
Rare Earth-Promoted Nickel Oxide Nanoparticles as Catalysts for N2O Direct Decomposition


Abstract
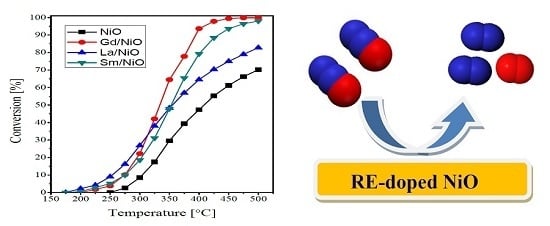
:
1. Introduction
2. Results
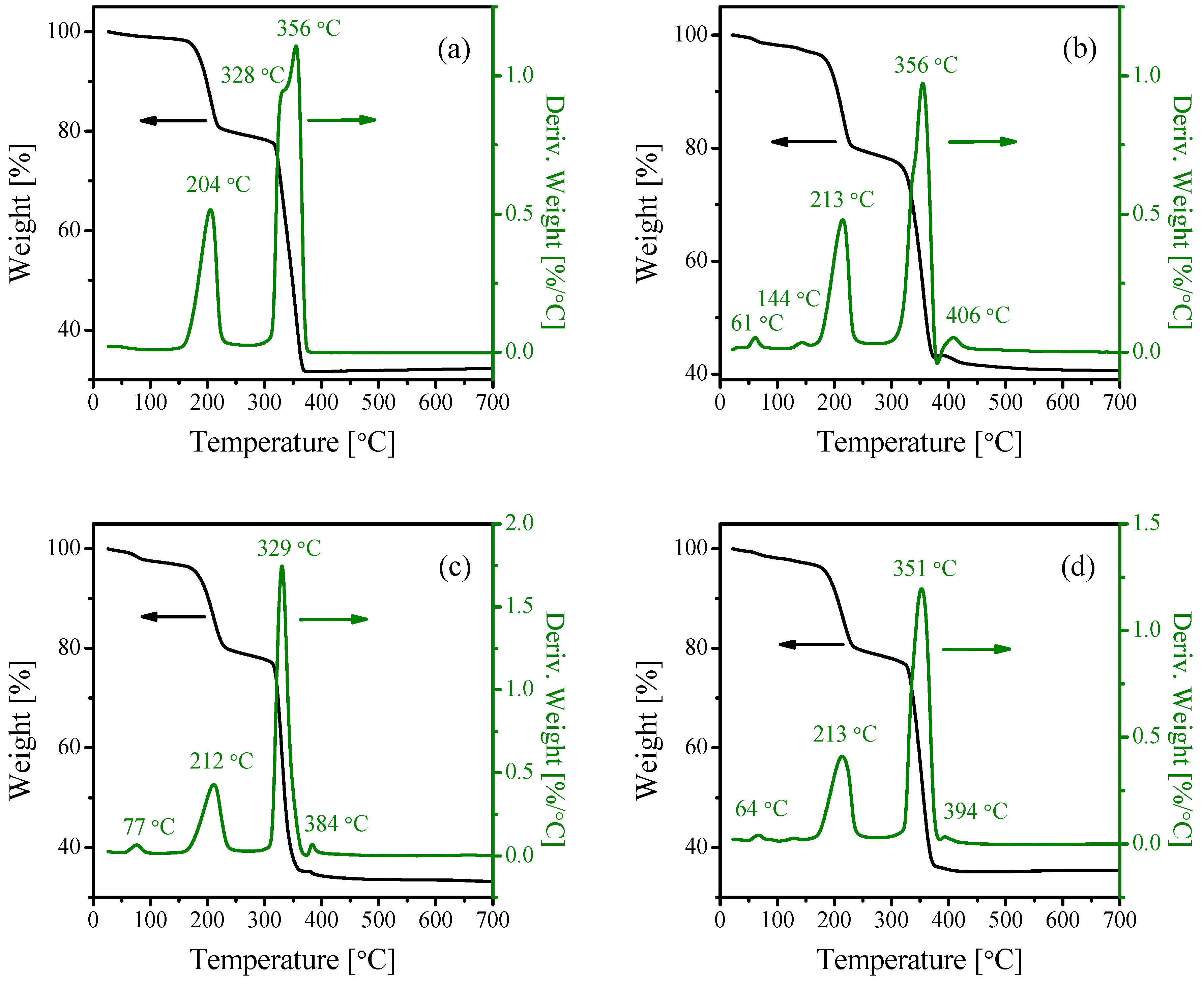
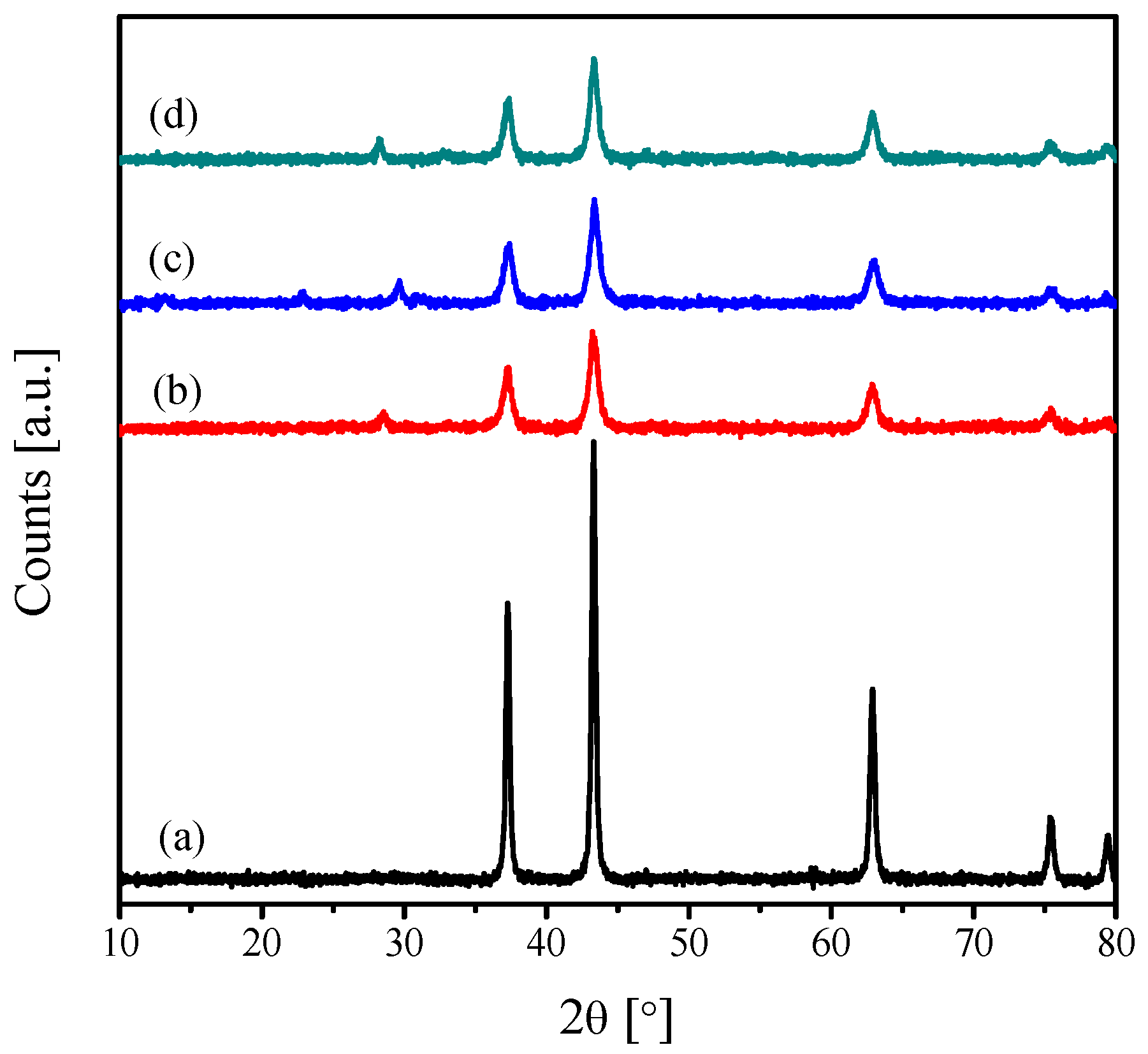
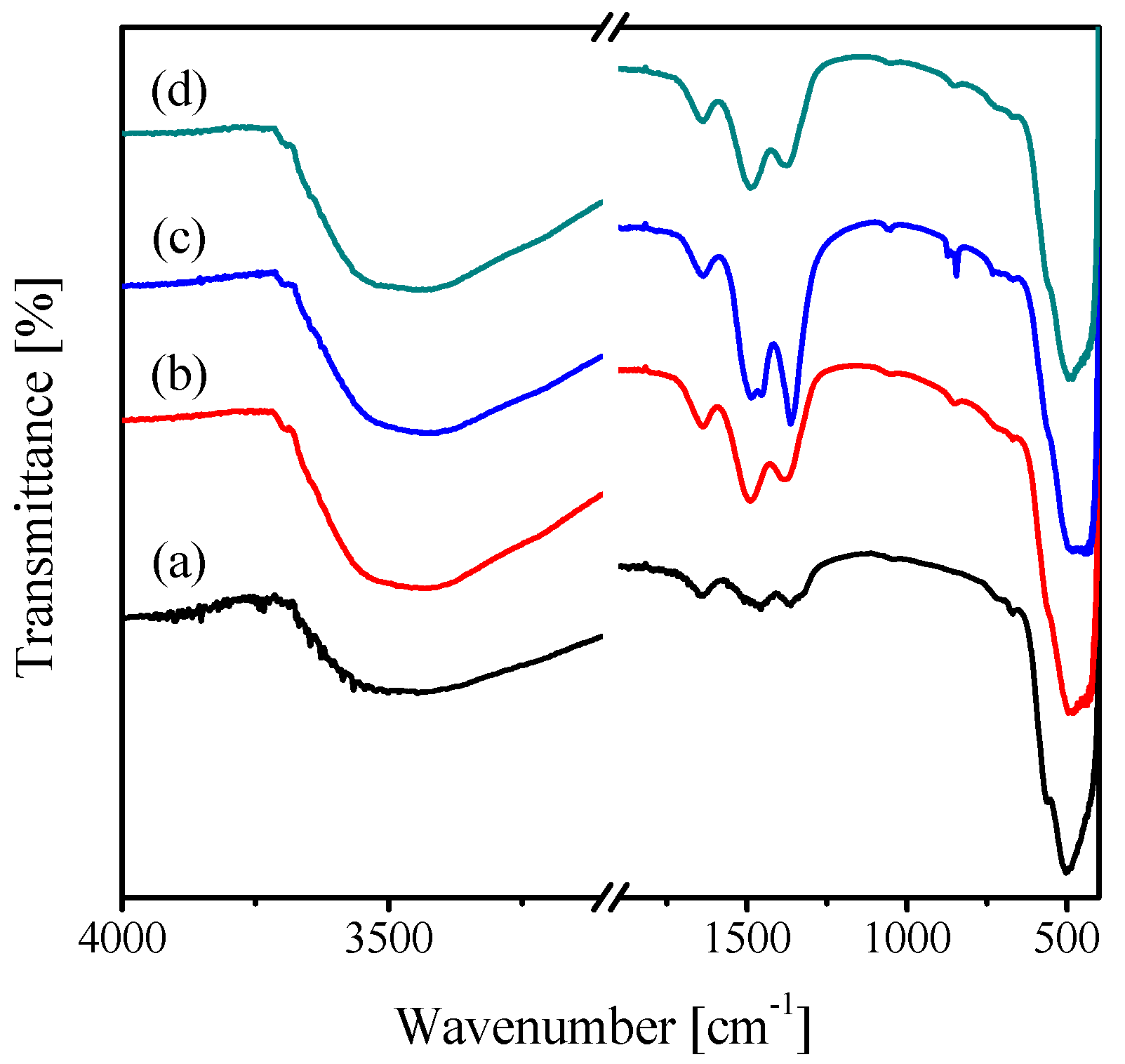
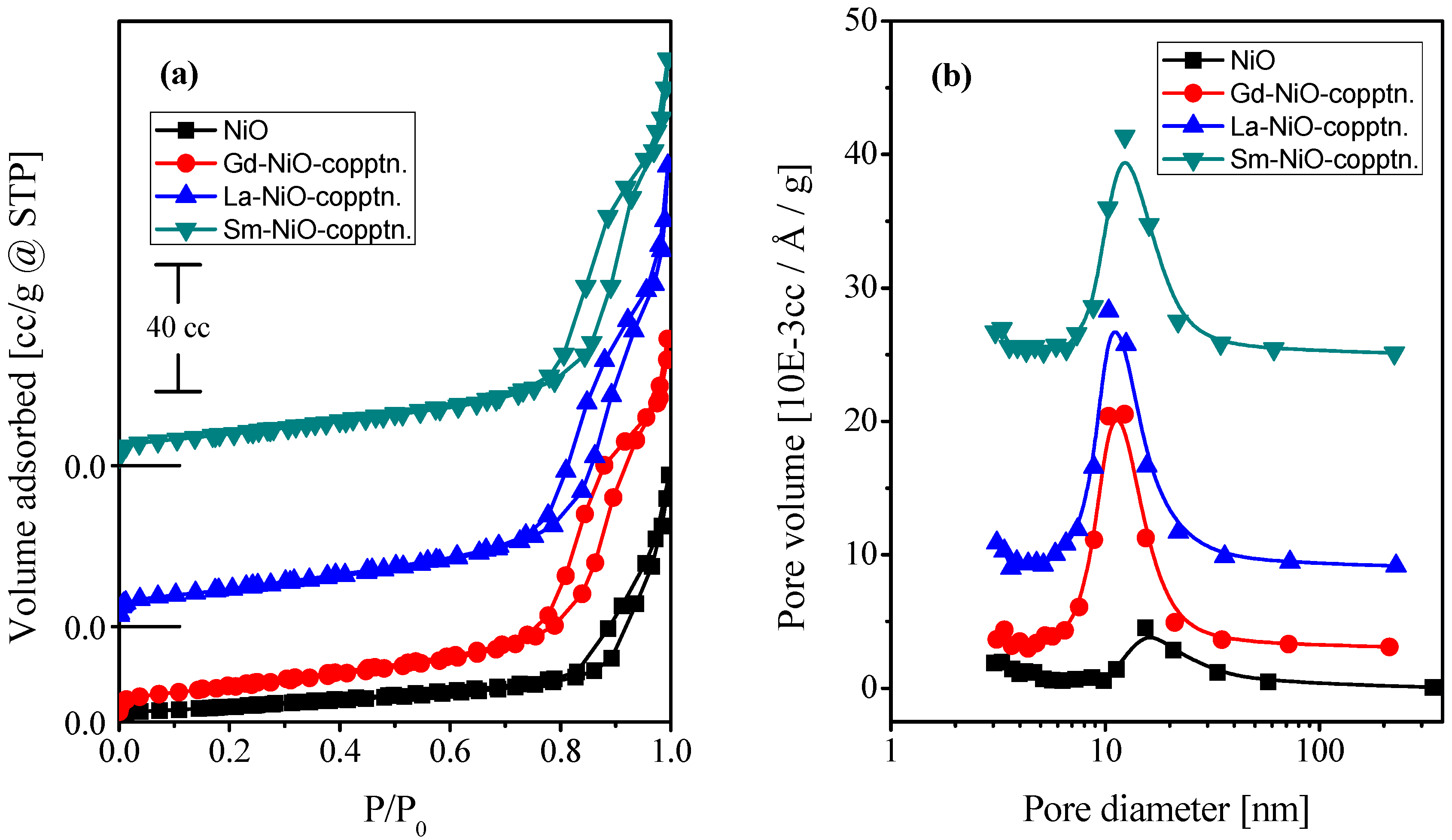
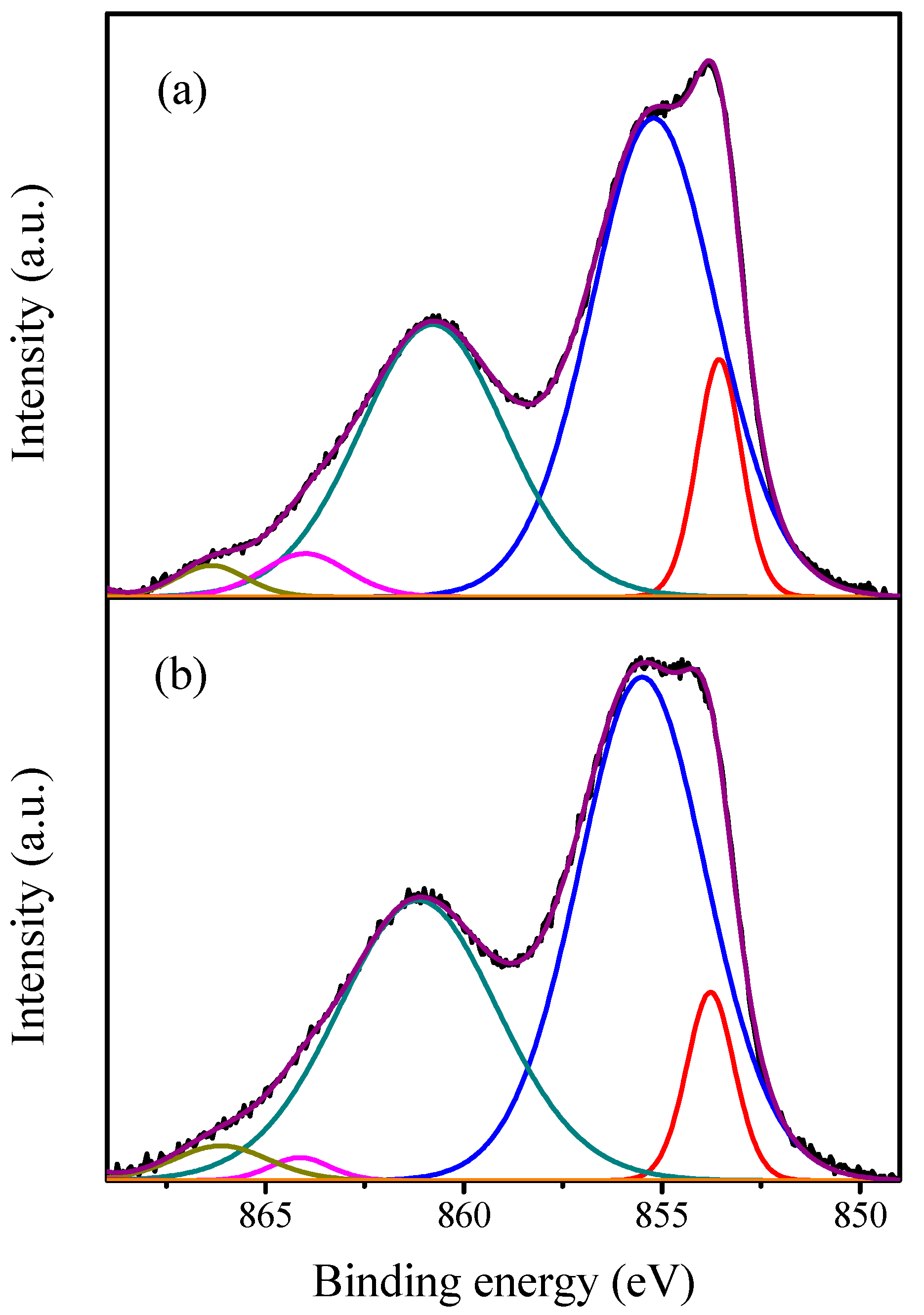
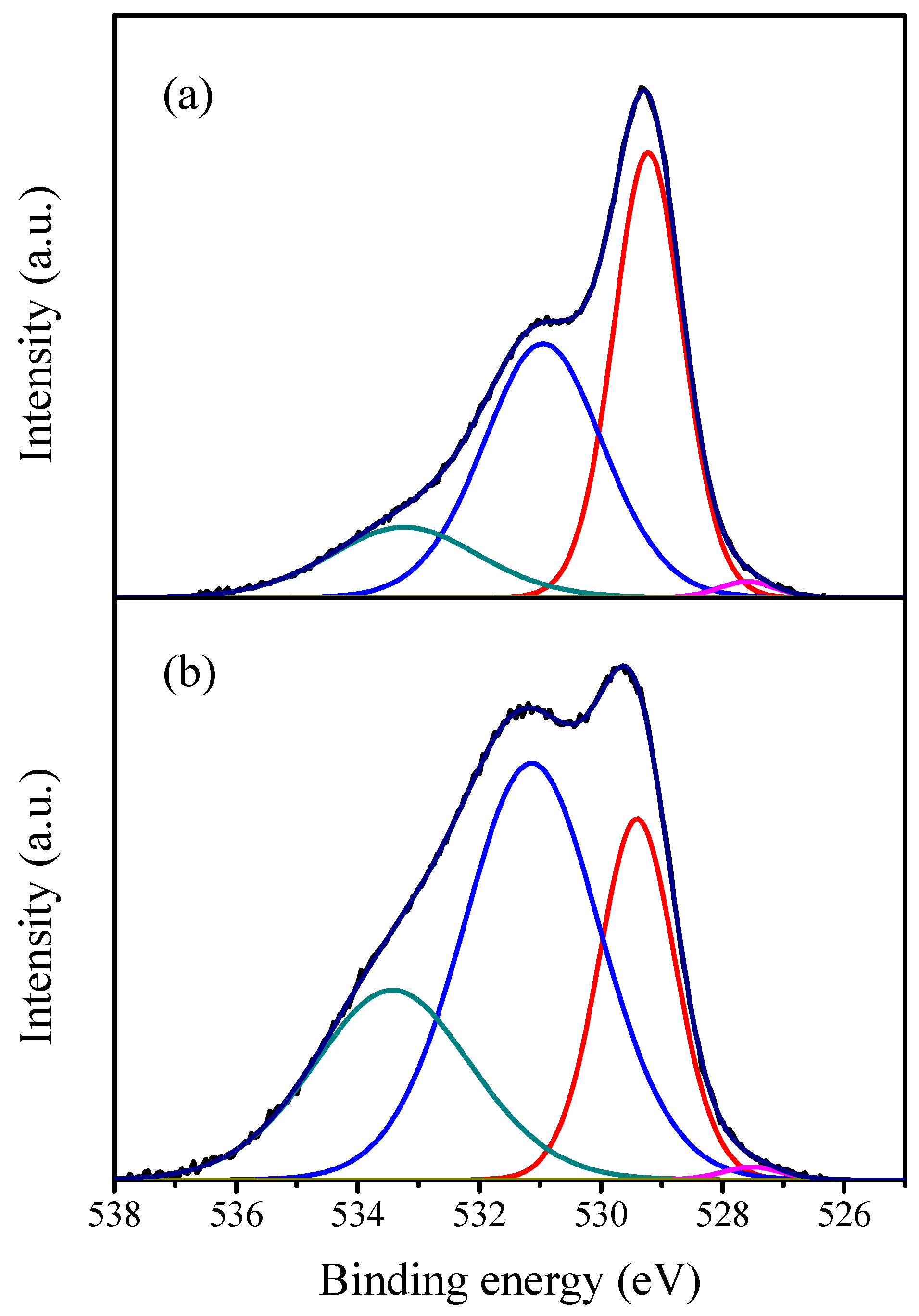
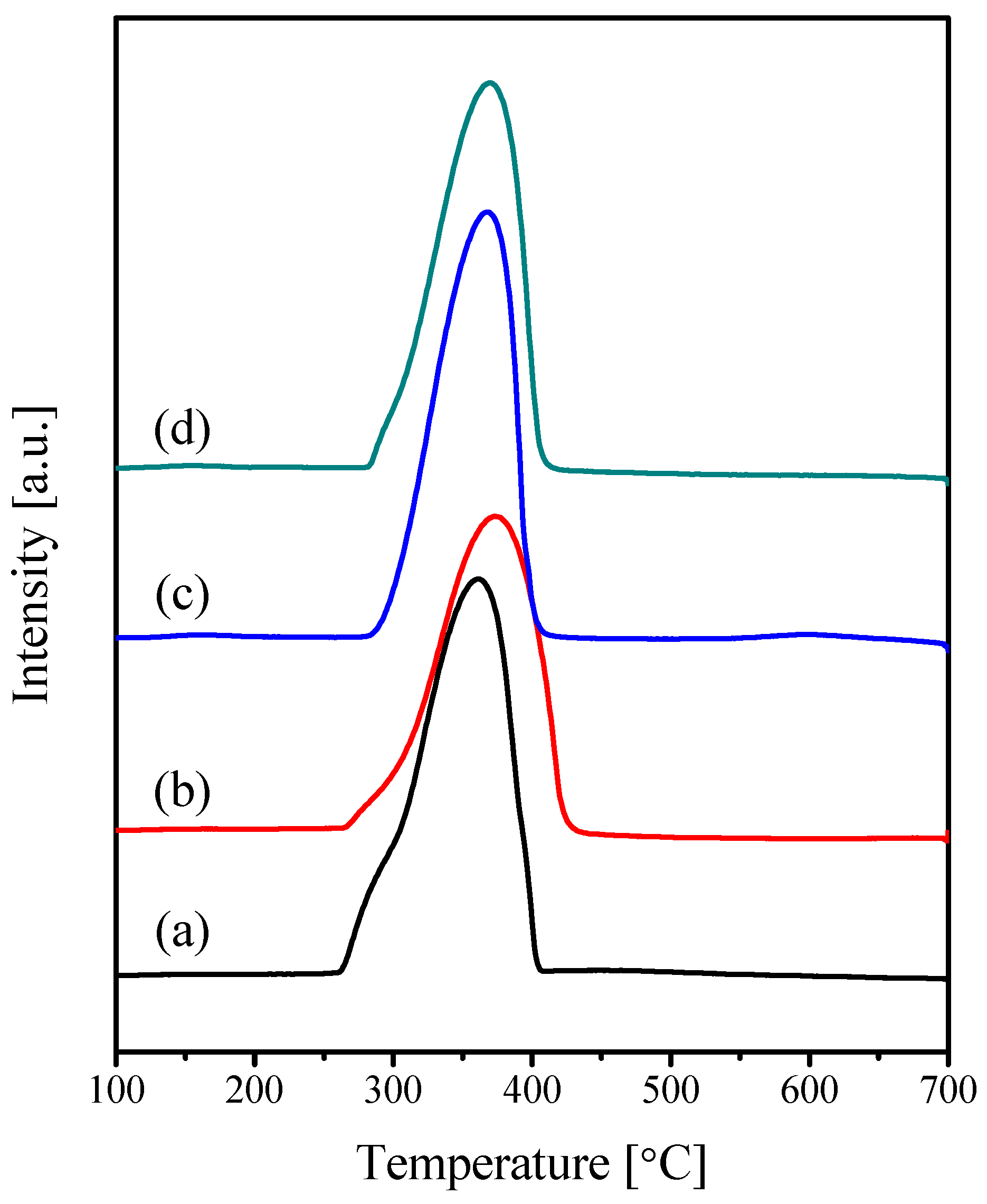
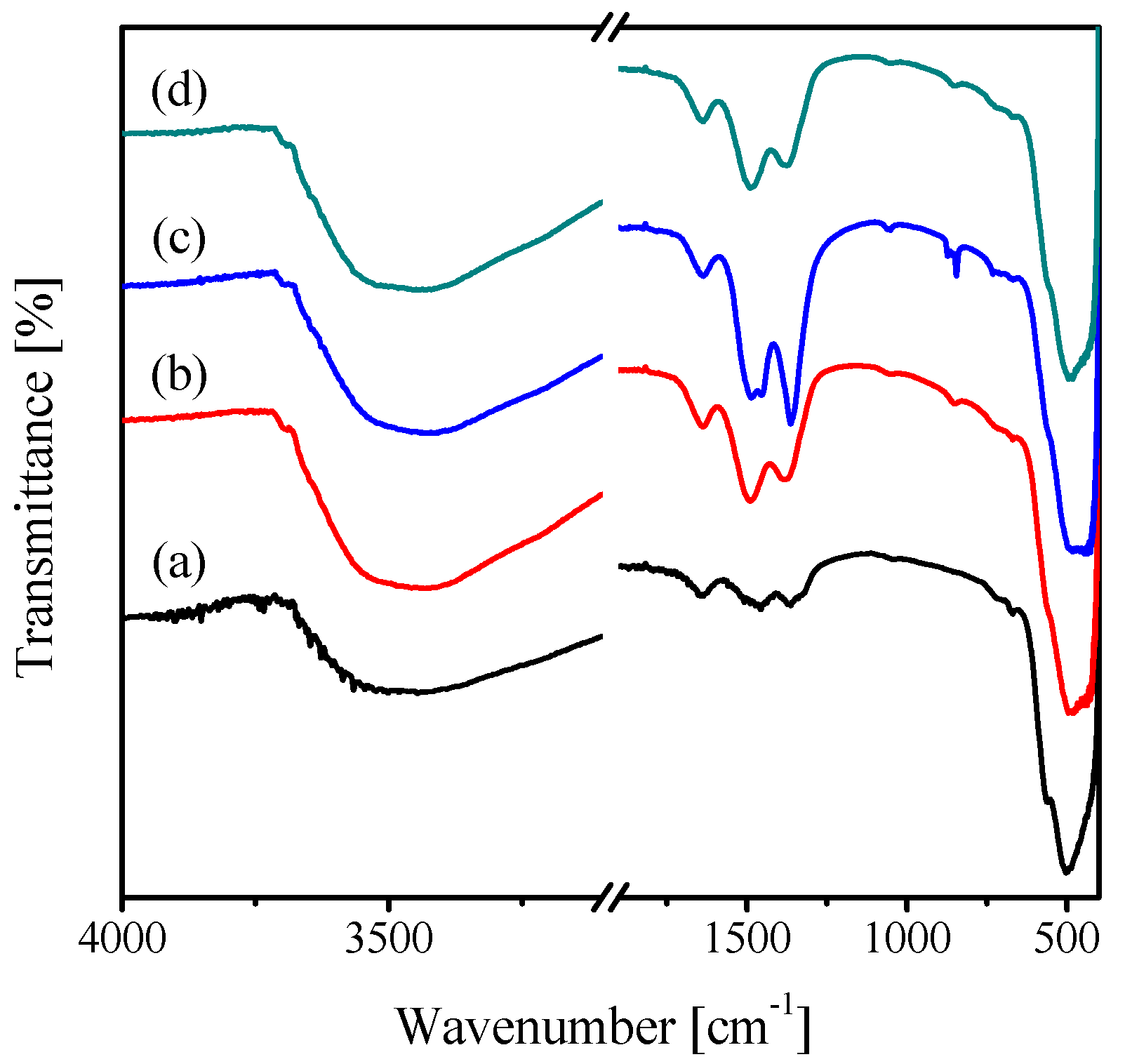
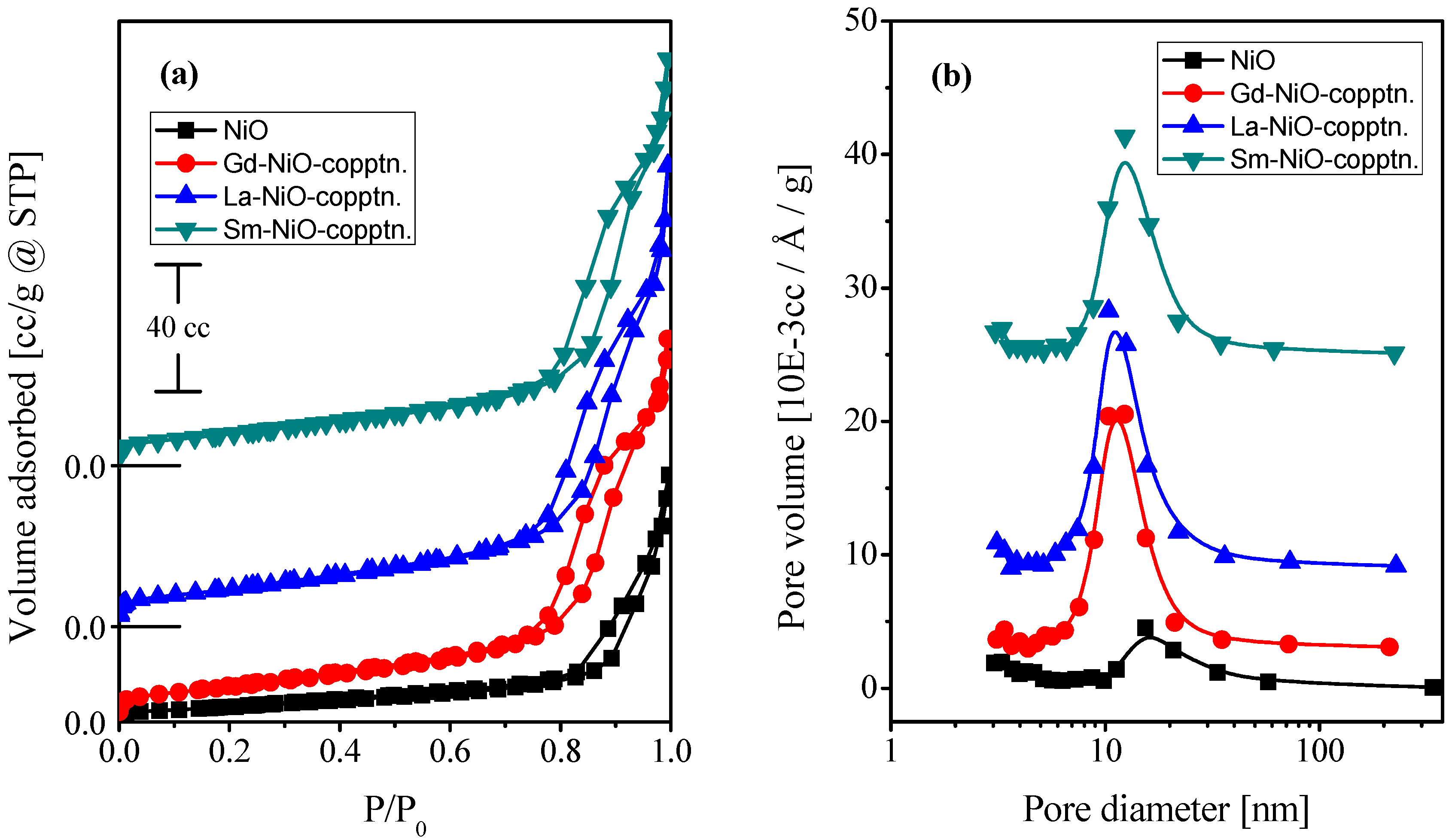
2.1. Characterization of the Catalysts
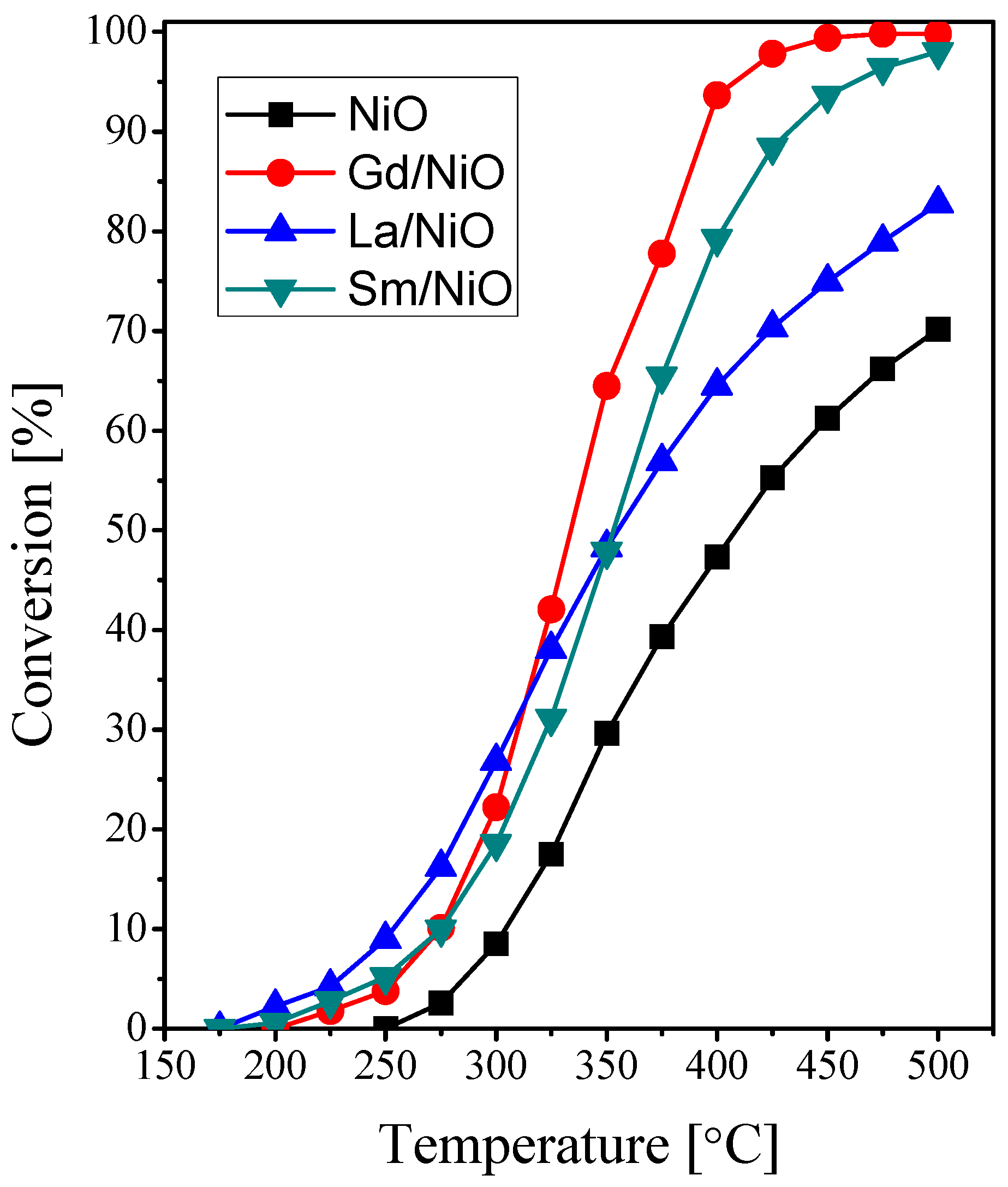
2.2. Catalytic Activity Measurements
3. Materials and Methods
3.1. Catalysts Preparation
3.2. Characterization Techniques
3.3. Catalytic Decomposition of N2O
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Centi, G.; Perathoner, S.; Vazzana, F.; Marella, M.; Tomaselli, M.; Mantegazza, M. Novel catalysts and catalytic technologies for N2O removal from industrial emissions containing O2, H2O and SO2. Adv. Environ. Res. 2000, 4, 325–338. [Google Scholar]
- Kapteijn, F.; Rodriguez-Mirasol, J.; Moulijn, J.A. Heterogeneous catalytic decomposition of nitrous oxide. Appl. Catal. B 1996, 9, 25–64. [Google Scholar]
- Pérez-Ramírez, J.; Kapteijn, F.; Schöffel, K.; Moulijn, J.A. Formation and control of N2O in nitric acid production where do we stand today? Appl. Catal. B 2003, 44, 117–151. [Google Scholar]
