
Adsorption and Oxidation Investigations over Pt/Al2O3 Catalyst: A Microcalorimetric Study


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Pt Dispersion
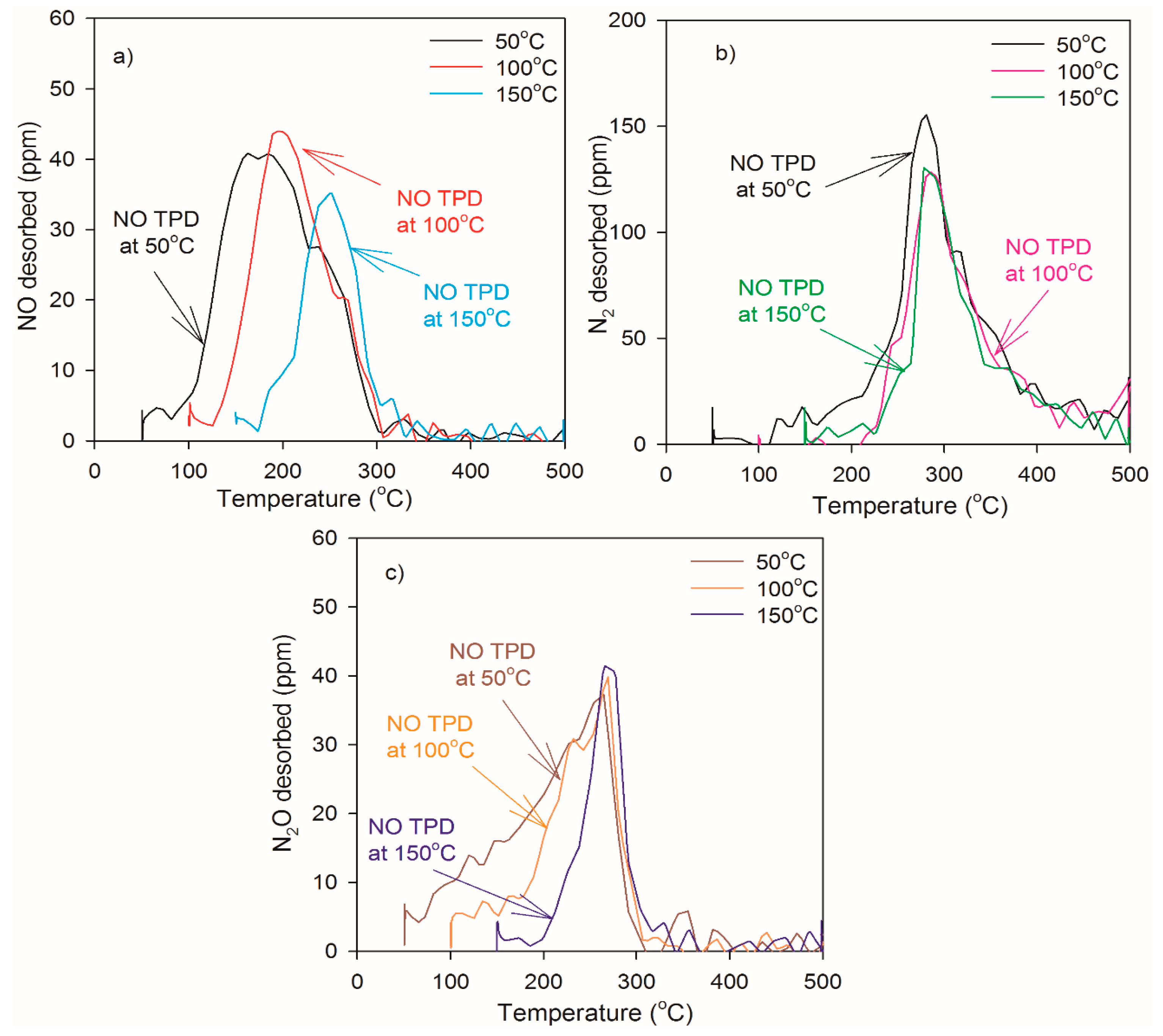
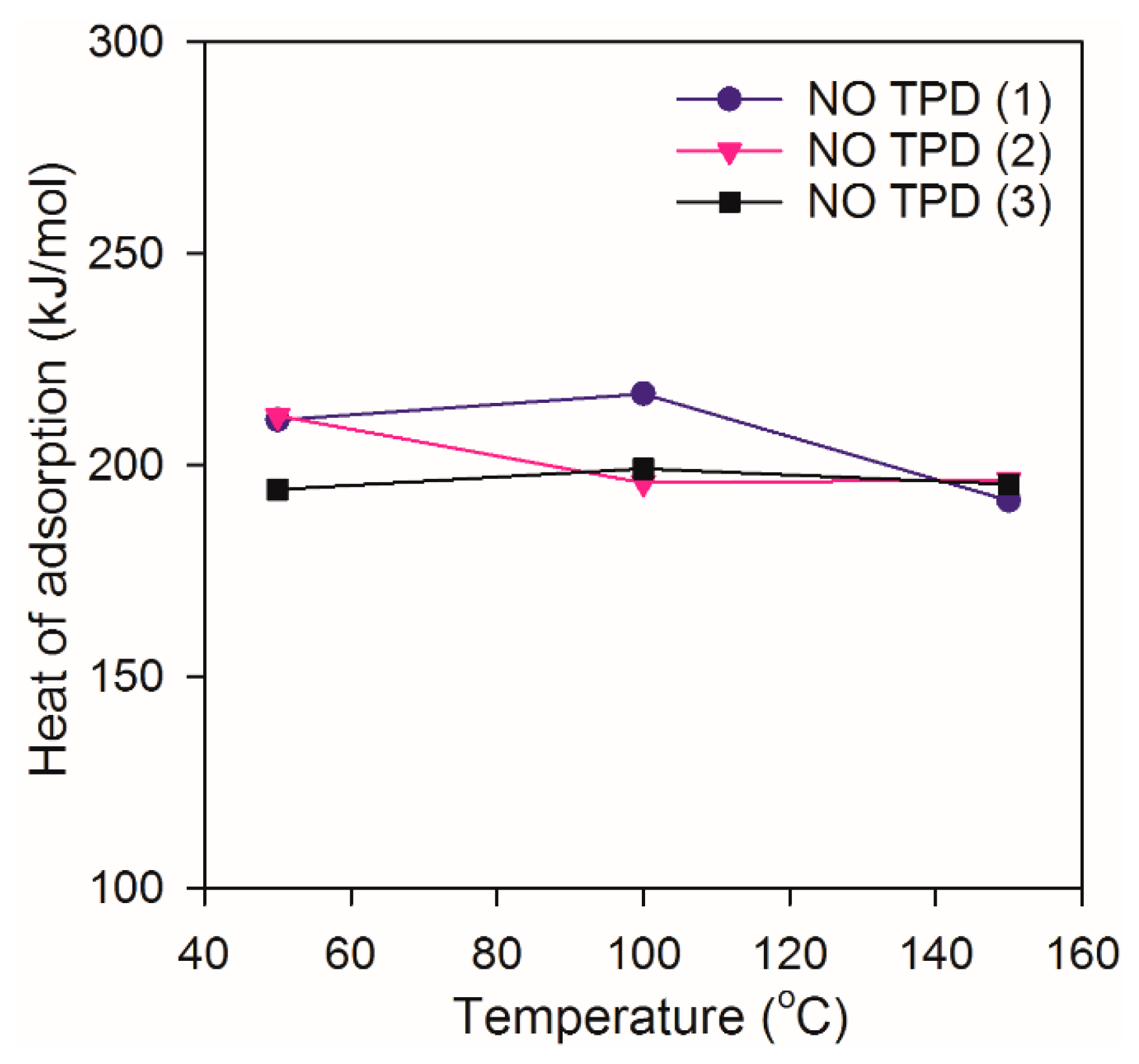
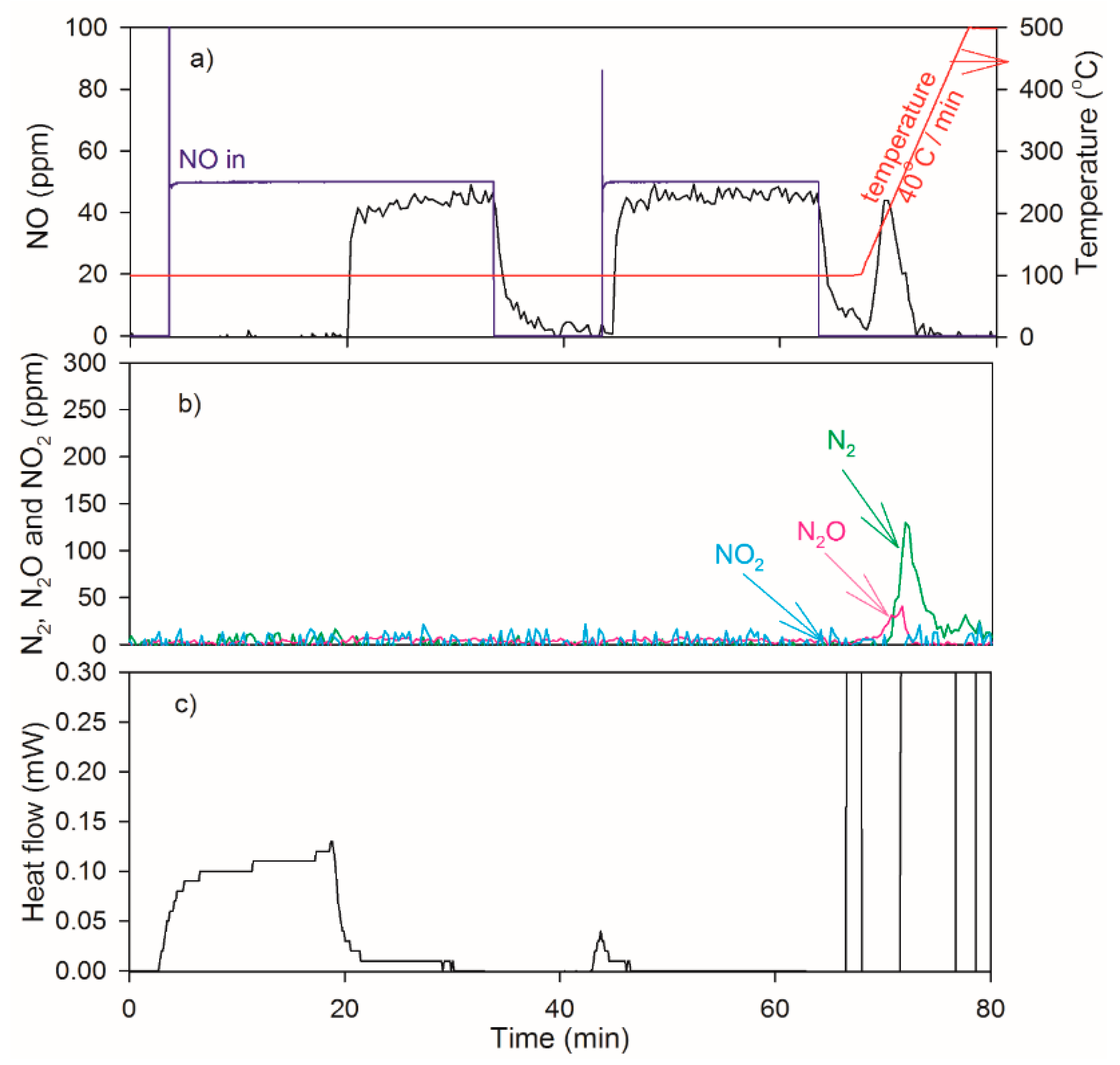
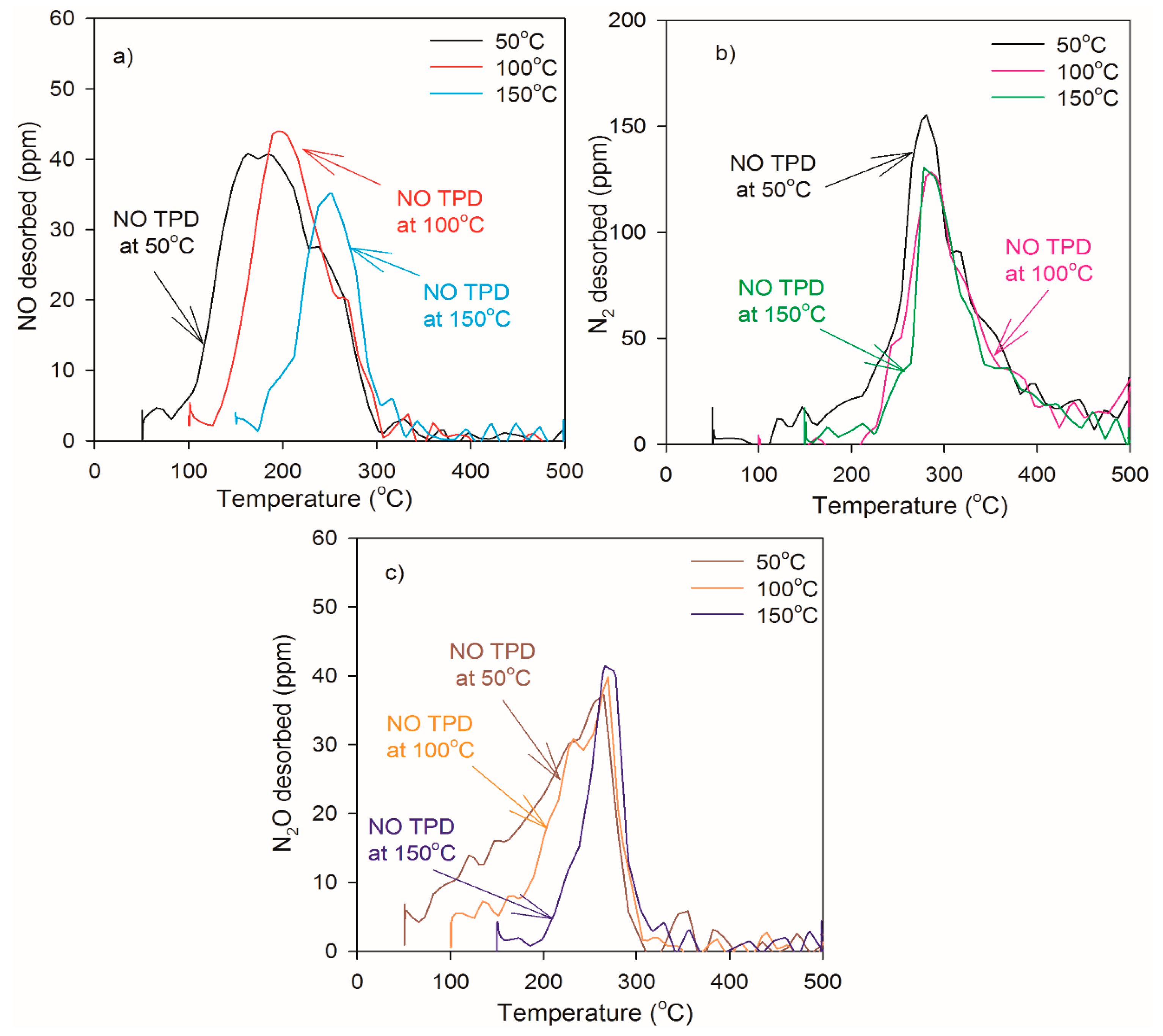
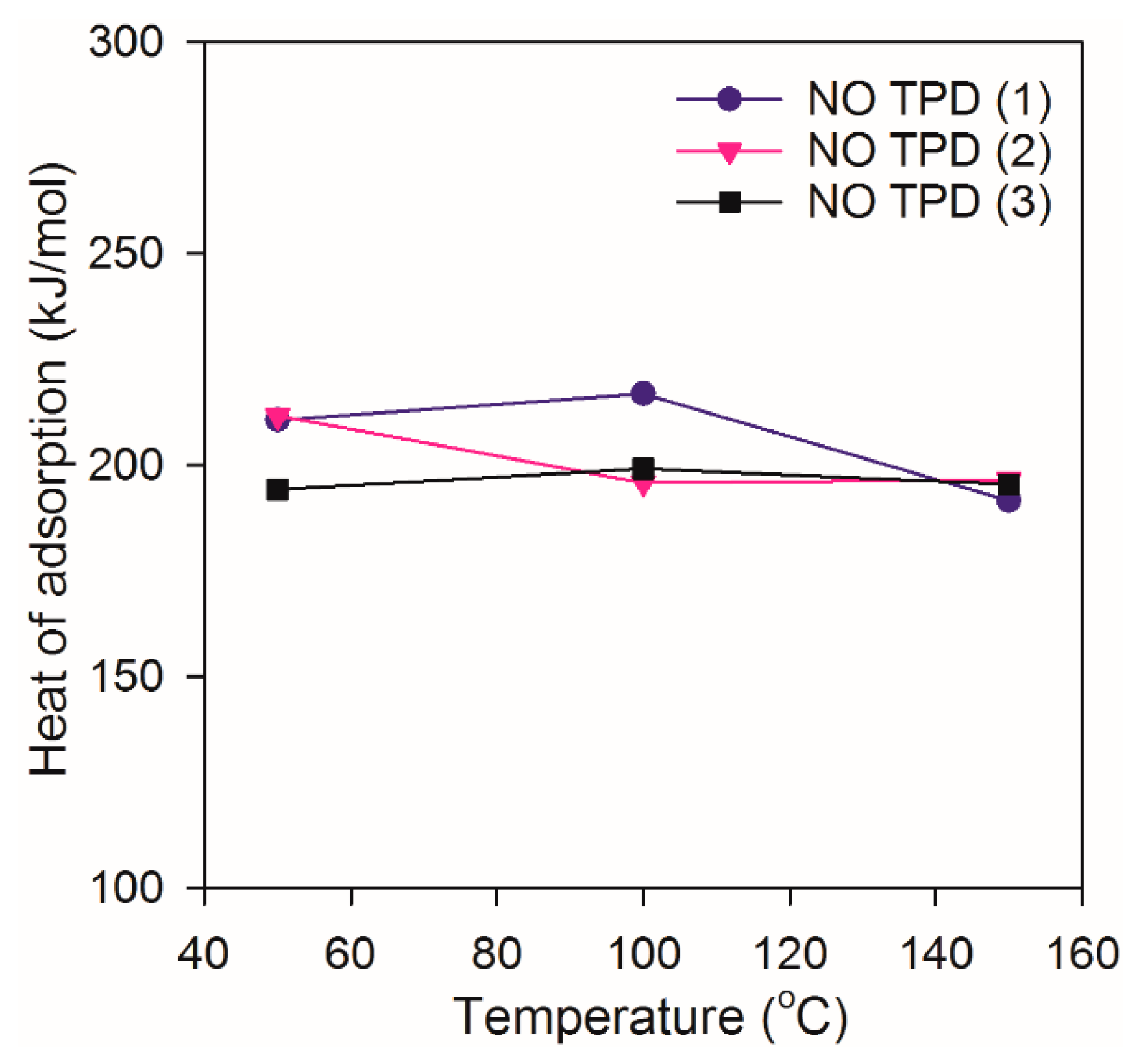
2.2. Heat of Adsorption during NO TPD Experiments
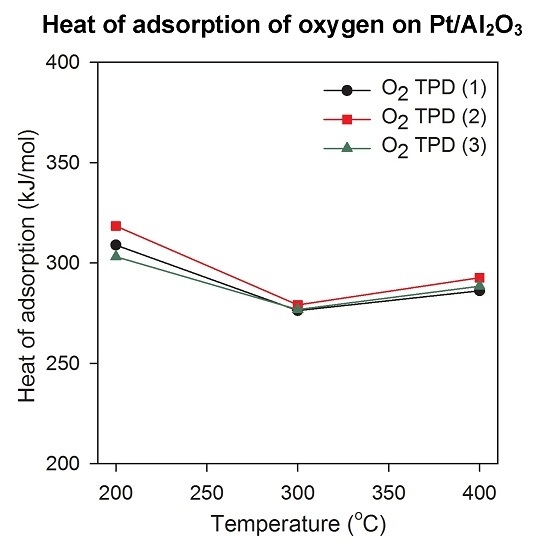
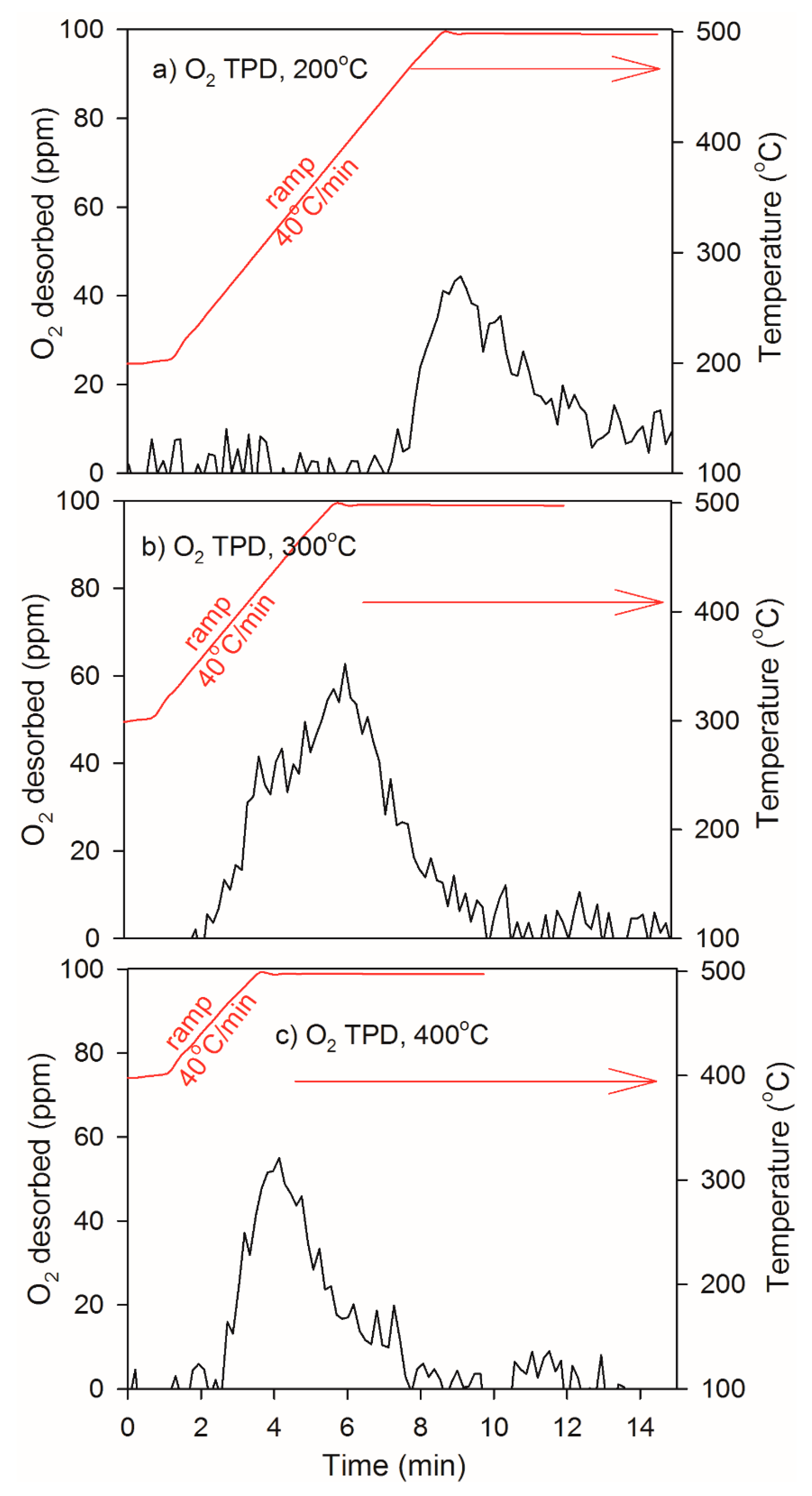
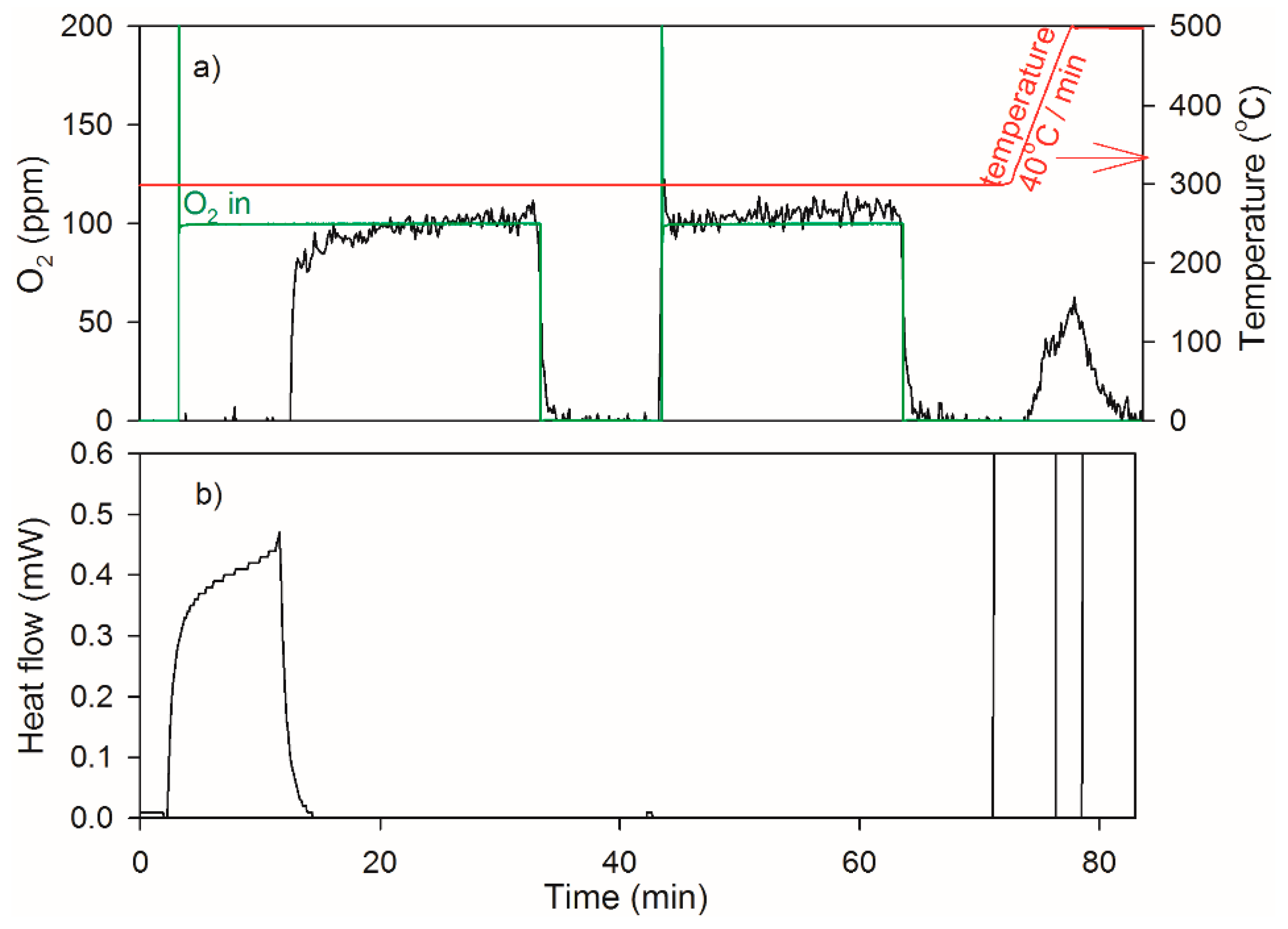
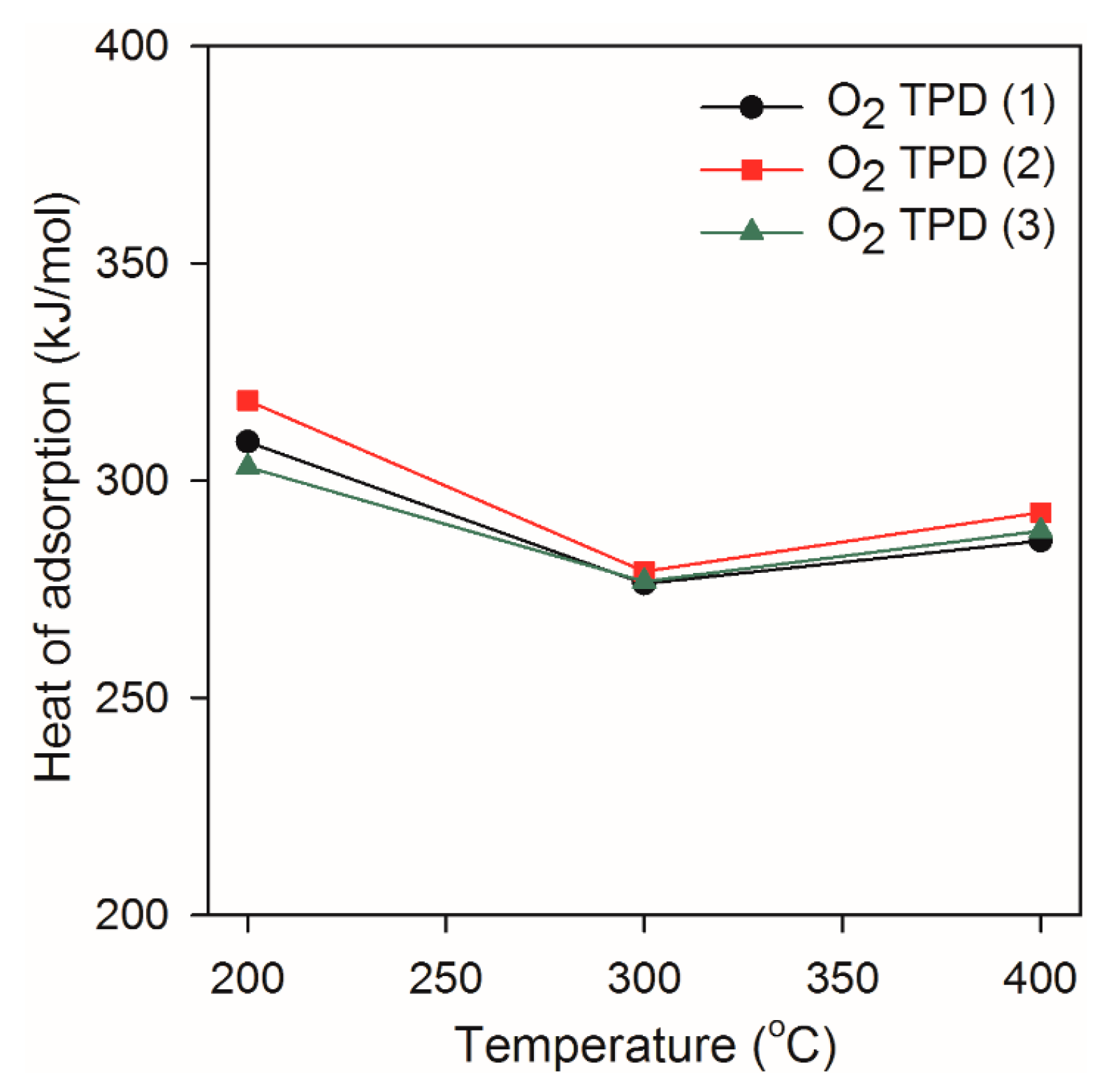
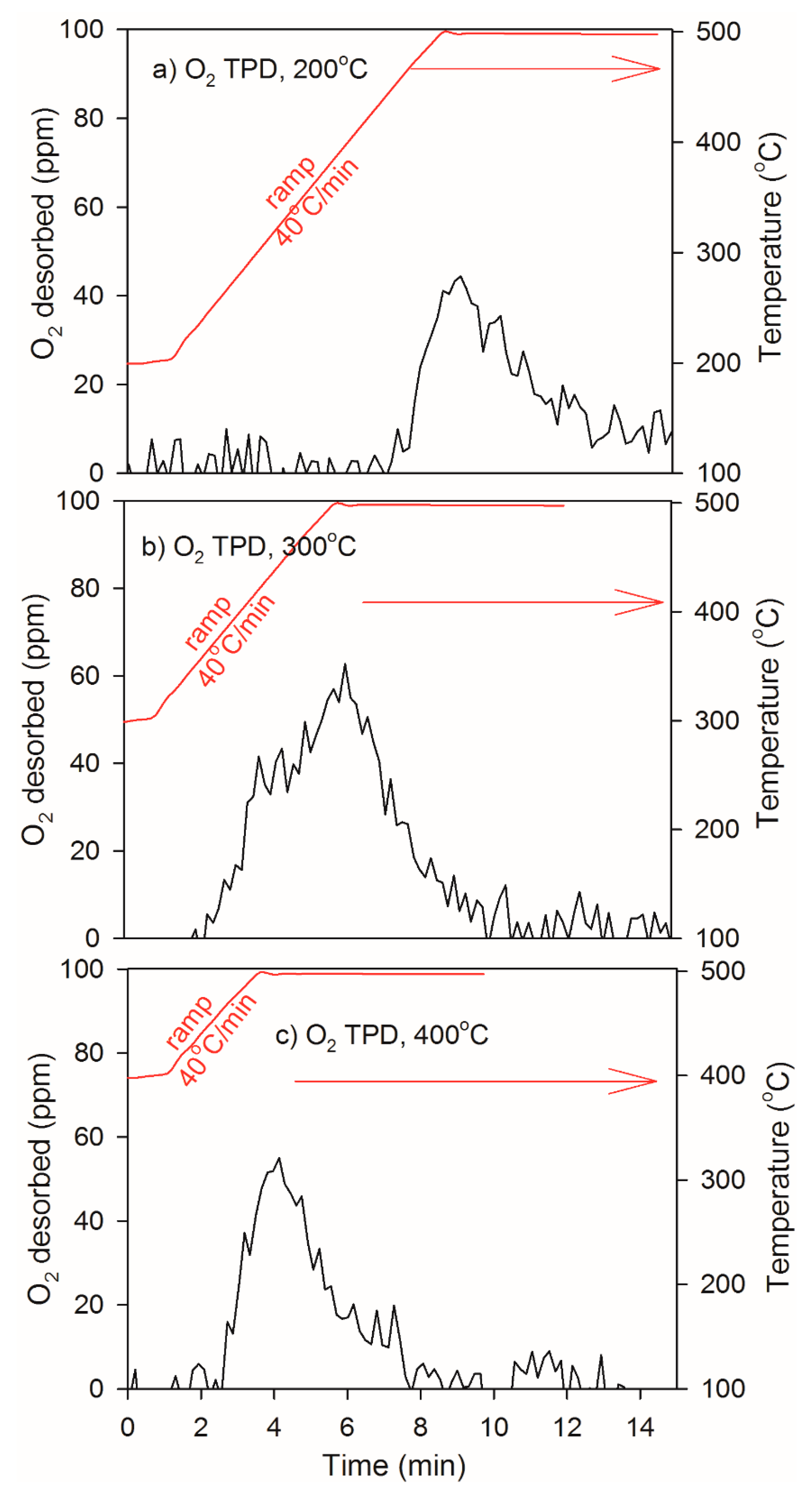
2.3. Heat of Adsorption during O2 TPD Experiments
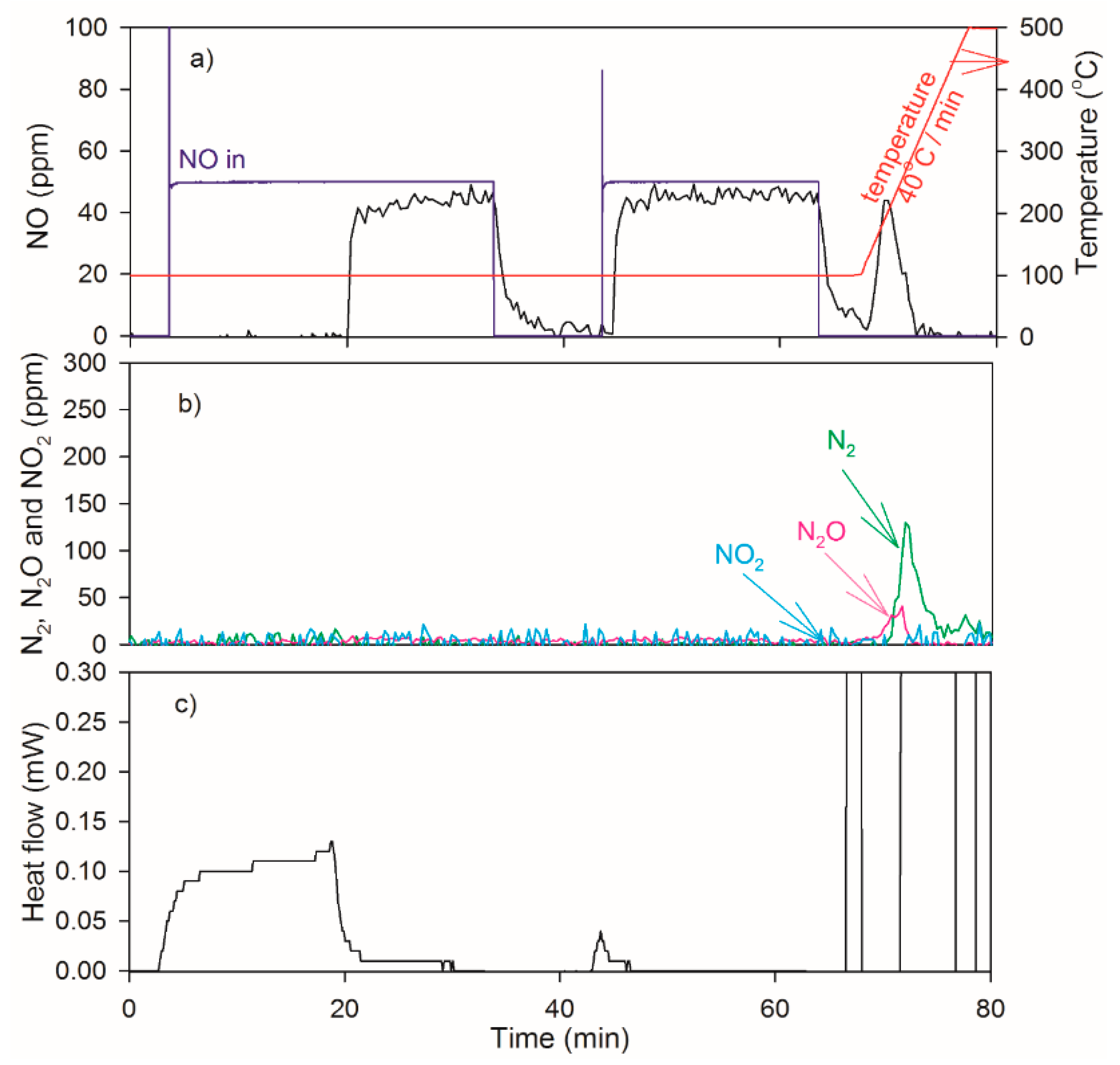
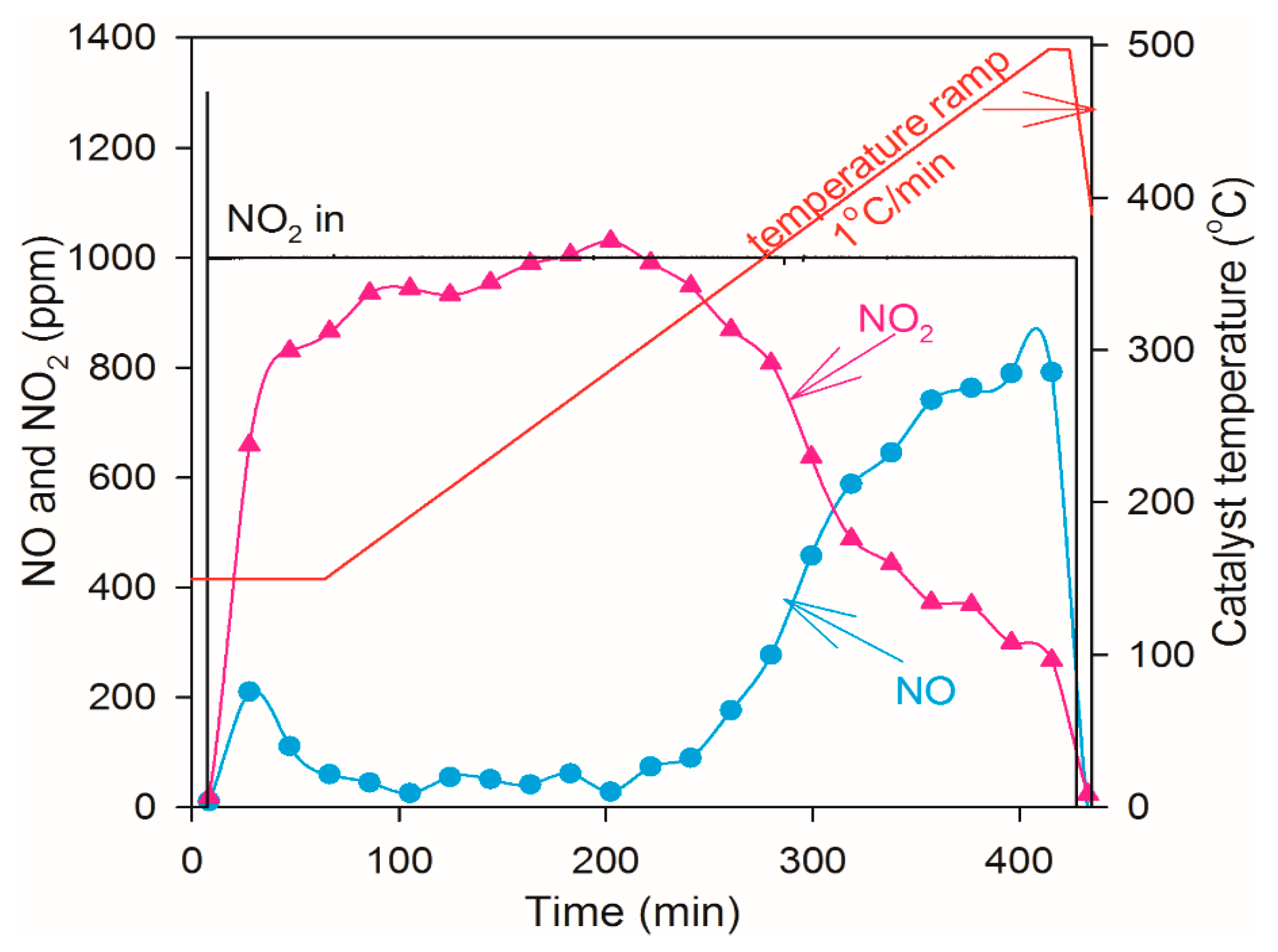
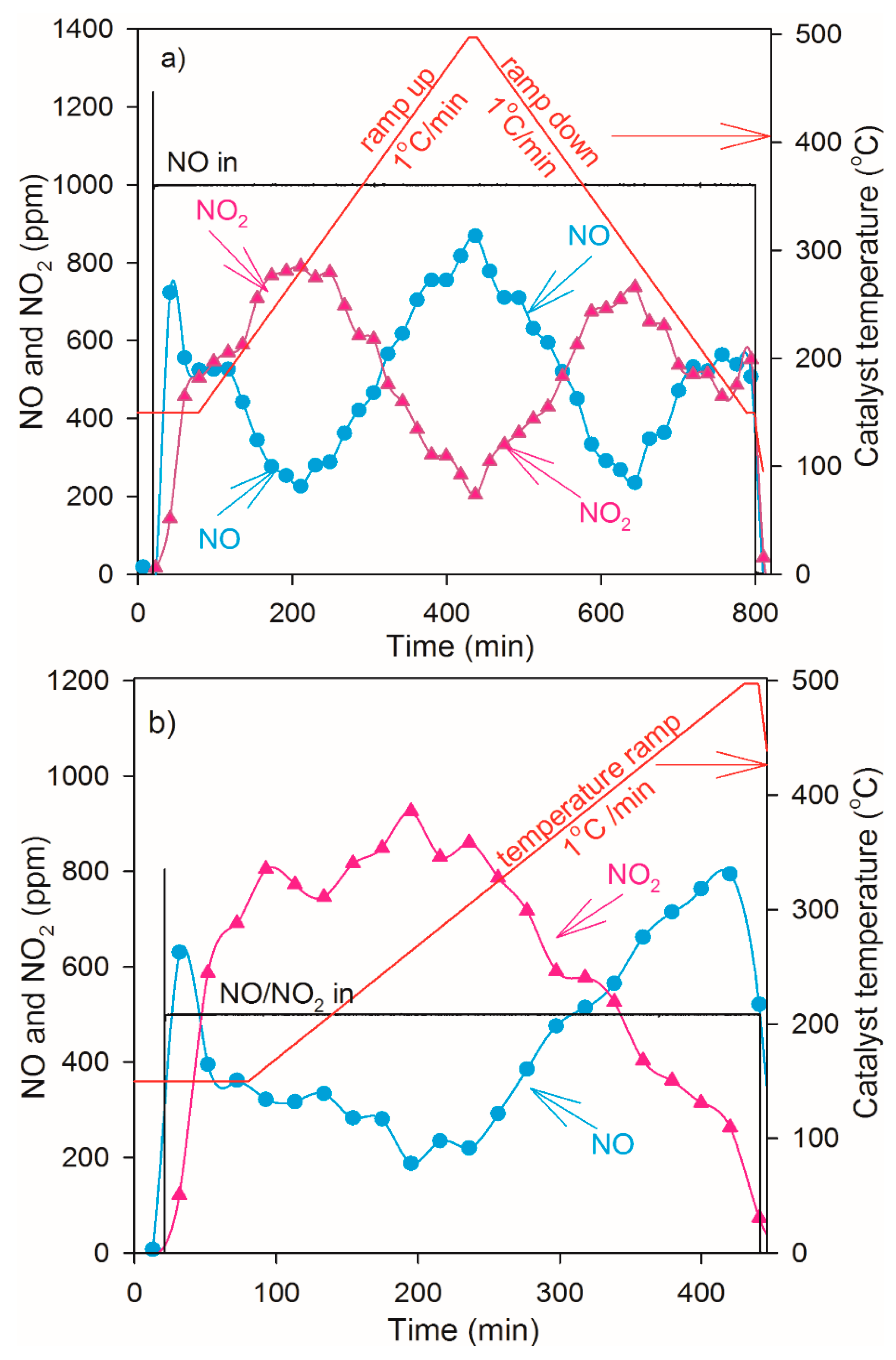
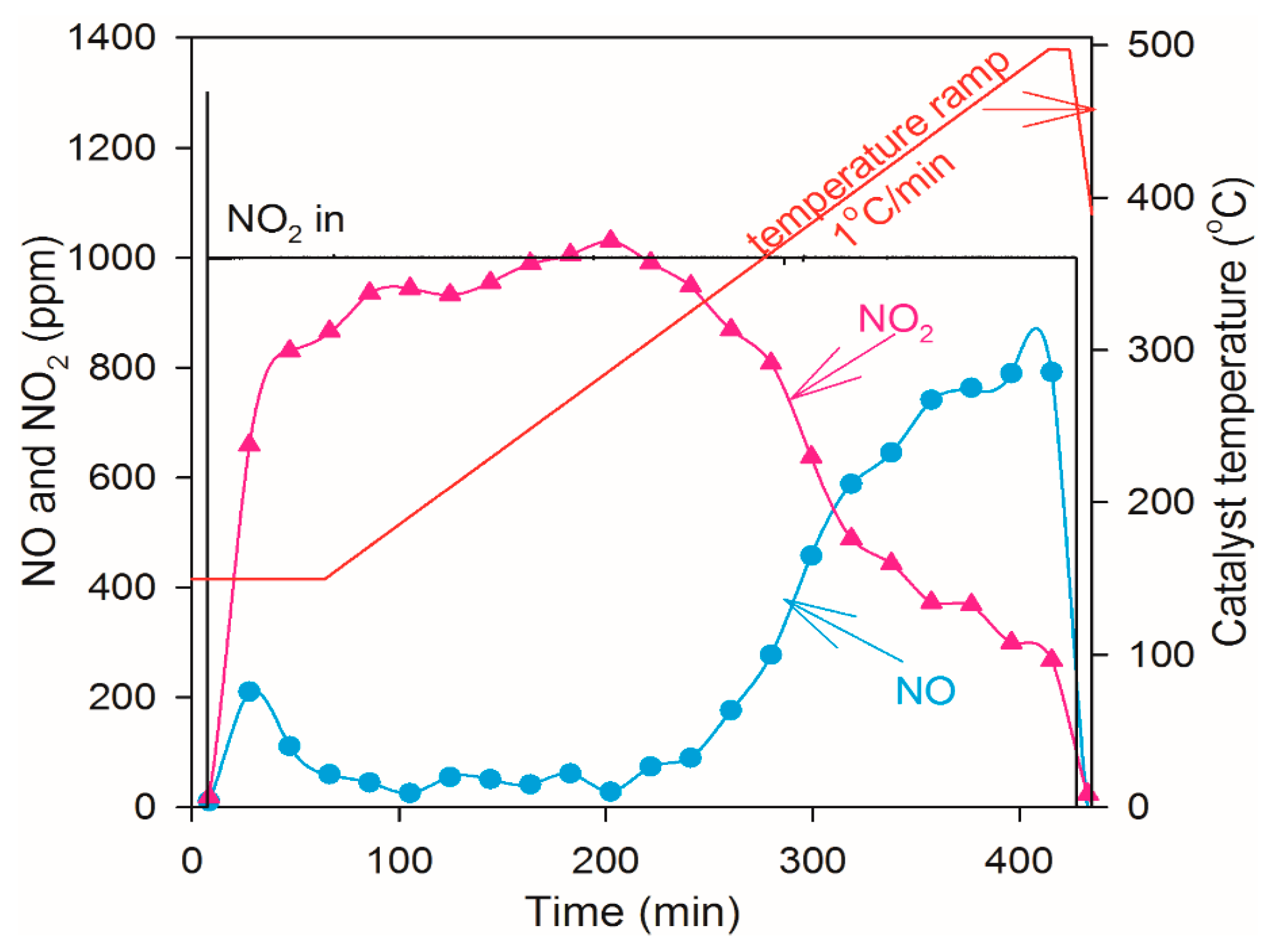
2.4. NO Oxidation and NO2 Dissociation Reaction
3. Experimental Section
3.1. Synthesis of 2 wt. % Pt/γ-Al2O3 Powder Catalyst
