
Steam Reforming of Bio-Ethanol to Produce Hydrogen over Co/CeO2 Catalysts Derived from Ce1−xCoxO2−y Precursors


Abstract
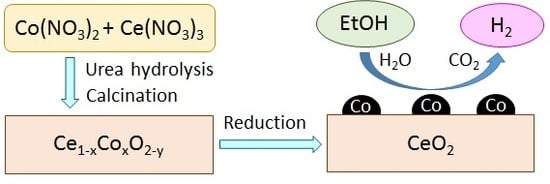
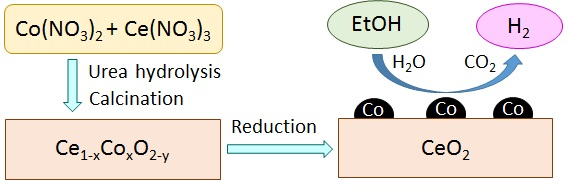
:
1. Introduction
2. Results and Discussion
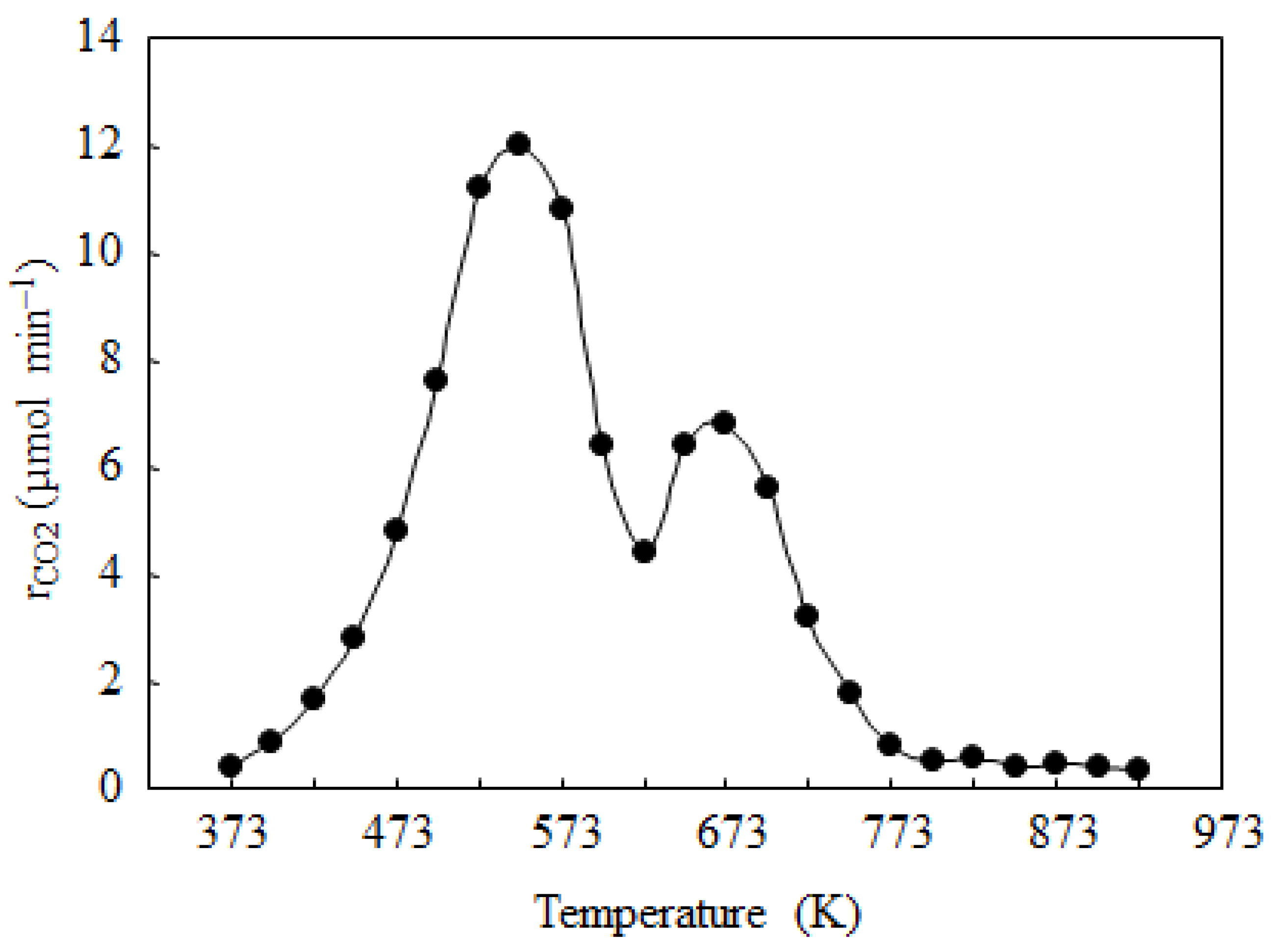
2.1. Catalyst Characterization
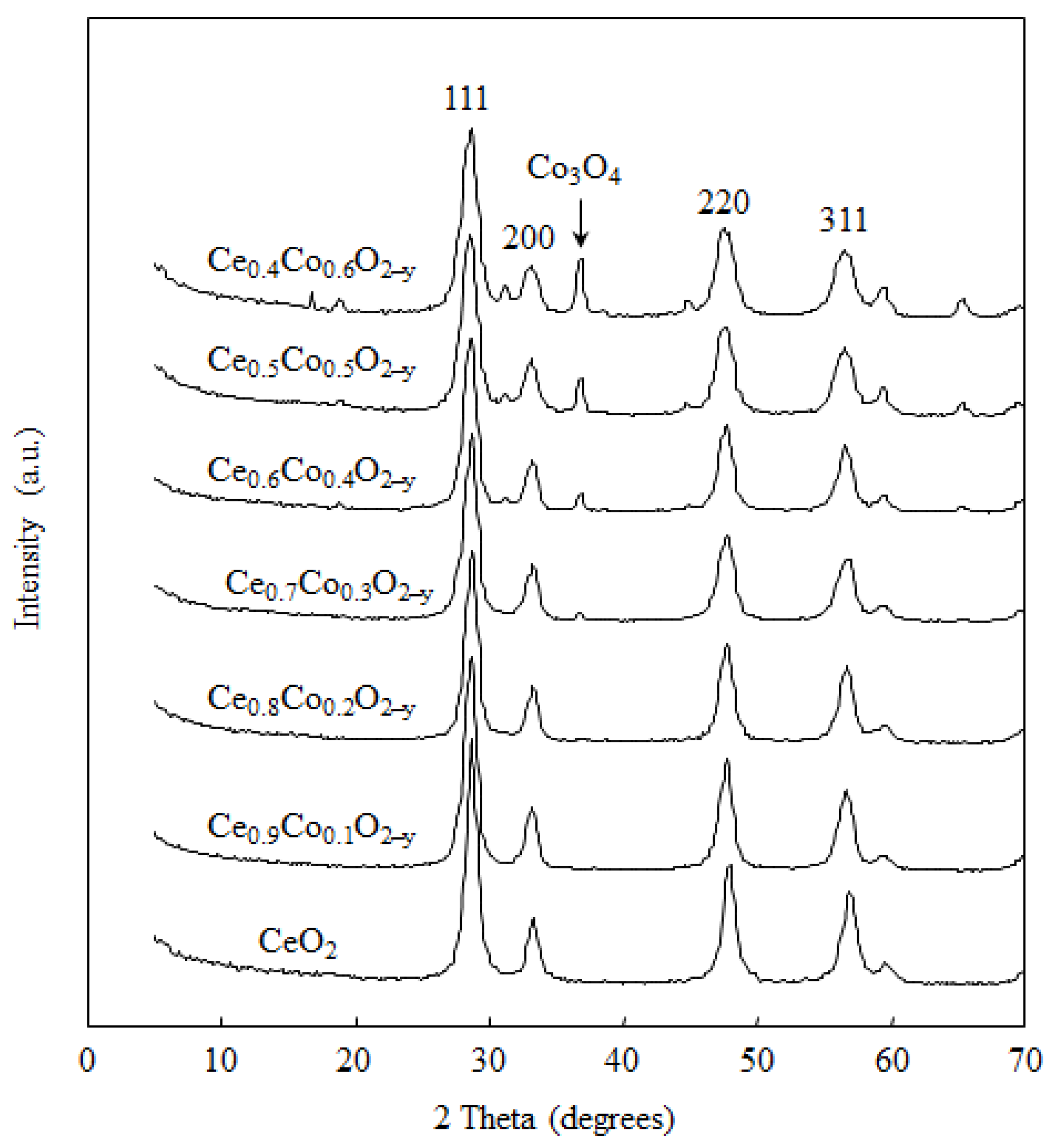
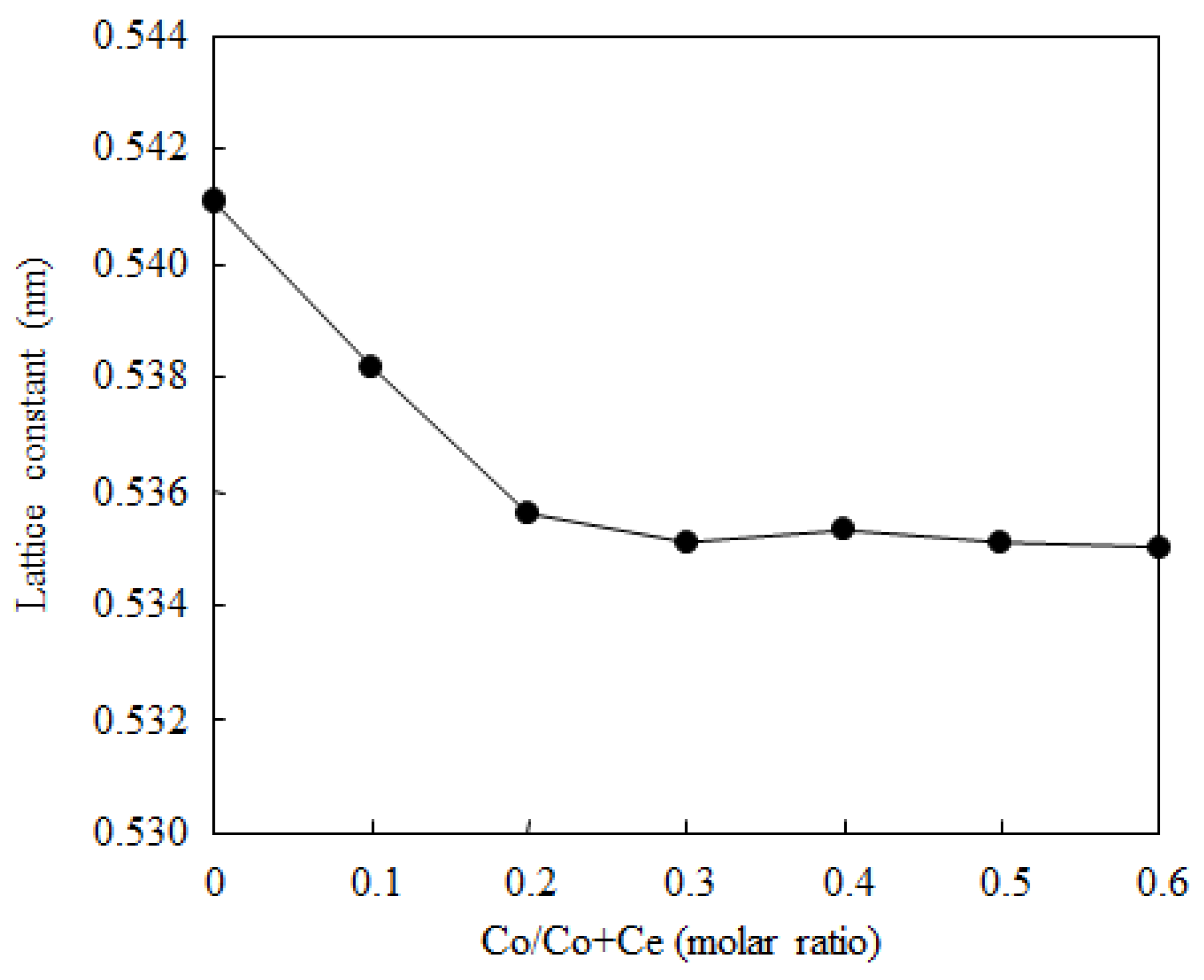

{kind=link}
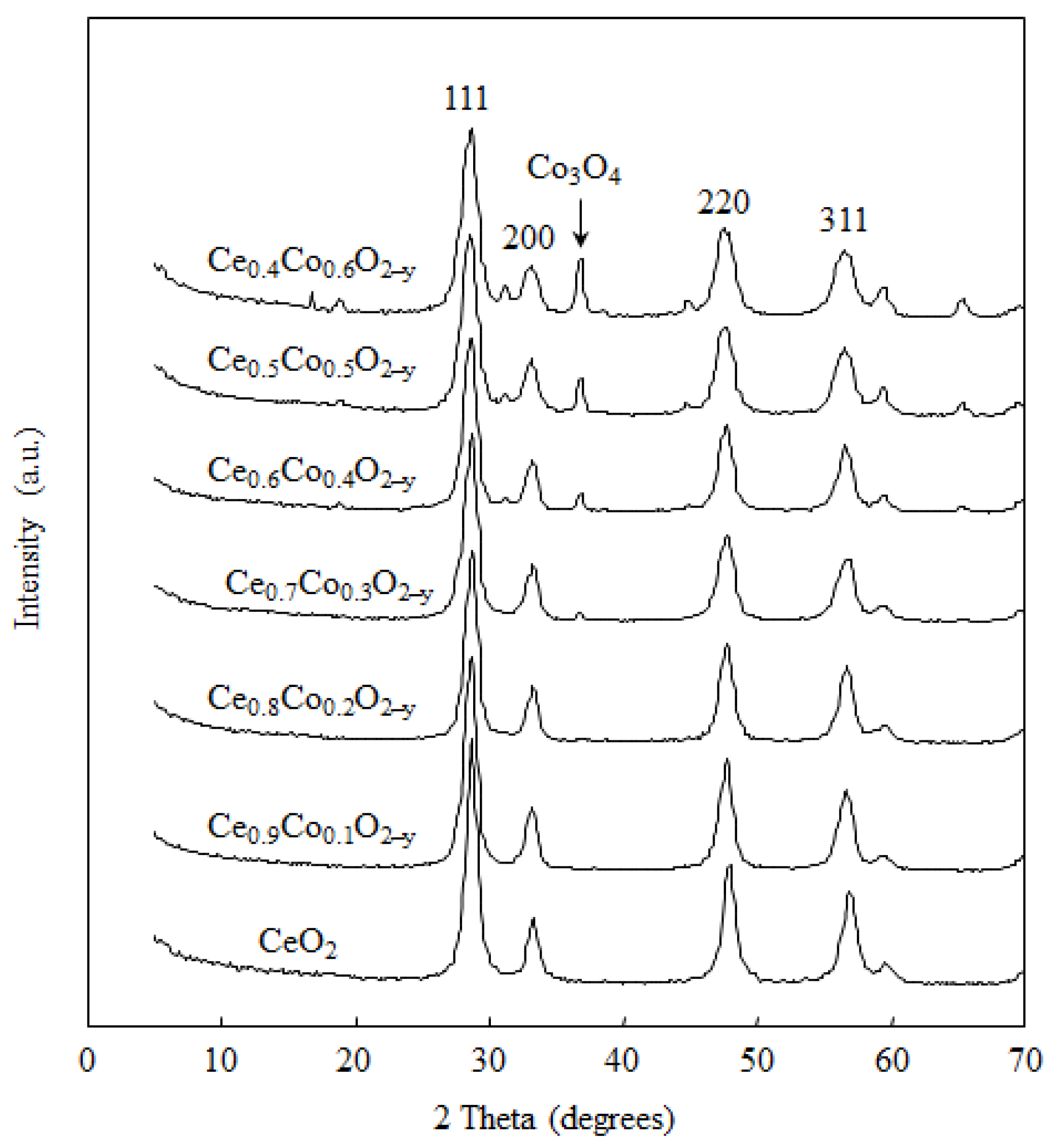{kind=link}
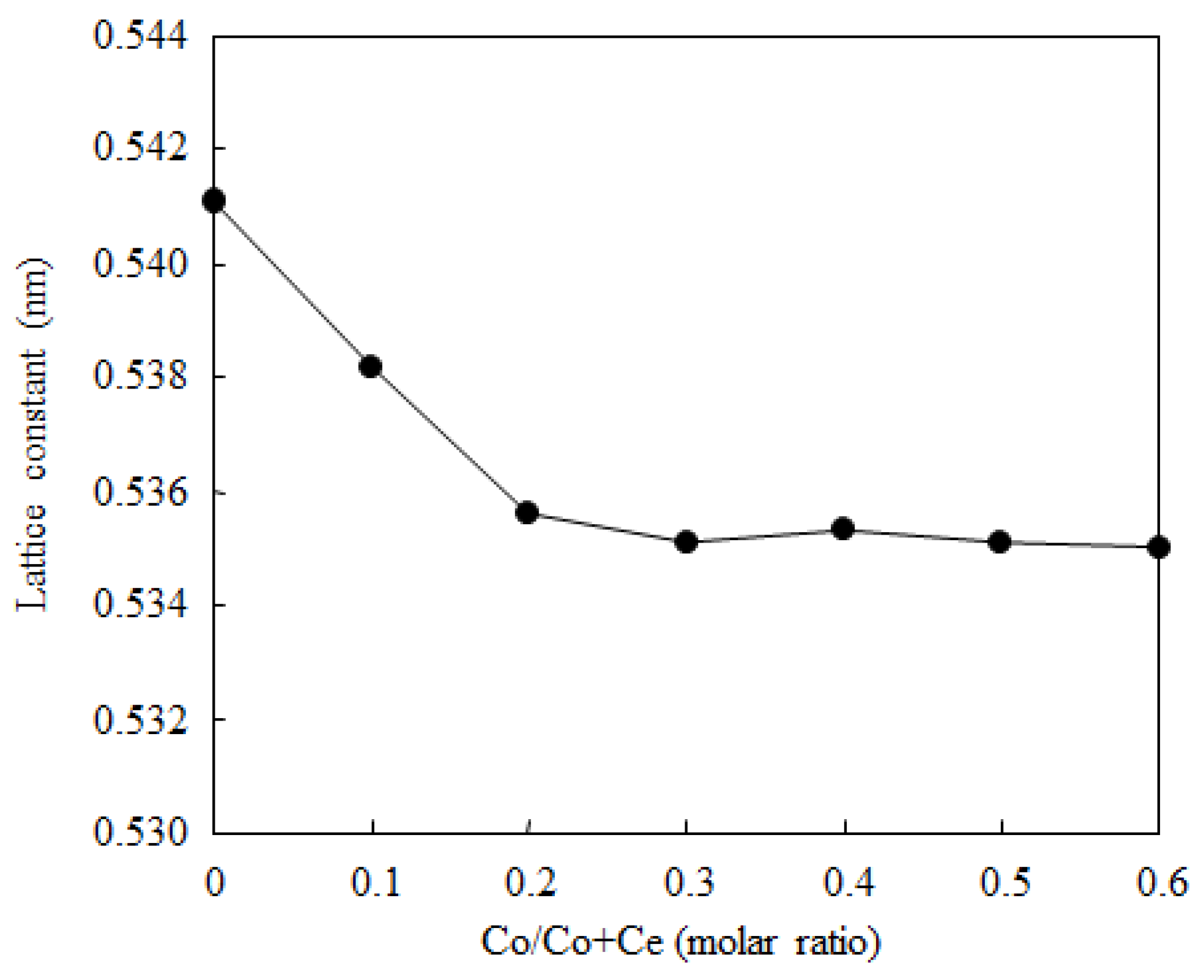{kind=link}
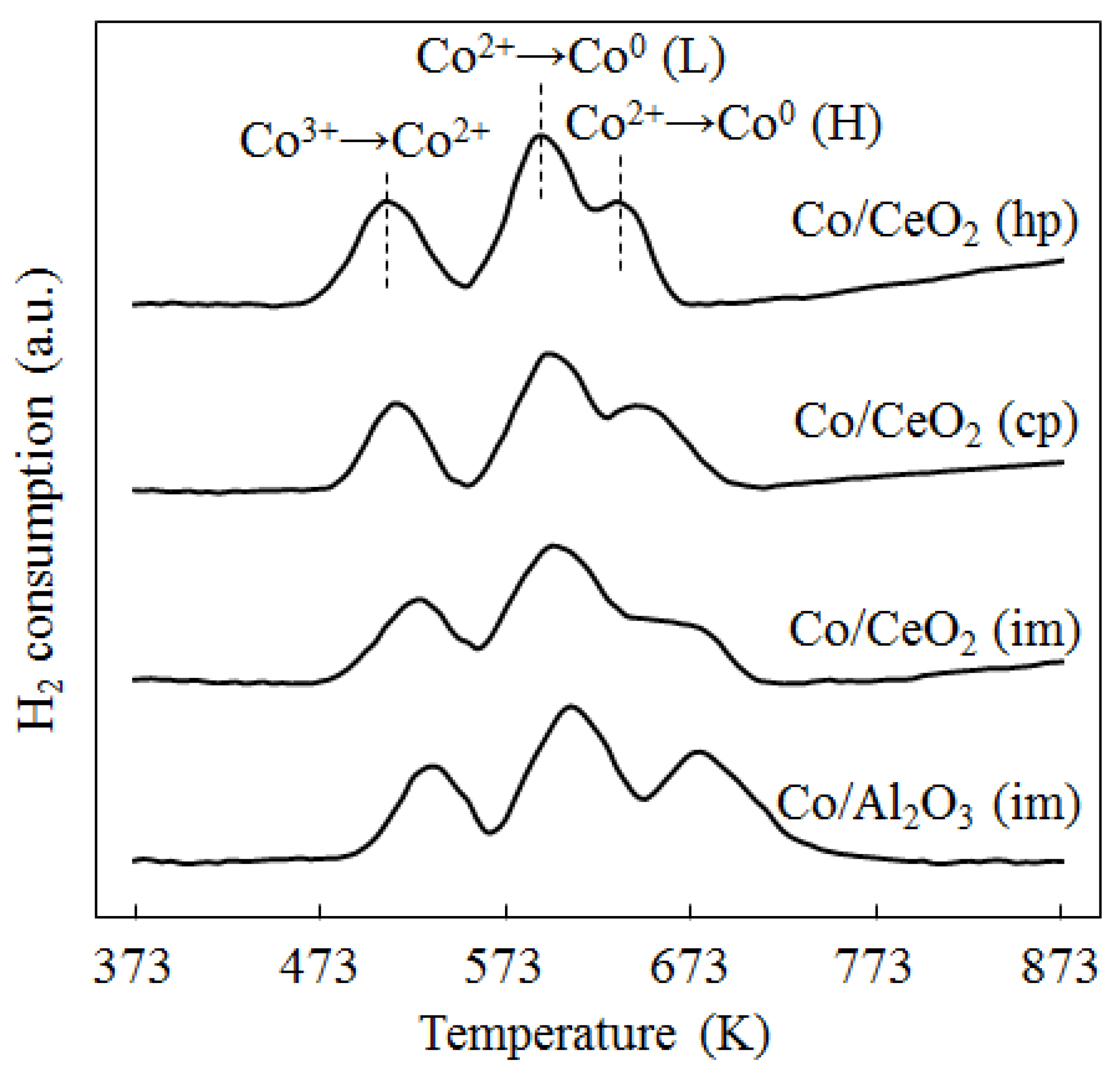{kind=link}
{kind=link}
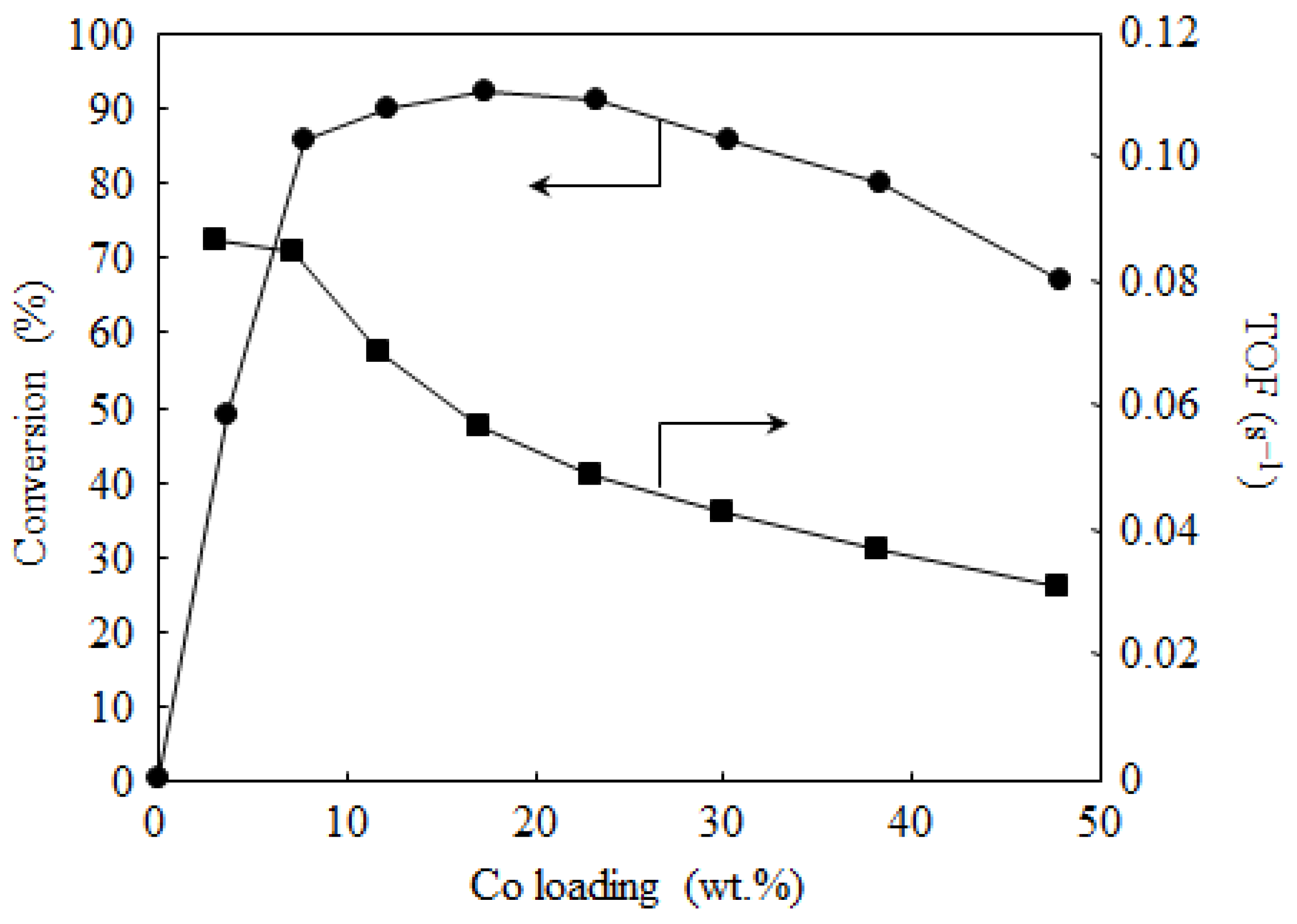{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
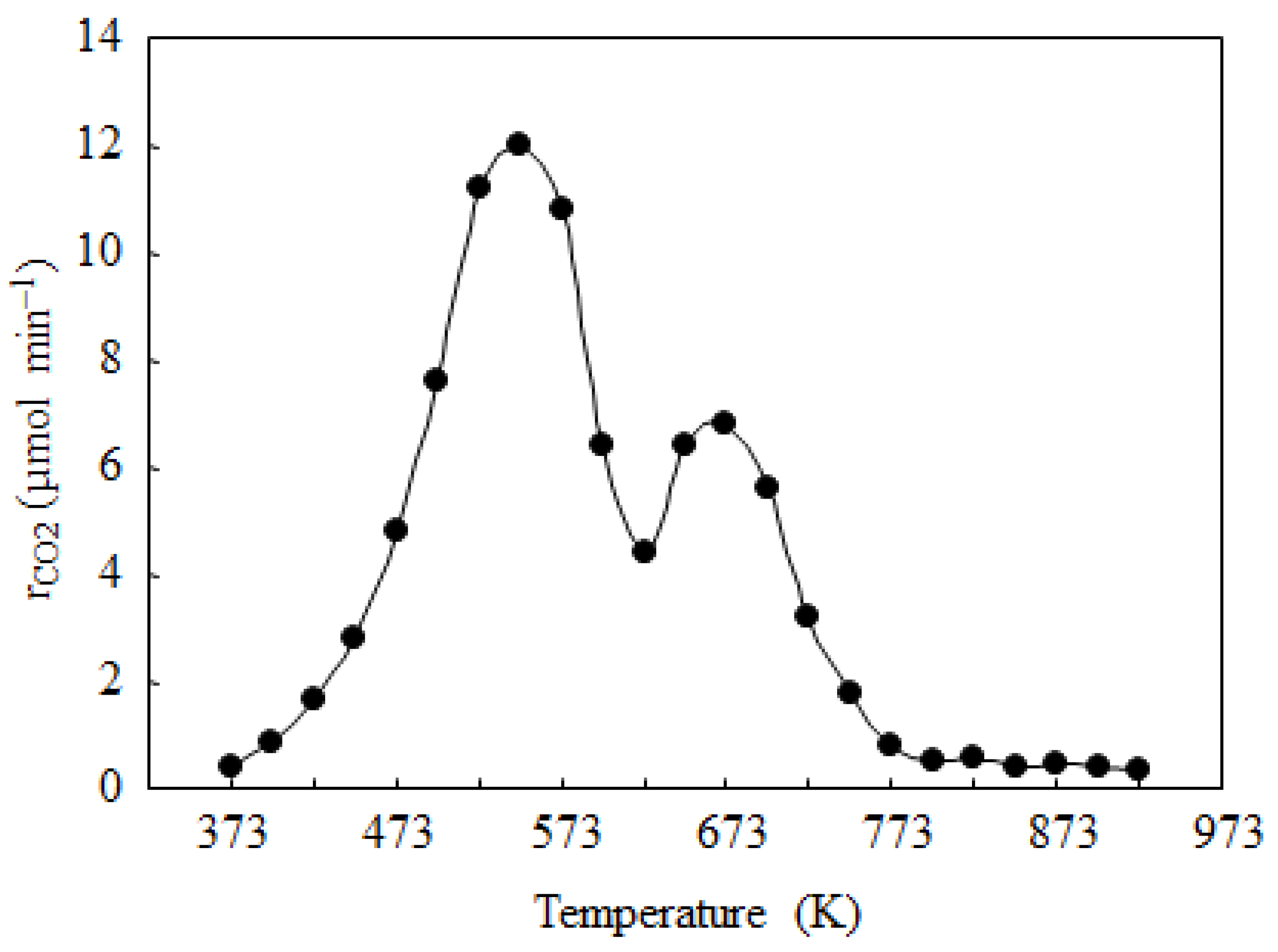{kind=link}
| Sample | BET Surface Area (m2·g−1) | Co Particle Size (nm) a | Co Dispersion Degree (%) a |
|---|---|---|---|
| Co/CeO2 (hp) | 135 | 7.6 | 12.6 |
| Co/CeO2 (cp) | 102 | 9.5 | 10.1 |
| Co/CeO2 (im) | 88 | 17.7 | 5.4 |
| Co/Al2O3 (im) | 147 | 14.3 | 6.7 |
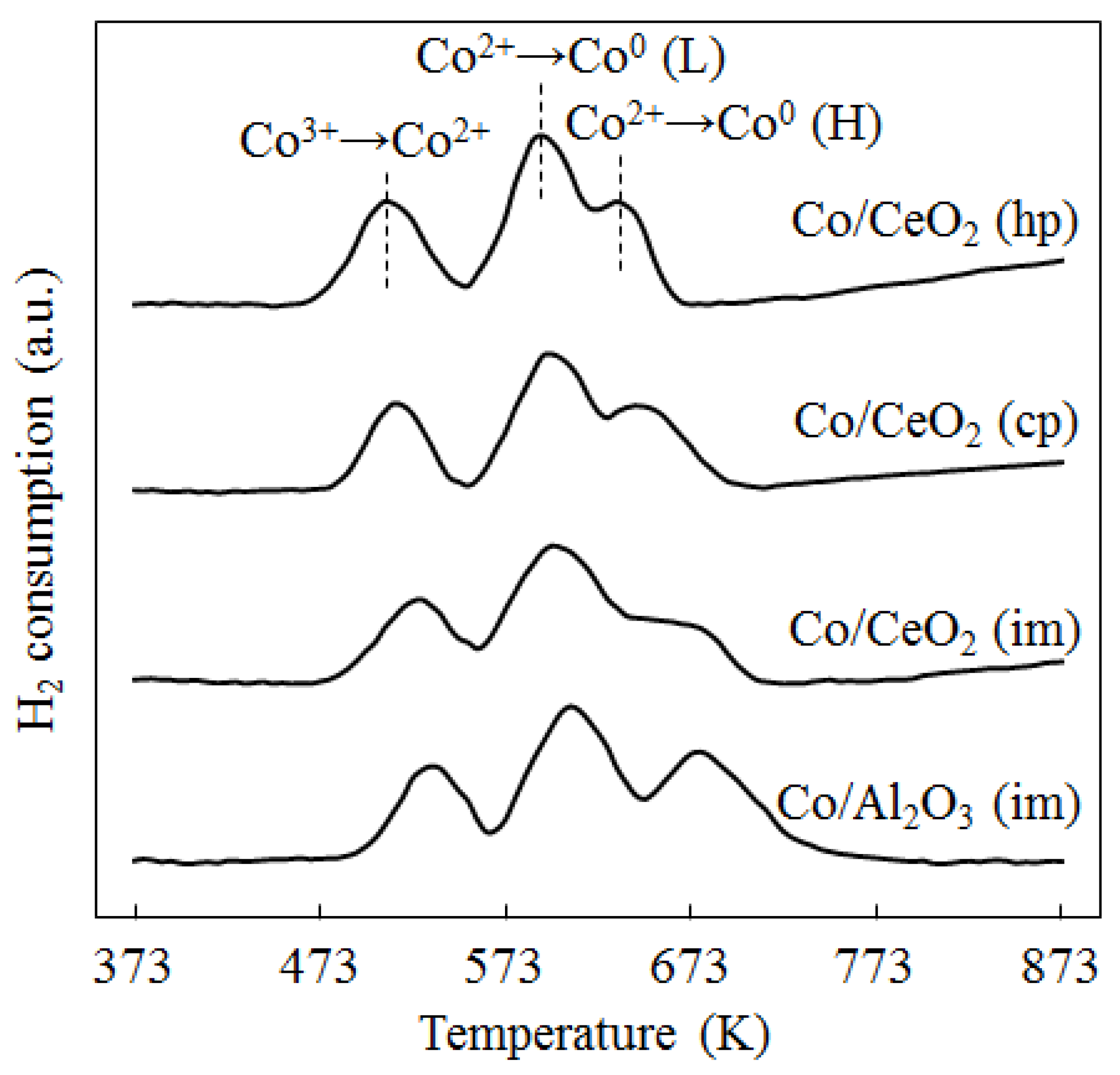

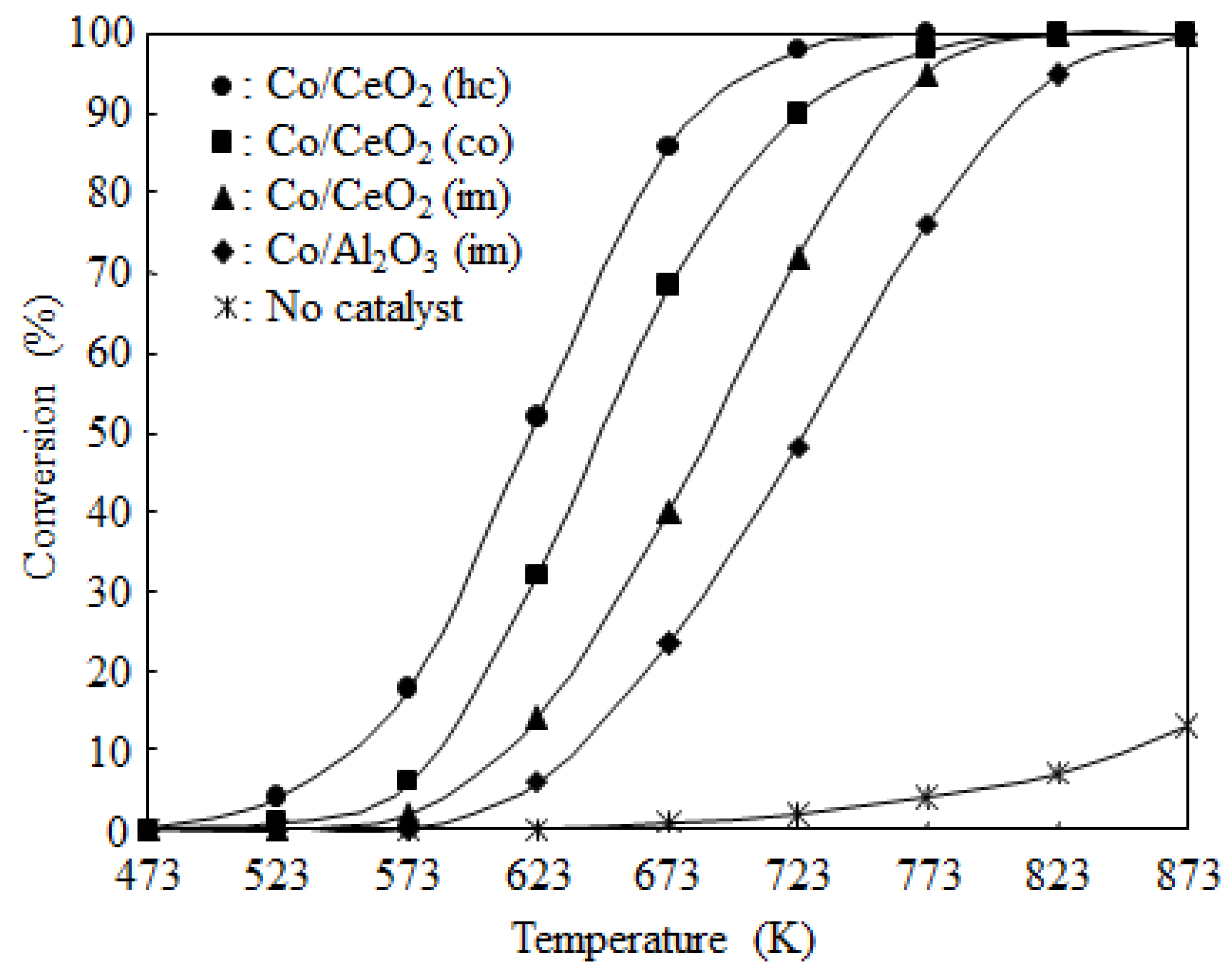
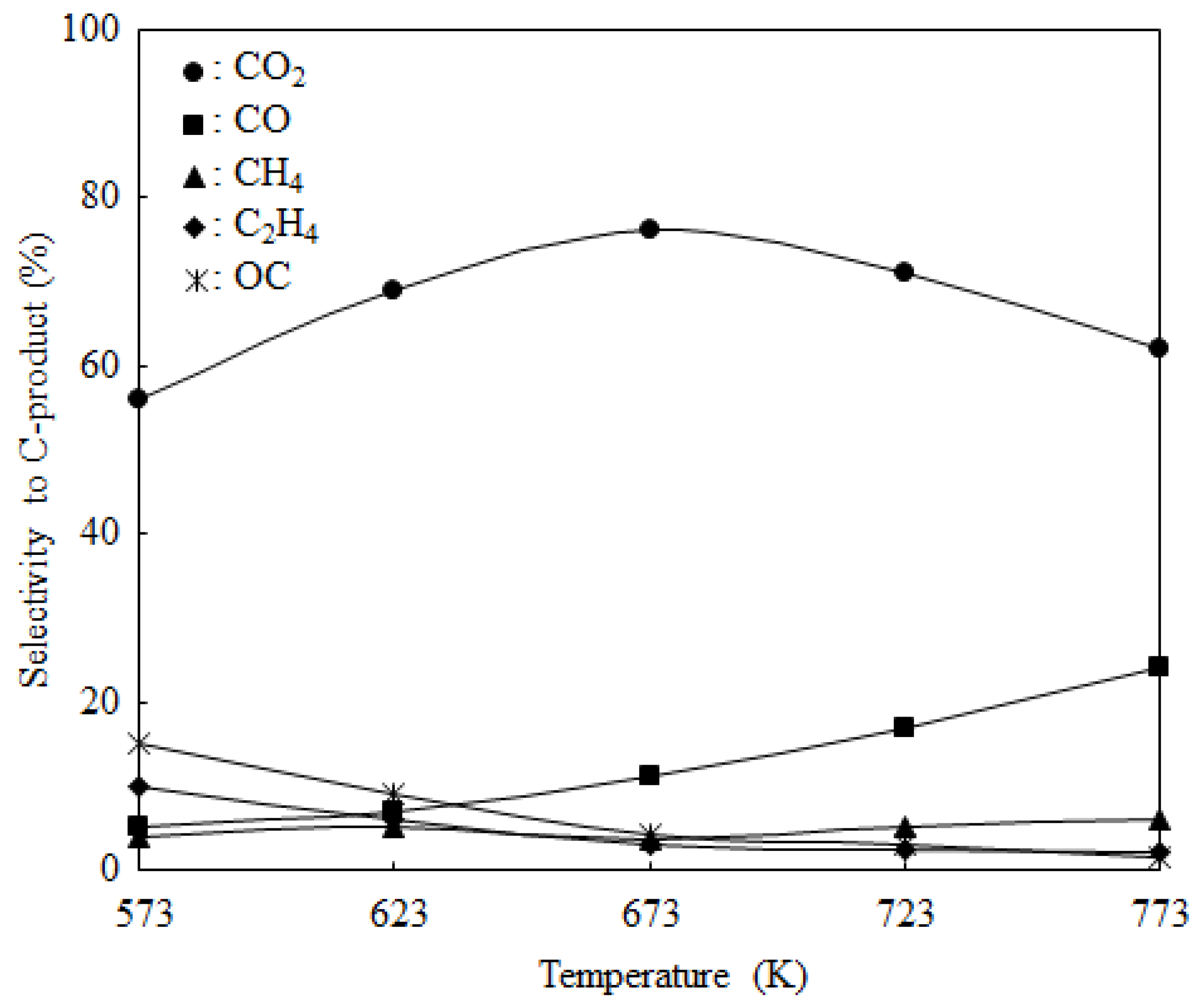
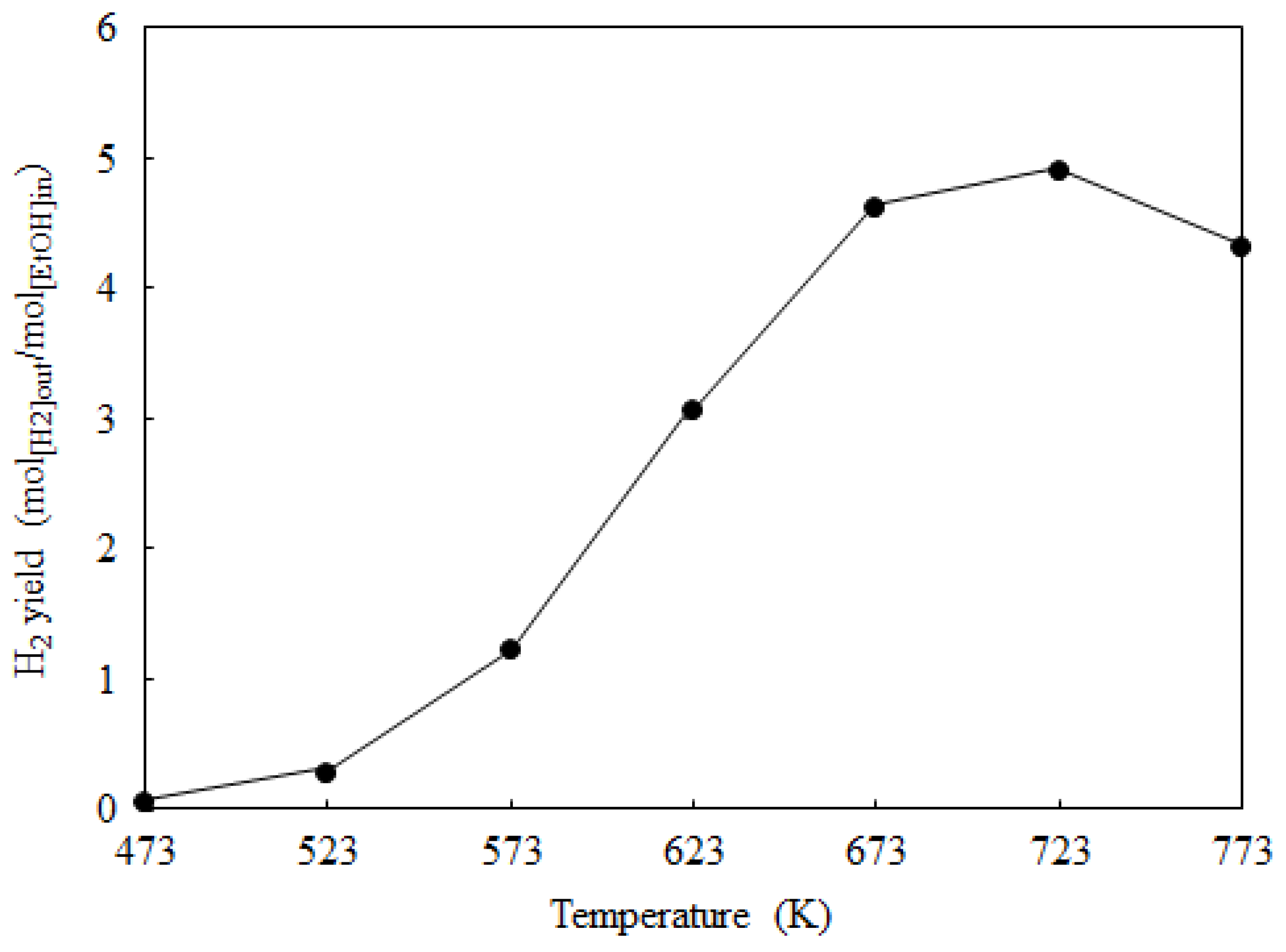
2.2. Catalyst Activity
| Catalyst b | Conversion of Ethanol (%) | Selectivity to C-products (%) | H2 Yield (Molar Ratio of [H2]out/[EtOH]in) | ||||
|---|---|---|---|---|---|---|---|
| CO2 | CO | CH4 | C2H4 | OC c | |||
| Co/Al2O3 (im) | 25.5 | 54.9 | 19.5 | 8.3 | 13.9 | 3.3 | 1.1 |
| Co/CeO2 (im) | 40.3 | 70.6 | 15.2 | 4.4 | 3.7 | 4.8 | 2.1 |
