
Heterogeneous Partial (amm)Oxidation and Oxidative Dehydrogenation Catalysis on Mixed Metal Oxides


Abstract
:1. Introduction and General Features of Partial Oxidation Reactions
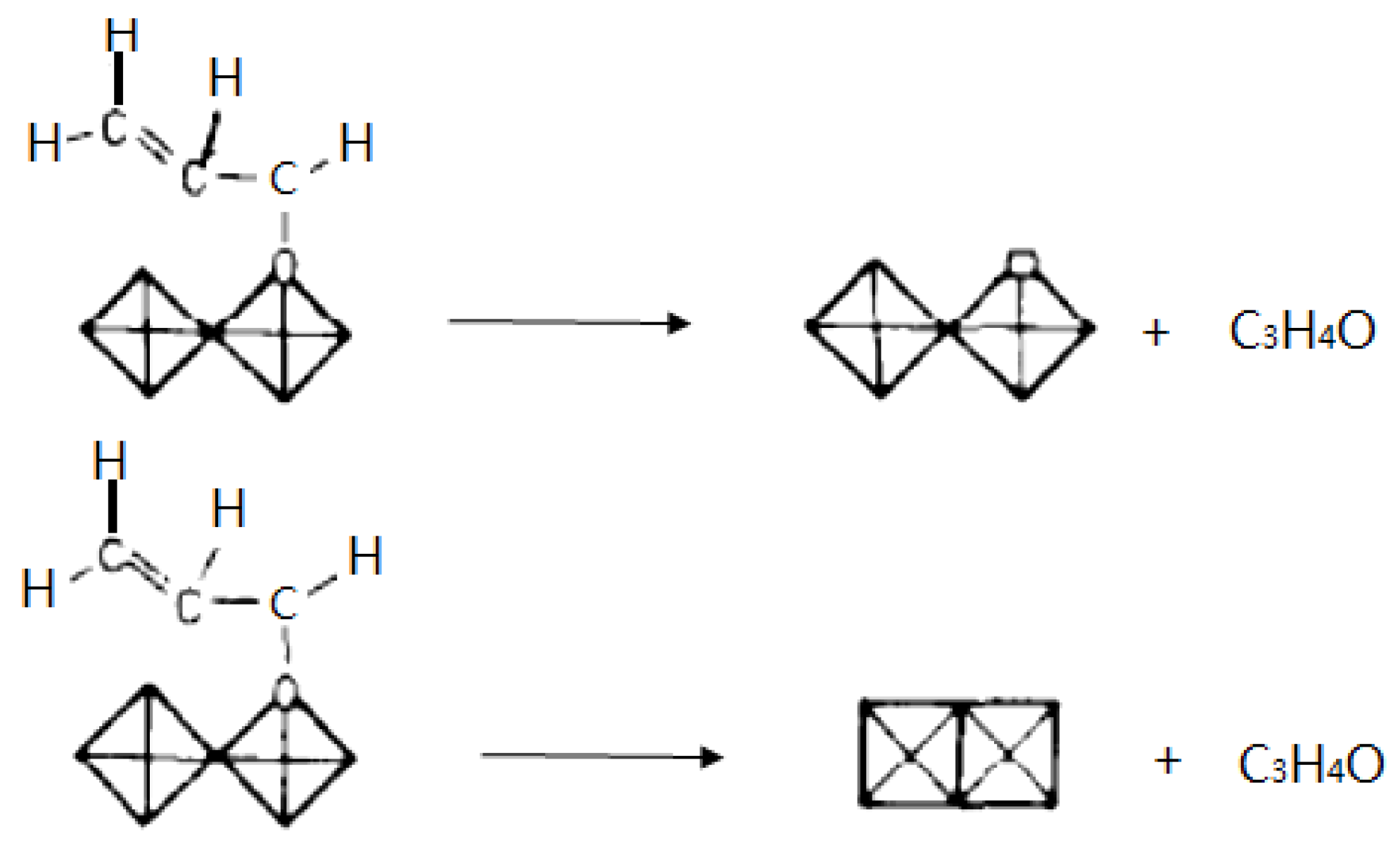

{kind=link}
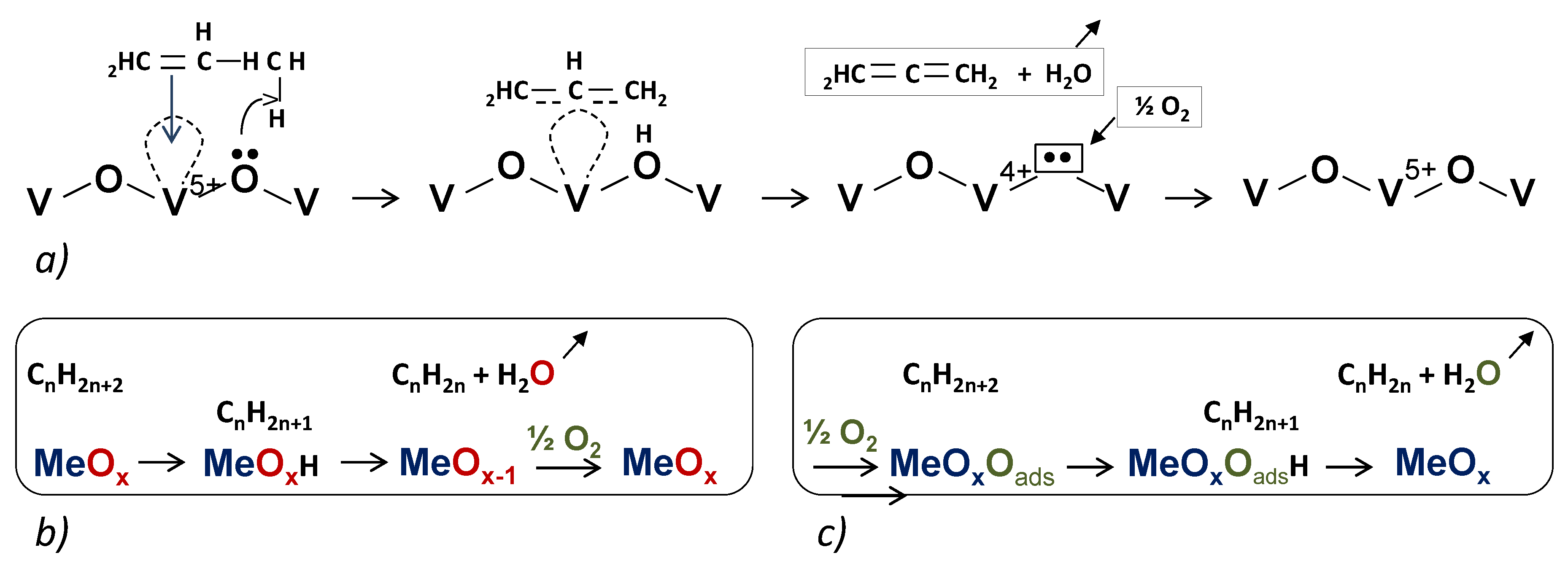{kind=link}
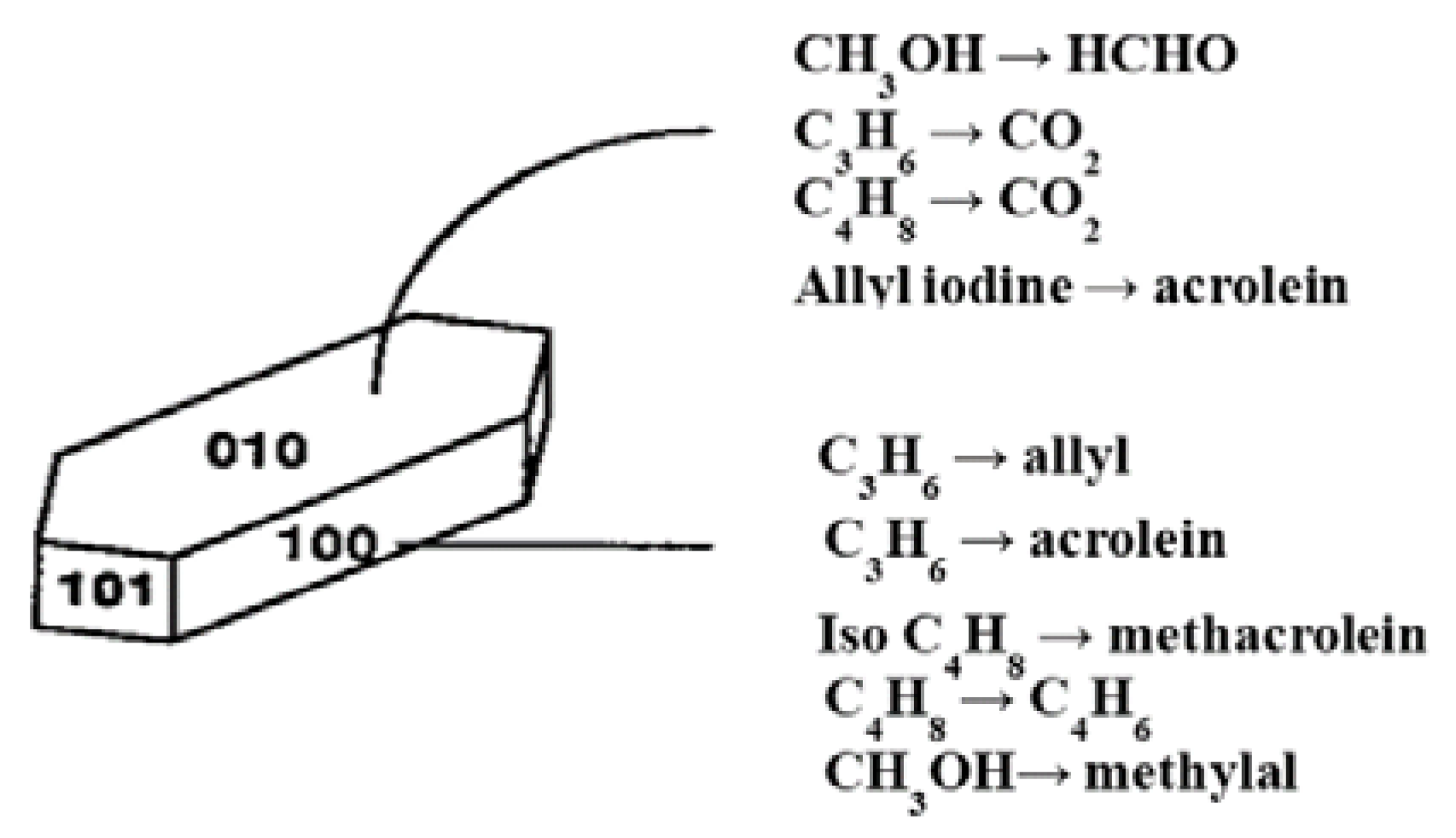{kind=link}
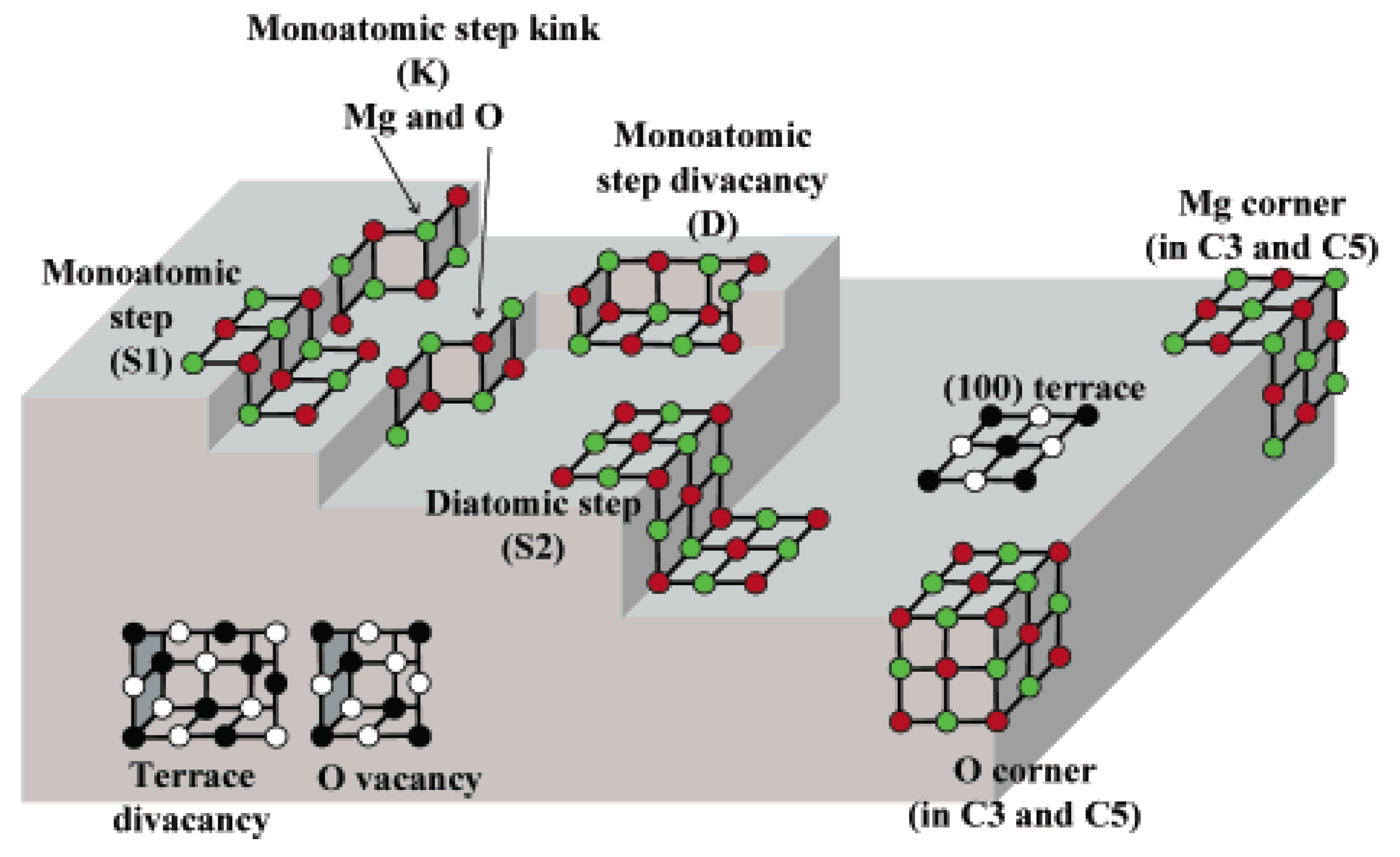{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
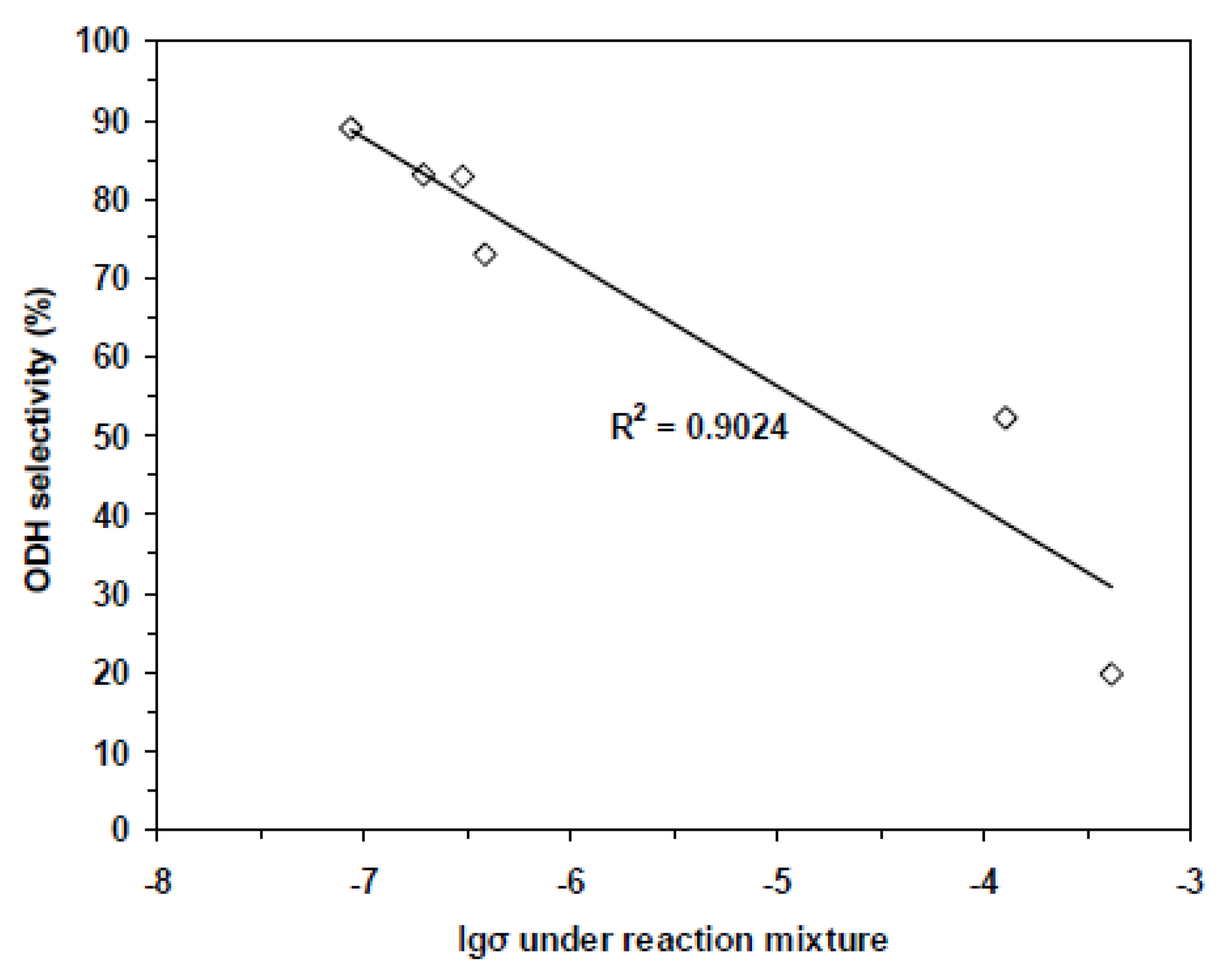{kind=link}
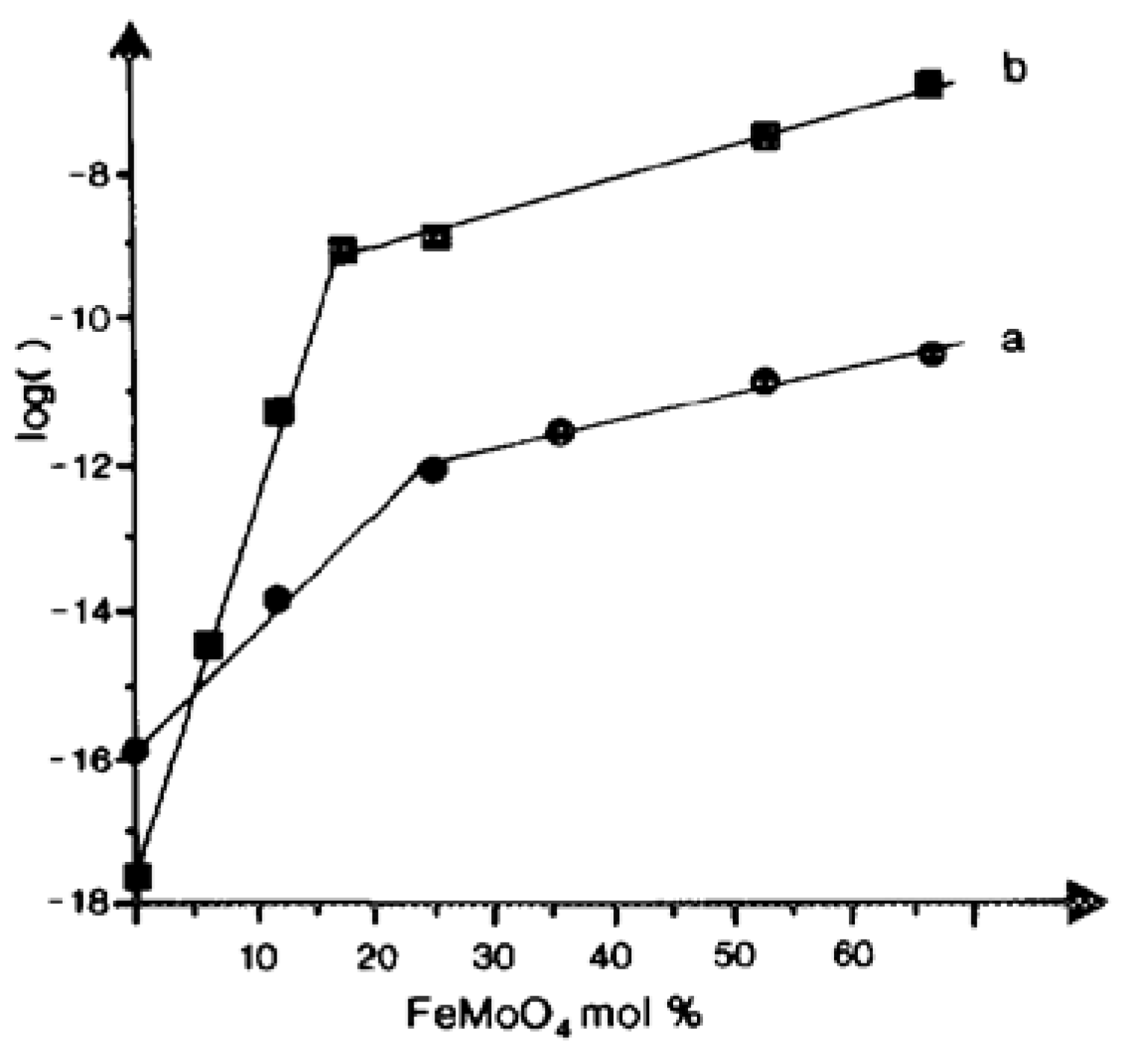{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Type | Reactants | Products | Nb of e− | Catalysts | Yield mol % | State |
|---|---|---|---|---|---|---|
| oxidative coupling | CH4 | ethane + ethylene | 2 | Li2O/MgO | 25 | NI |
| 4 | ||||||
| oxidative dehydrogenation | ethane | ethylene | 2 | Pt, mixed | 60 | NI |
| MoVTeNb-oxides | 75 | NI | ||||
| propane | propene | 2 | VMgO | 40 | NI | |
| n-butane | butene, butadiene | 2 | metal molybdates | 38 | NI | |
| 4 | ||||||
| ethylbenzene | styrene | 2 | FeO-AlPO4 | 70 | P | |
| methanol | formaldehyde | 4 | FeMO4 | 81 | I | |
| isobutyric acid | methacrylic acid | 2 | FePO4 | 75 | NI | |
| oxychloration | ethane, Cl2 | vinyl chloride | 4 | AgMnCoO | 15 | NI |
| ethene, Cl2 | 2 | CuPdCl | 90 | I | ||
| hydroxylation | benzene | phenol | 2 | Ti or Fe zeolite | - | I |
| selective oxidation | methane | CO + H2 | 2 | Pt or Ni | 90 | R |
| formaldehyde | 4 | MoSnPO | 16 | R | ||
| ethene | ethyloxide | 2 | Ag/Al2O3 | 8 | I | |
| acetaldehyde | 2 | V2O5 + PdCl2 | 50 | I | ||
| acetic acid | 4 | MoVNbO + Pd/Al2O3 | ||||
| ethane | acetic acid | 6 | MoVNb-O | 10 | P | |
| propene | acrolein | 4 | BiFeCoMoO | 86 | I | |
| propane | acrylic acid | 6 | MoVNb(Te,Sb)O | 8 | NI | |
| n-butane | maleic anhydride | 14 | (VO)2P2O7 | 70 | I | |
| i-butane | methacrylic acid | 8 | HPA, oxides | 11 | R | |
| i-butene | methacrolein | 6 | SnSbO | 65 | NI | |
| o-xylene | phthalic anhydride | 12 | V2O5/TiO2 | 82 | I | |
| acrolein | acrylic acid | 2 | VMoWO | 85 | I | |
| methacrolein | methacrylic acid | 2 | - | 95 | I | |
| t-butylic alcohol | methacrolein | 4 | BiMoFeCoO | 88 | I | |
| ammoxidation | Propene + NH3 | acrylonitrile | 6 | MoBiFeCoNiO | 80 | I |
| Propane + NH3 | 8 | VSbO, MoVO | 30 | I |
2. General Reaction Mechanisms

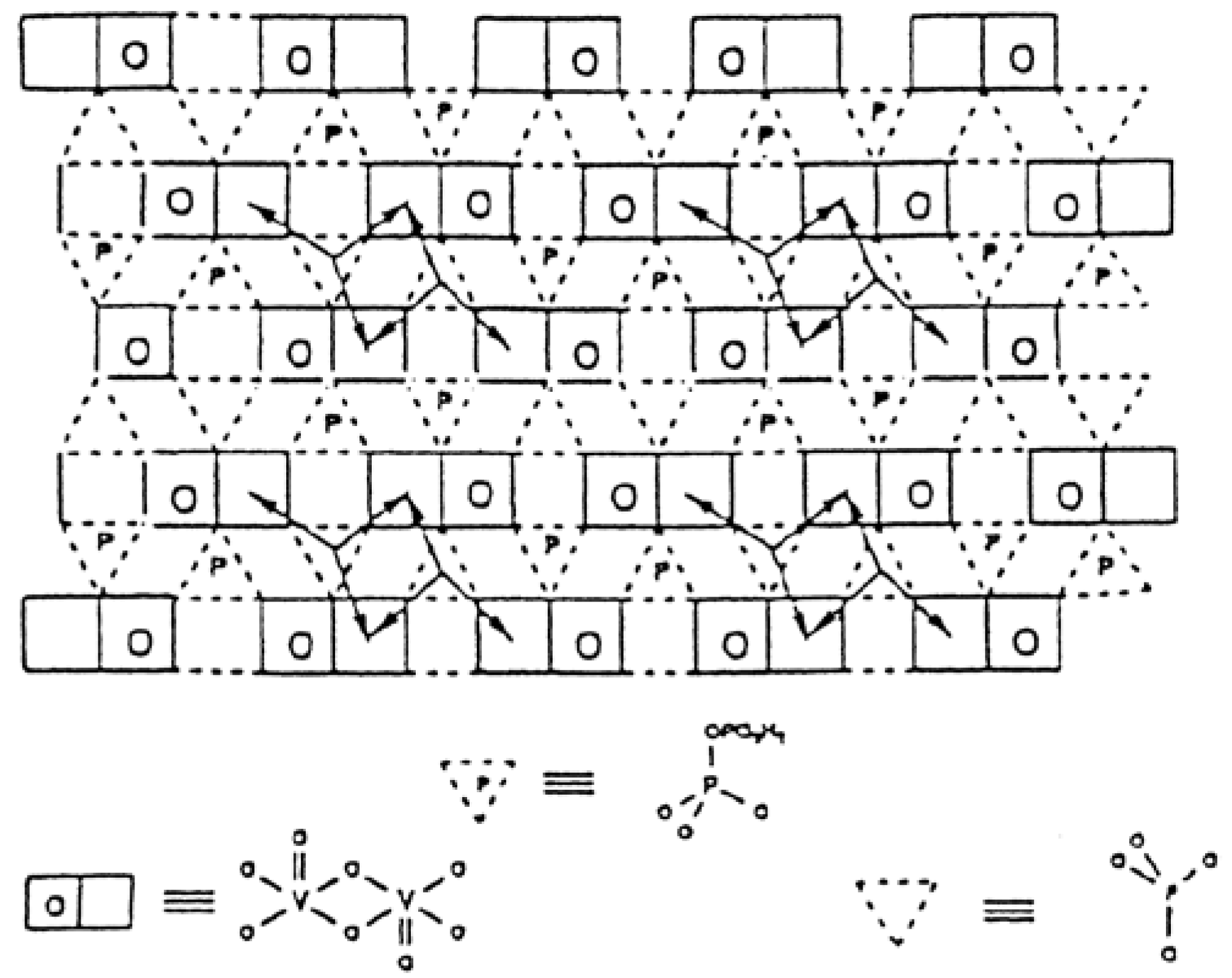
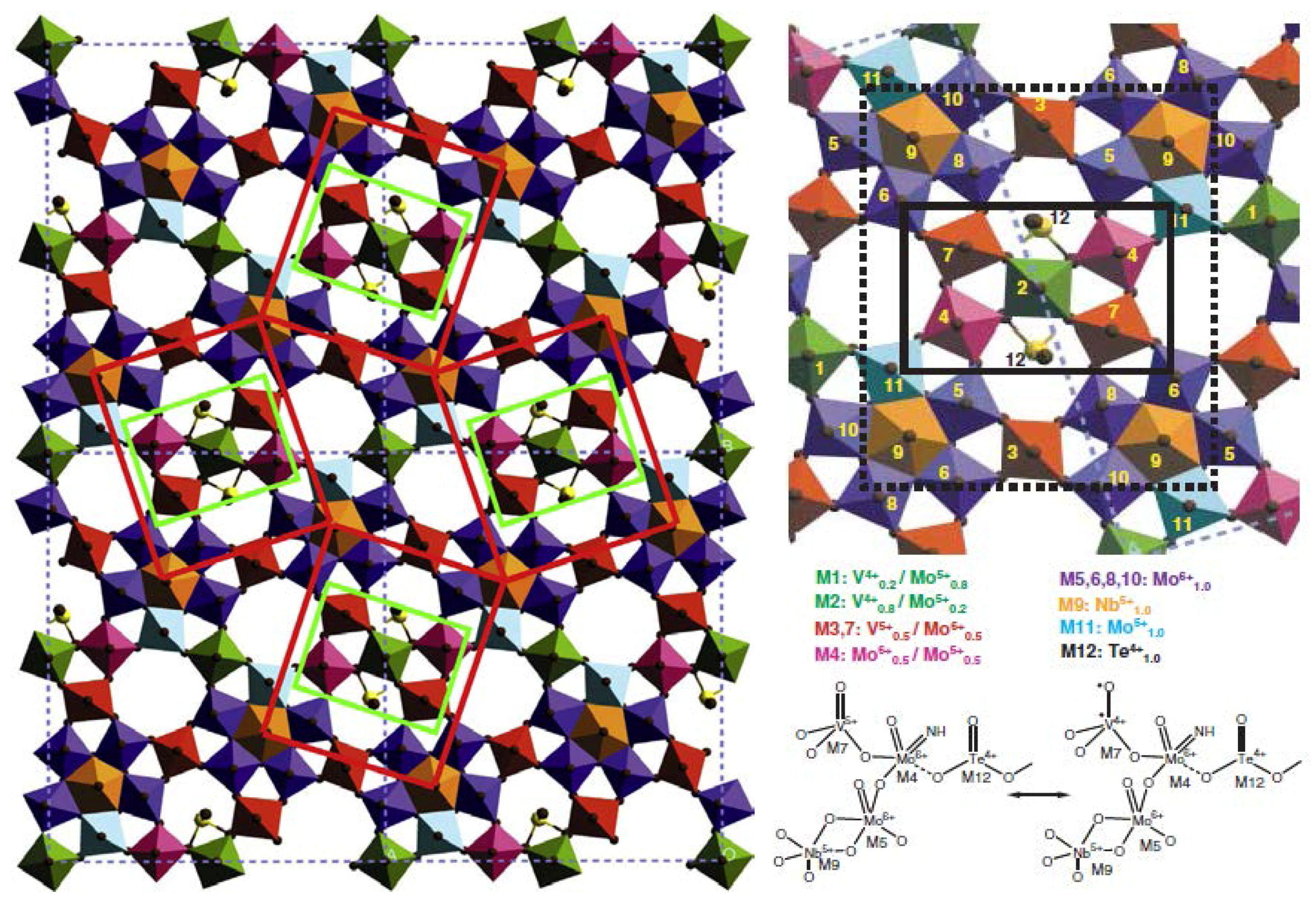
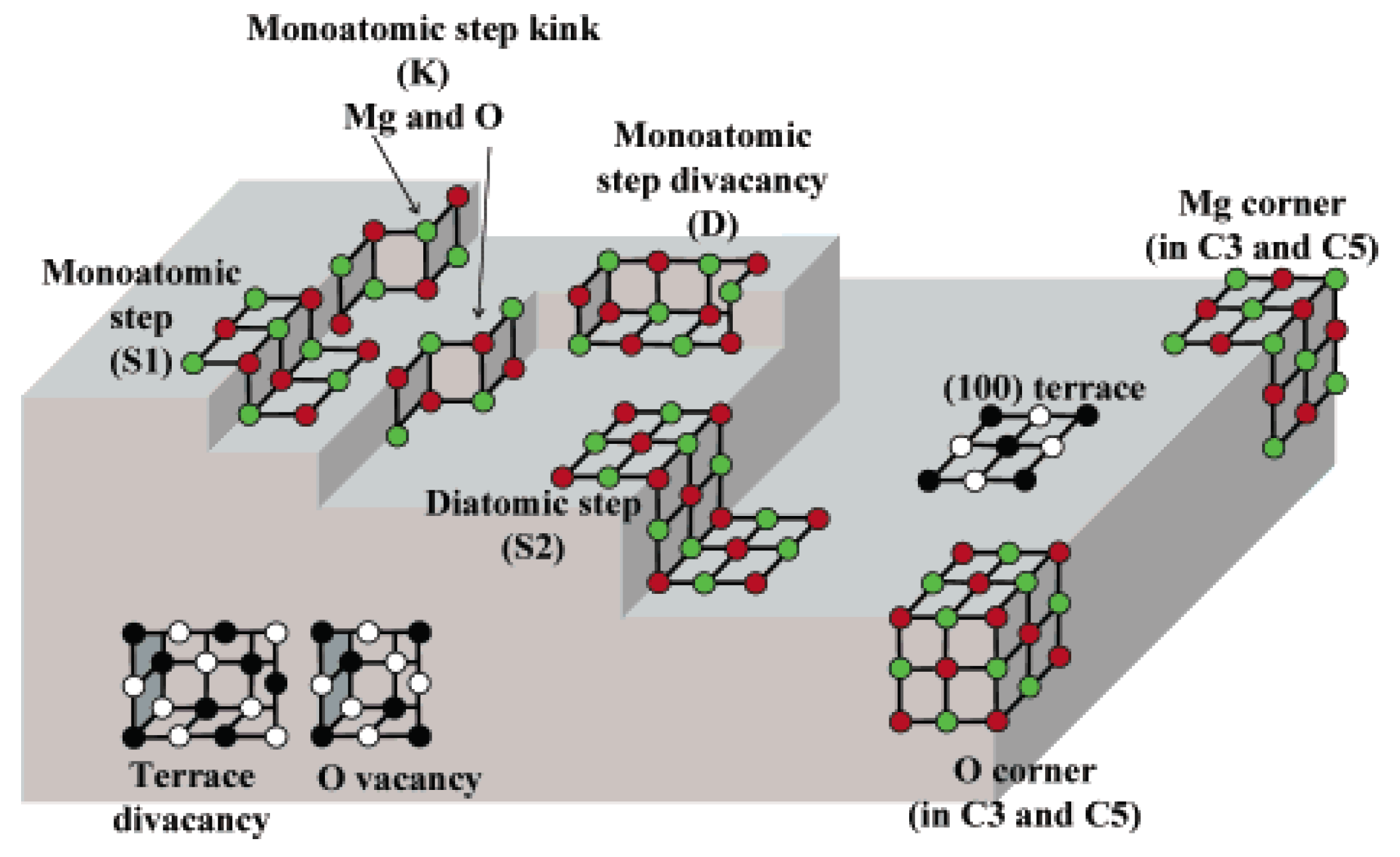
3. Structural Aspects of MxOy Oxides and Description of Active Sites in Some Partial Oxidation Reactions
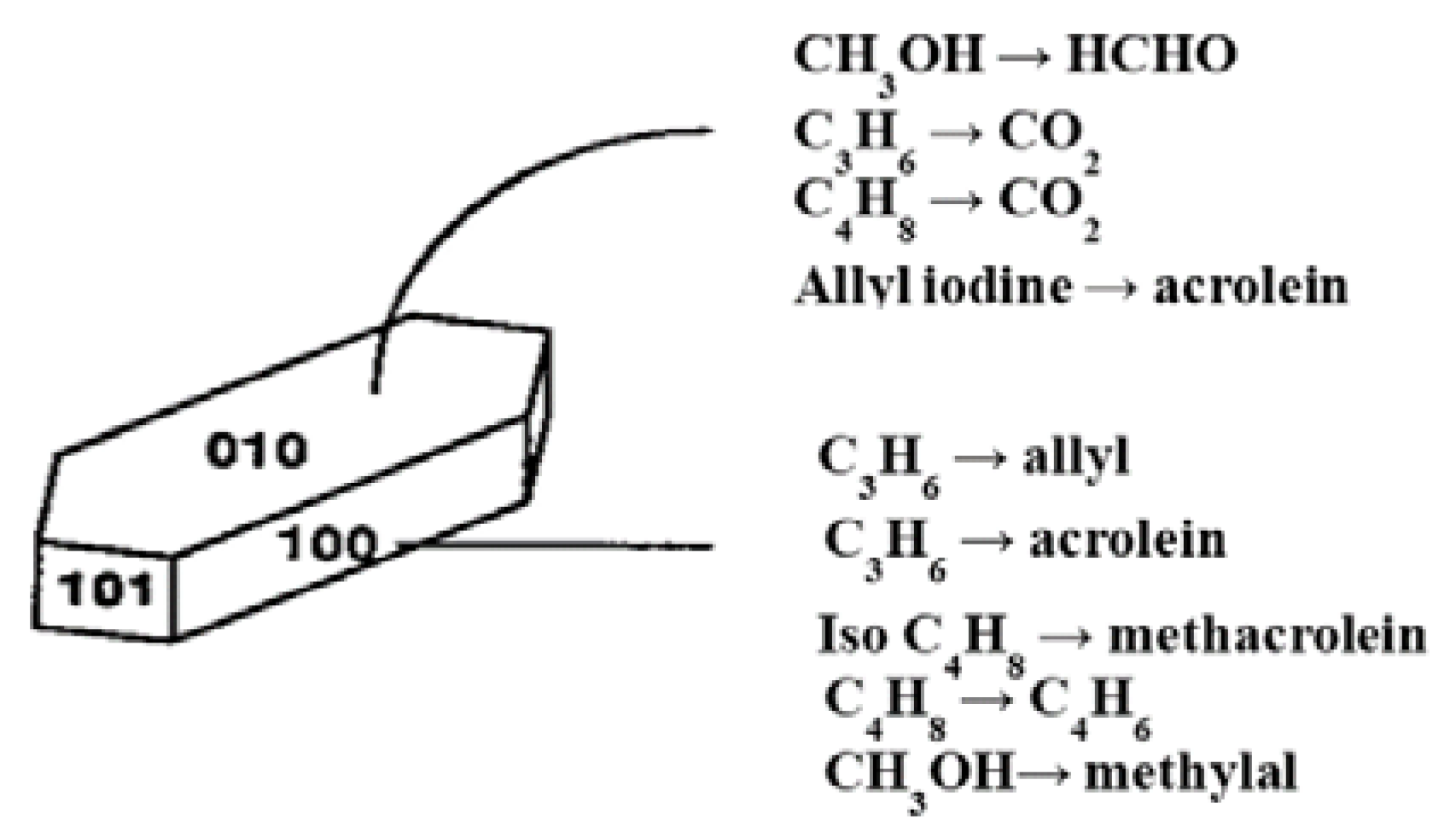
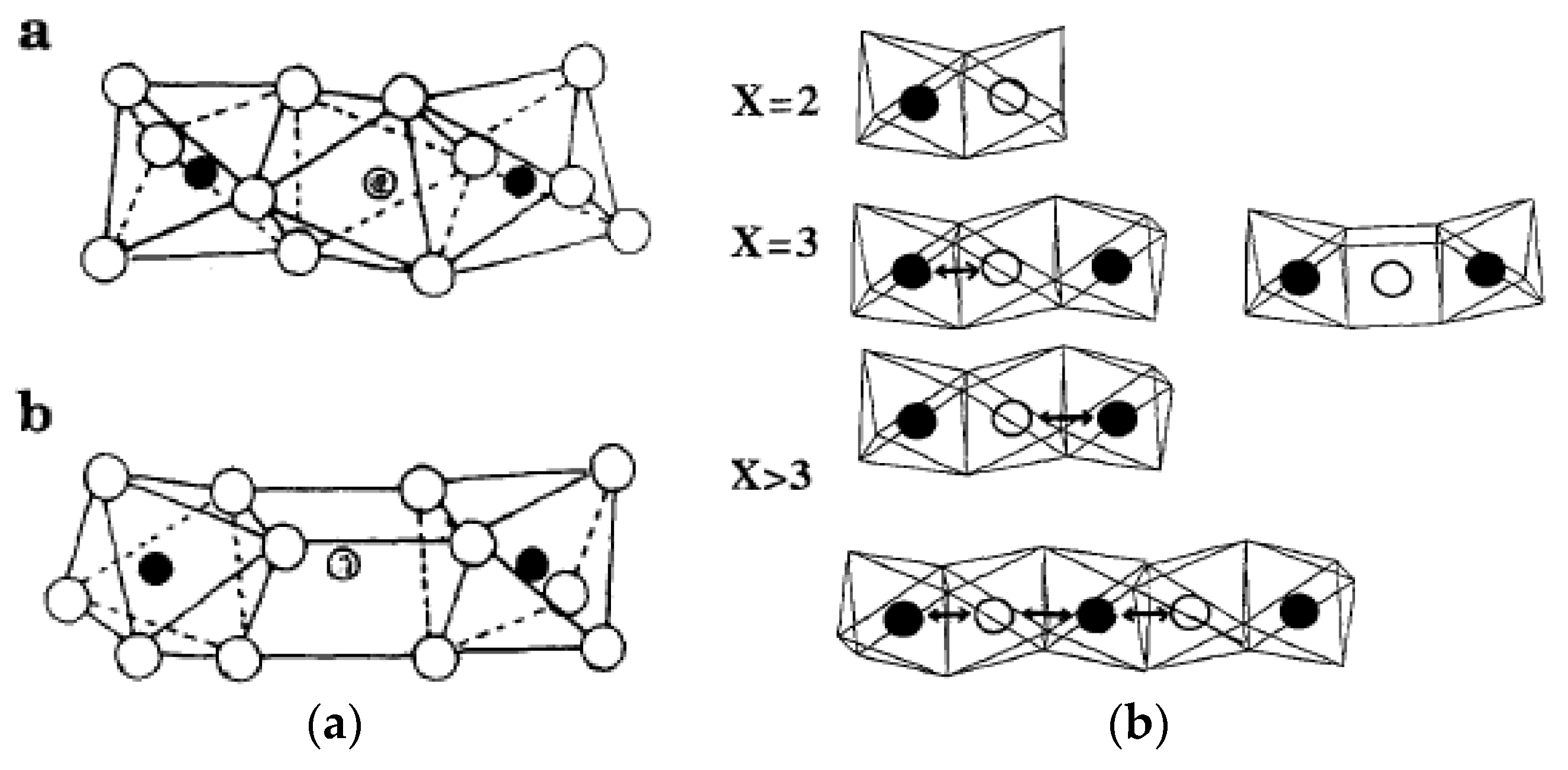
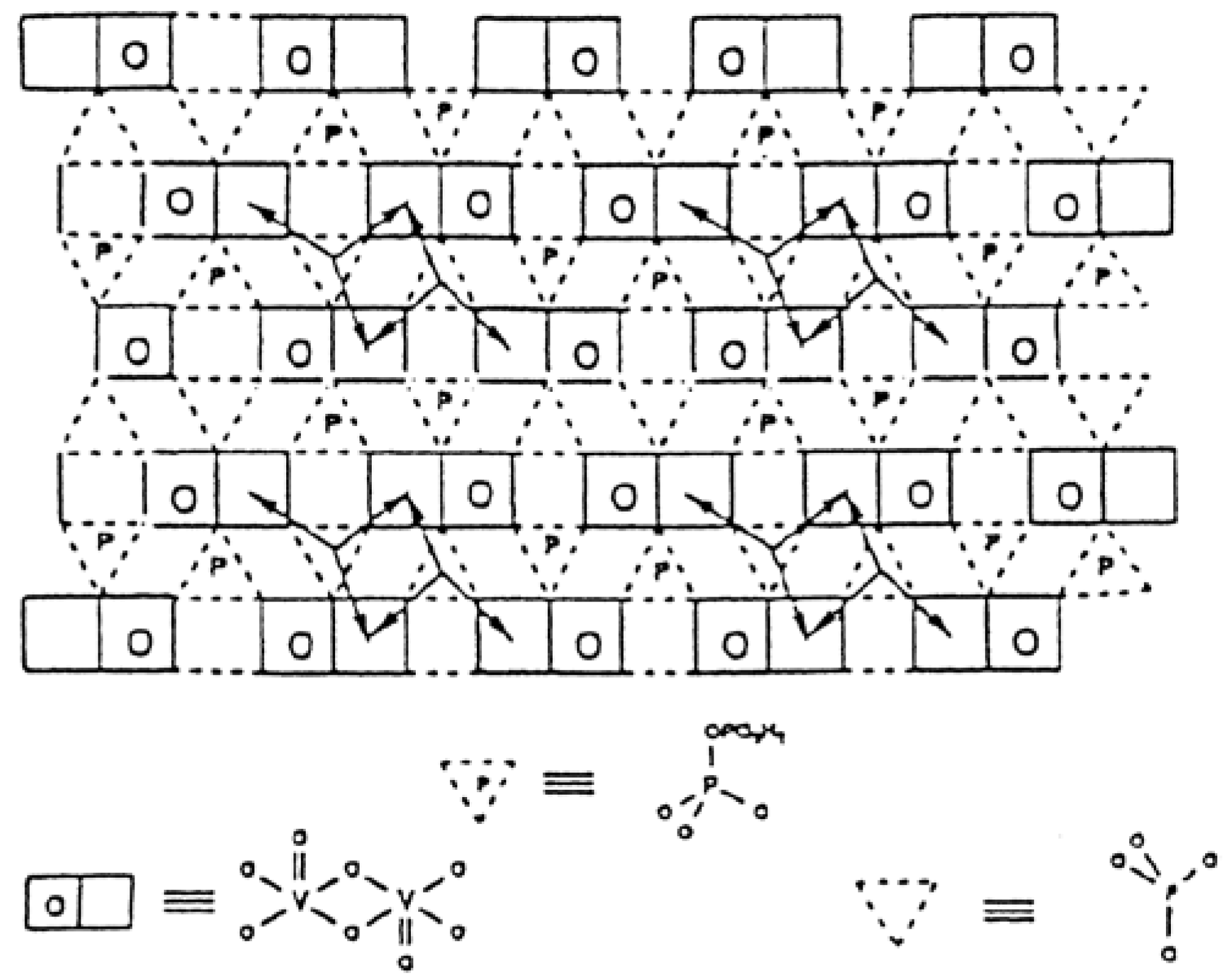
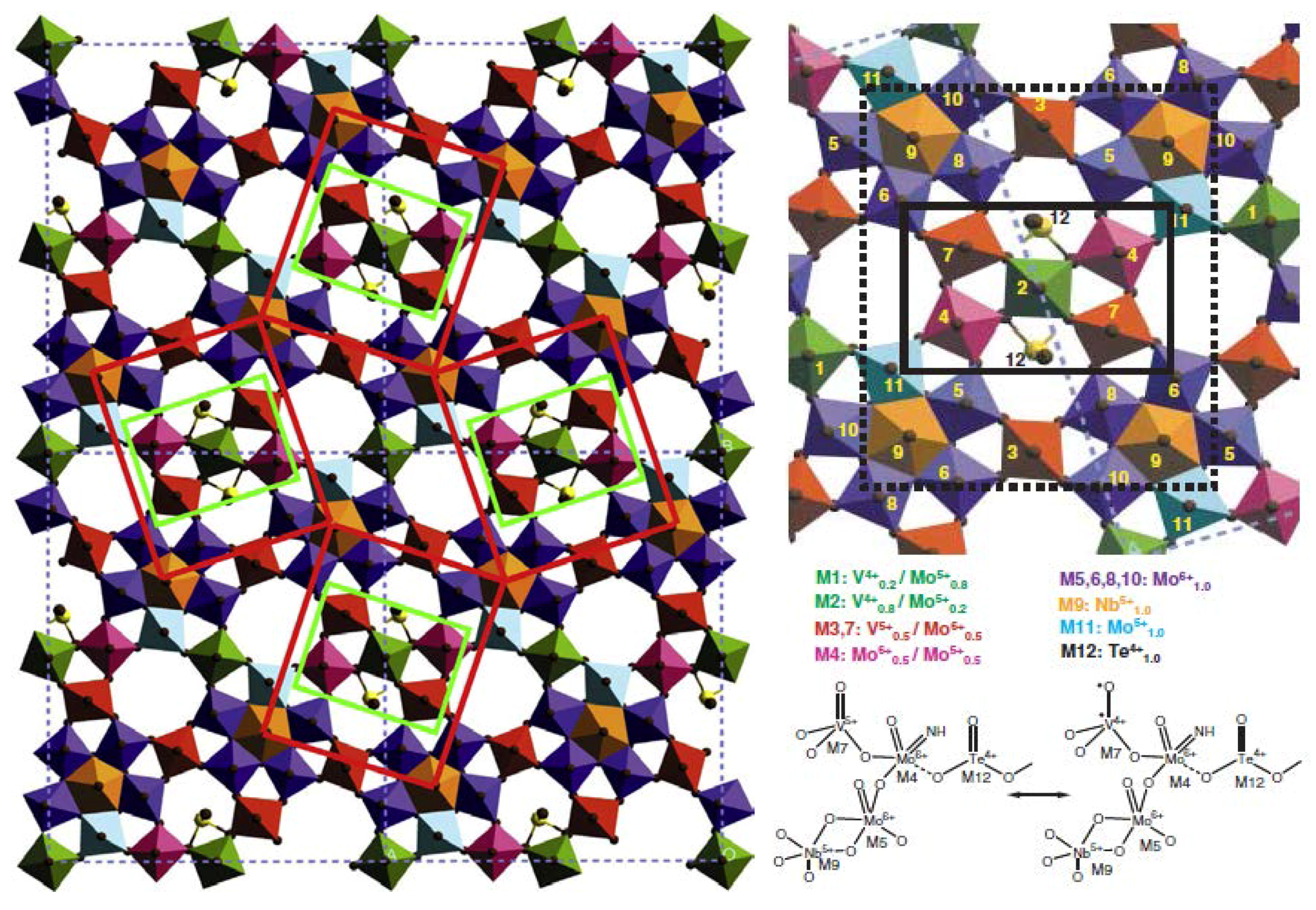
- Monomeric VO43− species exhibit an acid character and gives propene in isopropanol conversion, used as a test reaction, and total oxidation for o-xylene oxidation (coverage <0.43% in V2O5) as expected for acid sites;
- Polyvanadate species exhibit redox properties and give acetone in isopropanol conversion and phtalic anhydride in o-xylene conversion;
- V2O5 crystallites give low activity in o-xylene conversion and high selectivity in CO2.
4. Main Factors Acting in Partial Oxidation Reactions
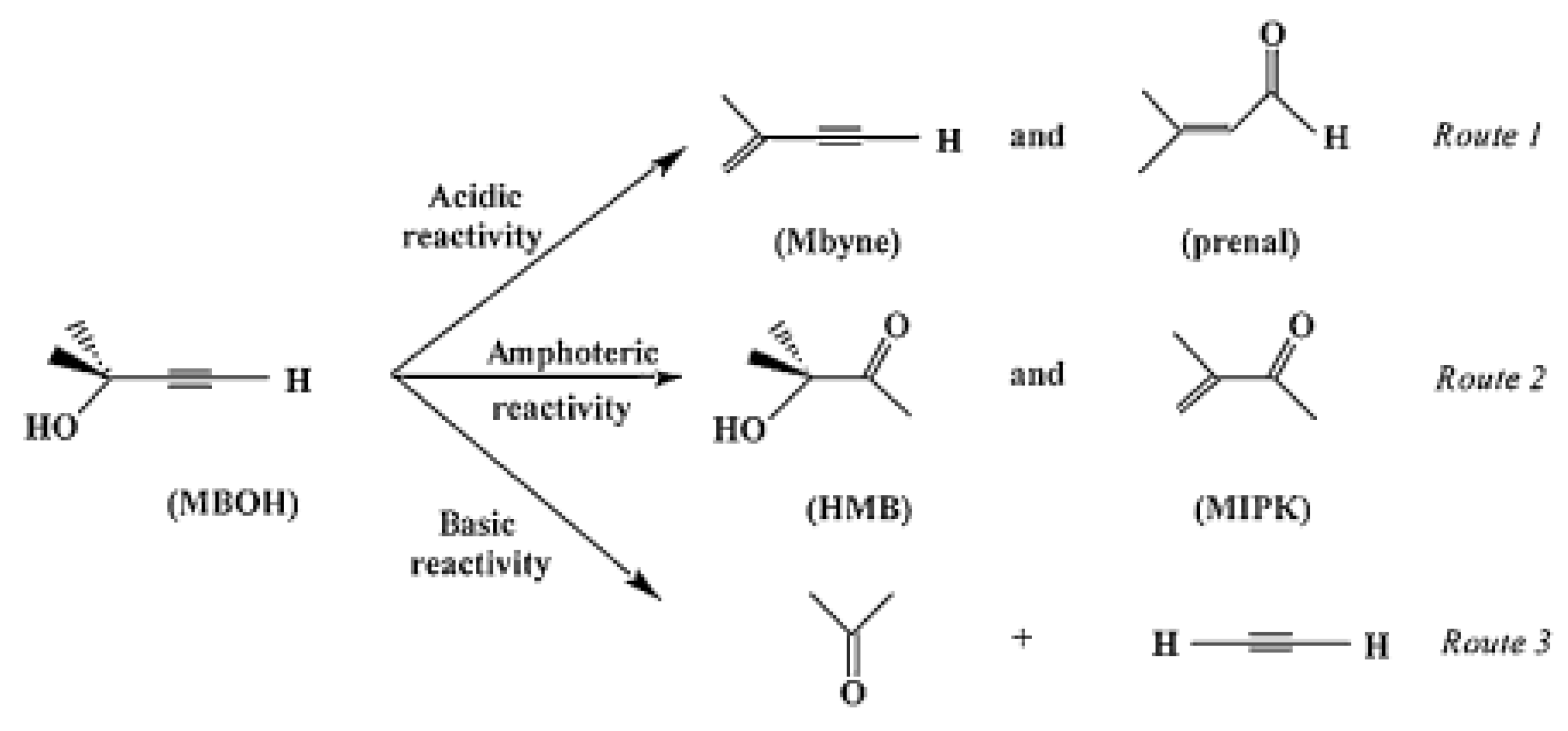
4.1. General Aspects of the Acid-Base Properties of Metal Oxides
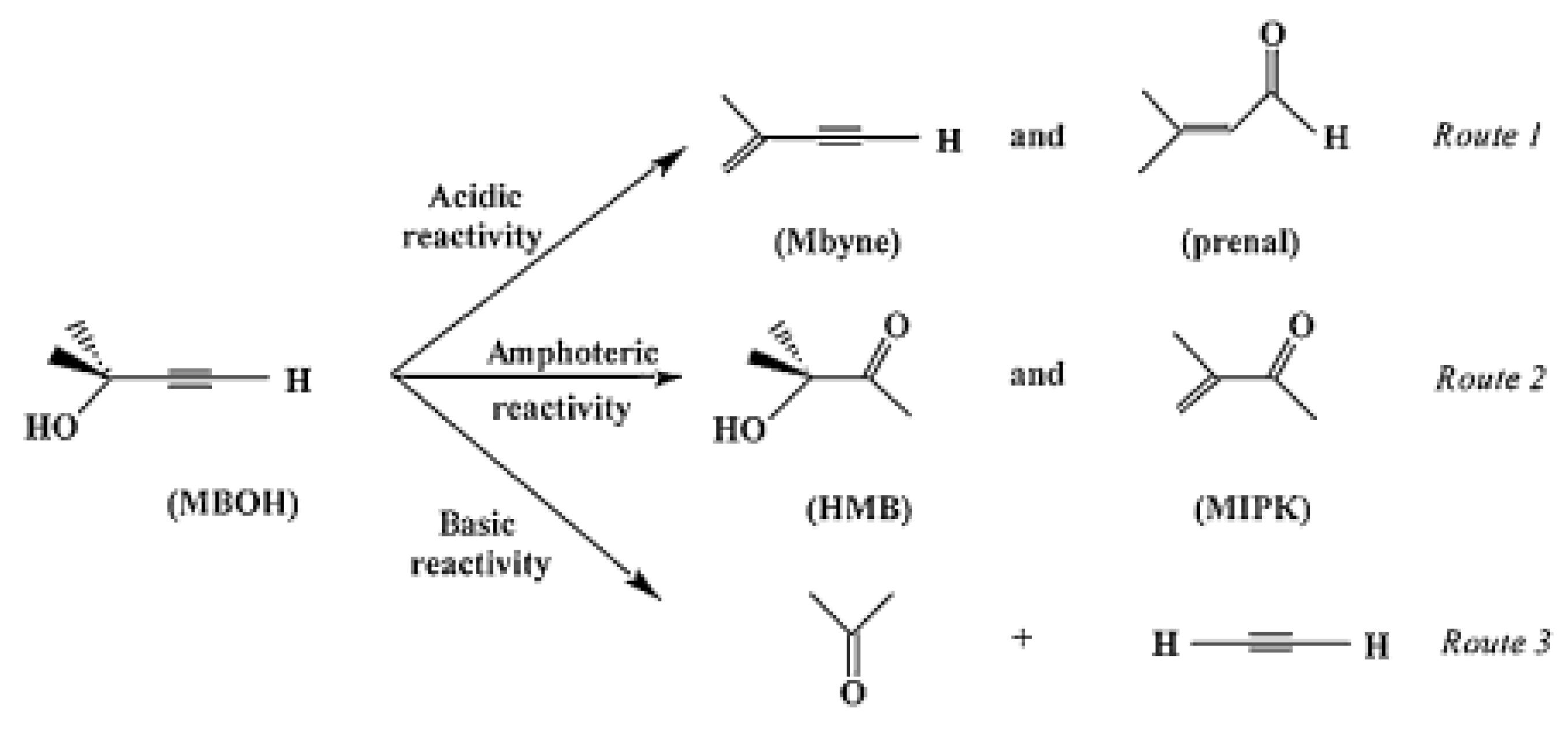
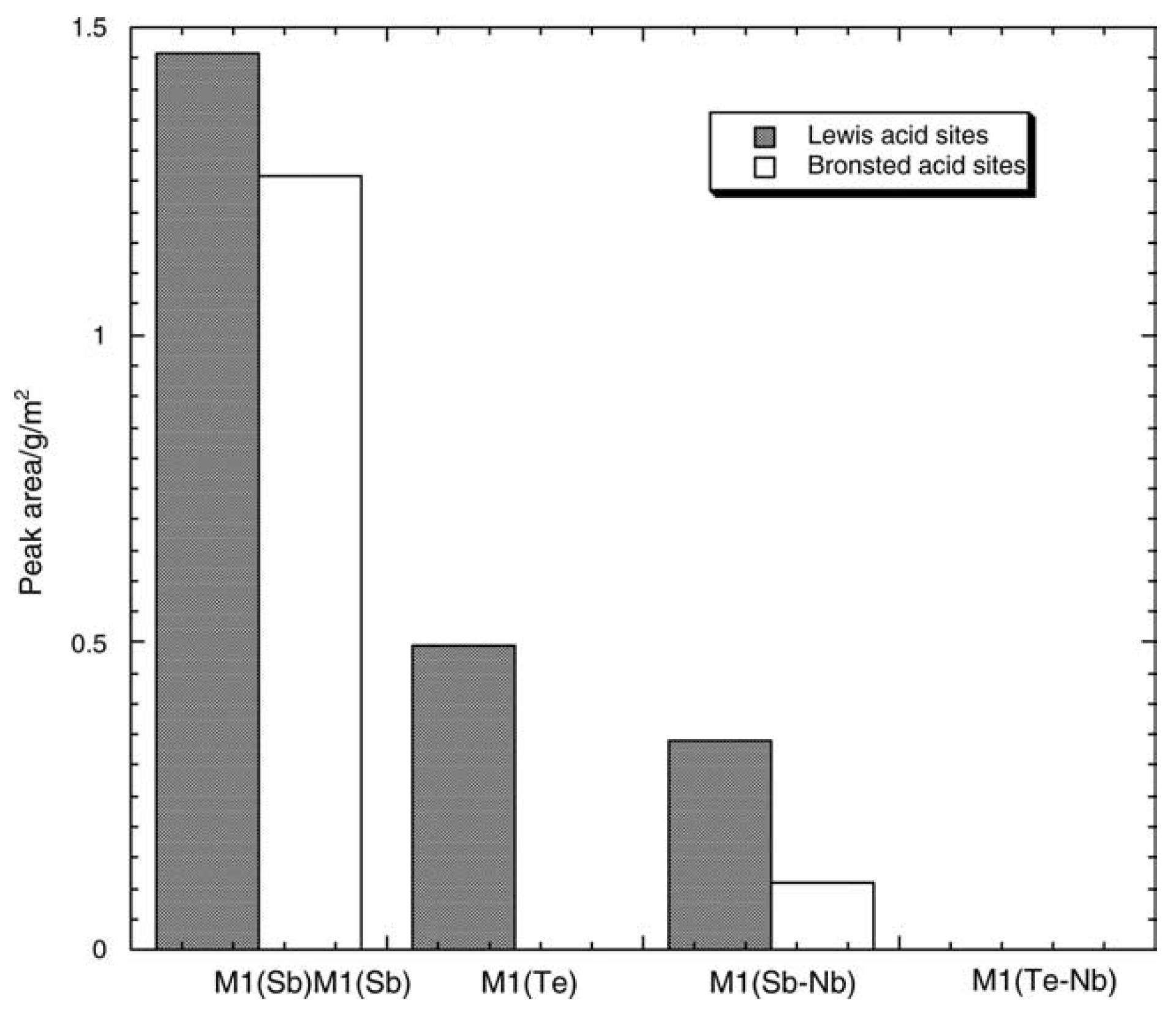
4.2. Importance of Electron Transfer Properties
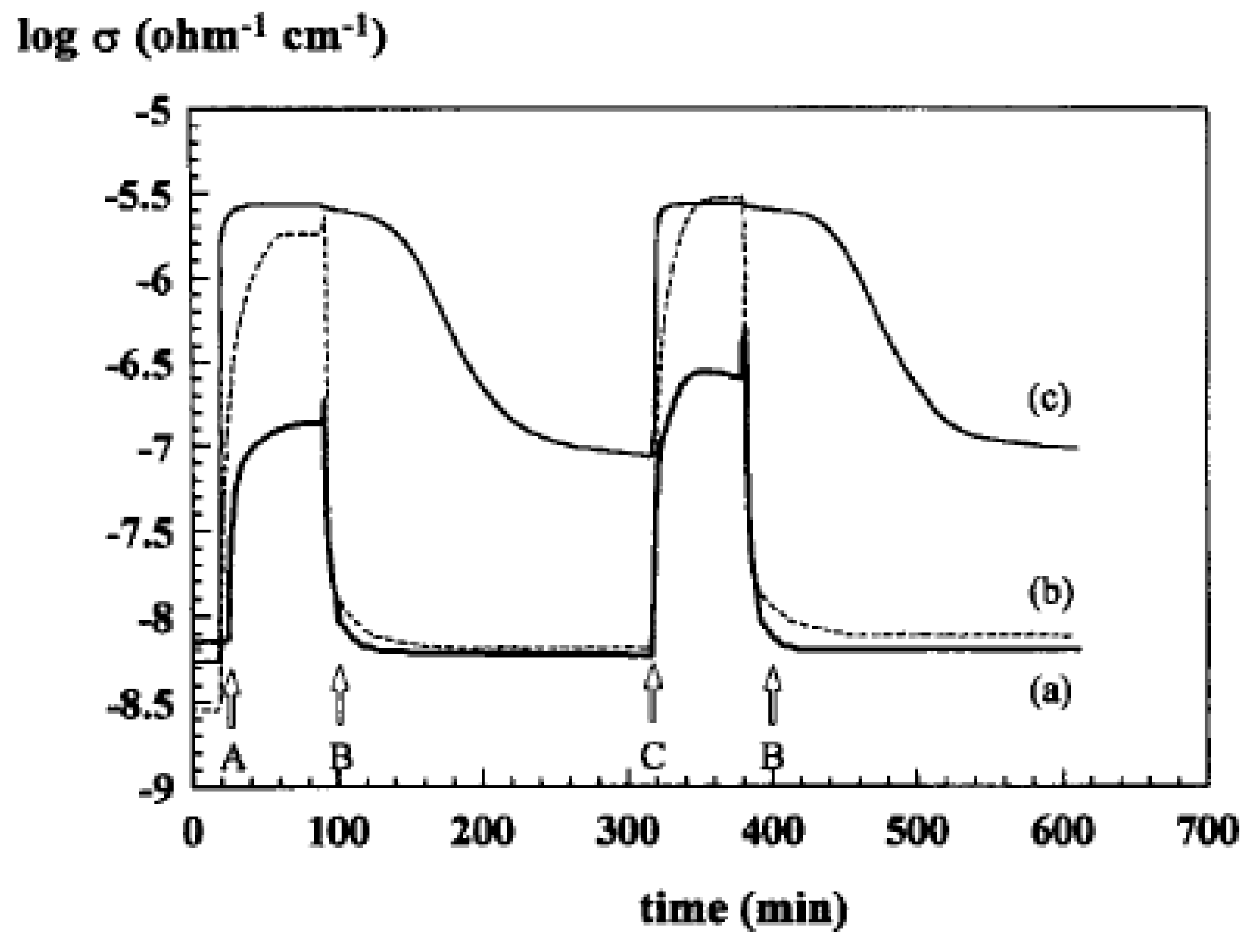
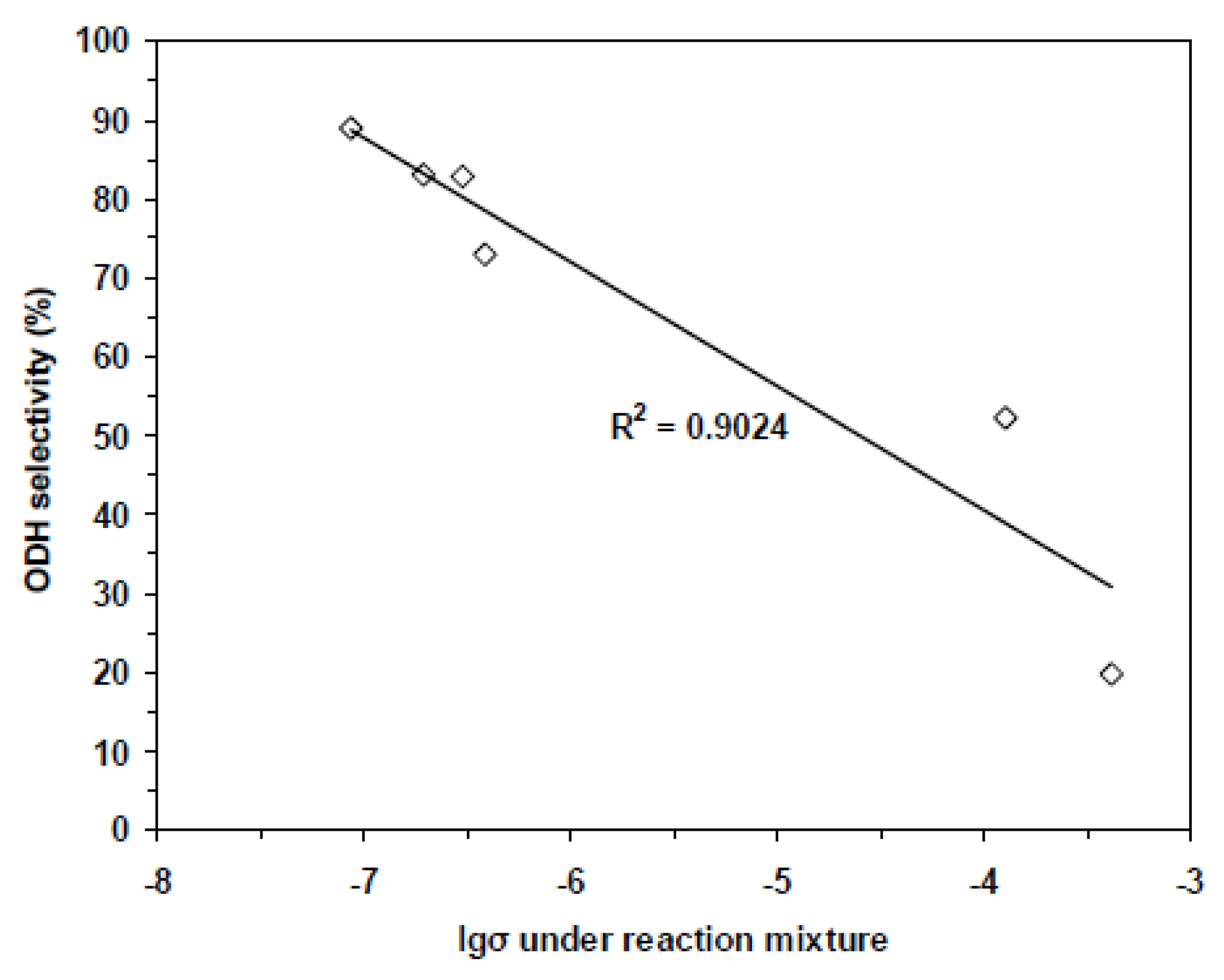
4.3. Synergy or Cooperation Effect in Mixtures of Phases [68,69]
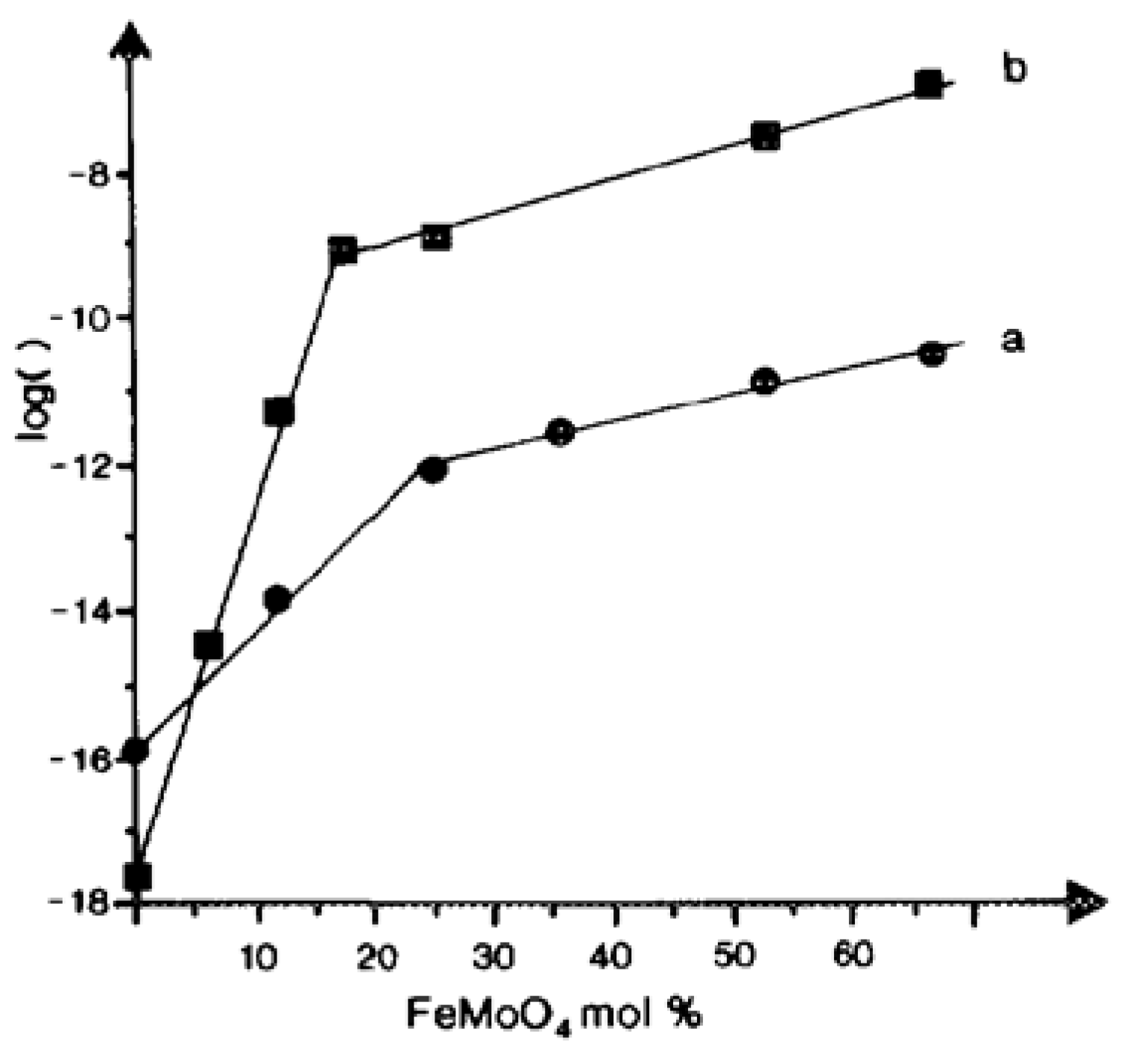
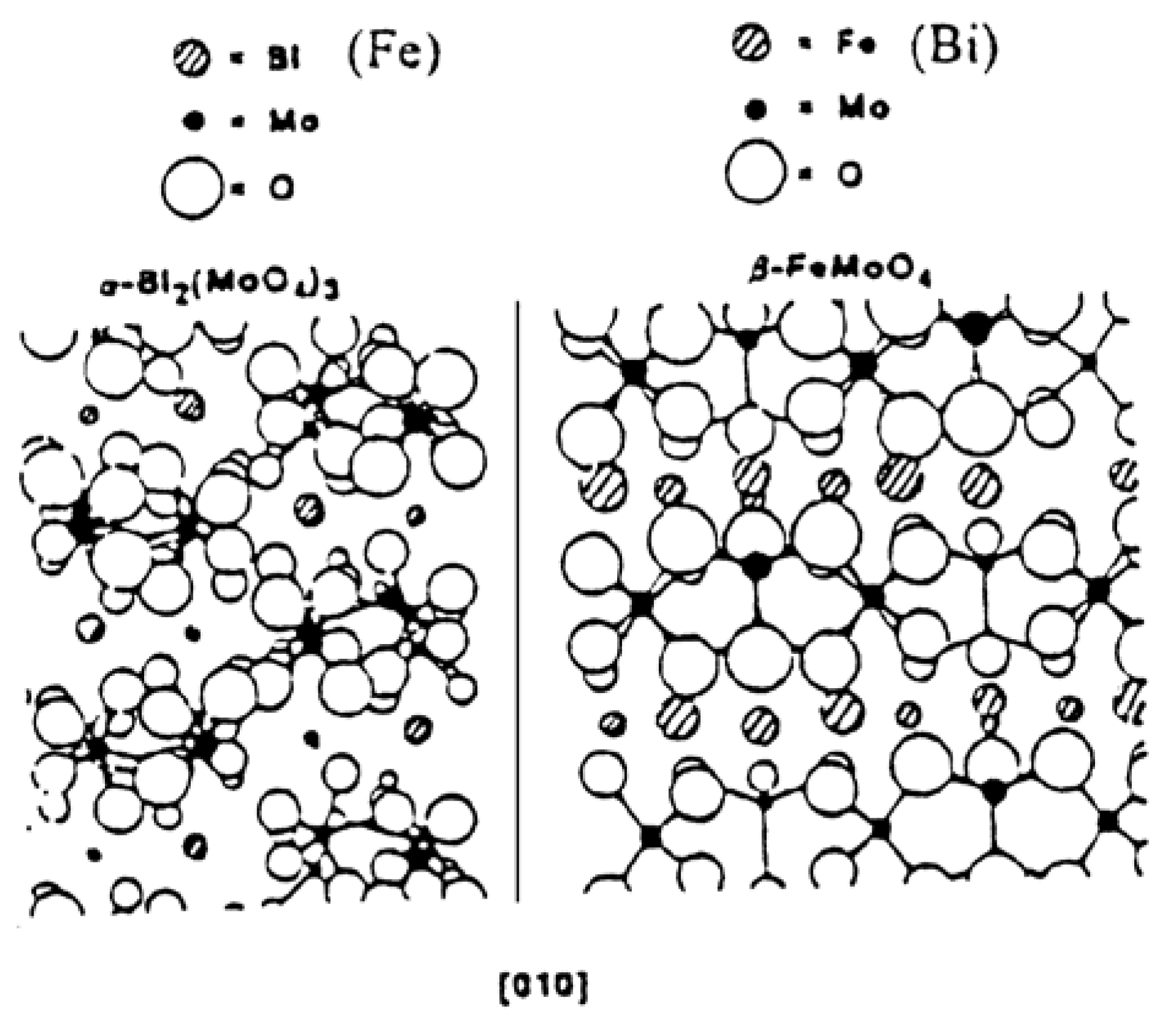
5. Changes in Catalyst Surface Structure and Chemical Composition during Catalytic Reaction
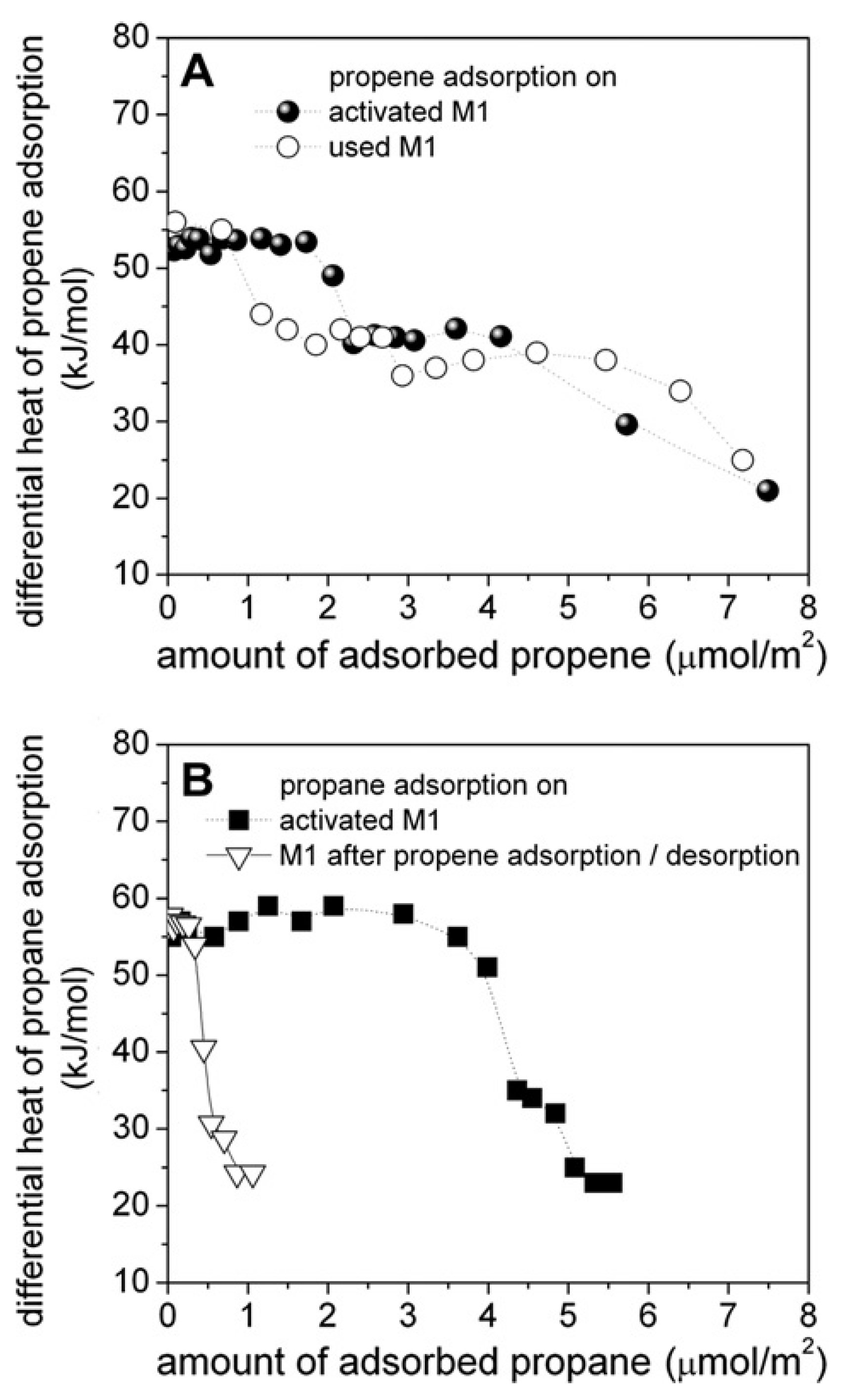
6. Dehydrogenation, Oxidative Dehydrogenation and Selective Oxidation of Light Paraffins
