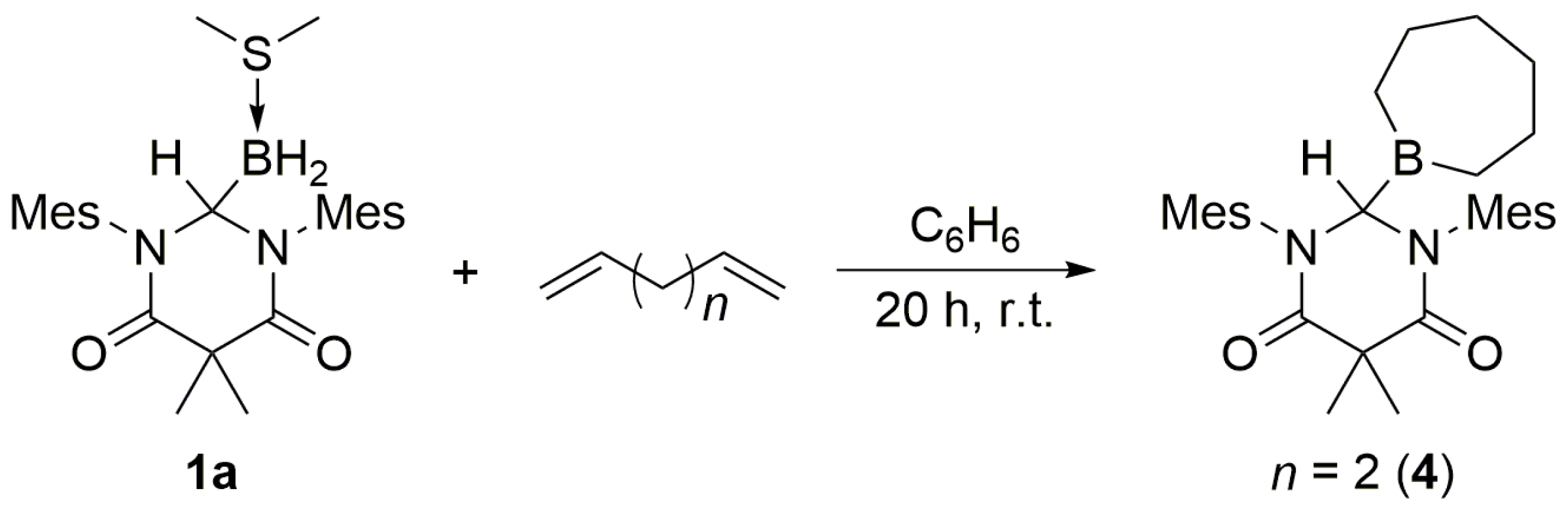
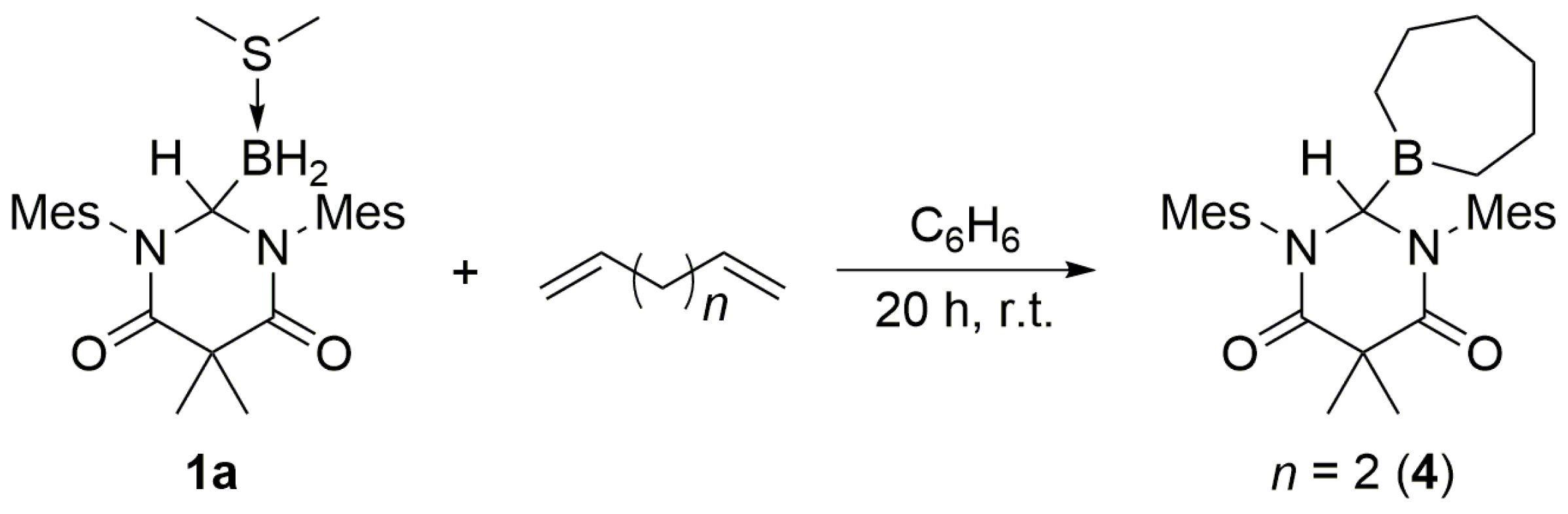
3. When an excess of 1,5-hexadiene was added to
1a, both double bonds underwent hydroboration, and
4 was obtained as the exclusive product (
Scheme 4).