| Co/CeO2 (cp) | 68.4 | 73.3 | 14.6 | 3.7 | 2.8 | 4.3 | 3.6 |
| Co/CeO2 (hp) | 85.9 | 76.2 | 12.1 | 3.5 | 3.1 | 4.2 | 4.7 |
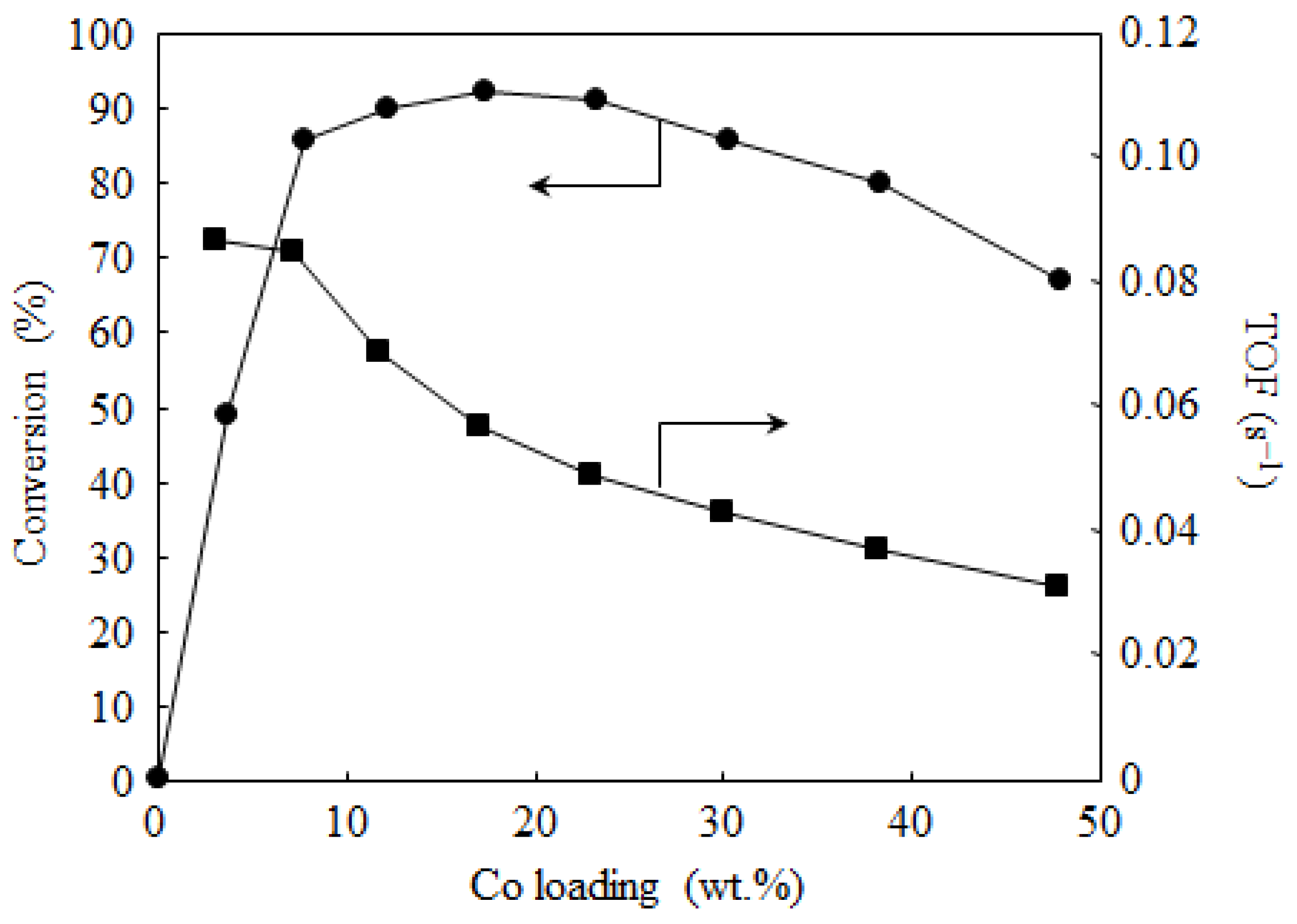
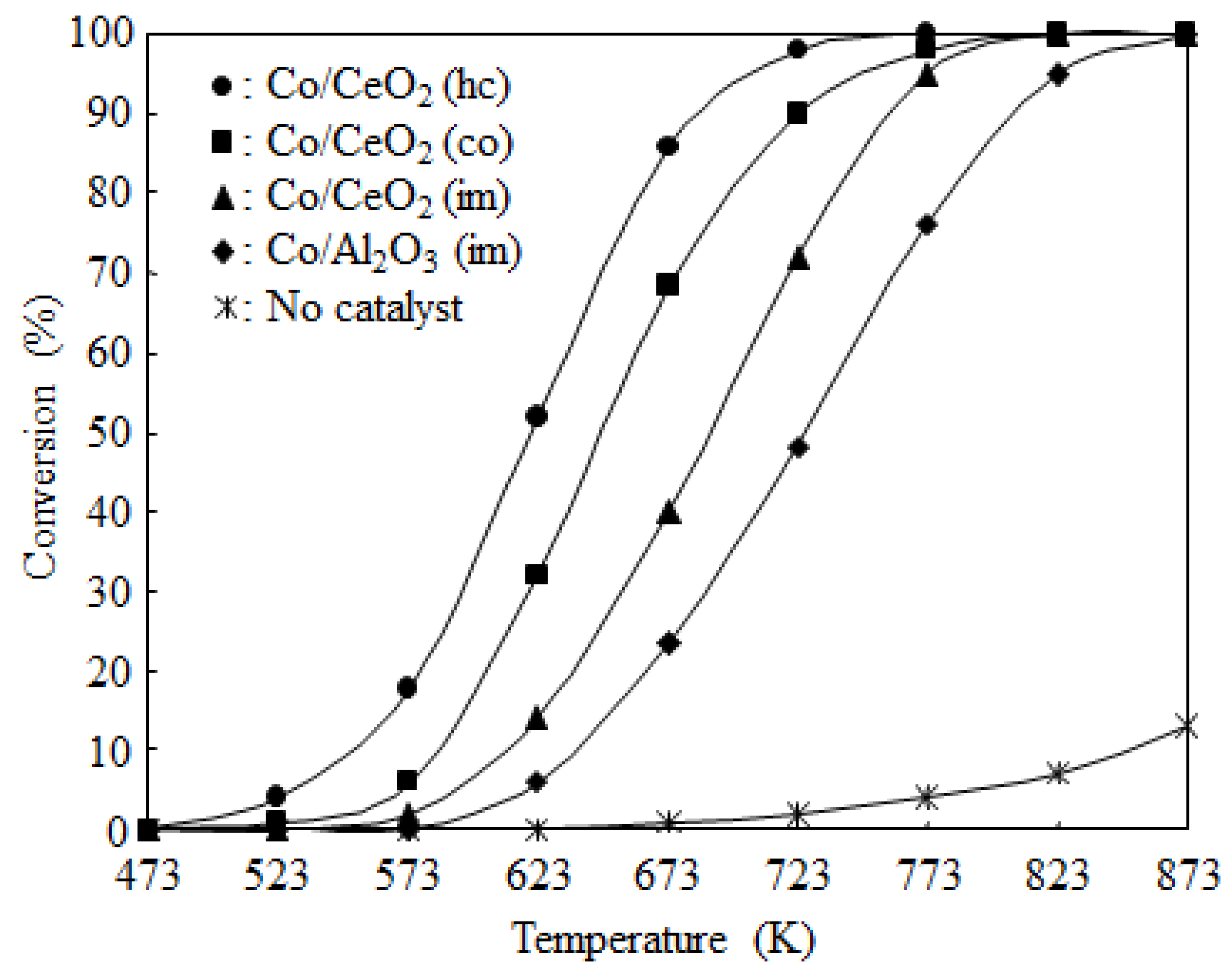
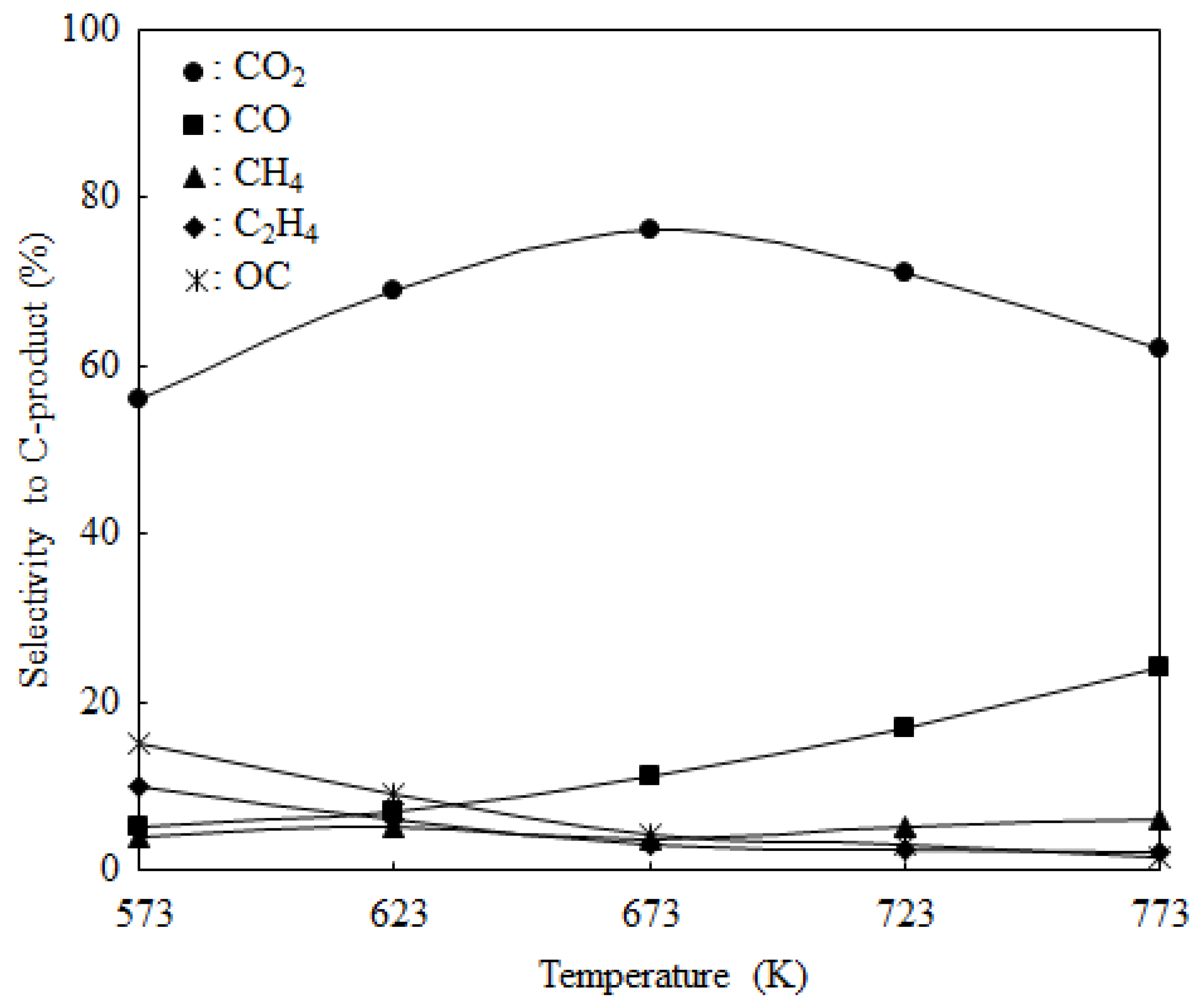
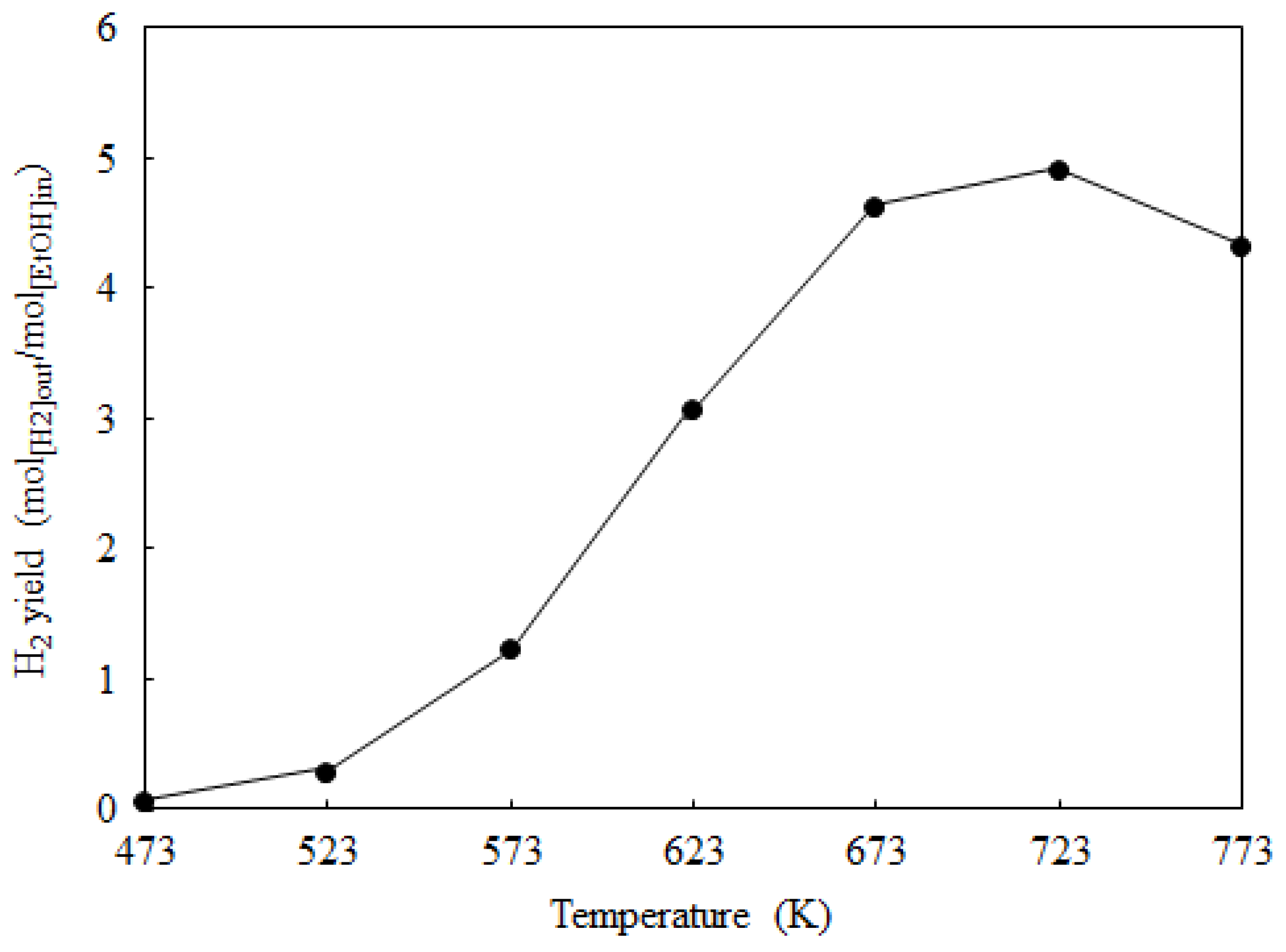
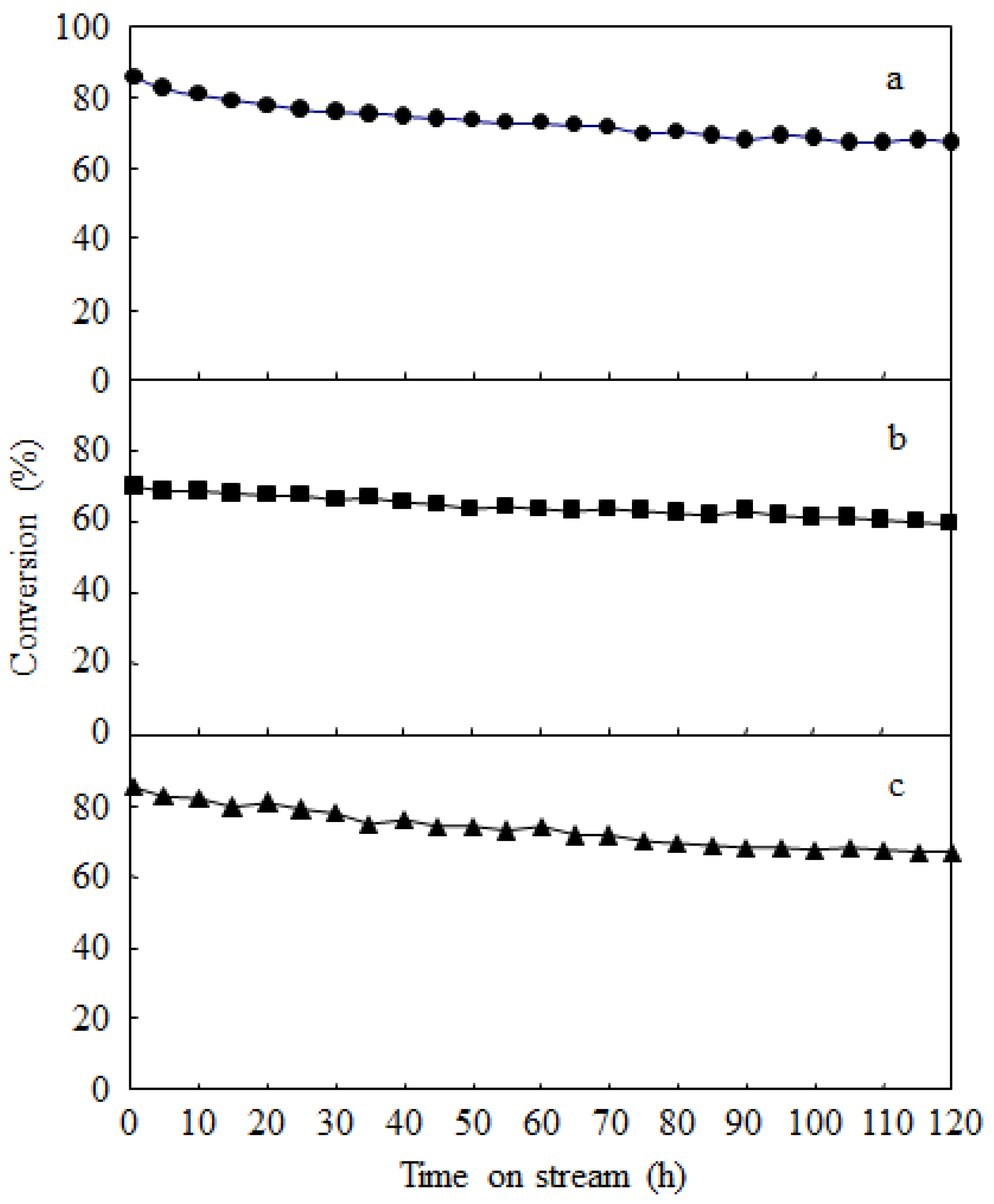
2.3. Catalyst Deactivation and Regeneration
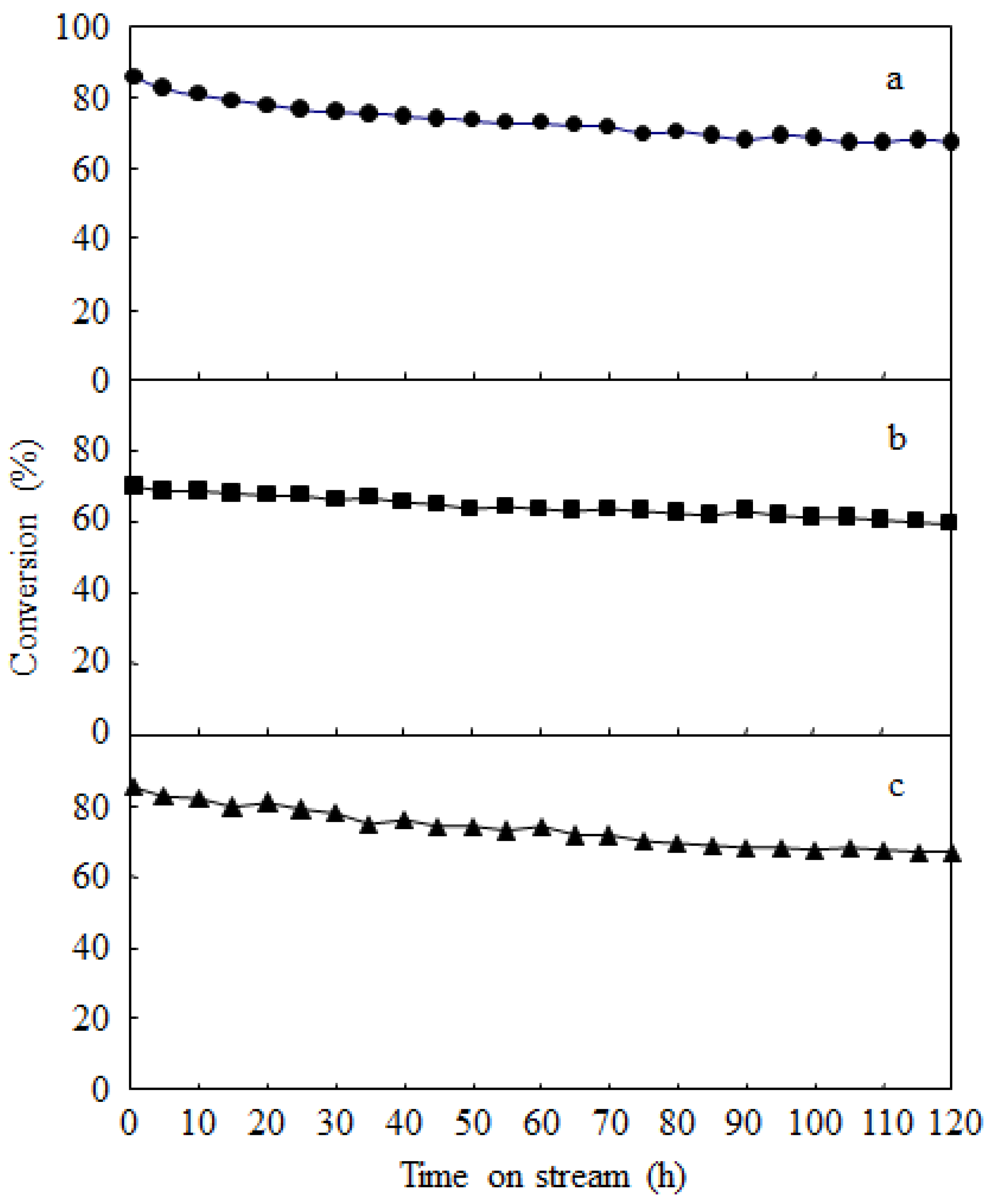
3. Experimental Section
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Catalytic Reaction
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Ioannides, T. Thermodynamic Analysis of Ethanol Processors for Fuel Cell Applications. J. Power Sources 2001, 92, 17–25. [Google Scholar] [CrossRef]
- Ni, M.; Leung, D.T.C.; Leung, K.H. A Review on Reforming Bio-Ethanol for Hydrogen Production. Int. J. Hydrogen Energy 2007, 32, 3238–3247. [Google Scholar] [CrossRef]
- Sun, J.; Qiu, X.; Wu, F.; Zhu, W.; Wang, W.; Hao, S. Hydrogen from Steam Reforming of Ethanol in Low and Middle Temperature Range for Fuel Cell Application. Int. J. Hydrogen Energy 2004, 29, 1075–1081. [Google Scholar] [CrossRef]
- Cifuentes, B.; Valero, M.F.; Conesa, J.A.; Cobo, M. Hydrogen Production by Steam Reforming of Ethanol on Rh–Pt Catalysts: Influence of CeO2, ZrO2, and La2O3 as Supports. Catalysts 2015, 5, 1872–1896. [Google Scholar] [CrossRef]
- Calles, J.A.; Carrero, A.; Vizcaíno, A.J.; Lindo, M. Effect of Ce and Zr Addition to Ni/SiO2 Catalysts for Hydrogen Production through Ethanol Steam Reforming. Catalysts 2015, 5, 58–76. [Google Scholar] [CrossRef]
