
Heterogeneous Asymmetric Oxidation Catalysis Using Hemophore HasApf. Application in the Chemoenzymatic Deracemization of sec-Alcohols with Sodium Borohydride

Abstract
:1. Introduction
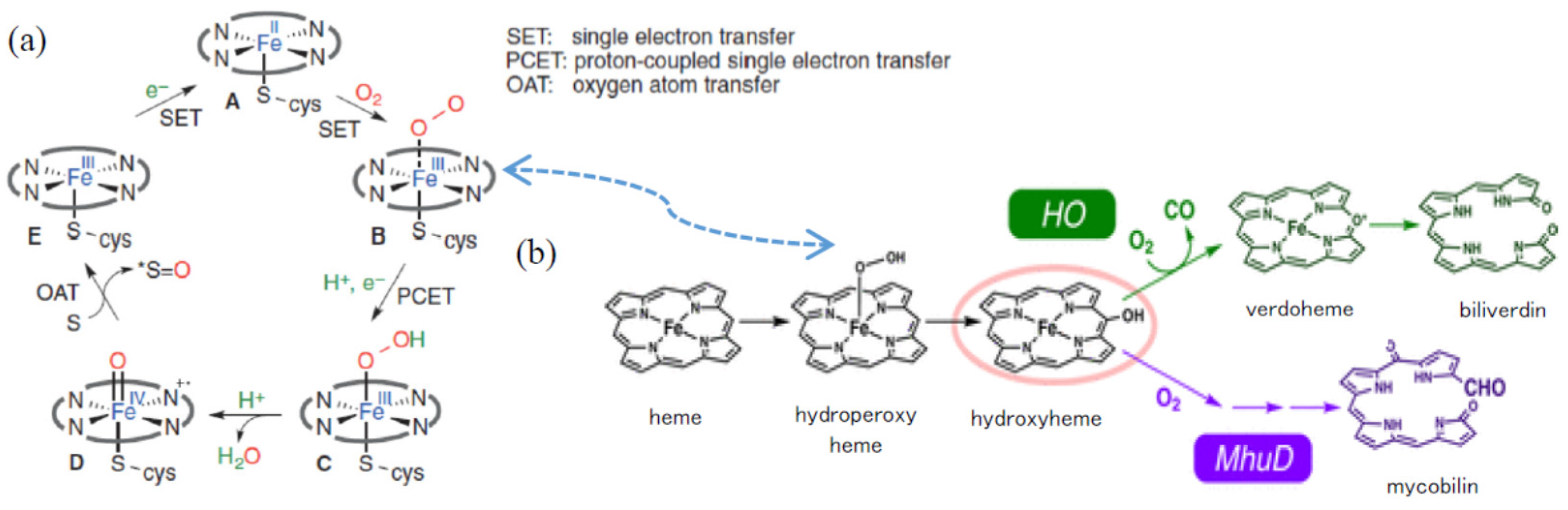
2. Results and Discussion
2.1. Characterization of HasApf
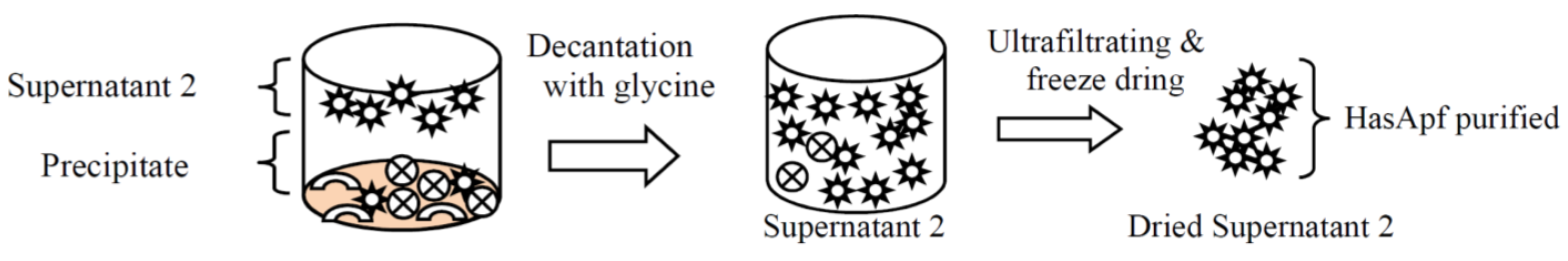
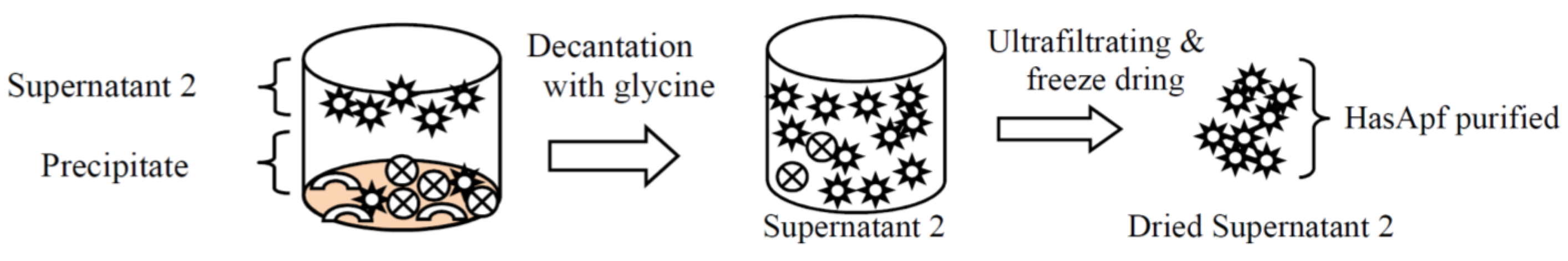
2.1.1. Purifications of HasApf from PP
2.1.2. Identification of HasApf from Supernatant 2 Filtrated by Vivaspin
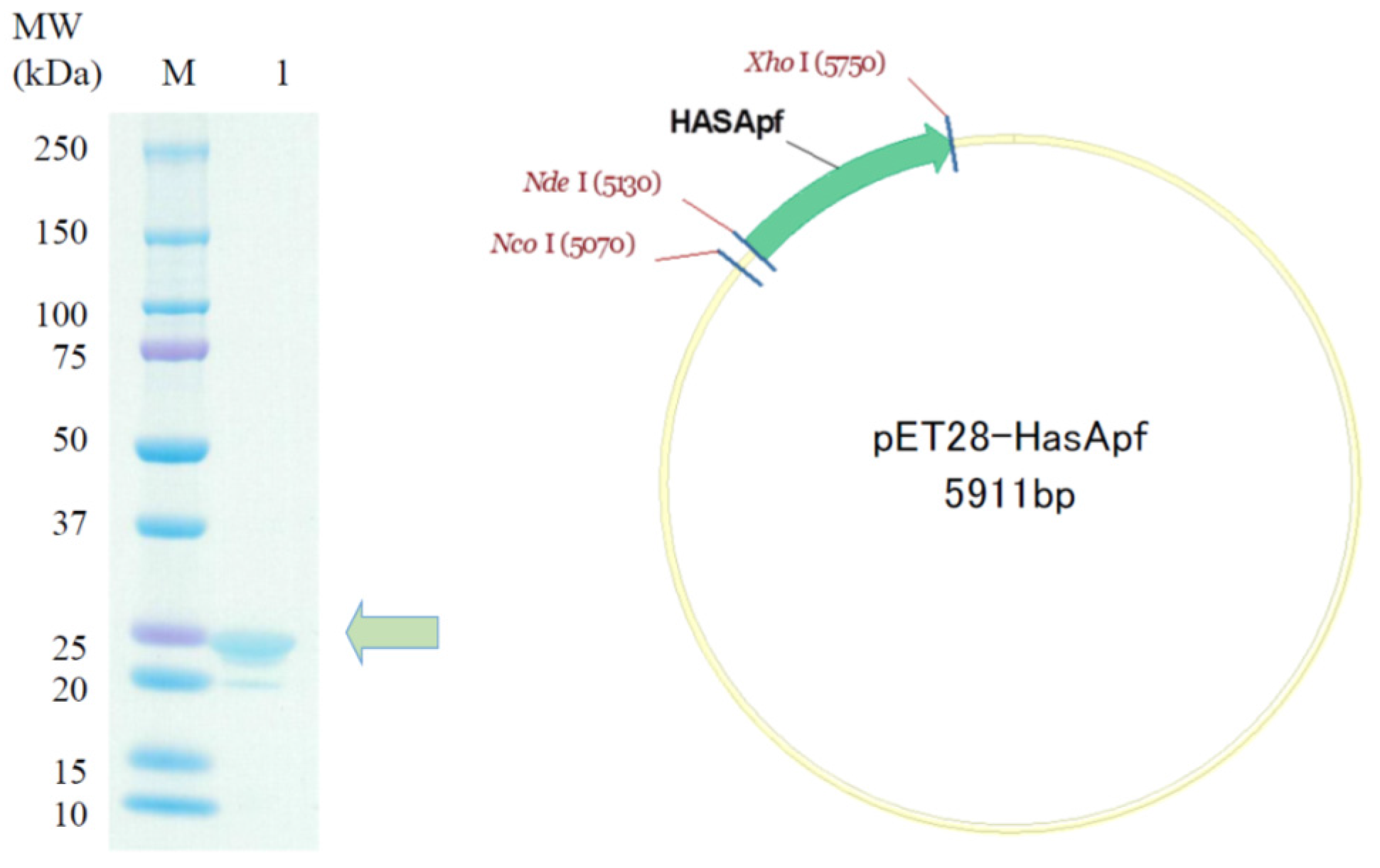
2.2. Preparation of HasApf Expressed by Escherichia coli Cells
HasApf Protein Expression and Purification
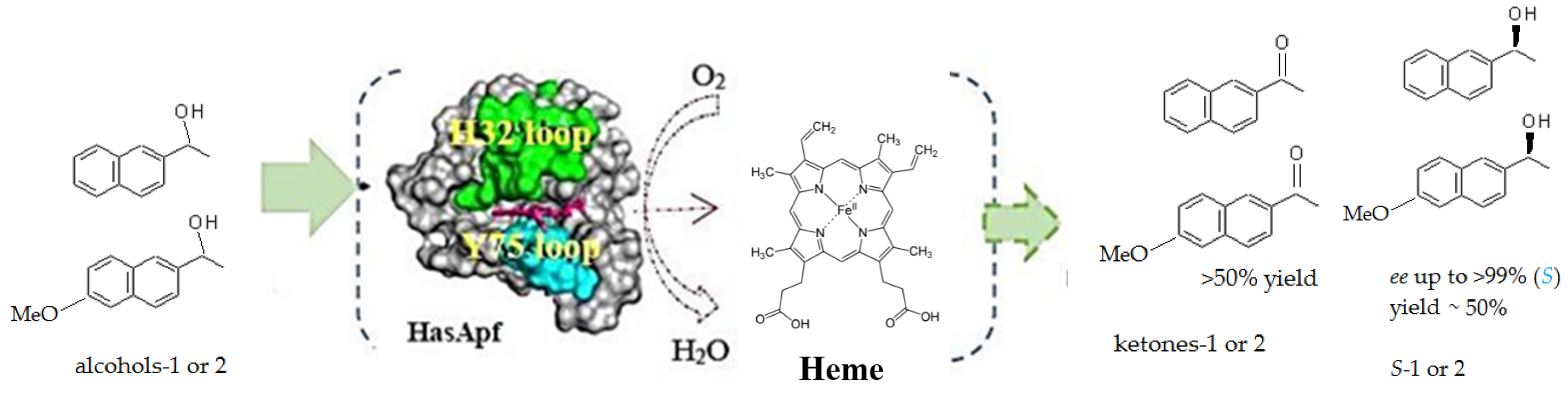
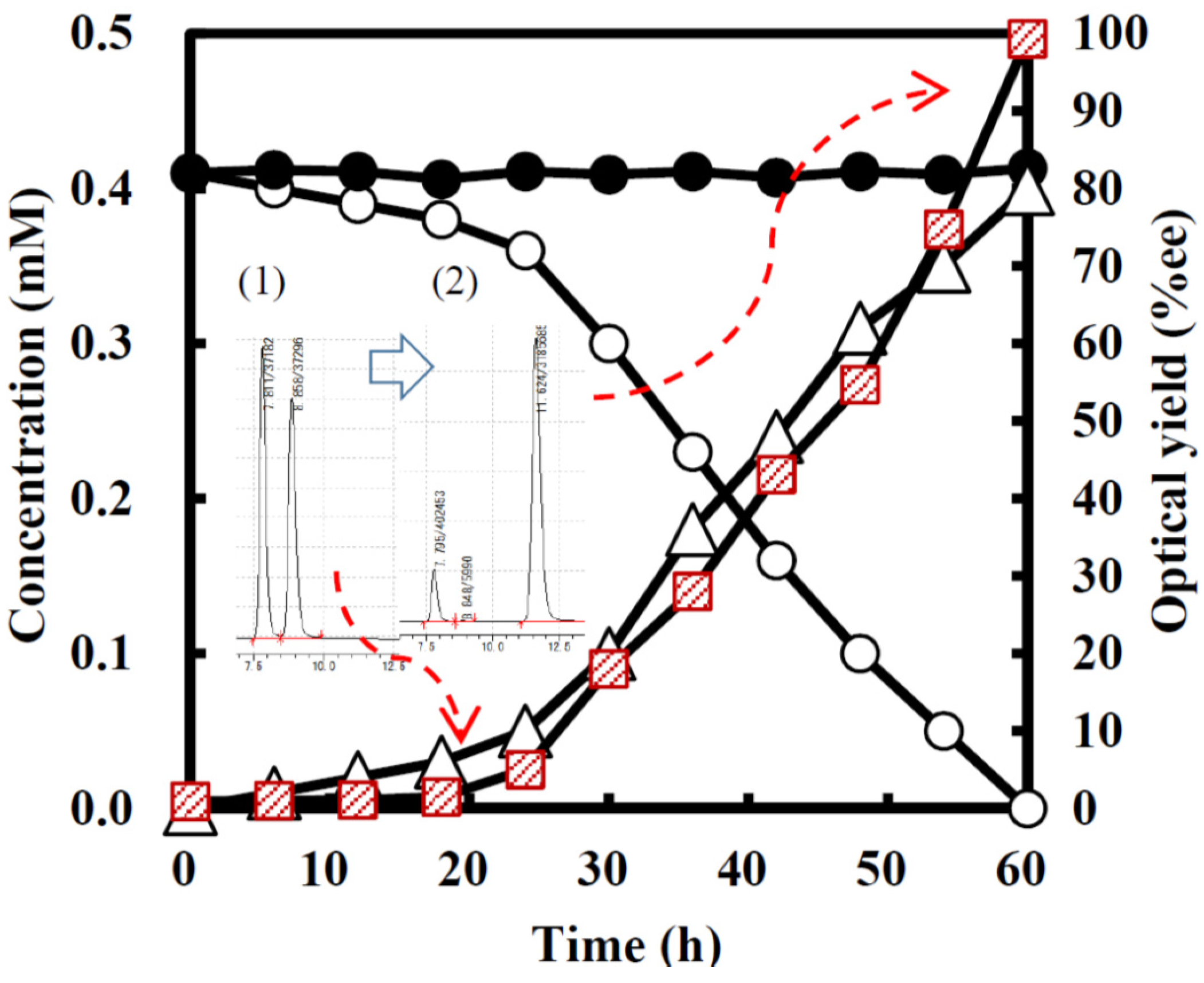
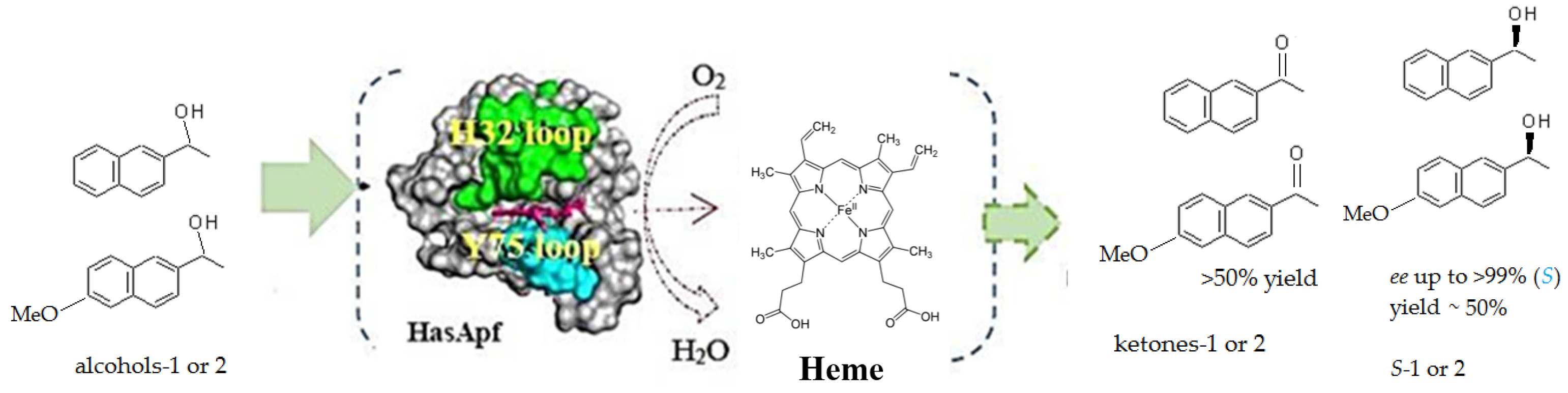
2.3. Kinetic Resolution and Physicochemical Analysis
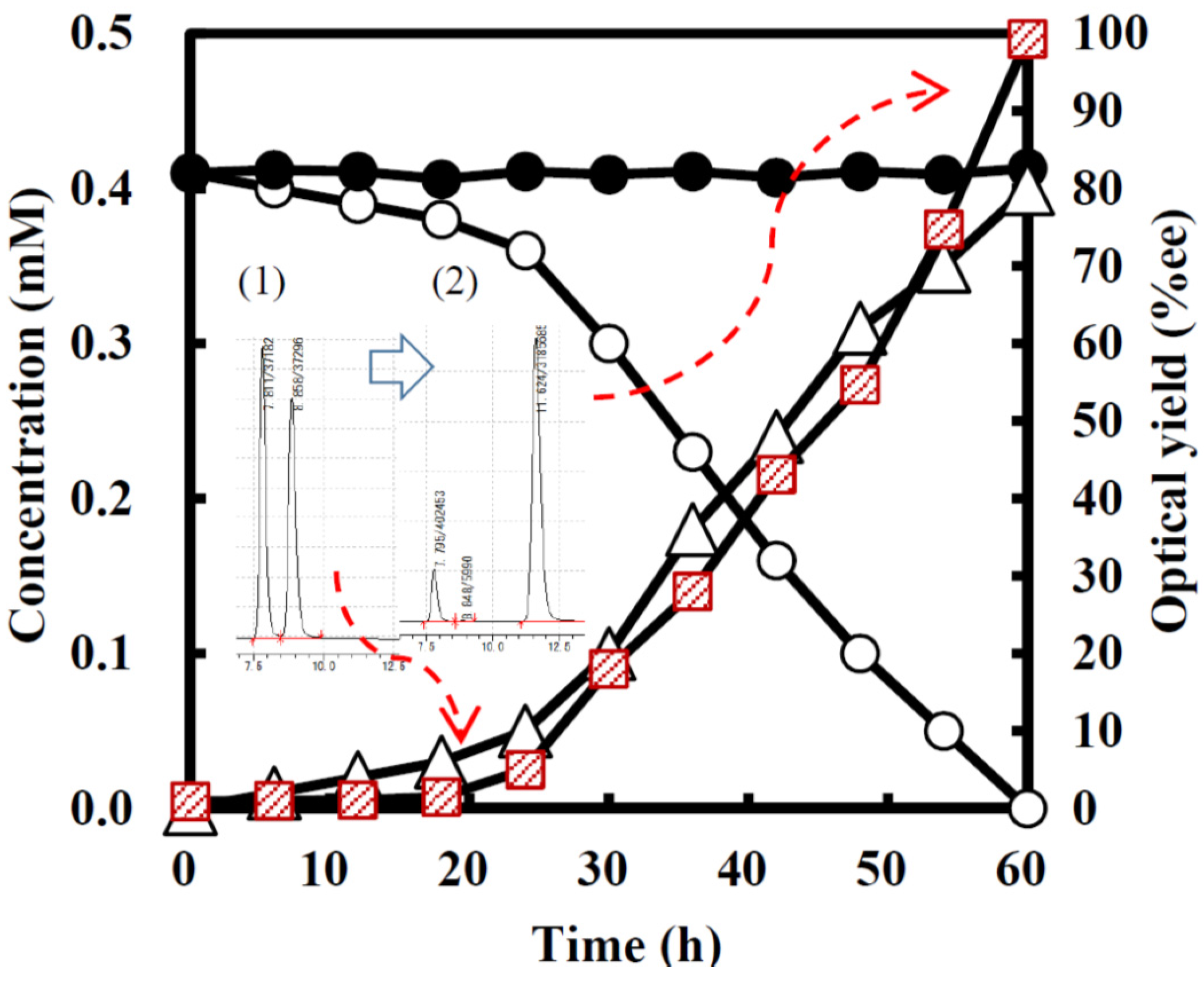
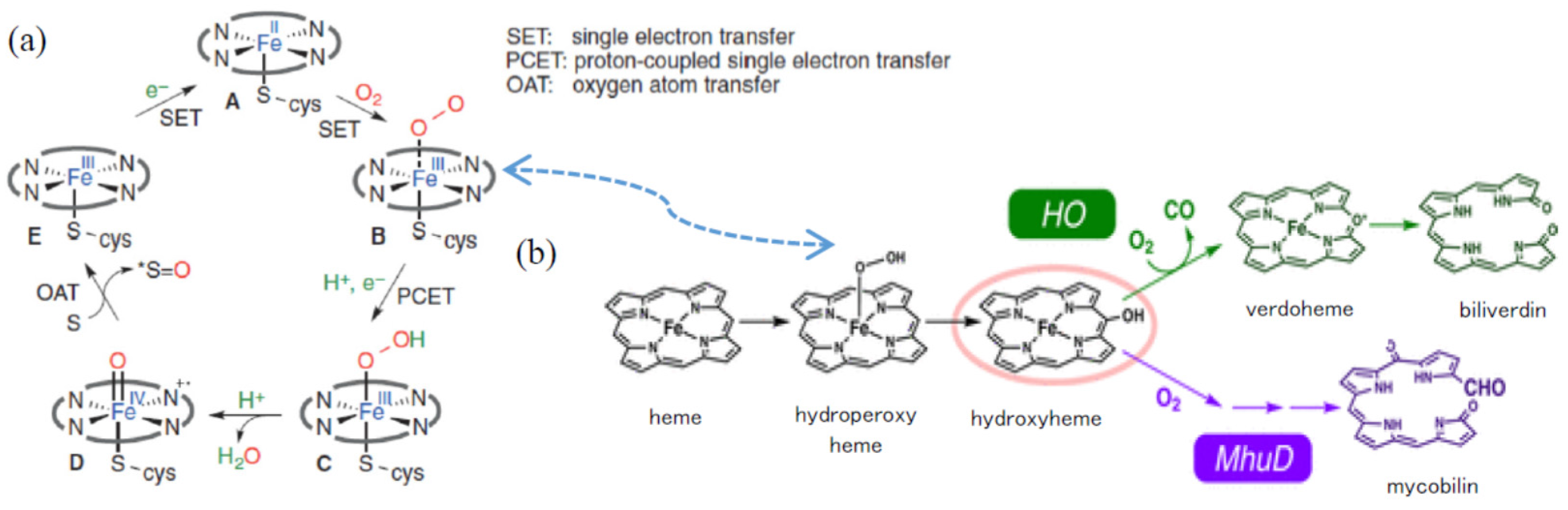
2.3.1. Asymmetric Oxidation of rac-1 with Bacterial HasApf
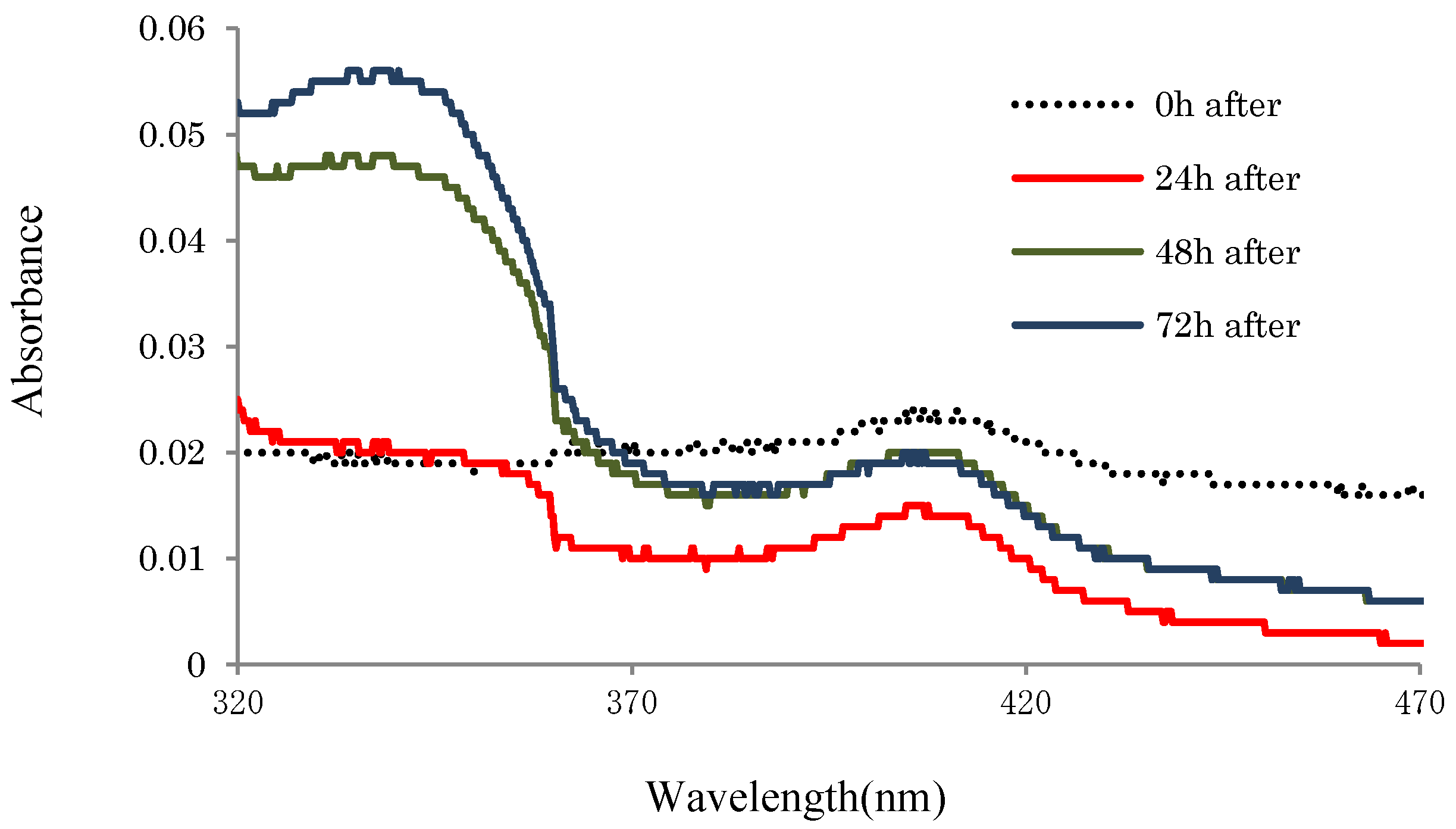
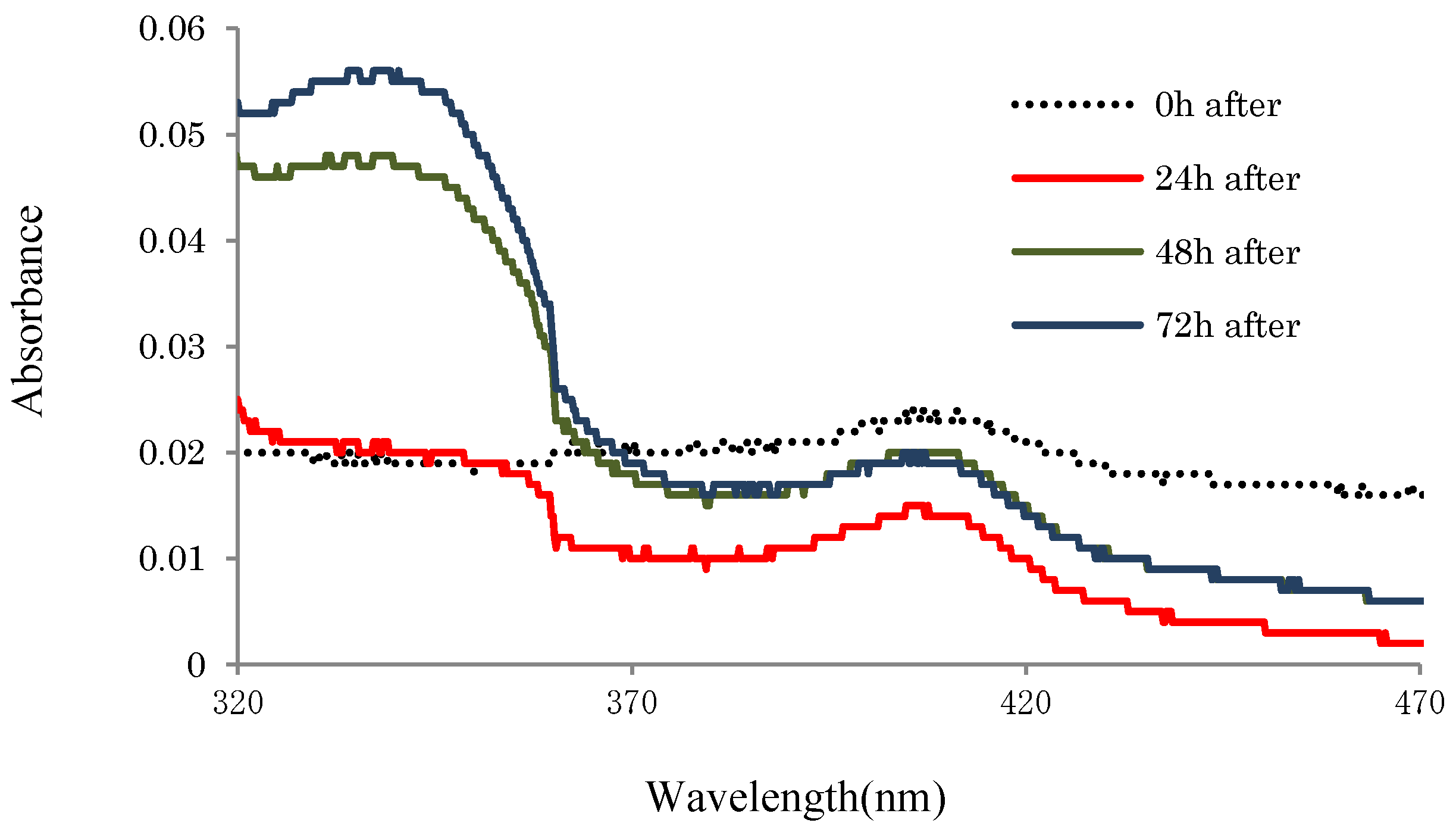
2.3.2. UV-Absorbances at Each of the HasApf Reaction Times
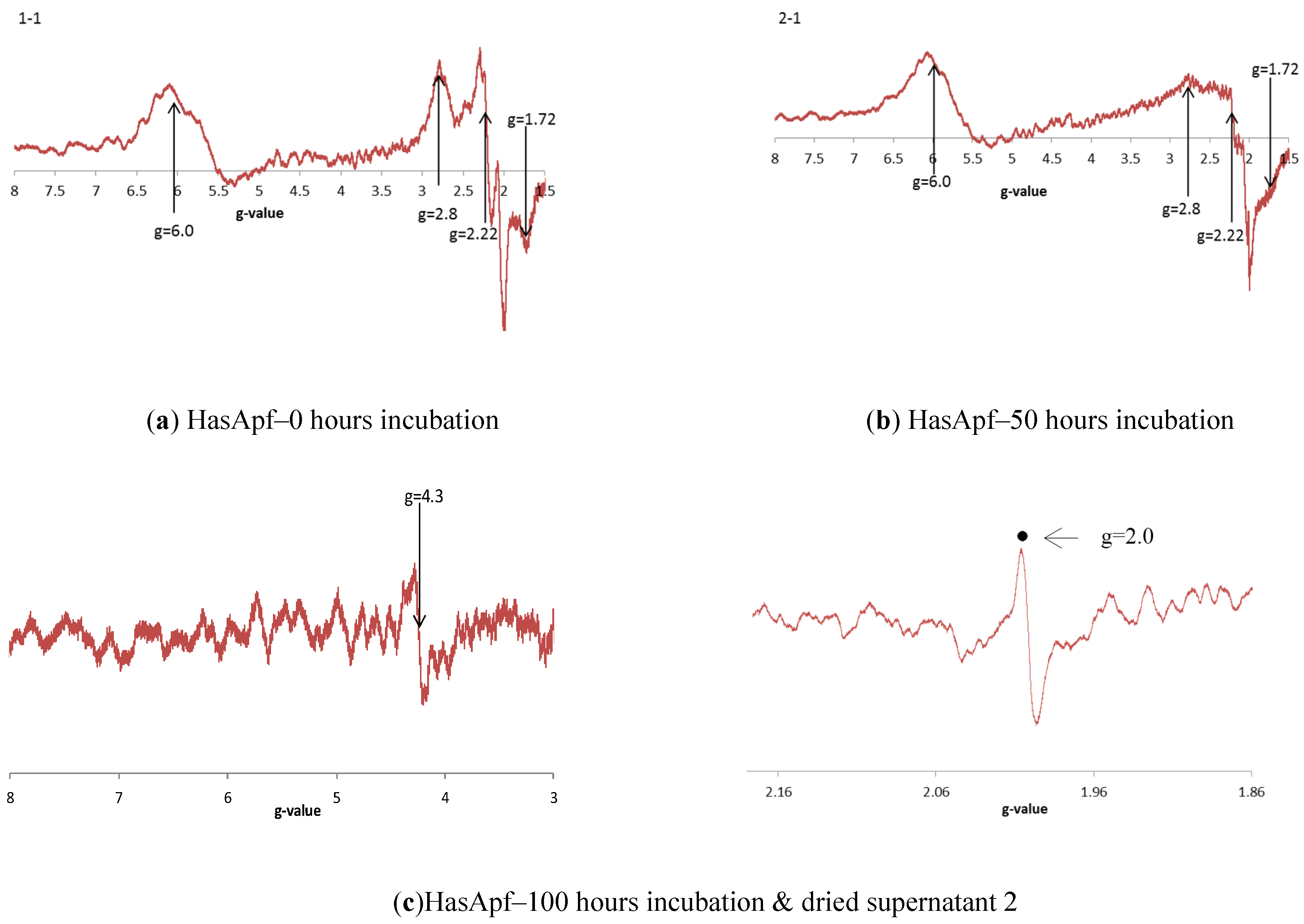
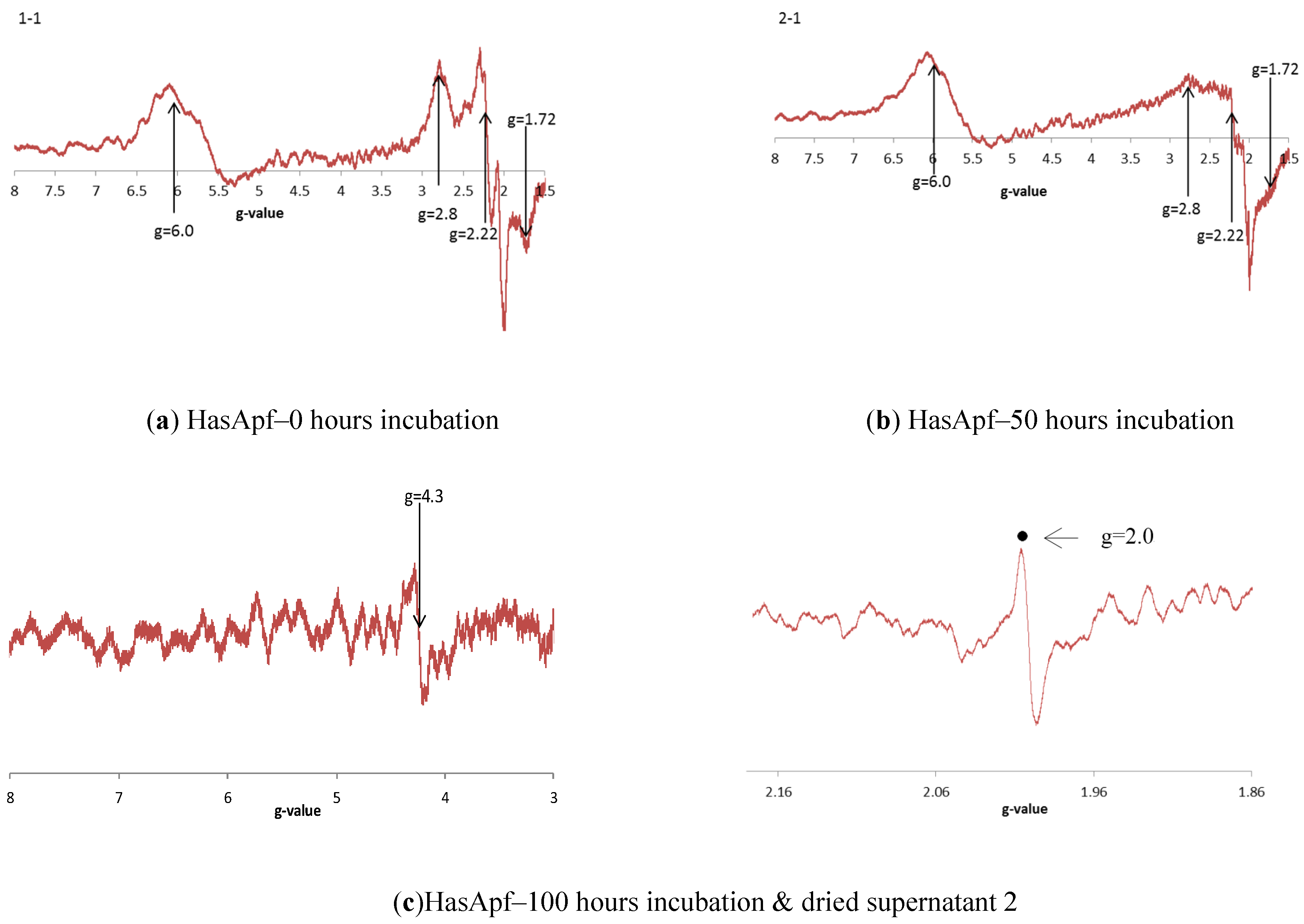
2.3.3. ESR Spectra at Each of the HASApf Reaction Times
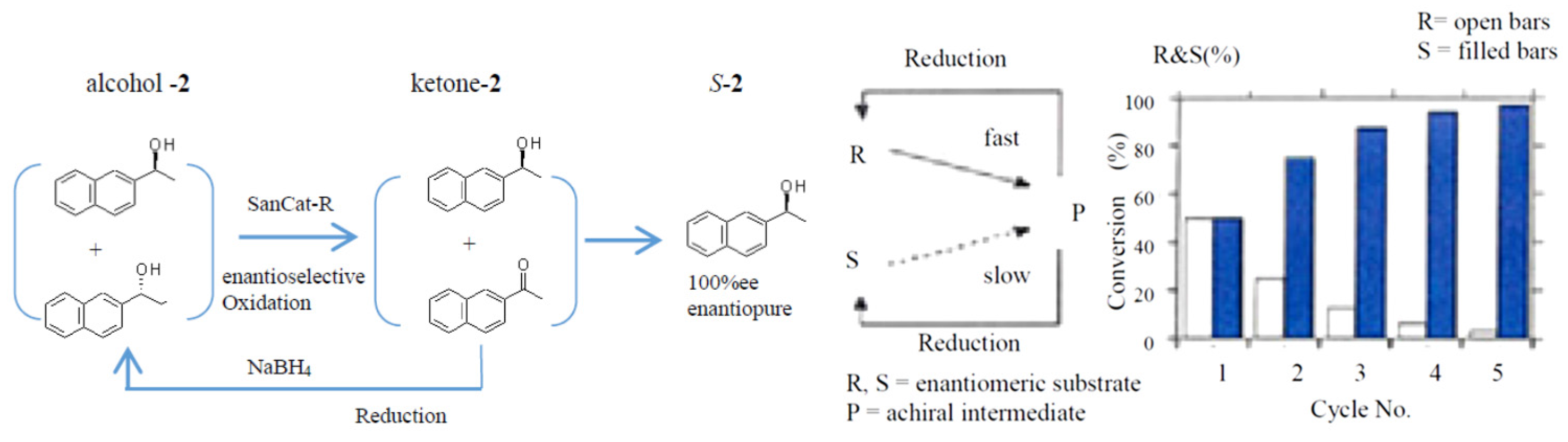
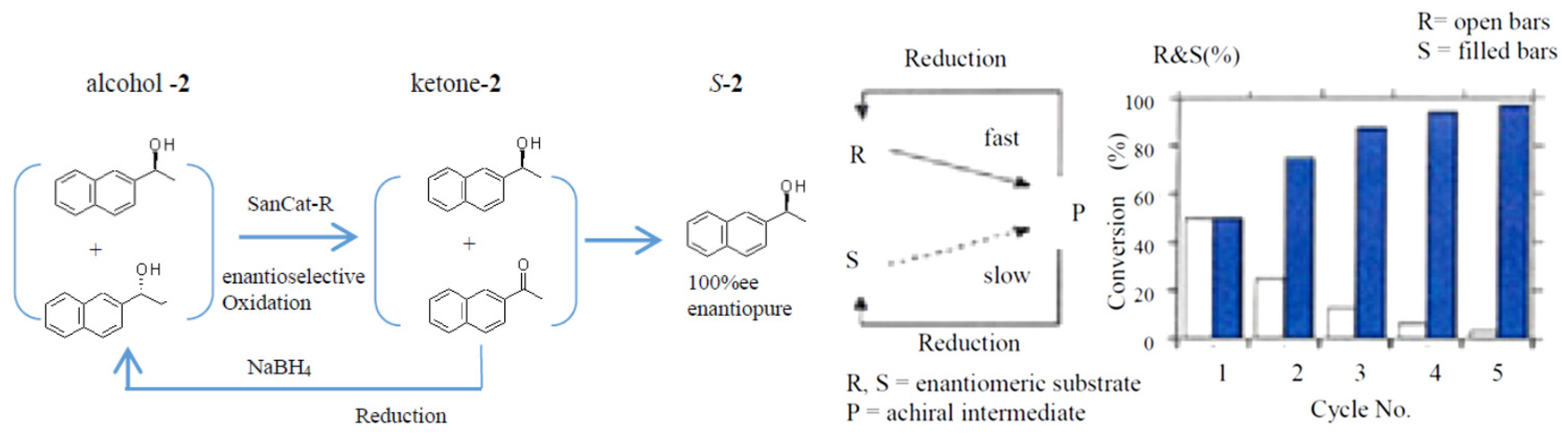
2.4. Application to a Chemoenzymatic Process
Cyclic Deracemization of rac-1 or -2 Using HasApf and NaBH4
3. Experimental Section
3.1. Catalyst Preparation
3.1.1. HasApf (Dried Precipitate 2) Preparation from PP(pea protein)
3.1.2. HasApf Protein Expression and Purification
3.1.3. SDS-PAGE of Expressed HasApf
3.1.4. General Procedure for Reactions Using (1) Expressed HasApf or (2) PEG-Precipitate 2
3.1.5. ESR Analyses and UV–vis Spectroscopy of Expressed HasApf
4. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Auld, D.S.; Bergman, T. The role of zinc for alcohol dehydrogenase structure and function. Cell Mol. Life Sci. 2008, 65, 3961–3970. [Google Scholar] [CrossRef] [PubMed]
- Sobolov, B.S.; Leonida, D.M.; Malik, B.A.; Voivodov, I.K.; McKinney, F.; Kim, J.; Fry, J.J. Cross-linked LDH crystals for lactate synthesis coupled to electroenzymatic regeneration of NADH. J. Org. Chem. 1996, 61, 2125–2128. [Google Scholar] [CrossRef]
- Liou, Y.; Charoenkwan, P.; Srinivasulu, Y.S.; Vasylenko, T.; Lai, S.; Lee, H.; Chen, Y.; Huang, H.; Ho, S. SCMHBP: Prediction and analysis of heme binding proteins using propensity scores of dipeptides. BMC Bioinform. 2014, 15, S4. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, S.; Nakahara, A.; Sato, T. Mechanism of Heme Uptake by Heme Acquisition System A. Chem. Lett. 2011, 40, 362–363. [Google Scholar] [CrossRef]
- Ozaki, S.; Sato, A.; Migita, T.C.; Uchida, T.; Ishimori, K. Spectroscopic Studies on HasA from Yersinia pseudotuberculosis. J. Inorg. Biochem. 2014, 138, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Krieg, S.; Huché, F.; Diederichs, K.; Izadi-Pruneyre, N.; Lecroisey, A.; Wandersman, P.C. Heme uptake across the outer membrane as revealed by crystal structures of the receptor-hemophore complex. Proc. Natl. Acad. Sci. USA 2009, 106, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Shoji, O.; Fujishiro, T.; Nakajima, H.; Kim, M.; Nagano, S.; Shiro, Y.; Watanabe, Y. Hydrogen Peroxide Dependent Monooxygenations by Tricking the Substrate Recognition of Cytochrome P450BSβ. Angew. Chem. Int. Ed. 2007, 46, 3656–3659. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd ed.; Kluwer Academic/Plenum: New York, NY, USA; Boston, MA, USA; Dorcrecht, The Netherlands; London, UK; Mosow, Russia, 2004. [Google Scholar]
- Machii, K.; Watanabe, Y. Acylperoxo-Iron (III) porphyrin complexes: A new entry of potent oxidants for the alkene epoxidation. J. Am. Chem. Soc. 1995, 117, 6691–6697. [Google Scholar] [CrossRef]