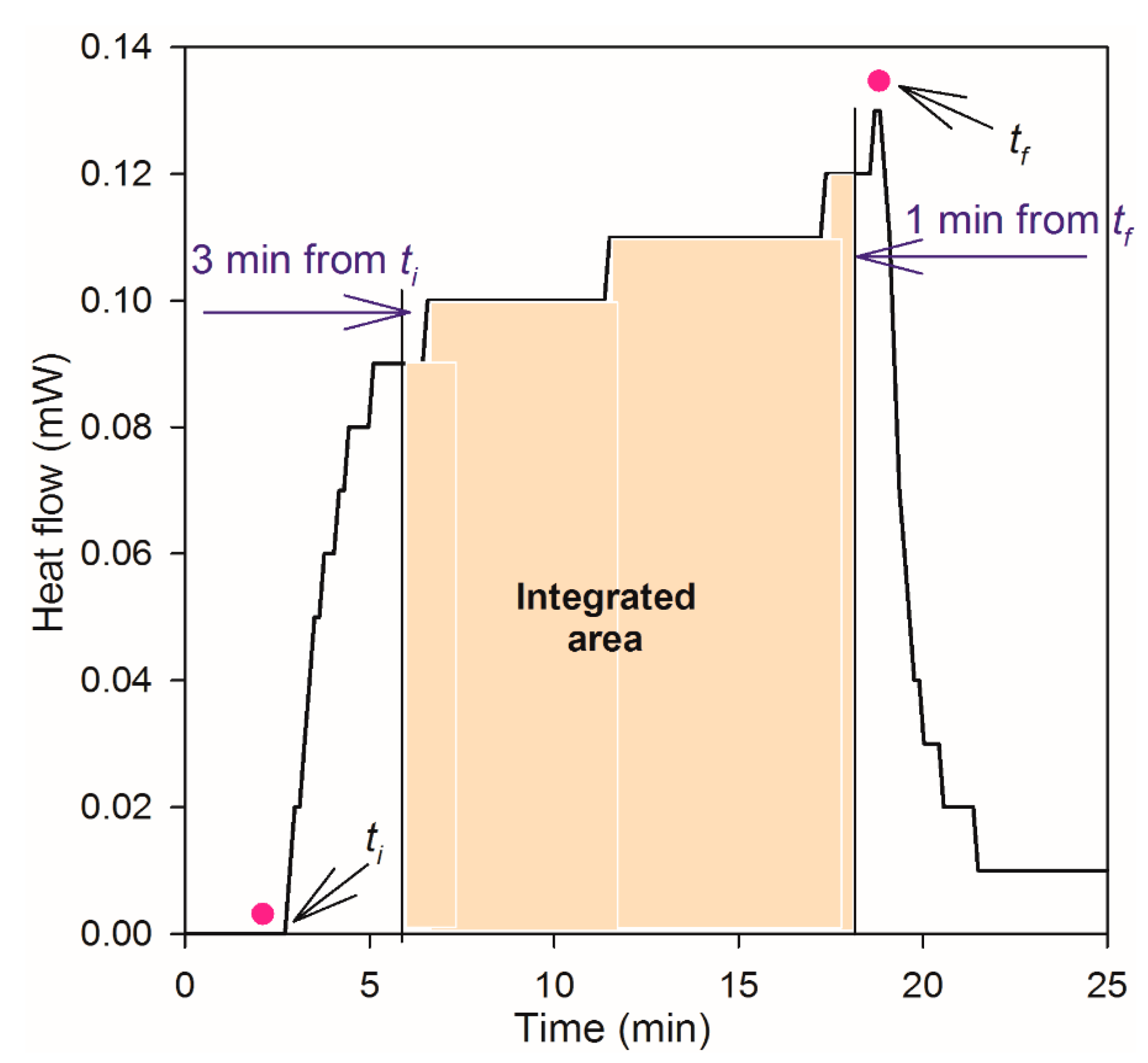
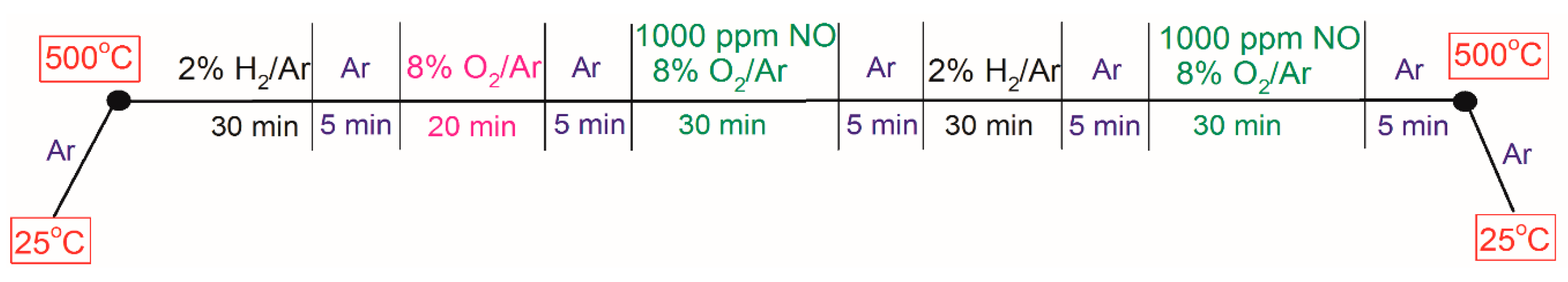
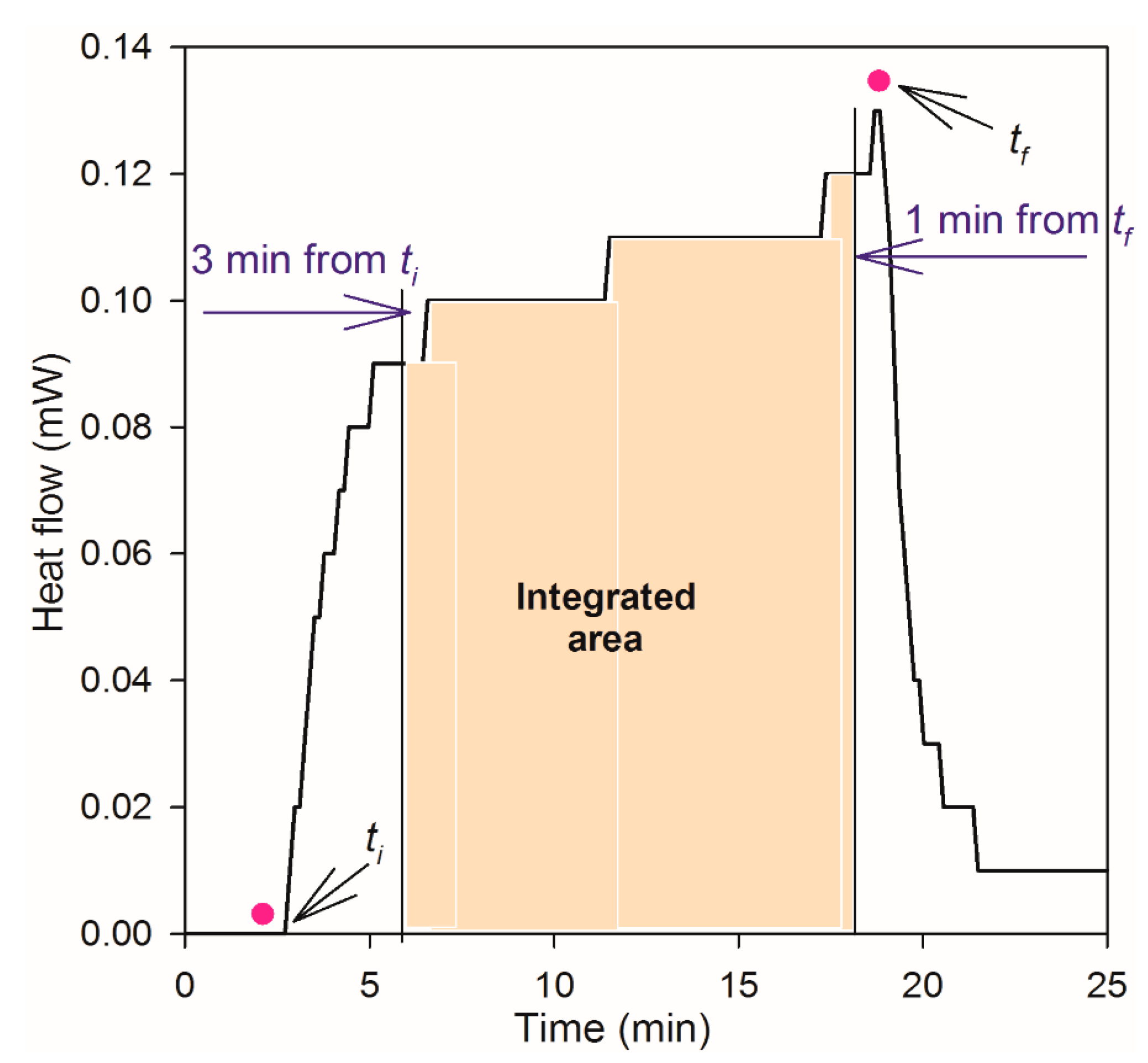
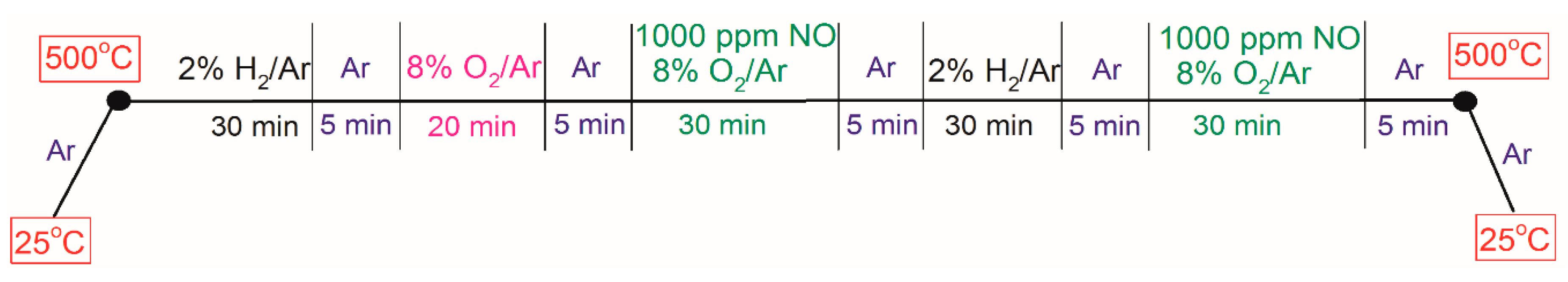
3.2. Microcalorimeter Measurements
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brandenberger, S.; Kröcher, O.; Tissler, A.; Althoff, R. The state of the art in selective catalytic reduction of NOx by ammonia using metal-exchanged zeolite catalysts. Catal. Rev. Sci. Eng. 2008, 50, 492–531. [Google Scholar] [CrossRef]
- Matsumoto, S. Catalytic reduction of nitrogen oxides in automotive exhaust containing excess oxygen by NOx storage-reduction catalyst. CATTECH 2000, 4, 102–109. [Google Scholar] [CrossRef]
- Weiss, B.M.; Iglesia, E. NO oxidation catalysis on Pt clusters: Elementary steps, structural requirements, and synergistic effects of NO2 adsorption sites. J. Phys. Chem. C 2009, 113, 13331–13340. [Google Scholar] [CrossRef]
