
Preparation of Cross-Linked Glucoamylase Aggregates Immobilization by Using Dextrin and Xanthan Gum as Protecting Agents


Abstract
:1. Introduction
2. Results and Discussion
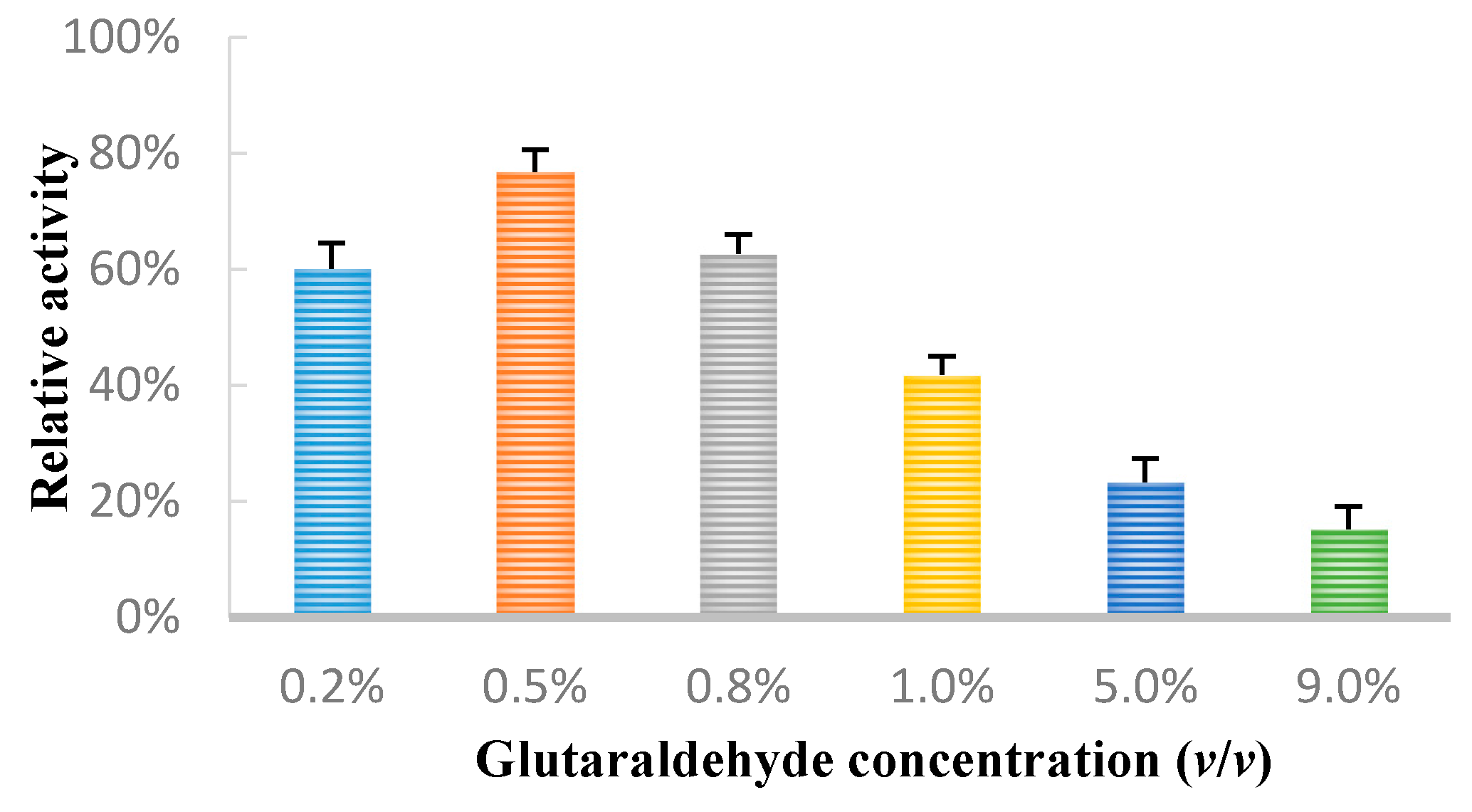
2.1. Glutaraldehyde Concentration
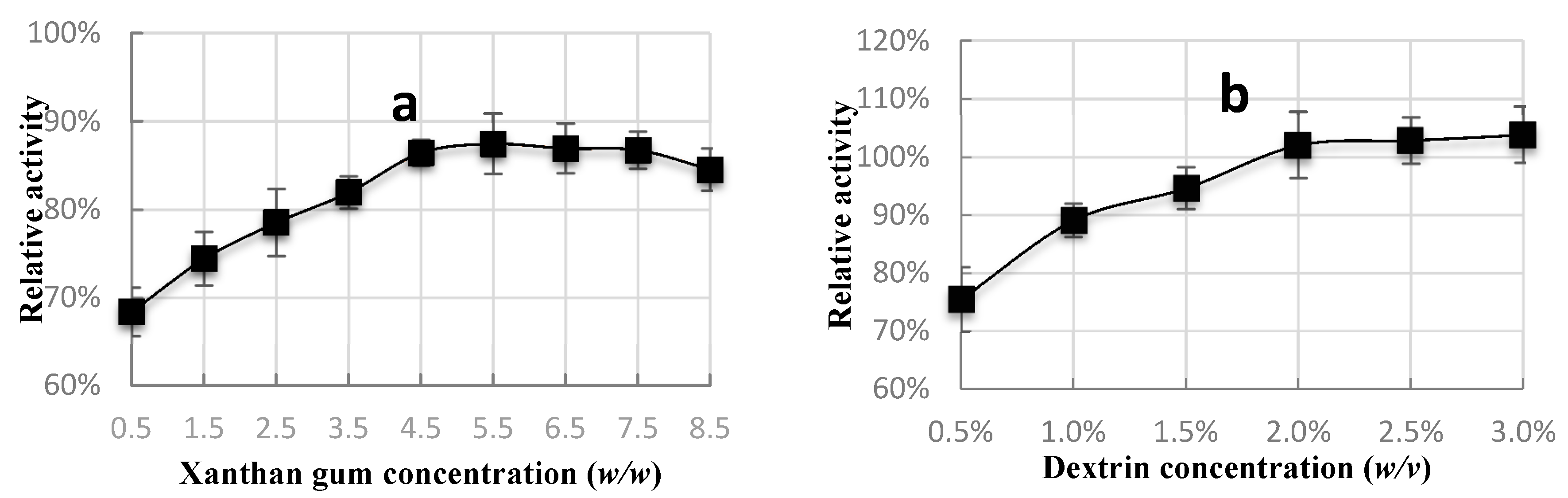
2.2. Protecting Agents Concentration
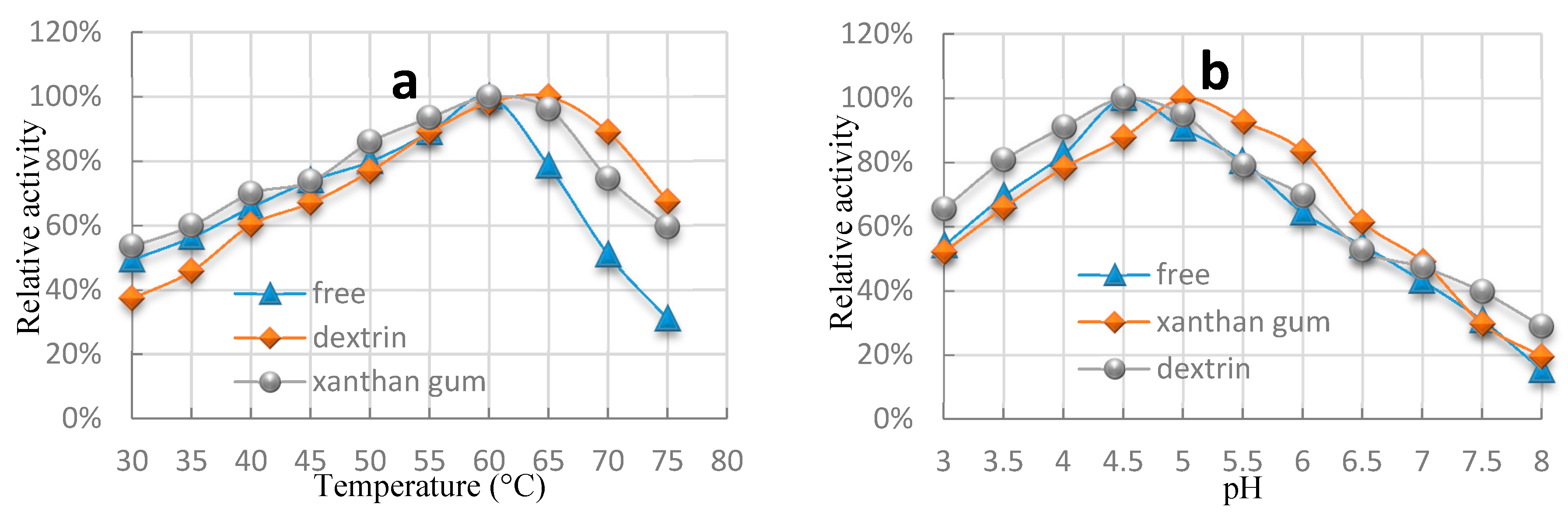
2.3. Dependence of Catalytic Activity on pH and Temperature
2.4. Thermostability of Immobilized Glucoamylase
2.5. Kinetic Constants
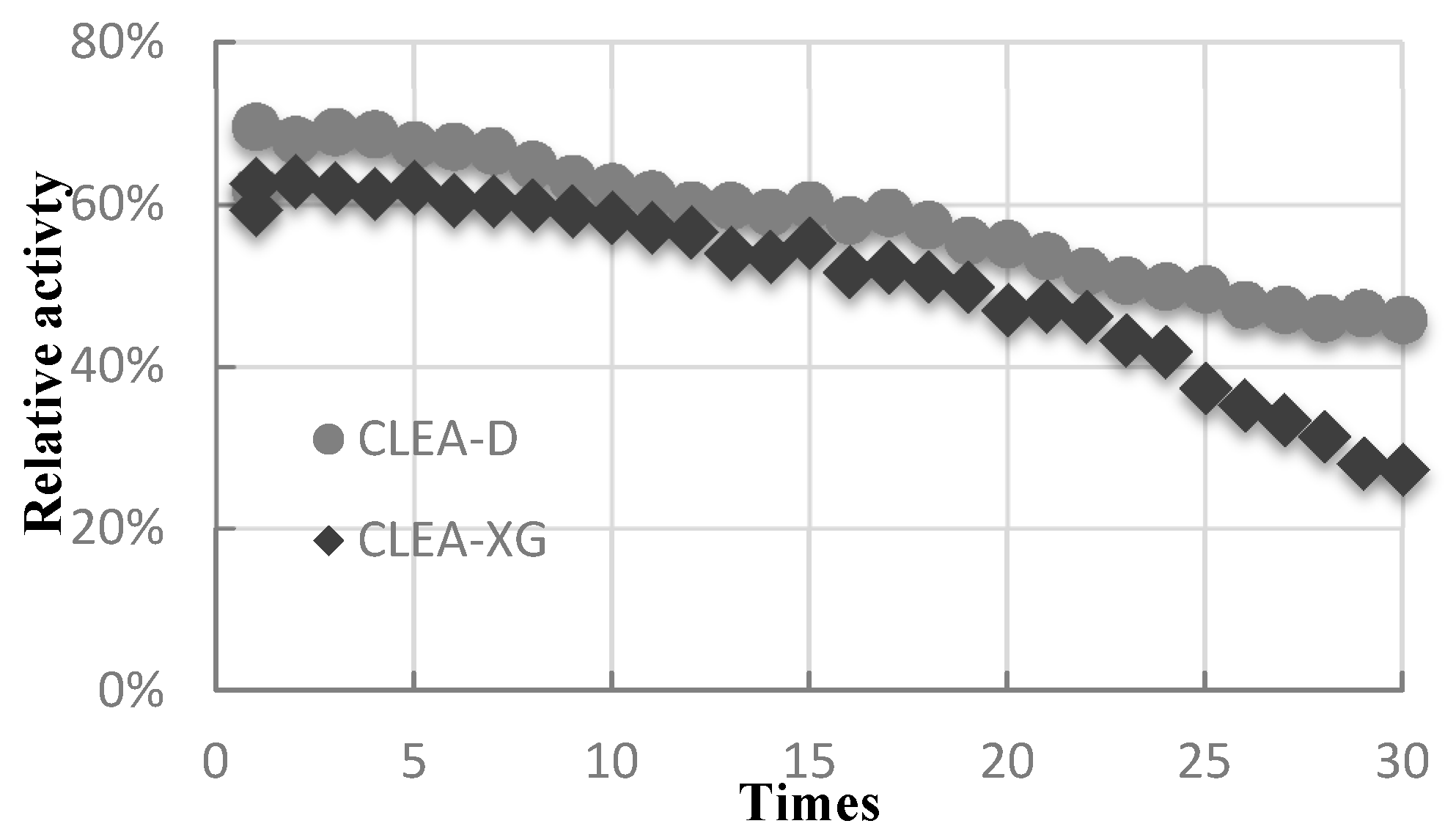
2.6. Recyclability and Storability
3. Experimental Section
3.1. Preparation
3.1.1. Glucoamylase
3.1.2. Glucoamylase Cross-Linked Enzyme Aggregates (CLEA)
3.2. Determination and Analysis
3.2.1. Activity Assay
3.2.2. Optimum Temperature, pH, and Kinetic Parameters Assay
3.2.3. Determination of Stability
3.2.4. Recoverability
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- He, J.; Zhang, W.N. Techno-economic evaluation of thermo-chemical biomass-to-ethano. Appl. Energy 2011, 88, 1224–1232. [Google Scholar] [CrossRef]
- Alvira, P.; Tomás-Pejó, E.; Ballesteros, M.; Negro, M.J. Pretreatment technologies for an efficient bioethanol production process based on enzymatic hydrolysis: A review. Bioresour. Technol. 2010, 101, 4851–4861. [Google Scholar] [CrossRef] [PubMed]
- Tester, R.F.; Qi, X.; Karkalas, J. Hydrolysis of native starches with amylases. Anim. Feed Sci. Technol. 2006, 130, 39–54. [Google Scholar] [CrossRef]
- Sheldon, R.A.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed]
- Liese, A.; Hilterhaus, L. Evaluation of immobilized enzymes for industrial applications. Chem. Soc. Rev. 2013, 42, 6236–6249. [Google Scholar] [CrossRef] [PubMed]