- Jepkorir, G.; Rodríguez, J.C.; Rui, H.; Im, W.; Lovell, S.; Battaile, K.P.; Alontaga, A.Y.; Yukl, E.T.; Moënne-Loccoz, P.; Rivera, M. Structural, NMR Spectroscopic, and Computational Investigation of Hemin Loading in the Hemophore HasAp from Pseudomonas aeruginosa. Am. Chem. Soc. 2010, 132, 9857–9872. [Google Scholar] [CrossRef] [PubMed]
- Giles, M.N.; Watts, B.A.; Giles, M.N.; Watts, B.A.; Giles, I.G.; Fry, H.F.; Littlechild, A.J.; Jacob, C. Metal and redox modulation of cysteine protein function. Chem. Biol. 2003, 10, 677–693. [Google Scholar] [CrossRef]
- Steinreiber, J.; Faber, K.; Griengl, H. De-racemization of enantiomers versus de-epimerization of diastereomers—Classification of dynamic kinetic asymmetric transformations (DYKAT). Chem. Eur. J. 2008, 14, 8060–8072. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.J. Deracemisation methods. Curr. Opin. Chem. Biol. 2010, 14, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Hein, J.E.; Hyunh, C.B.; Viedma, C.; Kellogg, R.M.; Blackmond, D.G. Pasteur’s tweezers revisited: On the mechanism of attrition-enhanced deracemization and resolution of chiral conglomerate solids. J. Am. Chem. Soc. 2012, 134, 12629–12636. [Google Scholar] [CrossRef] [PubMed]
- Carnell, A.J. Stereoinversions using microbial redox-reactions. Adv. Biochem. Eng./Biotechnol. 1999, 63, 57–72. [Google Scholar]
- Schober, M.; Toesch, M.; Knaus, T.; Strohmeier, G.A.; van Loo, B.; Fuchs, M.; Hollfelder, F.; Macheroux, P.; Faber, K. One-Pot Deracemization of sec-Alcohols: Enantioconvergent Enzymatic Hydrolysis of Alkyl Sulfates Using Stereocomplementary Sulfatases. Angew. Chem. Int. Ed. Engl. 2013, 52, 3277–3279. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, H. Chiral Resolution Function with Immobilized Food Proteins. Biotechnol. Prog. 2003, 19, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, H. Ability of different biomaterials to enantioselectively catalyze oxidation and reduction reactions. Biotechnol. Prog. 2004, 20, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, H. Treatment of germinated wheat to increase level of GABA and IP6 catalyzed by endogeneous enzymes. Biotechnol. Prog. 2005, 21, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, H.; Udagawa, K.; Kirimura, K. Cross-linked protein complex exhibiting asymmetric oxidation activities in the absence of added cofactor. Biotechnol. Prog. 2012, 28, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, H. The application of a cytochrome P450 enzyme eluted from encapsulated for the catalysis of enantioselective oxidation. RSC Adv. 2014, 4, 16333–16344. [Google Scholar] [CrossRef]
- Thakur, V.V.; Sudalai, A. Enantioselective synthesis of (S)-α-arylpropionic acids via Pd-catalyzed kinetic resolution of benzylic alcohols. Indian J. Chem. Sect. B 2005, 44B, 557–561. [Google Scholar] [CrossRef]
- Nagaoka, H. Application of a Heme-Binding Protein Eluted from Encapsulated Biomaterials to the Catalysis of Enantioselective Oxidation. ACS Catal. 2014, 4, 553–565. [Google Scholar] [CrossRef]
- Paulsen, I.T.; Press, C.M.; Ravel, J.; Kobayashi, D.Y.; Myers, G.S.; Mavrodi, D.V.; DeBoy, R.T.; Seshadri, R.; Ren, Q.; Madupu, R.; et al. Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat. Biotechnol. 2005, 23, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, H. An HASApf-Redoxin Complex causing Asymmetric Catalytic Oxidation via the Regenerative Formation of a Reactive Oxygen Species. Dalton Trans. 2015, 44, 13384–13393. [Google Scholar] [CrossRef] [PubMed]