- Mulla, S.S.; Chen, N.; Delgass, W.N.; Epling, W.S.; Ribeiro, F.H. NO2 inhibits the catalytic reaction of NO and O2 over Pt. Catal. Lett. 2005, 100, 267–270. [Google Scholar] [CrossRef]
- Venderbosch, R.H.; Prins, W.; van Swaaij, W.P.M. Platinum catalyzed oxidation of carbon monoxide as a model reaction in mass transfer measurements. Chem. Eng. Sci. 1998, 53, 3355–3366. [Google Scholar] [CrossRef]
- Bourane, A.; Bianchi, D. Oxidation of CO on a Pt/Al2O3 catalyst: From the surface elementary steps to light-off tests: I. Kinetic study of the oxidation of the linear CO species. J. Catal. 2001, 202, 34–44. [Google Scholar] [CrossRef]
- Twigg, M.V. Progress and future challenges in controlling automotive exhaust gas emissions. Appl. Catal. B 2007, 70, 2–15. [Google Scholar] [CrossRef]
- Getman, R.B.; Schneider, W.F.; Smeltz, A.D.; Delgass, W.N.; Ribeiro, F.H. Oxygen-coverage effects on molecular dissociations at a Pt metal surface. Phys. Rev. Lett. 2009, 102, 076101. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.P.; Yeh, C.T. Platinum-oxide species formed by oxidation of platinum crystallites supported on alumina. J. Mol. Catal. A Chem. 1996, 112, 295–302. [Google Scholar] [CrossRef]
