
Synthesis of New Chiral Benzimidazolylidene–Rh Complexes and Their Application in Asymmetric Addition Reactions of Organoboronic Acids to Aldehydes

Abstract
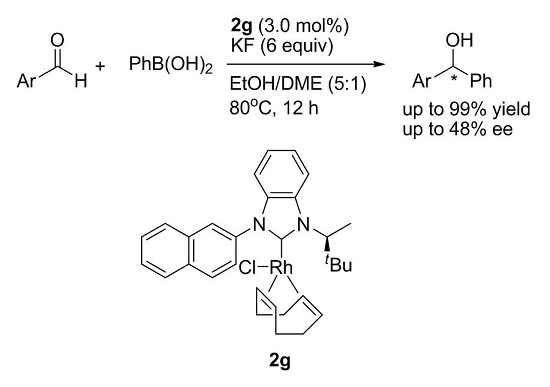
:
1. Introduction
2. Results
3. Materials and Methods
3.1. General
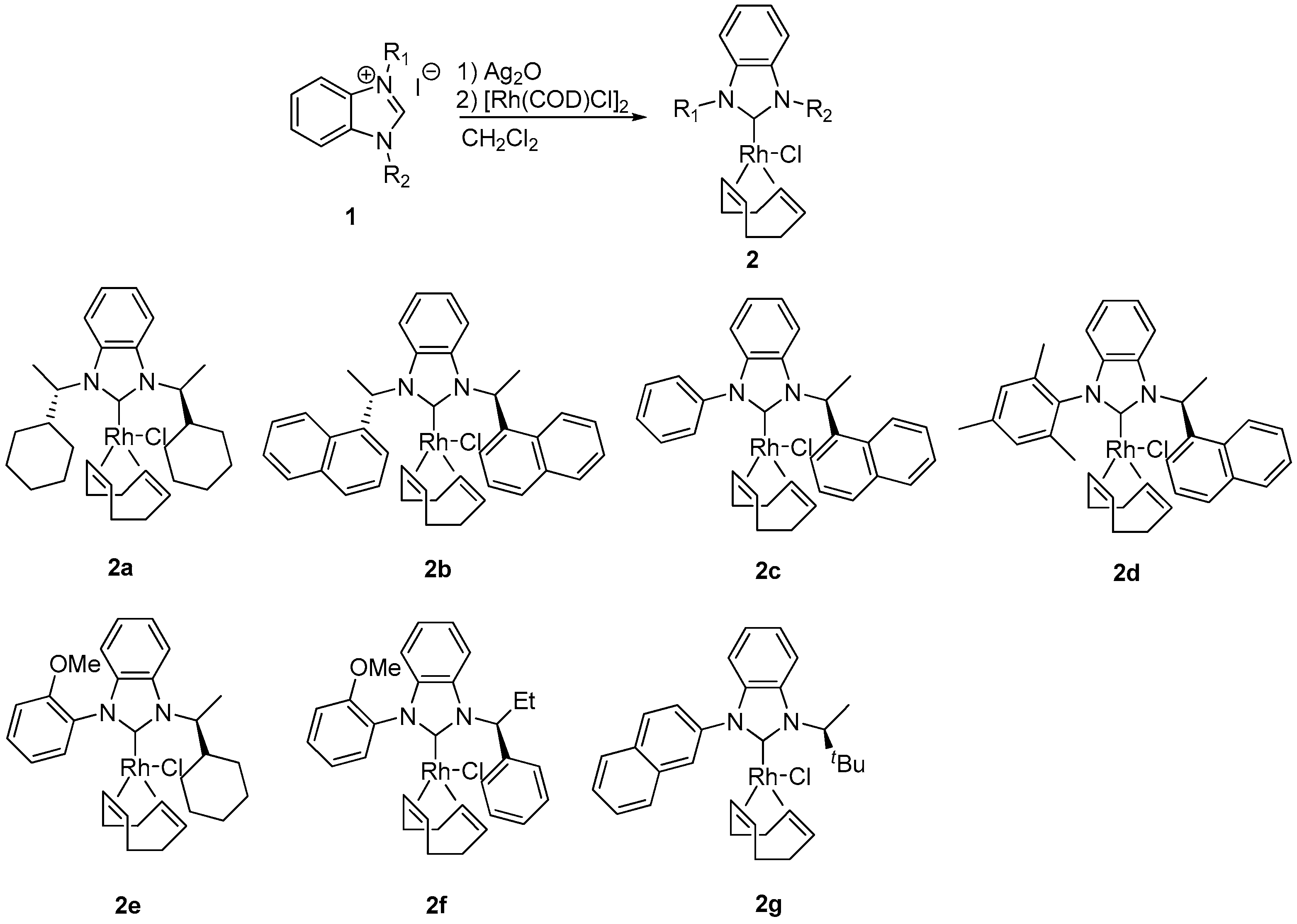
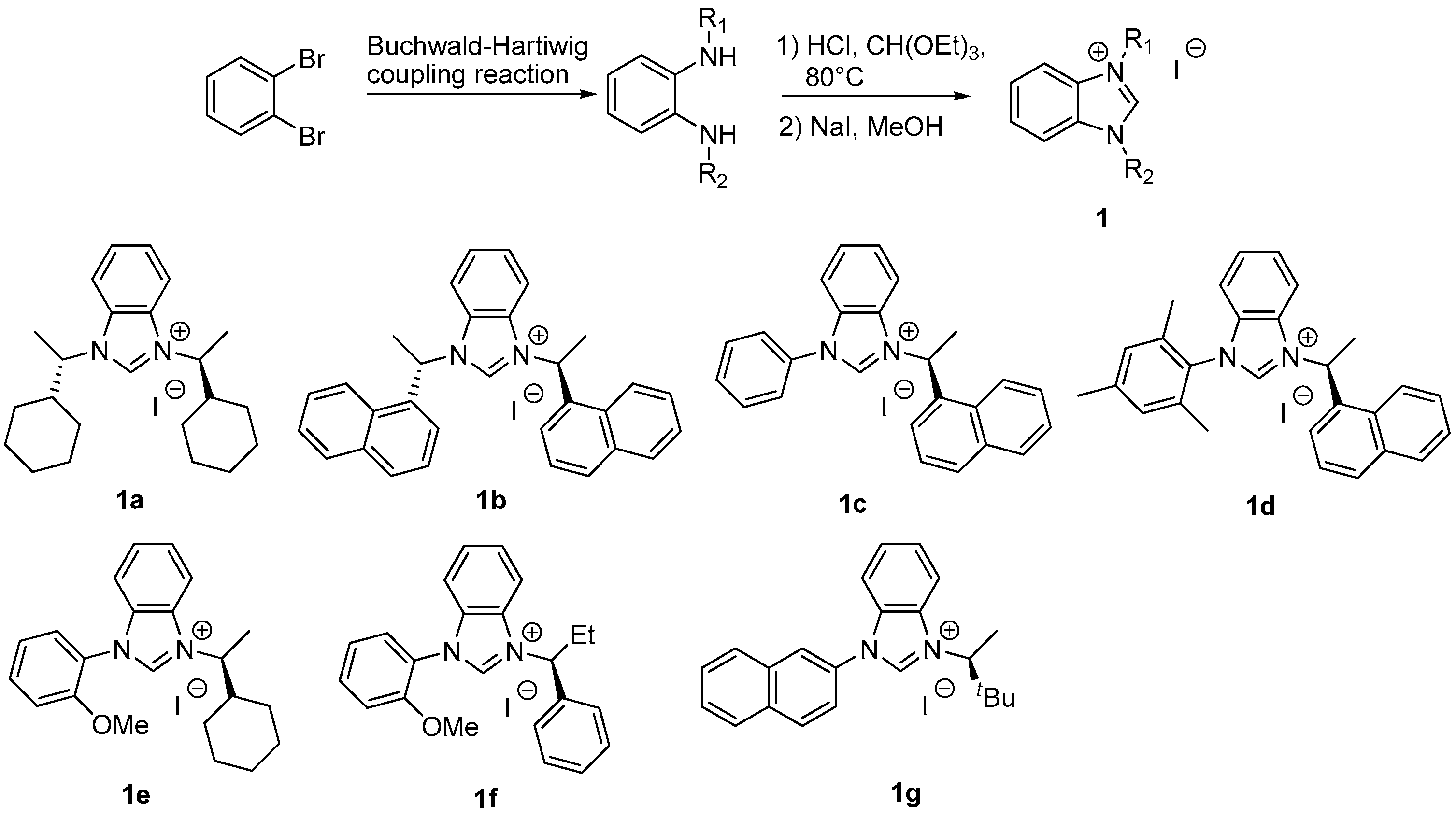
3.2. Preparation of Benzimidazolium Salt 1a–g
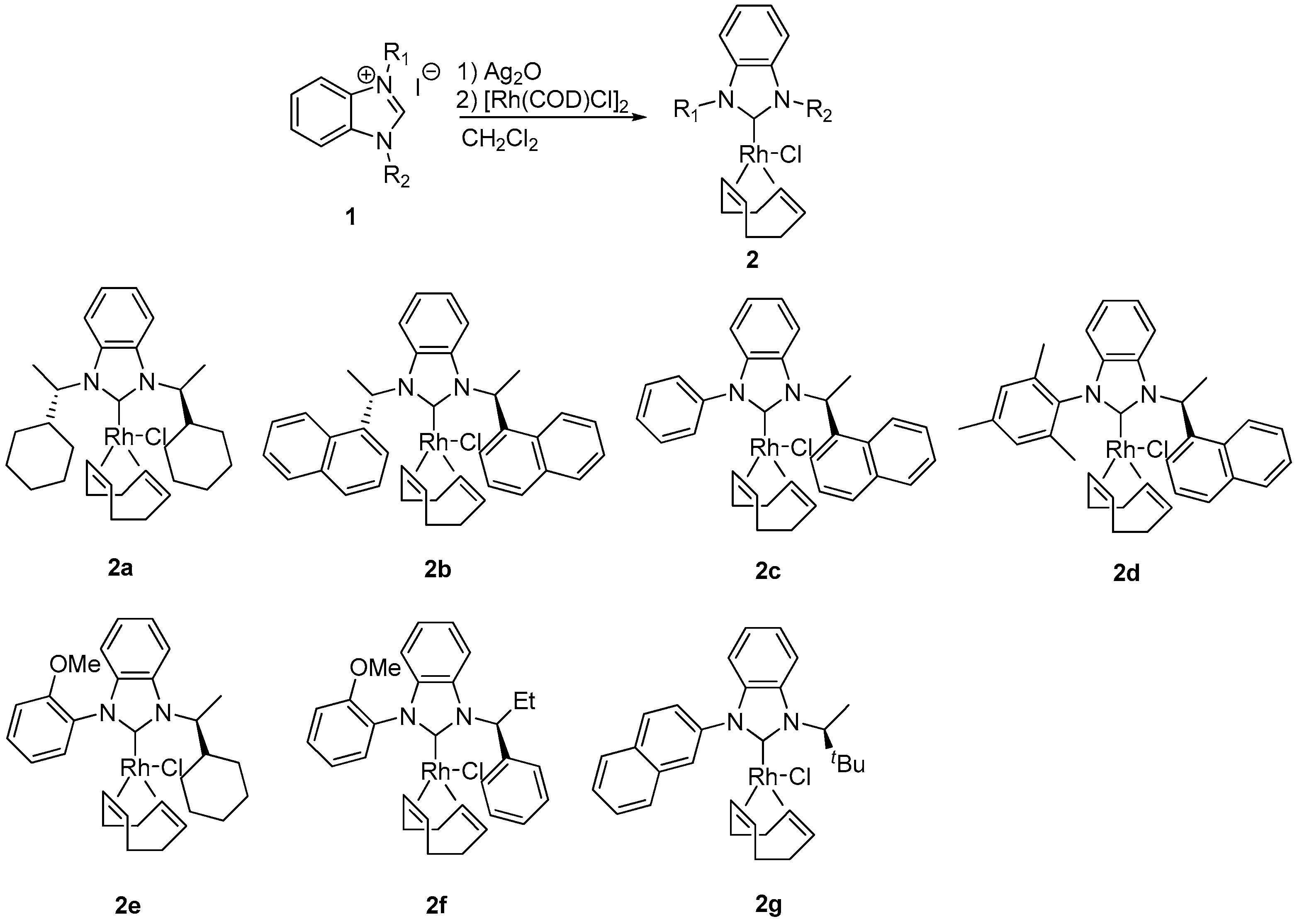
3.3. Preparation of NHC–Rh Complexes 2a–g
3.4. Representative Procedure for the Rh-Catalyzed Asymmetric Arylation of Aldehyde
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Herrmann, W.A.; Köcher, C. N-heterocyclic carbenes. Angew. Chem. Int. Ed. 1997, 36, 2162–2187. [Google Scholar] [CrossRef]
- Bourissou, D.; Guerret, O.; Gabbaï, F.P.; Bertrand, G. Stable carbenes. Chem. Rev. 2000, 100, 39–92. [Google Scholar] [CrossRef] [PubMed]
- Cavell, K.J.; McGuinness, D.S. Redox processes involving hydrocarbylmetal (N-heterocyclic carbene) complexes and associated imidazolium salts: Ramifications for catalysis. Coord. Chem. Rev. 2004, 248, 671–681. [Google Scholar] [CrossRef]
- Peris, E.; Crabtree, R.H. Recent homogeneous catalytic applications of chelate and pincer N-heterocyclic carbenes. Coord. Chem. Rev. 2004, 248, 2239–2246. [Google Scholar] [CrossRef]
- Crudden, C.M.; Allen, D.P. Stability and reactivity of N-heterocyclic carbene complexes. Coord. Chem. Rev. 2004, 248, 2247–2273. [Google Scholar] [CrossRef]
- Pugh, D.; Danopoulos, A.A. Metal complexes with ‘pincer’-type ligands incorporating N-heterocyclic carbene functionalities. Coord. Chem. Rev. 2007, 251, 610–641. [Google Scholar] [CrossRef]
