
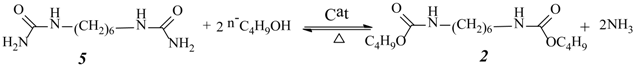
One-Pot Synthesis of Dialkyl Hexane-1,6-Dicarbamate from 1,6-Hexanediamine, Urea, and Alcohol over Zinc-Incorporated Berlinite (ZnAlPO4) Catalyst


Abstract
: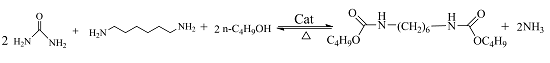
1. Introduction
2. Results and Discussion
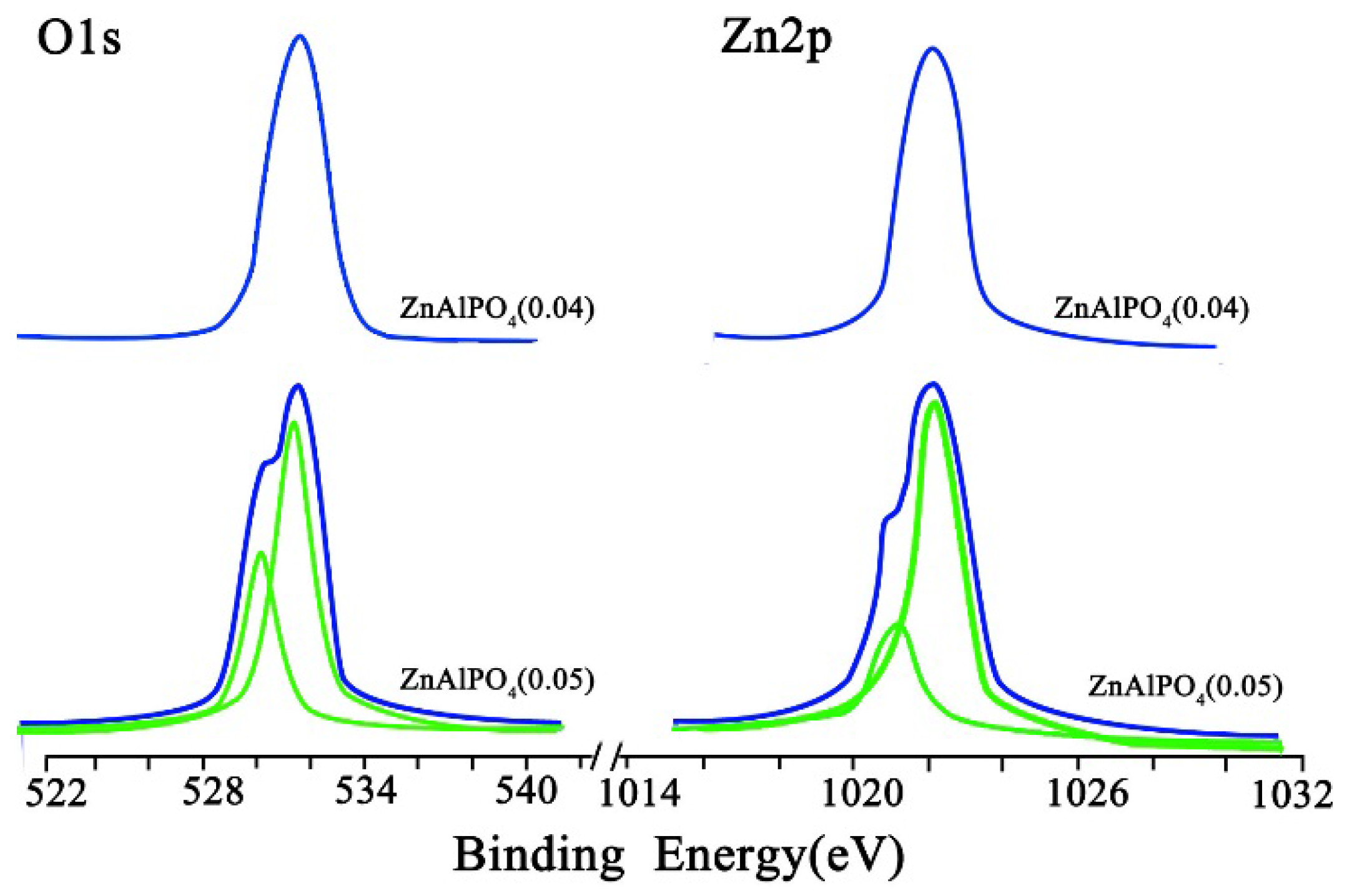
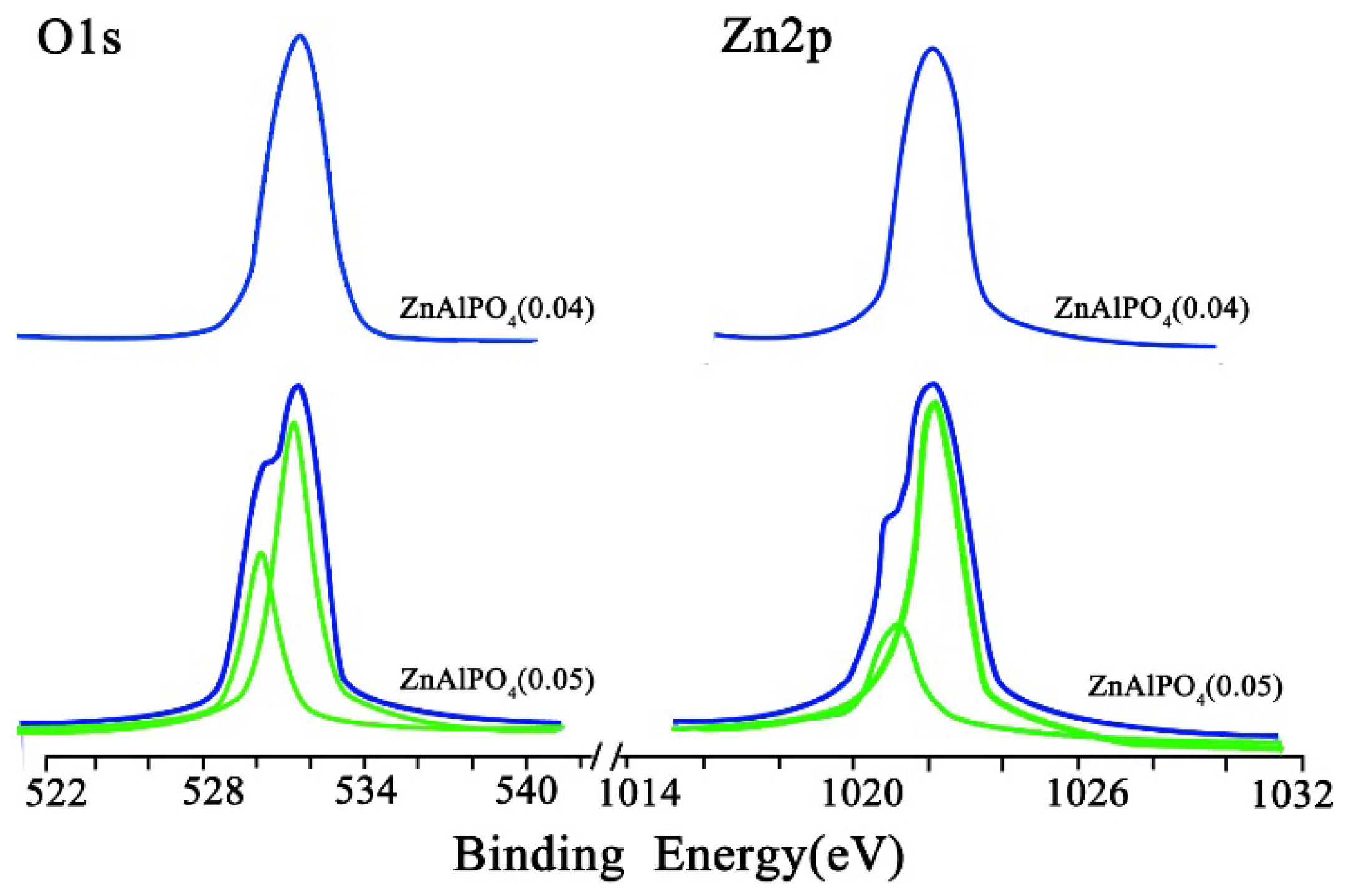
2.1. Catalyst Characterization
2.2. Catalytic Activity of Zn/AlPO4 Catalysts at Different Molar Ratios
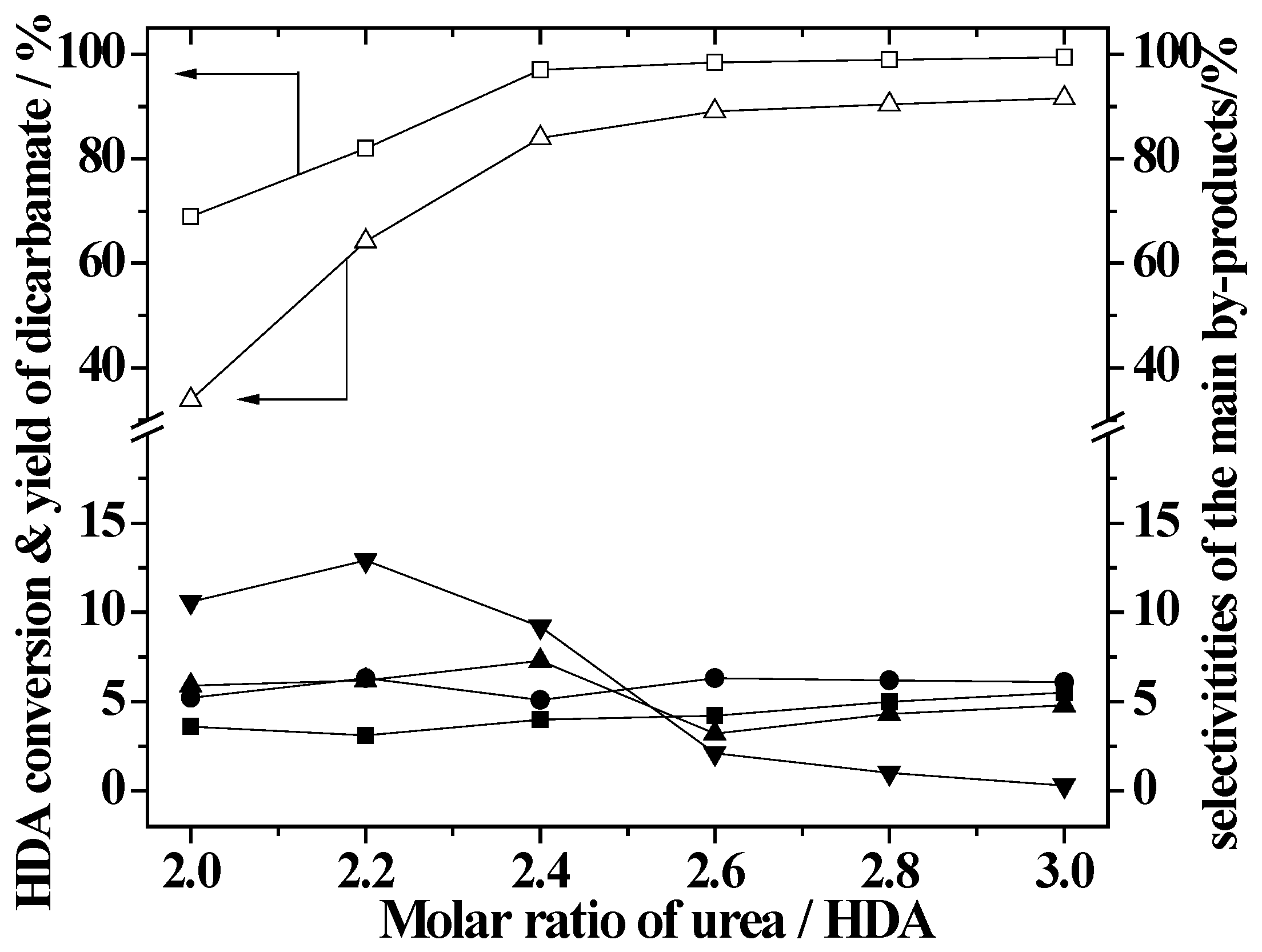
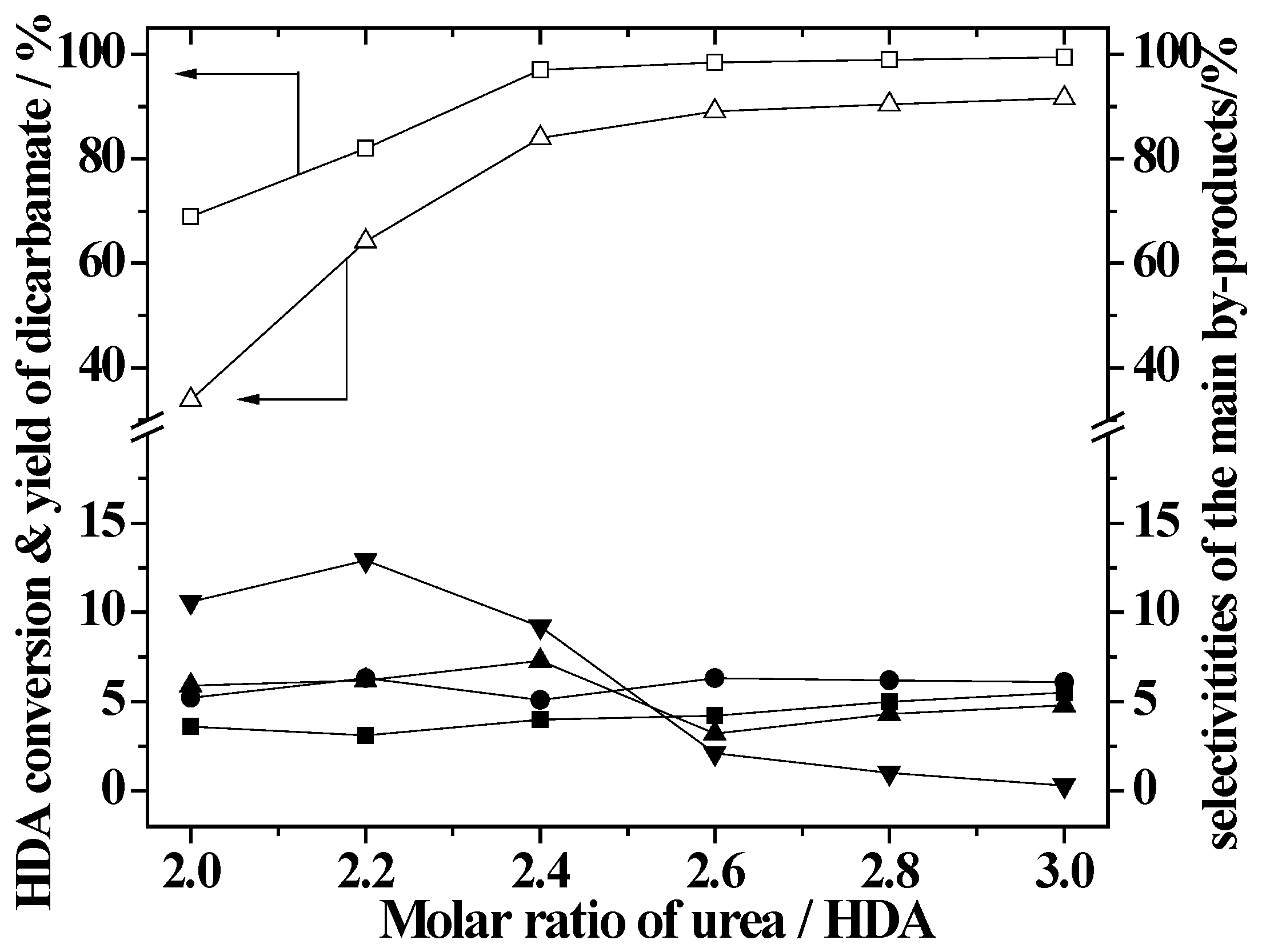
2.3. Effect of Urea/HDA Molar Ratio
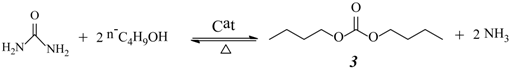
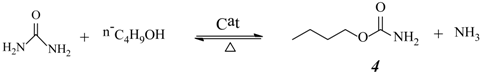
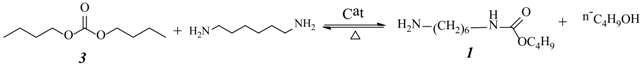
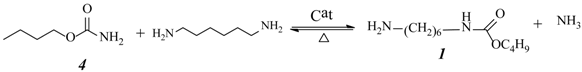
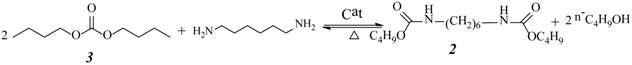
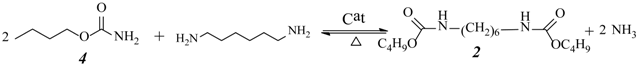
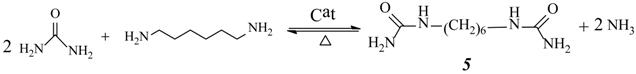
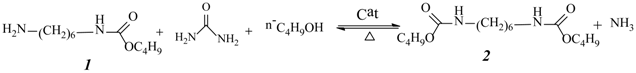
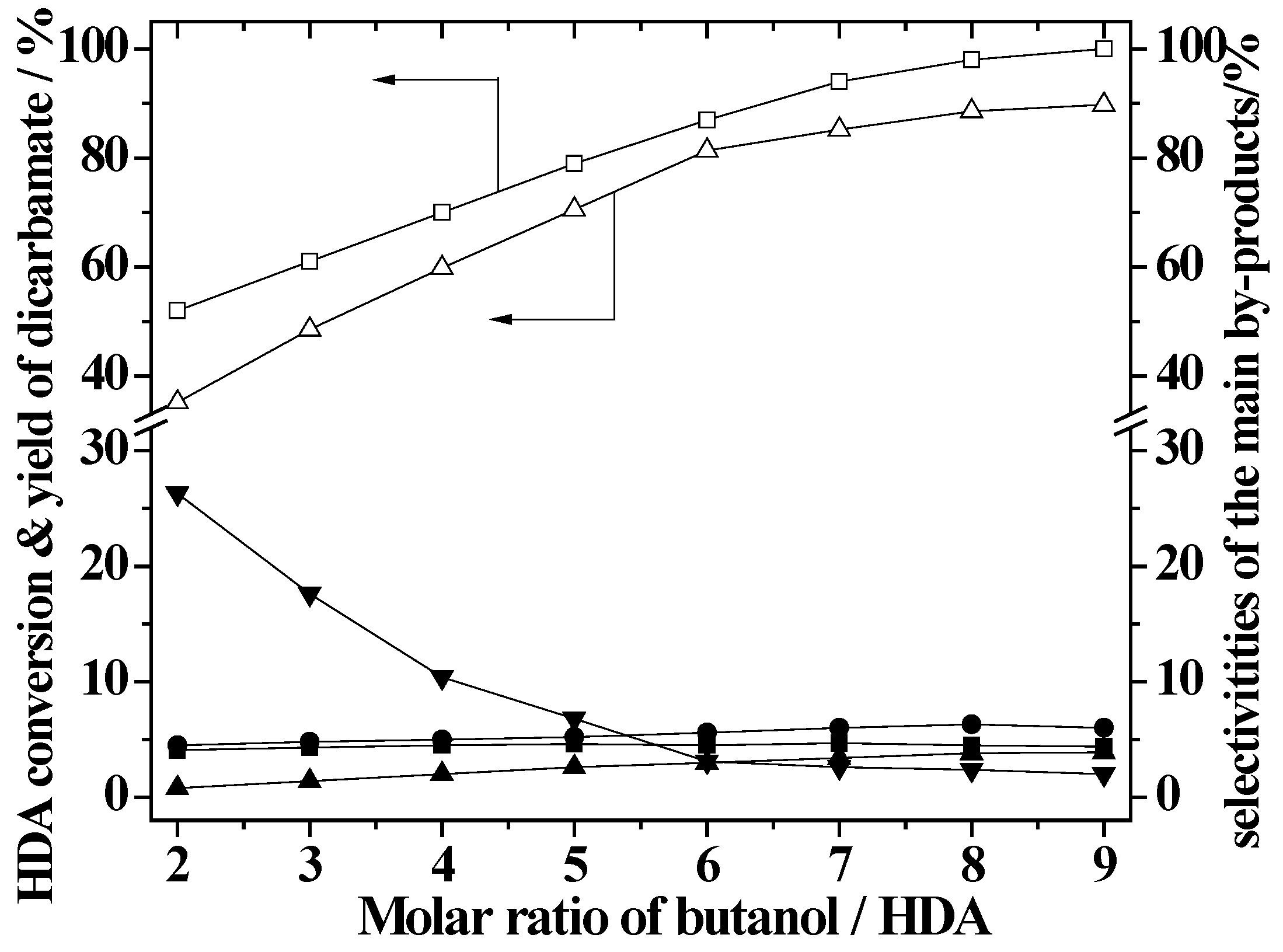
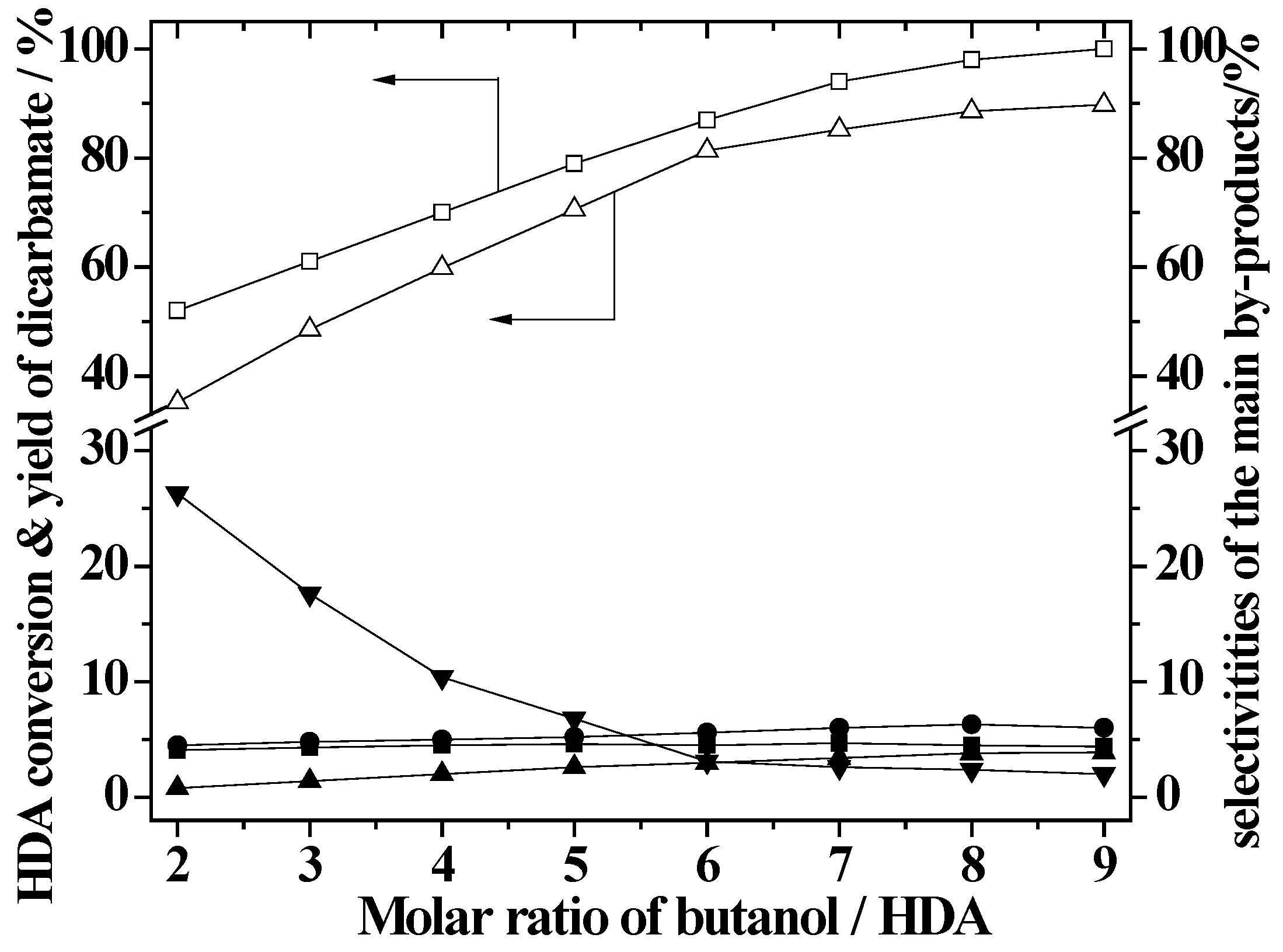
2.4. Effect of Butanol/HDA Molar Ratio
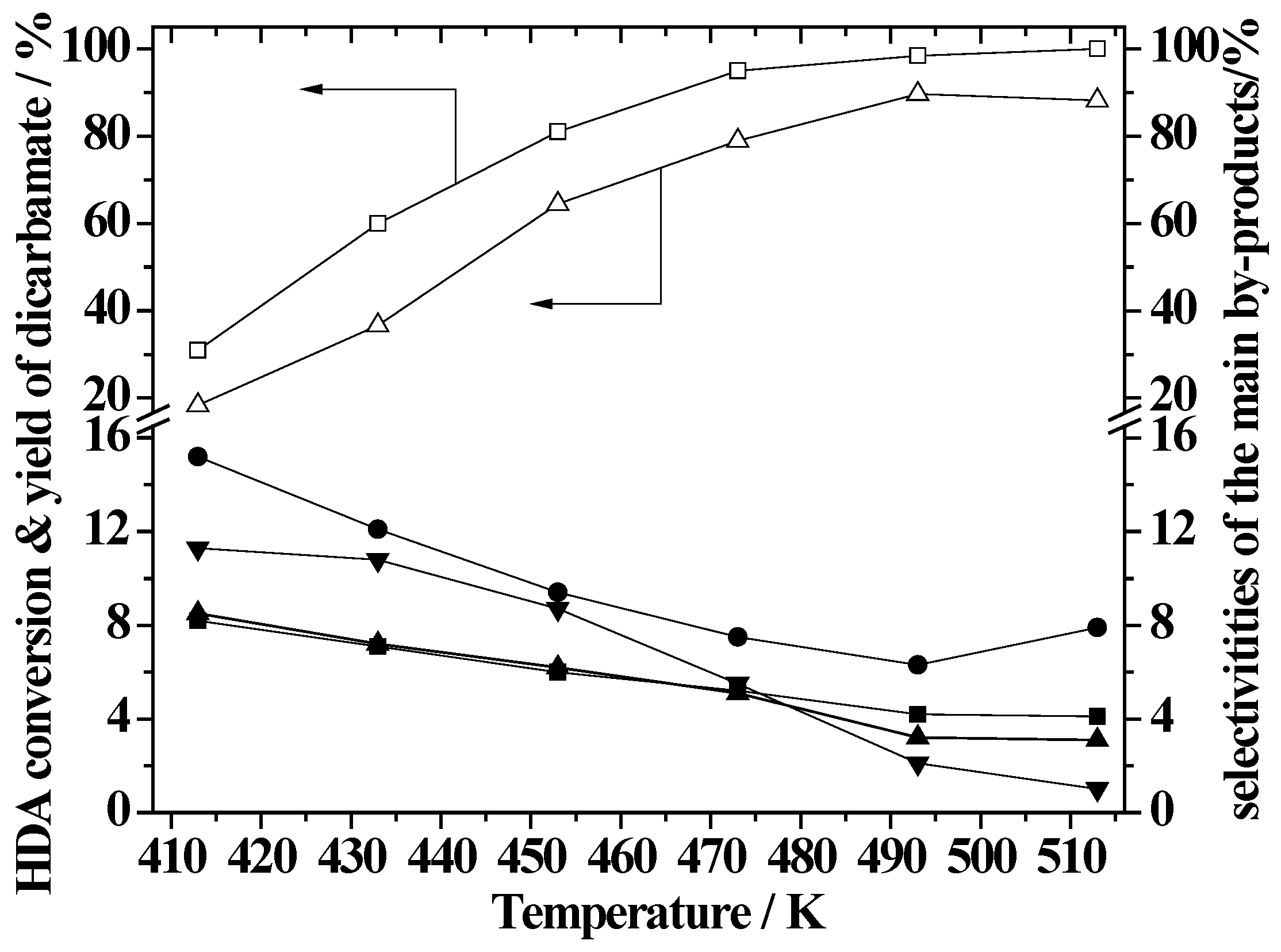
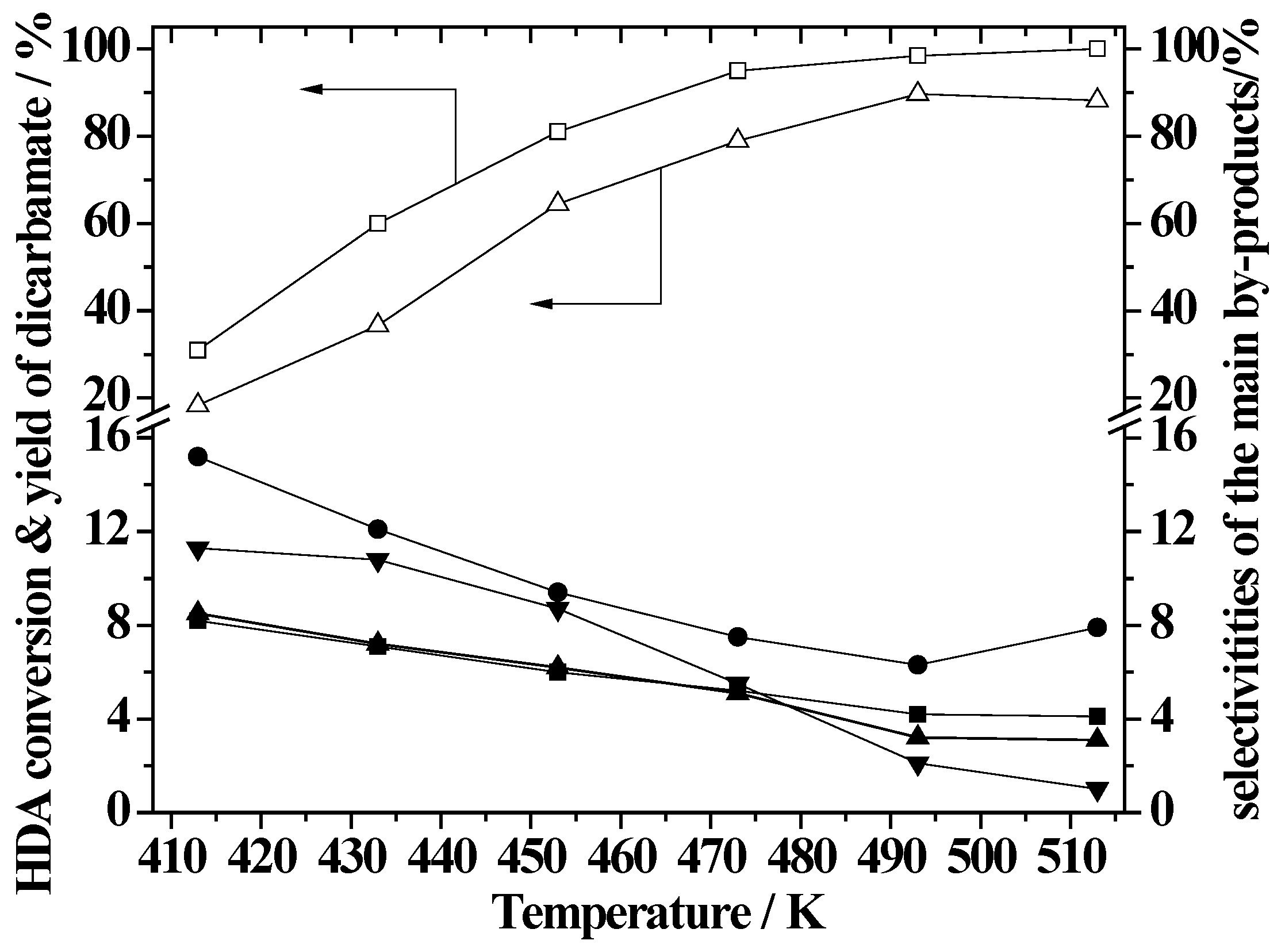
2.5. Effect of Reaction Temperature
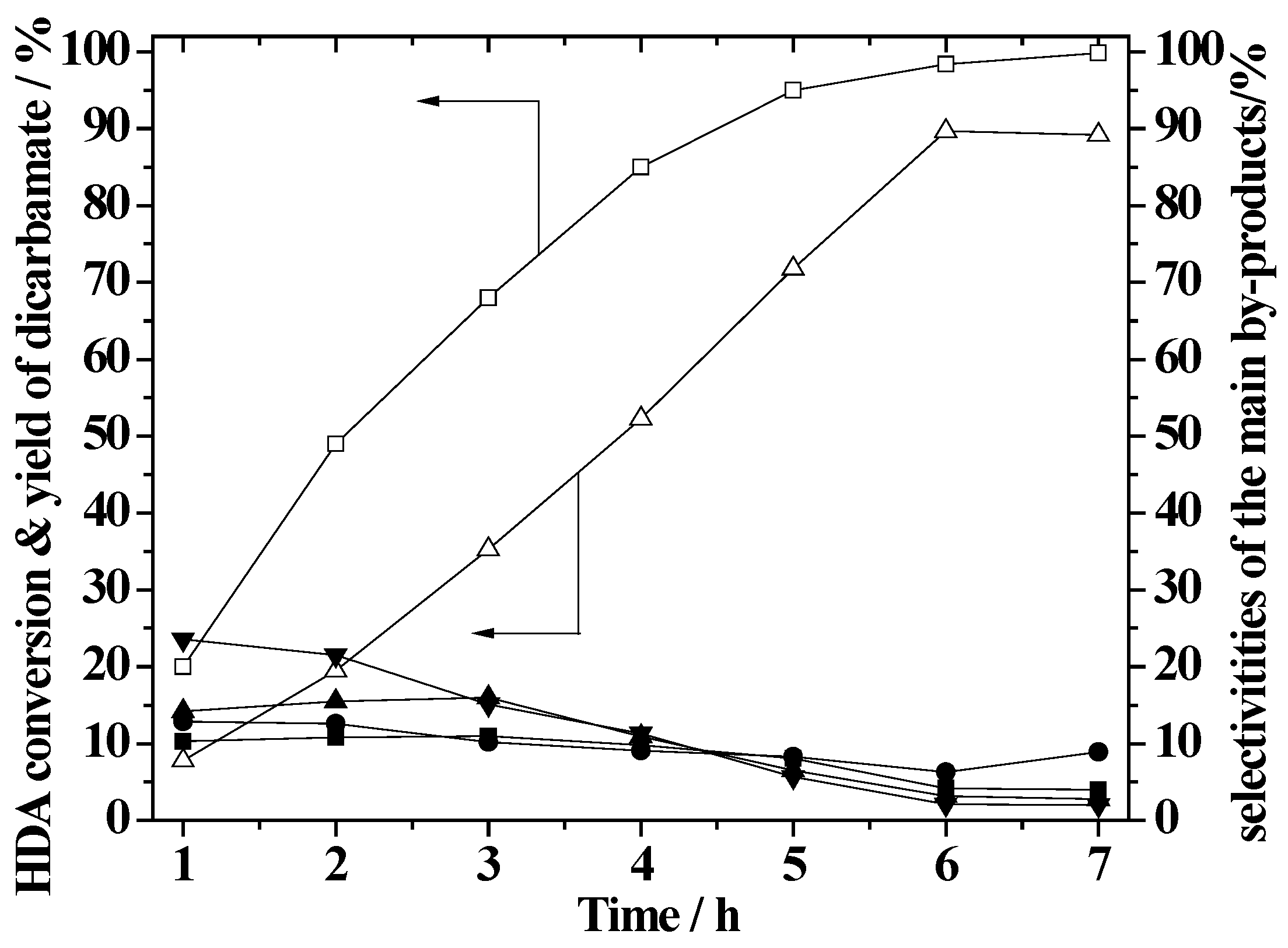
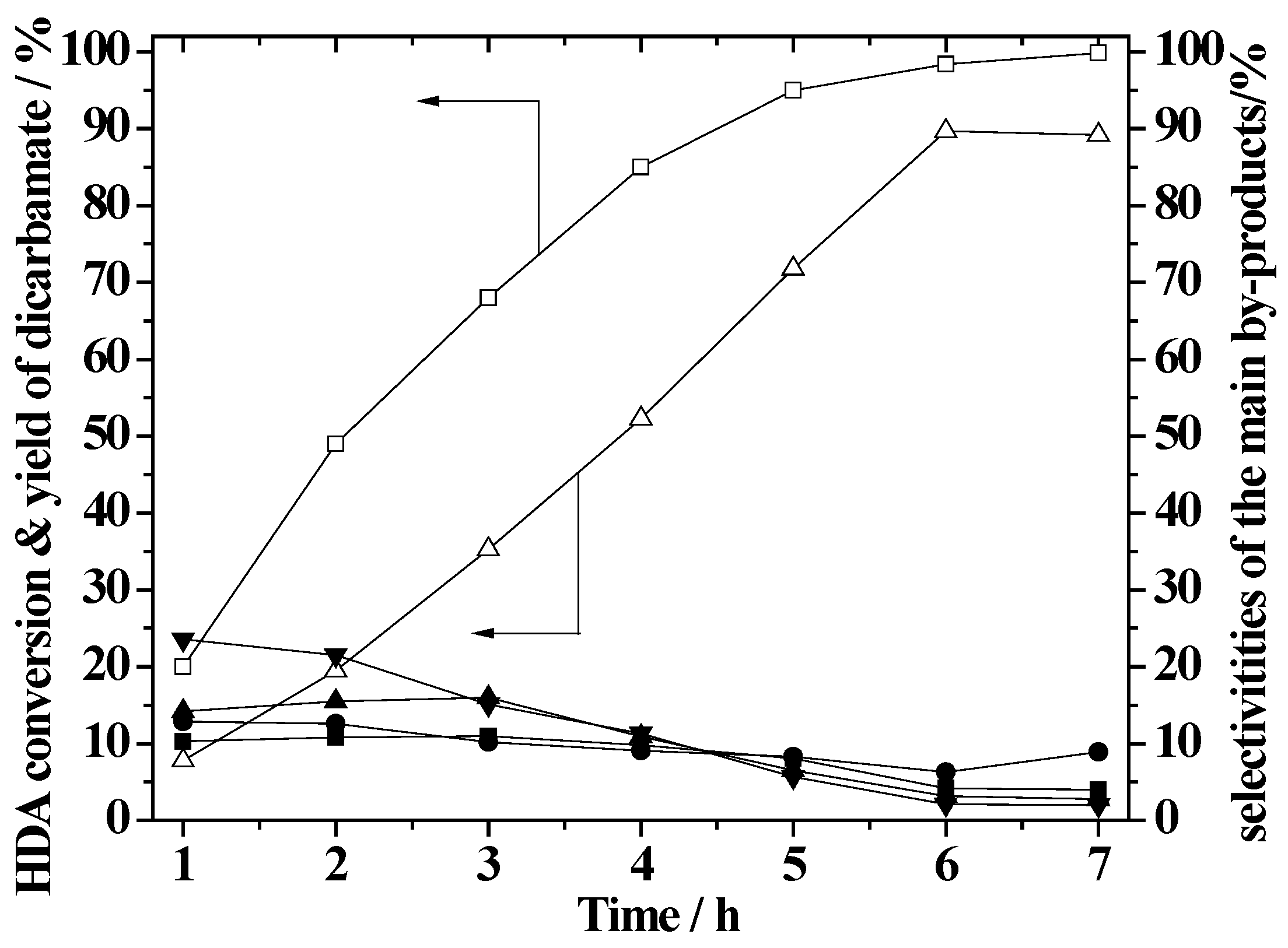
2.6. Effect of Reaction Time
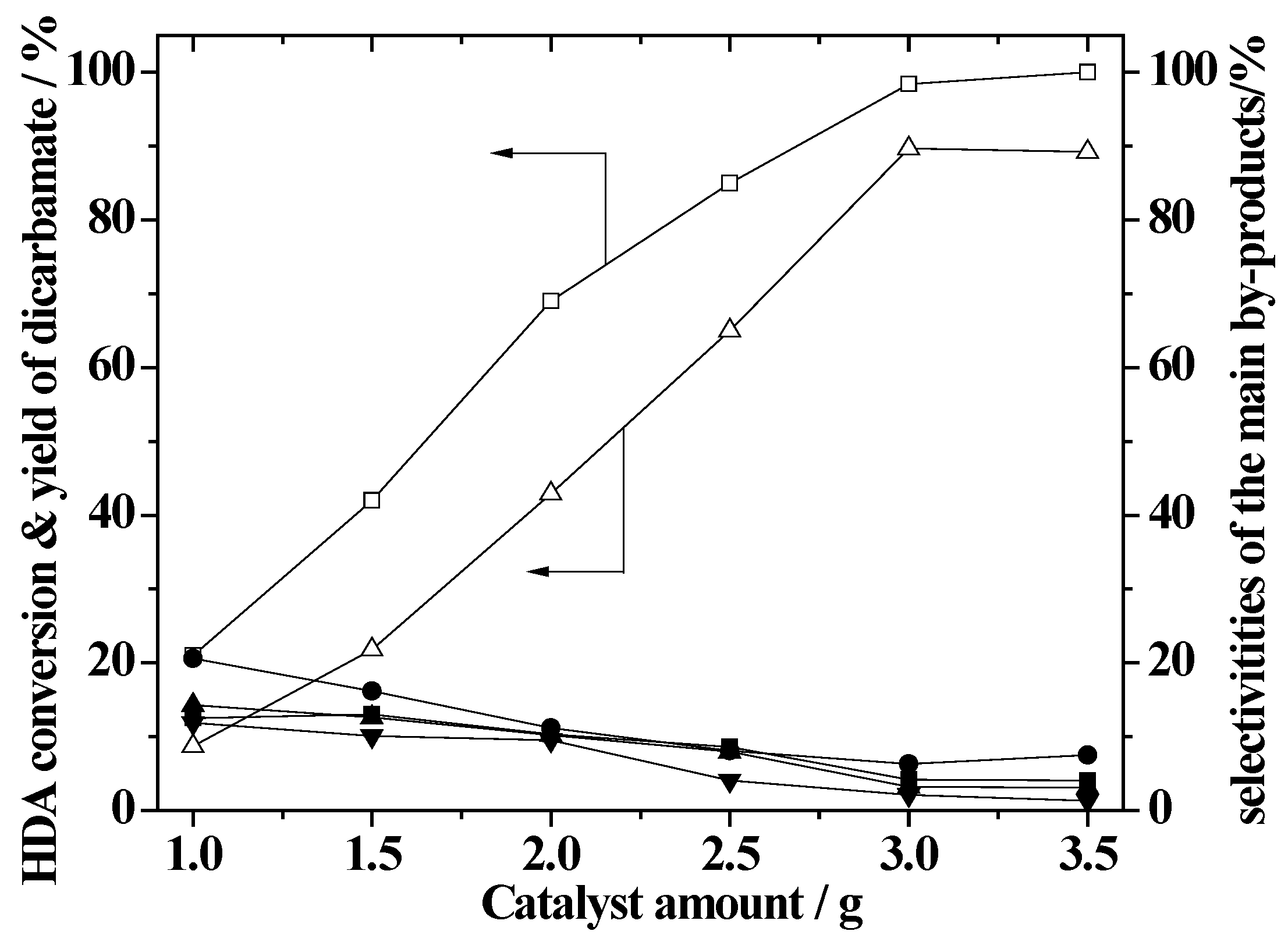
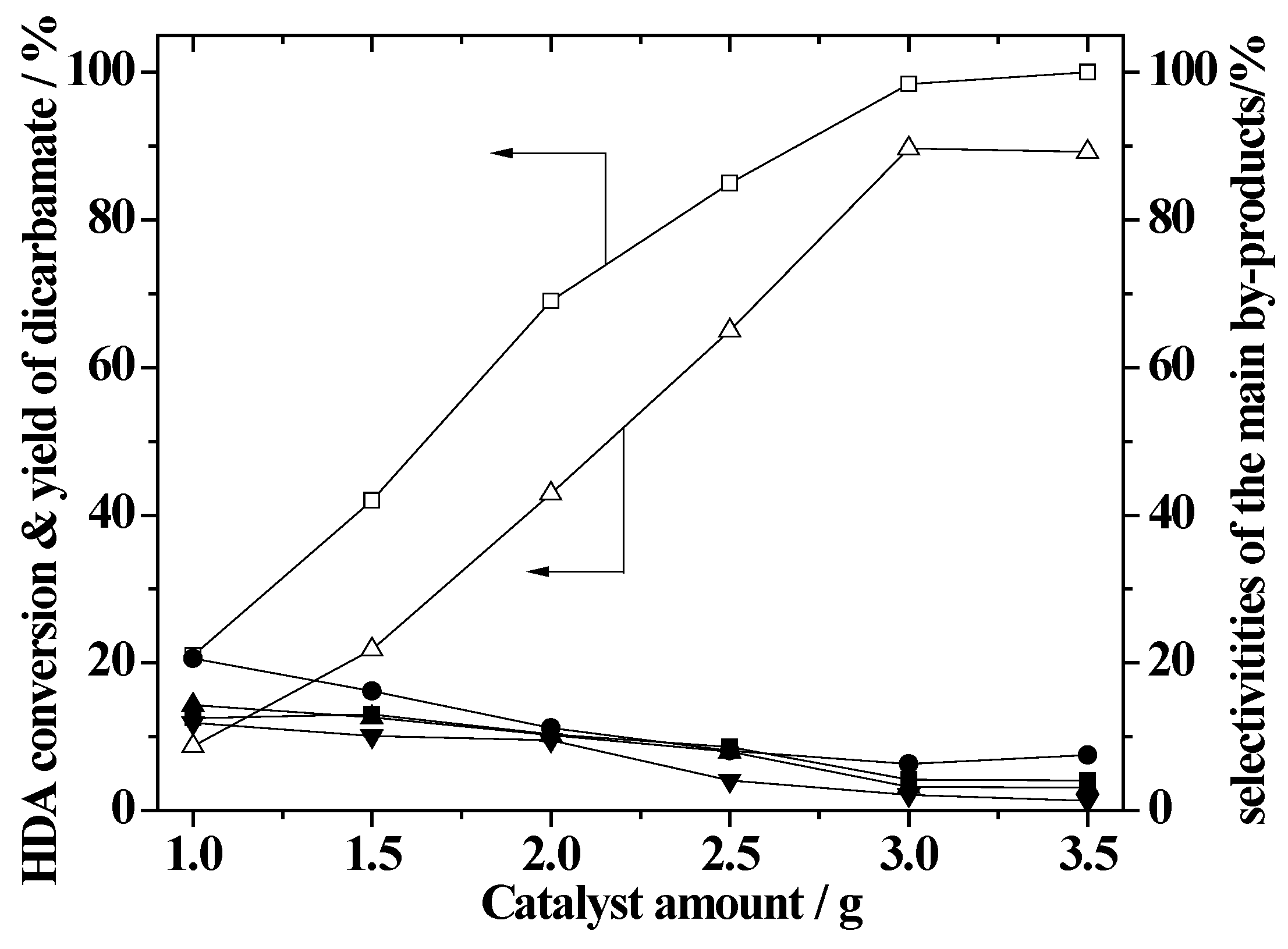
2.7. Effect of the Amount of ZnAlPO4 Catalyst