- Pérez-Ramírez, J. Prospects of N2O emission regulations in the European fertilizer industry. Appl. Catal. B 2007, 70, 31–35. [Google Scholar]
- Yan, L.; Ren, T.; Wang, X.; Ji, D.; Suo, J. Catalytic decomposition of N2O over MxCo1−xCo2O4 (M = Ni, Mg) spinel oxides. Appl. Catal. B 2003, 45, 85–90. [Google Scholar]
- Yan, L.; Ren, T.; Wang, X.; Gao, Q.; Ji, D.; Suo, J. Excellent catalytic performance of ZnxCo1−xCo2O4 spinel catalysts for the decomposition of nitrous oxide. Catal. Commun. 2003, 4, 505–509. [Google Scholar]
- Abu-Zied, B.M.; Soliman, S.A.; Abdellah, S.E. Enhanced direct N2O decomposition over CuxCo1−xCo2O4 (0.0 ≤ x ≤ 1.0) spinel-oxide catalysts. J. Ind. Eng. Chem. 2015, 21, 814–821. [Google Scholar]
- Abu-Zied, B.M.; Soliman, S.A.; Abdellah, S.E. Pure and Ni-substituted Co3O4 spinel catalysts for direct N2O decomposition. Chin. J. Catal. 2014, 35, 1105–1112. [Google Scholar]
- Stelmachowski, P.; Zasada, F.; Maniak, G.; Granger, P.; Inger, M.; Wilk, M.; Kotarba, A.; Sojka, Z. Optimization of multicomponent cobalt spinel catalyst for N2O abatement from nitric acid plant tail gases: Laboratory and pilot plant studies. Catal. Lett. 2009, 130, 637–641. [Google Scholar]
- Xue, L.; Zhang, C.; He, H.; Teraoka, Y. Catalytic decomposition of N2O over CeO2 promoted Co3O4 spinel catalyst. Appl. Catal. B 2007, 75, 167–174. [Google Scholar]