- Mata, J.A.; Poyatos, M.; Peris, E. Structural and catalytic properties of chelating bis- and tris-N-heterocyclic carbenes. Coord.Chem. Rev. 2007, 251, 841–859. [Google Scholar] [CrossRef]
- Sommer, W.J.; Weck, M. Supported N-heterocyclic carbene complexes in catalysis. Coord. Chem. Rev. 2007, 251, 860–873. [Google Scholar] [CrossRef]
- Hahn, F.E.; Jahnke, M.C. Heterocyclic carbenes: Synthesis and coordination chemistry. Angew. Chem. Int. Ed. 2008, 47, 3122–3172. [Google Scholar] [CrossRef] [PubMed]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.C.; Burgess, K. Chiral N-heterocyclic carbene-transition metal complexes in asymmetric catalysis. Tetrahedron 2003, 14, 951–961. [Google Scholar] [CrossRef]
- César, V.; Bellemin-Laponnaz, S.; Gade, L.H. Chiral N-heterocyclic carbenes as stereodirecting ligands in asymmetric catalysis. Chem. Soc. Rev. 2004, 33, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Douthwaite, R.E. Metal-mediated asymmetric alkylation using chiral N-heterocyclic carbenes derived from chiral amines. Coord. Chem. Rev. 2007, 251, 702–717. [Google Scholar] [CrossRef]
- Gade, L.H.; Bellemin-Laponnaz, S. Mixed oxazoline-carbenes as stereodirecting ligands for asymmetric catalysis. Coord. Chem. Rev. 2007, 251, 718–725. [Google Scholar] [CrossRef]
- Snead, D.R.; Seo, H.; Hong, S. Recent developments of chiral diaminocarbene-metal complexes for asymmetric catalysis. Curr. Org. Chem. 2008, 12, 1370–1387. [Google Scholar] [CrossRef]
- Wang, F.J.; Liu, L.J.; Wang, W.F.; Li, S.K.; Shi, M. Chiral NHC–metal-based asymmetric catalysis. Coord. Chem. Rev. 2012, 256, 804–853. [Google Scholar] [CrossRef]
- Meguro, K.; Aizawa, M.; Sohda, T.; Kawamatsu, Y.; Nagaoka, A. New 1,4-dihydropyridine derivatives with potent and long-lasting hypotensive effect. Chem. Pharm. Bull. 1985, 33, 3787–3797. [Google Scholar] [CrossRef]