2.8. The Influence of the Alcohol Type
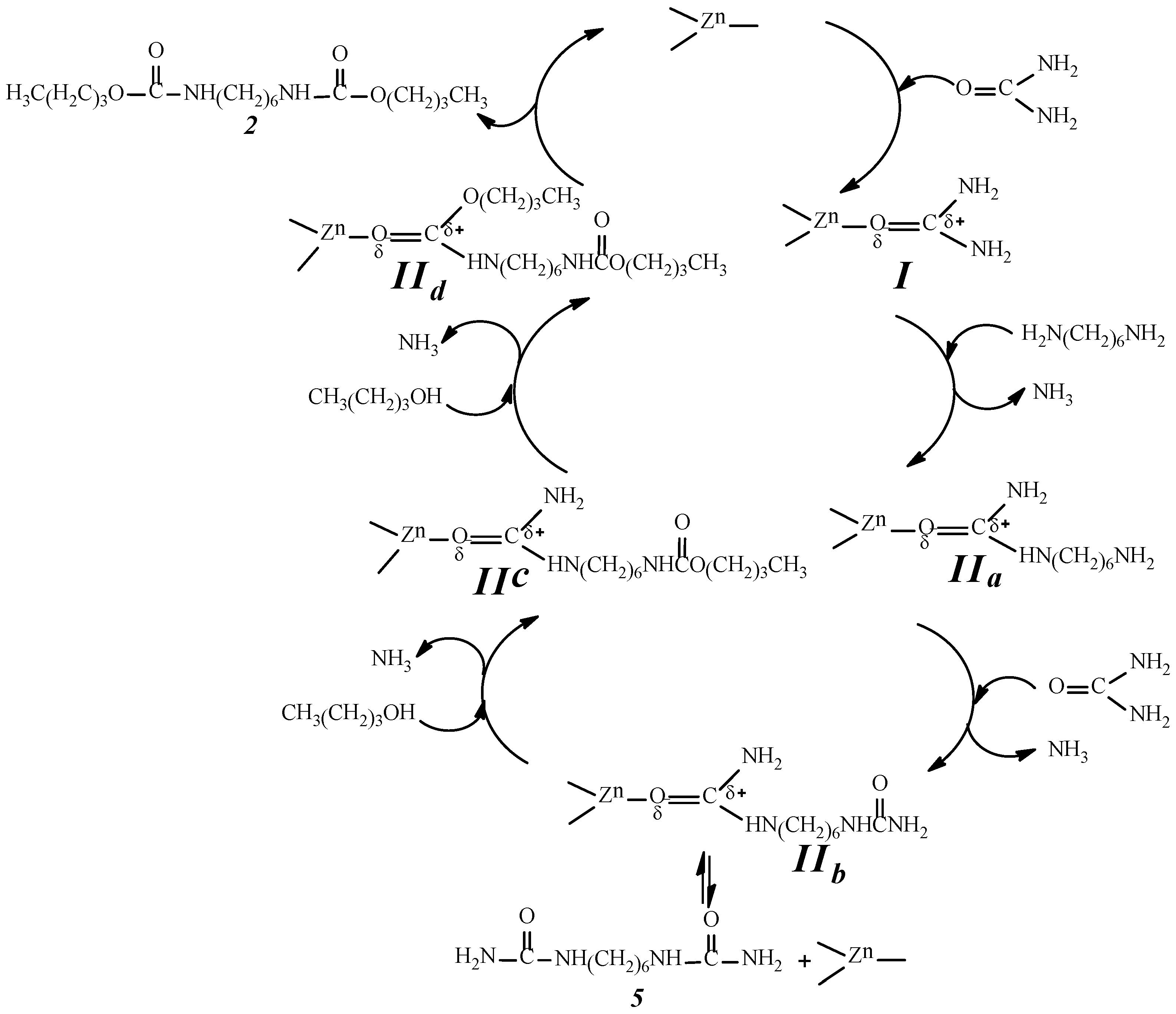
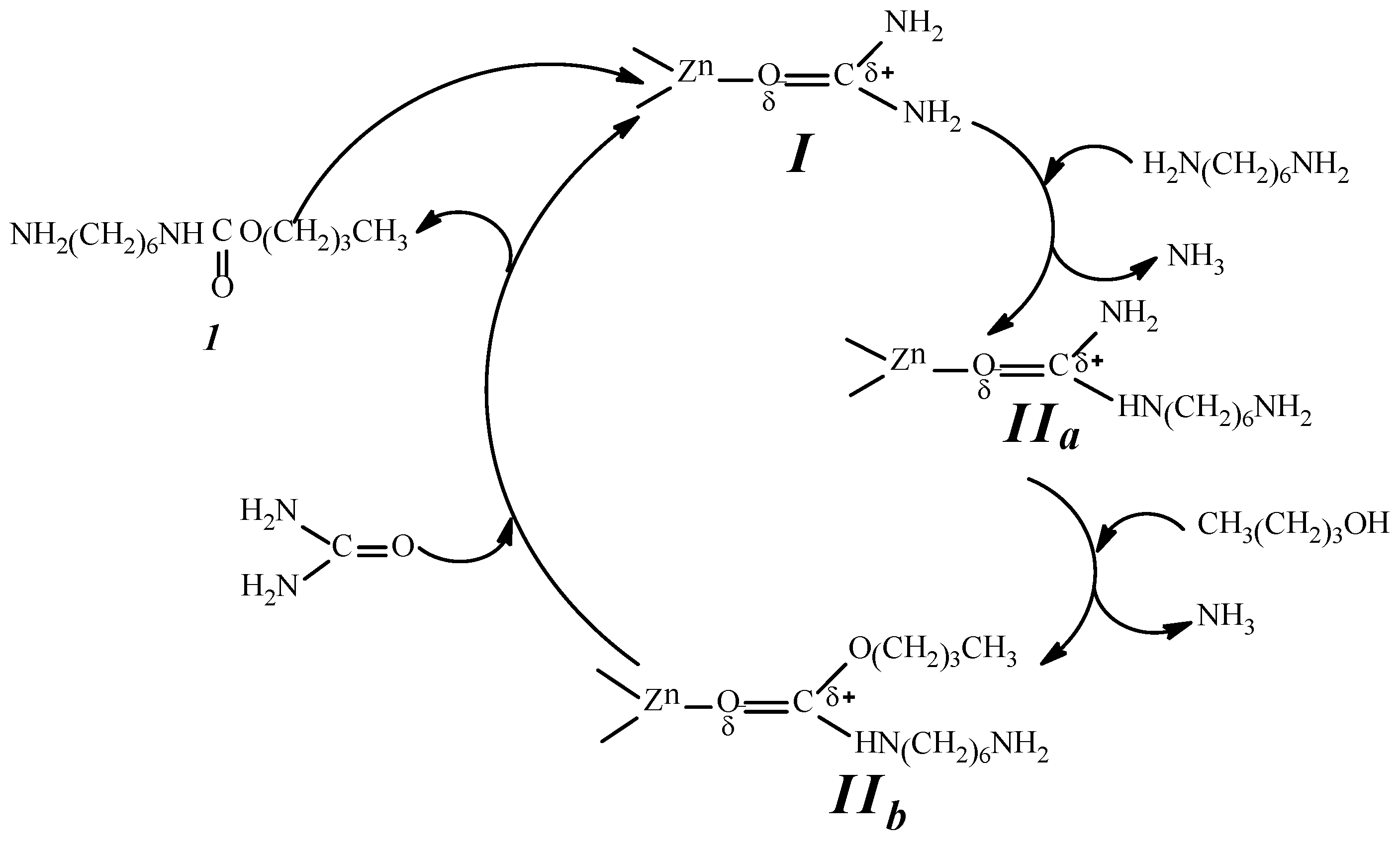
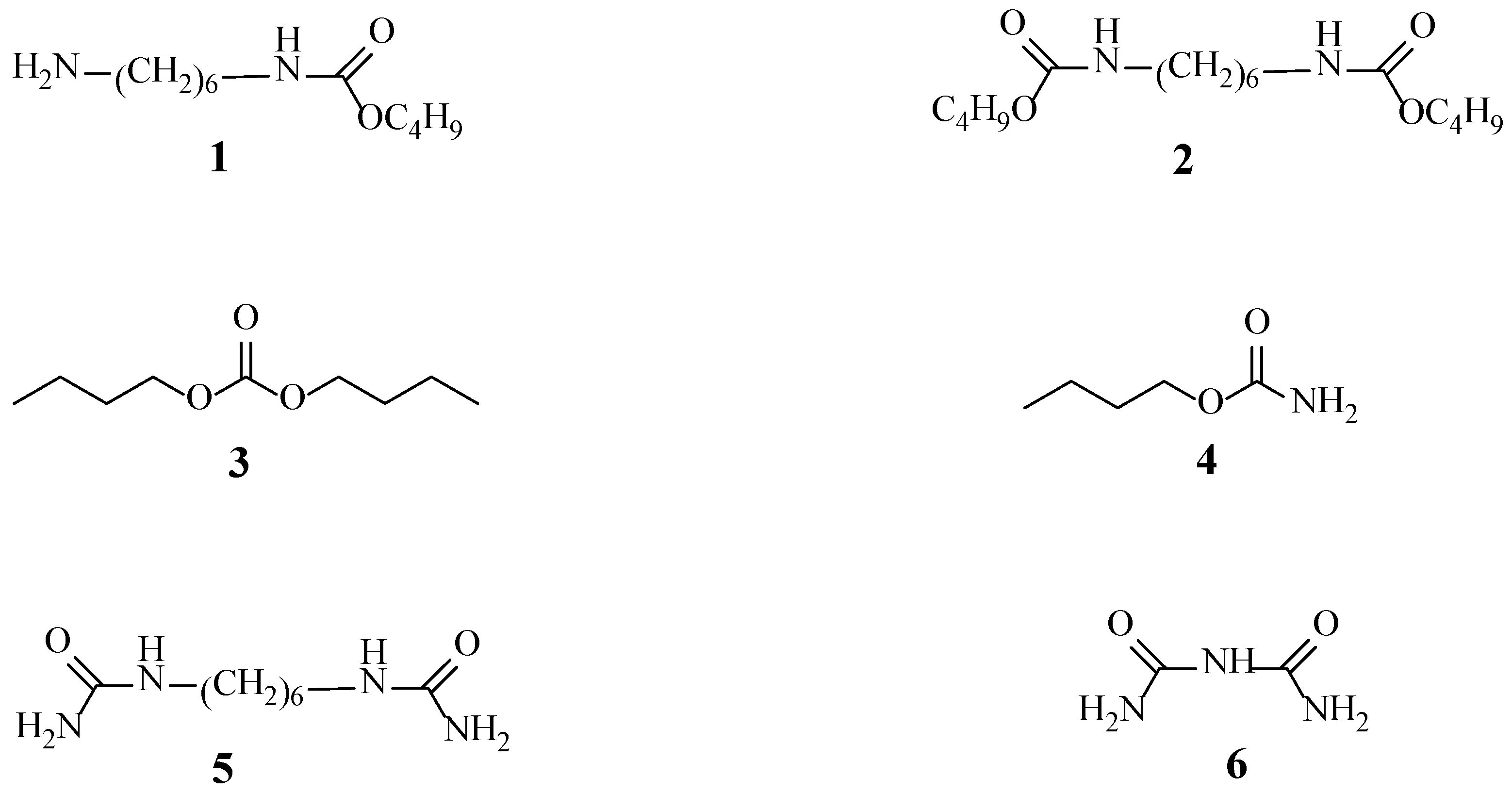
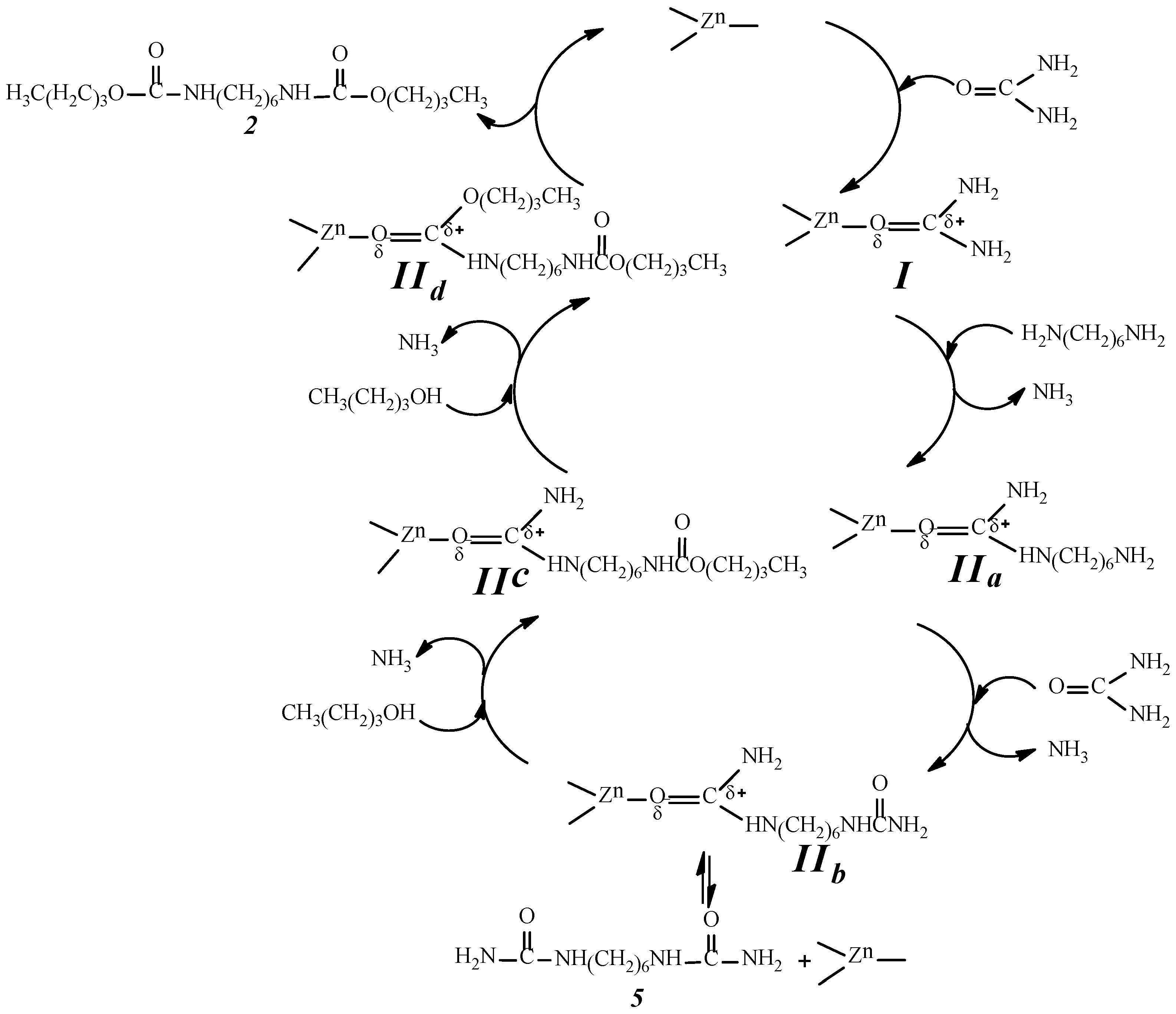
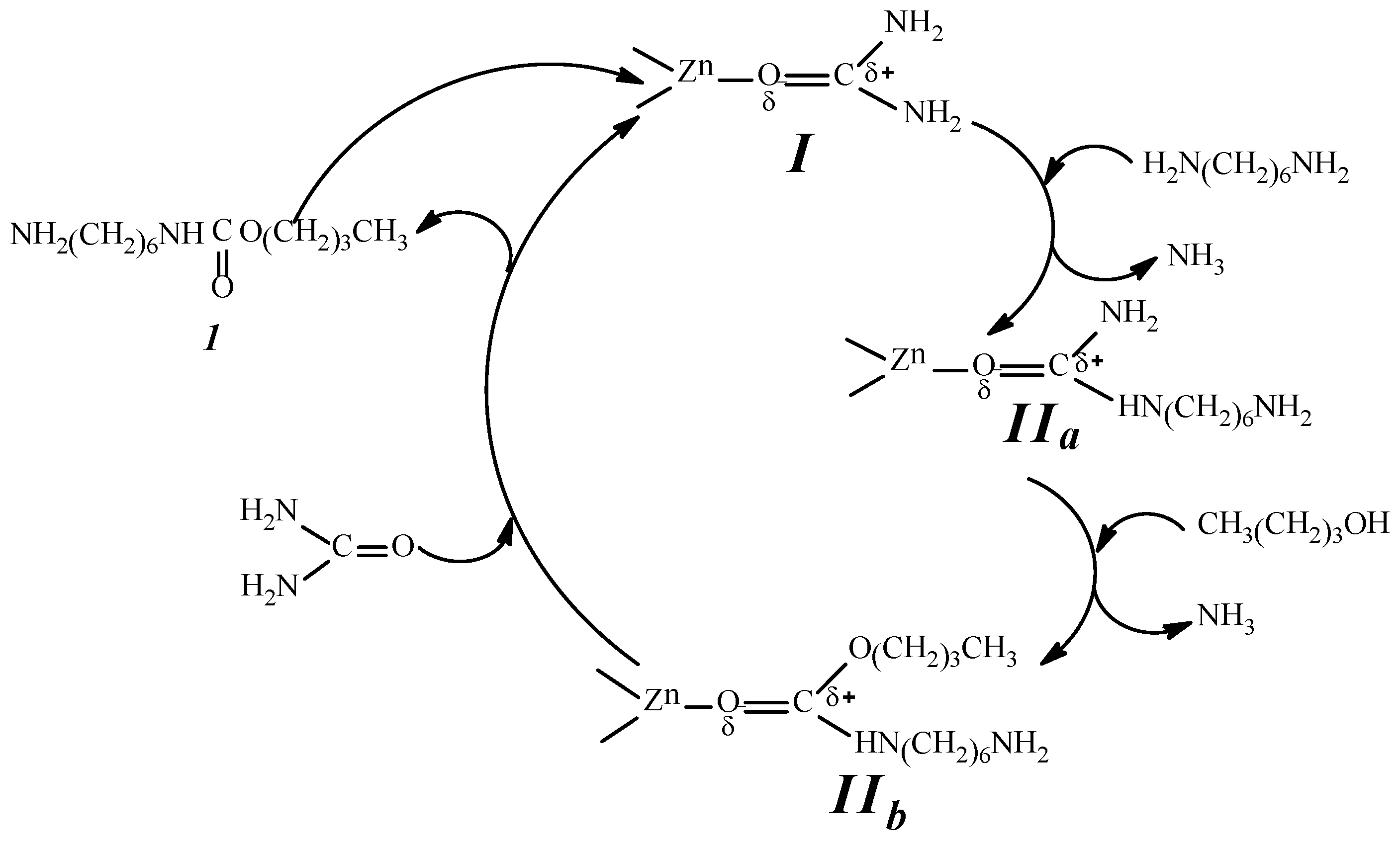
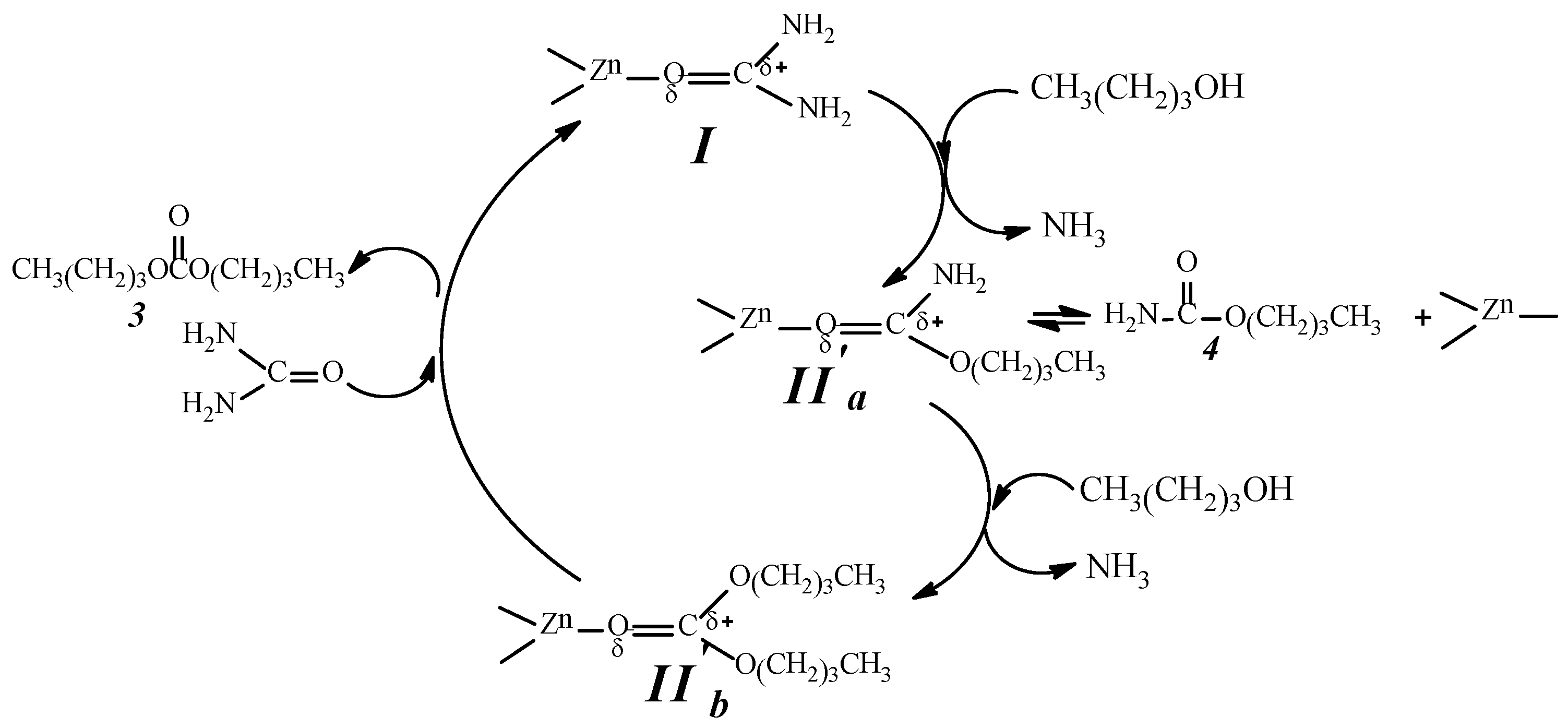
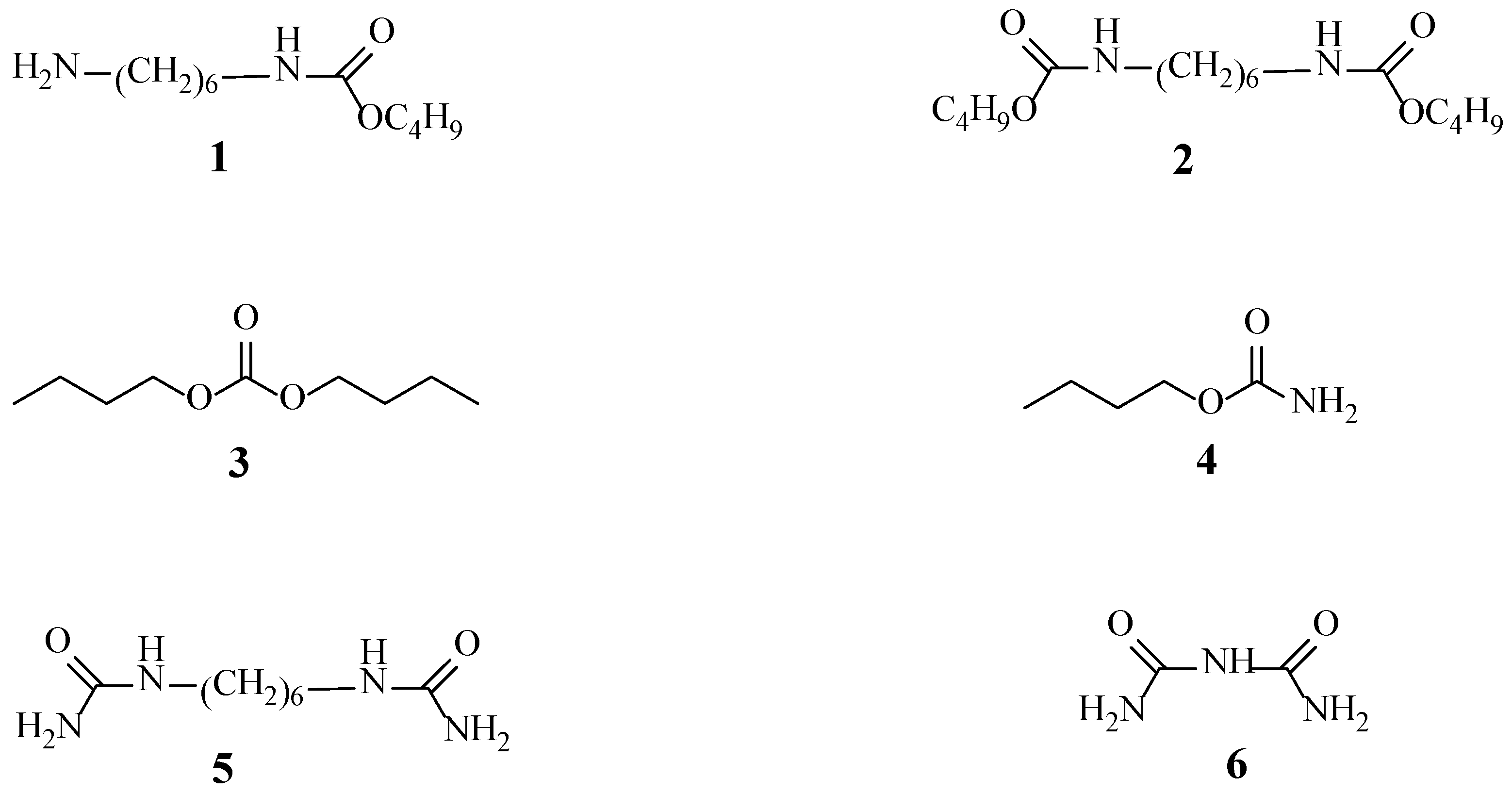
2.9. Analysis of the Reaction Mechanism
3. Experimental Section
3.1. Chemical Reagent
3.2. Preparation of Catalysts
3.3. Characterization of Catalysts
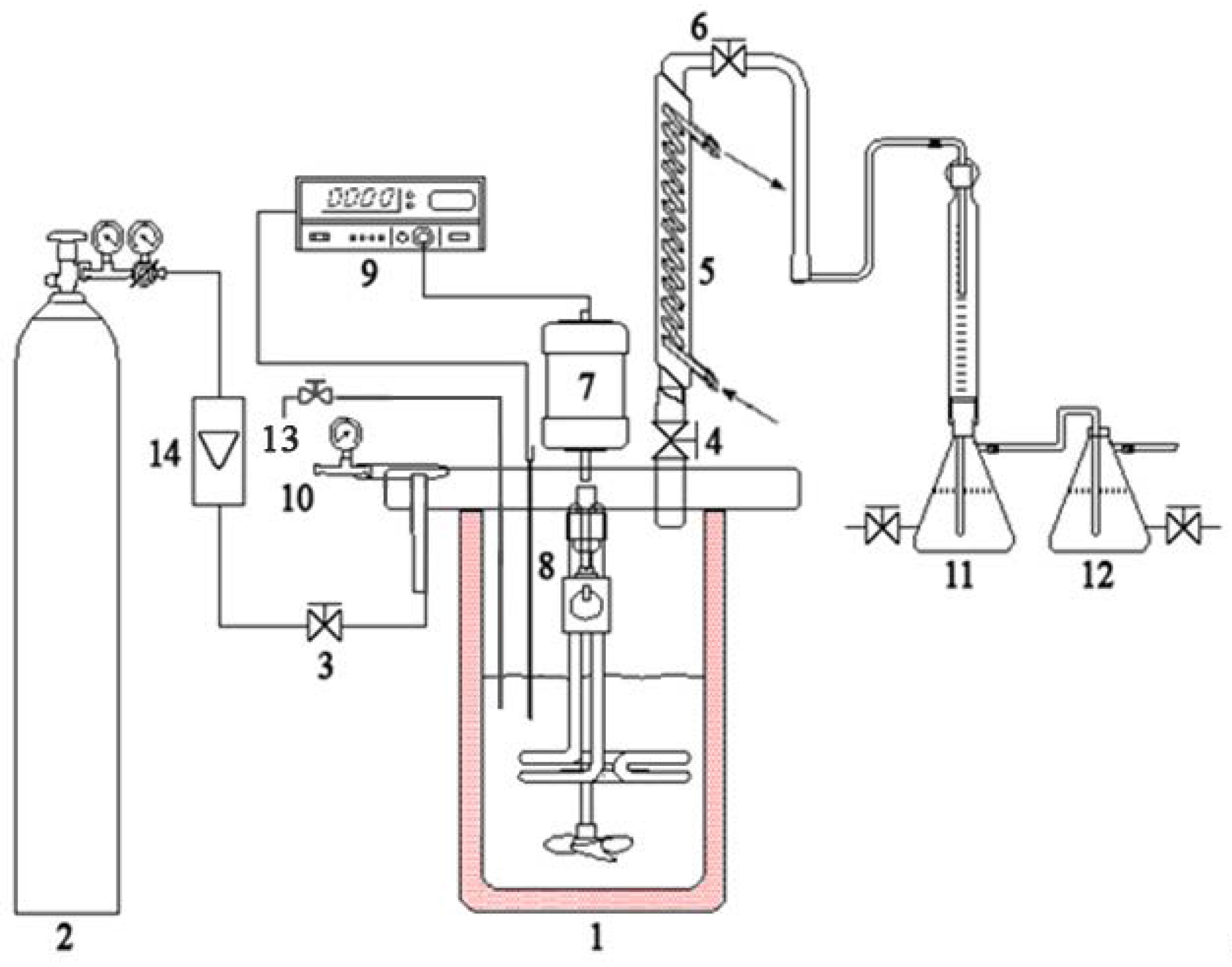
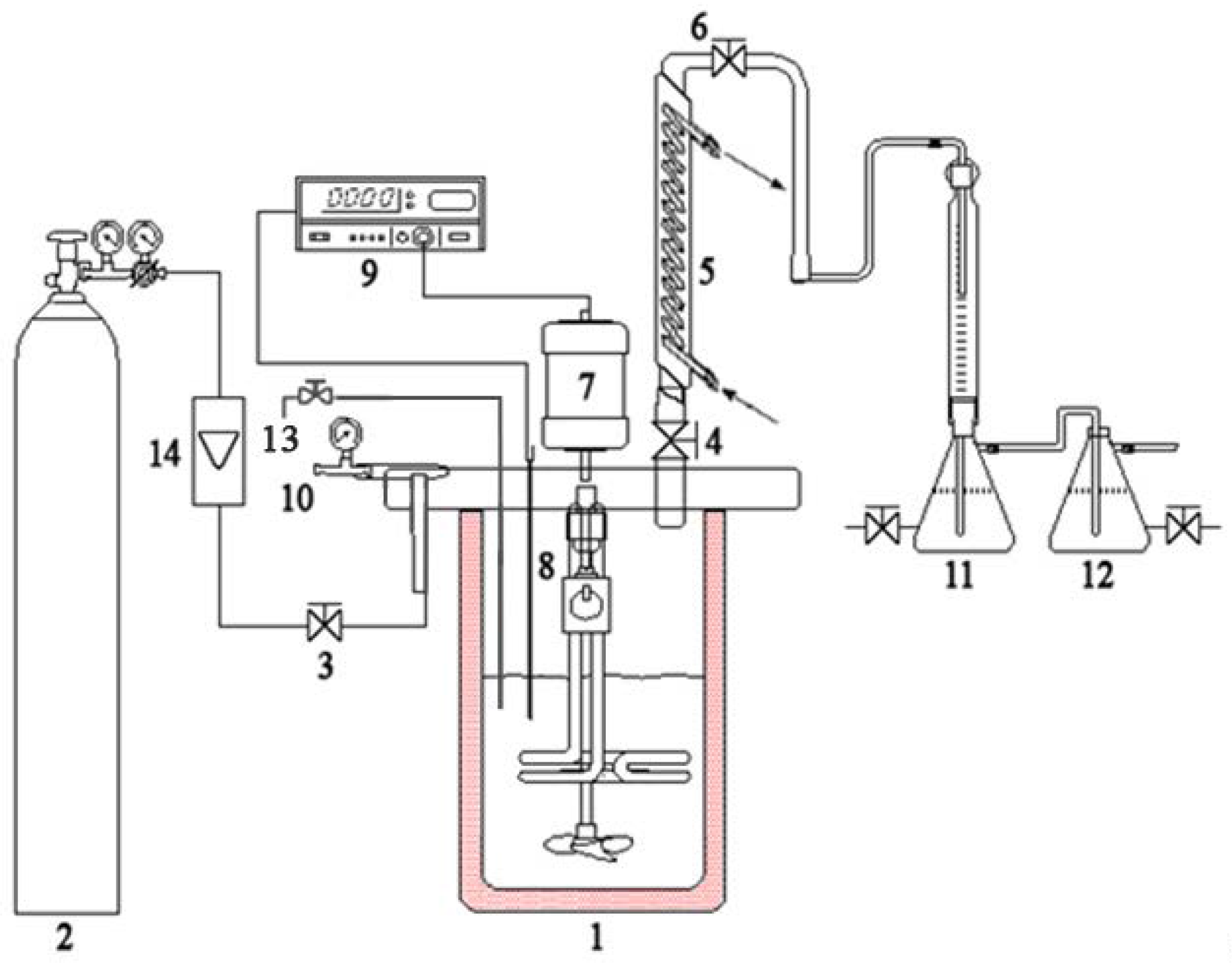
3.4. Apparatus and Procedure
3.5. Separation and Analysis of Intermediates and Products
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Greene, T.W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 3rd ed.; John Wiley & Sons: New York, NY, USA, 1999; p. 527. [Google Scholar]
- Vauthey, I.; Valot, F.; Gozzi, C.; Fache, F.; Lemaine, M. An environmentally benign access to carbamates and ureas. Tetrahedron. Lett. 2000, 41, 6347–6350. [Google Scholar] [CrossRef]
- Shapiro, S.L.; Bandurco, V.; Freedman, L. Reaction of t-Ethinyl Alcohols with Aryl Isocyanates. J. Org. Chem. 1961, 26, 3710–3712. [Google Scholar] [CrossRef]
- Ragaini, F.; Gasperini, M.; Cenini, S. Phosphorus Acids as Highly Efficient Promoters for the Palladium-Phenanthroline Catalyzed Carbonylation of Nitrobenzene to Methyl Phenylcarbamate. Adv. Synth. Catal. 2004, 346, 63–71. [Google Scholar] [CrossRef]