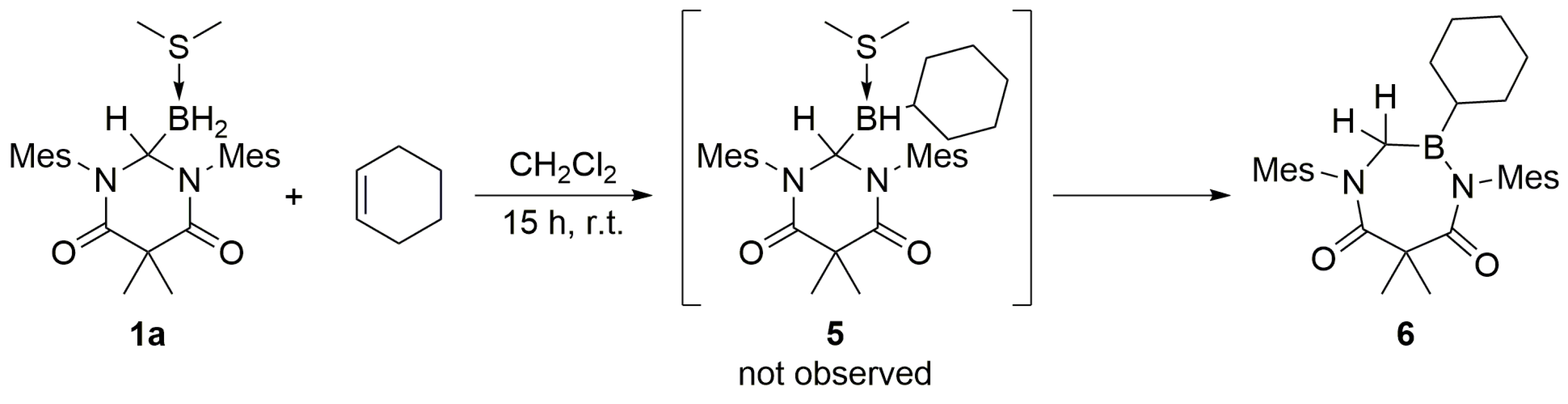
The ability of
1a to facilitate the hydroboration of internal olefins was also explored. When
1a was treated with an excess of cyclohexene, a product (
6) containing only one cyclohexyl moiety was obtained (
Scheme 5). The
1H NMR recorded for
6 showed a diagnostic singlet at δ 3.61 ppm (C
6D
6), which corresponded to the two hydrogen atoms at the former carbenoid center. As no reaction was observed between
2 and cyclohexene, we hypothesized that the initial formation of
5 (not observed) was rapidly followed by intramolecular ring-expansion. The hydroboration chemistry of
1a with internal olefins was further explored by independently treating
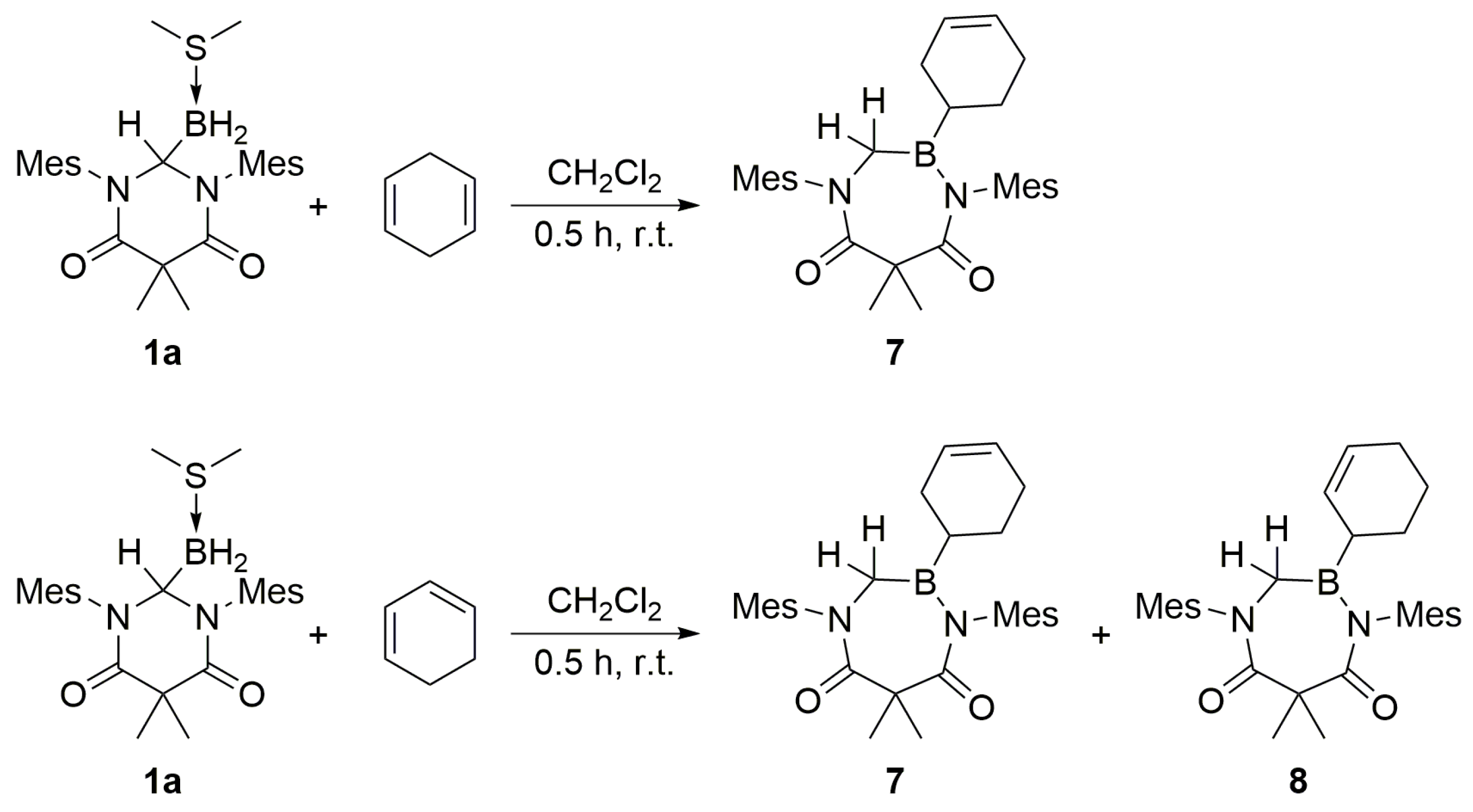
1a with 1,3- or 1,4-cyclohexadiene. Similar to the results obtained when cyclohexene was used as a substrate, the formation of ring-expanded products was observed (
Scheme 6). While the hydroboration of 1,4-cyclohexadiene readily produced
8 as a single product, as evidenced by the appearance of a doublet of triplets at δ 3.56 ppm (C
6D
6), the hydroboration of 1,3-cyclohexadiene yielded an equimolar mixture of
7 and
8. The mixture of isomers was supported by two distinct
1H NMR doublet of triplets at δ 3.56 ppm and δ 3.73 ppm (C
6D
6), which were assigned to the two hydrogen atoms attached to the former carbenoid centers in the respective compounds.