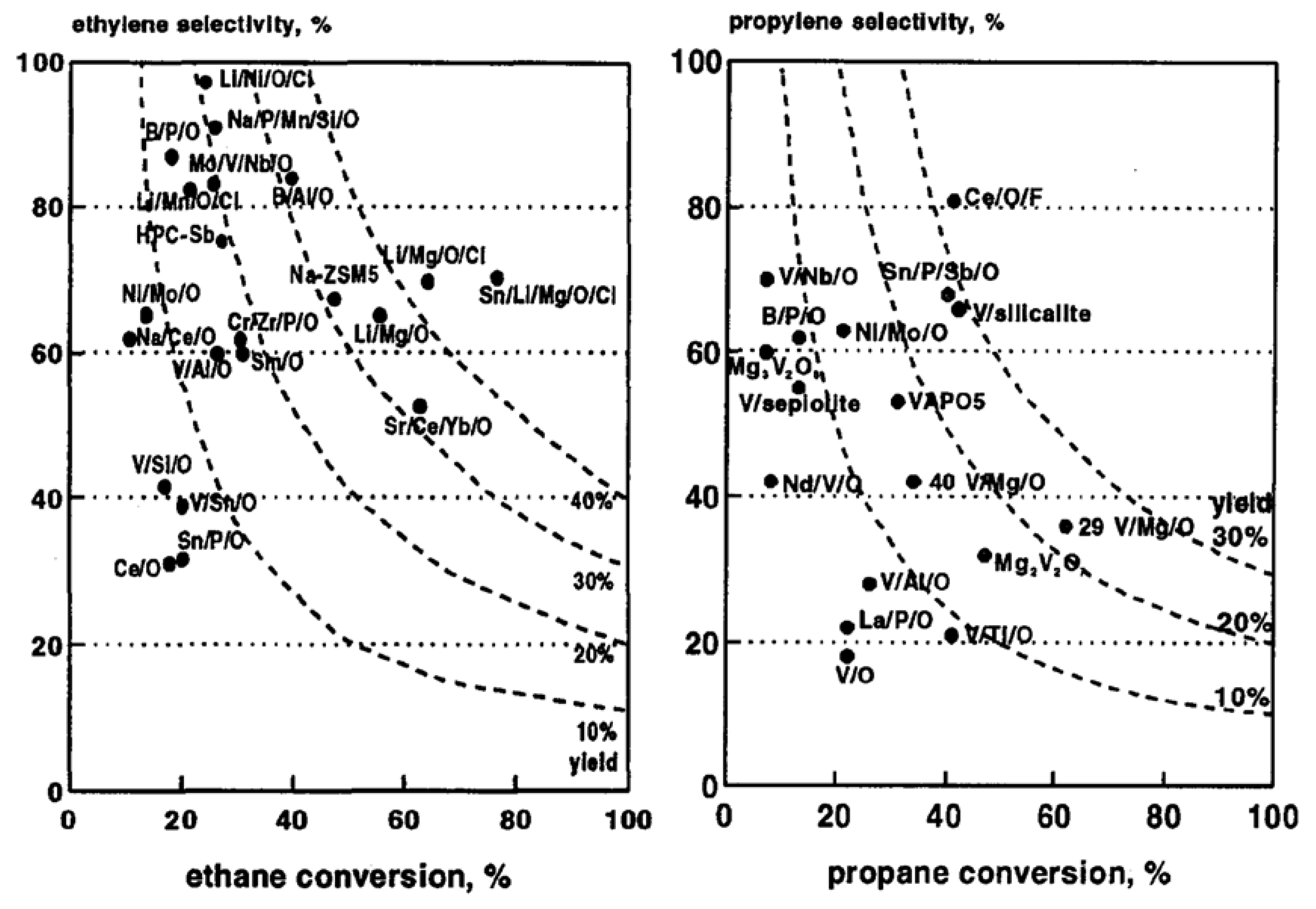
6.1. Case of Methane
6.2. Case of Ethane
6.3. Case of Propane
| Catalyst | T °C | Conversion % | Selectivity % | Yield % |
|---|---|---|---|---|
| Ethane → ethylene | ||||
| MoVTeNbO | 400 | 85 | 88 | 75 |
| NiNbO | 400 | 51 | 90 | 47 |
| VCoAPO-18 | 600 | 57 | 60 | 34 |
| Ethane → acetonitrile | ||||
| NbSbO/Al2O3 | 540 | 40 | 50 | 20 |
| Co-BEA | 476 | 47 | 57 | 26 |
| Ethane → acetic acid | ||||
| MoVSbNbReCaO | 277 | 14 | 78 | 11 |
| MoVNbPdOx | 300 | 5 | 82 | 4.1 |
| Propane → propylene | ||||
| VMgO | 540 | 62 | 38 | 24 |
| V-silicalite | 550 | 30 | 70 | 21 |
| Propane → acrylonitrile | ||||
| MoVTeNbO | 420 | 86 | 72 | 62 |
| VSbWOx/SiO2-Al2O3 | 500 | 67 | 60 | 40 |
| Propane → acrylic acid | ||||
| MoVTeNbO | 432 | 69 | 67 | 46 |
| MoVTeNbO A relevant | 380 | 80 | 61 | 49 |
6.4. Case of Butane
7. Role of Other Oxidants
8. General Conclusions and Perspectives
Conflicts of Interest
References
- Duprez, D.; Cavani, F. Handbook of Advanced Methods and Processes in Oxidation Catalysis; Imperial College Press: London, UK, 2014. [Google Scholar]
- Callahan, J.L.; Grasselli, R.K. A selectivity factor in vapor-phase hydrocarbon oxidation catalysis. AIChE J. 1963, 9, 755–760. [Google Scholar] [CrossRef]
- Ivars, F.; López Nieto, J.M. Light alkanes oxidation: Targets and current challenges. In Handbook of Advanced Methods and Processes in Oxidation Catalysis; Duprez, D., Cavani, F., Eds.; Imperial College Press: London, UK, 2014; pp. 767–834. [Google Scholar]
- Corma, A.; Garcia, H. Lewis acids as catalysts in oxidation reactions: From homogeneous to heterogeneous systems. Chem. Rev. 2002, 102, 3837–3892. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; van der Voort, P. Metal-organic frameworks as selective or chiral oxidation catalysts. Catal. Rev. Sci. Eng. 2014, 56, 1–56. [Google Scholar]
- Cavani, F.; Trifiro, F. The oxidative dehydrogenation of ethane and propane as an alternative way for the production of light olefins. Catal. Today 1995, 24, 307–313. [Google Scholar] [CrossRef]
- Védrine, J.C.; Millet, J.M.M. Heteropolyoxometallates catalysts for partial oxidation. In Metal Oxide Catalysis; Jackson, S.D., Hargreaves, J., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2009; Volume 2, pp. 561–594. [Google Scholar]
- Grasselli, R.K. Site isolation and phase cooperation: Two important concepts in selective oxidation catalysis: A retrospective. Catal. Today 2014, 238, 10–27. [Google Scholar] [CrossRef]
- Che, M.; Tench, A.J. Characterisation and reactivity of mononuclear oxygen species on oxide surfaces. Adv. Catal. 1982, 31, 77–133. [Google Scholar]
- Bielanski, A.; Haber, J. Oxygen in oxidation on transition metal oxides. Catal. Rev. Sci. Eng. 1979, 79, 1–31. [Google Scholar] [CrossRef]
- Haber, J.; Janas, J.; Schiavello, M.; Tilley, R.J.D. Tungsten oxides as catalysts in selective oxidation. J. Catal. 1983, 82, 395–403. [Google Scholar] [CrossRef]
- Bond, G.C.; Védrine, J.C. General Conclusions. Catal. Today 1994, 20, 171–178. [Google Scholar] [CrossRef]
- Hutchings, G.J. Vanadium phosphate: A new look at the active components of catalysts for the oxidation of butane to maleic anhydride. J. Mater. Chem. 2004, 14, 3385–3395. [Google Scholar] [CrossRef]
- Bordes-Richard, E.; Védrine, J.C. Catalyse sélective redox. Tech. Ingénieur 2013, 1215, 1–30. [Google Scholar]
- Grasselli, R.K.; Burrington, J.D. Selective oxidation and ammoxidation of propylene by heterogeneous catalysis. Adv. Catal. 1980, 30, 133–163. [Google Scholar]
- Mars, P.; van Krevelen, D.W. Oxidations carried out by means of vanadium oxide catalysts. Chem. Eng. Sci. 1954, 3, 41–49. [Google Scholar] [CrossRef]
- Vislovskiy, V.P.; Suleimanov, T.E.; Sinev, M.Y.; Tulenin, Yu.P.; Margolis, L.Y.; Cortés Corberán, V. On the role of heterogeneous and homogeneous processes in oxidative dehydrogenation of C3-C4 alkanes. Catal. Today 2000, 61, 287–293. [Google Scholar] [CrossRef]