- Shi, F.; Deng, Y. Polymer-Immobilized Gold Catalysts for the Efficient and Clean Syntheses of Carbamates and Symmetric Ureas by Oxidative Carbonylation of Aniline and Its Derivatives. J. Catal. 2002, 211, 548–552. [Google Scholar] [CrossRef]
- Kaminshkia, N.V.; Kostic, N.M. Alcoholysis of Urea Catalyzed by Palladium(II) Complexes. Inorg. Chem. 1998, 37, 4302–4312. [Google Scholar] [CrossRef]
- Lucas, N.; Amrute, A.P.; Palraj, K.; Shanbhag, G.V.; Vinu, A.; Halligudi, S.B. Non-phosgene route for the synthesis of methyl phenyl carbamate using ordered AlSBA-15 catalyst. J. Mol. Catal. A Chem. 2008, 295, 29–33. [Google Scholar] [CrossRef]
- Honda, M.; Sonehara, S.; Yasuda, H.; Nakagawa, Y.; Tomishige, K. Heterogeneous CeO2 catalyst for the one-pot synthesis of organic carbamates from amines, CO2 and alcohols. Green Chem. 2011, 13, 3406–3413. [Google Scholar] [CrossRef]
- Qin, F.; Li, Q.F.; Wang, J.W.; Feng, Y.L.; Kang, M.Q.; Zhu, Y.L.; Wang, X.K. One Pot Synthesis of Methyl N-Phenyl Carbamate from Aniline, Urea and Methanol. Catal. Lett. 2008, 126, 419–425. [Google Scholar] [CrossRef]
- Barzagli, F.; Mani, F.; Peruzzini, M. From greenhouse gas to feedstock: formation of ammonium carbamate from CO2 and NH3 in organic solvents and its catalytic conversion into urea under mild conditions. Green Chem. 2011, 13, 1267–1274. [Google Scholar] [CrossRef]
