
Tandem Reactions Combining Biocatalysts and Chemical Catalysts for Asymmetric Synthesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
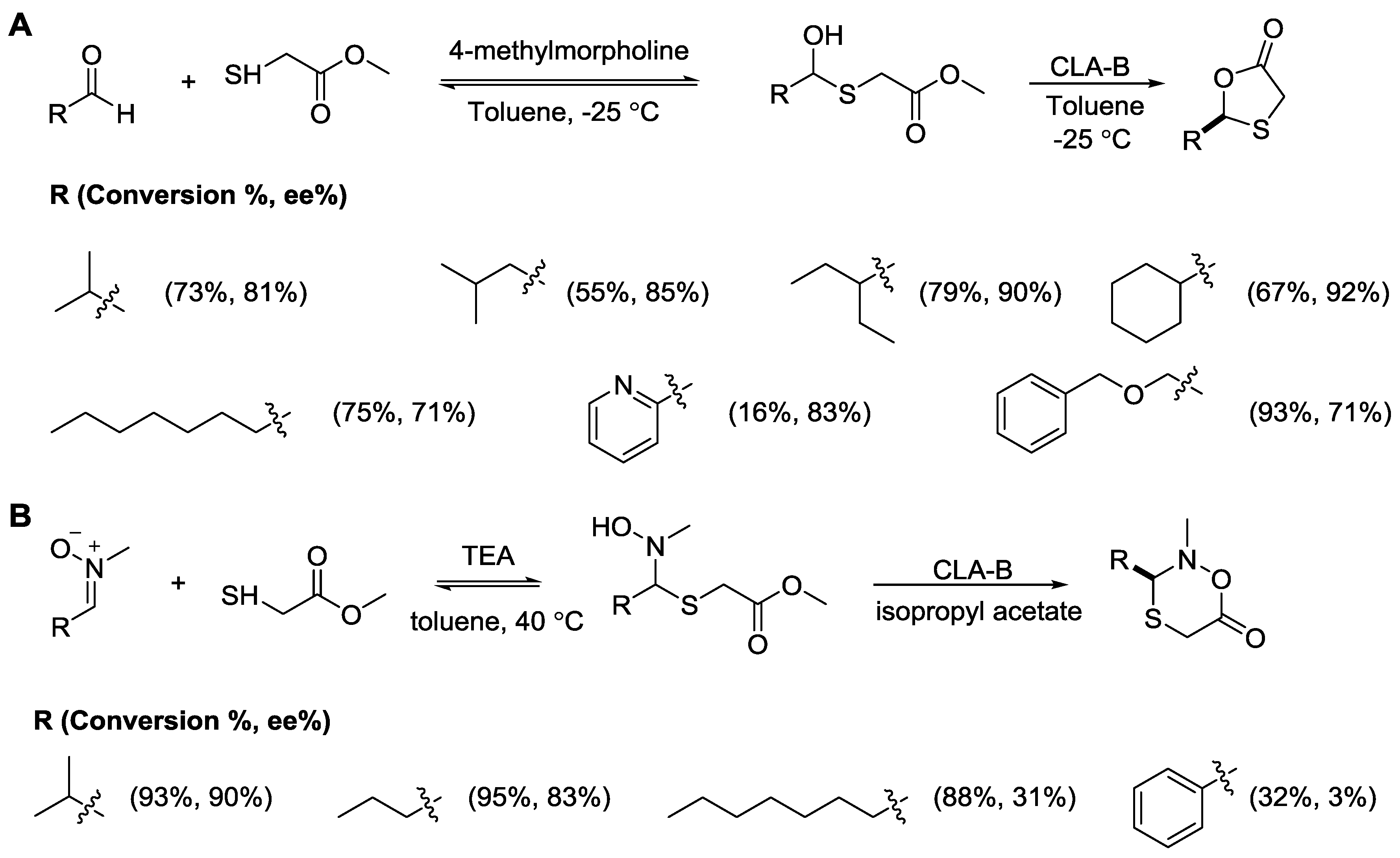{kind=link}
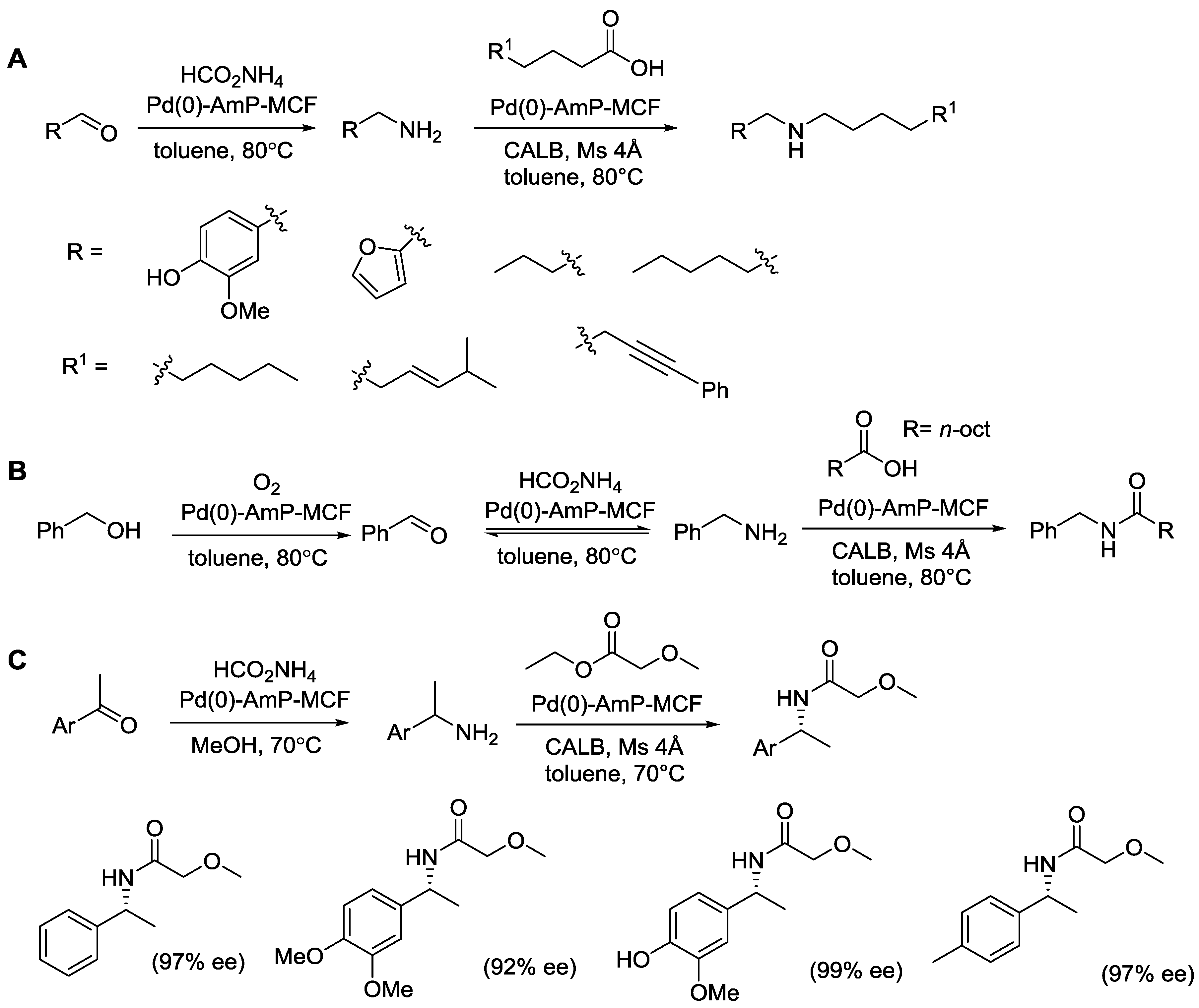{kind=link}
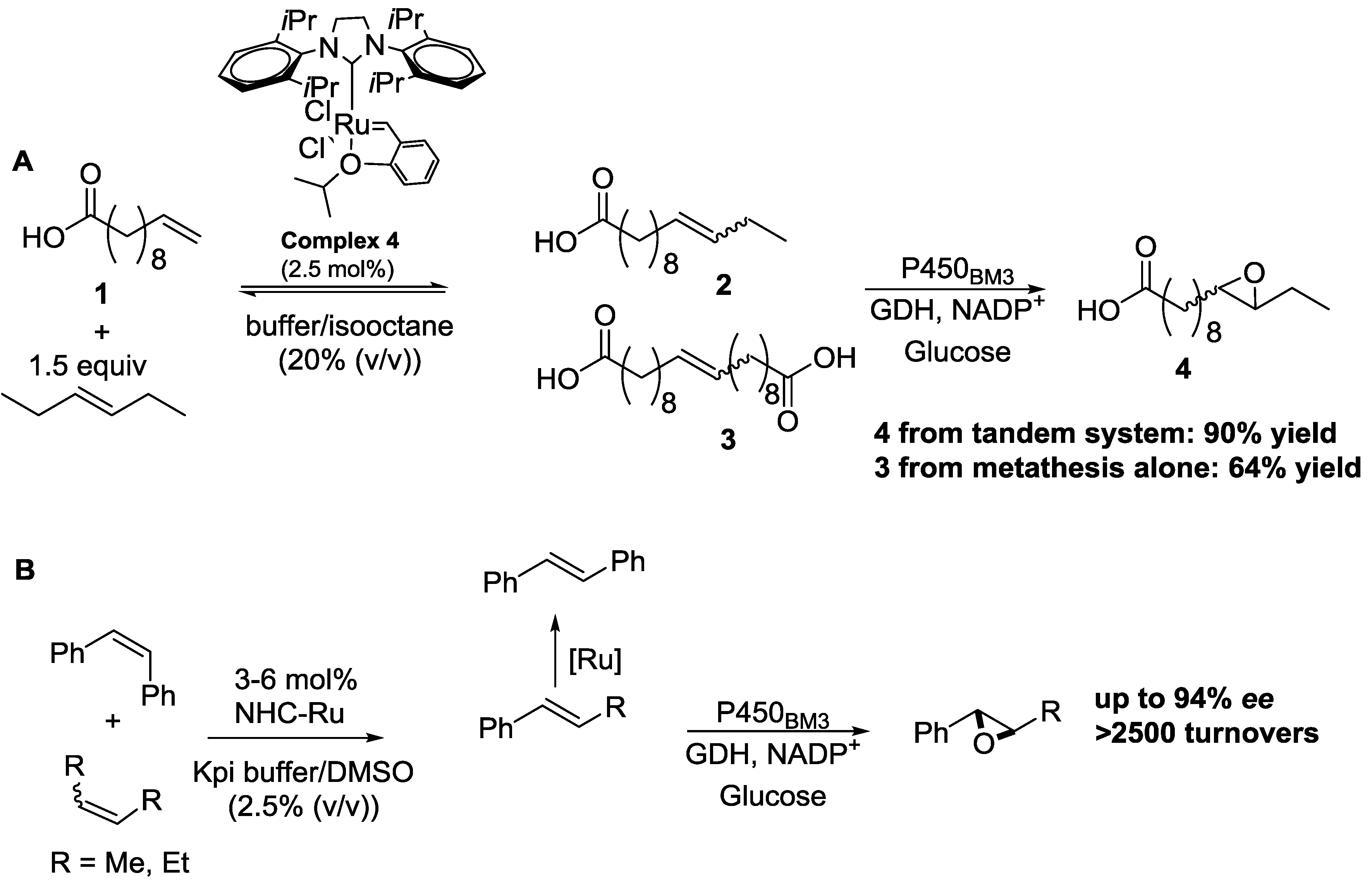{kind=link}
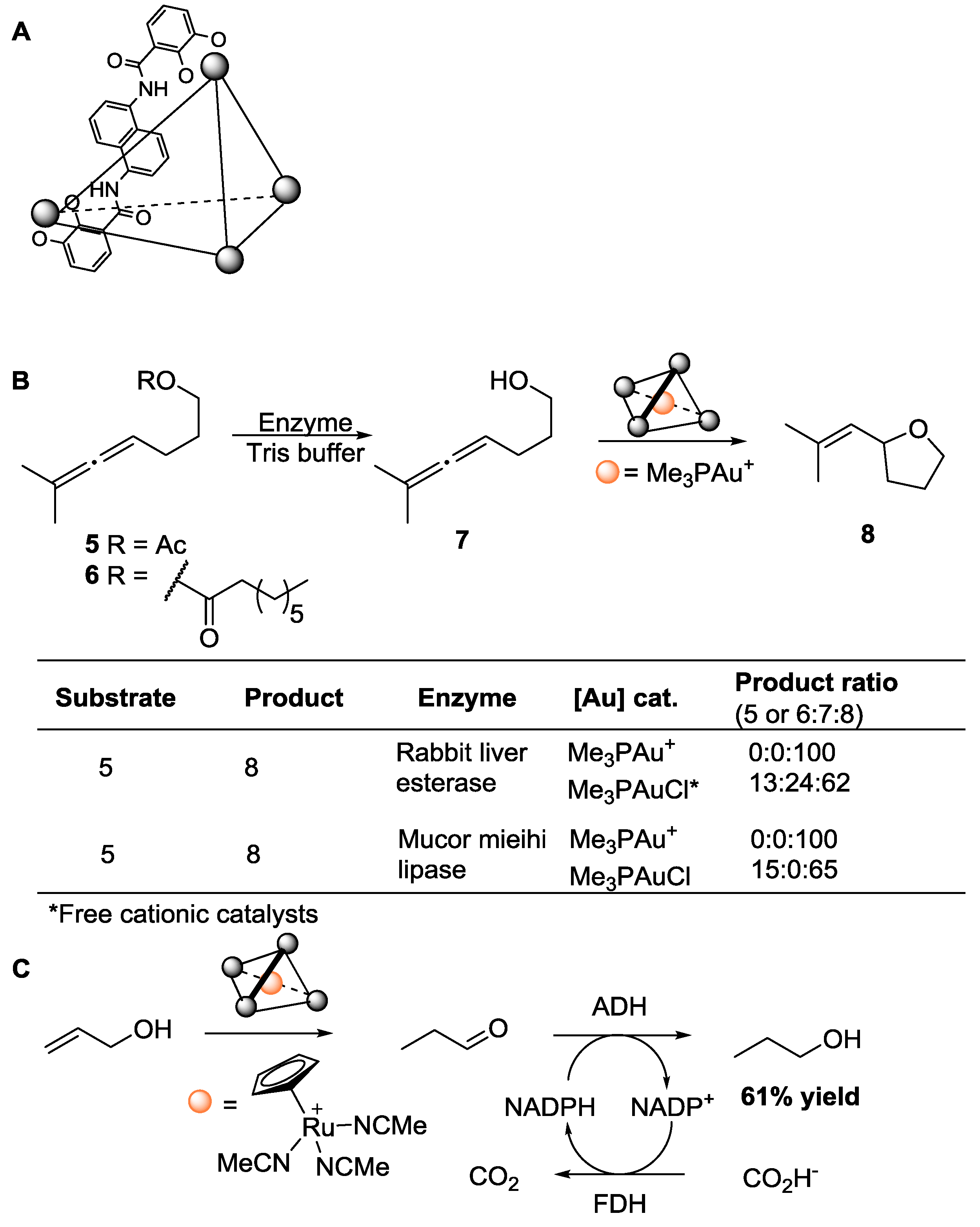{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
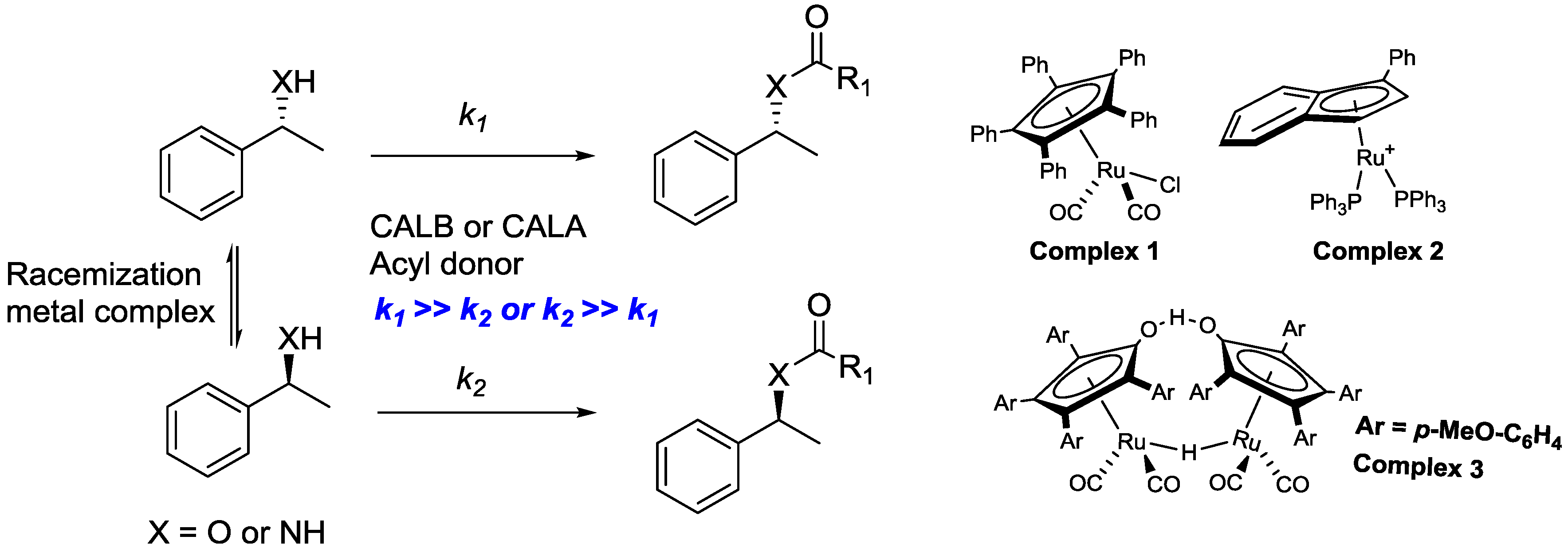
2. Dynamic Kinetic Resolution
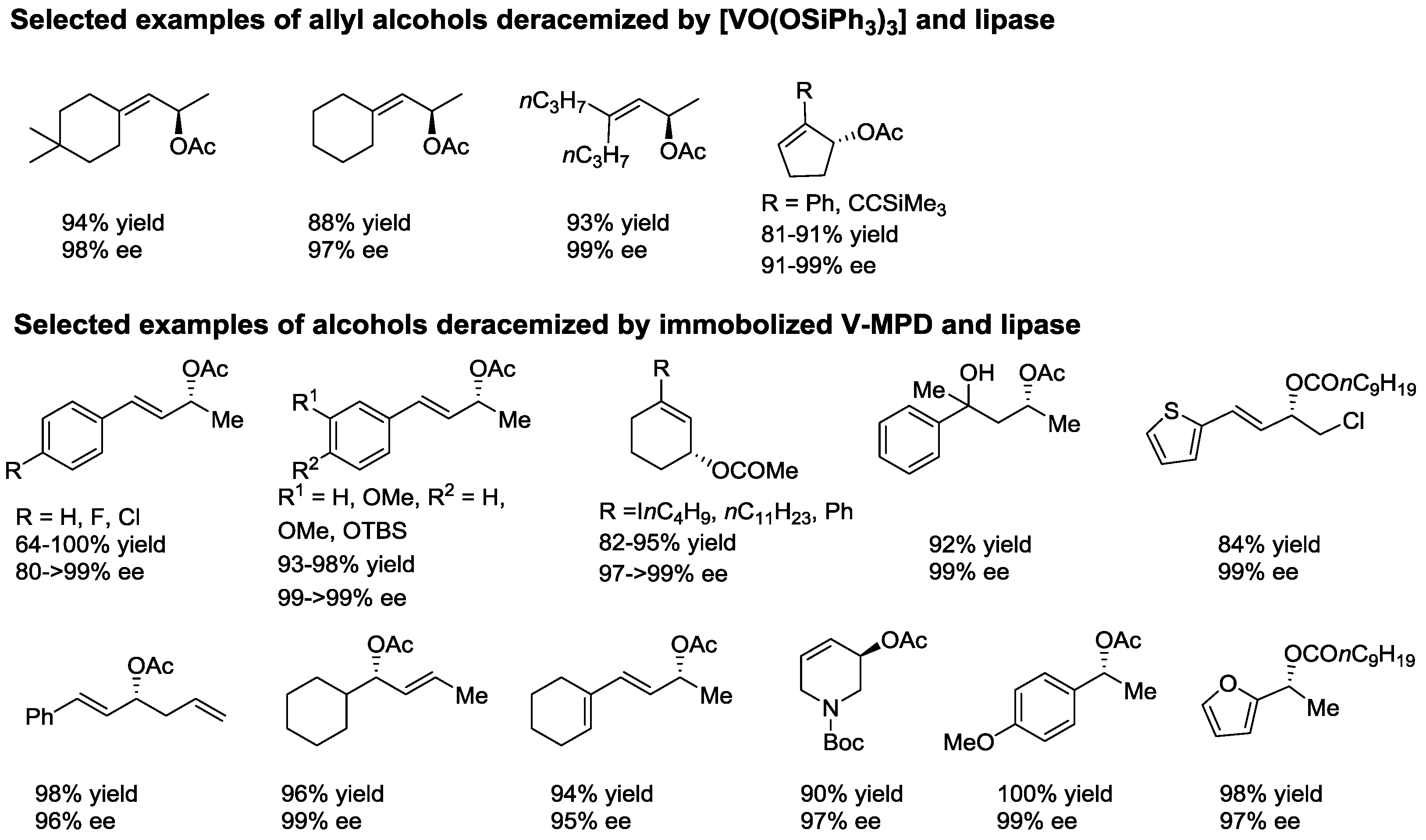
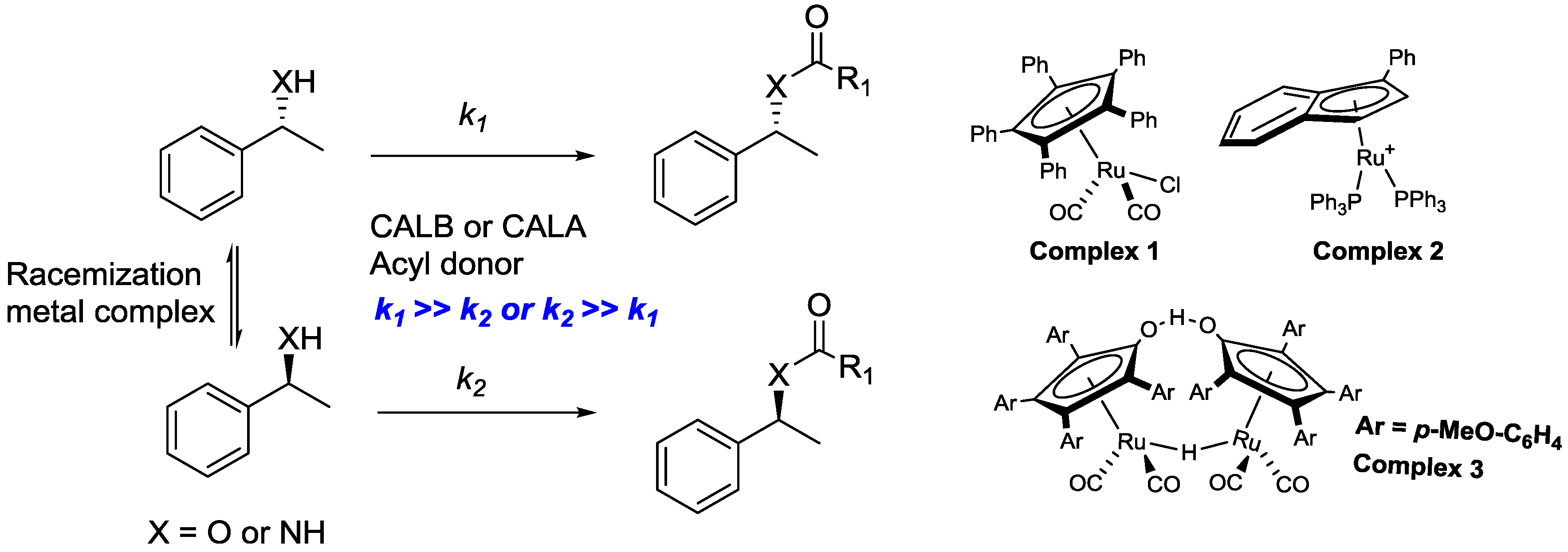
2.1. Dynamic Kinetic Resolution of Secondary Alcohols and Derivatives
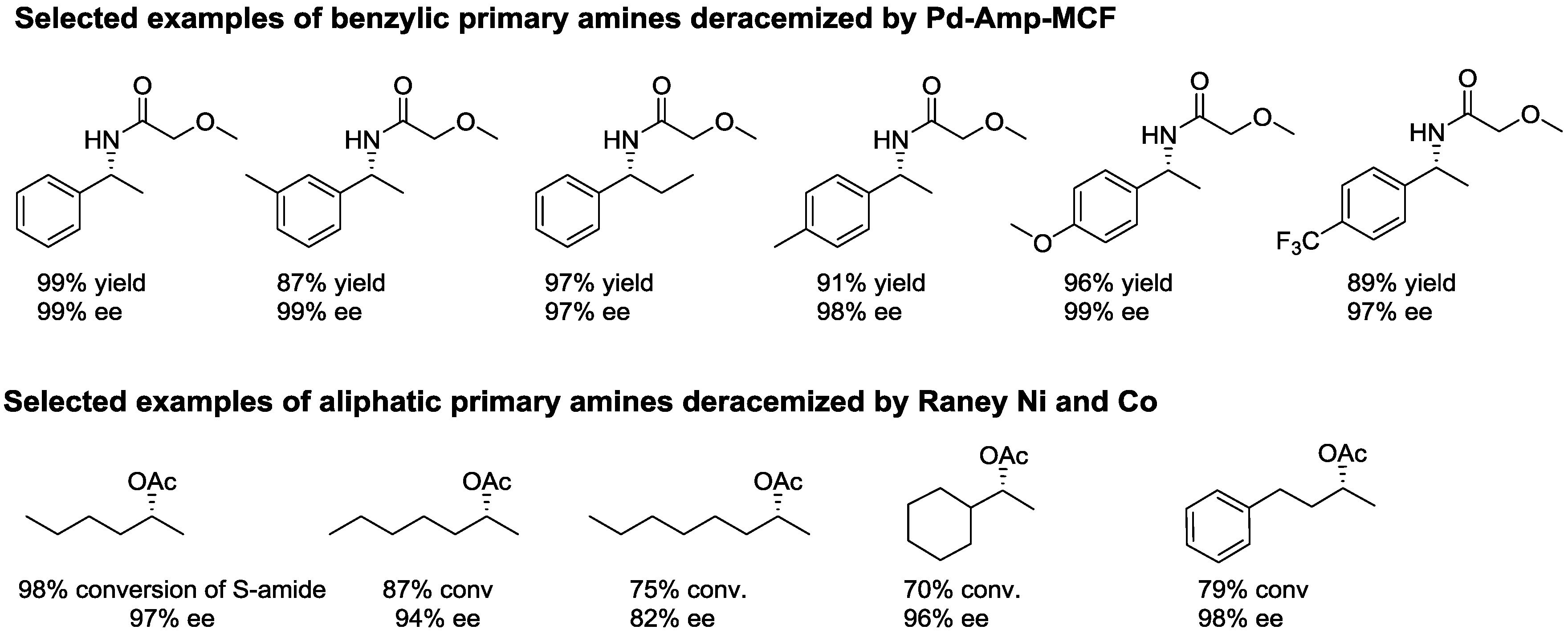
2.2. Dynamic Kinetic Resolution of Amines
2.3. Other Tandem Reactions by Transition Metal Catalysts and Lipases
3. One-Pot Chemoenzymatic Transformations
3.1. Concurrent Tandem Reactions by Transition-Metal Complexes and Enzymes
3.2. Coupling Visible-Light Photoredox Catalysis with Biocatalysts
3.3. Artificial Metalloenzymes for Selective Transformations
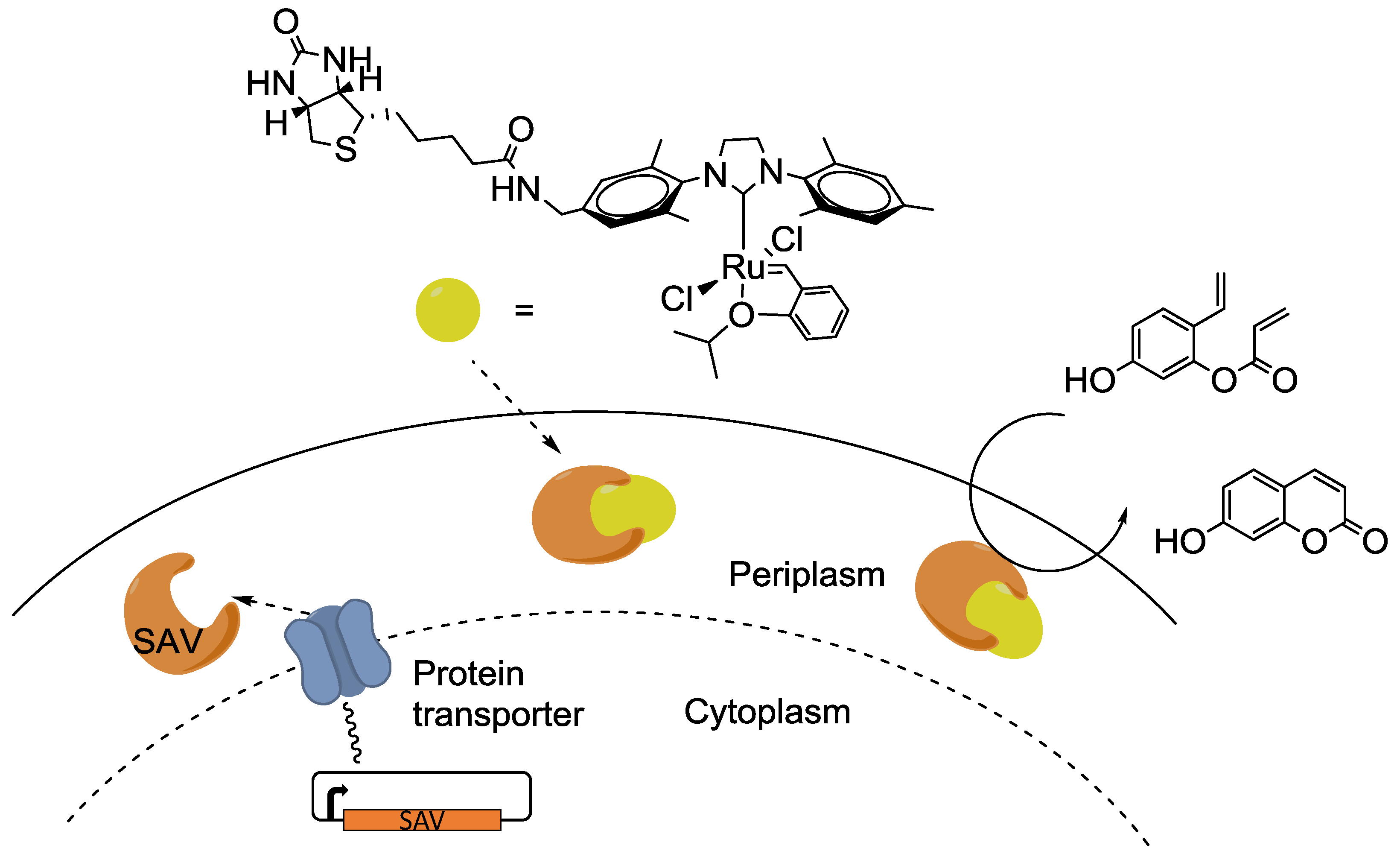
4. Interfacing the Transition-Metal Catalysis with Living Cells
5. Future Prospects and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sheldon, R.A. Multi-Step Enzyme Catalysis: Biotransformations and Chemoenzymatic Synthesis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; p. 256. [Google Scholar]
- Philip, J.P.; Clive, S.P.; Adrian, J.S. Tandem reactions in organic synthesis: Novel strategies for natural product elboration and the development of new synthetic methodology. Chem. Rev. 1996, 96, 195–206. [Google Scholar]
- Denard, C.A.; Hartwig, J.F.; Zhao, H. Multistep one-pot reactions combining biocatalysts and chemical catalysts for asymmetric synthesis. ACS Catal. 2013, 3, 2856–2864. [Google Scholar] [CrossRef]
- Wells, A.S.; Finch, G.L.; Michels, P.C.; Wong, J.W. Use of enzymes in the manufacture of active pharmaceutical ingredients—A science and safety-based approach to ensure patient safety and drug quality. Org. Process Res. Dev. 2012, 16, 1986–1993. [Google Scholar] [CrossRef]
- Winkler, C.K.; Tasnádi, G.; Clay, D.; Hall, M.; Faber, K. Asymmetric bioreduction of activated alkenes to industrially relevant optically active compounds. J. Biotechnol. 2012, 162, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Winkler, C.K.; Clay, D.; Davies, S.; O’Neill, P.; McDaid, P.; Debarge, S.; Steflik, J.; Karmilowicz, M.; Wong, J.W.; Faber, K. Chemoenzymatic asymmetric synthesis of pregabalin precursors via asymmetric bioreduction of β-cyanoacrylate esters using ene-reductases. J. Org. Chem. 2013, 78, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.A.; He, H.; Pham-Huy, C. Chiral drugs: An overview. Int. J. Biomed. Sci. 2006, 2, 85–100. [Google Scholar] [PubMed]