- Vargas, J.C.; Ivanova, S.; Thomas, S.; Roger, A.-C.; Pitchon, V. Influence of Gold on Ce–Zr–Co Fluorite-Type Mixed Oxide Catalysts for Ethanol Steam Reforming. Catalysts 2012, 2, 121–138. [Google Scholar] [CrossRef]
- Domínguez, M.; Taboada, E.; Molins, E.; Llorca, J. Co–Fe–Si Aerogel Catalytic Honeycombs for Low Temperature Ethanol Steam Reforming. Catalysts 2012, 2, 386–399. [Google Scholar] [CrossRef]
- Llorca, L.; Homs, N.; Sales, J.; Fierro, J.G.; Piscina, P.R. Effect of Sodium Addition on the Performance of Co–ZnO-based Catalysts for Hydrogen Production from Bioethanol. J. Catal. 2004, 222, 470–480. [Google Scholar] [CrossRef]
- Haga, F.; Nakajima, T.; Miya, H.; Mishima, S. Catalytic Properties of Supported Cobalt Catalysts for Steam Reforming of Ethanol. Catal. Lett. 1997, 48, 223–237. [Google Scholar] [CrossRef]
- Llorca, J.; Dalmon, J.A.; Piscina, P.R.; Homs, N. In Situ Magnetic Characterisation of Supported Cobalt Catalysts under Steam-Reforming of Ethanol. Appl. Catal. A 2003, 243, 261–269. [Google Scholar] [CrossRef]
- Tuti, S.; Pepe, F. On the Catalytic Activity of Cobalt Oxide for the Steam Reforming of Ethanol. Catal. Lett. 2008, 122, 196–203. [Google Scholar] [CrossRef]
- Urasaki, K.; Tokunaga, K.; Sekine, Y.; Kikuchi, E.; Matsukata, M. Hydrogen Production by Steam Reforming of Ethanol using Cobalt and Nickel Catalysts Supported on Strontium Titanate. Chem. Lett. 2005, 34, 668–669. [Google Scholar] [CrossRef]