- Germain, J.E. Catalytic Conversion of Hydrocarbons; Academic Press: New York, NY, USA, 1969. [Google Scholar]
- Bilde, J.; Janke, C.; Lorentz, C.; Delichère, P.; Popescu, I.; Marcu, I.C.; Loridant, S.; Brückner, A.; Millet, J.M.M. Molecular level insights into the structure of active sites of VAlO mixed oxides in propane ammoxidation. J. Phys. Chem. C 2013, 117, 22926–22938. [Google Scholar] [CrossRef]
- Jussola, J.A.; Mann, R.F.; Downie, J. The kinetics of the vapor-phase oxidation of o-xylene over a vanadium oxide catalyst. J. Catal. 1970, 17, 103–106. [Google Scholar] [CrossRef]
- Bordes, E. The role of structural chemistry of selective catalysts in heterogeneous mild oxidation of hydrocarbons. C. R. Acad. Sci. Ser. IIc: Chem. 2000, 2, 725–733. [Google Scholar] [CrossRef]
- Boudart, M. Effect of surface structure on catalytic activity. In Proceedings of the 6th International Congress on Catalysis, London, UK, 12–16 July 1976; Bond, G.C., Wells, P.B., Tompkins, F.C., Eds.; The Chemical Society: London, UK, 1977; pp. 1–9. [Google Scholar]
- Volta, J.C.; Desquesnes, W.; Moraweck, B.; Coudurier, G. A new method to obtain supported oriented oxides: MoO3 graphite catalyst for propylene oxidation to acrolein. Kinet. Catal. Lett. 1979, 12, 241–245. [Google Scholar] [CrossRef]
- Volta, J.C.; Tatibouët, J.M.; Phichitkul, C.; Germain, J.E. Structural-sensitive catalytic oxidation on MoO3 catalysts. In Proceedings of the 8th International Congress on Catalysis, Berlin, Germany, 2–6 July 1984; Verlag Chemie: Frankfurt am Main, Germany, 1984; Volume 4, pp. 451–458. [Google Scholar]
- Tatibouët, J.M.; Phichitkul, C.; Germain, J.E. Structure-sensitive catalytic oxidation of butenes on molybdenum trioxide crystallites. J. Catal. 1986, 99, 231–238. [Google Scholar] [CrossRef]
- Volta, J.C.; Portefaix, J.L. Structure sensitivity of mild oxidation reactions on oxide catalysts: A review. Appl. Catal. 1985, 18, 1–32. [Google Scholar] [CrossRef]
- Tatibouët, J.M.; Germain, J.E.; Volta, J.C. Structure-sensitive catalytic oxidation: Alcohols on graphite-supported molybdenum trioxide. J. Catal. 1983, 82, 240–248. [Google Scholar] [CrossRef]
- Haber, J. The concept of structure sensitivity in catalysis by oxides. Stud. Surf. Sci. Catal. 1989, 48, 447–467. [Google Scholar]
- Ziolkowski, J.; Bordes, E.; Courtine, P. Oxidation of butane and butene on the (100) face of (VO)2P2O7: A dynamic view in terms of crystallochemical model of active sites. J. Catal. 1990, 122, 126–150. [Google Scholar] [CrossRef]
- Ueda, W.; Oshihara, K. Selective oxidation of light alkanes over hydrothermally synthesized Mo–V–M–O (M = Al, Ga, Bi, Sb, and Te) oxide catalysts. Appl. Catal. A Gen. 2000, 200, 135–143. [Google Scholar] [CrossRef]
- Guliants, V.V.; Brongersma, H.H.; Knoester, A.; Gaffney, A.M.; Han, S. Surface active sites present in the orthorhombic M1 phases: Low energy ion scattering study of methanol and allyl alcohol chemisorption over Mo–V–Te–Nb–O and Mo–V–O catalysts. Top. Catal. 2006, 38, 41–50. [Google Scholar] [CrossRef]
- Celaya Sanfiz, A.; Hansen, T.W.; Sakthivel, A.; Trunschke, A.; Schlögl, R.; Knoester, A.; Brongersma, H.H.; Looi, M.H.; Hamid, S.B.A. How important is the (001) plane of M1 for selective oxidation of propane to acrylic acid? J. Catal. 2008, 258, 35–43. [Google Scholar] [CrossRef]
- Taylor, H.S. A theory of the catalytic surface. Proc. R. Soc. Lond. 1925, A108, 105–111. [Google Scholar] [CrossRef]
- Védrine, J.C. Revisiting active sites in heterogeneous catalysis: Their structure and their dynamic behaviour. Appl. Catal. A Gen. 2014, 474, 40–50. [Google Scholar] [CrossRef]