- Verho, O.; Backvall, J.E. Chemoenzymatic dynamic kinetic resolution: A powerful tool for the preparation of enantiomerically pure alcohols and amines. J. Am. Chem. Soc. 2015, 137, 3996–4009. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, A.S.; Miranda, L.S.; de Souza, R.O. Lipases: Valuable catalysts for dynamic kinetic resolutions. Biotechnol. Adv. 2015, 33, 372–393. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewicz, A.; Ahlsten, N.; Martin-Matute, B. Enantioselective synthesis of alcohols and amines by iridium-catalyzed hydrogenation, transfer hydrogenation, and related processes. Chemistry 2013, 19, 7274–7302. [Google Scholar] [CrossRef] [PubMed]
- Breuer, M.; Ditrich, K.; Habicher, T.; Hauer, B.; Kesseler, M.; Sturmer, R.; Zelinski, T. Industrial methods for the production of optically active intermediates. Angew. Chem. Int. Ed. 2004, 43, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.V.; Williams, J.M.J. Dynamic kinetic resolution with enzyme and palladium combinations. Tetrahedron Lett. 1996, 37, 1859–1862. [Google Scholar] [CrossRef]
- Pamies, O.; Backvall, J.E. Combination of enzymes and metal catalysts. A powerful approach in asymmetric catalysis. Chem. Rev. 2003, 103, 3247–3262. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Chung, Y.I.; Choi, Y.K.; Lee, H.K.; Kim, D.; Park, J. (S)-Selective dynamic kinetic resolution of secondary alcohols by the combination of subtilisin and an aminocyclopentadienylruthenium complex as the catalysts. J. Am. Chem. Soc. 2003, 125, 11494–11495. [Google Scholar] [CrossRef] [PubMed]
- Martin-Matute, B.; Backvall, J.E. Dynamic kinetic resolution catalyzed by enzymes and metals. Curr. Opin. Chem. Biol. 2007, 11, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Warner, M.C.; Backvall, J.E. Mechanistic aspects on cyclopentadienylruthenium complexes in catalytic racemization of alcohols. Acc. Chem. Res. 2013, 46, 2545–2555. [Google Scholar] [CrossRef] [PubMed]
- Martin-Matute, B.; Edin, M.; Bogar, K.; Backvall, J.E. Highly compatible metal and enzyme catalysts for efficient dynamic kinetic resolution of alcohols at ambient temperature. Angew. Chem. Int. Ed. 2004, 43, 6535–6539. [Google Scholar] [CrossRef] [PubMed]
- Martin-Matute, B.; Edin, M.; Bogar, K.; Kaynak, F.B.; Backvall, J.E. Combined ruthenium(II) and lipase catalysis for efficient dynamic kinetic resolution of secondary alcohols. Insight into the racemization mechanism. J. Am. Chem. Soc. 2005, 127, 8817–8825. [Google Scholar] [CrossRef] [PubMed]
- Lihammar, R.; Millet, R.; Backvall, J.E. Enzyme- and ruthenium-catalyzed dynamic kinetic resolution of functionalized cyclic allylic alcohols. J. Org. Chem. 2013, 78, 12114–12120. [Google Scholar] [CrossRef] [PubMed]