- Abu-Zied, B.M.; Soliman, S.A. Nitrous oxide decomposition over MCO3–Co3O4 (M = Ca, Sr, Ba) catalysts. Catal. Lett. 2009, 132, 299–310. [Google Scholar]
- Basahel, S.N.; Abd El-Maksod, I.H.; Abu-Zied, B.M.; Mokhtar, M. Effect of Zr4+ doping on the stabilization of ZnCo-mixed oxide spinel system and its catalytic activity towards N2O decomposition. J. Alloys Compd. 2010, 493, 630–635. [Google Scholar]
- Maniak, G.; Stelmachowski, P.; Kotarba, A.; Sojka, Z.; Rico-Pérez, V.; Bueno-López, A. Rationales for the selection of the best precursor for potassium doping of cobalt spinel based deN2O catalyst. Appl. Catal. B 2013, 136, 302–307. [Google Scholar]
- Abu-Zied, B.M. Nitrous oxide decomposition over alkali-promoted magnesium cobaltite catalysts. Chin. J. Catal. 2011, 32, 264–272. [Google Scholar]
- Franken, T.; Palkovits, R. Investigation of potassium doped mixed spinels CuxCo3−xO4 as catalysts for an efficient N2O decomposition in real reaction conditions. Appl. Catal. B 2015, 176, 298–305. [Google Scholar]
- Klyushina, A.; Pacultová, K.; Krejčová, S.; Słowik, G.; Jirátová, K.; Kovanda, F.; Ryczkowski, J.; Obalová, L. Advantages of stainless steel sieves as support for catalytic N2O decomposition over K-doped Co3O4. Catal. Today 2015, 257, 2–10. [Google Scholar]
- Amrousse, R.; Katsumi, T. Substituted ferrite MxFe1−xFe2O4 (M = Mn, Zn) catalysts for N2O catalytic decomposition processes. Catal. Commun. 2012, 26, 194–198. [Google Scholar]