- Du, J.F.; Li, W.; Li, L.; Wen, G.B.; Lin, Y.W.; Tan, X. Regulating the Coodination State of a Heme Protein by a Designed Distal Hydrogen-Bonding Network. ChemistryOpen 2015, 4, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bowler, B.E.; Lynch, K.; Eaton, S.S.; Eaton, G.R. Interspin Distances in Spin-Labeled Metmyoglobin Variants Determine by Saturation Recovery EPR. Biophys. J. 2000, 79, 1039–1052. [Google Scholar] [CrossRef]
- Gruber, C.C.; Lavandera, I.; Faber, K.; Kroutil, W. From a Racemate to a Single Enantiomer: Deracemization by Stereoinv-ersion. Adv. Synth. Catal. 2006, 348, 1789–1805. [Google Scholar] [CrossRef]
- Hori, H. Analysis of the principal g-tensors in single crystals of ferrimyoglobin complexes. Biochim. Biophys. Acta 1971, 251, 227–235. [Google Scholar] [CrossRef]
- Bizzari, A.R.; Cannistraro, S. Solvent modulation of the structural heterogeneity in iron(III) myoglobin samples: A low temperature EPR investigation. Eur. Biophys. J. 1993, 22, 259–267. [Google Scholar]
- Lee, D.S.; Yamada, H.; Sugimoto, H.; Matsunaga, I.; Ogura, H.; Ichihara, K.; Adachi, S.; Park, S.Y.; Shiro, Y. Substrate recognition and molecular mechanism of fatty acid hydroxylation by cytochrome P450 from Bacillus subtilis. Crystallographic, spectroscopic, and mutational studies. J. Biol. Chem. 2003, 278, 9761–9767. [Google Scholar] [CrossRef] [PubMed]
- Yosca, T.H.; Rittle, J.; Krest, C.M.; Onderko, E.L.; Beham, A.R.K.; Green, M.T. Iron(IV)hydroxide pKa and the Role of Thiolate Ligation in C–H Bond Activation by Cytochrome P450. Science 2013, 342, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Onoda, H.; Shoji, O.; Watanabe, Y. Acetate anion-triggered peroxygenation of non-native substrates by wild-type cytochrome P450s. Dalton Trans. 2015, 44, 15316–15323. [Google Scholar] [CrossRef] [PubMed]