- Norinder, J.; Bogar, K.; Kanupp, L.; Backvall, J.E. An enantioselective route to alpha-methyl carboxylic acids via metal and enzyme catalysis. Org. Lett. 2007, 9, 5095–5098. [Google Scholar] [CrossRef] [PubMed]
- Bogar, K.; Vidal, P.H.; Leon, A.R.; Backvall, J.E. Chemoenzymatic dynamic kinetic resolution of allylic alcohols: A highly enantioselective route to acyloin acetates. Org. Lett. 2007, 9, 3401–3404. [Google Scholar] [CrossRef] [PubMed]
- Traff, A.; Bogar, K.; Warner, M.; Backvall, J.E. Highly efficient route for enantioselective preparation of chlorohydrins via dynamic kinetic resolution. Org. Lett. 2008, 10, 4807–4810. [Google Scholar] [CrossRef] [PubMed]
- Leijondahl, K.; Boren, L.; Braun, R.; Backvall, J.E. Enzyme- and ruthenium-catalyzed dynamic kinetic asymmetric transformation of 1,5-diols. Application to the synthesis of (+)-Solenopsin A. J. Org. Chem. 2009, 74, 1988–1993. [Google Scholar] [CrossRef] [PubMed]
- Leijondahl, K.; Boren, L.; Braun, R.; Backvall, J.E. Enantiopure 1,5-diols from dynamic kinetic asymmetric transformation. Useful synthetic intermediates for the preparation of chiral heterocycles. Org. Lett. 2008, 10, 2027–2030. [Google Scholar] [CrossRef] [PubMed]
- Warner, M.C.; Shevchenko, G.A.; Jouda, S.; Bogar, K.; Backvall, J.E. Dynamic kinetic resolution of homoallylic alcohols: Application to the synthesis of enantiomerically pure 5,6-dihydropyran-2-ones and delta-lactones. Chemistry 2013, 19, 13859–13864. [Google Scholar] [CrossRef] [PubMed]
- Traff, A.; Lihammar, R.; Backvall, J.E. A chemoenzymatic dynamic kinetic resolution approach to enantiomerically pure (R)- and (S)-duloxetine. J. Org. Chem. 2011, 76, 3917–3921. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Salas, J.A.; Manzini, S.; Nolan, S.P. A cationic ruthenium complex for the dynamic kinetic resolution of secondary alcohols. Chemistry 2014, 20, 13132–13135. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Martinez-Castro, E.; Marcos, R.; Martin-Matute, B. Readily available ruthenium complex for efficient dynamic kinetic resolution of aromatic alpha-hydroxy ketones. Org. Lett. 2014, 16, 2256–2259. [Google Scholar] [CrossRef] [PubMed]
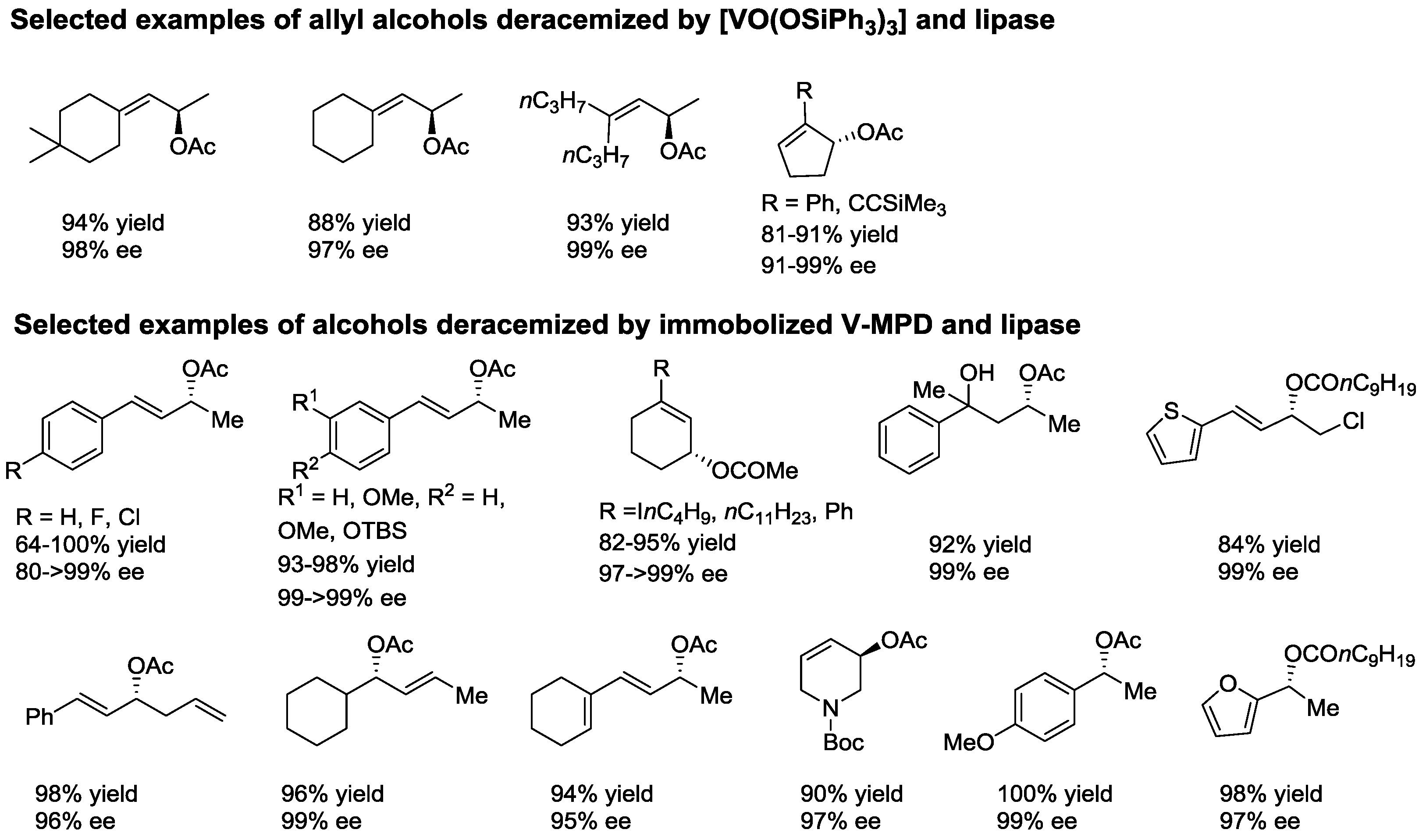
- Akai, S.; Tanimoto, K.; Kanao, Y.; Egi, M.; Yamamoto, T.; Kita, Y. A dynamic kinetic resolution of allyl alcohols by the combined use of lipases and [VO(OSiPh3)3]. Angew. Chem. Int. Ed. 2006, 45, 2592–2595. [Google Scholar] [CrossRef] [PubMed]
- Akai, S.; Hanada, R.; Fujiwara, N.; Kita, Y.; Egi, M. One-pot synthesis of optically active allyl esters via lipase-vanadium combo catalysis. Org. Lett. 2010, 12, 4900–4903. [Google Scholar] [CrossRef] [PubMed]