- Bichon, P.; Haugom, G.; Venvik, H.J.; Holmen, A.; Blekkan, E.A. Steam Reforming of Ethanol over Supported Co and Ni Catalysts. Top. Catal. 2008, 49, 38–45. [Google Scholar] [CrossRef]
- Sun, J.; Qiu, X.; Wu, F.; Zhu, W. H2 from Steam Reforming of Ethanol at Low Temperature over Ni/Y2O3, Ni/La2O3 and Ni/Al2O3 Catalysts for Fuel-Cell Application. Int. J. Hydrogen Energy 2005, 30, 437–445. [Google Scholar] [CrossRef]
- Deng, X.; Sun, J.; Yu, S.; Xi, J.; Zhu, W.; Qiu, X. Steam Reforming of Ethanol for Hydrogen Production over NiO/ZnO/ZrO2 Catalysts. Int. J. Hydrogen Energy 2008, 33, 1008–1013. [Google Scholar] [CrossRef]
- Biswas, P.; Kunzru, D. Steam Reforming of Ethanol for Production of Hydrogen over Ni/CeO2–ZrO2 Catalyst: Effect of Support and Metal Loading. Int. J. Hydrogen Energy 2007, 32, 969–980. [Google Scholar] [CrossRef]
- Biswas, P.; Kunzru, D. Steam Reforming of Ethanol on Ni–CeO2–ZrO2 Catalysts: Effect of Doping with Copper, Cobalt and Calcium. Catt. Lett. 2007, 118, 36–49. [Google Scholar] [CrossRef]
- Ye, J.L.; Wang, Y.Q.; Liu, Y.; Wang, H. Steam Reforming of Ethanol over Ni/CexTi1−xO2 Catalysts. Int. J. Hydrogen Energy 2008, 33, 6602–6611. [Google Scholar] [CrossRef]
- Zhang, B.; Cai, W.; Li, Y.; Xu, Y.; Shen, W. Hydrogen Production by Steam Reforming of Ethanol over an Ir/CeO2 Catalyst: Reaction Mechanism and Stability of the Catalyst. Int. J. Hydrog. Energy 2008, 33, 4377–4386. [Google Scholar] [CrossRef]
- Diagne, C.; Idriss, H.; Kiennemann, A. Hydrogen Production by Ethanol Reforming over Rh/CeO2–ZrO2 Catalysts. Catal. Commun. 2002, 3, 565–571. [Google Scholar] [CrossRef]
