Abstract
This paper presents an overview of heterogeneous partial (amm)oxidation and oxidative dehydrogenation (ODH) of hydrocarbons. The review has been voluntarily restricted to metal oxide-type catalysts, as the partial oxidation field is very broad and the number of catalysts is quite high. The main factors of solid catalysts for such reactions, designated by Grasselli as the “seven pillars”, and playing a determining role in catalytic properties, are considered to be, namely: isolation of active sites (known to be composed of ensembles of atoms), Me–O bond strength, crystalline structure, redox features, phase cooperation, multi-functionality and the nature of the surface oxygen species. Other important features and physical and chemical properties of solid catalysts, more or less related to the seven pillars, are also emphasized, including reaction sensitivity to metal oxide structure, epitaxial contact between an active phase and a second phase or its support, synergy effect between several phases, acid-base aspects, electron transfer ability, catalyst preparation and activation and reaction atmospheres, etc. Some examples are presented to illustrate the importance of these key factors. They include light alkanes (C1–C4) oxidation, ethane oxidation to ethylene and acetic acid on MoVTe(Sb)Nb-O and Nb doped NiO, propene oxidation to acrolein on BiMoCoFe-O systems, propane (amm)oxidation to (acrylonitrile) acrylic acid on MoVTe(Sb)Nb-O mixed oxides, butane oxidation to maleic anhydride on VPO: (VO)2P2O7-based catalyst, and isobutyric acid ODH to methacrylic acid on Fe hydroxyl phosphates. It is shown that active sites are composed of ensembles of atoms whose size and chemical composition depend on the reactants to be transformed (their chemical and size features) and the reaction mechanism, often of Mars and van Krevelen type. An important aspect is the fact that surface composition and surface crystalline structure vary with reaction on stream until reaching steady state, which makes characterisation of active and selective surface sites quite difficult. The use of oxidants other than O2, such as H2O2, N2O or CO2, is also briefly discussed. Based on such analysis and recent discoveries and process developments, our perspective is given.
1. Introduction and General Features of Partial Oxidation Reactions
In the chemical industry, hydrocarbons’ partial oxidation reactions correspond to important processes, playing a key role in numerous industrial, environmental and energy applications [1]. The majority of monomers and ~25% of all catalytic reactions are obtained by heterogeneous oxidation of hydrocarbons over mainly metal oxide catalysts, and even ~50% of bulk chemicals if one includes the synthesis of NO (NH3 oxidation over a Pt-based catalyst) and of SO3 (oxidation of SO2 over a V-based catalyst). For instance, C2 to C8 hydrocarbons lead to monomers such as ethylene, vinyl chloride, ethylene oxide, propene, acrolein, acrylic acid, acrylonitrile, butenes, methacrylic acid and methacrylates, maleic and phthalic anhydrides, etc. In the past, the majority of studies have dealt with olefin selective (amm)oxidation, in particular propene to acrolein over bismuth molybdate based catalysts and acrolein to (acrylonitrile)acrylic acid over Bi and V molybdates based catalysts by SOHIO group as early as in the 1960s [2], but also on light alkanes such as C2 to C5 activation by oxidative dehydrogenation (ODH) to olefins (C2= to C5=) or by oxidation to carboxylic acids on different oxides such as Bi molybdates, antimonates or vanadates, metal ions exchanged heteropolyacids of Keggin-type, etc. In the past 30 years, direct alkane selective oxidation has been the focus of many efforts in industry and academia [3], because of its higher abundance and lower price compared to the corresponding olefins. For instance, direct oxidation of ethane to acetic acid and/or ethylene on MoNbTeV-O or Nb doped NiO, direct (amm)oxidation of propane to (acrylonitrile)acrylic acid on MoNbTe(Sb)V–O, oxidation of butane to maleic anhydride on VPO catalysts, oxidative dehydrogenation of isobutyric acid to methacrylic acid on Fe hydroxyl-oxy-phosphates, etc., have led to the development of new industrial processes, as it will be discussed in the text below. So far, the most used solid catalysts for oxidation reactions are metals, metal oxides or metal complexes immobilized in zeolites, silica, alumina or polymeric resins [4] and, more recently, in metal organic frameworks (MOFs) for selective and chiral oxidation catalysis [5].
However, it is worth noting that alkanes’ ODH transformation is still a challenging task due to the low intrinsic chemical reactivity of the alkanes, which demands a high energy input to activate them. Note also that direct alkanes’ non oxidative dehydrogenation (DH) to olefins on noble metal supported catalysts is still employed industrially as being more efficient than alkane ODH although showing some major disadvantages, namely high endothermicity and high tendency to coking and, consequently, leading to short catalyst lifetime [6].
A main aspect of a partial oxidation reaction comprises the first C–H bond activation for oxidative dehydrogenation (ODH) or oxyfunctionalisation, considered as the rate determining step in the overall process. Usually, monomeric and dimeric V=O bonds are considered as active sites for light alkane activation in ODH reactions and larger V–O moieties for acrylic acid or acrylonitrile formation, as it will be discussed below (Section 3).
Complex metal oxides and multicomponent oxides catalysts represent the most important family of solid catalysts for partial heterogeneous oxidation catalysis as active phases or as supports. They include mixed metal oxides and hetero polyoxometallates [7]. The three main features of these oxides, which are essential for their application in catalysis, are (i) the coordination environment of surface atoms, (ii) the redox and, subsequently, acid-base and electron transfer properties and (iii) the oxidation states of the surface cations.
Seven key factors, designated as pillars by Grasselli [8], have been proposed to be satisfied for partial oxidation reactions to occur, namely: (i) nature of lattice oxygen anions: nucleophilic (selective) rather than electrophilic (total oxidation); (ii) redox properties of the metal oxide (removal of lattice oxygen and its rapid reinsertion); (iii) host structure (permits redox mechanism to take place without structure collapse); (iv) phase cooperation in multicomponent catalyst or supported catalyst (epitaxial growth and synergetic effects); (v) multifunctionality (e.g., α-H abstraction and O–/NH– insertion); (vi) active site isolation (to avoid a too high lattice O surface mobility and thus overoxidation); (vii) M-O bond strength (to be not too weak (total oxidation) nor too strong (inactivity) (Sabatier principle).
Point (i) of active oxygen species was discussed a long time ago in two review papers by Tench and Che [9], who described different types of oxygen species. For the redox mechanism one may write: O2 + e− → O2−; O2− + e− → O22− → 2O−; 2O− + 2e− → 2O2−. These oxygen species are more or less electrophilic (O2−, O22−, O−), nucleophilic (O2−) in the nomenclature proposed by Bielanski & Haber [10]. There have been extensive and controversial discussions about oxygen species in terms of their nature and location. Activation of molecular oxygen takes place by its reduction at the oxide surface which should present four free electrons per O2 molecule, whereas the highest oxidation state cations cannot activate oxygen. In other words, structural defects, encompassing lower oxidation state cations, have to be present.
Another general feature of such reactions in heterogeneous catalysis, which has appeared over the years, is that the oxide surface could be considered as living, as in a breathing motion, to allow the Mars and van Krevelen (MvK) mechanism to take place (vide infra). Such a property was clearly shown for iron phosphate catalysts used for isobutyric acid ODH to methacrylic acid (vide infra), implying that the oxide structure should be strong but mobile enough to allow the redox mechanism to occur, without structural collapse such as 2PO43− ↔ P2O74− + O2− as schematised in Figure 1.
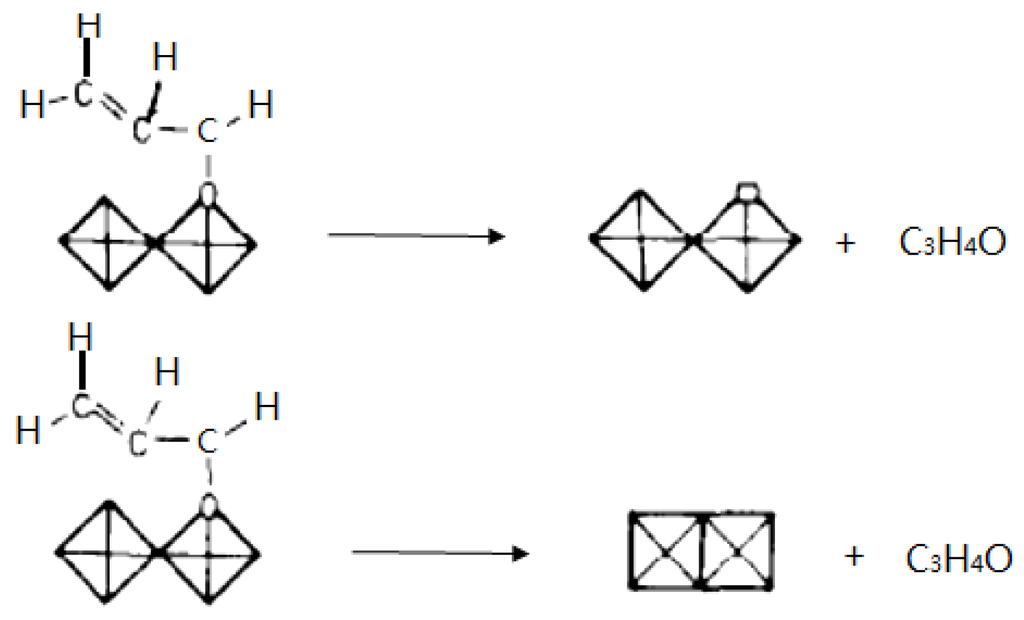
Figure 1.
Schematic representation of the mechanism of O insertion into a hydrocarbon molecule (left) and its consequences in creation of a reversible O vacancy in a metal oxide (right) P2O74− + O2− ↔ 2 PO43−. Reproduced from [11]. Copyright 2015, Academic Press.
It was also shown that such a property may suggest the existence of amorphous surface layers over a well crystallised underlying bulk phase, for example, for V2O5/TiO2 catalysts as observed by HR-TEM analysis [12], and for VPO catalysts in n-butane oxidation to maleic anhydride [13].

Table 1.
Main selective oxidation reactions in heterogeneous catalysis from [14]. Process: NI = not industrialised yet, I: industrialised; P: pilot, R: research.
| Reaction Type | Reactants | Products | Nb of e− | Catalysts | Yield mol % | State |
|---|---|---|---|---|---|---|
| oxidative coupling | CH4 | ethane + ethylene | 2 | Li2O/MgO | 25 | NI |
| 4 | ||||||
| oxidative dehydrogenation | ethane | ethylene | 2 | Pt, mixed | 60 | NI |
| MoVTeNb-oxides | 75 | NI | ||||
| propane | propene | 2 | VMgO | 40 | NI | |
| n-butane | butene, butadiene | 2 | metal molybdates | 38 | NI | |
| 4 | ||||||
| ethylbenzene | styrene | 2 | FeO-AlPO4 | 70 | P | |
| methanol | formaldehyde | 4 | FeMO4 | 81 | I | |
| isobutyric acid | methacrylic acid | 2 | FePO4 | 75 | NI | |
| oxychloration | ethane, Cl2 | vinyl chloride | 4 | AgMnCoO | 15 | NI |
| ethene, Cl2 | 2 | CuPdCl | 90 | I | ||
| hydroxylation | benzene | phenol | 2 | Ti or Fe zeolite | - | I |
| selective oxidation | methane | CO + H2 | 2 | Pt or Ni | 90 | R |
| formaldehyde | 4 | MoSnPO | 16 | R | ||
| ethene | ethyloxide | 2 | Ag/Al2O3 | 8 | I | |
| acetaldehyde | 2 | V2O5 + PdCl2 | 50 | I | ||
| acetic acid | 4 | MoVNbO + Pd/Al2O3 | ||||
| ethane | acetic acid | 6 | MoVNb-O | 10 | P | |
| propene | acrolein | 4 | BiFeCoMoO | 86 | I | |
| propane | acrylic acid | 6 | MoVNb(Te,Sb)O | 8 | NI | |
| n-butane | maleic anhydride | 14 | (VO)2P2O7 | 70 | I | |
| i-butane | methacrylic acid | 8 | HPA, oxides | 11 | R | |
| i-butene | methacrolein | 6 | SnSbO | 65 | NI | |
| o-xylene | phthalic anhydride | 12 | V2O5/TiO2 | 82 | I | |
| acrolein | acrylic acid | 2 | VMoWO | 85 | I | |
| methacrolein | methacrylic acid | 2 | - | 95 | I | |
| t-butylic alcohol | methacrolein | 4 | BiMoFeCoO | 88 | I | |
| ammoxidation | Propene + NH3 | acrylonitrile | 6 | MoBiFeCoNiO | 80 | I |
| Propane + NH3 | 8 | VSbO, MoVO | 30 | I |
Another important aspect to consider is the number of electrons necessary for a given reaction such as CH3–CH2–CH3 + O2− → CH3–CH=CH2 + H2O + 2e− or dissociation of molecular oxygen O2 into 2O2− requiring 4e− and their transfer ability within the material. In Table 1, some important partial oxidation reactions are reported as well as their state with respect to actual developments and number of electrons involved in the reaction. It then follows that electron mobility on the surface and even bulk catalyst layers may play an important role in determining catalytic properties. This feature could be determined via electrical conductivity measurements as it was observed in many cases (vide infra Section 4.2).
2. General Reaction Mechanisms
Many partial oxidation reaction mechanisms have been proposed in the literature [2,15], since the pioneering industrial work from Sohio scientists for propene oxidation/ammoxidation to acrolein/acrylonitrile on bismuth molybdate-based multicomponent catalysts (Bi2MO3O12/Co, FeMoO4) in the 1960s [2]. Propene chemisorption was suggested to occur on Mo centres, and the rate determining step was suggested to be the α-H atom abstraction by Bi-O leading to a radical-like α-allyl Mo complex. O or N insertion centres consist of coordinated unsaturated O=MoVl=O or HN=MoVI=NH sites, respectively. Reversible formation of α-O or N-allyl molybdenum complex occurs prior to subsequent (i.e., 2nd and 3rd) allylic H abstractions. The presence of Bi was suggested to facilitate the second H-abstraction, which has a lower activation energy Ea for oxidation than for ammoxidation.
For the partial oxidation reaction, the main reaction mechanism is the Mars and van Krevelen (MvK) one [16], schematised below. This mechanism concerns cations which can easily change their oxidation state such as Ce, Co, Cr, Cu, Fe, Mo, Ni, ,Ti, V, W, etc.
2[CatO] + R–CH → 2[Cat] + R–C–O + H2O
2[Cat] + O2 (gas) → 2[CatO]
In this scheme, [CatO] corresponds to the oxidised surface and [Cat] its reduced state; rred is the rate of catalyst reduction by a reactant and rox the rate of its re-oxidation by co-fed oxygen, R–CH and R–C–O the reactant and the product. The kinetic equation contains the concentration of reduced (θ) and oxidised (1-θ) sites of the catalyst. At the steady state, rred = rox or kred·pHC(1-θ) = kox·pO2θ with pHC, pO2 partial pressure of HC and O2 and kred, kox rate constants of reduction of catalyst (1st step) and rox rate of oxidation by O2 (2nd step). Their relative rate values are important for the selectivity and the reaction necessitates lattice oxygen anions, which are incorporated into the reactant molecule, whereas the corresponding vacancy created is replenished by gaseous oxygen in the re-oxidation step. If kred·pHC >> kox·pO2, reoxidation of the surface is the rate-determining step while if kred·pHC << kox·pO2, reduction of the surface is the rate-determining step. Thus, reoxidisability and reducibility of the catalyst will lead to different kinetic expressions. Lattice oxygen is introduced into the substrate or into H2O for ODH reaction. This process involves two active sites: an active cationic site and a site active for oxygen reduction. The process starts with the abstraction of a proton in α of the double bond by surface O2− and the sharing of electron between C=C and the empty d° orbital, by a two-electron transfer that reduces the transition metal cations. Reaction mechanisms in partial oxidation reactions may also include homogeneous phase reaction (radical-type mechanism), in addition to the usual heterogeneous one, even at relatively low temperatures such as below 300 °C while it occurs usually at high temperatures (600 °C and above) [17].