- Tardioli, P.W.; Vieira, M.F.; Vieira, A.M.S.; Zanin, G.M.; Betancor, L. Immobilization-stabilization of glucoamylase: Chemical modification of the enzyme surface followed by covalent attachment on highly activated glyoxyl-agarose supports. Process Biochem. 2011, 46. [Google Scholar] [CrossRef]
- Hamerska-Dudra, A.; Bryjak, J.; Trochimczuk, A.W. Immobilization of glucoamylase and trypsin on crosslinked thermosensitive carriers. Enzym. Microb. Technol. 2007, 41, 197–204. [Google Scholar] [CrossRef]
- Sheldon, R.A. Cross-linked enzyme aggregates (CLEAs): Stable and recyclable biocatalysts. Biochem. Soc. Trans. 2007, 35, 1583–1587. [Google Scholar] [CrossRef] [PubMed]
- Mateo, C.; Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzym. Microb. Technol. 2007, 40, 1451–1463. [Google Scholar] [CrossRef]
- Seung, H.S.; Suk, S.C.; Kyungmoon, P.; Young, J.Y. Novel hybrid immobilization of microorganisms and its applications to biological denitrification. Enzym. Microb. Technol. 2005, 37, 567–573. [Google Scholar] [CrossRef]
- Park, J.M.; Kim, M.; Park, J.Y.; Lee, D.H.; Lee, K.H.; Min, J.; Kim, Y.H. Immobilization of the cross-linked para-nitrobenzyl esterase of Bacillus subtilis aggregates onto magnetic beads. Process Biochem. 2010, 45, 259–263. [Google Scholar] [CrossRef]
- Milosavic, N.; Prodanovic, R.; Jovanovic, S.; Vujcic, Z. Immobilization of glucoamylase via its carbohydrate moiety on macroporouspoly (GMA-co-EGDMA). Enzym. Microb. Technol. 2007, 40, 1422–1426. [Google Scholar] [CrossRef]