- Stanchev, S.; Rakovska, R.; Berova, N.; Snatzke, G. Synthesis, absolute configuration and circular dichroism of some diarylmethane derivatives. Tetrahedron 1995, 6, 183–198. [Google Scholar] [CrossRef]
- Bolshan, Y.; Chen, C.; Chilenski, J.R.; Gosselin, F.; Mathre, D.J.; O’Shea, P.D.; Roy, A.; Tillyer, R.D. Nucleophilic displacement at benzhydryl centers: Asymmetric synthesis of 1,1-diarylalkyl derivatives. Org. Lett. 2004, 6, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Radin, N.S. Sphingolipids as coenzymes in anion transfer and tumor death. Bioorganic Medicinal Chem. 2004, 12, 6029–6037. [Google Scholar] [CrossRef]
- Fang, L.; Changduo, P.; Jiang, C. Progress in the addition of arylboronic acids to aromatic aldehydes and ketone. Chin. J. Org. Chem. 2010, 30, 633–639. [Google Scholar]
- Sakai, M.; Ueda, M.; Miyaura, N. Rhodium-catalyzed addition of organoboronic acids to aldehydes. Angew. Chem. Int. Ed. 1998, 37, 3279–3281. [Google Scholar] [CrossRef]
- Shintani, R.; Inoue, M.; Hayashi, T. Rhodium-catalyzed asymmetric addition of aryl- and alkenylboronic acids to isatins. Angew. Chem. Int. Ed. 2006, 45, 3353–3356. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.F.; Xie, J.H.; Shi, W.J.; Zhang, Q.; Zhou, Q.L. Enantioselective rhodium-catalyzed addition of arylboronic acids to aldehydes using chiral spiro monophosphite ligands. Org. Lett. 2006, 8, 1479–1481. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.F.; Xie, J.H.; Qiao, X.C.; Wang, L.X.; Zhou, Q.L. Enantioselective rhodium-catalyzed addition ofarylboronic acids to α-ketoesters. Angew. Chem. Int. Ed. 2008, 47, 4351–4353. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kurihara, K.; Miyaura, N. Me-bipam for enantioselective ruthenium(II)-catalyzed arylation of aldehydes with arylboronic acids. Angew. Chem. Int. Ed. 2009, 121, 4478–4480. [Google Scholar] [CrossRef]