Note that on MexOy oxide when Men+ is a transition metal cation in d0 electron configuration and containing an oxygen vacancy, olefins (C3=, C4=, etc.) and aromatics (benzene, xylenes, etc.) are efficiently activated, the cation acting as an electron acceptor [18] (see Figure 2a). For V-based catalysts, it is well accepted that the V5+ ion activates an alkane by H abstraction first and that a second H abstraction gives the allylic radical before insertion of oxygen. Catalytic activity was shown to be proportional to the total V concentration on the surface [19], while the higher selectivity was assigned to VO4 monomeric species or low-size oligomeric (VOx)n species. The MvK mechanism has also been found to be quite valid for alkane partial oxidation and ODH reactions. However, in some cases, power law model, rake model, Langmuir-Hinshelwood model (LH) (uniform surface with one type of site), Eley Rideal (ER) model were also considered with more or less success. A steady-state adsorption model (SSAM), which can be regarded as a surface variant of the MvK model, was developed by Downie et al. [20] and applied also to describe kinetics of the vapor-phase oxidation of o-xylene over the vanadium oxide catalyst.
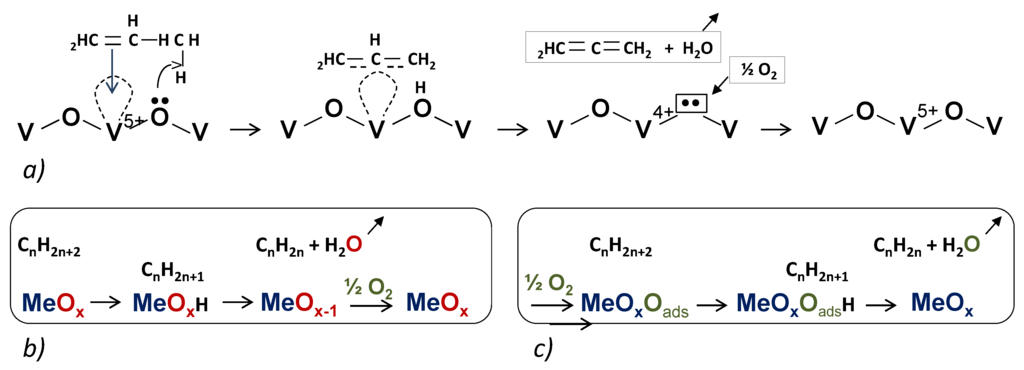
Figure 2.
Simplified schemes of: (a) propene adsorption O–V–O and formation of a π-allyl complex intermediate; (b) case of an alkane; (c) effect of adsorbed oxygen.
3. Structural Aspects of MxOy Oxides and Description of Active Sites in Some Partial Oxidation Reactions
It follows, from the general features described above, that the structural properties of MxOy oxides confer specific selectivity in oxidation reactions that depends on redox couples Mn+/M(n−p)+, length/strength/energy of M–O bonds, anionic defects (vacancies or anion excess, in particular O2−) and/or cationic (vacancies or cation excess) [21]. Surface and bulk mobility of oxygen species is also an important characteristic of the catalyst. We have seen above that partial oxidation catalysts generally contain a transition metal cation such as Ti, V, Cr, Mo, W, highly charged, small and polarising. In vanadyl (VO2+), molybdenyl (MoO22+) oxo-cations, multiple bonds are formed between the cation and oxygen. V5+ cations (ionic radius ri = 0.054 nm) float in an octahedral environment of six O2− (ri = 0.140 nm) and form V=O bond to be more stable. The V…O bond in trans V=O is longer, and its binding energy is weaker. Thus, this oxygen may easily be withdrawn, and the V coordination becomes five. Another striking feature is that octahedral environments of VO6 in V2O5 and in V2O4 are very close. Compared to V5+, O=V4+ and V4+…O bonds are longer (from 1.58 to 1.64 nm) and shorter (from 2.78 to 2.70 nm), and octahedron height changes very little. Therefore, at first approximation, one may expect an easy e− transfer: V5+ ↔ V4+ or Mo6+↔ Mo5+ couples (ri = 0.059 nm et 0.061 nm for Mo6+ and Mo5+) and thus improved catalytic properties. As a matter of fact, a catalyst will be more active as its structural changes are small, and one may expect such reactions to be structure sensitive as described by Boudart [22] for metallic catalysts. Structure sensitivity of metal oxides for oxidation reactions was first demonstrated by Volta [23] for propene partial oxidation. Volta has used a novel method to prepare MoO3 crystals with specific orientations. It was observed that propene was oxidised almost exclusively to acrolein on the (100) lateral face and CO2 on the apical (10-1), (101) and basal (010) faces. For the oxidation of isobutene and but-1-ene to 1-3-butadiene, the selectivities observed were completely different, namely (100) for methacrolein CH2=C(CH3)–CHO and COx from isobutene and (010) for 1–3 butadiene from but-1-ene, showing that the geometry of the reacting molecule and atomic arrangements at the oxide surface are important in partial oxidation reactions [24]. This is schematised in Figure 3 and was published by Tatibouët et al. [25] for butene oxidation.
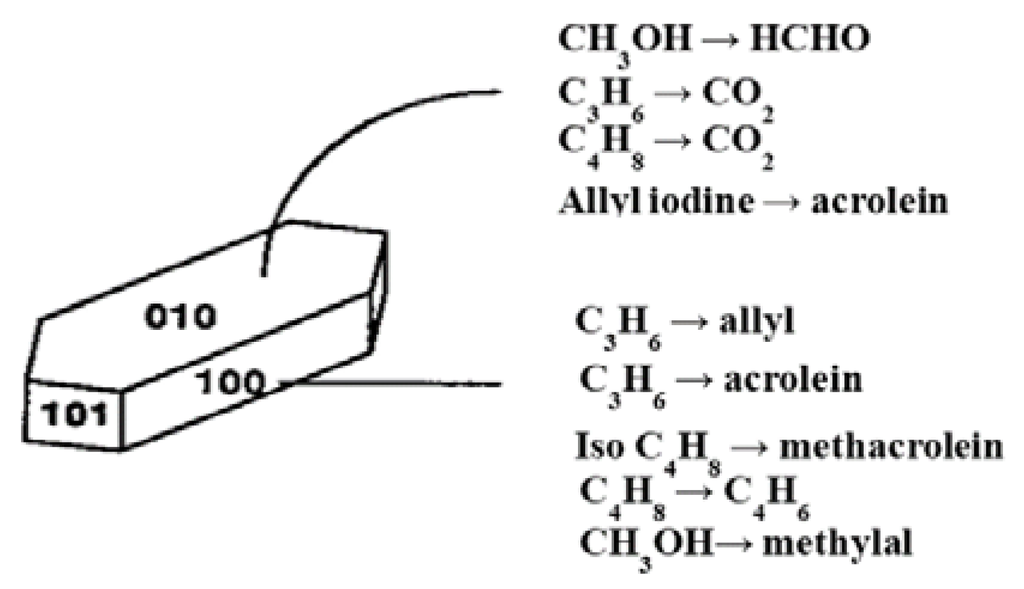
Figure 3.
Structural sensitivity of selective oxidation reactions of olefins or methanol on single crystals of MoO3 as plates (mainly (010) faces). Assignment of reactions to surfaces as indicated came from comparison of many sample shapes. Reproduced from [26]. Copyright 2015, Elsevier.
Structure sensitivity was also observed by Germain et al. [27] for the oxidation of methanol to formaldehyde and ethanol to acetaldehyde over MoO3 catalyst. However, in this case, the basal (010) face was observed to be selective for aldehyde formation. A review article [26] has summarized these findings. Later on, many selective oxidation reactions have been shown to be structure sensitive [28] such as n-butane to maleic anhydride on the (100) face of (VO)2P2O7 [29] or propane to acrylic acid on terminating (001) plane vs. lateral faces of needle shaped M1 phase (MoVTeNb-O) [30,31,32], etc. This structure sensitivity feature holds particularly true when the hydrocarbon molecule is large and the number of electrons involved in the reaction is high (see 4th column in Table 1), which implies that active sites for such reactions are quite complex.
In 1925, Taylor [33] proposed that “a chemical reaction is not catalysed over the entire solid surface of the catalyst but only at certain ‘active sites’ or centres”. He has also proposed that “chemisorption is an activated process, and may occur slowly”. Moreover, Taylor suggested that chemically active sites are “composed of an atom or an ensemble of atoms and could be sparse on the surface of a catalyst”. It follows that a catalyst surface can be poisoned by only a few molecules. Since that time, many descriptions of active sites have been proposed, in particular for metal oxides, as noted in [34].
For single oxides, e.g., MgO, surface sites could be Mg2+ or O2− ions with different coordination numbers depending on the defectuous surface structure as schematised in Figure 4.
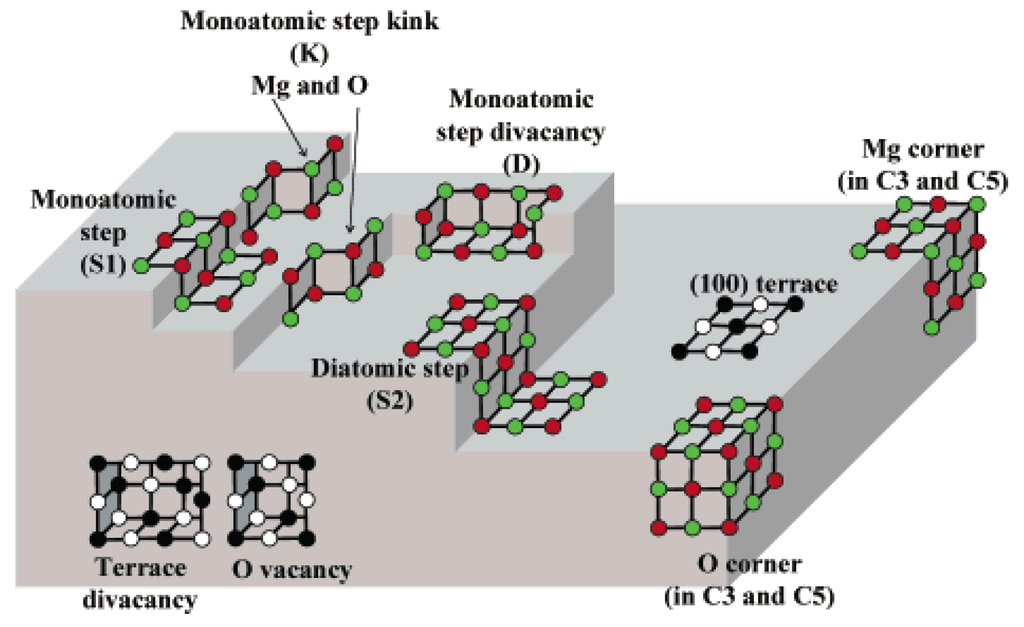
Figure 4.
Schematical representation of MgO surface. Reproduced from [35] Copyright 2016, ACS.
For large organic molecules and complex metal oxides used industrially, the active sites are usually ensembles of atoms in order to fulfill all requirements, to activate the reactants, to provide the number of electrons necessary for the reaction and thus to exhibit multifunctionality properties. In selective oxidation reactions, active sites have been shown to be isolated from each other to avoid overoxidation [2], which was assigned to quickly to lattice oxygen ions’ mobility. For instance, in the case of iron hydroxyphosphates, such as Fe23+Fe2+(P2O7)2 (+ H2O & 400 °C) ↔ Fe23+Fe2+(PO3OH)4 (+O2) → Fe2+x3+Fe1−x2+(PO3OH)4−x(PO4)x, and used for oxidative dehydrogenation of isobutyric acid to methacrylic acid, the most active and selective sites have been described [36,37] as ensembles of FeO6 octahedra trimers in αFe3P2O7, where electron transfer during the redox process occurs between Fe2+ and Fe3+ in αFe3P2O7, while in βFe3P2O7 no electron exchange was possible as calculated by extended Hückel theory [38] and shown in Figure 5, and, also, no catalytic property was observed, which certainly corresponds to low redox efficiency in the MvK mechanism.
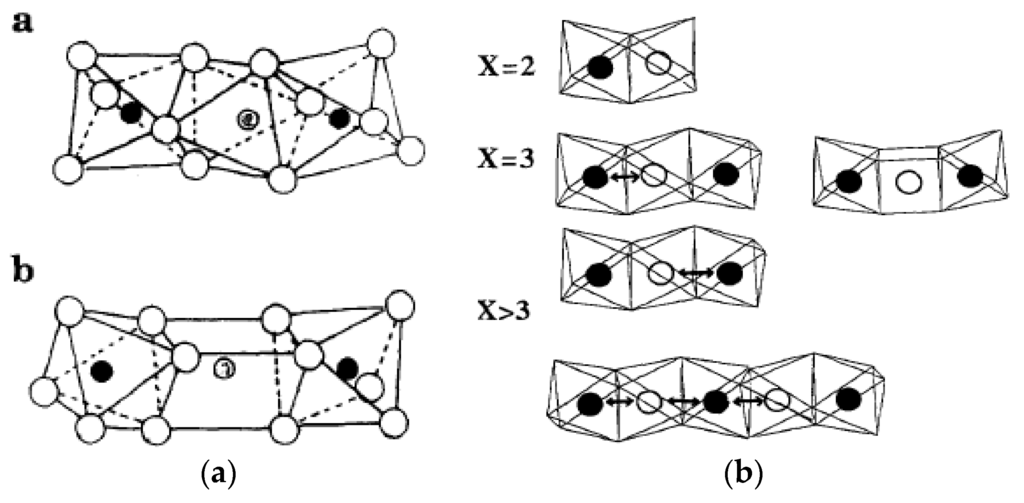
Figure 5.
Arrangements of FeO6 octhedra in trimeric clusters isolated one from the others by P2O7 groups: (a) αP2O7; (b) βFe3P2O7 grey circles: Fe2+, filled: Fe3+. Electron exchange between iron cations in FeO6 octahedra clusters calculated by extended Hückel molecular orbital theory. Reproduced from [36]. copyright 2016, Elsevier BV.
In fact, isobutyric acid (IBA) ODH to methacrylic acid and its esterification to methyl methacrylate (MMA) using FePO or (Cu)MoPV (heteropolyacid-type) or iron or nickel oxide based catalysts have been industrialized by different companies. There are some alternate methods to producing methyl methacrylate. Among them is the oxidative dehydrogenation of methyl isobutyrate, (MIB) which is formed by the carbonylation of propene in the presence of methanol [39].
In MIB ODH reactions, co-feeding water showed an increase in conversion of MIB over the catalysts. Along with higher conversion of MIB, the selectivity towards MMA was lower than in the case without co-feeding of water catalysts, due to the formation of the hydrolysis products of MMA and IBA. The mechanisms of formation of the products from MIB over the iron phosphate catalysts involves the Fe2+/Fe3+ redox couple, and the mechanism follows a similar pathway of Mars and van Krevelen mechanism.
Another example of active site description corresponds to (VO)2P2O7, designated as VPO catalyst, used industrially for butane oxidation to maleic anhydride. The active sites have been described as ensembles of four dimers of octahedral VO6 entities isolated by P2O7 groups as schematised in Figure 6 [40].
Many other examples of active sites described as isolated atoms or ensembles of atoms can be found in the literature. Let us take some of them. Panov et al., in their study of benzene direct oxidation to phenol by N2O on Fe-MFI catalysts, have shown that active sites are FeO dimers [41] inside the pores of the zeolite. Janas et al. [42] have studied Co(II) ions in SiBEA zeolite. At low Co content (0.8 wt. %), cobalt is present as lattice tetrahedral Co(II) species. For much higher Co content (11 wt. %), mainly extra-lattice octahedral Co(II) species and/or cobalt oxides are present as shown by DR UV-vis spectroscopy. It was then concluded that zeolite with isolated tetrahedral Co(II) species are active in selective catalytic reduction of NO by ethanol with selectivity towards N2 exceeding 85% for NO conversion from 30% to 70%. At variance, when extra-lattice octahedral Co(II) species and/or cobalt oxides are present, the full oxidation of ethanol and NO by O2 to CO2 and NO2, respectively, were observed to be the main reaction pathways. Isolated Co(II) cations in lattice position were thus suggested to be active sites for the reaction. Another example follows the pioneered works on metallic gold particles for CO oxidation at room temperature by Haruta [43], who has shown that small particles, in a given range of nm sizes, are active while bulk metal is inactive. This work has been extended to Co3O4 nanorods exhibiting (100) faces, for CO oxidation at low temperature [44] was also related to structure sensitivity face, although CO and O2 are rather small molecules.