- Kök, S.; Osman, B.; Kara, A.; Beşirli, N. Vinyl triazole carrying metal-chelated beads for the reversible immobilization of glucoamylase. J. Appl. Plolym. Sci. 2011, 120, 2563–2570. [Google Scholar] [CrossRef]
- Kovalenko, G.A.; Perminova, L.V. Immobilization of glucoamylase by adsorption on carbon supports and its application for heterogeneous hydrolysis of dextrin. Carbohydr. Res. 2008, 343, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Bayramoglu, G.; Yilmaz, M.; Arica, M.Y. Immobilization of a thermostable α-amylase onto reactive membranes: Kinetics characterization and application to continuous starch hydrolysis. Food Chem. 2004, 84, 591–599. [Google Scholar] [CrossRef]
- Cao, L.; van Langen, L.; Sheldon, R.A. Immobilised enzymes: Carrier-bound or carrier-free? Curr. Opin. Biotechnol. 2003, 14, 387–394. [Google Scholar] [CrossRef]
- Tischer, W.; Kasche, V. Immobilized enzymes: Crystals or carriers? Trends Biotechnol. 1999, 17, 326–335. [Google Scholar] [CrossRef]
- St Clair, N.L.; Navia, M.A. Cross-linked enzyme crystals as robust biocatalysts. J. Am. Chem. Soc. 1992, 114, 7314–7316. [Google Scholar] [CrossRef]
- Häring, D.; Schreier, P. Cross-linked enzyme crystals. Curr. Opin. Chem. Biol. 1999, 3, 35–38. [Google Scholar] [CrossRef]
- Schoevaart, R.; Wolbers, M.W.; Golubovic, M.; Ottens, M. Preparation, optimization, and structures of cross-linked enzyme aggregates (CLEAs). Biotechnol. Bioeng. 2004, 87, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.; Illanes, A.; Soler, L.; Henrıquez, M.J. Effect of the degree of cross-linking on the properties of different CLEAs of penicillin acylase. Process Biochem. 2009, 44, 322–326. [Google Scholar] [CrossRef]