- Prage, B.E.; Pawelzik, S.-C.; Busenlehner, S.L.; Kim, K.; Morgenstern, R.; Jakobsson, P.-J.; Armstrong, R.N. Location of Inhibitor Binding Sites in the Human Inducible Prostaglandin E Synthase, MPGES1. Biochemistry 2011, 50, 7684–7693. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.-L.; Carlsson, J.; Hogbom, M.; Hosler, J.P.; Gennis, R.B.; Brzezinski, P. Proton Uptake and pKa Changes in the Uncoupled Asn139Cys Variant of Cytochrome c Oxidase. Biochemistry 2013, 52, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Oldham, M.L.; Khare, D.; Quiocho, F.A.; Davidson, A.L.; Chen, J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature 2007, 450, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Ratliff, M.; Zhu, W.; Deshmukh, R.; Wilks, A.; Stojiljkovic, I. Homologues of Neisserial Heme Oxygenase in Gram-Negative Bacteria: Degradiation of Heme by the Product of thepiaA Gene of Pseudomonas aeruginosa. J. Bacteriol. 2001, 183, 6394–6403. [Google Scholar] [CrossRef] [PubMed]
- McDonald, A.R.; Bukowski, M.R.; Farquhar, E.R.; Jackson, T.A.; Koehntop, K.D.; Seo, M.S.; De Hont, R.F.; Stubna, A.; Halfen, J.A.; Muonck, E.; et al. Sulfur versus Iron Oxidation in an Iron-Thiolate Model Complex. J. Am. Chem. Soc. 2010, 132, 17118–17129. [Google Scholar] [CrossRef] [PubMed]
- Unno, M.; Ardèvol, A.; Rovira, C.; Ikeda-Saito, M. Structures of the Substrate-Free and the Product-Bound Forms of HmuO, a Heme Oxygenase from Corynebacterium diphtheriae: X-ray crystallography and molecular dynamics investigation. J. Biol. Chem. 2013, 288, 34443–34458. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Nambu, S.; Ono, Y.; Goulding, C.W.; Tsumoto, K.; Ikeda, M. Heme degradation by Staphylococcus aureus IsdG and IsdI liberates formaldehyde rather than carbon monoxide. Biochemistry 2013, 52, 3025–3027. [Google Scholar] [CrossRef] [PubMed]
- Ishimori, K.; Watanabe, Y. Unique Heme Environmental Structures in Heme-regulated Proteins Using Heme as the Signaling Molecule. Chem. Lett. 2014, 43, 1680–1689. [Google Scholar] [CrossRef]