- Egi, M.; Sugiyama, K.; Saneto, M.; Hanada, R.; Kato, K.; Akai, S. A mesoporous-silica-immobilized oxovanadium cocatalyst for the lipase-catalyzed dynamic kinetic resolution of racemic alcohols. Angew. Chem. Int. Ed. 2013, 52, 3654–3658. [Google Scholar] [CrossRef] [PubMed]
- Nieguth, R.; ten Dam, J.; Petrenz, A.; Ramanathan, A.; Hanefeld, U.; Ansorge-Schumacher, M.B. Combined heterogeneous bio- and chemo-catalysis for dynamic kinetic resolution of (rac)-benzoin. RSC Adv. 2014, 4, 45495–45503. [Google Scholar] [CrossRef]
- Li, X.; Yan, Y.; Wang, W.; Zhang, Y.; Tang, Y. Activity modulation of core and shell in nanozeolite@enzyme bi-functional catalyst for dynamic kinetic resolution. J. Colloid Interface Sci. 2015, 438, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, X.; Wang, Z.; Tang, Y.; Zhang, Y. Enhancement of (stereo)selectivity in dynamic kinetic resolution using a core-shell nanozeolite@enzyme as a bi-functional catalyst. Chem. Commun. 2014, 50, 9501–9504. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Oh, Y.; Choi, Y.K.; Choi, E.; Kim, K.; Park, J.; Kim, M.-J. Dynamic kinetic resolution of diarylmethanols with an activated lipoprotein lipase. ACS Catal. 2015, 5, 683–689. [Google Scholar] [CrossRef]
- Wikmark, Y.; Svedendahl Humble, M.; Backvall, J.E. Combinatorial library based engineering of candida antarctica lipase a for enantioselective transacylation of sec-alcohols in organic solvent. Angew. Chem. Int. Ed. 2015, 54, 4284–4288. [Google Scholar] [CrossRef] [PubMed]
- Goossen, L.J.; Paetzold, J. Pd-catalyzed decarbonylative olefination of aryl esters: Towards a waste-free heck reaction. Angew. Chem. Int. Ed. 2002, 41, 1237–1241. [Google Scholar] [CrossRef]
- Thalen, L.K.; Zhao, D.; Sortais, J.B.; Paetzold, J.; Hoben, C.; Backvall, J.E. A chemoenzymatic approach to enantiomerically pure amines using dynamic kinetic resolution: Application to the synthesis of norsertraline. Chemistry 2009, 15, 3403–3410. [Google Scholar] [CrossRef] [PubMed]
- Thalen, L.K.; Backvall, J.E. Development of dynamic kinetic resolution on large scale for (+/−)-1-phenylethylamine. Beilstein J. Org. Chem. 2010, 6, 823–829. [Google Scholar] [CrossRef] [PubMed]
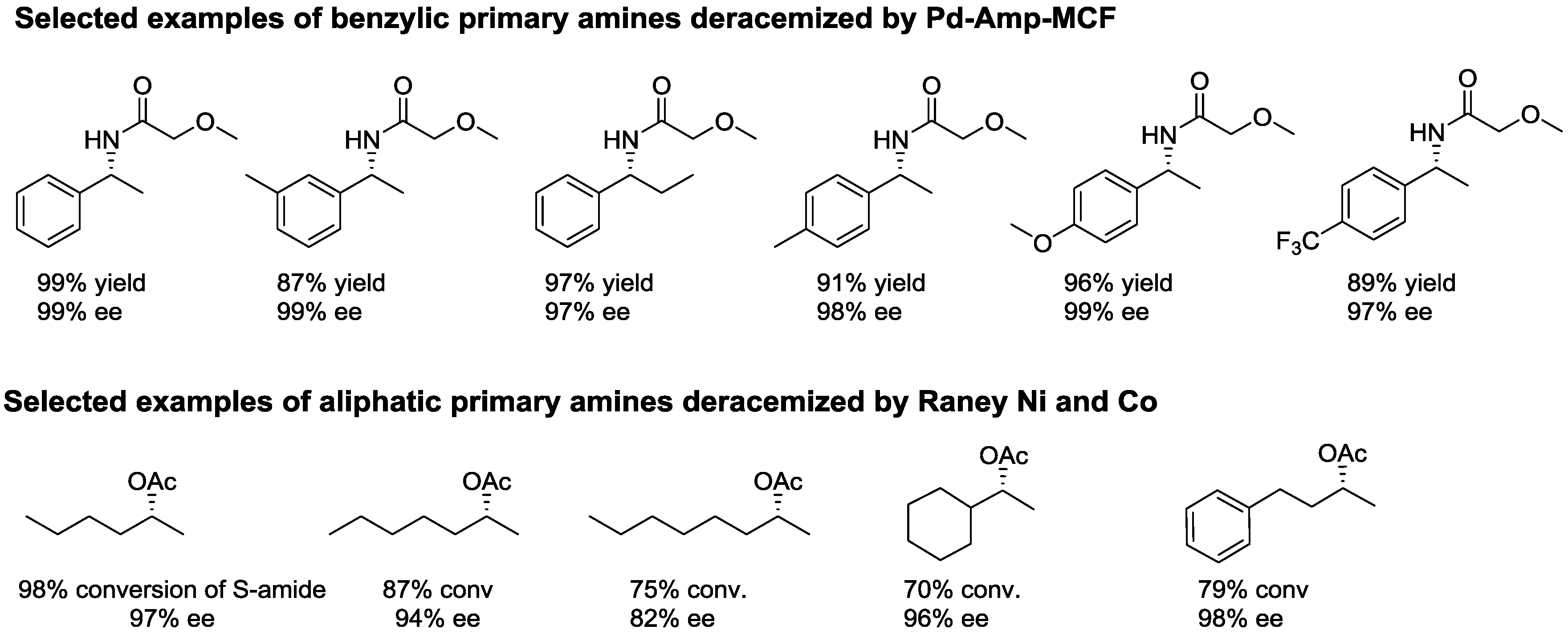
- Gustafson, K.P.; Lihammar, R.; Verho, O.; Engstrom, K.; Backvall, J.E. Chemoenzymatic dynamic kinetic resolution of primary amines using a recyclable palladium nanoparticle catalyst together with lipases. J. Org. Chem. 2014, 79, 3747–3751. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Jia, G.; Zhang, Y.; Li, C. Modification of supported Pd catalysts by alkalic salts in the selective racemization and dynamic kinetic resolution of primary amines. Catal. Sci. Technol. 2014, 4, 464–471. [Google Scholar] [CrossRef]