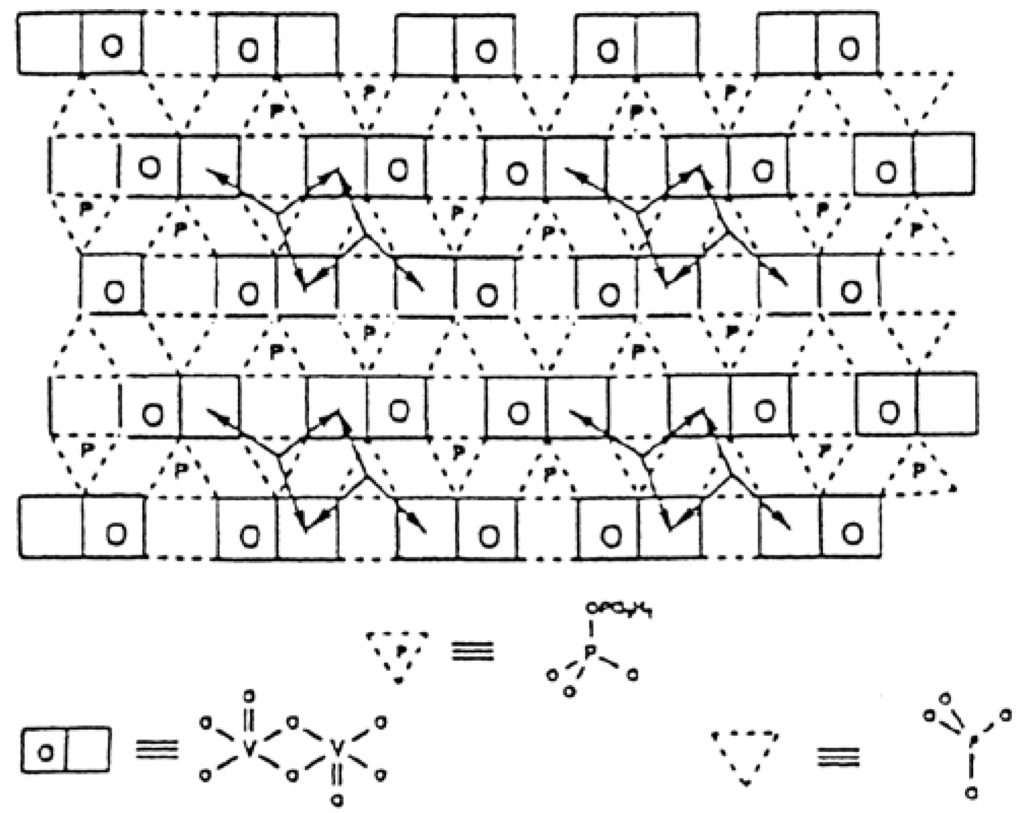
Figure 6.
Schematic representation of a surface structure of a type of polytype of (VO)2P2O7. The arrows represent the facile pathways for surface oxygen mobility. This illustrates the site isolation principle by surface P2O7 groups which constitute a barrier to oxygen diffusion. Reproduced from [8] Copyright 2016, Elsevier BV.
Such an ensemble of atoms as active sites has also been proposed for enzymatic oxidation/hydroxylation reactions using cytochrome P-450 enzyme [45]. The major challenge in mimicking cytochrome-P450 enzymes is the development of catalysts able to induce insertion of one O atom from O2 on the substrate and a 2e− reduction of the other O atom to water. In nature, this is carried out by NAD(P)H reductase protein. To overcome this problem, addition of several reductants such as zinc dust or aldehydes to the metalloporphyrins’ oxidation catalytic systems or use of high temperatures and pressures have been reported. For instance, porphyrin-like complexes, such as N-bridged di-iron oxo phthalocyanine associating cytochrome P-450 and a monooxygenase enzyme and using H2O2 [46] has been shown to oxidise methane [47] into formic acid and secondarily to formaldehyde and methanol in specific conditions (40 °C, 3.2 MPa, CH3CN as solvent), while monomeric Fe complexes were found inactive. However, transformation of metalloporphyrins as valuable oxidation catalysts for large scale purposes is still a challenging issue, mainly due to porphyrin stability, cost and reutilization. This could depend on the development of efficient procedures for the immobilization of porphyrins into/onto solid supports such as mesoporous aluminosilicates, polymers’ double layered hydroxides, which unfortunately generally require the use of polluting oxidants and O2 with co-reductants.
Let us take another and more recent example. In the past 15 years, a tremendous amount of work has been performed and published on a complex mixed oxide based on MoVTe(Sb)Nb-O, with V/Mo, Nb/Mo and Te(Sb)/Mo ratios variable but preferentially close to 0.3, 0.1 and 0.2, respectively, able to (amm)oxidise directly propane to (acrylonitrile) acrylic acid in the presence of water vapour [48]. In the absence of water vapour, mainly propylene is formed. The highest acrylonitrile yield was found for V0.3Te0.23Nb0.12MoOx supported on silica after calcination under nitrogen at 620 °C [49]. In fact, this catalyst family was discovered by researchers of Union Carbide, who published their work in 1978 [50] on ethane oxidation to ethylene and acetic acid. This material was shown to be Mo-based bronzes, the preparation and activation in air or N2 of which was observed to greatly influence their structure and catalytic properties [51]. The best catalysts for (amm)oxidation were further observed to contain a phase designated M1 phase, often associated with another phase designated M2. It was suggested [52] that the reaction needs either four monomeric or two dimeric units of redox storage to deal with one molecule of oxygen, according to:
4V4+–OH + OH− → 4V5+=O + 4e− + 4H2O
O2 + 2H2O + 4e− → 4OH−
4 V4+–OH + O2 → 4V5+=O + 2H2O
O=V–O–V=O + C3H6 → O=V–O–V–OC3H5 + H+ + e−
O=V–O–V=OC3H5 + H+ + e− → HO–V–O–V–OC3H5
HO–V–O–V–OC3H5 + 3OH− → HO–V–O–V–OC3H4O + 2H2O + 3e−
This oxidation leads to acrolein being still bounded to the active site, as the electrons released from the organic substrate do not reduce the V centre. Active sites were suggested [53] to be isolated ensembles of atoms as schematised in Figure 7.
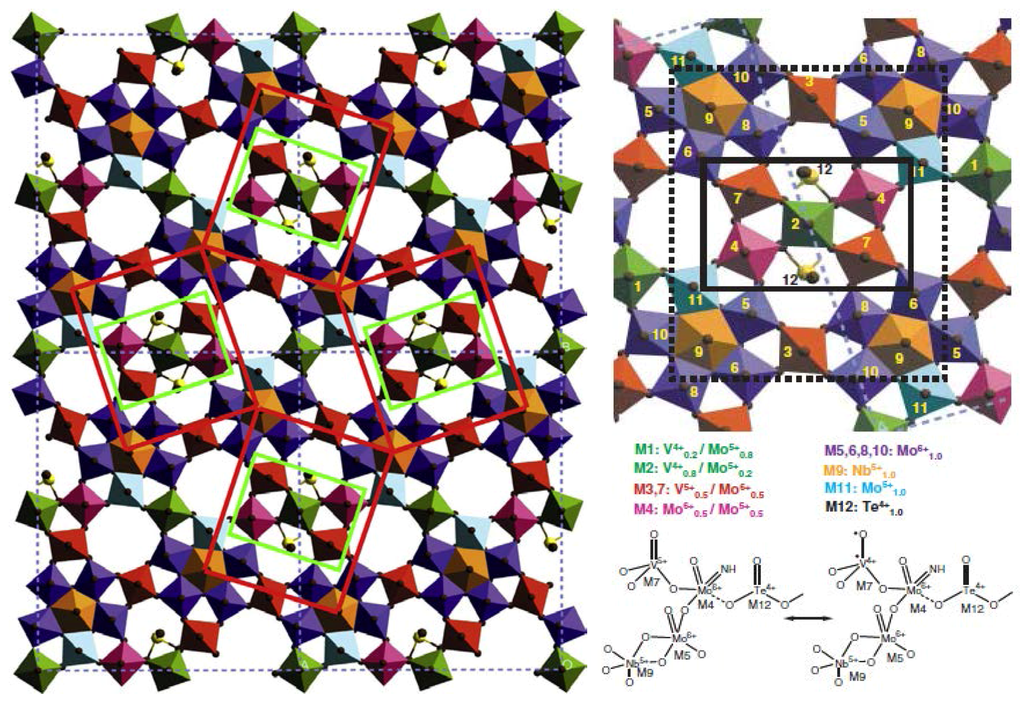
Figure 7.
Active site (left) in Mo7.8V1.2NbTe0.94O28.9 (M1): 2 × 2 unit cell structure model of M1 in (001) projection showing four isolated catalytically active centres. The site sites are isolated from each other by four Nb-bipyramids that are surrounded by five MOn-octahedra. ChemDraw depiction (right). Adapted from figures 15 & 16 in [8], copyright 2016, Elsevier BV.
In a study of V2O5/TiO2 catalyst studied by the Eurocat oxide group, it was found that: [54]
- Monomeric VO43− species exhibit an acid character and gives propene in isopropanol conversion, used as a test reaction, and total oxidation for o-xylene oxidation (coverage <0.43% in V2O5) as expected for acid sites;
- Polyvanadate species exhibit redox properties and give acetone in isopropanol conversion and phtalic anhydride in o-xylene conversion;
- V2O5 crystallites give low activity in o-xylene conversion and high selectivity in CO2.
This shows that the size of the active sites is quite important for catalytic partial oxidation reactions depending also on the compound size and the multifunctionality necessary for the reaction. Such concept has been largely discussed in the literature for many years and monomeric isolated species or low-size oligomeric vanadium oxide species were suggested for light alkane oxidation and ammoxidation reactions (vide infra) as suggested in [3] (Figure 24-3, p. 779).
4. Main Factors Acting in Partial Oxidation Reactions
4.1. General Aspects of the Acid-Base Properties of Metal Oxides
Metal oxides are composed of redox metal cation and lattice oxygen anions which are Lewis acid and basic sites, respectively. The acid-base characteristics of the oxide have three major effects, namely on reactant molecule activation, on the rates of competitive pathways of transformation and on the rate of adsorption and desorption of reactants and products.
It is known [55,56] that during a reaction, an acid surface would favour desorption of an acid intermediate while a basic surface would favour desorption of a basic intermediate, which then would avoid further over-oxidation of the adsorbed intermediate to CO2. Thus, acidic products, such as carboxylic acids, should favourably be formed on acid surfaces while basic products, such as olefins in ODH reactions, should favourably be formed on basic surfaces. This is why the presence and the strength of Lewis acid sites (cations) and of basic sites (O2− or OH−) are important and should be characterised carefully. For such a characterisation [57], one usually uses FTIR or microcalorimetry of adsorbed probe molecules (basic NH3, pyridine or acid CO2, SO2, respectively) or test reactions such as isopropanol conversion which gives propene by dehydration on acid sites or acetone by dehydrogenation on basic sites in the absence of air in the feed or on redox sites in the presence of air [58]. Another test reaction has been proposed for acid base characterisation, namely 2-methyl but-3-en-1-yn-2-ol (MBOH) conversion, which follows three different pathways depending on the acid-base properties of the catalyst, as schematised in Figure 8.
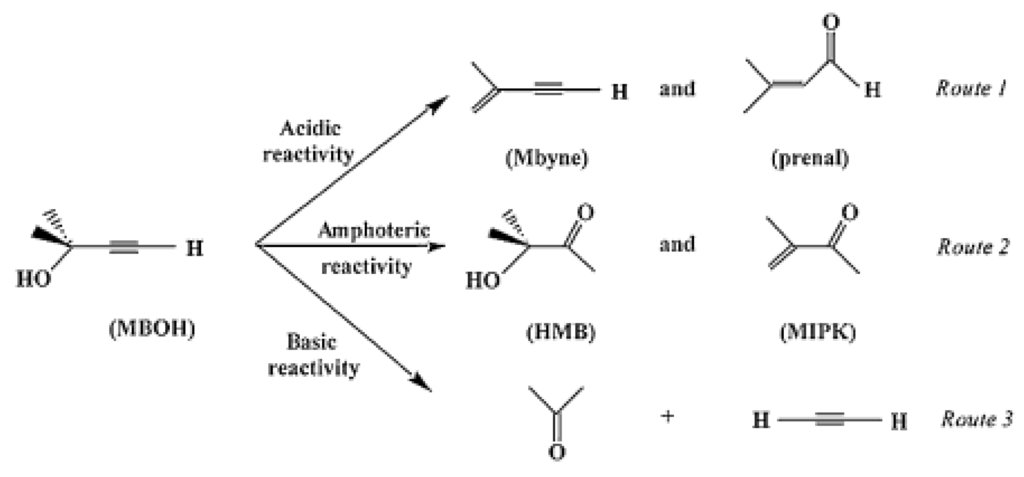
Figure 8.
Scheme of 2-methyl but-3-en-1-yn-2-ol (MBOH) reaction pathways for an acid-base catalyst Reproduced from [59] copyright 2016, Taylor & Francis.
An oxygen element has a highly electronegative character and its bond with metals is ionic, thus more basic, or covalent with non-metals thus less basic. The electronegativity of a cation depends on its oxidation state and increases as the oxidation state increases. Oxides of metals at high oxidation state, such as V5+ or Mo6+, are characterized by a covalent metal to oxygen bond and behave as acid oxides, whereas the same elements at lower oxidation state have a more ionic character and behave more as basic oxides. Note that metal cations, active for oxidation reactions, have several oxidation states and are able to easily change their oxidation state and, thus, their acid base character under certain catalytic reaction conditions.
The strength and nature of acid/basic sites are important parameters as they are expected to participate in the molecule activation as for the first H abstraction. Let us take as an example the MoV(Nb)Te(Sb)–O based M1 and M2 phases, mentioned above. Their acidity was determined for [60] with pyridine as a probe molecule as shown in Figure 9. Both Brønsted and Lewis acid sites were detected and it was observed that a too strong acidity favours total oxidation, as expected, and, thus, that a moderate acidity is favourable for better selectivity to acrylic acid/acrylonitrile.
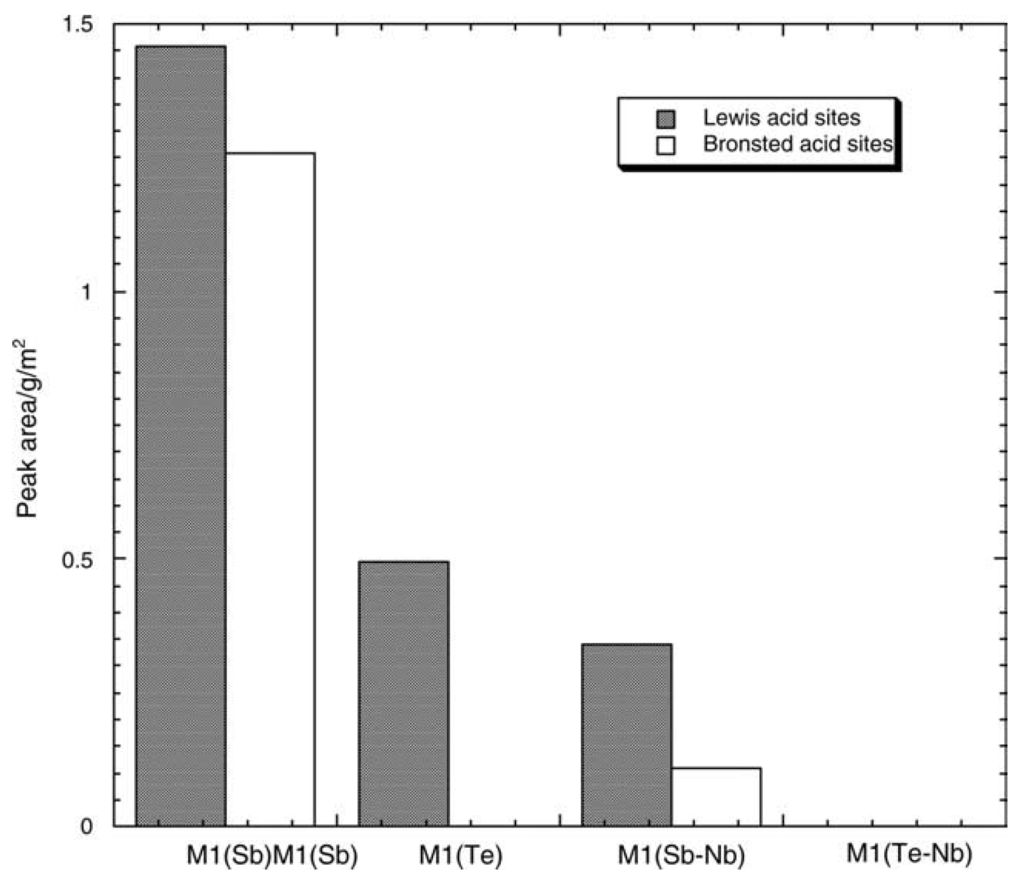
Figure 9.