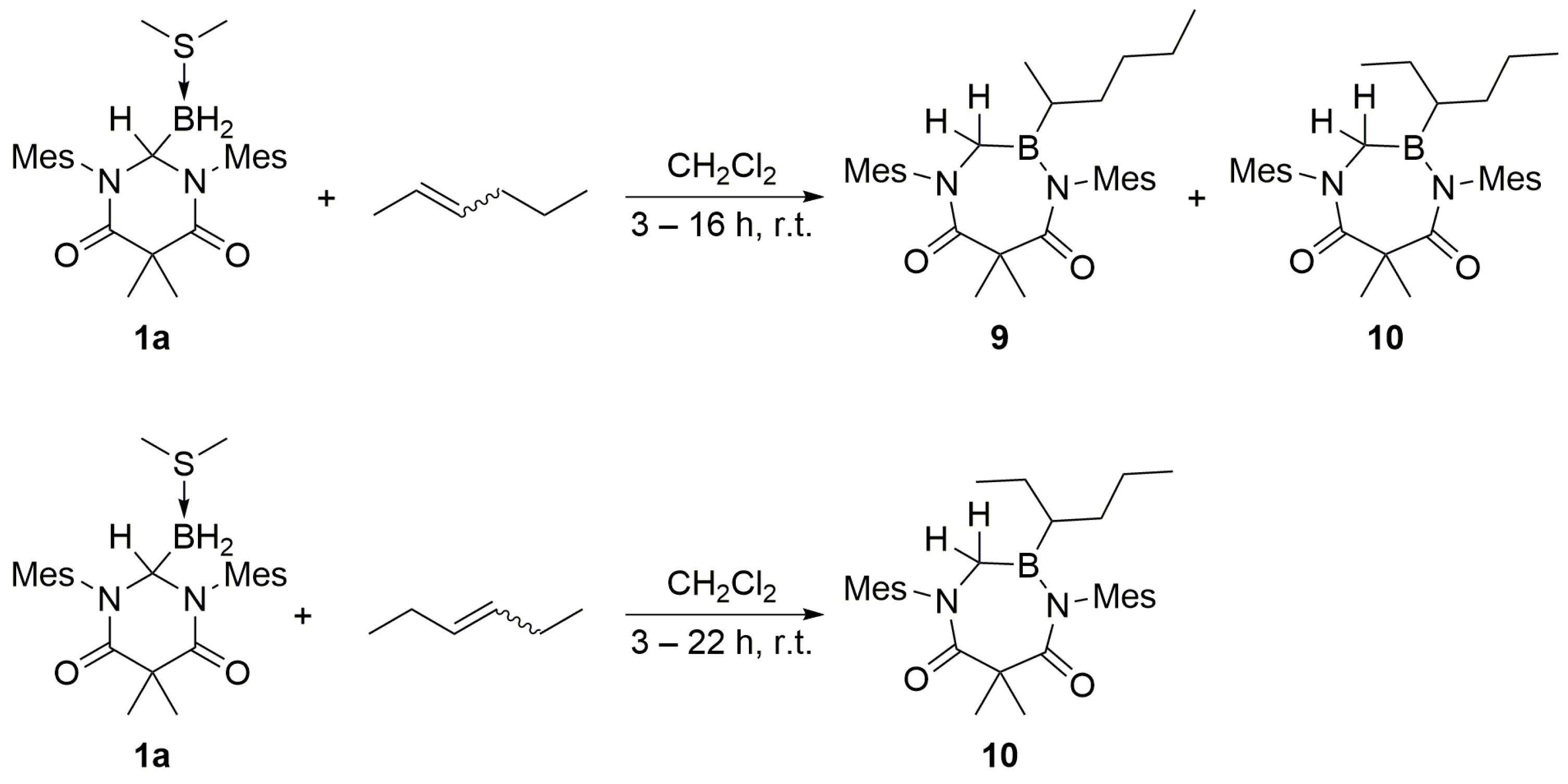
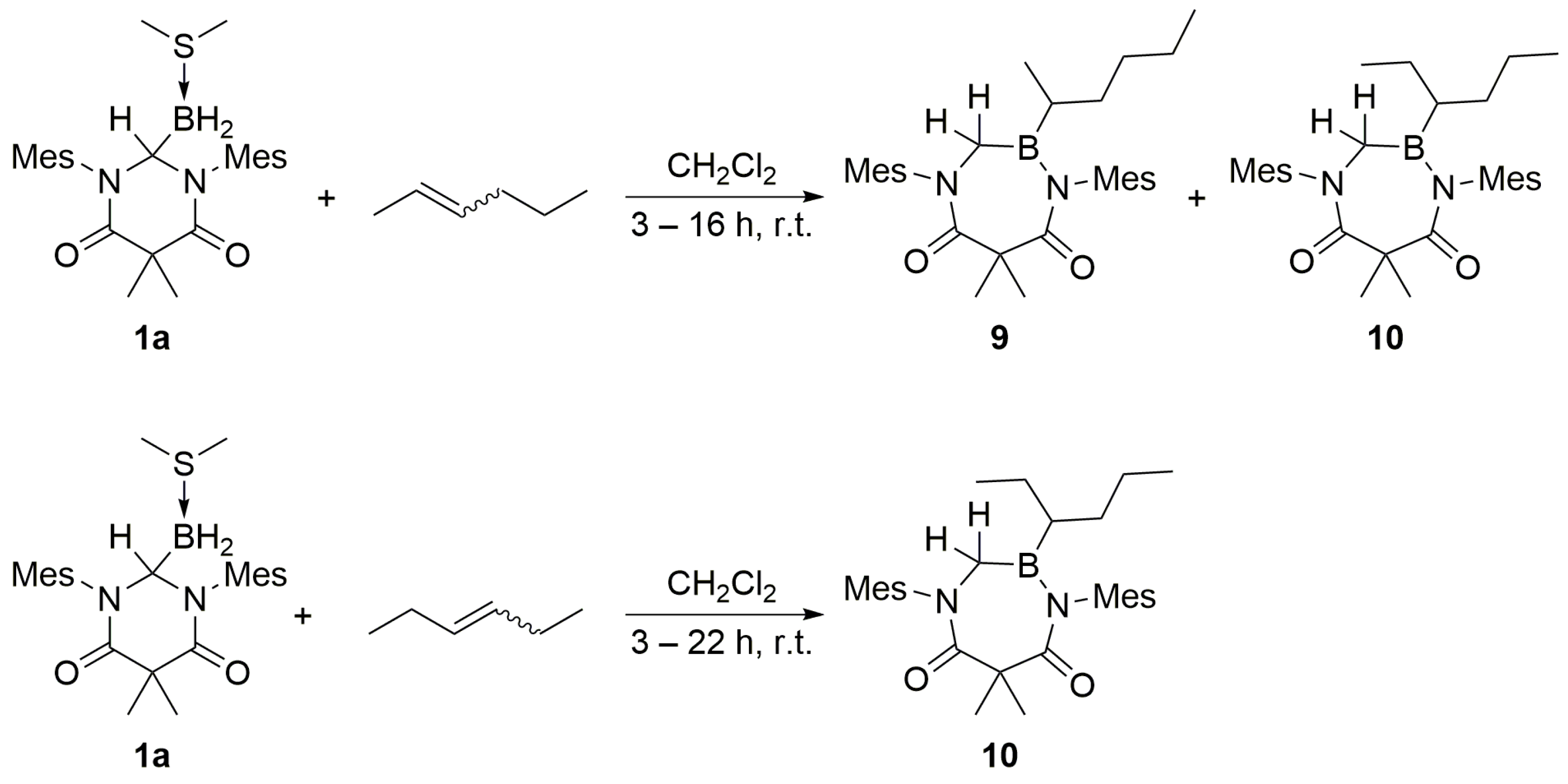
Acyclic internal olefins were also studied as hydroboration substrates. Upon the addition of
cis- or
trans-2-hexene to a CH
2Cl
2 solution of
1a, the appearance of overlapping multiplets at δ 3.67 ppm was observed in a 1:2 ratio in the corresponding
1H NMR spectrum (C
6D
6) recorded for the respective products
9 and
10 (
Scheme 7). Over time or upon heating the reaction mixture to 55 °C, the two multiplets converted to a single multiplet resonance at δ 3.67 ppm (see