- Ma, G.; Xu, Z.; Zhang, P.; Liu, J.; Hao, X.; Ouyang, J.; Liang, P.; You, S.; Jia, X. A novel synthesis of rasagiline via a chemoenzymatic dynamic kinetic resolution. Org. Process Res. Dev. 2014, 18, 1169–1174. [Google Scholar] [CrossRef]
- Mavrynsky, D.; Leino, R. An approach to chemoenzymatic DKR of amines in soxhlet apparatus. J. Organomet. Chem. 2014, 760, 161–166. [Google Scholar]
- De Miranda, A.S.; de Souza, R.O.M.A.; Miranda, L.S.M. Ammonium formate as a green hydrogen source for clean semi-continuous enzymatic dynamic kinetic resolution of (+/−)-α-methylbenzylamine. RSC Adv. 2014, 4, 13620–13625. [Google Scholar] [CrossRef]
- Xia, B.; Cheng, G.; Lin, X.; Wu, Q. Dynamic double kinetic resolution of amines and alcohols under the cocatalysis of Raney nickel/Candida antarctica lipase B: From concept to application. Eur. J. Org. Chem. 2014, 2014, 2917–2923. [Google Scholar] [CrossRef]
- Parvulescu, A.N.; Jacobs, P.A.; De Vos, D.E. Heterogeneous raney nickel and cobalt catalysts for racemization and dynamic kinetic resolution of amines. Adv. Synth. Catal. 2008, 350, 113–121. [Google Scholar] [CrossRef]
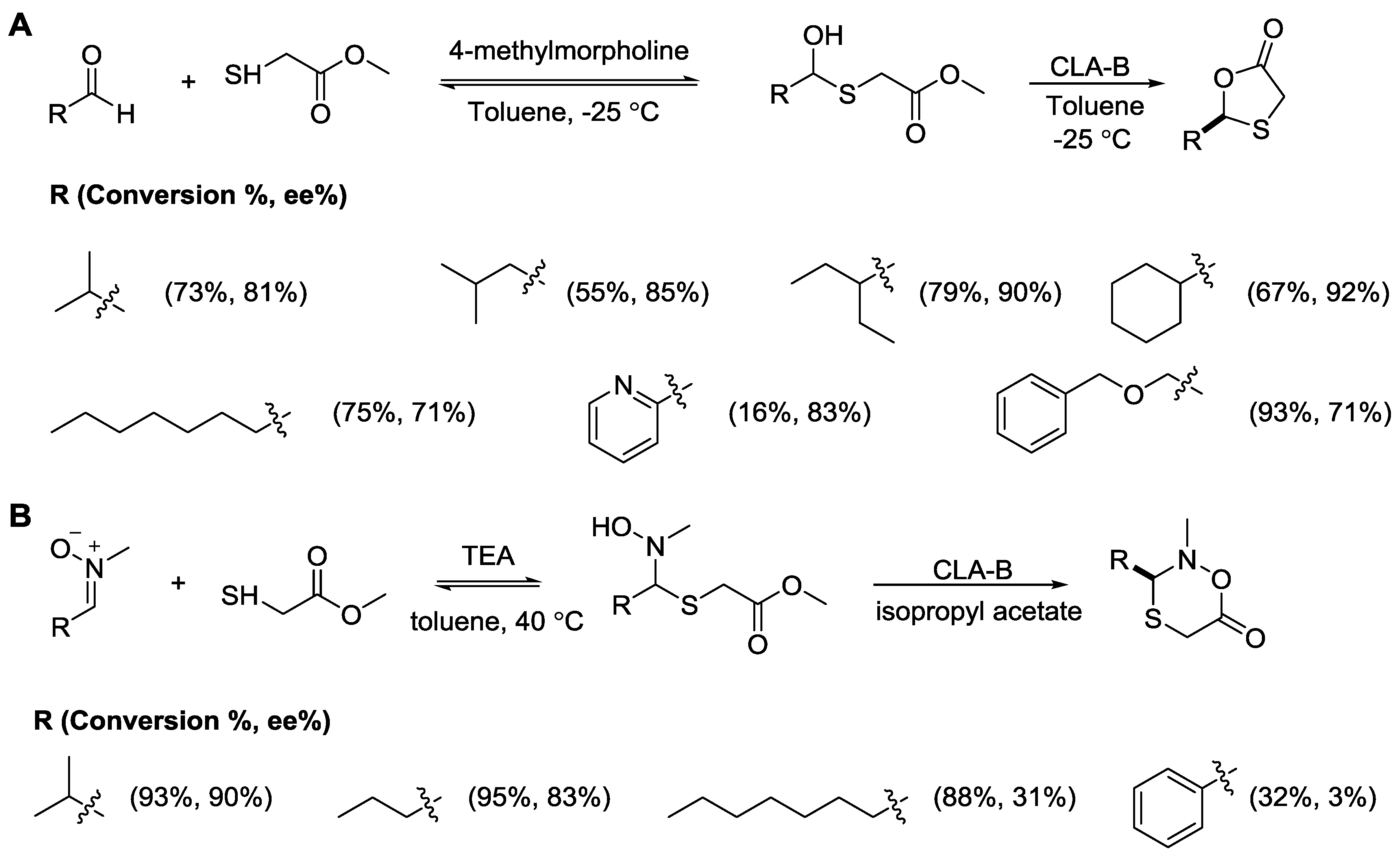
- Zhang, Y.; Schaufelberger, F.; Sakulsombat, M.; Liu, C.; Ramström, O. Asymmetric synthesis of 1,3-oxathiolan-5-one derivatives through dynamic covalent kinetic resolution. Tetrahedron 2014, 70, 3826–3831. [Google Scholar] [CrossRef]
- Hu, L.; Zhang, Y.; Ramstrom, O. Lipase-catalyzed asymmetric synthesis of oxathiazinanones through dynamic covalent kinetic resolution. Org. Biomol. Chem. 2014, 12, 3572–3575. [Google Scholar] [CrossRef] [PubMed]
- Palo-Nieto, C.; Afewerki, S.; Anderson, M.; Tai, C.-W.; Berglund, P.; Córdova, A. Integrated heterogeneous metal/enzymatic multiple relay catalysis for eco-friendly and asymmetric synthesis. ACS Catal. 2016, 6, 3932–3940. [Google Scholar] [CrossRef]
- Kohler, V.; Turner, N.J. Artificial concurrent catalytic processes involving enzymes. Chem. Commun. 2015, 51, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Groger, H.; Hummel, W. Combining the ‘two worlds’ of chemocatalysis and biocatalysis towards multi-step one-pot processes in aqueous media. Curr. Opin. Chem. Biol. 2014, 19, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Nirio, M.; Akira, S. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 2457–2483. [Google Scholar]
- James, M.T.; Xun-tian, J. The wacker reactions and related alkene oxidation. Curr. Org. Chem. 2003, 7, 369–396. [Google Scholar]
- Costabile, C.; Cavallo, L. Origin of enantioselectivity in the asymmetric Ru-catalyzed metathesis of olefins. J. Am. Chem. Soc. 2004, 126, 9592–9600. [Google Scholar] [CrossRef] [PubMed]
- Jay, C.C.; Deryn, E.F. Ruthenium-catalyzed ring-closing matethesis: Recent advances, limitations and opportunities. Curr. Org. Chem. 2006, 10, 185–202. [Google Scholar]