Comparison of the Brønsted and Lewis relative acidity of the M1 phases of MoVTe(Sb)NbO, expressed as area under the pyridine peaks at 1540 and 1450 cm−1 normalized by surface unit in the spectra recorded after desorption at 100 °C, From [60], Copyright 2015, Elsevier BV.
In another example in the same family, namely K-free and K-MoVSbO bronzes of formula (SbO)2M20O56, thus of M1-type structure [61], the selectivity to acrylic acid was observed to be higher than for the starting sample while, when P was added to M1 phase [62], acrylic acid selectivity was also improved. It was also observed that Sb samples had medium strength acidity and Te samples even lower strength and that Nb insertion decreases both acidities, while WOx added to MoVTeNb-O increases propane to acrylic acid by improving propene formation [63]. In a recent study [64] on Bi promoted MoVTeNb-Ox catalyst for propane ammoxidation, it was suggested that Bi suppressed acrylonitrile’s further transformation and decreases the acidity of the catalyst.
Acidic properties were also studied [65] using NH3 TPD and acid-base titration with a standardized NH4OH solution (−log [NH3] = 6.7) for Mo1V0.3Sb0.35Nb0.08On mixed oxide, calcined under N2. This compound was composed mainly of Sb4Mo10O11 (isomorphous of M2 phase) and (VxNbyMo1−x−y)5O14 (M1-type) phases. Low Lewis acidity and no Brønsted acidity were observed for such sample.
4.2. Importance of Electron Transfer Properties
In the first step of a redox mechanism surface, oxygen anions O2− are extracted to be inserted in products while cations are reduced according to: Mn+ + pe− → M(n−p)+. For example: V5+2O5 + 2e− → V4+2O4 + □ (where □ represents an oxygen vacancy) or V2O5 → V2O4 + ½ O2 as usually written for an oxide reduction. In the absence of oxygen in the feed, these vacancies are filled via O2− diffusion from the bulk, as this occurs in ionic conductors and the solid is more and more reduced. In general, O2 is added to the reactants to promote reoxidation as diffusion of O2− ions is too slow. The catalyst surface permits reduction of O2 according to: O2 + 4e− + 2□surf → 2O2−surf. The electron transfer property of a solid catalyst is thus an important aspect of the Mars and van Krevelen mechanism, which implies high mobility of lattice oxygen anions and of electrons to ensure a fast reaction.
Some typical reactions have already been listed above in Table 1, including the number of e− involved in them. The majority of selective catalysts are semiconductors due to defects present naturally on their surface. They could be n-type (n: negative or e− donors in the valence band) or p-type (p: positive or e− hole acceptor in the conduction band). Note that insertion of dopants may allow to modulate semi conductivity. N-type semi conduction is favourable to O2 activation to 2O2−, while p-type and/or, if the cation is more electropositive, other oxygen species can be formed at the surface according to:
O2 + e− → O2−; O2− + e → O22−; O22− → 2 O−; O− + e− → O2−
Such electrophilic species are more stable for electropositive cations such as Mg2+, Ca2+, La3+, Ce4+, etc. and would give carbon oxides for the oxidation of an unsaturated hydrocarbon. On the contrary, O2− is selective in ethylene epoxidation on Ag0, and O− species are very reactive at low temperatures for aldehydes to acids, benzene to phenol on titanosilicate or alkane activation using N2O instead of O2.
Under an oxygen atmosphere, the electron trapped by adsorbed O2 is provided by the valence band of the oxide and the electrical conductivity increases with the consumption of oxygen. Conversely, exposure to a reducing gas creates oxygen vacancies, releases electrons in the valence band and creates new positive holes with a decrease of electrical conductivity. This is why electrical conductivity measurement has been used for characterising redox properties of metal oxides. In Figure 10 and Figure 11, the variations of electrical conductivity are shown respectively vs. the local atmosphere indicated in the caption for VMgO catalysts with different V content, used for propane ODH to propene [66] (Figure 10) and in relation to selectivity for ethane to ethene on Nb doped NiO catalysts [67] (Figure 11).
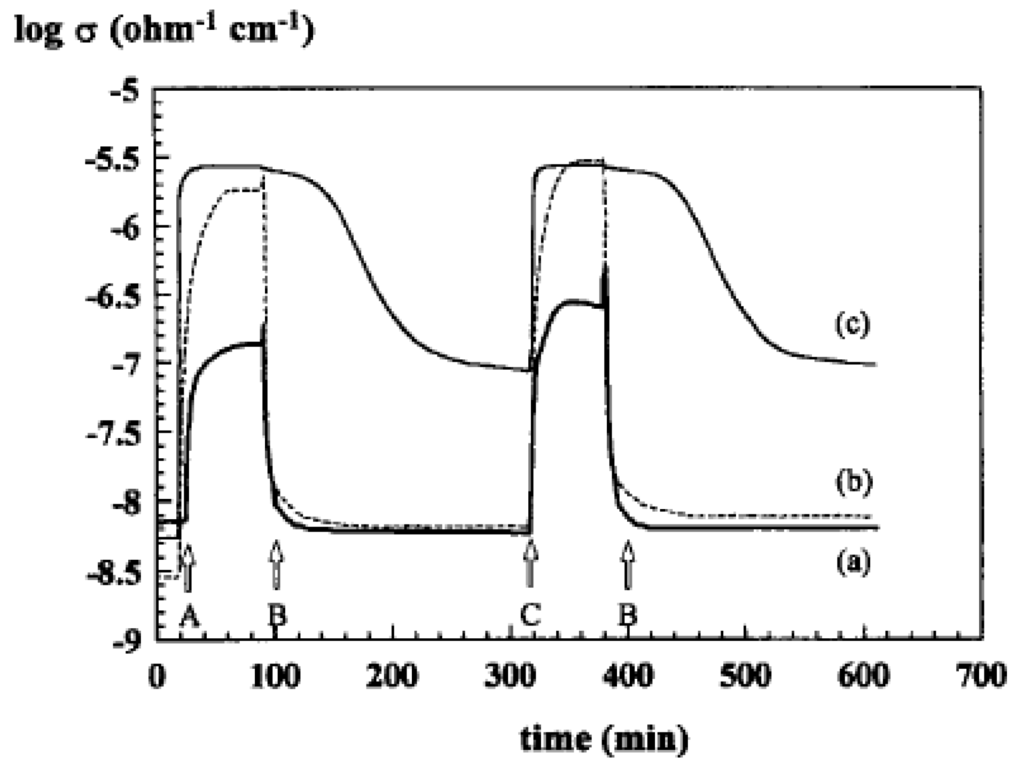
Figure 10.
Variations of electrical conductivity during redox cycles [66] Copyright 2015, Elsevier BV: (A) at 500 °C for 14V/VMgO, (B) at 800 °C for 14 V/MgO and (C) for 45 V/MgO. (a) C3H8/O2 = 1.2/1; (b) vacuum; and then O2; (c) C3.
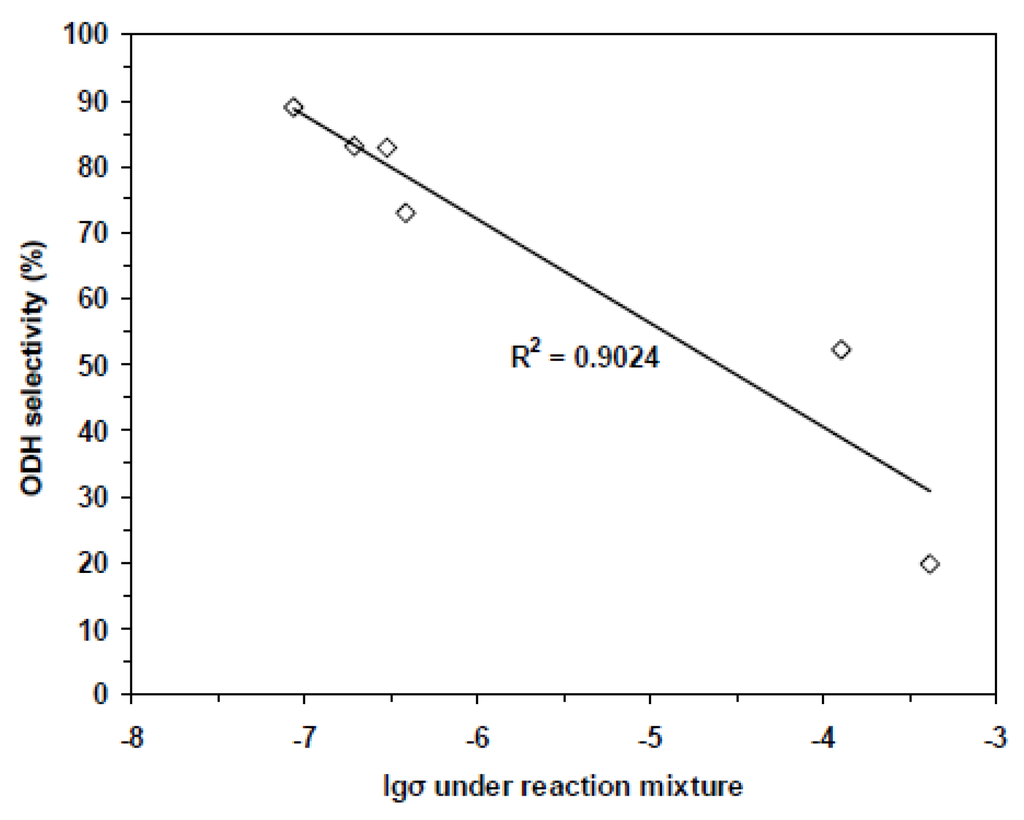
Figure 11.
Variation of ethylene selectivity at ethane isoconversion at 350 °C vs. electrical conductivity for NiO and Nb-doped NiO catalysts (From [67] copyright 2015, RSC Great Britain).
4.3. Synergy or Cooperation Effect in Mixtures of Phases [68,69]
Synergy effects have been very often met in heterogeneous catalysis. They occur when the following conditions are met: (i) bi- or multi-phasic systems, and (ii) catalytic properties are drastically enhanced compared to that of each single component. One of the most striking examples came from the bismuth molybdate-type catalysts developed at SOHIO in the 1960s for propene oxidation/ammoxidation in acrolein/acrylonitrile [2]. It was further shown that the different phases α, β and γ are active for propene oxidation to acrolein, the α-phase being the best [70]. Moreover, industrial catalysts, designated as multicomponent catalysts, contain also other molybdates, in particular Co(Fe)MoO4. Using electrical conductivity measurements and Mössbauer spectroscopy [71], it was shown that the synergy effect was in fact due to the enhanced electrical conductivity of the Co(Fe) molybdate support due to the presence of both FeII and FeIII in a CoII molybdate which facilitates the redox Mars and van Krevelen mechanism [72] (Figure 12). It was then suggested that intimate contact between the active Bi molybdate phase and its support Co(Fe)MoO4, as illustrated in Figure 13, allows an improved redox mechanism by an easy electron transfer Fe2+ ↔ e− + Fe3+. Such synergy effect holds true for many other systems, like, for instance, V2O5/TiO2-anatase or TiO2-B for o-xylene to phthalic anhydride [73,74], or ammoxidation of toluene [75]. It was observed for many mixed metal oxides (Ti, V, Mo, Nb, Te, Fe, Co, Bi, Sb, P), whose structures allow for local elastic constraints by distortion and connection of MOn coordinated polyhedra. The effect was interpreted as being related to phase cooperation between different phases due to a close crystallographic adaptation of lattices at their mutual interface as schematized in Figure 13, and is designated as coherent interfaces [76].
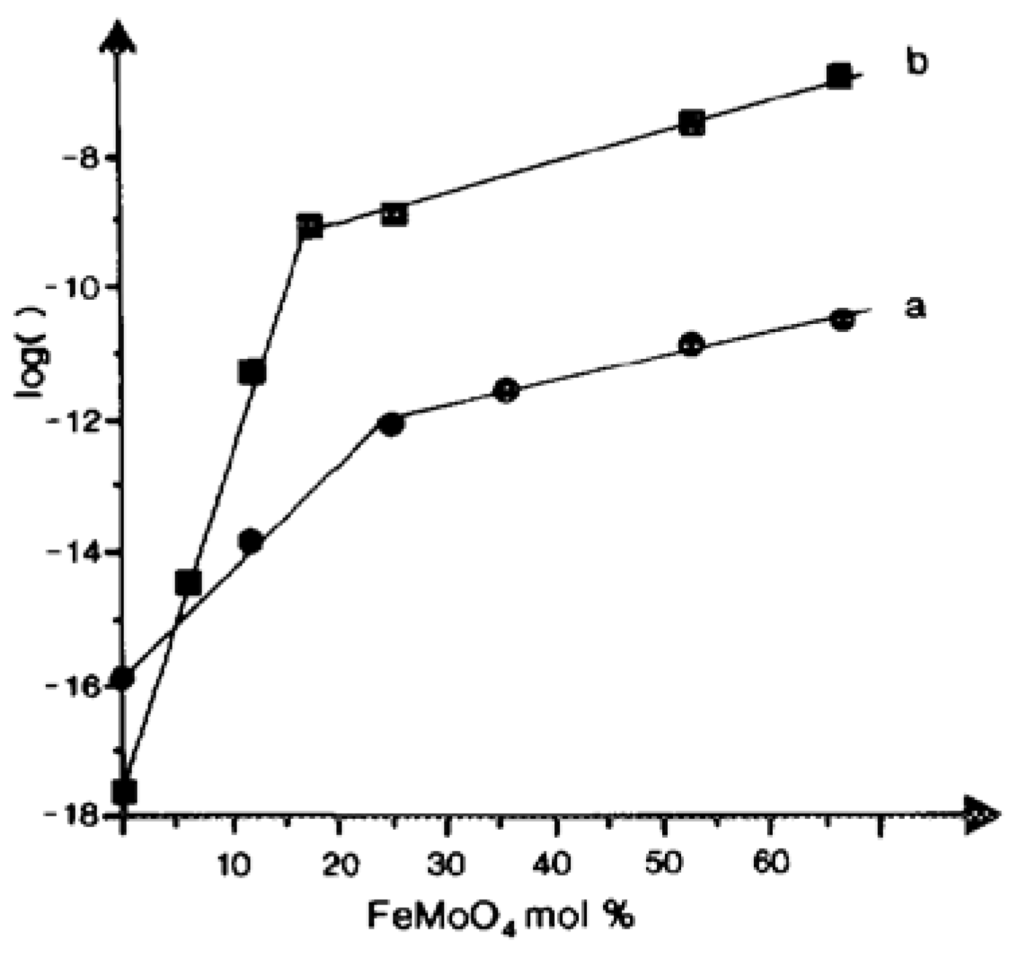
Figure 12.
(a) Log rate of acrolein formation in propene oxidation on Bi2Mo3O12:FexCo1−xMoO4 (0.043:1) at 380 °C; (b) log σ of FexCo1−xMoO4 solid solution vs. its iron molybdate content. Reproduced from [72] Copyright 2015, Elsevier BV.