Supplementary Materials, Figure S17). Similar results were reported by Curran and co-workers, who attributed the phenomena to “chain walking” of a carbene–BH
3 adduct [
19,
20,
21,
22,
23,
24,
25,
26]. Additionally, when
cis- or
trans-3-hexene was introduced to
1a, compound
10 was obtained as the sole product, as indicated by the presence of a single multiplet at δ 3.67 ppm (C
6D
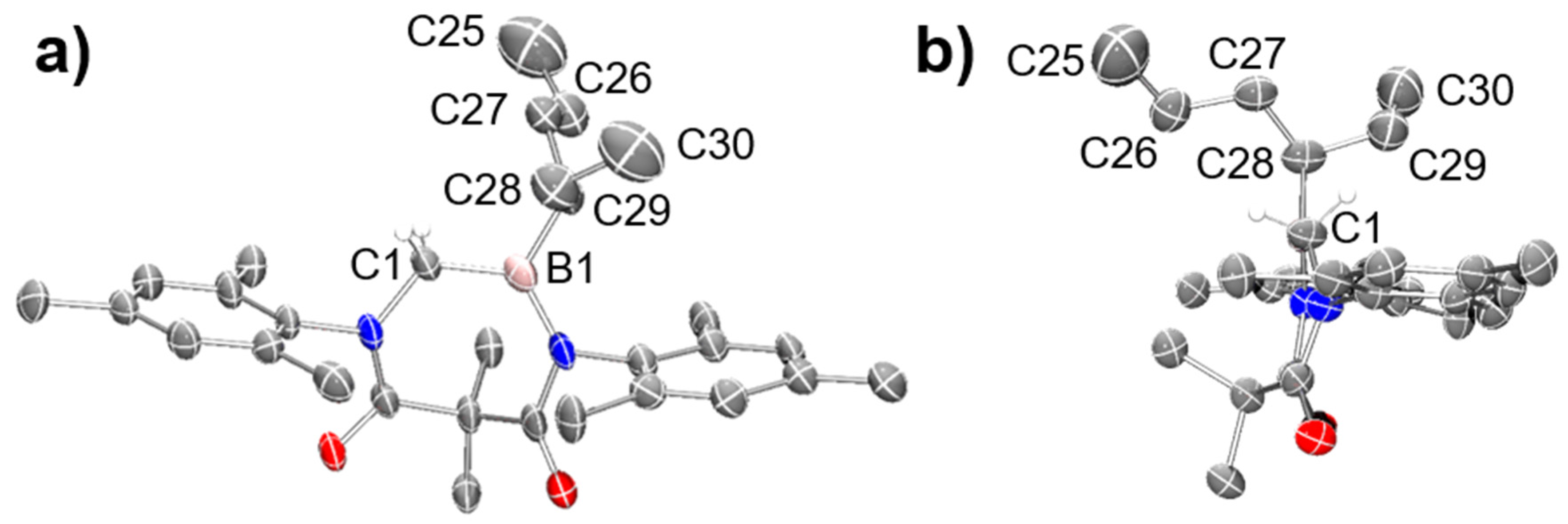
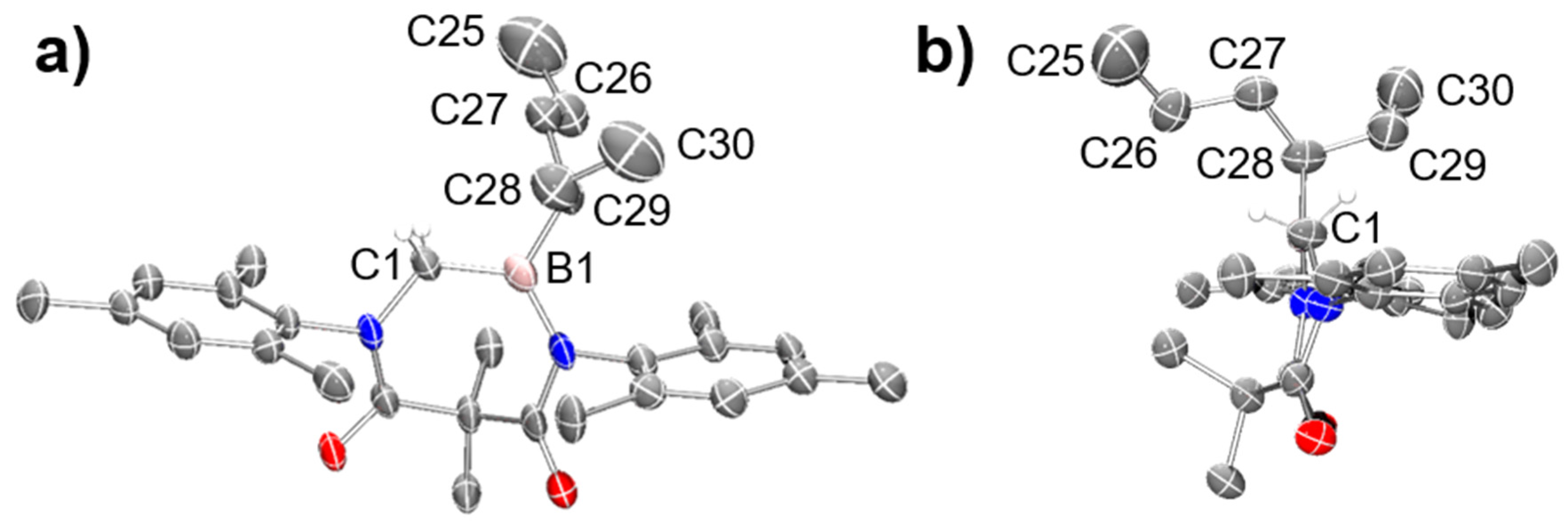
6). The structure of
10 was also unambiguously confirmed using single crystal X-ray crystallography (
Figure 1).