- Gauchot, V.; Kroutil, W.; Schmitzer, A.R. Highly recyclable chemo-/biocatalyzed cascade reactions with ionic liquids: One-pot synthesis of chiral biaryl alcohols. Chemistry 2010, 16, 6748–6751. [Google Scholar] [CrossRef] [PubMed]
- Boffi, A.; Cacchi, S.; Ceci, P.; Cirilli, R.; Fabrizi, G.; Prastaro, A.; Niembro, S.; Shafir, A.; Vallribera, A. The heck reaction of allylic alcohols catalyzed by palladium nanoparticles in water: Chemoenzymatic synthesis of (R)-(−)-rhododendrol. ChemCatChem 2011, 3, 347–353. [Google Scholar] [CrossRef]
- Schnapperelle, I.; Hummel, W.; Groger, H. Formal asymmetric hydration of non-activated alkenes in aqueous medium through a “chemoenzymatic catalytic system”. Chemistry 2012, 18, 1073–1076. [Google Scholar] [CrossRef] [PubMed]
- Denard, C.A.; Huang, H.; Bartlett, M.J.; Lu, L.; Tan, Y.C.; Zhao, H.M.; Hartwig, J.F. Cooperative tandem catalysis by an organometallic complex and a metalloenzyme. Angew. Chem. Int. Ed. 2014, 53, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Denard, C.A.; Bartlett, M.J.; Wang, Y.; Lu, L.; Hartwig, J.F.; Zhao, H. Development of a one-pot tandem reaction combining ruthenium-catalyzed alkene metathesis and enantioselective enzymatic oxidation to produce aryl epoxides. ACS Catal. 2015, 5, 3817–3822. [Google Scholar] [CrossRef]
- Wang, Z.J.; Clary, K.N.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. A supramolecular approach to combining enzymatic and transition metal catalysis. Nat. Chem. 2013, 5, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Kaphan, D.M.; Levin, M.D.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. A supramolecular microenvironment strategy for transition metal catalysis. Science 2015, 350, 1235–1238. [Google Scholar] [CrossRef] [PubMed]
- Heidlindemann, M.; Rulli, G.; Berkessel, A.; Hummel, W.; Gröger, H. Combination of asymmetric organo- and biocatalytic reactions in organic media using immobilized catalysts in different compartments. ACS Catal. 2014, 4, 1099–1103. [Google Scholar] [CrossRef]
- Sperl, J.M.; Carsten, J.M.; Guterl, J.-K.; Lommes, P.; Sieber, V. Reaction design for the compartmented combination of heterogeneous and enzyme catalysis. ACS Catal. 2016, 6, 6329–6334. [Google Scholar] [CrossRef]
- Metzner, R.; Hummel, W.; Wetterich, F.; König, B.; Gröger, H. Integrated biocatalysis in multistep drug synthesis without intermediate isolation: A de novo approach toward a rosuvastatin key building block. Org. Process Res. Dev. 2015, 19, 635–638. [Google Scholar] [CrossRef]
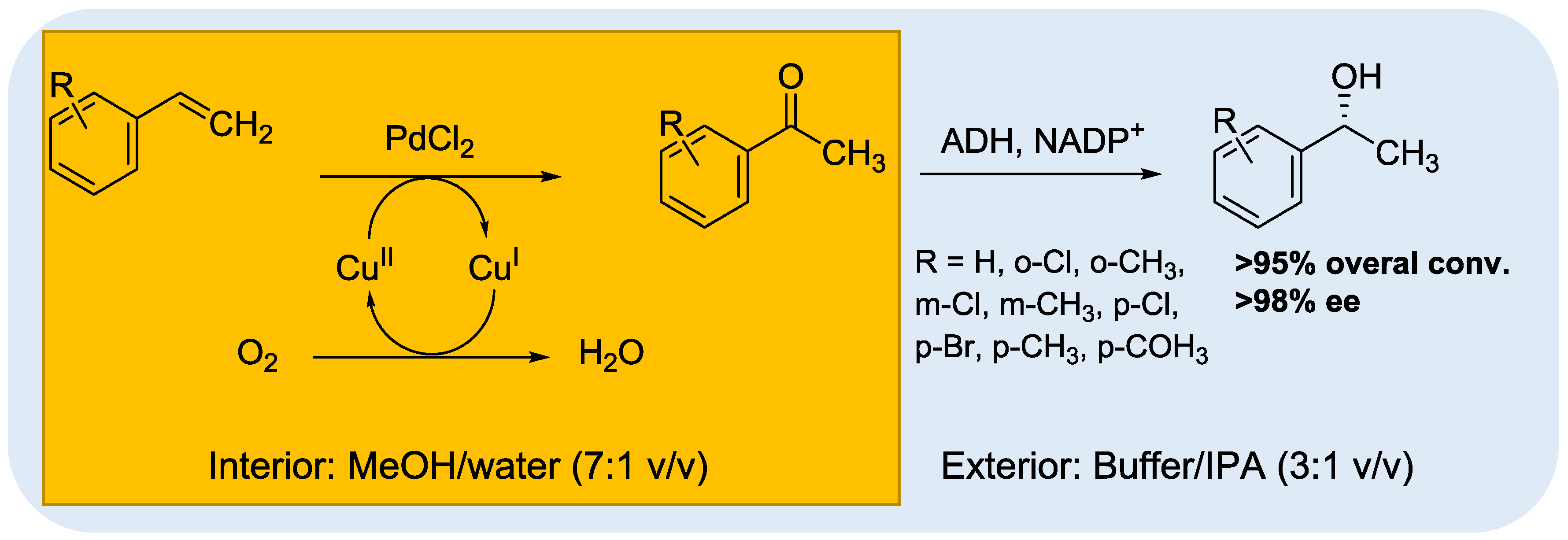
- Sato, H.; Hummel, W.; Groger, H. Cooperative catalysis of noncompatible catalysts through compartmentalization: Wacker oxidation and enzymatic reduction in a one-pot process in aqueous media. Angew. Chem. Int. Ed. 2015, 54, 4488–4492. [Google Scholar] [CrossRef] [PubMed]