- Scott, M.; Goeffroy, M.; Chiu, W.; Blackford, M.A.; Idriss, H. Hydrogen Production from Ethanol over Rh–Pd/CeO2 Catalysts. Top. Catal. 2008, 51, 13–21. [Google Scholar] [CrossRef]
- Platon, A.; Roh, H.S.; King, D.L.; Wang, Y. Deactivation Studies of Rh/Ce0.8Zr0.2O2 Catalysts in Low Temperature Ethanol Steam Reforming. Top. Catal. 2007, 46, 374–379. [Google Scholar] [CrossRef]
- Roh, H.S.; Wang, Y.; King, D.L. Selective Production of H2 from Ethanol at Low Temperatures over Rh/ZrO2–CeO2 Catalysts. Top. Catal. 2008, 49, 32–37. [Google Scholar] [CrossRef]
- Aupretre, F.; Descorme, C.; Duprez, D. Bio-Ethanol Catalytic Steam Reforming over Supported Metal Catalysts. Catal. Commun. 2002, 3, 263–267. [Google Scholar] [CrossRef]
- Cavallaro, S. Ethanol Steam Reforming on Rh/Al2O3 Catalysts. Energy Fuels 2000, 14, 1195–1199. [Google Scholar] [CrossRef]
- Shishido, T.; Yamamoto, Y.; Morioka, H.; Takaki, H.; Takehira, K. Active Cu/ZnO and Cu/ZnO/Al2O3 Catalysts Prepared by Homogeneous Precipitation Method in Steam Reforming of Methanol. Appl. Catal. A 2004, 263, 249–453. [Google Scholar] [CrossRef]
- Shishido, T.; Yamamoto, M.; Li, D.; Tian, Y.; Morioka, H.; Honda, M.; Sano, T.; Takehira, K. Water-Gas Shift Reaction over Cu/ZnO and Cu/ZnO/Al2O3 Catalysts Prepared by Homogeneous Precipitation. Appl. Catal. A 2006, 303, 62–71. [Google Scholar] [CrossRef]