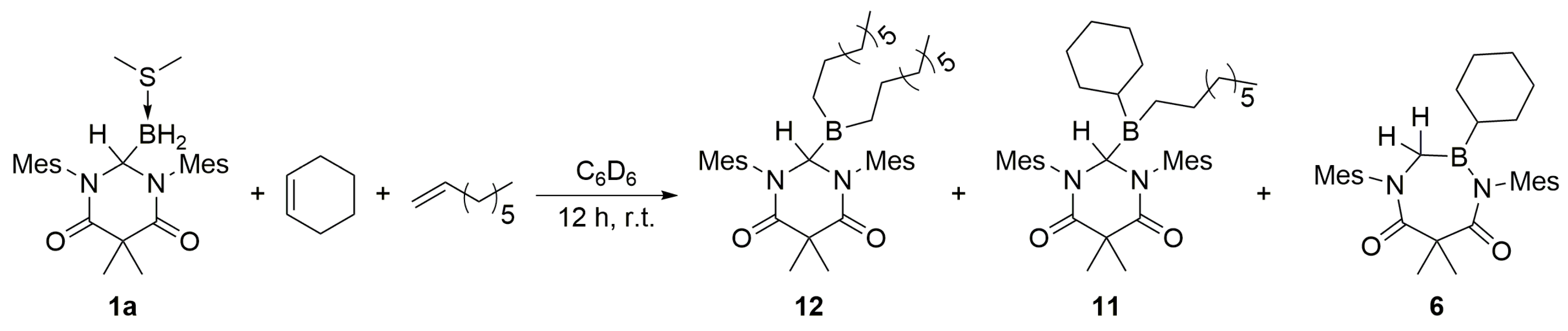
Finally, efforts were directed toward the determination of conditions which facilitate a stepwise hydroboration of an internal olefin and a terminal olefin to obtain the corresponding mixed product. Upon the initial addition of excess cyclohexene to a benzene solution of
1a, the ring-expanded compound
6 was observed as the exclusive product via
1H NMR spectroscopy analysis of the crude reaction mixture. No reaction was observed upon the subsequent addition of 1-octene. In a separate experiment, a stoichiometric mixture of cyclohexene and 1-octene was added in excess to a benzene solution of
1a. The product of this reaction was determined by
1H NMR spectroscopy and mass spectrometry to contain a nearly equimolar mixture of the mixed product
11, the ring-expanded product
6, and
12 [
29] (
Scheme 8). We surmise that after the initial hydroboration of cyclohexene, the respective product did not enable the hydroboration of 1-octene, but instead underwent intramolecular ring-expansion to yield
6. These results indicated that
1a facilitated the hydroboration of two olefins, but only when the first reaction was with a terminal olefin. Furthermore, a
1H NMR spectrum recorded after the addition of 0.5 equiv of 1-hexene to a CD
2Cl
2 solution of
1a exhibited singlets at δ 6.08 ppm and δ 5.69 ppm in a 1:3 ratio, which were assigned to the methine groups of
3 and residual
1a, respectively. Based on these results, we concluded that hydroboration preceded intramolecular ring-expansion.
3. Materials and Methods
3.1. General Information
All procedures were performed in a nitrogen-filled glove box unless otherwise noted. Solvents were dried and degassed by a Vacuum Atmospheres Company solvent purification system (Vacuum Atmosphere Co., Hawthorne, CA, USA) and stored over 4 Å molecular sieves in a nitrogen-filled glove box. Unless otherwise specified, reagents were purchased from commercial sources and used without further purification.
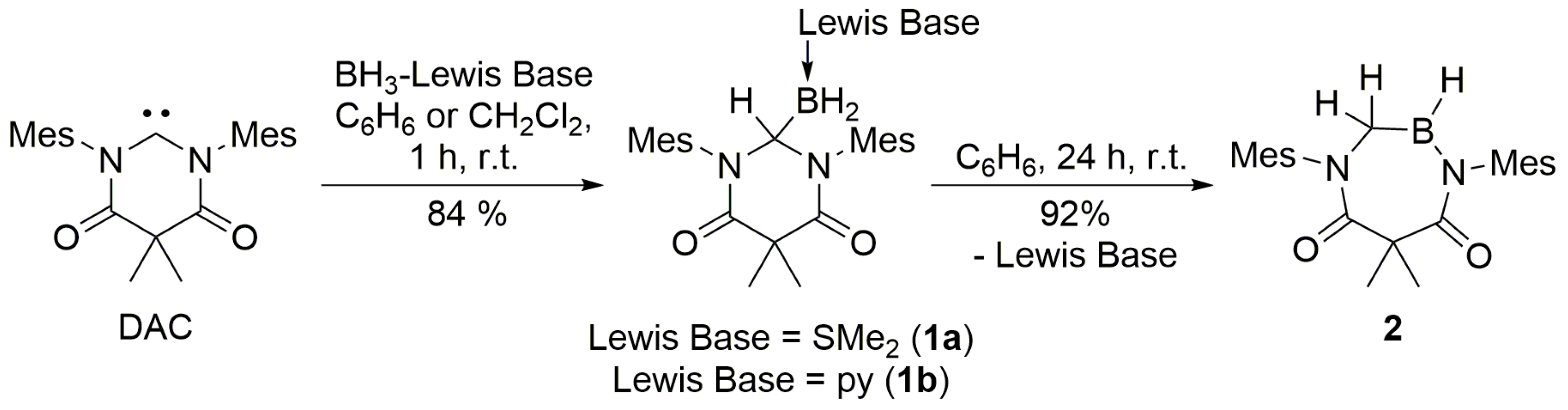
N,N’-dimesityl-4,6-diketo-5,5-dimethylpyrimidin-2-ylidene, as well as adducts
1a and
1b, were synthesized according to previously-reported procedures [
29,
30]. The hydroboration reactions described herein are unoptimized. Melting points were obtained using a MPA100 OptiMelt automated melting point apparatus (Stanford Research Systems Inc., Sunnyvale, CA, USA) and are uncorrected. NMR spectra were recorded on a MR 400, Inova 500, or DirectDrive 600 MHz spectrometer (Varian, Inc., Palo Alto, CA, USA), MR 400 MHz spectrometer (Agilent Technologies, Santa Clara, CA, USA) or an Ascend™ 400 MHz spectrometer (Bruker Co., Fällanden, Switzerland via Bruker Korea Co., Ltd., Seongnam-si, Korea). Chemical shifts (δ) are given in ppm and are referenced to the residual solvent (
1H: C
6D
6, 7.16 ppm;
13C: C
6D
6, 128.06 ppm). Linear predictions were applied to all
11B NMR spectra to remove the signals that corresponded to the boron found in the glass of the NMR tubes used in the corresponding experiments; the
11B NMR spectrum of C
6D
6 as a “blank” was also collected as a reference. Infrared (IR) spectra were recorded on a Nicolet iS5 system (Thermo Fisher Scientific, Waltham, MA, USA) equipped with an iD3 attenuated total reflectance (ATR) attachment (diamond crystal) or in a KBr pellet. High resolution mass spectra (HRMS) were obtained with a Autospec-Ultima mass spectrometer (Waters Co., Milford, MA, USA) using chemical ionization (CI) or a 6530 QTOF mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) using electrospray ionization (ESI). Low resolution mass spectra (LRMS) were obtained with a 6130 single quadrupole mass spectrometer equipped with an 1200 LC system (Agilent Technologies, Santa Clara, CA, USA). Elemental analyses were performed with a 2000 Organic Elemental Analyzer (Thermo Fisher Scientific, Waltham, MA, USA).
