
Mechanism-Guided Discovery of an Esterase Scaffold with Promiscuous Amidase Activity †


Abstract
:
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Sequence/Plasmid
4.2. Strains and Growth Media
4.3. Mutagenesis
4.4. Protein Expression
4.5. Protein Extraction and Purification
4.6. Size Exclusion Chromatography
4.7. Active Site Titration
4.8. Enzyme Activity and Kinetic Data
4.9. Molecular Dynamics Simulations
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DFT | density functional theory |
| SEC | size exclusion chromatography |
| TS | transition state |
| MD | molecular dynamics |
Appendix
A. Codon optimized sequence of patatin with an N-terminal His6-tag
B. The patatin protein sequence used in the present work
References
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Reetz, M.T. Biocatalysis in organic chemistry and biotechnology: Past, present, and future. J. Am. Chem. Soc. 2013, 135, 12480–12496. [Google Scholar] [CrossRef] [PubMed]
- Both, P.; Busch, H.; Kelly, P.P.; Mutti, F.G.; Turner, N.J.; Flitsch, S.L. Whole-cell biocatalysts for stereoselective C–H amination reactions. Angew. Chem. Int. Ed. 2016, 55, 1511–1513. [Google Scholar] [CrossRef] [PubMed]
- Siegel, J.B.; Smith, A.L.; Poust, S.; Wargacki, A.J.; Bar-Even, A.; Louw, C.; Shen, B.W.; Eiben, C.B.; Tran, H.M.; Noor, E.; et al. Computational protein design enables a novel one-carbon assimilation pathway. Proc. Natl. Acad. Sci. USA 2015, 112, 3704–3709. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.R.; Lohman, D.C.; Wolfenden, R. Catalytic proficiency: The extreme case of S–O cleaving sulfatases. J. Am. Chem. Soc. 2012, 134, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Qin, X.; Liu, J.; Gui, C.; Wang, B.; Li, J.; Ju, J. Deciphering the biosynthetic origin of l-allo-isoleucine. J. Am. Chem. Soc. 2016, 138, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Peter, D.M.; Schada von Borzyskowski, L.; Kiefer, P.; Christen, P.; Vorholt, J.A.; Erb, T.J. Screening and engineering the synthetic potential of carboxylating reductases from central metabolism and polyketide biosynthesis. Angew. Chem. Int. Ed. 2015, 54, 13457–13461. [Google Scholar] [CrossRef] [PubMed]
- Nestl, B.M.; Hammer, S.C.; Nebel, B.A.; Hauer, B. New generation of biocatalysts for organic synthesis. Angew. Chem. Int. Ed. 2014, 53, 3070–3095. [Google Scholar] [CrossRef] [PubMed]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Syren, P.-O.; Lindgren, E.; Hoeffken, H.W.; Branneby, C.; Maurer, S.; Hauer, B.; Hult, K. Increased activity of enzymatic transacylation of acrylates through rational design of lipases. J. Mol. Catal. B Enzym. 2010, 65, 3–10. [Google Scholar] [CrossRef]
- Schmidt, S.; Scherkus, C.; Muschiol, J.; Menyes, U.; Winkler, T.; Hummel, W.; Groeger, H.; Liese, A.; Herz, H.-G.; Bornscheuer, U.T. An enzyme cascade synthesis of ε-caprolactone and its oligomers. Angew. Chem. Int. Ed. 2015, 54, 2784–2787. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.C.Y.; Keasling, J.D. Production of isoprenoid pharmaceuticals by engineered microbes. Nat. Chem. Biol. 2006, 2, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Coelho, P.S.; Wang, Z.J.; Ener, M.E.; Baril, S.A.; Kannan, A.; Arnold, F.H.; Brustad, E.M. A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins in vivo. Nat. Chem. Biol. 2013, 9, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Renata, H.; Wang, Z.J.; Arnold, F.H. Expanding the enzyme universe: Accessing non-natural reactions by mechanism-guided directed evolution. Angew. Chem. Int. Ed. 2015, 54, 3351–3367. [Google Scholar] [CrossRef] [PubMed]