- Chizallet, C.; Costentin, G.; Che, M.; Delbecq, F.; Sautet, P. Revisiting acido-basicity of the MgO surface by periodic density functional theory calculations: role of surface topology and ion coordination on water dissociation. J. Phys. Chem. B 2006, 110, 15878–15886. [Google Scholar] [CrossRef] [PubMed]
- Védrine, J.C.; Coudurier, G.; Millet, J.M.M. Molecular design of active sites in partial oxidation reactions on metallic oxides. Catal. Today 1997, 33, 3–13. [Google Scholar] [CrossRef]
- Millet, J.M.M. FePO catalysts for the selective oxidative dehydrogenation of isobutyric acid into methacrylic acid. Catal. Rev. Sci. Eng. 1998, 40, 1–38. [Google Scholar] [CrossRef]
- Robert, V.; Borshch, S.A.; Bigot, B. Molecular orbital model of electron delocalization in mixed-valence trimeric clusters. J. Phys. Chem. 1996, 100, 580–584. [Google Scholar] [CrossRef]
- Haynes, A.; Maitlis, P.M.; Morris, G.E.; Sunley, G.J.; Adams, H.; Badger, P.W.; Bowers, C.M.; Cook, D.B.; Elliott, P.I.P.; Ghaffar, T.; et al. Promotion of Iridium-Catalyzed Methanol Carbonylation: Mechanistic Studies of the Cativa Process. J. Am. Chem. Soc. 2004, 126, 2847–2861. [Google Scholar] [PubMed]
- Agashar, P.A.; de Caul, L.; Grasselli, R.K. A molecular level mechanism of n-butane oxidation to maleic anhydride over vanadyl pyrophosphate. Catal. Lett. 1994, 23, 339–346. [Google Scholar] [CrossRef]
- Panov, G.I.; Kharitonov, A.S.; Sobolev, V.I. Oxidative hydroxylation using dinitrogen monoxide: A possible route for organic synthesis over zeolites. Appl. Catal. A 1993, 98, 1–20. [Google Scholar] [CrossRef]
- Janas, J.; Shishido, T.; Che, M.; Dzwigaj, S. Role of tetrahedral Co(II) sites of CoSiBEA zeolite in the selective catalytic reduction of NO: XRD, UV-vis, XAS and catalysis study. Appl. Catal. B 2009, 89, 196–203. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Haruta, M.; Shen, W. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 2009, 458, 746–749. [Google Scholar]
- Ogliaro, F.; Harris, N.; Cohen, S.; Filatov, M.; de Visser, S.P.; Saik, S. A model “rebound” mechanism of hydroxylation by cytochrome P450: Stepwise and effectively concerned pathways and their reactivity patterns. J. Am. Chem. Soc. 2000, 122, 8977–8989. [Google Scholar] [CrossRef]
- Afanasiev, P.; Kudrik, E.V.; Millet, J.M.M.; Bouchu, D.; Sorokin, A. High-valent di iron species generated from N-bridged diiron phthalocyanine and H2O2. J. Chem. Soc. Dalton Trans. 2011, 40, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Kudrik, E.V.; Afanasiev, P.; Alvarez, L.X.; Dubourdeaux, P.; Clémancey, M.; Latour, J.M.; Blondin, G.; Bouchu, D.; Albrieux, F.; Nefedov, S.E.; et al. An N-bridged high-valent diiron-oxo species on a porphyrin platform that can oxidize methane. Nat. Chem. 2012, 4, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Ushikubo, T.; Oshima, K.; Kayou, A.; Vaarkamp, M.; Hatano, M. Ammoxidation of propane over catalysts comprising mixed oxides of Mo and V. J. Catal. 1997, 169, 394–396. [Google Scholar] [CrossRef]
- Oshama, K.; Kayo, A.; Umezawa, T.K.; Kiyono, K.; Sawaki, I. Synthesis of MoVTe(Sb)-O composite oxide catalyst via reduction of polyoxometalates in an aqueous medium. EP 529 853, 1992. [Google Scholar]
- Thorsteinson, E.M.; Wilson, T.P.; Young, F.G.; Kasai, P.H. The oxidative dehydrogenation of ethane over catalysts containing mixed oxides of molybdenum and vanadium. J. Catal. 1978, 52, 116–132. [Google Scholar] [CrossRef]
- Botella, P.E.; García-González, E.; López Nieto, J.M.; González-Calbet, J.M. MoVTeNbO multifunctional catalysts: Correlation between constituent crystalline phases and catalytic performance. Solid State Sci. 2005, 7, 507–519. [Google Scholar] [CrossRef]
- Schlögl, R. Active sites for propane oxidation: Some generic considerations. Top. Catal. 2011, 54, 627–638. [Google Scholar] [CrossRef]
- Grasselli, R.K.; Burrington, J.D.; Buttrey, D.J.; de Santo, P., Jr.; Lugmair, C.G.; Volpe, A.F., Jr.; Weingand, T. Multifunctionality of active centers in (amm)oxidation catalysts: From Bi–Mo–Ox to Mo–V–Nb–(Te, Sb)–Ox. Top. Catal. 2003, 23, 5–22. [Google Scholar] [CrossRef]