- Morikawa, S.; Michigami, K.; Amii, H. Novel axially chiral phosphine ligand with a fluoro alcohol moiety for Rh-catalyzed asymmetric arylation of aromatic aldehydes. Org. Lett. 2010, 12, 2520–2523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xu, Q.; Zhang, X.C.; Zhang, T.; Shi, M. Axially chiral C2-symmetric N-heterocyclic carbene (NHC) palladium complexes-catalyzed asymmetric arylation of aldehydes with arylboronic acids. Tetrahedron 2010, 21, 1928–1935. [Google Scholar] [CrossRef]
- Shintani, R.; Takatsu, K.; Hayashi, T. Copper-catalyzed asymmetric addition of arylboronates to isatins: a catalytic cycle involving alkox Copper intermediates. Chem. Commun. 2010, 46, 6822–6824. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gu, P.; Shi, M.; McDowell, P.; Li, G.G. Catalytic asymmetric addition of arylboronic acids to isatins using C2-symmetric cationic N-heterocyclic carbenes (NHCs) Pd2+ diaqua complexes as catalysts. Org. Lett. 2011, 13, 2314–2317. [Google Scholar] [CrossRef] [PubMed]
- Arao, T.; Sato, K.; Kondo, K.; Aoyama, T. Function of an N-heterocyclic carbene ligand based on concept of chiral mimetic. Chem. Pharm. Bull. 2006, 54, 1576–1581. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.S.; Ma, Y.D.; Liu, X.; Duan, W.Z.; Qu, B.; Song, C. Planar chiral imidazolium salts based on [2.2]paracyclophane in the asymmetric rhodium-catalyzed 1,2-addition of arylboronic acids to aldehydes. Tetrahedron 2010, 21, 292–298. [Google Scholar] [CrossRef]