- Seitz, M.; Syren, P.-O.; Steiner, L.; Klebensberger, J.; Nestl, B.M.; Hauer, B. Synthesis of heterocyclic terpenoids by promiscuous squalene-hopene cyclases. ChemBioChem 2013, 14, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Khersonsky, O.; Tawfik, D.S. Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu. Rev. Biochem. 2010, 79, 471–505. [Google Scholar] [PubMed]
- Hult, K.; Berglund, P. Enzyme promiscuity: Mechanism and applications. Trends Biotechnol. 2007, 25, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Ollis, D.L.; Cheah, E.; Cygler, M.; Dijkstra, B.; Frolow, F.; Franken, S.M.; Harel, M.; Remington, S.J.; Silman, I.; Schrag, J.; et al. The alpha/beta hydrolase fold. Protein Eng. 1992, 5, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Rauwerdink, A.; Kazlauskas, R.J. How the same core catalytic machinery catalyzes 17 different reactions: The serine-histidine-aspartate catalytic triad of α/β-hydrolase fold enzymes. ACS Catal. 2015, 5, 6153–6176. [Google Scholar] [CrossRef]
- Brieke, C.; Peschke, M.; Haslinger, K.; Cryle, M.J. Sequential in vitro cyclization by cytochrome P450 enzymes of glycopeptide antibiotic precursors bearing the x-domain from nonribosomal peptide biosynthesis. Angew. Chem. Int. Ed. 2015, 54, 15715–15719. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.L.; Crock, J.; Bohlmann, J.; Croteau, R. Sesquiterpene synthases from grand fir (Abies grandis). Comparison of constitutive and wound-induced activities, and cdna isolation, characterization, and bacterial expression of δ-selinene synthase and γ-humulene synthase. J. Biol. Chem. 1998, 273, 2078–2089. [Google Scholar] [CrossRef] [PubMed]
- Steinkeller, G.; Gruber, C.C.; Pavkov-Keller, T.; Binter, A.; Steiner, K.; Winkler, C.; Łyskowski, A.; Schwamberger, O.; Oberer, M.; Schwab, H.; et al. Identification of promiscuous ene-reductase activity by mining structural databases using active site constellations. Nat. Commun. 2014, 5, 4150. [Google Scholar] [CrossRef]