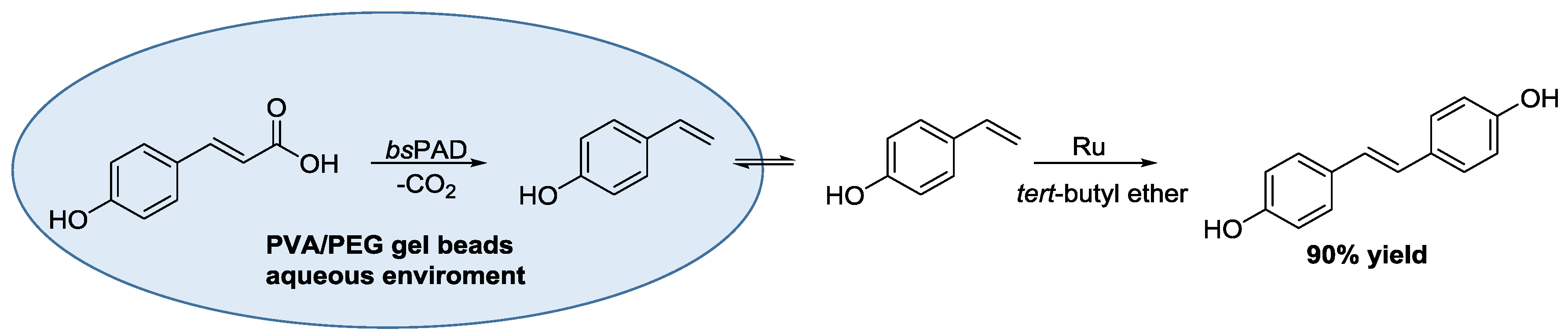
- Gómez Baraibar, Á.; Reichert, D.; Mügge, C.; Seger, S.; Gröger, H.; Kourist, R. A one-pot cascade reaction combining an encapsulated decarboxylase with a metathesis catalyst for the synthesis of bio-based antioxidants. Angew. Chem. Int. Ed. 2016, 55, 14823–14827. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.; Zhao, J.; Chen, X. Cooperative photoredox catalysis. Chem. Soc. Rev. 2016, 45, 3026–3038. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.H.; Nguyen, D.; Dwaraknath, S.; Mahadevan, S.; Chavez, G.; Nguyen, A.; Dao, T.; Mullen, S.; Nguyen, T.A.; Cheruzel, L.E. An efficient light-driven P450 BM3 biocatalyst. J. Am. Chem. Soc. 2013, 135, 14484–14487. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, S.H.; Cha, G.S.; Choi, D.S.; Nam, D.H.; Lee, J.H.; Lee, J.K.; Yun, C.H.; Jeong, K.J.; Park, C.B. Cofactor-free light-driven whole-cell cytochrome P450 catalysis. Angew. Chem. Int. Ed. 2015, 54, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Baeg, J.O.; Park, N.J.; Yadav, R.K. A photocatalyst/enzyme couple that uses solar energy in the asymmetric reduction of acetophenones. Angew. Chem. Int. Ed. 2012, 51, 11624–11628. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Ryu, J.; Kim, J.H.; Lee, S.H.; Park, C.B. Biocatalyzed artificial photosynthesis by hydrogen-terminated silicon nanowires. ChemSusChem 2012, 5, 2129–2132. [Google Scholar] [CrossRef] [PubMed]
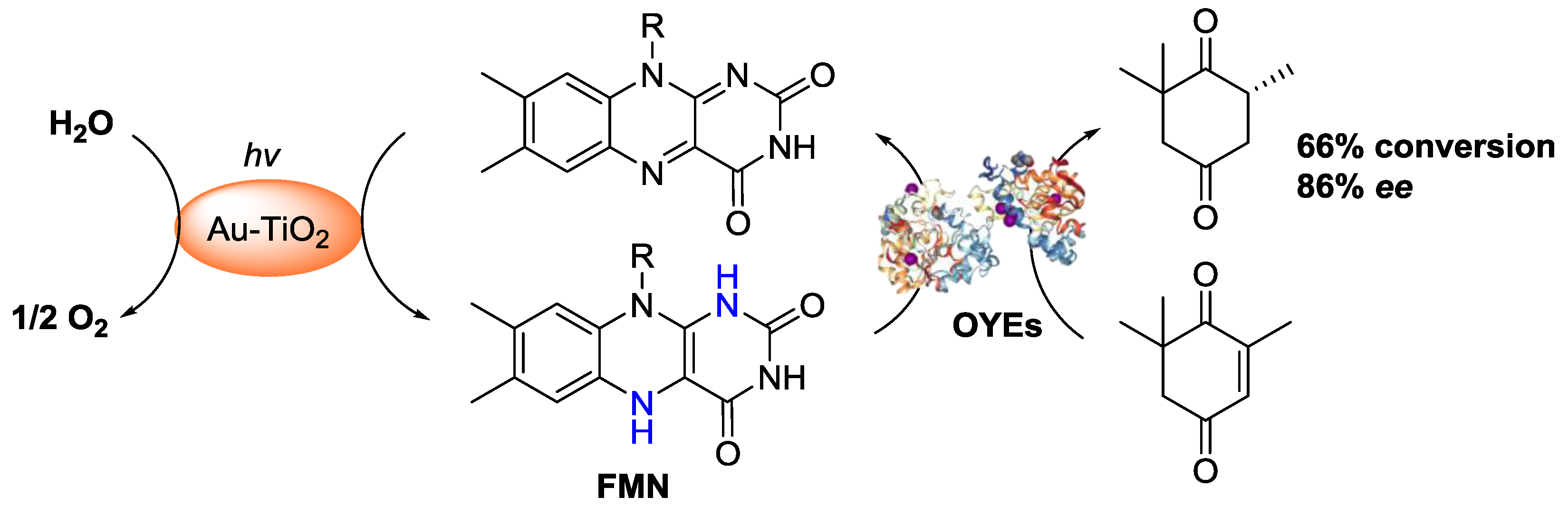
- Mifsud, M.; Gargiulo, S.; Iborra, S.; Arends, I.W.; Hollmann, F.; Corma, A. Photobiocatalytic chemistry of oxidoreductases using water as the electron donor. Nat. Commun. 2014, 5, 3145. [Google Scholar] [CrossRef] [PubMed]
- Peers, M.K.; Toogood, H.S.; Heyes, D.J.; Mansell, D.; Coe, B.J.; Scrutton, N.S. Light-driven biocatalytic reduction of α,β-unsaturated compounds by ene reductases employing transition metal complexes as photosensitizers. Catal. Sci. Technol. 2016, 6, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Heinisch, T.; Ward, T.R. Artificial metalloenzymes based on the biotin-streptavidin technology: Challenges and opportunities. Acc. Chem. Res. 2016, 49, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Rosati, F.; Roelfes, G. Artificial metalloenzymes. ChemCatChem 2010, 2, 916–927. [Google Scholar] [CrossRef]
- Nastri, F.; Chino, M.; Maglio, O.; Bhagi-Damodaran, A.; Lu, Y.; Lombardi, A. Design and engineering of artificial oxygen-activating metalloenzymes. Chem. Soc. Rev. 2016, 45, 5020–5054. [Google Scholar] [CrossRef] [PubMed]