- Michalczak, H.W.; Kohlstruk, S.; Krecainski, M.; Grund, G.; Lomoelder, R. Process for the Continuous Preparation of (Cyclo)Aliphatic Diisocyanates. US Patent 8,816,125, 26 August 2014. [Google Scholar]
- Bohmholdt, G.; Disteldorf, J.; Kirchner, P.; Michalczak, H.W. From Diamines, Urea, Alcohols and by-Products from Thermal Cracking. US Patent 5,087,739, 11 February 1992. [Google Scholar]
- Wilmes, O.; Konig, E.; Nachtkamp, K.; Kysela, E. Process for the Production of Diurethanes and Their Use for the Production of Diisocyanates. US Patent 5,744,633, 28 April 1998. [Google Scholar]
- Otterbach, A.; Schwarz, H.V.; Mattner, O.; Merger, F.; Schwarz, W.; Brandt, E. Multistep, Continuous Preparation of Organic Polyisocyanates. US Patent 5,386,053, 31 January 1995. [Google Scholar]
- Wang, P.X.; Ma, Y.B.; Liu, S.M.; Zhou, F.; Yang, B.Q.; Deng, Y.Q. N-Substituted carbamate synthesis using urea as carbonyl source over TiO2-Cr2O3/SiO2 catalyst. Green Chem. 2015, 17, 3964–3971. [Google Scholar] [CrossRef]
- Wang, G.R.; Li, X.; Wang, Y.J.; Zhao, X.Q. Mechanism for Synthesis of Dibutyl Toluene-2,4-Dicarbamate via Urea Route Catalyzed by γ-Al2O3. Ind. Eng. Chem. Res. 2014, 53, 2130–2136. [Google Scholar] [CrossRef]
- Sun, D.L.; Deng, J.R.; Chao, Z.S. Catalysis over zinc-incorporated berlinite (ZnAlPO4) of the methoxycarbonylation of 1,6-hexanediamine with dimethyl carbonate to form dimethylhexane-1,6-dicarbamate. Chem. Cent. J. 2007. [Google Scholar] [CrossRef] [PubMed]