- Duan, W.Z.; Ma, Y.D.; Qu, B.; Zhao, L.; Chen, J.Q.; Song, C. Synthesis of new alkoxy/sulfonate-substituted carbene precursors derived from [2.2]paracyclophane and their application in the asymmetric arylation of aldehydes. Tetrahedron 2012, 23, 1369–1375. [Google Scholar] [CrossRef]
- Duan, W.Z.; Ma, Y.D.; He, F.Y.; Zhao, L.; Chen, J.Q.; Song, C. Synthesis of novel planar chiral Ag and Rh N-heterocyclic carbine complexes derived from [2.2]paracyclophane and their application in ultrasound assisted asymmetric addition reactions of organoboronic acids to aldehydes. Tetrahedron 2013, 24, 241–248. [Google Scholar] [CrossRef]
- Chen, J.Q.; Yang, S.B.; Chen, Z.; Song, C.; Ma, Y.D. Synthesis of novel macrocyclic planar chiral carbene–Ag complexes derived from [2.2]paracyclophane for Rh-catalyzed asymmetric 1,2-additions of arylboronic acids to aromatic aldehydes. Tetrahedron 2015, 26, 288–295. [Google Scholar] [CrossRef]
- He, W.P.; Zhou, B.H.; Zhou, Y.L.; Li, X.R.; Fan, L.M.; Shou, H.W.; Li, J. Synthesis of new benzimidazolium salts and their application in the asymmetric arylation of aldehydes. Tetrahedron Lett. 2016, 57, 3152–3155. [Google Scholar] [CrossRef]
- He, W.P.; Zhao, W.; Zhou, B.H.; Liu, H.F.; Li, X.R.; Li, L.L.; Li, J.; Shi, J.Y. Synthesis of C2-symmetric benzimidazolium salts and their application in palladium-catalyzed enantioselective intramolecular α-arylation of amides. Molecules 2016, 21, 742. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.X.; Liu, Y.; Zhang, L.; Jia, Y.E.; Wang, P.; Zhuo, F.F.; An, X.T.; Da, C.S. Aryl bromides as inexpensive starting materials in the catalytic enantioselective arylation of aryl aldehydes: The additive TMEDA enhances the enantioselectivity. J. Org. Chem. 2014, 79, 10696–10702. [Google Scholar] [CrossRef] [PubMed]