3.2. DAC–BH3 SMe2 + 1-Hexene to Obtain 3
An excess of 1-hexene (0.1 mL, 0.8056 mmol, 4.6 equiv.) was added dropwise to a stirred solution of 1a (79.6 mg, 0.1759 mmol) in dichloromethane (2.0 mL). The resulting mixture was stirred for 17 h at ambient temperature, whereupon the volatiles were removed under reduced pressure to afford 3 as a white solid (0.0961 g, 0.1720 mmol, 98% yield). mp = 120–123 °C. 1H NMR (C6D6, 499.86 MHz): δ 0.98 (br m, 26H), 1.59 (s, 3H), 2.01 (overlapping s, 9H), 2.08 (s, 6H), 2.41 (s, 6H), 6.09 (s, 1H), 6.58 (overlapping s, 2H), 6.66 (overlapping s, 2H). 13C NMR (C6D6, 125.70 MHz): δ 14.28, 19.45, 19.47, 20.78, 21.26, 22.84, 22.97, 24.34, 31.94, 48.40, 67.49, 129.76, 129.91, 135.24, 136.02, 138.01, 138.66, 172.26. 11B NMR (C6D6, 128.39 MHz): δ 78.42 (s). IR (KBr): ν = 3447.0, 2956.3, 2920.9, 2856.1, 1692.8, 1658.4, 1606.3, 1481.4, 1464.9, 1456.9, 1410.5, 1357.2, 1321.5, 1211.5, 1165.5 cm−1. HRMS (ESI): [M + H]+ calcd for C36H56N2O2B: 559.4435; found: 559.4442. Anal. Calcd for C36H55N2O2B: C, 77.40; H, 9.92; N, 5.01; found: C, 77.05; H, 10.11; N, 5.12.
3.3. DAC–BH3 SMe2 + 1,5-Hexadiene to Obtain 4
An excess of 1,5-hexadiene (ca. 0.03 mL) was added dropwise to a stirred solution of 1a (10.8 mg, 0.0237 mmol) in benzene (1.0 mL). The mixture was stirred for 20 h at ambient temperature, whereupon the volatiles were removed under reduced pressure to afford 4 as a white solid (0.0106 g, 0.0224 mmol, 95% yield). mp = 173–176 °C. 1H NMR (C6D6, 499.86 MHz): δ 0.41–1.07 (br m, 12H), 1.57 (t, J = 5.0 Hz, 3H), 2.00 (overlapping s, 9H), 2.07 (s, 6H), 2.38 (t, J = 4.5 Hz, 6H), 5.98 (s, 1H), 6.57 (s, 2H), 6.66 (s, 2H). 13C NMR (C6D6, 125.70 MHz): δ 19.16, 19.30, 19.40, 19.47, 20.77, 21.30, 22.88, 22.95, 32.69, 33.31, 48.36, 66.41, 129.49, 129.88, 135.15, 136.04, 138.06, 138.76, 172.24. 11B NMR (C6D6, 160.37 MHz): δ 78.98 (s). IR (KBr): ν = 3427.4, 2976.9, 2918.2, 2859.3, 1684.9, 1656.9, 1606.3, 1482.0, 1459.5, 1441.0, 1410.2, 1372.3, 1358.4, 1311.52, 1187.8, 1099.1, 1012.4, 849.6, 725.4, 569.5, 511.7 cm−1. HRMS (ESI): [M + H]+ calcd for C30H42N2O2B: 473.3339; found: 473.3345. Anal. Calcd for C30H41N2O2B: C, 76.26; H, 8.75; N, 5.93; found: C, 76.08; H, 8.39; N, 5.93.
3.4. DAC–BH3 SMe2 + Cyclohexene to Obtain 6
An excess of cyclohexene (ca. 0.05 mL) was added dropwise to a stirred solution of 1a (80.6 mg, 0.1781 mmol) in dichloromethane (1.0 mL). The mixture was stirred for 15 h at ambient temperature, whereupon the volatiles were removed under reduced pressure to afford 6 as a white solid (0.0780 g, 0.1651 mmol, 93% yield). mp = 81–84 °C. 1H NMR (C6D6, 499.86 MHz): δ 0.74 (br m, 4H), 1.32 (br m, 2H), 1.40 (br m, 4H), 1.77 (s, 6H), 2.05 (s, 6H), 2.09 (s, 3H), 2.13 (s, 3H), 2.16 (s, 6H), 3.61 (s, 2H), 6.76 (s, 2H), 6.81 (s, 2H). 13C NMR (C6D6, 125.70 MHz): δ 17.82, 19.06, 20.87, 20.92, 26.02, 26.41, 27.40, 29.39, 31.78, 47.39, 129.70, 129.87, 134.26, 134.41, 136.70, 136.84, 138.92, 142.24, 168.38, 176.76. 11B NMR (C6D6, 160.37 MHz): δ 53.35 (s). IR (KBr): ν = 3445.4, 2919.8, 2847.9, 2372.4, 2280.5, 1694.6, 1645.2, 1620.3, 1607.6, 1485.7, 1458.2, 1398.7, 1382.1, 1367.63, 1343.7, 1243.7, 1197.6, 1168.4, 1091.0, 755.9, 688.0 cm−1. HRMS (ESI): [M + H]+ calcd for C30H41N2O2B: 473.33390; found: 473.33470.