- Höhne, M.; Schätzle, S.; Jochens, H.; Robins, K.; Bornscheuer, U.T. Rational assignment of key motifs for function guides in silico enzyme identification. Nat. Chem. Biol. 2010, 6, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Schellenberger, V.; Braune, K.; Hofmann, H.-J.; Jakubke, H.-D. The specificity of chymotrypsin. Eur. J. Biochem. 1991, 199, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Syren, P.-O.; Hult, K. Amidases have a hydrogen bond that facilitates nitrogen inversion, but esterases have not. ChemCatChem 2011, 3, 853–860. [Google Scholar] [CrossRef]
- Syren, P.-O. The solution of nitrogen inversion in amidases. FEBS J. 2013, 280, 3069–3083. [Google Scholar] [CrossRef] [PubMed]
- Ekici, O.D.; Paetzel, M.; Dalbey, R.E. Unconventional serine proteases: Variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 2008, 17, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Hendil-Forssell, P.; Martinelle, M.; Syren, P.-O. Exploring water as building bricks in enzyme engineering. Chem. Commun. 2015, 51, 17221–17224. [Google Scholar] [CrossRef] [PubMed]
- Devamani, T.; Rauwerdink, A.M.; Lunzer, M.; Jones, B.J.; Mooney, J.L.; Tan, M.A.O.; Zhang, Z.-J.; Xu, J.-H.; Dean, A.M.; Kazlauskas, R.J. Catalytic promiscuity of ancestral esterases and hydroxynitrile lyases. J. Am. Chem. Soc. 2016, 138, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.J.; Thornton, J.W. Analyzing protein structure and function using ancestral gene reconstruction. Curr. Opin. Struct. Biol. 2010, 20, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Risso, V.A.; Gavira, J.A.; Mejia-Carmona, D.F.; Gaucher, E.A.; Sanchez-Ruiz, J.M. Hyperstability and substrate promiscuity in laboratory resurrections of precambrian β-lactamases. J. Am. Chem. Soc. 2013, 135, 2899–2902. [Google Scholar] [CrossRef] [PubMed]