- Bhanage, B.M.; Fujita, S.; Ikushima, Y.; Arai, M. Transesterification of urea and ethylene glycol to ethylene carbonate as an important step for urea based dimethyl carbonate synthesis. Green Chem. 2003, 5, 429–432. [Google Scholar] [CrossRef]
- Wang, M.H.; Zhao, N.; Sun, Y.H. Synthesis of Dimethyl Carbonate from Urea and Methanol over ZnO. Ind. Eng. Chem. Res. 2005, 44, 7596–7599. [Google Scholar] [CrossRef]
- Merger, F.; Towae, F. Process for the Preparation of N-aryl di- or Polyurethanes. US Patent 4,611,079, 9 September 1986. [Google Scholar]
- Wagner, C.D.; Zatko, D.A.; Raymond, R.H.; Chem, A. Use of the oxygen KLL auger lines in identification of surface chemical states by electron spectroscopy for chemical analysis. Anal. Chem. 1980, 52, 1445–1451. [Google Scholar] [CrossRef]
- Sun, D.L.; Luo, J.Y.; Wen, R.Y.; Deng, J.R.; Chao, Z.S. Phosgene-free synthesis of hexamethylene-1,6-diisocyanate by the catalytic decomposition of dimethylhexane-1,6-dicarbamate over zinc-incorporated berlinite (ZnAlPO4). J. Hazard. Mater. 2014, 266, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Rokita, M.; Handke, M.; Mozgawa, W. Spectroscopic studies of polymorphs of AlPO4 and SiO2. J. Mol. Struct. 1998, 450, 213–217. [Google Scholar] [CrossRef]