- Caricato, M.; Sharma, A.K.; Coluccini, C.; Pasini, D. Nanostructuring with chirality: Binaphthyl-based synthons for the production of functional oriented nanomaterials. Nanoscale 2014, 6, 7165–7174. [Google Scholar] [CrossRef]
- Caricato, M.; Delforge, A.; Bonifazi, D.; Dondi, D.; Mazzanti, A.; Pasini, D. Chiral nanostructuring of multivalent macrocycles in solution and on surfaces. Org. Biomol. Chem. 2015, 13, 3593–3601. [Google Scholar] [CrossRef] [PubMed]


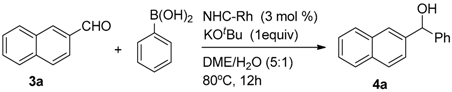
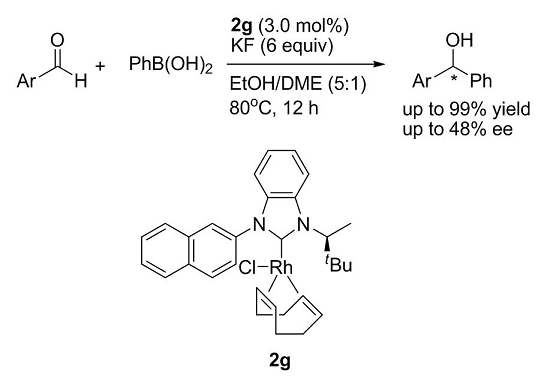
| Entry a | Ligand | Yield (%) b | ee (%) c |
|---|---|---|---|
| 1 | 2a | 99 | 3 |
| 2 | 2b | 99 | 1 |
| 3 | 2c | 99 | 6 |
| 4 | 2d | 98 | 17 |
| 5 | 2e | 99 | 3 |
| 6 | 2f | 98 | 3 |
| 7 | 2g | 99 | 18 |
| 8 | no catalyst | – | – |
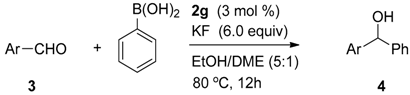
| Entry a | Base | Solvent | Temperature (°C) | Yield (%) b | ee (%) c |
|---|---|---|---|---|---|
| 1 | NaOtBu | DME/H2O (5:1) | 80 | 99 | 18 |
| 2 | LiOtBu | DME/H2O (5:1) | 80 | 42 | 29 |
| 3 | LiOH | DME/H2O (5:1) | 80 | 99 | 17 |
| 4 | KF (1 equiv.) | DME/H2O (5:1) | 80 | 70 | 32 |
| 5 | KF (3 equiv.) | DME/H2O (3:1) | 80 | 80 | 32 |
| 6 | KF (6 equiv.) | DME/H2O (5:1) | 80 | 99 | 32 |
| 7 | KF (6 equiv.) | DME/H2O (10:1) | 80 | 99 | 14 |
| 8 | KF (6 equiv.) | DME/H2O (3:1) | 80 | 99 | 18 |
| 9 | KF (6 equiv.) | Toluene/H2O (5:1) | 80 | 99 | 22 |
| 10 | KF (6 equiv.) | MeOH/DME (5:1) | 80 | 99 | 25 |
| 11 | KF (6 equiv.) | t-BuOH/MeOH (5:1) | 80 | 99 | 24 |
| 12 | KF (6 equiv.) | MeOH | 80 | 99 | 21 |
| 13 | KF (6 equiv.) | i-PrOH | 80 | 99 | 34 |
| 14 | KF (6 equiv.) | t-BuOH/EtOH (5:1) | 80 | 99 | 25 |
| 15 | KF (6 equiv.) | DME | 80 | 93 | 9 |
| 16 | KF (6 equiv.) | EtOH/DME (5:1) | 80 | 99 | 35 |
| 17 | KF (6 equiv.) | EtOH | 80 | 99 | 32 |