- Rydel, T.J.; Williams, J.M.; Krieger, E.; Moshiri, F.; Stallings, W.C.; Brown, S.M.; Pershing, J.C.; Purcell, J.P.; Alibhai, M.F. The crystal structure, mutagenesis, and activity studies reveal that patatin is a lipid acyl hydrolase with a Ser-Asp catalytic dyad. Biochemistry 2003, 42, 6696–6708. [Google Scholar] [CrossRef] [PubMed]
- Syren, P.-O.; Hendil-Forssell, P.; Aumailley, L.; Besenmatter, W.; Gounine, F.; Svendsen, A.; Martinelle, M.; Hult, K. Esterases with an introduced amidase-like hydrogen bond in the transition state have increased amidase specificity. ChemBioChem 2012, 13, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Syren, P.-O.; Le Joubioux, F.; Ben Henda, Y.; Maugard, T.; Hult, K.; Graber, M. Proton shuttle mechanism in the transition state of lipase-catalyzed n-acylation of amino alcohols. ChemCatChem 2013, 5, 1842–1853. [Google Scholar] [CrossRef]
- Deslongchamps, P. Stereoelectronic control in the cleavage of tetrahedral intermediates in the hydrolysis of esters and amides. Tetrahedron 1975, 31, 2463–2490. [Google Scholar] [CrossRef]
- Bachovchin, D.A.; Cravatt, B.F. The pharmacological landscape and therapeutic potential of serine hydrolases. Nat. Rev. Drug Discov. 2012, 11, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Dessen, A.; Tang, J.; Schmidt, H.; Stahl, M.; Clark, J.D.; Seehra, J.; Somers, W.S. Crystal structure of human cytosolic phospholipase A2 reveals a novel topology and catalytic mechanism. Cell 1999, 97, 349–360. [Google Scholar] [CrossRef]
- Hackenschmidt, S.; Moldenhauer, E.J.; Behrens, G.A.; Gand, M.; Pavlidis, I.V.; Bornscheuer, U.T. Enhancement of promiscuous amidase activity of a bacillus subtilis esterase by formation of a π-π network. ChemCatChem 2014, 6, 1015–1020. [Google Scholar] [CrossRef]