- Védrine, J.C. Eurocat oxide V2O5/TiO2. Catal Today 1994, 20, 1–178. [Google Scholar]
- Védrine, J.C. The role of redox, acid-base and collective properties and of crystalline state of heterogeneous catalysts in the selective oxidation of hydrocarbons. Top. Catal. 2002, 21, 97–106. [Google Scholar] [CrossRef]
- Tielens, F.; Dzwigaj, S. Probing acid-base sites in vanadium redox zeolites by DFT calculation and compared with FTIR results. Catal. Today 2010, 152, 66–69. [Google Scholar] [CrossRef]
- Védrine, J.C. Acid-base characterization of heterogeneous catalysts: An up-to-date overview. Res. Chem. Intermed. 2015. [Google Scholar] [CrossRef]
- Noller, H.; Tomke, K. Transition states of catalytic dehydration and dehydrogenation of alcohols. J. Mol. Catal. 1979, 6, 375–392. [Google Scholar] [CrossRef]
- Lauron-Pernot, H. Evaluation of surface acido-basic properties of inorganic-based solids by model alcohol catalytic reaction networks. Catal. Rev. Sci. Eng. 2006, 348, 315–361. [Google Scholar] [CrossRef]
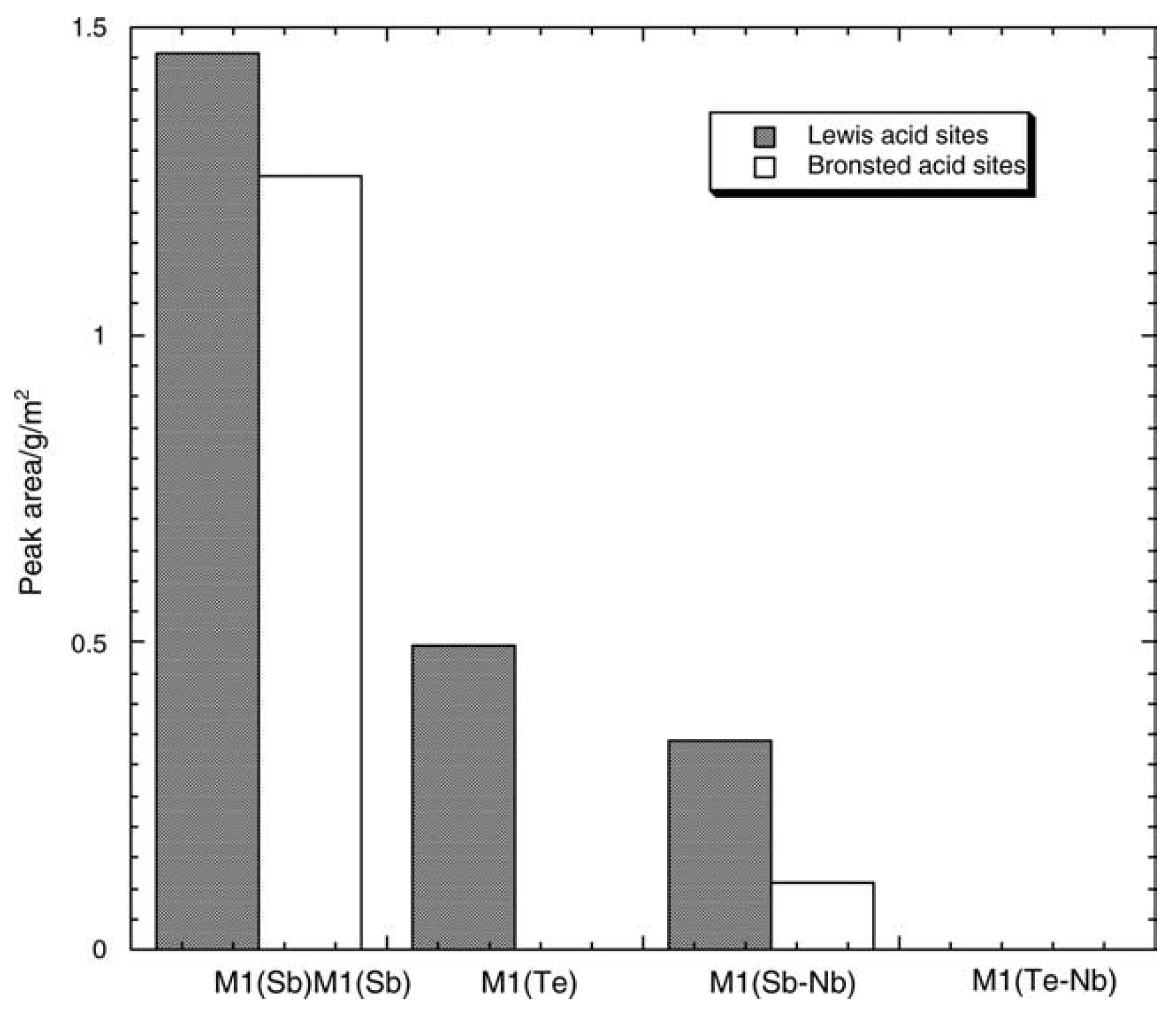
- Baca, M.; Pigamo, A.; Dubois, J.L.; Millet, J.M.M. Fourier transform infrared spectroscopic study of surface acidity by pyridine adsorption on the M1 active phase of the MoVTe(Sb)NbO catalysts used in propane oxidation. Catal. Commun. 2005, 6, 215–220. [Google Scholar] [CrossRef]
- Ivars-Barceló, F.; Millet, J.M.M.; Blasco, T.; Concepción, P.; Valente, J.S.; López Nieto, J.M. Understanding effects of activation-treatments in K-free and K-MoVSbO bronze catalysts for propane partial oxidation. Catal. Today 2014, 238, 41–48. [Google Scholar] [CrossRef]
- Grasselli, R.K.; Lugmair, C.G.; Volpe, A.E.; Andersson, A.; Burrington, J.D. Enhancement of acrylic acid yields in propane and propylene oxidation by selective P doping of MoV(Nb)TeO-based M1 and M2 catalysts. Catal. Today 2010, 157, 33–38. [Google Scholar] [CrossRef]
- Jo, B.Y.; Kum, S.S.; Moon, S.H. Performance of WOx-added Mo–V–Te–Nb–O catalysts in the partial oxidation of propane to acrylic acid. Appl. Catal. A 2010, 378, 76–82. [Google Scholar] [CrossRef]
- Andrushkevich, T.V.; Popova, G.Y.; Chesalov, Y.A.; Ischenko, E.V.; Khramov, M.I.; Kaichev, V.V. Propane ammoxidation on Bi promoted MoVTeNbOx oxide catalysts: Effect of mixture composition. Appl. Catal. A 2015, 506, 109–117. [Google Scholar] [CrossRef]
- Novakova, E.N.; Védrine, J.C. Propane selective oxidation to propene and oxygenates on metal oxides. In Metal Oxides: Chemistry and Applications; Fierro, J.L.G., Ed.; CRC: Boca Raton, FL, USA; Taylor & Francis: Baton Rouge, LA, USA, 2006; pp. 444–446. [Google Scholar]
- Pantazidis, A.; Burrows, A.; Kiely, C.J.; Mirodatos, C. Direct evidence of active surface reconstruction during oxidative dehydrogenation of propane over VMgO catalyst. J. Catal. 1998, 177, 325–334. [Google Scholar] [CrossRef]
- Popescu, I.; Skoufa, Z.; Heracleous, E.; Lemonidou, A.; Marcu, I.C. A study of electrical conductivity measurements of the semi conductive and redox properties of Nb-doped NiO catalysts in correlation with the oxidative dehydrogenation of ethane. Phys. Chem. Chem. Phys. 2015, 17, 8138–8147. [Google Scholar] [CrossRef] [PubMed]
- Courtine, P.; Bordes, E. Synergistic effects in multicomponent catalysts for selective oxidation. Stud. Surf. Sci. Catal. 1997, 110, 177–184. [Google Scholar]
- Bordes, E. Synergistic effects in selective oxidation catalysis: Does phase cooperation result in site isolation? Top. Catal. 2001, 15, 131–137. [Google Scholar] [CrossRef]
- Carson, D.; Coudurier, G.; Védrine, J.C.; Laarif, A.; Theobald, F. Synergy effects in the catalytic properties of Bi molybdate. J. Chem. Soc. Faraday Trans. I 1983, 79, 1921–1929. [Google Scholar] [CrossRef]