- Lin, H.Y.; Yang, B.L.; Sun, J.J.; Wang, X.P.; Wang, D.P. Kinetics studies for the synthesis of dimethyl carbonate from urea and methanol. Chem. Eng. J. 2004, 21, 103–110. [Google Scholar] [CrossRef]
- Sun, J.J.; Yang, B.L.; Lin, H.Y. A Semi-continuous Process for the Synthesis of Methyl Carbamate from Urea and Methanol. Chem. Eng. Technol. 2004, 27, 435–439. [Google Scholar] [CrossRef]
- Friederichs, W.; Penninger, S.; Wershofen, S. Process for the Preparation of Highly Pure Aromatic Diurethanes and/or Polyurethanes. US Patent 5,399,736, 21 March 1995. [Google Scholar]
- Sun, D.L.; Xie, S.J.; Deng, J.R.; Huang, C.J.; Ruckenstein, E.; Chao, Z.S. CH3COONa as an effective catalyst for methoxycarbonylation of 1,6-hexanediamine by dimethyl carbonate to dimethylhexane-1,6-dicarbamate. Green Chem. 2010, 12, 483–490. [Google Scholar] [CrossRef]
- Wang, D.P.; Yang, B.; Zhai, X.W.; Zhou, L.G. Synthesis of diethyl carbonate by catalytic alcoholysis of urea. Fuel Process Technol. 2007, 88, 807–812. [Google Scholar] [CrossRef]
- Shivarkar, A.B.; Gupte, S.P.; Chaudhari, R.V. Carbamate synthesis via transfunctionalization of substituted ureas and carbonates. J. Mol. Catal. A 2004, 223, 85–92. [Google Scholar] [CrossRef]
- Carloni, S.; Devos, D.E.; Jacobs, P.A.; Maggi, R.; Sartori, G.; Raffaella, S. Catalytic Activity of MCM-41–TBD in the Selective Preparation of Carbamates and Unsymmetrical Alkyl Carbonates from Diethyl Carbonate. J. Catal. 2002, 205, 199–204. [Google Scholar] [CrossRef]