- Hayakawa, T.; Harihara, H.; Andersen, A.G.; York, A.; Suzuki, K.; Yasuda, H.; Takehira, K. A Sustainable Catalyst for the Partial Oxidation of Methane to Syngas: Ni/Ca1−xSrxTiO3, Prepared in Situ from Perovskite Precursors. Angew. Chem. Int. Ed. 1996, 35, 192–195. [Google Scholar] [CrossRef]
- Liu, Y.; Suzuki, K.; Hamakawa, S.; Hayakawa, T.; Murata, K.; Ishii, T.; Kumagai, M. Catalytic Methanol Decomposition at Low Temperature over Pd Catalyst Derived from Mesoporous Silica Carried Pd-Hydrotalcite. Chem. Lett. 2000, 29, 486–487. [Google Scholar] [CrossRef]
- Liu, Y.; Suzuki, K.; Hamakawa, S.; Hayakawa, T.; Murata, K.; Ishii, T.; Kumagai, M. Highly Active Methanol Decomposition Catalyst Derived from Pd-Hydrotalcite Dispersed on Mesoporous Silica. Catal. Lett. 2000, 66, 205–213. [Google Scholar] [CrossRef]
- Liu, Y.; Hayakawa, T.; Suzuki, K.; Hamakawa, S. Production of Hydrogen by Steam Reforming of Methanol over Cu/CeO2 Catalysts Derived from Ce1−xCuxO2−x Precursors. Catal. Commun. 2001, 2, 195–200. [Google Scholar] [CrossRef]
- Liu, Y.; Hayakawa, T.; Suzuki, K.; Hamakawa, S.; Tsunoda, T.; Ishii, T.; Kumagai, M. High Active Copper/Ceria Catalysts for the Steam Reforming of Methanol. Appl. Catal. A Gen. 2002, 223, 137–145. [Google Scholar] [CrossRef]
- Liu, Y.; Hayakawa, T.; Ishii, T.; Kumagai, M.; Yasuda, H.; Suzuki, K.; Hamakawa, S.; Murata, K. Methanol Decomposition to Synthesis Gas at Low Temperature over Palladium Support on Ceria-Zirconia Solid Solutions. Appl. Catal. A 2001, 210, 301–314. [Google Scholar] [CrossRef]