| 18 | KF (6 equiv.) | Dioxane | 80 | 94 | 17 |
| 19 | KF (6 equiv.) | i-PrOH/DME (5:1) | 80 | 99 | 34 |
| 20 | KF (6 equiv.) | EtOH/DME (5:1) | 50 | – | – |
| 21 | KF (6 equiv.) | i-PrOH | 50 | 43 | 33 |
| 22 | KF (6 equiv.) | i-PrOH/DME (5:1) | 50 | 47 | 36 |

| Entry a | Ar1 | Yield (%) b | ee (%) c |
|---|---|---|---|
| 1 | 1-Naphthyl 3b | 97 4b | 43 |
| 2 | 2-MeOPh 3c | 99 4c | 37 |
| 3 | 4-MeOPh 3d | 85 4d | 46 |
| 4 | 4-CF3Ph 3e | 94 4e | 40 |
| 5 | 3,4-DiMePh 3f | 99 4f | 28 |
| 6 | 4-EtPh 3g | 99 4g | 36 |
| 7 | 2-FPh 3h | 93 4h | 38 |
| 8 | 3,5-DiFPh 3i | 88 4i | 28 |
| 9 | 4-NO2Ph 3j | 94 4j | 28 |
| 10 | 2-thienyl 3k | 99 4k | 18 |
| 11 | 2-furyl 3l | 98 4l | 19 |
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, W.; Zhou, B.; Li, J.; Shi, J. Synthesis of New Chiral Benzimidazolylidene–Rh Complexes and Their Application in Asymmetric Addition Reactions of Organoboronic Acids to Aldehydes. Catalysts 2016, 6, 132. https://doi.org/10.3390/catal6090132
He W, Zhou B, Li J, Shi J. Synthesis of New Chiral Benzimidazolylidene–Rh Complexes and Their Application in Asymmetric Addition Reactions of Organoboronic Acids to Aldehydes. Catalysts. 2016; 6(9):132. https://doi.org/10.3390/catal6090132
Chicago/Turabian StyleHe, Weiping, Bihui Zhou, Jie Li, and Jianyou Shi. 2016. "Synthesis of New Chiral Benzimidazolylidene–Rh Complexes and Their Application in Asymmetric Addition Reactions of Organoboronic Acids to Aldehydes" Catalysts 6, no. 9: 132. https://doi.org/10.3390/catal6090132
APA StyleHe, W., Zhou, B., Li, J., & Shi, J. (2016). Synthesis of New Chiral Benzimidazolylidene–Rh Complexes and Their Application in Asymmetric Addition Reactions of Organoboronic Acids to Aldehydes. Catalysts, 6(9), 132. https://doi.org/10.3390/catal6090132