- Wang, C.B.; Yeh, C.T. Effects of particle size on the progressive oxidation of nanometer platinum by dioxygen. J. Catal. 1998, 178, 450–456. [Google Scholar] [CrossRef]
- Wang, C.B.; Yeh, C.T. Oxidation behavior of alumina-supported platinum metal catalysts. Appl. Catal. A 2001, 209, 1–9. [Google Scholar] [CrossRef]
- Mukerji, R.J.; Bolina, A.S.; Brown, W.A. An investigation of the effect of pre-dosed O atoms on the adsorption of NO on Pt{211}. Surf. Sci. 2003, 547, 27–44. [Google Scholar] [CrossRef]
- Rienks, E.D.L.; Bakker, J.W.; Baraldi, A.; Carabineiro, S.A.C.; Lizzit, S.; Weststrate, C.J.; Nieuwenhuys, B.E. Interaction of nitric oxide with Pt(100). A fast X-ray photoelectron spectroscopy study. Surf. Sci. 2002, 516, 109–117. [Google Scholar] [CrossRef]
- Weststrate, C.J.; Bakker, J.W.; Rienks, E.D.L.; Vinod, C.P.; Lizzit, S.; Petaccia, L.; Baraldi, A.; Nieuwenhuys, B.E. Synchrotron XPS and desorption study of the NO chemistry on a stepped Pt surface. Surf. Sci. 2006, 600, 1991–2001. [Google Scholar] [CrossRef]
- Cardona-Martinez, N.; Dumesic, J.A. Applications of adsorption microcalorimetry to the study of heterogeneous catalysis. In Advances in Catalysis; Eley, D.D., Pines, H., Weisz, P.B., Eds.; Academic Press Inc.: Amsterdam, The Netherlands, 1992; ISBN 978-0-12-007838-7. Volume 38, pp. 149–244. [Google Scholar]
- Spiewak, B.E.; Dumesic, J.A. Applications of adsorption microcalorimetry for the characterization of metal-based catalysts. Thermochim. Acta 1998, 312, 95–104. [Google Scholar] [CrossRef]
- Wartnaby, C.E.; Stuck, A.; Yeo, Y.Y.; King, D.A. Microcalorimetric heats of adsorption for CO, NO, and oxygen on Pt{110}. J. Phys. Chem. 1996, 100, 12483–12488. [Google Scholar] [CrossRef]
- Brown, W.A.; Kose, R.; King, D.A. Femtomole adsorption calorimetry on single-crystal surfaces. Chem. Rev. 1998, 98, 797–832. [Google Scholar] [CrossRef] [PubMed]