- Uchida, T.; Sekine, Y.; Ikeda-Saito, M.; Ishimori, K. A heme degradation enzyme, HutZ, from Vibrio cholerae. Chem. Commun. 2012, 48, 6741–6743. [Google Scholar] [CrossRef] [PubMed]
- Nambu, S.; Matsui, T.; Celia, W.; Goulding, C.W.; Takahashi, S.; Ikeda-Saito, M. A new way to degrade heme: The Mycobacteriaum tuberoculosis enzyme MhuD catalyzes heme degradation without generating CO. J. Biol. Chem. 2013, 288, 10101–10109. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.K.; Lee, K.T.; Mohamed, A.R. Homogeneous, heterogeneous and enzymatic catalysis for transesterification of high free fatty acid oil (waste cooking oil) to biodiesel: A review. Biotechnol. Adv. 2010, 28, 500–518. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.Y.; Liu, J.; Hahn, M.; Qiao, S.; Middelberg, A.P.J.; He, L. Encapsulation of lipase in mesoporous silica yolk–shell spheres with enhanced enzyme stability. RSC Adv. 2013, 3, 22008–22013. [Google Scholar] [CrossRef]
- Protein Sequence Based on a BLAST Query Sequence Analysis of Cycle No. for YP 262445.1 is Available on the Internet on NCBI resource: http://www.ncbi.nlm.nih.gov/protein/70732682 (accessed on 17 December 2015).


| Process (each fraction) | Materials | Bacteria (CFU/g) | Resolutions | Outcomes | ||||
| rac-2/ catalyst | Times | Result | ||||||
| 1 | PPg | powder | 210 f | Detected types: 1. aerobic spore- bearing bacterium (Geobacillus, Alicyclobacillus, Bacillus, Paenibacillus) 2. catalase-and gram-positive coccus bacterium (Staphylococcus, Kocuria, Micrococcus) | ||||
| 2 | PPg gel under aeration | Na+alginate | NDa | |||||
| Ca2+Cl2 | ||||||||
| D.W.j | ||||||||
| PP gel | ∞ | |||||||
| 3 | PPg gel suspension | solution | 1.2 mM/5 mLb | 10 h | N.R.d | HasApf weakly activated | ||
| 4 | Supernatant 1 | solution | ||||||
| 5a | Precipitate 1 dried | powder | NDa | 1.2 mM/30 mgc | 15 h | |||
| 6 | Precipitate 2 dried | powder | C.R.e | HasApf highly activated by buffered glycine | ||||