- Shalviri, A.; Liu, Q.; Abdekhodaie, M.J.; Wu, X.Y. Novel modified starch–xanthan gum hydrogels for controlled drug delivery: Synthesis and characterization. Carbohydr. Polym. 2010, 79, 898–907. [Google Scholar] [CrossRef]
- García-Ochoa, F.; Santos, V.E.; Casas, J.A.; Gómez, E. Xanthan gum: Production, recovery, and properties. Biotechnol. Adv. 2000, 18, 549–579. [Google Scholar] [CrossRef]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Glutaraldehyde in bio-catalysts design: A useful crosslinker and a versatile tool in enzyme immobilization. RSC Adv. 2014, 4, 1583–1600. [Google Scholar] [CrossRef]
- Aytar, B.S.; Bakir, U. Preparation of cross-linked tyrosinase aggregates. Process Biochem. 2008, 43, 125–131. [Google Scholar] [CrossRef]
- Matijosyte, I.; Arends, I.W.C.E.; de Vries, S.; Sheldon, R.A. Preparation and use of cross-linked enzyme aggregates (CLEAs) of laccases. J. Mol. Catal. B Enzym. 2010, 62, 142–148. [Google Scholar] [CrossRef]
- Shah, S.; Sharma, A.; Gupta, M.N. Preparation of cross-linked enzyme aggregates by using bovine serum albumin as a proteic feeder. Anal. Biochem. 2006, 351, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Guo, C.; Liu, H.Z.; Liu, C.Z. Reversible immobilization of glucoamylase by metal affinity adsorption on magnetic chelator particles. J. Mol. Catal. B Enzym. 2007, 48, 1–7. [Google Scholar] [CrossRef]
- Kumari, A.; Kayastha, A.M. Immobilization of soybean (Glycine max) α-amylase onto Chitosan and Amberlite MB-150 beads: Optimization and characterization. J. Mol. Catal. B Enzym. 2011, 69, 8–14. [Google Scholar] [CrossRef]