- Lenski, R.E.; Wiser, M.J.; Ribeck, N.; Blount, Z.D.; Nahum, J.R.; Morris, J.J.; Zaman, L.; Turner, C.B.; Wade, B.D.; Maddamsetti, R.; et al. Sustained fitness gains and variability in fitness trajectories in the long-term evolution experiment with escherichia coli. Proc. R. Soc. B 2015, 282, 20152292. [Google Scholar] [CrossRef] [PubMed]
- Keeling, C.I.; Weisshaar, S.; Ralph, S.G.; Jancsik, S.; Hamberger, B.; Dullat, H.K.; Bohlmann, J. Transcriptome mining, functional characterization, and phylogeny of a large terpene synthase gene family in spruce (Picea spp.). BMC Plant Biol. 2011, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Powles, S.B.; Yu, Q. Evolution in action: Plants resistant to herbicides. Annu. Rev. Plant Biol. 2010, 61, 317–347. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.E.; Marchetti, R.V.; Cowan, A.I.; Howitt, S.M.; Broeer, S.; Kirk, K. Chloroquine transport via the malaria parasite’s chloroquine resistance transporter. Science 2009, 325, 1680–1682. [Google Scholar] [CrossRef] [PubMed]
- Hammer, S.C.; Marjanovic, A.; Dominicus, J.M.; Nestl, B.M.; Hauer, B. Squalene hopene cyclases are protonases for stereoselective bronsted acid catalysis. Nat. Chem. Biol. 2015, 11, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Constable, D.J.C.; Dunn, P.J.; Hayler, J.D.; Humphrey, G.R.; Leazer, J.L., Jr.; Linderman, R.J.; Lorenz, K.; Manley, J.; Pearlman, B.A.; Wells, A.; et al. Key green chemistry research Areas-A perspective from pharmaceutical manufacturers. Green Chem. 2007, 9, 411–420. [Google Scholar] [CrossRef]
- Thayer, A.M. Making peptides at large scale. Chem. Eng. News 2011, 89, 21–25. [Google Scholar]