- Amrousse, R.; Tsutsumi, A.; Bachar, A.; Lahcene, D. N2O catalytic decomposition over nano-sized particles of Co-substituted Fe3O4 substrates. Appl. Catal. A 2013, 450, 253–260. [Google Scholar]
- Wang, J.; Feng, M.; Zhang, H.-J.; Xu, X.-F. Catalytic decomposition of N2O over Mg-Fe mixed oxides. J. Fuel Chem. Technol. 2014, 42, 1464–1469. [Google Scholar]
- Pasha, N.; Lingaiah, N.; Reddy, P.S.S.; Sai Prasad, P.S. Direct decomposition of N2O over cesium-doped CuO catalysts. Catal. Lett. 2009, 127, 101–106. [Google Scholar]
- Abu-Zied, B.M.; Bawaked, S.M.; Kosa, S.A.; Schwieger, W. Effect of Some Rare Earth Oxides Doping on the Morphology, Crystallite Size, Electrical Conductivity and N2O Decomposition Activity of CuO Catalyst. Int. J. Electrochem. Sci. 2016, 11, 1568–1580. [Google Scholar]
- Abu-Zied, B.M.; Bawaked, S.M.; Kosa, S.A.; Schwieger, W. Impact of Gd-, La-, Nd- and Y-doping on the textural, electrical conductivity and N2O decomposition activity of CuO Catalyst. Int. J. Electrochem. Sci. 2016, 11, 2230–2246. [Google Scholar]
- Pasha, N.; Lingaiah, N.; Reddy, P.S.S.; Sai Prasad, P.S. An investigation into the effect of Cs promotion on the catalytic activity of NiO in the direct decomposition of N2O. Catal. Lett. 2007, 118, 64–68. [Google Scholar] [CrossRef]
- Abu-Zied, B.M.; Asiri, A.M. The role of alkali promoters in enhancing the direct N2O decomposition reactivity over NiO catalysts. Chin. J. Catal. 2015, 36, 1837–1845. [Google Scholar]