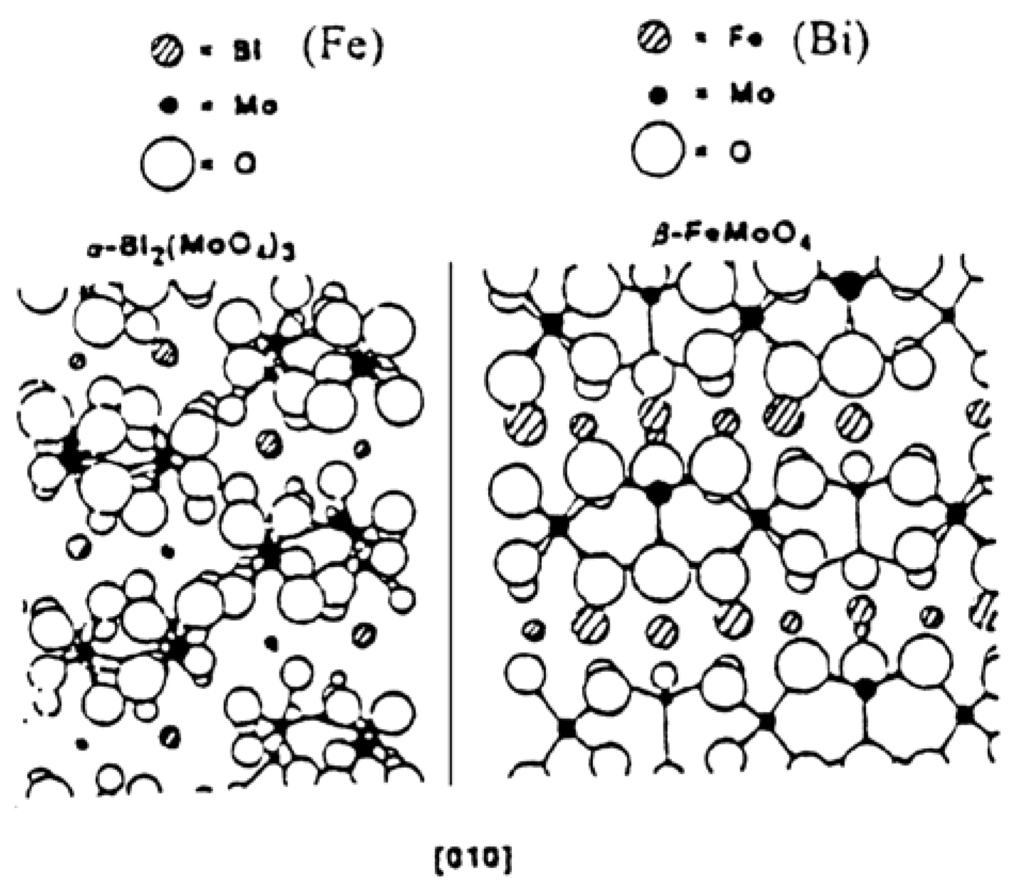
Figure 13.
Epitaxial match of (010) face of Bi2Mo3O12 and β-FeMoO4. (From [77] copyright 2016, Elsevier BV.).
Similar cooperation effects have been observed for multicomponent antimoniates (MCA) such as FeSb7.7Ox or W0.25TeFe10Sb25Ox or KCsNiMgFeMnBiCrMo-O with phase cooperation FeSbO4 + Sb2O4(5) or FeSbO4+Sb2O4(5) + FeMoOy, for VPO catalysts [78] and for Mo7.8V1.2NbTe0.94O28.(M1) + Mo4.67V1.33Te1.82O19.82 (M2) [79]. Delmon et al. [80] have given an explanation for this synergy effect, what they designated as “remote control effect”, which corresponds to “a multiplying phenomenon, i.e., that a spillover species creates and/or regenerates catalytic sites which work many times”. In such a mechanism, one phase, designated as donor phase, dissociates oxygen to form a surface mobile species, which spills over to the other phase, which is the acceptor active phase. In a multicomponent catalyst system, “donor” and “acceptor” sites are present as it was shown for metal oxides [79] or sulfides [81] able to provide oxygen/sulfide ions that spill over from one oxide/sulfide to the other.
Recently, a new way of preparing bismuth molybdates by mild hydrothermal synthesis and new mixed Bi, Ce molybdates were found to be quite active for propylene oxidation [82] and ammoxidation [83], respectively, even more than the classical α-Bi2Mo3O12 phase. The catalyst BixCe2−xMo3O12, e.g., BiCe1.5Mo3O12 of solid solution nature has a scheelite structure, i.e., ABO4, where A2+ is a large cation 8-coordinated to oxygen and B6+ a small cation tetrahedrally coordinated to oxygen. Substitution of a divalent cation by a trivalent one such as Bi results in a defect scheelite A2□(BO4)3 where □ indicates a vacancy. The active site has been described as Bi cation surrounded by two Ce cations, which readily undergo Ce3+ ↔ Ce4+ redox phenomenon, and are located next to a vacant cation site, thus able to accommodate a lone pair of electrons.
5. Changes in Catalyst Surface Structure and Chemical Composition during Catalytic Reaction
This is a general aspect of dynamic structure “which is constantly under going changes in its chemical composition and geometrical structure during reaction” and which has been known for a very long time for many reactions in heterogeneous catalysis, as reminded by Tim from BASF in his introductory lecture to 8th ICC in Berlin (p. 21 in [84]). For instance, in oxidation catalysis, it is well known that VPO catalyst necessitates many hours on stream in n-butane oxidation to maleic anhydride before optimal performance is reached, whereas excess of P at the surface vs. P:V = 1:1 stoichiometry was detected by LEIS and XPS techniques. This phenomenon corresponds to a steady state of the catalyst surface in redox conditions, with often demonstrates some elemental migration toward or away from the surface. This may characterise elemental segregation or the formation of a sublayer of different composition than the bulk as shown by Millet et al. [85] for MoVTeNb-O catalyst used for propane oxidation to acrylic acid or ammoxidation to acrylonitrile. These authors have observed [86] that the surface composition of the M1 phase differs from the bulk which implies that the catalytically active sites are no part of the M1 crystal structure but part of all terminating planes. It follows that acrylic acid formation may be related to surface depletion in Mo6+ and enrichment in V5+ sites. In the presence of steam in the feed, the active sites for acrylic acid formation consist of V5+ oxo-species in close vicinity to Te4+ sites in a Te/V ratio of 1.4. The active sites formed under propane oxidation conditions are embedded in a thin layer enriched in V, Te, and Nb on the surface of the M1 phase. It follows that structure sensitivity properties as described above have to be considered with caution.
In a study using microcalorimetry, Schlögl et al. have shown that on activated catalyst (calcined) the adsorption energy of propene and propane are approximately constant vs. coverage (57 kJ·mol−1) but after reaction the energy decreases and is heterogeneous in strength vs. coverage. The first part is thus characteristic of “single site heterogeneous catalyst” (SSHC site as defined by Thomas [87]), i.e., sites of similar strength and structure and thus of similar selectivity and activity. However, under reaction conditions, the surface is modified and the sites are heterogeneous in strength (not satisfying the SSHC principle anymore) and thus heterogeneous in their activity and/or selectivity. Moreover, in-situ XPS analysis has shown that depletion of Mo and that V enrichment did occur as discussed in the previous paragraph. This is shown in Figure 14.
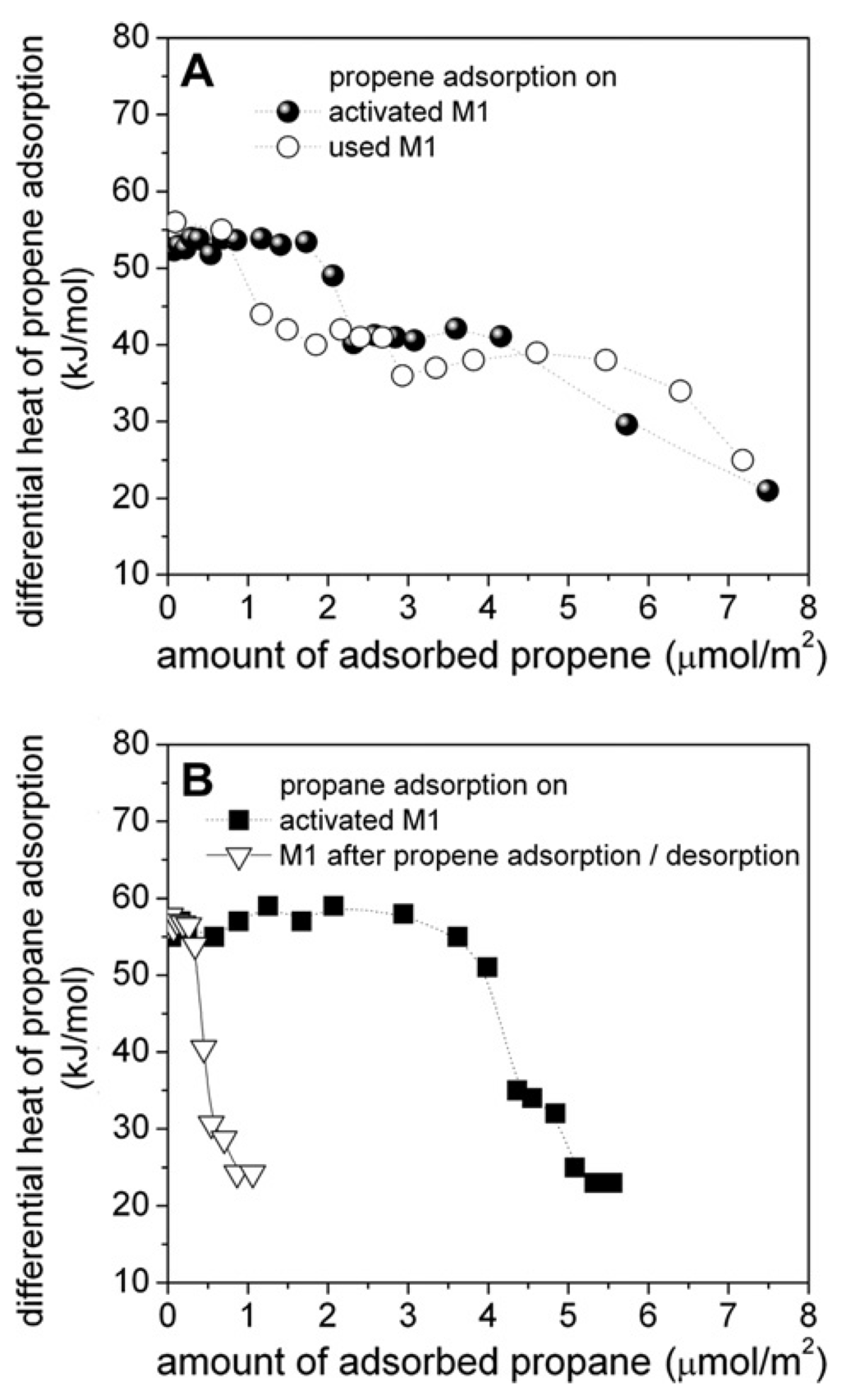
Figure 14.
(A) Heat of propene adsorption on M1 MoVTeNb oxide before (filled circles) and after propane oxidation (open circles; reaction conditions of propane oxidation: 400 °C, C3/O2/H2O/N2 = 3/6/40/51). (B) Heat of propane adsorption on activated (fresh) M1 MoVTeNb oxide, (filled squares) and after adsorption and subsequent desorption of propene at 40 °C (open triangles). The differential heat of adsorption is plotted as a function of coverage normalized to the specific surface area (from [88] copyright 2015, Elsevier BV).
This phenomenon indicates that under steady state conditions, as usually met for partial oxidation reactions, which necessitate hours on stream before steady state is reached, surface active sites are different from the starting surface and are the best sites for the reaction in terms of selectivity [66]. It also shows that caution has to be taken when catalyst characterisation is done on the starting material and that in-situ characterisation, i.e., under real working conditions and after reaction steady state has been reached, is necessary to better characterise the catalyst surface and the active catalytic sites.
6. Dehydrogenation, Oxidative Dehydrogenation and Selective Oxidation of Light Paraffins
Low molecular weight alkenes, such as ethene, propene, and butenes, can be formed via non-oxidative dehydrogenation of the corresponding alkane. However, direct dehydrogenation (DH) reactions are endothermic, thermodynamically limited reactions and lead to the formation of coke deposits and of lower molecular weight alkanes, which decrease alkene yields but are commercially well established and still in use.
There are various processes for light alkanes’ DH such as Catofin (Lummus), Olefex (UOP), Linde-BASF, Snamprogetti-Yarsintez, and Star (Philipps petroleum) with chromia-alumina and fixed bed, supported Pt in moving bed, chromia-alumina and fixed bed, chromia-alumina and fluid bed and supported Pt and moving bed, respectively. However, endothermicity, on one side—which thus necessitates heat supply and high coke formation, due to thermal cracking, as well as frequent regenerations—and, on the other side, thermodynamic olefin yield limitation make the process unappealing and open the way to oxidative dehydrogenation (ODH) processes [89].
Oxidative dehydrogenation (ODH) of light alkanes provides another way to obtain alkenes, as the reaction is exothermic and avoids the thermodynamic constraints of direct DH approaches by forming water as a by-product. In addition, carbon deposition during ODH is eliminated, leading to stable catalytic activity. Compared to the conventional steam cracking process of alkanes’ dehydrogenation to olefins and catalytic direct DH processes, ODH can allow for a less expensive process, lowering greenhouse gas emissions and saving energy. However, the yield of alkenes obtained by ODH on most catalysts is limited by alkene oxidation to CO and CO2. In fact, at present, no catalysts have been found to be active and selective enough to convert light alkanes to olefins by ODH to permit an industrial process to be acceptable.
Ethane, propane, butane or pentane ODH reactions are governed by the C–H bond strengths, which equal 376 kJ·mol−1 for C–C, 420 for C–H primary, 401 C–H secondary, 390 for C–H tertiary, 361 for C–H allyl and 445 for C–H vinyl. For instance, as ethane contains only primary C–H bonds and its dehydrogenated product ethylene contains only vinylic C–H bonds, its activation requires the highest temperature vs. C3, C4 or C5, but the reaction is the most selective in terms of the formation of alkene. In Figure 15, we reported literature data for such ODH reactions, where it clearly appears that selectivity decreases with an increase in conversion, due to higher oxidability of the olefins vs. the starting alkane. This is why no new process for the production of light olefins from their corresponding alkane has been commercialised [90].
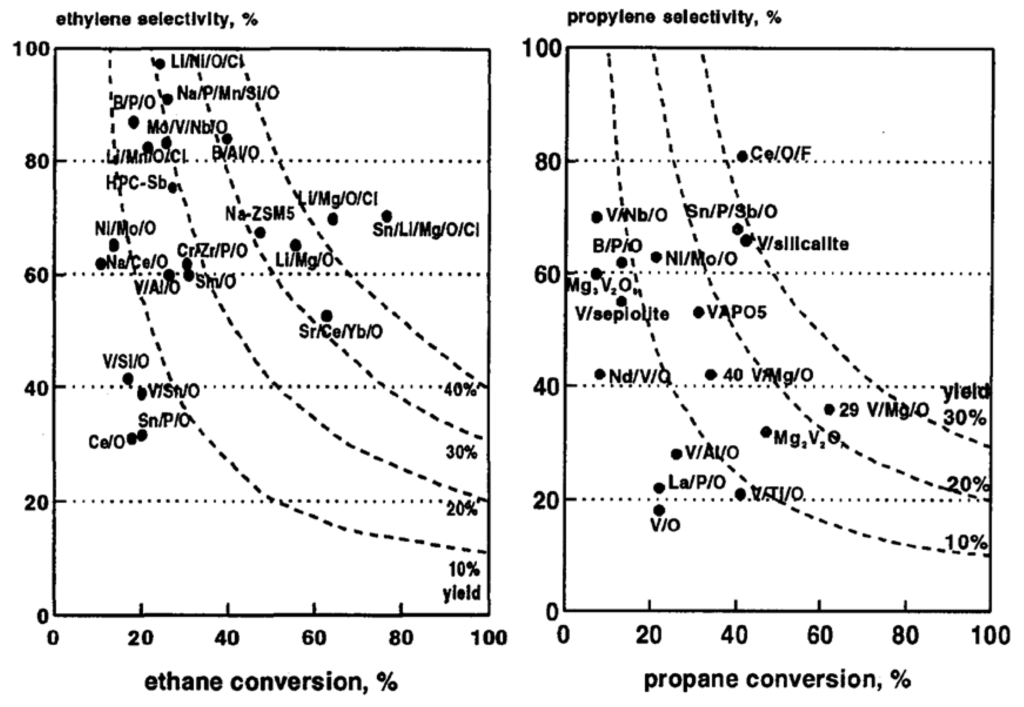
Figure 15.