- Ku, B.; Lee, K.-H.; Park, W.S.; Yang, C.-S.; Ge, J.; Lee, S.-G.; Cha, S.-S.; Shao, F.; Heo, W.D.; Jung, J.U.; et al. Vipd of legionella pneumophila targets activated Rab5 and Rab22 to interfere with endosomal trafficking in macrophages. PLoS Pathog. 2012, 8, e1003082. [Google Scholar] [CrossRef] [PubMed]
- Bergeret, F.; Gavalda, S.; Chalut, C.; Malaga, W.; Quemard, A.; Pedelacq, J.-D.; Daffe, M.; Guilhot, C.; Mourey, L.; Bon, C. Biochemical and structural study of the atypical acyltransferase domain from the mycobacterial polyketide synthase Pks13. J. Biol. Chem. 2012, 287, 33675–33690. [Google Scholar] [CrossRef] [PubMed]
- Kourist, R.; Bartsch, S.; Fransson, L.; Hult, K.; Bornscheuer, U.T. Understanding promiscuous amidase activity of an esterase from bacillus subtilis. ChemBioChem 2008, 9, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, L. Serine protease mechanism and specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Magnusson, A.O.; Rotticci-Mulder, J.C.; Santagostino, A.; Hult, K. Creating space for large secondary alcohols by rational redesign of candida antarctica lipase B. ChemBioChem 2005, 6, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Janes, L.E. Screening Methods to Identify Stereoselective Hydrolases for Synthetic Applications. Ph.D. Thesis, Department of Chemistry, McGill University, Montreal, QC, Canada, 1998. [Google Scholar]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins Struct. Funct. Bioinform. 2004, 57, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Wijeyesakere, S.J.; Richardson, R.J.; Stuckey, J.A. Crystal Structure of Patatin-17 in Complex with Aged and Non-Aged Organophosphorus Compounds. PLoS ONE 2014, 9, e108245. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.E.M.; Vriend, G. Fast empirical Pka prediction by ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]

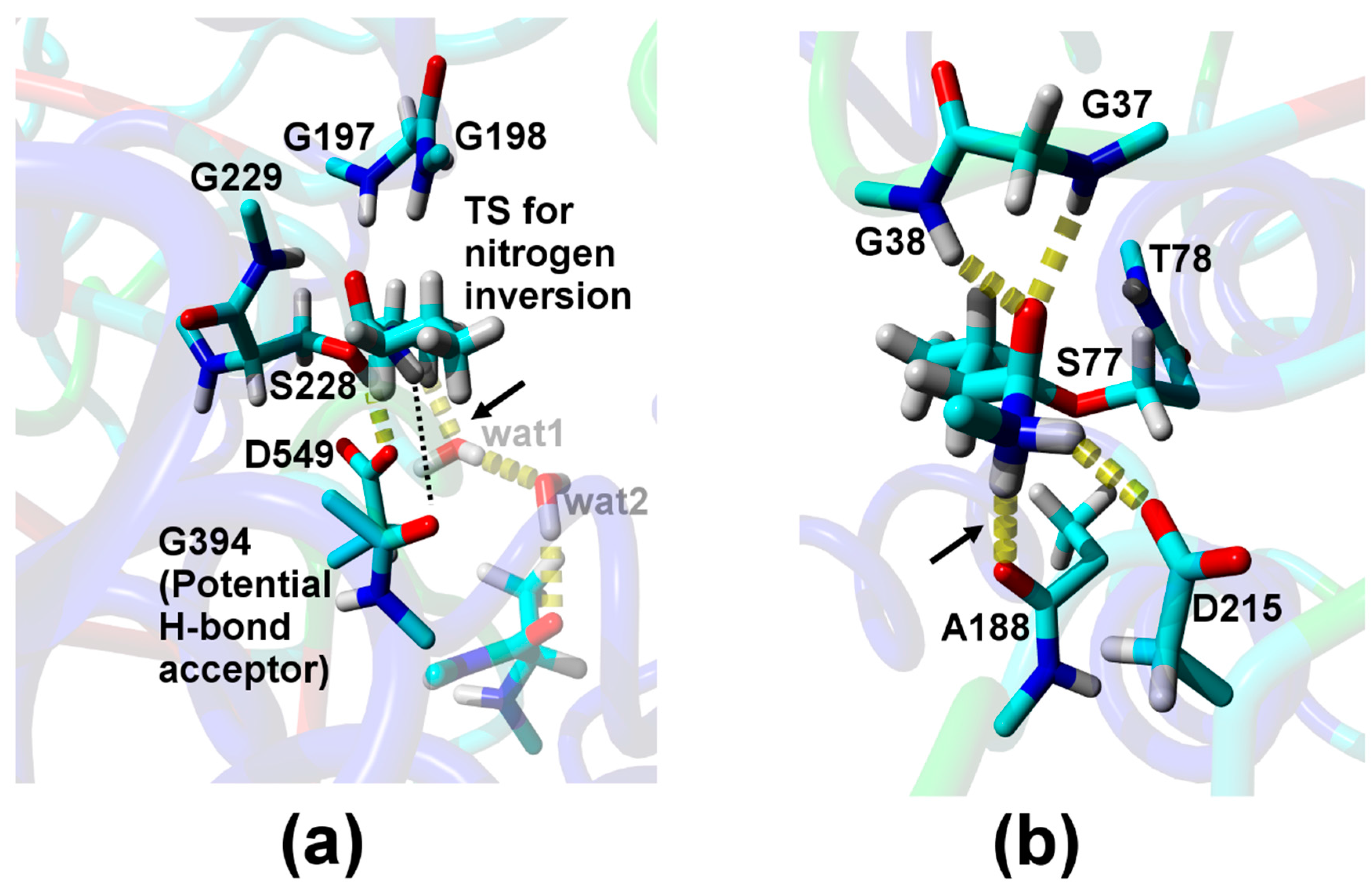