- Wu, H.-P.; Li, W.-J.; Guo, L.; Pan, Y.-F.; Xu, X.-F. Effect of promoter species and precursors on catalytic activity of alkali metal promoted NiAl mixed oxides for N2O decomposition. J. Fuel Chem. Technol. 2011, 39, 550–555. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, X.; Zhang, X.; Tursun, M.; Yu, H. Promotion by Co of a NiO-BaCO3 catalyst for N2O decomposition. Chin. J. Catal. 2015, 36, 344–347. [Google Scholar]
- Zhang, F.; Wang, X.; Zhang, X.; Turxun, M.; Yu, H.; Zhao, J. The catalytic activity of NiO for N2O decomposition doubly promoted by barium and cerium. Chem. Eng. J. 2014, 256, 365–371. [Google Scholar] [CrossRef]
- Barakat, A.; Al-Noaimi, M.; Suleiman, M.; Aldwayyan, A.S.; Hammouti, B.; Ben Hadda, T.; Haddad, S.F.; Boshaala, A.; Warad, I. One step synthesis of NiO nanoparticles via solid-state thermal decomposition at low-temperature of novel aqua(2,9-dimethyl-1,10-phenanthroline)NiCl2 complex. Int. J. Mol. Sci. 2013, 14, 23941–23954. [Google Scholar] [CrossRef] [PubMed]
- El-Kemary, M.; Nagy, N.; El-Mehasseb, I. Nickel oxide nanoparticles: Synthesis and spectral studies of interactions with glucose. Mater. Sci. Semicond. Process. 2013, 16, 1747–1752. [Google Scholar] [CrossRef]
- Abu-Zied, B.M.; Soliman, S.A. Thermal decomposition of praseodymium acetate as a precursor of praseodymium oxide catalyst. Thermochim. Acta 2008, 470, 91–97. [Google Scholar] [CrossRef]