Best yields in olefin selectivity vs. conversion for ethane and propane ODH. Reproduced from [6]. Copyright 2015, Elsevier BV.
6.1. Case of Methane
Because of its natural high abundance on earth, methane oxidative transformation to C2-hydrocarbons, methanol or formaldehyde has led to intense research efforts worldwide [91] by partial oxidation (POM) to methanol or formaldehyde (MoOx or VOx species) or oxidative coupling (OCM) to ethane and ethylene or by halogenation and oxihalogenation, but no industrialisation processes have been developed yet. An alternative process is the transformation of methane to syngas, a mildly exothermal reaction with H2/CO = 2 as an alternative to the well-known steam reforming process. Another possibility is to transform methane to hydrogen and benzene and secondarily naphthalene, anthracene and higher aromatics on Mo/MFI or Zn-MFI catalysts at very high temperatures (>700 °C) but yield in aromatics has been very low and deactivation has been very high due to the large amount of aromatics formed [92].
6.2. Case of Ethane
Ethene is one of the most important building blocks in the chemical industry and is used to synthesize polymers. It was surprisingly shown that NiO presents high catalytic activity for ethane conversion and ethylene selectivity [93] and that the method of preparation of the catalysts governs its catalytic activity [94,95]. NiO prepared by the sol-gel method [67] starts converting ethane at ∼275 °C, and the conversion increases up to 350 °C (23% conversion and 47% selectivity in ethylene), while with NiO prepared by the precipitation method [67], the yield decreases to ∼8% and to 3% with the NiO prepared by a microemulsion method, i.e., both conversion and selectivity in the catalytic oxidation of ethane on NiO catalyst depend on the way in which the catalyst is prepared. When Nb was deposited on NiO, the catalytic activity for ethane conversion and ethylene selectivity were improved. This phenomenon was assigned to electron transfer enhancement played by Nb, leading to easier activation of the C-H bond of ethane (see above Section 4.2 & Figure 11 from [67]). Other promoters such as Zr, which acts as a Lewis acid, but also Mg, Li, Ga, Ti, Ta,[96] or alumina have also been proposed for NiO catalyst to increase ethylene yield.
However, ethane oxidation was also found to give acetic acid in addition to ethylene. The main processes for synthesis of acetic acid include methanol carbonylation on Rh or Ir-based catalysts (originally by Monsanto, now also by Celanes, BP-Cativa, etc.), liquid phase oxidation of acetaldehyde, n-butane and naphta and, more recently, ethylene oxidation in gas phase (Showa Denko), butene liquid phase oxidation (Wacker) and gas phase ethane oxidation. The later reaction C2H6 + (3/2) O2 → CH3COOH + H2O is only viable if ethane is easily available on the industrial platform and cheap. Different classes of catalysts have been proposed but the highest performing one is a mixed (VNbMo)5O14 based oxide described by Thorsteinson et al. [50] with a d-spacing close to 0.40 nm The best composition was claimed to be Mo0.73V0.18Nb0.09Ox with 10% ethane conversion at 286 °C and almost total selectivity to ethylene. At 200 °C, catalysts based on (MoVNb)5O14 microdomains in MoO3 were found to give 45% selectivity both in acetic acid and ethylene, at 25% ethane conversion. Even better yields in ethylene (75%) have been obtained at 400 °C over the same type of material further on [97]. It was suggested that ethane first adsorbs on Mo6+ or V5+ to form an ethoxide species which transforms to ethylene by β-elimination, but also possibly oxidises to acetaldehyde by α-elimination, transformed to surface acetate and then to acetic acid by its hydrolysis. MoVNb-O catalyst family thus appears as the most promising possibility at present for ethane to ethylene and acetic acid, the problem being that both products are formed, and limiting acetic acid formation, if desired, is difficult. However, when adding Pd, the selectivity to acid acetic may reach 80% [98,99], and even more when adding water in the feed [100]. The same effect holds true for propane oxidative dehydrogenation by Pt-H3PMo12O40 [101] with O2 and H2 and for propane ammoxidation to acrylonitrile. High transformation to acetic acid was claimed by Saudi Basic on MoVNbP-O; MoVLaPd-O; MoVNbPd-O under a pressure of 1 to 2 MPa. The technology developed by Saudi Basic is now commercial [102], using the catalyst presenting multifunctionality properties. Ethylene as well as acetic acid are rather stable compounds and do not transform to CO2 in the catalytic conditions chosen, with unconverted ethane and ethylene being recycled. Other systems have also been proposed such as MoVAl-O with Fe, Ga, Ti starting from the Anderson-type polyoxometallates, supported Keggin-type MoVP-O polyoxometallates and titania supported vanadium oxide or V(Mo)P-O. A recent review paper [103] summarizes the present situation.
6.3. Case of Propane
A large number of catalysts, mostly V based, have been investigated for propane ODH but propene yield varies in the 8% to 20% range. The upper yield was attained with VMgO, Mg2V2O7 or MgVxSbyOz, or with orthovanadate on hydrotalcite [104], while V2O5 supported on Al2O3, Bi2O3, La2O3, Nb2O5, SiO2, Sm2O3 or TiO2 gives inferior performance. Co and Ni molybdates were also investigated with different dopants such as Fe, Mg, Ni or P and gave yields near 15% with high productivity and no production of undesirable oxidised products. The major problem is that, in addition to alkane activation, such metal oxide catalysts may possess also O– or NH– insertion properties and thus may give undesired oxygenates or nitriles in addition to the olefin. The use of Pt based catalyst and of short contact times may be a solution to get mainly the olefin, which is known to be a reaction intermediate. At variance, one may add a catalyst known to transform the olefin to unsaturated carboxylic acids or nitriles for complete transformation of alkanes to carboxylic acids or nitriles, but both operating temperature and chemical compatibility of both catalysts need to be optimised.
For propane ammoxidation, it has been shown that the mixed oxides’ surfaces are nitrided [19]. For instance, AlO3(NH)VO5.6 was suggested to be an active site, while insertion of nitrogen in the bulk was observed to enhance electrical conductivity and, thus, electron transfer ability in the redox mechanism.
Another aspect in ODH reaction is the nature and surface density of surface oxygen species. For instance, in the case of propane ODH, both propylene and ethylene can be formed depending on the reaction mechanism by dehydrogenation or cracking. Comparing non reducible (rare earth/alkaline earth oxides) and reducible (V based oxides) catalysts, it was shown [105] that the ratio propylene over ethylene depends on the strength and mainly the density of oxidising active sites (DOS), with high DOS favouring propylene. The oxidising sites are those active for H-atom abstraction, usually V5+=O and or bridged V5+–O–M (M = metal cation in the next cationic position, i.e., V or another metal atom from the structure or support).
The best results obtained for ethane and propane partial oxidation or ammoxidation are given in Table 2.
Table 2.
Summary of the best results in the literature for partial oxidation of C2–C3 alkanes from [3] p. 772 and references herein.
| Catalyst | T °C | Conversion % | Selectivity % | Yield % |
|---|---|---|---|---|
| Ethane → ethylene | ||||
| MoVTeNbO | 400 | 85 | 88 | 75 |
| NiNbO | 400 | 51 | 90 | 47 |
| VCoAPO-18 | 600 | 57 | 60 | 34 |
| Ethane → acetonitrile | ||||
| NbSbO/Al2O3 | 540 | 40 | 50 | 20 |
| Co-BEA | 476 | 47 | 57 | 26 |
| Ethane → acetic acid | ||||
| MoVSbNbReCaO | 277 | 14 | 78 | 11 |
| MoVNbPdOx | 300 | 5 | 82 | 4.1 |
| Propane → propylene | ||||
| VMgO | 540 | 62 | 38 | 24 |
| V-silicalite | 550 | 30 | 70 | 21 |
| Propane → acrylonitrile | ||||
| MoVTeNbO | 420 | 86 | 72 | 62 |
| VSbWOx/SiO2-Al2O3 | 500 | 67 | 60 | 40 |
| Propane → acrylic acid | ||||
| MoVTeNbO | 432 | 69 | 67 | 46 |
| MoVTeNbO A relevant | 380 | 80 | 61 | 49 |
6.4. Case of Butane
Here, ODH reaction for n-and iso-butanes to butene and isobutene on catalysts of the same type as described for ethane or propane such as VMgO or metal molybdates is examined. Another example corresponds to the catalytic conversion of n-butane to butenes by oxidative dehydrogenation (ODH). It is well known that supported vanadium pentoxide is a promising catalyst for the ODH of n-butane. MgO supported vanadium was reported as a very selective catalyst in propane and n-butane ODH, while V/γ-A12O3 was observed to exhibit a good selectivity to ethene for ethane ODH but a poor selectivity in the ODH of n-butane. The acid-base character of the support was suggested to lead to this different behaviour. On MgO, a support with basic properties, the interaction between vanadium species and the support leads to the formation of vanadate compounds. In the case of more acidic supports, such as SiO2 or A12O3, a weak interaction is expected leading to less dispersed vanadium species on the surface favouring the formation of V2O5 crystallites.
However, the most important process concerns butane partial oxidation on (VO)2P2O7 catalyst, designated as VPO, and which substituted the old benzene oxidation process in the 1980s. A description of active sites as ensembles of four dimers of octahedral VO6 entities isolated by P2O7 groups (Section 3 and Figure 6) and surface composition during catalytic reaction (Section 5) has already been given above. In such a reaction, maleic anhydride is relatively stable to further oxidation which permits high yields. This is also why, in a moving bed technology, developed in Asturias, Spain by Du Pont Co, the catalyst, encapsulated by porous silica to limit attrition of catalyst grains, that maleic anhydride was further transformed to less stable tetrahydrofurane (THF) by reduction on Pd based catalyst (see below Section 8).
7. Role of Other Oxidants
Instead of molecular oxygen as the oxidant, carbon dioxide can also be used as a non-traditional oxygen source or oxidant [106], for instance, according to:
CH3-CH3 + CO2 → CH2 = CH2 + CO + H2O
However, in addition to ethylene, CO and water, propane and methane can be formed as a function of the catalysts used. The use of CO2 has the advantage of producing CO and thus recovering some of the fuel capacity of the disappearing hydrogen while O2 produces water. In addition, utilisation of CO2 should be an alternative to their valorisation. CO2 is less aggressive than oxygen, and it is less likely to favour secondary reactions by reacting with the ethylene produced by dehydrogenation. The amount of oxygen in the ODH feed with O2 is limited by the danger of explosion. The use of CO2 as an oxidant can overcome the high energy consumption for the endothermic pyrolysis and prevents deep oxidation, but unfortunately, oxidative dehydrogenation of ethane with CO2 remains an endothermic reaction and requires addition of heat for its activation [102,107]. However, the low chemical reactivity of CO2 leads to a difficult activation, which usually results in higher reaction temperatures. This drawback makes the use of CO2 as a selective oxidant well suited for the activation of ethane, which also requires high temperatures. However, the first coupling of a dehydrogenation reaction with a hydrogen-consuming reaction, known as the Deacon Process [108], was performed in 1868 and can be viewed as an oxidative dehydrogenation of HCl (2HCl + 1/2O2 → Cl2 + H2O). The use of CO2 to improve catalytic conversion and selectivity in methane oxidative coupling and ethane ODH was reported in 1993 [109] on Li+/MgO catalyst. Several metal oxides have been used as catalysts [110,111], and the most effective catalysts with high ethane conversion (>60%) and ethylene selectivity (>90%) contained Cr or Ce elements.
Other oxidants instead of molecular oxygen and CO2 have been studied, including sulfur oxide, N2O, H2O2, organic peroxides, hypochlorite, bromine, iodine and their compounds. The advantage of using these oxidants is that high selectivity for dehydrogenation could be obtained. The corrosive nature of the halogen and sulfur oxide gases and the potential environmental concern over their use for N2O or their synthesis for H2O2 have deferred commercialisation of these processes.
8. General Conclusions and Perspectives
In this review, we have limited ourselves to studying heterogeneous partial oxidation reactions on metallic mixed oxides, and we have presented the main features and requirements for better activity and overall better selectivity. The majority of the processes described are rather old, namely more than half a century old, although many of them have been greatly improved over the years either by adjusting their preparation and activation procedures or by chemical engineering in reactors and process improvements. For environmental reasons, some processes had to be abandoned due to the need to use environmentally friendly reactants or solvents such as sulfuric acid, cyanic acid, etc. Although many challenges have been solved, more or less completely, improvements are still possible. One of the most important challenges remains alkane upgrading to olefins [112] or carboxylic acids/aldehydes, in particular methane. The most researched possibility is to discover single phase structures, containing the desired catalytic functionalities, without the need to use two separate catalysts in two separate reactors. There is much room to further optimise the structure of known catalysts. Reactions such as simultaneous dehydration and selective oxidation or ammoxidation of biomass derived intermediates such as glycerin, lactic acid or 3-hydroxypropionic acid to acrylic acid or acrylonitrile, respectively, are important perspectives to follow up.
It is clear that partial oxidation solid catalysts are very complex as they must have many functionalities and behave most often as “living” or “breathing” materials at their surface, leading to element migration toward or from the surface to the bulk during catalyst activation or under reaction conditions up until reaching steady state. This migration phenomenon was observed using surface techniques such as XPS and LEIS, showing that surface structure varied from the bulk structure and was even observed to be amorphous using XRD (epitaxial close contact being maintained between surface and bulk layers). It has been shown that oxidation reactions are structure sensitive, i.e., necessitate ensembles of atoms in specific structural arrangements as active sites, isolated from each other to avoid over-oxidation to CO2. Acid-base and overall redox and electron conductivity properties are quite important to activate reactant molecules and to allow the redox mechanism to occur. Note that a too strong acidity could be deleterious and is often neutralized with low amounts of a base such as alkaline ions. Redox, a phenomenon which is crucial to permitting the Mars and van Krevelen mechanisms to function, is an important property which could be enhanced by higher electrical conductivity of the surface or by using semi-conducting materials or semi-conducting supports. Such oxidation reactions obey the new approach of nanocatalysts, with control of the size, shape and chemical composition of the catalysts as highlighted recently in a review article devoted to selectivity in heterogeneous catalysis [113].
In the present stage, the most important trends concern oxidation of ethane to ethylene or acetic acid and propane (amm)oxidation on MoVTe(Sb)Nb-O catalysts (Mo5O14 based catalysts with Te, Sb or Nb incorporated into the structure, leading to M1 and M2 phases). A desirable aim/challenge is to substitute Te, which is easily reducible, volatile and poisonous, by any other similarly selective element such as Bi or Sb. Note that Bi has already been tried but without success, while Sb has interesting properties.
As we have described above, catalytic cracking of alkanes gives many by-products and is not necessarily optimised. In the case of propane dehydrogenation, propene is formed as the main product. The drawback is that the dehydrogenation equilibrium favours propene only at high temperatures or low pressure, increasing the overall cost of the process. The need for cryogenic separation of the unconverted propane and produced propene also increases the process costs. Improvements of the dehydrogenation process to make it commercially more attractive can be done by shifting the equilibrium by removal of one of the reaction products. This can be done either physically by means of a membrane, or chemically by in-situ catalytic oxidation using for instance a transition metal oxide. This approach has the advantage of an exothermic oxidation which facilitates the dehydrogenation. However, mixing H2, O2 and hydrocarbons at high temperatures creates a highly dangerous mixture and a high level of risk of explosion, which can be reduced by using oxygen-selective membranes.