- Liu, Y.; Hayakawa, T.; Tsunoda, T.; Suzuki, K.; Hamakawa, S.; Murata, K.; Shiozaki, R.; Ishii, T.; Kumagai, M. Steam Reforming of Methanol over Cu/CeO2 Catalysts Studied in Comparison with Cu/ZnO and Cu/Zn(Al)O Catalysts. Top. Catal. 2003, 22, 205–213. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Hanaoka, T.; Inaba, M.; Sakanishi, K. Syntheses of New Peroxo-Polyoxometalates Intercalated Layered Double Hydroxides for Propene Epoxidation by Molecular Oxygen in Methanol. J. Catal. 2007, 248, 277–287. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M.; Takahara, I.; Okabe, K. Synthesis of Mixed Alcohols from Syngas over Cs-Modified Cu/Ce1−xZrxO2 Catalysts. J. Jpn. Pet. Inst. 2010, 53, 153–159. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M.; Takahara, I.; Okabe, K. Synthesis of Ethanol from Syngas over Rh/Ce1−xZrxO2 Catalysts. Catal. Today 2011, 164, 308–314. [Google Scholar]
- Liu, Y.; Murata, K.; Inaba, M.; Takahara, I.; Okabe, K. Mixed Alcohols Synthesis from Syngas over Cs- and Ni-Modified Cu/CeO2 Catalysts. Fuel 2013, 104, 62–69. [Google Scholar] [CrossRef]
- Liu, Y.; Hanaoka, T.; Miyazawa, T.; Murata, K.; Okabe, K.; Sakanishi, K. Fischer-Tropsch Synthesis in Slurry-phase Reactors over Mn- and Zr-Modified Co/SiO2 Catalysts. Fuel Process. Technol. 2009, 90, 901–908. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Okabe, K.; Hanaoka, T.; Sakanishi, K. Synthesis of Zr-Grafted SBA-15 as an Effective Support for Cobalt Catalyst in Fischer-Tropsch Synthesis. Chem. Lett. 2008, 37, 984–985. [Google Scholar] [CrossRef]