- Fiorin, V.; Borthwick, D.; King, D.A. Microcalorimetry of O2 and NO on flat and stepped platinum surfaces. Surf. Sci. 2009, 603, 1360–1364. [Google Scholar] [CrossRef]
- Dawody, J.; Eurenius, L.; Abdulhamid, H.; Skoglundh, M.; Olsson, E.; Fridell, E. Platinum dispersion measurements for Pt/BaO/Al2O3, NOx storage catalysts. Appl. Catal. A 2005, 296, 157–168. [Google Scholar] [CrossRef]
- Wilken, N.; Kamasamudram, K.; Currier, N.W.; Li, J.; Yezerets, A.; Olsson, L. Heat of adsorption for NH3, NO2 and NO on Cu-Beta zeolite using microcalorimeter for NH3 SCR applications. Catal. Today 2010, 151, 237–243. [Google Scholar] [CrossRef]
- Olsson, L.; Fridell, E. The influence of Pt oxide formation and Pt dispersion on the reactions NO2 ⇄ NO + 1/2 O2 over Pt/Al2O3 and Pt/BaO/Al2O3. J. Catal. 2002, 210, 340–353. [Google Scholar] [CrossRef]
- Olsson, L.; Wijayanti, K.; Leistner, K.; Kumar, A.; Joshi, S.Y.; Kamasamudram, K.; Currier, N.W.; Yezerets, A. A multi-site kinetic model for NH3-SCR over Cu/SSZ-13. Appl. Catal. B Environ. 2015, 174–175, 212–224. [Google Scholar] [CrossRef]
- Olsson, L.; Persson, H.; Fridell, E.; Skoglundh, M.; Andersson, B. A kinetic study of NO oxidation and NOx storage on Pt/Al2O3 and Pt/BaO/Al2O3. J. Phys. Chem B 2001, 105, 6895–6906. [Google Scholar] [CrossRef]
- Olsson, L.; Westerberg, B.; Persson, H.; Fridell, E.; Skoglundh, M.; Andersson, B. A kinetic study of oxygen adsorption/desorption and NO oxidation over Pt/Al2O3 catalysts. J. Phys. Chem. B. 1999, 103, 10433–10439. [Google Scholar] [CrossRef]
- Auvray, X.P.; Olsson, L. Effect of enhanced support acidity on the sulfate storage and the activity of Pt/γ-Al2O3 for NO oxidation and propylene oxidation. Catal. Lett. 2014, 144, 22–31. [Google Scholar] [CrossRef]