Another approach is based on the reactors used for the reaction chosen. Usually, fixed beds are preferred as thermal control is easier, but considering the size of the tubes, secondary reactions may occur if the first intermediate product (e.g., the olefin) is easily converted. Moreover, safety reasons may impose a low reactant over oxygen ratio (to be out of explosion limits). This is why fluidized or moving bed reactors have been developed, as it was the case by DuPont Co in Asturias, Spain, for butane to maleic anhydride on VPO catalysts [114]. Further developments of chemical engineering and reactors remain an attractive possibility for the future, to circumvent problems in selective oxidation reactions [115]. A possibility resides in using membrane reactors to control the relative feed rate of oxygen and of the organic compound, mainly with the aim of increasing organic compound concentration (limited by explosion limits in fixed be reactors) and, thus, providing higher production rates [116]. For instance, a VPO catalyst was deposited as a thin layer on mesoporous MFI membrane [117] or packed in a tube of porous alumina. Metal oxide nanoparticles entrapped in porous materials may well be interesting for selective oxidation reactions, such as alkane upgrading ones, but up until now, and to our knowledge, no success was mentioned. A conventional fixed bed reactor could also be used with a porous metallic membrane immersed in a gas-solid fluid bed reactor [118].
Conflicts of Interest
The author declares no conflict of interest.
References
- Duprez, D.; Cavani, F. Handbook of Advanced Methods and Processes in Oxidation Catalysis; Imperial College Press: London, UK, 2014. [Google Scholar]
- Callahan, J.L.; Grasselli, R.K. A selectivity factor in vapor-phase hydrocarbon oxidation catalysis. AIChE J. 1963, 9, 755–760. [Google Scholar] [CrossRef]
- Ivars, F.; López Nieto, J.M. Light alkanes oxidation: Targets and current challenges. In Handbook of Advanced Methods and Processes in Oxidation Catalysis; Duprez, D., Cavani, F., Eds.; Imperial College Press: London, UK, 2014; pp. 767–834. [Google Scholar]
- Corma, A.; Garcia, H. Lewis acids as catalysts in oxidation reactions: From homogeneous to heterogeneous systems. Chem. Rev. 2002, 102, 3837–3892. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; van der Voort, P. Metal-organic frameworks as selective or chiral oxidation catalysts. Catal. Rev. Sci. Eng. 2014, 56, 1–56. [Google Scholar]
- Cavani, F.; Trifiro, F. The oxidative dehydrogenation of ethane and propane as an alternative way for the production of light olefins. Catal. Today 1995, 24, 307–313. [Google Scholar] [CrossRef]
- Védrine, J.C.; Millet, J.M.M. Heteropolyoxometallates catalysts for partial oxidation. In Metal Oxide Catalysis; Jackson, S.D., Hargreaves, J., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2009; Volume 2, pp. 561–594. [Google Scholar]
- Grasselli, R.K. Site isolation and phase cooperation: Two important concepts in selective oxidation catalysis: A retrospective. Catal. Today 2014, 238, 10–27. [Google Scholar] [CrossRef]
- Che, M.; Tench, A.J. Characterisation and reactivity of mononuclear oxygen species on oxide surfaces. Adv. Catal. 1982, 31, 77–133. [Google Scholar]
- Bielanski, A.; Haber, J. Oxygen in oxidation on transition metal oxides. Catal. Rev. Sci. Eng. 1979, 79, 1–31. [Google Scholar] [CrossRef]
- Haber, J.; Janas, J.; Schiavello, M.; Tilley, R.J.D. Tungsten oxides as catalysts in selective oxidation. J. Catal. 1983, 82, 395–403. [Google Scholar] [CrossRef]
- Bond, G.C.; Védrine, J.C. General Conclusions. Catal. Today 1994, 20, 171–178. [Google Scholar] [CrossRef]
- Hutchings, G.J. Vanadium phosphate: A new look at the active components of catalysts for the oxidation of butane to maleic anhydride. J. Mater. Chem. 2004, 14, 3385–3395. [Google Scholar] [CrossRef]
- Bordes-Richard, E.; Védrine, J.C. Catalyse sélective redox. Tech. Ingénieur 2013, 1215, 1–30. [Google Scholar]
- Grasselli, R.K.; Burrington, J.D. Selective oxidation and ammoxidation of propylene by heterogeneous catalysis. Adv. Catal. 1980, 30, 133–163. [Google Scholar]
- Mars, P.; van Krevelen, D.W. Oxidations carried out by means of vanadium oxide catalysts. Chem. Eng. Sci. 1954, 3, 41–49. [Google Scholar] [CrossRef]
- Vislovskiy, V.P.; Suleimanov, T.E.; Sinev, M.Y.; Tulenin, Yu.P.; Margolis, L.Y.; Cortés Corberán, V. On the role of heterogeneous and homogeneous processes in oxidative dehydrogenation of C3-C4 alkanes. Catal. Today 2000, 61, 287–293. [Google Scholar] [CrossRef]
- Germain, J.E. Catalytic Conversion of Hydrocarbons; Academic Press: New York, NY, USA, 1969. [Google Scholar]
- Bilde, J.; Janke, C.; Lorentz, C.; Delichère, P.; Popescu, I.; Marcu, I.C.; Loridant, S.; Brückner, A.; Millet, J.M.M. Molecular level insights into the structure of active sites of VAlO mixed oxides in propane ammoxidation. J. Phys. Chem. C 2013, 117, 22926–22938. [Google Scholar] [CrossRef]
- Jussola, J.A.; Mann, R.F.; Downie, J. The kinetics of the vapor-phase oxidation of o-xylene over a vanadium oxide catalyst. J. Catal. 1970, 17, 103–106. [Google Scholar] [CrossRef]
- Bordes, E. The role of structural chemistry of selective catalysts in heterogeneous mild oxidation of hydrocarbons. C. R. Acad. Sci. Ser. IIc: Chem. 2000, 2, 725–733. [Google Scholar] [CrossRef]
- Boudart, M. Effect of surface structure on catalytic activity. In Proceedings of the 6th International Congress on Catalysis, London, UK, 12–16 July 1976; Bond, G.C., Wells, P.B., Tompkins, F.C., Eds.; The Chemical Society: London, UK, 1977; pp. 1–9. [Google Scholar]
- Volta, J.C.; Desquesnes, W.; Moraweck, B.; Coudurier, G. A new method to obtain supported oriented oxides: MoO3 graphite catalyst for propylene oxidation to acrolein. Kinet. Catal. Lett. 1979, 12, 241–245. [Google Scholar] [CrossRef]
- Volta, J.C.; Tatibouët, J.M.; Phichitkul, C.; Germain, J.E. Structural-sensitive catalytic oxidation on MoO3 catalysts. In Proceedings of the 8th International Congress on Catalysis, Berlin, Germany, 2–6 July 1984; Verlag Chemie: Frankfurt am Main, Germany, 1984; Volume 4, pp. 451–458. [Google Scholar]
- Tatibouët, J.M.; Phichitkul, C.; Germain, J.E. Structure-sensitive catalytic oxidation of butenes on molybdenum trioxide crystallites. J. Catal. 1986, 99, 231–238. [Google Scholar] [CrossRef]
- Volta, J.C.; Portefaix, J.L. Structure sensitivity of mild oxidation reactions on oxide catalysts: A review. Appl. Catal. 1985, 18, 1–32. [Google Scholar] [CrossRef]
- Tatibouët, J.M.; Germain, J.E.; Volta, J.C. Structure-sensitive catalytic oxidation: Alcohols on graphite-supported molybdenum trioxide. J. Catal. 1983, 82, 240–248. [Google Scholar] [CrossRef]
- Haber, J. The concept of structure sensitivity in catalysis by oxides. Stud. Surf. Sci. Catal. 1989, 48, 447–467. [Google Scholar]
- Ziolkowski, J.; Bordes, E.; Courtine, P. Oxidation of butane and butene on the (100) face of (VO)2P2O7: A dynamic view in terms of crystallochemical model of active sites. J. Catal. 1990, 122, 126–150. [Google Scholar] [CrossRef]
- Ueda, W.; Oshihara, K. Selective oxidation of light alkanes over hydrothermally synthesized Mo–V–M–O (M = Al, Ga, Bi, Sb, and Te) oxide catalysts. Appl. Catal. A Gen. 2000, 200, 135–143. [Google Scholar] [CrossRef]
- Guliants, V.V.; Brongersma, H.H.; Knoester, A.; Gaffney, A.M.; Han, S. Surface active sites present in the orthorhombic M1 phases: Low energy ion scattering study of methanol and allyl alcohol chemisorption over Mo–V–Te–Nb–O and Mo–V–O catalysts. Top. Catal. 2006, 38, 41–50. [Google Scholar] [CrossRef]
- Celaya Sanfiz, A.; Hansen, T.W.; Sakthivel, A.; Trunschke, A.; Schlögl, R.; Knoester, A.; Brongersma, H.H.; Looi, M.H.; Hamid, S.B.A. How important is the (001) plane of M1 for selective oxidation of propane to acrylic acid? J. Catal. 2008, 258, 35–43. [Google Scholar] [CrossRef]
- Taylor, H.S. A theory of the catalytic surface. Proc. R. Soc. Lond. 1925, A108, 105–111. [Google Scholar] [CrossRef]
- Védrine, J.C. Revisiting active sites in heterogeneous catalysis: Their structure and their dynamic behaviour. Appl. Catal. A Gen. 2014, 474, 40–50. [Google Scholar] [CrossRef]
- Chizallet, C.; Costentin, G.; Che, M.; Delbecq, F.; Sautet, P. Revisiting acido-basicity of the MgO surface by periodic density functional theory calculations: role of surface topology and ion coordination on water dissociation. J. Phys. Chem. B 2006, 110, 15878–15886. [Google Scholar] [CrossRef] [PubMed]
- Védrine, J.C.; Coudurier, G.; Millet, J.M.M. Molecular design of active sites in partial oxidation reactions on metallic oxides. Catal. Today 1997, 33, 3–13. [Google Scholar] [CrossRef]
- Millet, J.M.M. FePO catalysts for the selective oxidative dehydrogenation of isobutyric acid into methacrylic acid. Catal. Rev. Sci. Eng. 1998, 40, 1–38. [Google Scholar] [CrossRef]
- Robert, V.; Borshch, S.A.; Bigot, B. Molecular orbital model of electron delocalization in mixed-valence trimeric clusters. J. Phys. Chem. 1996, 100, 580–584. [Google Scholar] [CrossRef]
- Haynes, A.; Maitlis, P.M.; Morris, G.E.; Sunley, G.J.; Adams, H.; Badger, P.W.; Bowers, C.M.; Cook, D.B.; Elliott, P.I.P.; Ghaffar, T.; et al. Promotion of Iridium-Catalyzed Methanol Carbonylation: Mechanistic Studies of the Cativa Process. J. Am. Chem. Soc. 2004, 126, 2847–2861. [Google Scholar] [PubMed]
- Agashar, P.A.; de Caul, L.; Grasselli, R.K. A molecular level mechanism of n-butane oxidation to maleic anhydride over vanadyl pyrophosphate. Catal. Lett. 1994, 23, 339–346. [Google Scholar] [CrossRef]
- Panov, G.I.; Kharitonov, A.S.; Sobolev, V.I. Oxidative hydroxylation using dinitrogen monoxide: A possible route for organic synthesis over zeolites. Appl. Catal. A 1993, 98, 1–20. [Google Scholar] [CrossRef]
- Janas, J.; Shishido, T.; Che, M.; Dzwigaj, S. Role of tetrahedral Co(II) sites of CoSiBEA zeolite in the selective catalytic reduction of NO: XRD, UV-vis, XAS and catalysis study. Appl. Catal. B 2009, 89, 196–203. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Haruta, M.; Shen, W. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 2009, 458, 746–749. [Google Scholar]
- Ogliaro, F.; Harris, N.; Cohen, S.; Filatov, M.; de Visser, S.P.; Saik, S. A model “rebound” mechanism of hydroxylation by cytochrome P450: Stepwise and effectively concerned pathways and their reactivity patterns. J. Am. Chem. Soc. 2000, 122, 8977–8989. [Google Scholar] [CrossRef]
- Afanasiev, P.; Kudrik, E.V.; Millet, J.M.M.; Bouchu, D.; Sorokin, A. High-valent di iron species generated from N-bridged diiron phthalocyanine and H2O2. J. Chem. Soc. Dalton Trans. 2011, 40, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Kudrik, E.V.; Afanasiev, P.; Alvarez, L.X.; Dubourdeaux, P.; Clémancey, M.; Latour, J.M.; Blondin, G.; Bouchu, D.; Albrieux, F.; Nefedov, S.E.; et al. An N-bridged high-valent diiron-oxo species on a porphyrin platform that can oxidize methane. Nat. Chem. 2012, 4, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Ushikubo, T.; Oshima, K.; Kayou, A.; Vaarkamp, M.; Hatano, M. Ammoxidation of propane over catalysts comprising mixed oxides of Mo and V. J. Catal. 1997, 169, 394–396. [Google Scholar] [CrossRef]
- Oshama, K.; Kayo, A.; Umezawa, T.K.; Kiyono, K.; Sawaki, I. Synthesis of MoVTe(Sb)-O composite oxide catalyst via reduction of polyoxometalates in an aqueous medium. EP 529 853, 1992. [Google Scholar]
- Thorsteinson, E.M.; Wilson, T.P.; Young, F.G.; Kasai, P.H. The oxidative dehydrogenation of ethane over catalysts containing mixed oxides of molybdenum and vanadium. J. Catal. 1978, 52, 116–132. [Google Scholar] [CrossRef]
- Botella, P.E.; García-González, E.; López Nieto, J.M.; González-Calbet, J.M. MoVTeNbO multifunctional catalysts: Correlation between constituent crystalline phases and catalytic performance. Solid State Sci. 2005, 7, 507–519. [Google Scholar] [CrossRef]
- Schlögl, R. Active sites for propane oxidation: Some generic considerations. Top. Catal. 2011, 54, 627–638. [Google Scholar] [CrossRef]
- Grasselli, R.K.; Burrington, J.D.; Buttrey, D.J.; de Santo, P., Jr.; Lugmair, C.G.; Volpe, A.F., Jr.; Weingand, T. Multifunctionality of active centers in (amm)oxidation catalysts: From Bi–Mo–Ox to Mo–V–Nb–(Te, Sb)–Ox. Top. Catal. 2003, 23, 5–22. [Google Scholar] [CrossRef]
- Védrine, J.C. Eurocat oxide V2O5/TiO2. Catal Today 1994, 20, 1–178. [Google Scholar]
- Védrine, J.C. The role of redox, acid-base and collective properties and of crystalline state of heterogeneous catalysts in the selective oxidation of hydrocarbons. Top. Catal. 2002, 21, 97–106. [Google Scholar] [CrossRef]
- Tielens, F.; Dzwigaj, S. Probing acid-base sites in vanadium redox zeolites by DFT calculation and compared with FTIR results. Catal. Today 2010, 152, 66–69. [Google Scholar] [CrossRef]
- Védrine, J.C. Acid-base characterization of heterogeneous catalysts: An up-to-date overview. Res. Chem. Intermed. 2015. [Google Scholar] [CrossRef]
- Noller, H.; Tomke, K. Transition states of catalytic dehydration and dehydrogenation of alcohols. J. Mol. Catal. 1979, 6, 375–392. [Google Scholar] [CrossRef]
- Lauron-Pernot, H. Evaluation of surface acido-basic properties of inorganic-based solids by model alcohol catalytic reaction networks. Catal. Rev. Sci. Eng. 2006, 348, 315–361. [Google Scholar] [CrossRef]
- Baca, M.; Pigamo, A.; Dubois, J.L.; Millet, J.M.M. Fourier transform infrared spectroscopic study of surface acidity by pyridine adsorption on the M1 active phase of the MoVTe(Sb)NbO catalysts used in propane oxidation. Catal. Commun. 2005, 6, 215–220. [Google Scholar] [CrossRef]