- Fan, L.; Fujimoto, K. Reaction Mechanism of Methanol Synthesis from Carbon Dioxide and Hydrogen on Ceria-Supported Palladium Catalysts with SMSI Effect. J. Catal. 1997, 172, 238–242. [Google Scholar] [CrossRef]
- Sun, S.; Tsubaki, N.; Fujimoto, K. The Catalytic Performance and Characterization of Fischer-Tropsch Synthesis Co/SiO2 Catalysts Prepared from Mixed Cobalt Salts. Appl. Catal. A 2000, 202, 121–131. [Google Scholar] [CrossRef]
- Sun, S.; Fujimoto, K.; Yoneyama, Y.; Tsubaki, N. Fischer-Tropsch Synthesis Using Co/SiO2 Catalysts Prepared from Mixed Precursors and Addition Effect of Noble Metal Salts. Fuel 2002, 81, 1583–1591. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Murata, K.; Inaba, M. Steam Reforming of Bio-Ethanol to Produce Hydrogen over Co/CeO2 Catalysts Derived from Ce1−xCoxO2−y Precursors. Catalysts 2016, 6, 26. https://doi.org/10.3390/catal6020026
Liu Y, Murata K, Inaba M. Steam Reforming of Bio-Ethanol to Produce Hydrogen over Co/CeO2 Catalysts Derived from Ce1−xCoxO2−y Precursors. Catalysts. 2016; 6(2):26. https://doi.org/10.3390/catal6020026
Chicago/Turabian StyleLiu, Yanyong, Kazuhisa Murata, and Megumu Inaba. 2016. "Steam Reforming of Bio-Ethanol to Produce Hydrogen over Co/CeO2 Catalysts Derived from Ce1−xCoxO2−y Precursors" Catalysts 6, no. 2: 26. https://doi.org/10.3390/catal6020026
APA StyleLiu, Y., Murata, K., & Inaba, M. (2016). Steam Reforming of Bio-Ethanol to Produce Hydrogen over Co/CeO2 Catalysts Derived from Ce1−xCoxO2−y Precursors. Catalysts, 6(2), 26. https://doi.org/10.3390/catal6020026