- Zhao, W.B.; Peng, W.C.; Wang, D.F.; Zhao, N.; Li, J.P.; Xiao, F.K.; Wei, W.; Sun, Y.H. Zinc oxide as the precursor of homogenous catalyst for synthesis of dialkyl carbonate from urea and alcohols. Catal. Commun. 2009, 10, 655–658. [Google Scholar]



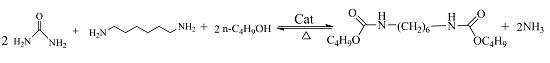{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalysts | χHDA b (%) | Si c (%) | 2 d Yield (%) | ||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||||
| 1 | None e | 36.8 | 13.9 | 21.3 | 26.9 | 31.7 | 5.9 | 7.8 |
| 2 | ZnAlPO4(0) | 37.5 | 14.2 | 21.9 | 27.4 | 31.2 | 5.8 | 7.8 |
| 3 | ZnAlPO4(0.01) | 51.7 | 18.8 | 37.1 | 20.4 | 20.1 | 4.7 | 19.2 |
| 4 | ZnAlPO4(0.02) | 68.3 | 19.6 | 52.2 | 16.1 | 10.0 | 4.5 | 35.7 |
| 5 | ZnAlPO4(0.03) | 83.9 | 17.4 | 67.7 | 8.3 | 9.7 | 3.7 | 56.8 |
| 6 | ZnAlPO4(0.04) | 98.4 | 6.3 | 90.5 | 3.2 | 4.2 | 2.1 | 89.1 |
| 7 | ZnAlPO4(0.05) | 98.9 | 7.2 | 89.7 | 3.5 | 4.5 | 2.6 | 88.7 |
| 8 | ZnAlPO4(0.06) | 98.0 | 11.0 | 81.2 | 5.1 | 6.6 | 3.2 | 79.6 |
| 9 | ZnAlPO4(0.07) | 96.2 | 13.9 | 74.3 | 7.6 | 7.2 | 3.9 | 71.5 |
| 10 | ZnAlPO4(0.08) | 93.1 | 16.2 | 68.4 | 9.3 | 8.1 | 4.6 | 63.7 |
| 11 | ZnAlPO4(0.15) | 87.9 | 22.0 | 23.1 | 28.0 | 23.2 | 5.1 | 20.3 |
| 12 | ZnO | 88.1 | 22.3 | 22.4 | 29.1 | 23.0 | 5.2 | 19.7 |
| Entry | Alcohol | X b | Sc c | Yc d |
|---|---|---|---|---|