- Sanjay, G.; Sugunan, S. Glucoamylase immobilized on montmorillonite: Synthesis, characterization and starch hydrolysis activity in a fixed bed reactor. Catal. Commun. 2005, 6, 525–530. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; van Langen, L.M.; van Rantwijk, F.; Sheldon, R.A. A new, mild cross-linking methodology to prepare cross-linked enzyme aggregates. Biotechnol. Bioeng. 2004, 86, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Tao, D.; Lin, Z.; Yu, H.; Xin, T. Preparation of cross-linked aggregates of aminoacylase from Aspergillus melleus by using bovine serum albumin as an inert additive. Bioresour. Technol. 2010, 101, 6569–6571. [Google Scholar]
- Germain, P.; Crichton, R.R. Characterization of a chemically modified β-amylase immobilized on porous silica. J. Chem. Technol. Biotechnol. 1988, 41, 297–315. [Google Scholar] [CrossRef]
- Cao, L.Q. Immobilised enzymes: Science or art? Curr. Opin. Chem. Biol. 2005, 9, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Tanriseven, A.; Ölҫer, Z. A novel method for the immobilization of glucoamylase onto polyglutaraldehyde-activated gelatin. Biochem. Eng. J. 2008, 39, 430–434. [Google Scholar] [CrossRef]
- Abraham, T.E.; Joseph, J.R.; Bai, L.; Bindhu, V.; Jayakumar, K.K. Crosslinked enzyme crystals of glucoamylase as a potent catalyst for biotransformations. Carbohydr. Res. 2004, 339, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Galan, C.; Berenguer-Murcia, A.; Fernandez-Lafuente, R.; Rodrigues, R.C. Potential of different enzyme immobilization strategies to improve enzyme performance. Adv. Synth. Catal. 2011, 353, 2885–2904. [Google Scholar] [CrossRef]
- Sangeetha, K.; Abraham, T.E. Preparation and characterization of cross-linked enzyme aggregates (CLEA) of Subtilisin for controlled release applications. Int. J. Biol. Macromol. 2008, 43, 314–319. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | 60 °C | 70 °C | ||