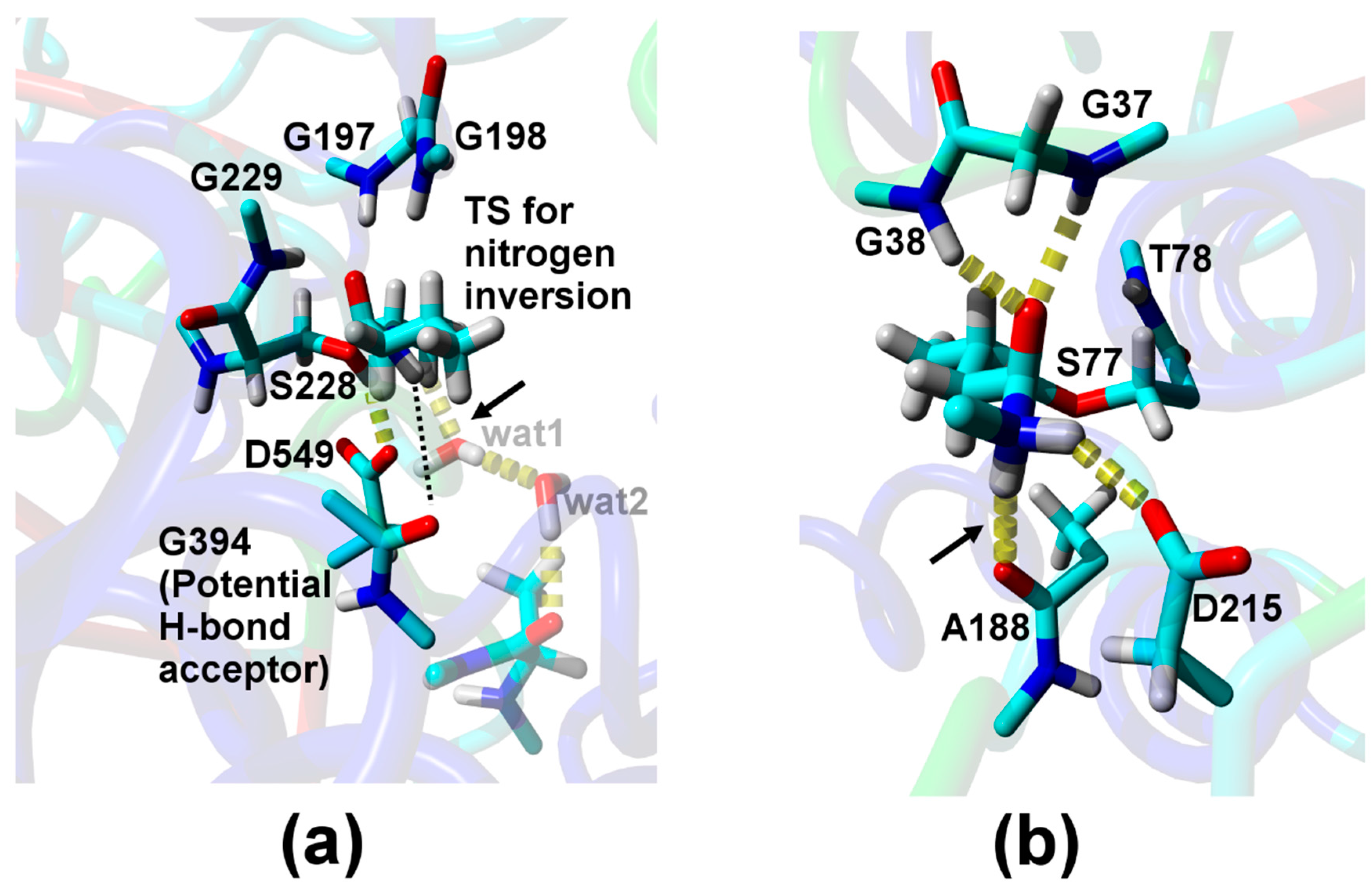
{kind=link}
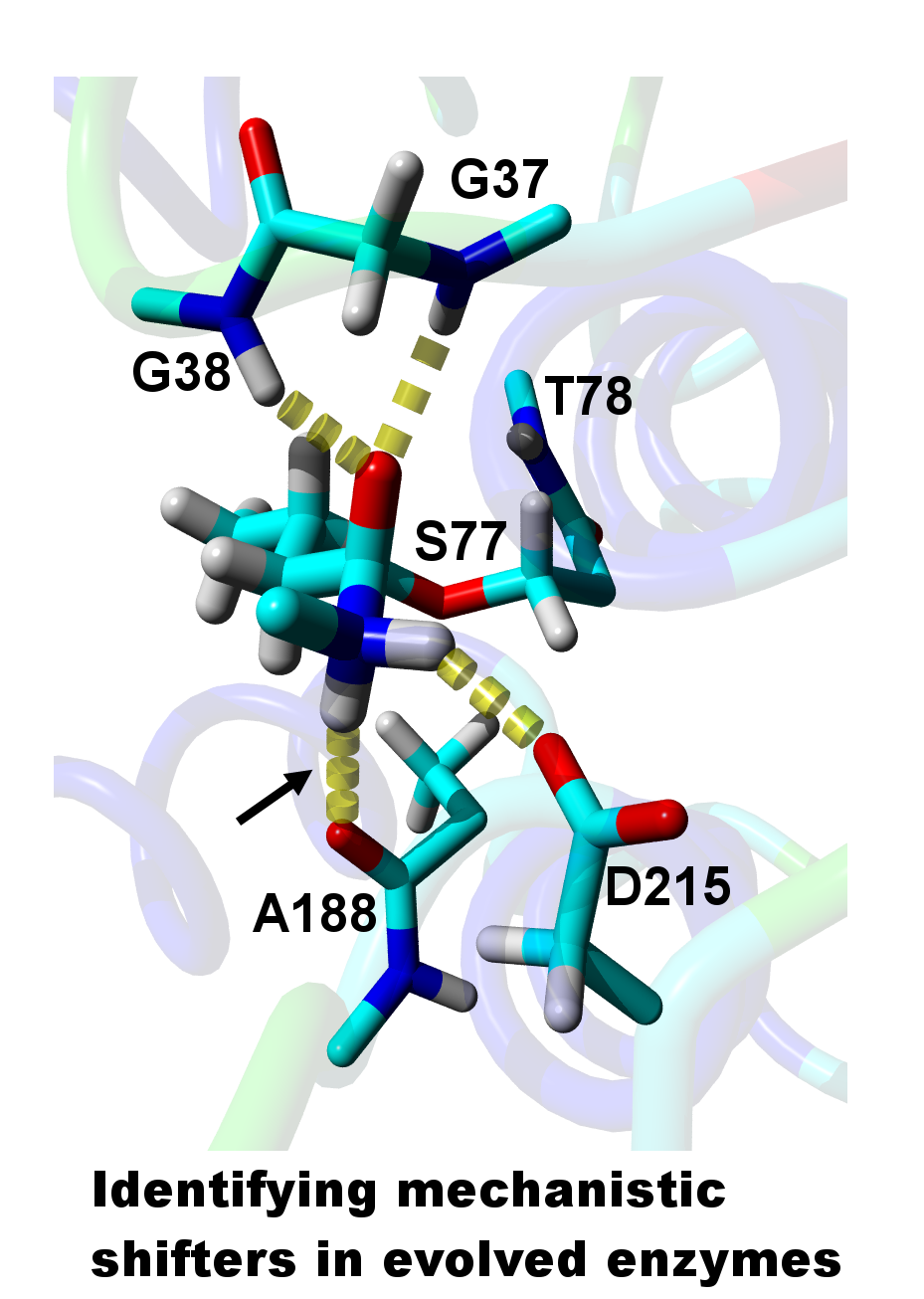
{kind=link}
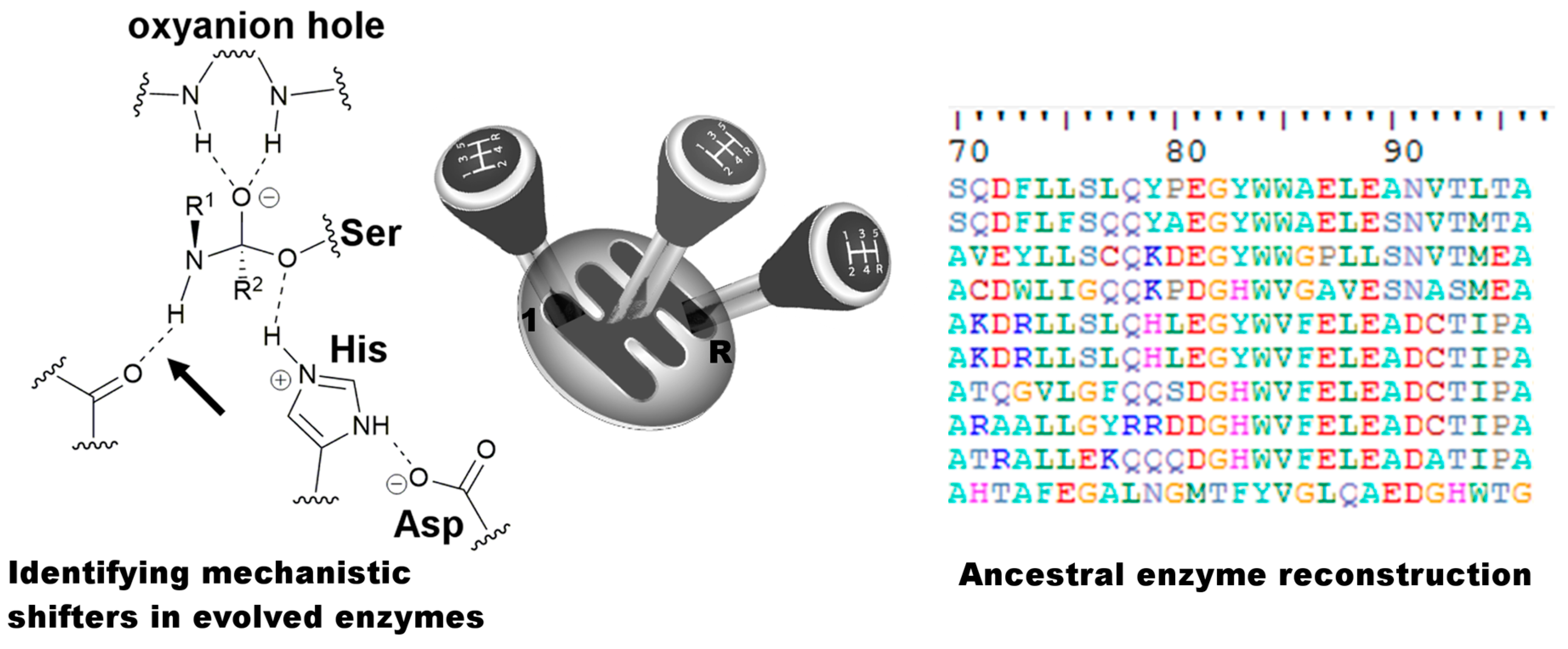
{kind=link}
| Relative Probability [%] b | NH–Oacceptor Distance [Å] c | NH–Oacceptor Angle [°] d |
|---|---|---|
| Direct H-bond to A188 e | ||
| 2.0 | 2.2 (4.1) | 161 (136) |
| H-bond via one water f | ||
| 0.1 | 2.1 (2.9) | 136 (100) |
| Biocatalyst | kcat/KM, Amide (s−1·M−1) | kcat/KM, Ester (s−1·M−1) | Relative Amidase over Esterase Activity | References |
|---|---|---|---|---|
| S. cardiophyllum patatin | ≈0.45 (V0 = 0.00045 s−1) [a] | 20,900 | 2.0 × 10−5 (3.0 × 10−5) [b] | This work |
| B. subtilis esterase | 0.46 | 4200 | 1.1 × 10−4 | [39] |
| H. insolens cutinase | 0.09 | 770,000 | 1.2 × 10−7 | [34] |
| C. antarctica lipase B wild type | 0.08 | 32,000 | 2.5 × 10−6 | [34] |
| C. antarctica lipase B I189A | 1.8 | 12,343 | 1.5 × 10−4 | [29] |
| Proteases | - | - | - | - |
| Chymotrypsin | - | - | 1.0 × 10−4 [c] | [25] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kürten, C.; Carlberg, B.; Syrén, P.-O. Mechanism-Guided Discovery of an Esterase Scaffold with Promiscuous Amidase Activity. Catalysts 2016, 6, 90. https://doi.org/10.3390/catal6060090
Kürten C, Carlberg B, Syrén P-O. Mechanism-Guided Discovery of an Esterase Scaffold with Promiscuous Amidase Activity. Catalysts. 2016; 6(6):90. https://doi.org/10.3390/catal6060090
Chicago/Turabian StyleKürten, Charlotte, Bengt Carlberg, and Per-Olof Syrén. 2016. "Mechanism-Guided Discovery of an Esterase Scaffold with Promiscuous Amidase Activity" Catalysts 6, no. 6: 90. https://doi.org/10.3390/catal6060090
APA StyleKürten, C., Carlberg, B., & Syrén, P.-O. (2016). Mechanism-Guided Discovery of an Esterase Scaffold with Promiscuous Amidase Activity. Catalysts, 6(6), 90. https://doi.org/10.3390/catal6060090