| 1 e | MeOH | 89.8 | 82.5 | 74.1 |
| 2 | EtOH | 79.7 | 88.2 | 70.3 |
| 3 | n-PrOH | 77.3 | 88.5 | 68.4 |
| 4 | n-BuOH | 76.9 | 87.5 | 67.3 |
| 5 | n-PeOH | 75.4 | 86.9 | 65.5 |
| 6 | 2-BuOH | 70.2 | 87.5 | 61.4 |
| 7 | 2-PrOH | 69.1 | 85.5 | 59.1 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, D.-L.; Mai, J.-J.; Deng, J.-R.; Idem, R.; Liang, Z.-W. One-Pot Synthesis of Dialkyl Hexane-1,6-Dicarbamate from 1,6-Hexanediamine, Urea, and Alcohol over Zinc-Incorporated Berlinite (ZnAlPO4) Catalyst. Catalysts 2016, 6, 28. https://doi.org/10.3390/catal6020028
Sun D-L, Mai J-J, Deng J-R, Idem R, Liang Z-W. One-Pot Synthesis of Dialkyl Hexane-1,6-Dicarbamate from 1,6-Hexanediamine, Urea, and Alcohol over Zinc-Incorporated Berlinite (ZnAlPO4) Catalyst. Catalysts. 2016; 6(2):28. https://doi.org/10.3390/catal6020028
Chicago/Turabian StyleSun, Da-Lei, Ji-Jin Mai, Jian-Ru Deng, Raphael Idem, and Zhi-Wu Liang. 2016. "One-Pot Synthesis of Dialkyl Hexane-1,6-Dicarbamate from 1,6-Hexanediamine, Urea, and Alcohol over Zinc-Incorporated Berlinite (ZnAlPO4) Catalyst" Catalysts 6, no. 2: 28. https://doi.org/10.3390/catal6020028
APA StyleSun, D.-L., Mai, J.-J., Deng, J.-R., Idem, R., & Liang, Z.-W. (2016). One-Pot Synthesis of Dialkyl Hexane-1,6-Dicarbamate from 1,6-Hexanediamine, Urea, and Alcohol over Zinc-Incorporated Berlinite (ZnAlPO4) Catalyst. Catalysts, 6(2), 28. https://doi.org/10.3390/catal6020028