3.5. DAC–BH3 SMe2 + 1,4-Cyclohexadiene to Obtain 7
An excess of 1,4-cyclohexadiene (ca. 0.04 mL) was added to a stirred solution of 1a (20 mg, 0.0442 mmol) in dichloromethane (0.5 mL). The mixture was stirred for 30 min at ambient temperature, whereupon the volatiles were removed under reduced pressure to afford 7 as a white solid (0.0187g, 0.03975 mmol, 90% yield). mp = 153–159 °C. 1H NMR (C6D6, 499.86 MHz): δ 0.96 (s, 2H), 1.57 (s, 4H), 1.77 (s, 3H), 1.80 (s, 3H), 2.03 (s, 6H), 2.05 (s, 3H), 2.14 (s, 6H), 2.15 (s, 3H), 3.56 (dt, Jd = 35.00 Hz, Jt = 21.00 Hz, 2H), 5.43 (t, J = 1.5 Hz, 1H), 5.60 (m, 1H), 6.70 (m, 1H), 6.72 (m, 1H), 6.81 (s, 2H). 13C NMR (C6D6, 125.70 MHz): δ 17.80, 17.84, 19.02, 19.06, 20.84, 20.92, 25.34, 25.77, 26.00, 26.03, 26.07, 26.76, 27.43, 47.07, 124.61, 126.86, 129.68, 129.79, 129.85, 129.86, 134.20, 134.26, 134.28, 134.47, 136.80, 136.85, 138.77, 142.21, 168.31, 176.69. 11B NMR (C6D6, 160.37 MHz): δ 52.87 (s). IR (KBr): ν = 3016.7, 2916.6, 1700.13, 1683.9, 1668.4, 1652.5, 1506.1, 1484.0, 1457.2, 1398.0, 1367.9, 1344.8, 1305.3, 1256.5, 1235.3, 1207.5, 1164.1, 1095.3, 850.4 cm−1. HRMS (ESI): [M + H]+ calcd for C30H39N2O2B: 472.32130; found: 472.32120. Anal. Calcd for C30H39N2O2B: C, 76.59; H, 8.36; N, 5.95; found: C, 76.64; H, 8.32; N, 6.02.
3.6. DAC–BH3 SMe2 + 1,3-Cyclohexadiene to Obtain a Mixture of 7 + 8
An excess of 1,3-cyclohexadiene (ca. 0.04 mL) was added to a stirred solution of 1a (20 mg, 0.0442 mmol) in dichloromethane (0.5 mL). The mixture was stirred for 30 min at ambient temperature, whereupon the volatiles were removed under reduced pressure to afford a mixture of 7 + 8 as a white solid in quantitative yield. mp = 72–96 °C. 1H NMR (C6D6, 499.86 MHz): δ 0.91–1.70 (br m, 14H), 1.76 (s, 3H), 1.77 (s, 3H), 1.80 (overlapping s, 6H), 2.03 (s, 6H), 2.05 (s, 6H), 2.08 (overlapping s, 6H), 2.10 (s, 3H), 2.13 (s, 3H), 2.14 (s, 6H), 2.15 (s, 6H), 3.56 (dt, Jd = 34.49 Hz, Jt = 20.99 Hz, 2H), 3.73 (dt, Jd = 35.99 Hz, Jt = 14.50 Hz, 2H), 5.35 (m, 1H), 5.49 (m, 1H), 5.67 (m, 1H), 5.85 (m, 1H), 6.73 (m, 6H), 6.81 (s, 2H). 13C NMR (C6D6, 125.70 MHz): δ 17.78, 17.80, 17.84, 19.02, 19.03, 19.05, 19.13, 20.85, 20.87, 20.90, 20.92, 22.33, 22.59, 22.79, 22.93, 24.69, 25.22, 25.34, 25.40, 25.45, 25.77, 26.00, 26.03, 26.05, 26.71, 27.43, 29.40, 30.01, 47.07, 47.64, 53.34, 53.44, 124.73, 129.82, 129.91, 134.13, 134.20, 134.26, 134.28, 134.47, 134.50, 136.76, 136.80, 136.85, 136.86, 138.77, 138.91, 142.16, 142.20, 168.24, 168.31, 176.69, 176.78. 11B NMR (C6D6, 160.37 MHz): δ 52.81 (br s). IR (KBr): ν = 3014.0, 2920.1, 2856.7, 1700.2, 1652.5, 1484.0, 1464.9, 1397.7, 1367.2, 1305.6, 1239.8, 1164.0, 1095.3, 850.5 cm−1. HRMS (ESI): [M + H]+ calcd for C30H39N2O2B: 471.31830; found: 471.31870. Anal. Calcd for C30H39N2O2B: C, 76.59; H, 8.36; N, 5.95; found: C, 76.70; H, 8.39; N, 5.57.
3.7. DAC–BH3 SMe2 + cis-2-Hexene to Obtain a Mixture of 9 + 10
An excess of cis-2-hexene (ca. 0.05 mL) was added to a stirred solution of 1a (84.4 mg, 0.1865 mmol) in dichloromethane (2.0 mL). The mixture was stirred for 16 h at ambient temperature, whereupon the volatiles were removed under reduced pressure to afford products 9 and 10 as a white solid in 1:2 ratio (0.0765 g, 0.1612 mmol, 86% yield). mp = 60–63 °C. 1H NMR (C6D6, 499.86 MHz): δ 0.79 (m, 7H), 1.16 (m, 6H), 1.64 (s, 3H), 1.70 (s, 3H), 2.01 (m, 12H), 2.10 (s, 12H), 2.12 (s, 3H), 2.14 (s, 3H), 2.25 (m, 9H), 2.31 (m, 6H), 3.70 (m, 2H), 6.86–6.91 (s, 2H), 6.95 (s, 4H). 13C NMR (C6D6, 125.70 MHz): δ 14.03, 16.47, 17.80, 17.86, 19.01, 20.85, 20.90, 22.71, 22.96, 24.61, 24.73, 25.93, 26.01, 26.04, 26.11, 31.34, 33.45, 46.27, 53.35, 129.53, 129.60, 129.70, 129.88, 134.19, 134.22, 134.67, 136.74, 136.83, 138.76, 142.32, 168.37, 176.57. 11B NMR (C6D6, 160.37 MHz): δ 53.72 (br s). IR (KBr): ν = 3480.4, 2922.6, 2869.5, 2857.6, 1699.5, 1695.0, 1652.2, 1608.7, 1483.7, 1464.0, 1397.7, 1381.4, 1366.5, 1262.7, 1241.6, 1229.0, 1199.1, 1164.0 cm−1. HRMS (CI): [M + H]+ calcd for C30H44N2O2B: 475.3496; found: 475.3492. Anal. Calcd for C30H43N2O2B: C, 75.94; H, 9.13; N, 5.90; found: C, 75.83; H, 9.26; N, 5.76.