- Ponceblanc, H.; Millet, J.M.M.; Coudurier, G.; Herrmann, J.M.; Védrine, J.C. Study of multiphasic molybdates based catalysts. Part I—Electrical conductivity study of valence states and solubility limits in mixed iron and cobalt molybdates. J. Catal. 1993, 142, 373–380. [Google Scholar] [CrossRef]
- Millet, J.M.M.; Ponceblanc, H.; Coudurier, G.; Herrmann, J.M.; Védrine, J.C. Study of multiphasic molybdates based catalysts. Part II—Synergy effect between bismuth and mixed iron and cobalt molybdates in mild oxidation of propene. J. Catal. 1993, 142, 381–391. [Google Scholar] [CrossRef]
- Wainwright, M.S.; Foster, N.R. Catalysts, Kinetics and Reactor Design in Phthalic Anhydride Synthesis. Catal. Rev. Sci. Eng. 1979, 19, 211–292. [Google Scholar] [CrossRef]
- Papachryssanthou, J.; Bordes, E.; Véjux, A.; Courtine, P.; Marchand, R.; Tournoux, M. TiO2(B), a new support for V2O5 in the oxidation of O-xylene. Catal. Today 1987, 1, 219–227. [Google Scholar] [CrossRef]
- Sanati, M.; Wallenberg, R.; Andersson, A.; Jasen, S.; To, Y. Vanadia catalysts on anatase, rutile, and TiO2(B) for the ammoxidation of toluene: An ESR and high-resolution electron microscopy characterization. J. Catal. 1991, 132, 128–144. [Google Scholar] [CrossRef]
- Véjux, A.; Courtine, P. Interfacial reactions between V2O5 and TiO2 (anatase): Role of the structural properties. J. Solid State Chem. 1978, 23, 93–103. [Google Scholar] [CrossRef]
- Grasselli, R.K. Advances and future trends in selective oxidation and ammoxidation catalysis. Catal. Today 1999, 49, 141–154. [Google Scholar] [CrossRef]
- Volta, J.C. Vanadium phosphorus oxides, a reference catalyst for mild oxidation of light alkanes. C.R. Acad. Sci. IIc: Chem. 2000, 3, 717–723. [Google Scholar] [CrossRef]
- Korovchenko, P.; Shiju, N.R.; Dozier, A.K.; Graham, U.M.; Guerrero-Pérez, M.O.; Guliants, V.V. M1 to M2 phase transformation and phase cooperation in bulk mixed metal Mo–V–M–O (M = Te,Nb) catalysts for selective ammoxidation of propane. Top. Catal. 2007, 50, 43–51. [Google Scholar] [CrossRef]
- Weng, L.T.; Delmon, B. Phase cooperation and remote control effects in selective oxidation catalysts. Appl. Catal. A 1992, 81, 141–213. [Google Scholar] [CrossRef]
- Karroua, M.; Grange, P.; Delmon, B. Existence of synergy between “CoMoS” and Co9S8: New proof of remote control in hydrodesulfurization. Appl. Catal. 1989, 50, L5–L10. [Google Scholar] [CrossRef]
- Schuh, K.; Kleist, W.; Høj, M.; Trouillet, V.; Beato, P.; Degn Jensen, A.; Grunwaldt, J.D. Bismuth molybdate catalysts prepared by mild hydrothermal synthesis: Influence of pH on the selective oxidation of propylene. Catalysts 2015, 5, 1554–1573. [Google Scholar] [CrossRef]
- Brazdil, J.F.; Toft, M.A.; Lin, S.Y.; McKenna, S.T.; Zajac, G.; Kaduk, J.; Golab, T. Characterization of bismuth-cerium-molybdate selective propylene ammoxidation catalysts. Appl. Catal. A 2015, 495, 115–123. [Google Scholar] [CrossRef]
- Timm, B. The ammonia synthesis and heterogeneous catalysis. A historical review. In Proceedings of the 8th International Congress on Catalysis, Berlin, Germany; Dechema: Frankfurt am Main, Germany; Verlag Chemie: Frankfurt am Main, Germany, 1984; Volume 1, pp. 7–30. [Google Scholar]
- Mehlomakulu, B.; Nguyen, T.T.N.; Delichère, P.; van Teen, E.; Millet, J.M.M. Identification of the active species in oxidation reactions on mixed oxide catalysts: Supra-surface or bulk-surface species. J. Catal. 2012, 289, 1–10. [Google Scholar] [CrossRef]
- Schlögl, R. Concepts in selective oxidation of small alkane molecules. In Modern Heterogeneous Oxidation Catalysis; Mizuno, N., Ed.; Wiley-VCH: Weinheim, Germany, 2009; pp. 1–42. [Google Scholar]
- Thomas, J.M. The societal significance of catalysis and the growing practical importance of single-site heterogeneous catalysts. Proc. Roy. Soc. A 2012, 468, 1884–1903. [Google Scholar]