- Andonova, S.; Marchionni, V.; Borelli, M.; Nedyalkova, R.; Lietti, L.; Olsson, L. Mechanistic investigations of the promoting role of Rh on the NSR performance of NOx storage bao-based catalysts. Appl. Catal. B Environ. 2013, 132–133, 266–281. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihai, O.; Creaser, D.; Olsson, L. Adsorption and Oxidation Investigations over Pt/Al2O3 Catalyst: A Microcalorimetric Study. Catalysts 2016, 6, 73. https://doi.org/10.3390/catal6050073
Mihai O, Creaser D, Olsson L. Adsorption and Oxidation Investigations over Pt/Al2O3 Catalyst: A Microcalorimetric Study. Catalysts. 2016; 6(5):73. https://doi.org/10.3390/catal6050073
Chicago/Turabian StyleMihai, Oana, Derek Creaser, and Louise Olsson. 2016. "Adsorption and Oxidation Investigations over Pt/Al2O3 Catalyst: A Microcalorimetric Study" Catalysts 6, no. 5: 73. https://doi.org/10.3390/catal6050073
APA StyleMihai, O., Creaser, D., & Olsson, L. (2016). Adsorption and Oxidation Investigations over Pt/Al2O3 Catalyst: A Microcalorimetric Study. Catalysts, 6(5), 73. https://doi.org/10.3390/catal6050073