- Abu-Zied, B.M.; Bawaked, S.M.; Kosa, S.A.; Schwieger, W. Effect of Pr, Sm, and Tb doping on the morphology, crystallite size, and N2O decomposition activity of Co3O4 nanorods. J. Nanomater. 2015, 2015, 580582:1–580582:10. [Google Scholar] [CrossRef]
- Leofanti, G.; Padovan, M.; Tozzola, G.; Venturelli, B. Surface area and pore texture of catalysts. Catal. Today 1998, 41, 207–219. [Google Scholar] [CrossRef]
- Piumetti, M.; Hussain, M.; Fino, D.; Russo, N. Mesoporous silica supported Rh catalysts for high concentration N2O decomposition. Appl. Catal. B 2015, 165, 158–168. [Google Scholar] [CrossRef]
- Babu, G.A.; Hayakawa, Y.; Ravi, G. Microwave synthesis and magnetic investigations of surfactant assisted NiO nanostructures. Mater. Lett. 2015, 149, 54–57. [Google Scholar] [CrossRef]
- Kaichev, V.V.; Gladky, A.Y.; Prosvirin, I.P.; Saraev, A.A.; Hävecker, M.; Knop-Gericke, A.; Schlögl, R.; Bukhtiyarov, V.I. In situ XPS study of self-sustained oscillations in catalytic oxidation of propane over nickel. Surf. Sci. 2013, 609, 113–118. [Google Scholar] [CrossRef]
- Guil-López, R.; La Parola, V.; Peña, M.A.; Fierro, J.L.G. Evolution of the Ni-active centres into ex hydrotalcite oxide catalysts during the COx-free hydrogen production by methane decomposition. Int. J. Hydrogen Energy 2012, 37, 7042–7055. [Google Scholar] [CrossRef]