- Hävecker, M.; Wrabetz, S.; Kröhnert, J.; Csepei, L.I.; D’Alnoncourt, R.N.; Kolen’ko, Y.V.; Girgsdies, F.; Schlögl, R.; Trunschke, A. Surface chemistry of phase-pure M1 MoVTeNb oxide during operation in selective oxidation of propane to acrylic acid. J. Catal. 2012, 285, 48–60. [Google Scholar] [CrossRef]
- Kung, H. Oxidative dehydrogenation of light C2 to C4 alkanes. Adv. Catal. 1994, 40, 1–38. [Google Scholar]
- Blasco, T.; López-Nieto, J.M. Oxidative dehydrogenation of short chain alkanes on supported vanadium oxide catalysts. Appl. Catal. A 1997, 157, 117–142. [Google Scholar] [CrossRef]
- Tabaka, K.; Teng, Y.; Takemoto, T.; Suzuki, E.; Bañares, M.A.; Peña, M.A.; Fierro, J.L.G. Activation of methane by oxygen and nitrogen oxides. Catal. Rev. 2002, 44, 1–58. [Google Scholar] [CrossRef]
- Wang, L.H.; Tao, L.X.; Xie, M.S.; Xu, G.F.; Huang, J.H.; Xu, Y. Dehydrogenation and aromatization of methane under non-oxidizing conditions. Catal. Lett. 1993, 21, 35–41. [Google Scholar] [CrossRef]
- Heracleous, E.; Lemonidou, A. Ni-Nb-O mixed oxides as highly active and selective catalysts for ethane production via ethane oxidative dehydrogenation. Part I: Characterization and catalytic performance. J. Catal. 2006, 237, 162–174. [Google Scholar] [CrossRef]
- Wu, Y.; Gao, J.; He, Y.; Wu, T. Preparation and characterization of Ni–Zr–O nanoparticles and its catalytic behavior for ethane oxidative dehydrogenation. Appl. Surf. Sci. 2012, 258, 4922–4928. [Google Scholar] [CrossRef]
- Wu, Y.; He, Y.; Wu, T.; Weng, W.; Wan, H. Effect of synthesis method on the physical and catalytic property of nanosized NiO. Mater. Lett. 2007, 61, 2679–2682. [Google Scholar] [CrossRef]
- Popescu, I.; Heracleous, E.; Skoufa, Z.; Lemonidou, A.; Marcu, I.C. Study of electrical conductivity measurements of semiconductive and redox properties of M-doped NiO (M = Li, Mg, Al, Ga, Ti, Nb) catalysts for the oxidative dehydrogenation of ethane. Phys. Chem. Chem. Phys. 2014, 16, 4962–4970. [Google Scholar] [CrossRef] [PubMed]
- López-Nieto, J.M.; Botella, P.; Vázquez, M.L.; Dejoz, A. The selective oxidative dehydrogenation of ethane over hydrothermally synthesised MoVTeNb catalysts. Chem. Commun. 2002, 1906–1907. [Google Scholar]
- Soliman, M.; Al-Zeghayer, Y.; Al-Awadi, A.S.; Al-Mayman, S. Economics of acetic acid production by partial oxidation of ethane. APCBEE Procedia 2012, 3, 200–208. [Google Scholar] [CrossRef]
- Linke, D.; Wolf, D.; Zey, B.S.; Baerns, M.; Timpe, O.; Schlögl, R.; Zeyβ, S.; Dingerdissen, U. Catalytic partial oxidation of ethane to acetic acid over Mo1V0.25Nb0.12Pd0.0005Ox, I: Catalyst performance and reaction mechanism. J. Catal. 2002, 205, 16–31. [Google Scholar] [CrossRef]
- Rahman, F.; Loughlin, K.F.; Al-Saleh, M.A.; Saeed, M.R.; Tukur, N.M.; Hossain, M.M.; Karim, K.; Mamedov, A. Kinetics and mechanism of partial oxidation of ethane to ethylene and acetic acid over MoV type catalysts. Appl. Catal. A 2010, 375, 17–25. [Google Scholar] [CrossRef]
- Kutnetsova, N.I.; Popova, G.Y.; Kuznetsova, L.I.; Zaikovskii, V.I.; Koscheev, S.V.; Andrushkevich, T.V.; Lisitsyn, A.S.; Likholobov, V.A.; Han, S. Improving the performance of Pt-H3PMo12O40 catalysts in the selective dehydrogenation of propane with O2 and H2. Catal. Today 2015, 245, 179–185. [Google Scholar] [CrossRef]
- Mamedov, E.; Karim, K. Selective oxidation at SABIC: Innovative catalysts and technologies. In Handbook of Advanced Methods and Processes in Oxidation Catalysis; Duprez, D., Cavani, F., Eds.; Imperial College Press: London, UK, 2014; pp. 291–301. [Google Scholar]
- Gärtner, C.A.; van Veen, A.C.; Lercher, J.A. Oxidative dehydrogenation of ethane: Common principles and mechanistic aspects. ChemCatChem 2013, 5, 3196–3217. [Google Scholar] [CrossRef]
- Valverde, J.A.; Echavarria, A.; Eon, J.G.; Faro, A.C.; Palacio, L.A. V-Mg-Al catalyst from hydrotalcite for oxidative dehydrogenation of propane. React. Kinet. Mech. Catal. 2014, 11, 679–696. [Google Scholar] [CrossRef]