| 6a | AGh-Precipitate 2 | powder | ||||||
| 6b | PEGi-Precipitate 2 | powder | ||||||
| 7 | Supernatant 2 | solution | ∞ | 1.2 mM/5 mLb | 10 h | |||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cycle No. | Deracemization: PEG–Precipitate 2 (1.0 g)/rac-1 (100 mg)/Deionized Water (400 mL) | |||||||
|---|---|---|---|---|---|---|---|---|
| NaBH4 | Time (h) | Ketone % | Alcohol % a | (R)-Isomer % | (S)-Isomer % | Compd | OP/%ee b | |
| 1st | 0 | 23 | 49 | 51 | 1.2 | 98.8 | (S)-1 c | 97.6 |
| 1.0 | 99.0 | (S)-2 d | 98.0 | |||||
| 2nd | 50 mg | 28 | 32 | 68 | 0.8 | 99.2 | (S)-1 | 98.4 |
| 0.7 | 99.3 | (S)-2 | 98.6 | |||||
| 3rd | 50 mg | 33 | 17 | 83 | 0.4 | 99.6 | (S)-1 | 99.2 |
| 0.2 | 99.8 | (S)-2 | 99.4 | |||||
| 4th | 50 mg | 40 | 4 | 96 | 0.2 | 99.8 | (S)-1 | 99.4 |
| 0.1 | 99.9 | (S)-2 | 99.8 | |||||
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagaoka, H. Heterogeneous Asymmetric Oxidation Catalysis Using Hemophore HasApf. Application in the Chemoenzymatic Deracemization of sec-Alcohols with Sodium Borohydride. Catalysts 2016, 6, 38. https://doi.org/10.3390/catal6030038
Nagaoka H. Heterogeneous Asymmetric Oxidation Catalysis Using Hemophore HasApf. Application in the Chemoenzymatic Deracemization of sec-Alcohols with Sodium Borohydride. Catalysts. 2016; 6(3):38. https://doi.org/10.3390/catal6030038
Chicago/Turabian StyleNagaoka, Hiroyuki. 2016. "Heterogeneous Asymmetric Oxidation Catalysis Using Hemophore HasApf. Application in the Chemoenzymatic Deracemization of sec-Alcohols with Sodium Borohydride" Catalysts 6, no. 3: 38. https://doi.org/10.3390/catal6030038
APA StyleNagaoka, H. (2016). Heterogeneous Asymmetric Oxidation Catalysis Using Hemophore HasApf. Application in the Chemoenzymatic Deracemization of sec-Alcohols with Sodium Borohydride. Catalysts, 6(3), 38. https://doi.org/10.3390/catal6030038