- Rodríguez, J.L.; Valenzuela, M.A.; Poznyak, T.; Lartundo, L.; Chairez, I. Reactivity of NiO for 2,4-D degradation with ozone: XPS studies. J. Hazard. Mater. 2013, 262, 472–481. [Google Scholar]
- Biesinger, M.C.; Payne, B.P.; Lau, L.W.M.; Gerson, A.; Smart, R.St.C. X-ray photoelectron spectroscopic chemical state quantification of mixed nickel metal, oxide and hydroxide systems. Surf. Interf. Anal. 2009, 41, 324–332. [Google Scholar] [CrossRef]
- Payne, B.P.; Biesinger, M.C.; McIntyre, N.S. Use of oxygen/nickel ratios in the XPS characterisation of oxide phases on nickel metal and nickel alloy surfaces. J. Electron. Spectrosc. Relat. Phenom. 2012, 185, 159–166. [Google Scholar] [CrossRef]
- Abu-Zied, B.M.; Asiri, A.M. An investigation of the thermal decomposition of silver acetate as a precursor for nano-sized Ag-catalyst. Thermochim. Acta 2014, 581, 110–117. [Google Scholar] [CrossRef]
- Yu, F.; Xu, X.; Peng, H.; Yu, H.; Dai, Y.; Liu, W.; Ying, J.; Sun, Q.; Wang, X. Porous NiO nano-sheet as an active and stable catalyst for CH4 deep oxidation. Appl. Catal. A 2015, 507, 109–118. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhou, D.; Wu, Y.; Wu, T. Oxidative dehydrogenation of ethane over RE-NiO (RE = La, Nd, Sm, Gd) catalysts. J. Rare Earths 2013, 31, 669–673. [Google Scholar] [CrossRef]
- Asano, K.; Ohnishi, C.; Iwamoto, S.; Shioya, Y.; Inoue, M. Potassium-doped Co3O4 catalyst for direct decomposition of N2O. Appl. Catal. B 2008, 78, 242–249. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Crystallite Size (nm) | SBET (m2·g−1) | Vp (cc·g−1) | Pd (nm) |