|---|---|---|---|---|
| ki (min−1) | t1/2 (min) | ki (min−1) | t1/2 (min) | |
| CLEA-XG | 1.49 × 10−4 | 463 ± 15 | 1.67 × 10−3 | 415 ± 18 |
| CLEA-D | 9.86 × 10−4 | 703 ± 21 | 2.61 × 10−3 | 266 ± 14 |
| free | 4.33 × 10−3 | 160 ± 16 | 1.65 × 10−2 | 42 ± 9 |
| Types | Km (μg·L−1) | Vm (μg·min−1·mL−1) | Effectiveness Factor η |
|---|---|---|---|
| Free | 46 ± 3 | 3.3 × 103 | - |
| CLEA-D | 55 ± 5 | 3.1 × 103 | 0.94 |
| CLEA-XG | 70 ± 3 | 3.3 × 103 | 0.99 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.-D.; Wu, J.; Jia, D.-C.; Wan, Y.-H.; Yang, N.; Qiao, M. Preparation of Cross-Linked Glucoamylase Aggregates Immobilization by Using Dextrin and Xanthan Gum as Protecting Agents. Catalysts 2016, 6, 77. https://doi.org/10.3390/catal6060077
Li X-D, Wu J, Jia D-C, Wan Y-H, Yang N, Qiao M. Preparation of Cross-Linked Glucoamylase Aggregates Immobilization by Using Dextrin and Xanthan Gum as Protecting Agents. Catalysts. 2016; 6(6):77. https://doi.org/10.3390/catal6060077
Chicago/Turabian StyleLi, Xiao-Dong, Jia Wu, Dong-Chen Jia, Yong-Hu Wan, Na Yang, and Min Qiao. 2016. "Preparation of Cross-Linked Glucoamylase Aggregates Immobilization by Using Dextrin and Xanthan Gum as Protecting Agents" Catalysts 6, no. 6: 77. https://doi.org/10.3390/catal6060077
APA StyleLi, X.-D., Wu, J., Jia, D.-C., Wan, Y.-H., Yang, N., & Qiao, M. (2016). Preparation of Cross-Linked Glucoamylase Aggregates Immobilization by Using Dextrin and Xanthan Gum as Protecting Agents. Catalysts, 6(6), 77. https://doi.org/10.3390/catal6060077