- Ivars-Barceló, F.; Millet, J.M.M.; Blasco, T.; Concepción, P.; Valente, J.S.; López Nieto, J.M. Understanding effects of activation-treatments in K-free and K-MoVSbO bronze catalysts for propane partial oxidation. Catal. Today 2014, 238, 41–48. [Google Scholar] [CrossRef]
- Grasselli, R.K.; Lugmair, C.G.; Volpe, A.E.; Andersson, A.; Burrington, J.D. Enhancement of acrylic acid yields in propane and propylene oxidation by selective P doping of MoV(Nb)TeO-based M1 and M2 catalysts. Catal. Today 2010, 157, 33–38. [Google Scholar] [CrossRef]
- Jo, B.Y.; Kum, S.S.; Moon, S.H. Performance of WOx-added Mo–V–Te–Nb–O catalysts in the partial oxidation of propane to acrylic acid. Appl. Catal. A 2010, 378, 76–82. [Google Scholar] [CrossRef]
- Andrushkevich, T.V.; Popova, G.Y.; Chesalov, Y.A.; Ischenko, E.V.; Khramov, M.I.; Kaichev, V.V. Propane ammoxidation on Bi promoted MoVTeNbOx oxide catalysts: Effect of mixture composition. Appl. Catal. A 2015, 506, 109–117. [Google Scholar] [CrossRef]
- Novakova, E.N.; Védrine, J.C. Propane selective oxidation to propene and oxygenates on metal oxides. In Metal Oxides: Chemistry and Applications; Fierro, J.L.G., Ed.; CRC: Boca Raton, FL, USA; Taylor & Francis: Baton Rouge, LA, USA, 2006; pp. 444–446. [Google Scholar]
- Pantazidis, A.; Burrows, A.; Kiely, C.J.; Mirodatos, C. Direct evidence of active surface reconstruction during oxidative dehydrogenation of propane over VMgO catalyst. J. Catal. 1998, 177, 325–334. [Google Scholar] [CrossRef]
- Popescu, I.; Skoufa, Z.; Heracleous, E.; Lemonidou, A.; Marcu, I.C. A study of electrical conductivity measurements of the semi conductive and redox properties of Nb-doped NiO catalysts in correlation with the oxidative dehydrogenation of ethane. Phys. Chem. Chem. Phys. 2015, 17, 8138–8147. [Google Scholar] [CrossRef] [PubMed]
- Courtine, P.; Bordes, E. Synergistic effects in multicomponent catalysts for selective oxidation. Stud. Surf. Sci. Catal. 1997, 110, 177–184. [Google Scholar]
- Bordes, E. Synergistic effects in selective oxidation catalysis: Does phase cooperation result in site isolation? Top. Catal. 2001, 15, 131–137. [Google Scholar] [CrossRef]
- Carson, D.; Coudurier, G.; Védrine, J.C.; Laarif, A.; Theobald, F. Synergy effects in the catalytic properties of Bi molybdate. J. Chem. Soc. Faraday Trans. I 1983, 79, 1921–1929. [Google Scholar] [CrossRef]
- Ponceblanc, H.; Millet, J.M.M.; Coudurier, G.; Herrmann, J.M.; Védrine, J.C. Study of multiphasic molybdates based catalysts. Part I—Electrical conductivity study of valence states and solubility limits in mixed iron and cobalt molybdates. J. Catal. 1993, 142, 373–380. [Google Scholar] [CrossRef]
- Millet, J.M.M.; Ponceblanc, H.; Coudurier, G.; Herrmann, J.M.; Védrine, J.C. Study of multiphasic molybdates based catalysts. Part II—Synergy effect between bismuth and mixed iron and cobalt molybdates in mild oxidation of propene. J. Catal. 1993, 142, 381–391. [Google Scholar] [CrossRef]
- Wainwright, M.S.; Foster, N.R. Catalysts, Kinetics and Reactor Design in Phthalic Anhydride Synthesis. Catal. Rev. Sci. Eng. 1979, 19, 211–292. [Google Scholar] [CrossRef]
- Papachryssanthou, J.; Bordes, E.; Véjux, A.; Courtine, P.; Marchand, R.; Tournoux, M. TiO2(B), a new support for V2O5 in the oxidation of O-xylene. Catal. Today 1987, 1, 219–227. [Google Scholar] [CrossRef]
- Sanati, M.; Wallenberg, R.; Andersson, A.; Jasen, S.; To, Y. Vanadia catalysts on anatase, rutile, and TiO2(B) for the ammoxidation of toluene: An ESR and high-resolution electron microscopy characterization. J. Catal. 1991, 132, 128–144. [Google Scholar] [CrossRef]
- Véjux, A.; Courtine, P. Interfacial reactions between V2O5 and TiO2 (anatase): Role of the structural properties. J. Solid State Chem. 1978, 23, 93–103. [Google Scholar] [CrossRef]
- Grasselli, R.K. Advances and future trends in selective oxidation and ammoxidation catalysis. Catal. Today 1999, 49, 141–154. [Google Scholar] [CrossRef]
- Volta, J.C. Vanadium phosphorus oxides, a reference catalyst for mild oxidation of light alkanes. C.R. Acad. Sci. IIc: Chem. 2000, 3, 717–723. [Google Scholar] [CrossRef]
- Korovchenko, P.; Shiju, N.R.; Dozier, A.K.; Graham, U.M.; Guerrero-Pérez, M.O.; Guliants, V.V. M1 to M2 phase transformation and phase cooperation in bulk mixed metal Mo–V–M–O (M = Te,Nb) catalysts for selective ammoxidation of propane. Top. Catal. 2007, 50, 43–51. [Google Scholar] [CrossRef]
- Weng, L.T.; Delmon, B. Phase cooperation and remote control effects in selective oxidation catalysts. Appl. Catal. A 1992, 81, 141–213. [Google Scholar] [CrossRef]
- Karroua, M.; Grange, P.; Delmon, B. Existence of synergy between “CoMoS” and Co9S8: New proof of remote control in hydrodesulfurization. Appl. Catal. 1989, 50, L5–L10. [Google Scholar] [CrossRef]
- Schuh, K.; Kleist, W.; Høj, M.; Trouillet, V.; Beato, P.; Degn Jensen, A.; Grunwaldt, J.D. Bismuth molybdate catalysts prepared by mild hydrothermal synthesis: Influence of pH on the selective oxidation of propylene. Catalysts 2015, 5, 1554–1573. [Google Scholar] [CrossRef]
- Brazdil, J.F.; Toft, M.A.; Lin, S.Y.; McKenna, S.T.; Zajac, G.; Kaduk, J.; Golab, T. Characterization of bismuth-cerium-molybdate selective propylene ammoxidation catalysts. Appl. Catal. A 2015, 495, 115–123. [Google Scholar] [CrossRef]
- Timm, B. The ammonia synthesis and heterogeneous catalysis. A historical review. In Proceedings of the 8th International Congress on Catalysis, Berlin, Germany; Dechema: Frankfurt am Main, Germany; Verlag Chemie: Frankfurt am Main, Germany, 1984; Volume 1, pp. 7–30. [Google Scholar]
- Mehlomakulu, B.; Nguyen, T.T.N.; Delichère, P.; van Teen, E.; Millet, J.M.M. Identification of the active species in oxidation reactions on mixed oxide catalysts: Supra-surface or bulk-surface species. J. Catal. 2012, 289, 1–10. [Google Scholar] [CrossRef]
- Schlögl, R. Concepts in selective oxidation of small alkane molecules. In Modern Heterogeneous Oxidation Catalysis; Mizuno, N., Ed.; Wiley-VCH: Weinheim, Germany, 2009; pp. 1–42. [Google Scholar]
- Thomas, J.M. The societal significance of catalysis and the growing practical importance of single-site heterogeneous catalysts. Proc. Roy. Soc. A 2012, 468, 1884–1903. [Google Scholar]
- Hävecker, M.; Wrabetz, S.; Kröhnert, J.; Csepei, L.I.; D’Alnoncourt, R.N.; Kolen’ko, Y.V.; Girgsdies, F.; Schlögl, R.; Trunschke, A. Surface chemistry of phase-pure M1 MoVTeNb oxide during operation in selective oxidation of propane to acrylic acid. J. Catal. 2012, 285, 48–60. [Google Scholar] [CrossRef]
- Kung, H. Oxidative dehydrogenation of light C2 to C4 alkanes. Adv. Catal. 1994, 40, 1–38. [Google Scholar]
- Blasco, T.; López-Nieto, J.M. Oxidative dehydrogenation of short chain alkanes on supported vanadium oxide catalysts. Appl. Catal. A 1997, 157, 117–142. [Google Scholar] [CrossRef]
- Tabaka, K.; Teng, Y.; Takemoto, T.; Suzuki, E.; Bañares, M.A.; Peña, M.A.; Fierro, J.L.G. Activation of methane by oxygen and nitrogen oxides. Catal. Rev. 2002, 44, 1–58. [Google Scholar] [CrossRef]
- Wang, L.H.; Tao, L.X.; Xie, M.S.; Xu, G.F.; Huang, J.H.; Xu, Y. Dehydrogenation and aromatization of methane under non-oxidizing conditions. Catal. Lett. 1993, 21, 35–41. [Google Scholar] [CrossRef]
- Heracleous, E.; Lemonidou, A. Ni-Nb-O mixed oxides as highly active and selective catalysts for ethane production via ethane oxidative dehydrogenation. Part I: Characterization and catalytic performance. J. Catal. 2006, 237, 162–174. [Google Scholar] [CrossRef]
- Wu, Y.; Gao, J.; He, Y.; Wu, T. Preparation and characterization of Ni–Zr–O nanoparticles and its catalytic behavior for ethane oxidative dehydrogenation. Appl. Surf. Sci. 2012, 258, 4922–4928. [Google Scholar] [CrossRef]
- Wu, Y.; He, Y.; Wu, T.; Weng, W.; Wan, H. Effect of synthesis method on the physical and catalytic property of nanosized NiO. Mater. Lett. 2007, 61, 2679–2682. [Google Scholar] [CrossRef]
- Popescu, I.; Heracleous, E.; Skoufa, Z.; Lemonidou, A.; Marcu, I.C. Study of electrical conductivity measurements of semiconductive and redox properties of M-doped NiO (M = Li, Mg, Al, Ga, Ti, Nb) catalysts for the oxidative dehydrogenation of ethane. Phys. Chem. Chem. Phys. 2014, 16, 4962–4970. [Google Scholar] [CrossRef] [PubMed]
- López-Nieto, J.M.; Botella, P.; Vázquez, M.L.; Dejoz, A. The selective oxidative dehydrogenation of ethane over hydrothermally synthesised MoVTeNb catalysts. Chem. Commun. 2002, 1906–1907. [Google Scholar]
- Soliman, M.; Al-Zeghayer, Y.; Al-Awadi, A.S.; Al-Mayman, S. Economics of acetic acid production by partial oxidation of ethane. APCBEE Procedia 2012, 3, 200–208. [Google Scholar] [CrossRef]
- Linke, D.; Wolf, D.; Zey, B.S.; Baerns, M.; Timpe, O.; Schlögl, R.; Zeyβ, S.; Dingerdissen, U. Catalytic partial oxidation of ethane to acetic acid over Mo1V0.25Nb0.12Pd0.0005Ox, I: Catalyst performance and reaction mechanism. J. Catal. 2002, 205, 16–31. [Google Scholar] [CrossRef]
- Rahman, F.; Loughlin, K.F.; Al-Saleh, M.A.; Saeed, M.R.; Tukur, N.M.; Hossain, M.M.; Karim, K.; Mamedov, A. Kinetics and mechanism of partial oxidation of ethane to ethylene and acetic acid over MoV type catalysts. Appl. Catal. A 2010, 375, 17–25. [Google Scholar] [CrossRef]
- Kutnetsova, N.I.; Popova, G.Y.; Kuznetsova, L.I.; Zaikovskii, V.I.; Koscheev, S.V.; Andrushkevich, T.V.; Lisitsyn, A.S.; Likholobov, V.A.; Han, S. Improving the performance of Pt-H3PMo12O40 catalysts in the selective dehydrogenation of propane with O2 and H2. Catal. Today 2015, 245, 179–185. [Google Scholar] [CrossRef]
- Mamedov, E.; Karim, K. Selective oxidation at SABIC: Innovative catalysts and technologies. In Handbook of Advanced Methods and Processes in Oxidation Catalysis; Duprez, D., Cavani, F., Eds.; Imperial College Press: London, UK, 2014; pp. 291–301. [Google Scholar]
- Gärtner, C.A.; van Veen, A.C.; Lercher, J.A. Oxidative dehydrogenation of ethane: Common principles and mechanistic aspects. ChemCatChem 2013, 5, 3196–3217. [Google Scholar] [CrossRef]
- Valverde, J.A.; Echavarria, A.; Eon, J.G.; Faro, A.C.; Palacio, L.A. V-Mg-Al catalyst from hydrotalcite for oxidative dehydrogenation of propane. React. Kinet. Mech. Catal. 2014, 11, 679–696. [Google Scholar] [CrossRef]
- Kondratenko, E.V.; Sinev, M.Y. Effect of nature and surface density of oxygen species on product distribution in the oxidative dehydrogenation of propane over oxide catalysts. Appl. Catal. A 2007, 325, 353–361. [Google Scholar] [CrossRef]
- Valenzuela, R.X.; Bueno, G.; Cortés Corberán, V.; Xu, Y.; Chen, C.G. Selective oxidehydrogenation of ethane with CO2 over CeO2-based catalysts. Catal. Today 2000, 61, 43–48. [Google Scholar] [CrossRef]
- Cortés Corberán, V. Novel approaches for the improvement of selectivity in the oxidative activation of light alkanes. Catal. Today 2005, 99, 33–41. [Google Scholar] [CrossRef]
- Deacon, H. Improvement in the manufacture of chlorine. US Patent 85370A, 29 December 1868. [Google Scholar]
- Wang, D.J.; Xu, M.T.; Shi, C.L.; Lunsford, J.H. Effect of carbon dioxide on the selectivities obtained during the partial oxidation of methane and ethane over Li+/MgO catalysts. Catal. Lett. 1993, 18, 323–328. [Google Scholar] [CrossRef]
- Wang, S.; Murata, K.; Hayakawa, T.; Hamakawa, S.; Suzuki, K. Dehydrogenation of ethane with carbon dioxide over supported chromium oxide catalysts. Appl. Catal A 2000, 196, 1–8. [Google Scholar] [CrossRef]
- Xu, L.Y.; Liu, J.X.; Yang, H.; Xu, Y.; Wang, Q.X.; Lin, L.W. Regeneration behaviors of Fe/Si-2 and Fe–Mn/Si-2 catalysts for C2H6 dehydrogenation with CO2 to C2H4. Catal. Lett. 1999, 62, 185–189. [Google Scholar] [CrossRef]
- Cavani, F.; Ballarini, N.; Cericola, A. Oxidative dehydrogenation of ethane and propane: How far from commercial implementation? Catal. Today 2007, 127, 113–131. [Google Scholar] [CrossRef]
- Somorjai, G.A.; Park, J.Y. Molecular factor of catalytic selectivity. Angew. Chem. Int. Ed. 2008, 47, 9212–9228. [Google Scholar] [CrossRef] [PubMed]
- Contractor, R.M.; Garnett, D.I.; Horowitz, H.S.; Bergna, H.E.; Patience, G.S. A new commercial scale process for n-butane oxidation to maleic anhydride using a circulating fluidized bed reactor. Stud. Surf. Sci. Catal. 1994, 82, 233–242. [Google Scholar]
- Groppi, G.; Beretta, A.; Troconi, E. Structured catalytic reactors for selective oxidations. In Handbook of Advanced Methods and Processes in Oxidation Catalysis; Duprez, D., Cavani, F., Eds.; Imperial College Press: London, UK, 2014; pp. 943–997. [Google Scholar]
- Menéndez, M. Membrane reactors as tools for improved catalytic oxidation process. In Handbook of Advanced Methods and Processes in Oxidation Catalysis; Duprez, D., Cavani, F., Eds.; Imperial College Press: London, UK, 2014; pp. 921–942. [Google Scholar]
- Mallada, R.; Menéndez, M.; Santamaria, J. Use of membrane reactors for the oxidation of butane to maleic anhydride under high butane concentration. Catal. Today 2000, 56, 191–197. [Google Scholar] [CrossRef]
- Bordes-Richard, E.; Shekari, A.; Patience, G.S. Vanadium phosphorus oxide catalyst for n-butane selective oxidation: From catalyst synthesis to the industrial process. In Handbook of Advanced Methods and Processes in Oxidation Catalysis; Duprez, D., Cavani, F., Eds.; Imperial College Press: London, UK, 2014; pp. 549–585. [Google Scholar]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).