- Kondratenko, E.V.; Sinev, M.Y. Effect of nature and surface density of oxygen species on product distribution in the oxidative dehydrogenation of propane over oxide catalysts. Appl. Catal. A 2007, 325, 353–361. [Google Scholar] [CrossRef]
- Valenzuela, R.X.; Bueno, G.; Cortés Corberán, V.; Xu, Y.; Chen, C.G. Selective oxidehydrogenation of ethane with CO2 over CeO2-based catalysts. Catal. Today 2000, 61, 43–48. [Google Scholar] [CrossRef]
- Cortés Corberán, V. Novel approaches for the improvement of selectivity in the oxidative activation of light alkanes. Catal. Today 2005, 99, 33–41. [Google Scholar] [CrossRef]
- Deacon, H. Improvement in the manufacture of chlorine. US Patent 85370A, 29 December 1868. [Google Scholar]
- Wang, D.J.; Xu, M.T.; Shi, C.L.; Lunsford, J.H. Effect of carbon dioxide on the selectivities obtained during the partial oxidation of methane and ethane over Li+/MgO catalysts. Catal. Lett. 1993, 18, 323–328. [Google Scholar] [CrossRef]
- Wang, S.; Murata, K.; Hayakawa, T.; Hamakawa, S.; Suzuki, K. Dehydrogenation of ethane with carbon dioxide over supported chromium oxide catalysts. Appl. Catal A 2000, 196, 1–8. [Google Scholar] [CrossRef]
- Xu, L.Y.; Liu, J.X.; Yang, H.; Xu, Y.; Wang, Q.X.; Lin, L.W. Regeneration behaviors of Fe/Si-2 and Fe–Mn/Si-2 catalysts for C2H6 dehydrogenation with CO2 to C2H4. Catal. Lett. 1999, 62, 185–189. [Google Scholar] [CrossRef]
- Cavani, F.; Ballarini, N.; Cericola, A. Oxidative dehydrogenation of ethane and propane: How far from commercial implementation? Catal. Today 2007, 127, 113–131. [Google Scholar] [CrossRef]
- Somorjai, G.A.; Park, J.Y. Molecular factor of catalytic selectivity. Angew. Chem. Int. Ed. 2008, 47, 9212–9228. [Google Scholar] [CrossRef] [PubMed]
- Contractor, R.M.; Garnett, D.I.; Horowitz, H.S.; Bergna, H.E.; Patience, G.S. A new commercial scale process for n-butane oxidation to maleic anhydride using a circulating fluidized bed reactor. Stud. Surf. Sci. Catal. 1994, 82, 233–242. [Google Scholar]
- Groppi, G.; Beretta, A.; Troconi, E. Structured catalytic reactors for selective oxidations. In Handbook of Advanced Methods and Processes in Oxidation Catalysis; Duprez, D., Cavani, F., Eds.; Imperial College Press: London, UK, 2014; pp. 943–997. [Google Scholar]
- Menéndez, M. Membrane reactors as tools for improved catalytic oxidation process. In Handbook of Advanced Methods and Processes in Oxidation Catalysis; Duprez, D., Cavani, F., Eds.; Imperial College Press: London, UK, 2014; pp. 921–942. [Google Scholar]
- Mallada, R.; Menéndez, M.; Santamaria, J. Use of membrane reactors for the oxidation of butane to maleic anhydride under high butane concentration. Catal. Today 2000, 56, 191–197. [Google Scholar] [CrossRef]
- Bordes-Richard, E.; Shekari, A.; Patience, G.S. Vanadium phosphorus oxide catalyst for n-butane selective oxidation: From catalyst synthesis to the industrial process. In Handbook of Advanced Methods and Processes in Oxidation Catalysis; Duprez, D., Cavani, F., Eds.; Imperial College Press: London, UK, 2014; pp. 549–585. [Google Scholar]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Védrine, J.C. Heterogeneous Partial (amm)Oxidation and Oxidative Dehydrogenation Catalysis on Mixed Metal Oxides. Catalysts 2016, 6, 22. https://doi.org/10.3390/catal6020022
Védrine JC. Heterogeneous Partial (amm)Oxidation and Oxidative Dehydrogenation Catalysis on Mixed Metal Oxides. Catalysts. 2016; 6(2):22. https://doi.org/10.3390/catal6020022
Chicago/Turabian StyleVédrine, Jacques C. 2016. "Heterogeneous Partial (amm)Oxidation and Oxidative Dehydrogenation Catalysis on Mixed Metal Oxides" Catalysts 6, no. 2: 22. https://doi.org/10.3390/catal6020022
APA StyleVédrine, J. C. (2016). Heterogeneous Partial (amm)Oxidation and Oxidative Dehydrogenation Catalysis on Mixed Metal Oxides. Catalysts, 6(2), 22. https://doi.org/10.3390/catal6020022