|---|---|---|---|---|
| NiO | 45 | 17 | 0.120 | 15.423 |
| Gd/NiO | 22 | 37 | 0.188 | 12.34 |
| La/NiO | 25 | 41 | 0.226 | 10.377 |
| Sm/NiO | 25 | 37 | 0.200 | 12.351 |
| Catalyst | Ni0 | NiO | ||
|---|---|---|---|---|
| BE (eV) | Area (%) | BE (eV) | Area (%) | |
| NiO | 853.55 | 14.96 | 855.21 | 85.04 |
| Gd/NiO | 853.53 | 12.15 | 855.13 | 87.84 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abu-Zied, B.M.; Bawaked, S.M.; Kosa, S.A.; Schwieger, W. Rare Earth-Promoted Nickel Oxide Nanoparticles as Catalysts for N2O Direct Decomposition. Catalysts 2016, 6, 70. https://doi.org/10.3390/catal6050070
Abu-Zied BM, Bawaked SM, Kosa SA, Schwieger W. Rare Earth-Promoted Nickel Oxide Nanoparticles as Catalysts for N2O Direct Decomposition. Catalysts. 2016; 6(5):70. https://doi.org/10.3390/catal6050070
Chicago/Turabian StyleAbu-Zied, Bahaa M., Salem M. Bawaked, Samia A. Kosa, and Wilhelm Schwieger. 2016. "Rare Earth-Promoted Nickel Oxide Nanoparticles as Catalysts for N2O Direct Decomposition" Catalysts 6, no. 5: 70. https://doi.org/10.3390/catal6050070
APA StyleAbu-Zied, B. M., Bawaked, S. M., Kosa, S. A., & Schwieger, W. (2016). Rare Earth-Promoted Nickel Oxide Nanoparticles as Catalysts for N2O Direct Decomposition. Catalysts, 6(5), 70. https://doi.org/10.3390/catal6050070