3.8. DAC–BH3 SMe2 + trans-2-Hexene to Obtain a Mixture of 9 + 10
An excess of trans-2-hexene (ca. 0.05 mL) was added to a stirred solution of 1a (89.4 mg, 0.1976 mmol) in dichloromethane (2.0 mL). The mixture was stirred for 3 h at ambient temperature, whereupon the volatiles were removed under reduced pressure to afford a mixture of 9 + 10 as a white solid (0.0860 g, 0.1812 mmol, 92% yield). mp = 64–67 °C. 1H NMR (C6D6, 499.86 MHz): δ 0.63 (m, 3H), 0.72 (t, J = 7.25 Hz, 3H), 0.79–1.18 (m, 7H), 1.79 (s, 6H), 2.07–2.12 (m, 12H), 2.16 (m, 3H), 3.66 (m, 2H), 6.76 (s, 2H), 6.79 (s, 2H). 13C NMR (C6D6, 125.70 MHz): δ 14.03, 16.47, 17.80, 17.86, 19.02, 20.85, 20.91, 22.71, 22.98, 24.61, 24.73, 25.94, 26.02, 26.04, 26.11, 31.34, 33.45, 46.27, 53.35, 129.53, 129.60, 129.70, 129.88, 134.20, 134.23, 134.67, 136.73, 136.82, 138.79, 142.34, 168.33, 176.58. 11B NMR (C6D6, 160.37 MHz): δ 53.72 (bs). IR (KBr): ν = 3480.4, 2954.6, 2922.6, 2869.5, 2857.6, 1699.5, 1695.0, 1652.2, 1608.7, 1483.7, 1464.0, 1397.7, 1381.4, 1366.5, 1262.7, 1241.6, 1229.0, 1199.1, 1164.0 cm−1. HRMS (CI): [M + H]+ calcd for C30H44N2O2B: 475.3496; found: 475.3503. Anal. Calcd for C30H43N2O2B: C, 75.94; H, 9.13; N, 5.90; found: C, 75.54; H, 9.08; N, 5.74.
3.9. DAC–BH3 SMe2 + cis-3-Hexene to Obtain 10
An excess of cis-3-hexene (ca. 0.05 mL) was added to a stirred solution of 1a (57.1 mg, 0.1262 mmol) in dichloromethane (2.0 mL). The mixture was stirred for 3 h at ambient temperature, whereupon the volatiles were removed under reduced pressure to afford 10 as a white solid (0.0581 g, 0.1224 mmol, 97% yield). Single crystals suitable for single crystal X-ray diffraction were grown using a slow evaporation of saturated benzene solution of product 10. mp = 60–64 °C. 1H NMR (C6D6, 499.86 MHz): δ 0.63 (m, 3H), 0.72 (t, J = 7.25 Hz, 3H), 0.80–1.17 (m, 7H), 1.80 (s, 6H), 2.08–2.12 (m, 12H), 2.16 (m, 3H), 3.67 (dt, Jd = 31.24 Hz, Jt = 20.99 Hz, 2H), 6.77 (s, 2H), 6.79 (s, 2H). 13C NMR (C6D6, 125.70 MHz): δ 14.02, 16.47, 17.81, 17.86, 19.01, 20.84, 20.90, 22.72, 22.98, 24.61, 24.73, 25.94, 26.01, 26.04, 26.11, 31.34, 33.46, 46.26, 53.36, 129.53, 129.60, 129.70, 129.88, 134.20, 134.23, 134.68, 136.73, 136.83, 138.80, 142.34, 168.36, 176.58. 11B NMR (C6D6, 160.37 MHz): δ 55.26 (br s). HRMS (CI): [M + H]+ calcd for C30H44N2O2B: 475.3496; found: 475.3498. Anal. Calcd for C30H43N2O2B: C, 75.94; H, 9.13; N, 5.90; found: C, 76.16; H, 9.10; N, 5.87.
3.10. DAC–BH3 SMe2 + trans-3-Hexene to Obtain 10
An excess of trans-3-hexene (ca. 0.04 mL) was added to a stirred solution of 1a (12.03 mg, 0.0266 mmol) in dichloromethane (2.0 mL). The mixture was stirred for 22 h at ambient temperature whereupon the volatiles were removed under reduced pressure to afford 10 as a white solid in quantitative yield. mp = 58–62 °C. 1H NMR (C6D6, 499.86 MHz): δ 0.63 (m, 3H), 0.72 (t, J = 7.25 Hz, 3H), 0.78–1.19 (m, 7H), 1.79 (s, 6H), 2.08–2.12 (m, 12H), 2.16 (m, 3H), 3.67 (dt, Jd = 31.24 Hz, Jt = 20.74 Hz, 2H), 6.76 (s, 2H), 6.79 (s, 2H). 13C NMR (C6D6, 125.70 MHz): δ 14.02, 16.46, 17.79, 17.84, 19.01, 20.85, 20.90, 22.71, 22.97, 24.60, 24.72, 25.92, 25.99, 26.02, 26.09, 31.33, 33.44, 46.28, 53.33, 129.57, 129.61, 129.71, 129.87, 134.15, 134.18, 134.65, 136.76, 136.86, 138.76, 142.27, 168.45, 176.53. 11B NMR (C6D6, 160.37 MHz): δ 53.71 (br s). HRMS (CI): [M + H]+ calcd for C30H44N2O2B: 475.3496; found: 475.3497. Anal. Calcd for C30H43N2O2B: C, 75.94; H, 9.13; N, 5.90; found: C, 76.24; H, 9.10; N, 5.79.
3.11. DAC–BH3 SMe2 + 1-Octene + Cyclohexene
An excess 1:1 molar mixture of 1-octene and cyclohexene (ca. 0.03 mL) was added dropwise to a stirred solution of 1a (10 mg, 0.0221 mmol) in dichloromethane (0.7 mL). The mixture was stirred for 12 h at ambient temperature, whereupon the crude reaction mixture was analyzed by 1H NMR spectrometry and low resolution mass spectrometry. LRMS for 6 (ESI): [M + H]+ calcd for C30H42N2O2B: 473.3; found: 473.3. LRMS for 11 (ESI): [M + H]+ calcd for C38H58N2O2B: 585.5; found: 585.5. LRMS for 12 (ESI): [M + H]+ calcd for C40H64N2O2B: 615.5; found: 615.5.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}