- Petrik, I.D.; Liu, J.; Lu, Y. Metalloenzyme design and engineering through strategic modifications of native protein scaffolds. Curr. Opin. Chem. Biol. 2014, 19, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.; Roelfes, G. Artificial metalloenzymes for enantioselective catalysis. Curr. Opin. Chem. Biol. 2014, 19, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Cangelosi, V.M.; Zastrow, M.L.; Tegoni, M.; Plegaria, J.S.; Tebo, A.G.; Mocny, C.S.; Ruckthong, L.; Qayyum, H.; Pecoraro, V.L. Protein design: Toward functional metalloenzymes. Chem. Rev. 2014, 114, 3495–3578. [Google Scholar] [CrossRef] [PubMed]
- Drienovská, I.; Rioz-Martínez, A.; Draksharapu, A.; Roelfes, G. Novel artificial metalloenzymes by in vivo incorporation of metal-binding unnatural amino acids. Chem. Sci. 2015, 6, 770–776. [Google Scholar] [CrossRef]
- Heinisch, T.; Pellizzoni, M.; Durrenberger, M.; Tinberg, C.E.; Kohler, V.; Klehr, J.; Haussinger, D.; Baker, D.; Ward, T.R. Improving the catalytic performance of an artificial metalloenzyme by computational design. J. Am. Chem. Soc. 2015, 137, 10414–10419. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.C. Metallopeptide catalysts and artificial metalloenzymes containing unnatural amino acids. Curr. Opin. Chem. Biol. 2015, 25, 27–35. [Google Scholar] [CrossRef] [PubMed]
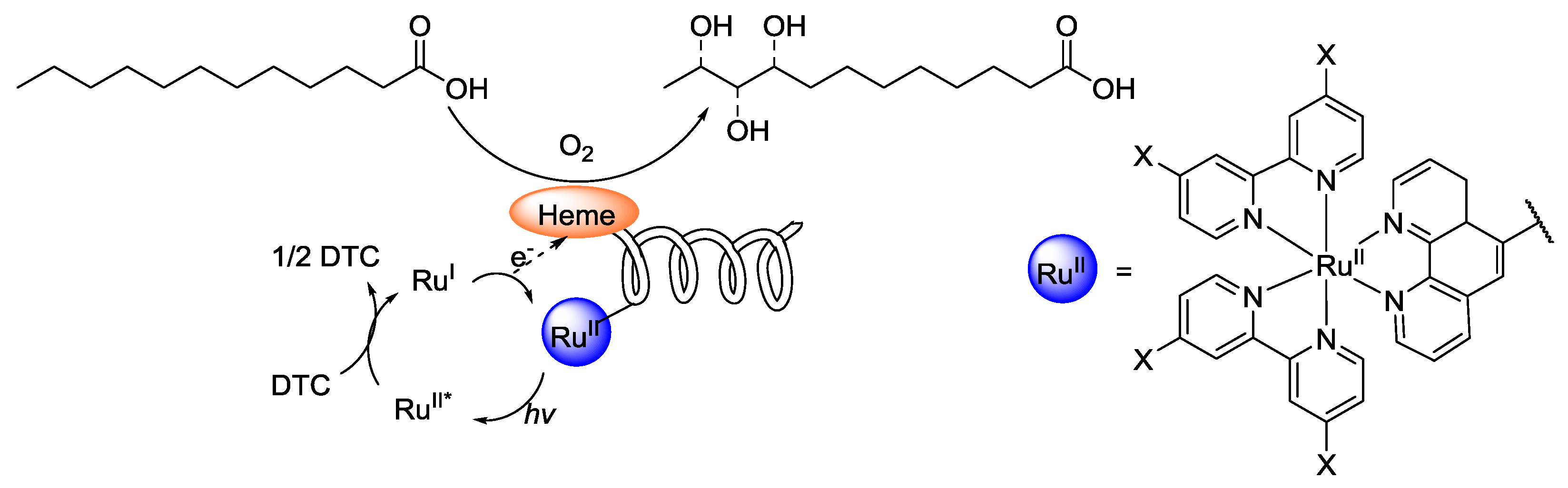
- Key, H.M.; Dydio, P.; Clark, D.S.; Hartwig, J.F. Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 2016, 534, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Jeschek, M.; Reuter, R.; Heinisch, T.; Trindler, C.; Klehr, J.; Panke, S.; Ward, T.R. Directed evolution of artificial metalloenzymes for in vivo metathesis. Nature 2016, 537, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Wilson, Y.M.; Durrenberger, M.; Nogueira, E.S.; Ward, T.R. Neutralizing the detrimental effect of glutathione on precious metal catalysts. J. Am. Chem. Soc. 2014, 136, 8928–8932. [Google Scholar] [CrossRef] [PubMed]
- Kohler, V.; Wilson, Y.M.; Durrenberger, M.; Ghislieri, D.; Churakova, E.; Quinto, T.; Knorr, L.; Haussinger, D.; Hollmann, F.; Turner, N.J.; et al. Synthetic cascades are enabled by combining biocatalysts with artificial metalloenzymes. Nat. Chem. 2013, 5, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, J.R. Microbial reduction of metals and radionuclides. FEMS Microbiol. Rev. 2003, 27, 411–425. [Google Scholar] [CrossRef]
- De Windt, W.; Aelterman, P.; Verstraete, W. Bioreductive deposition of palladium (0) nanoparticles on shewanella oneidensis with catalytic activity towards reductive dechlorination of polychlorinated biphenyls. Environ. Microbiol. 2005, 7, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Raab, R.M.; Tyo, K.; Stephanopoulos, G. Metabolic engineering. Adv. Biochem. Eng. Biotechnol. 2005, 100, 1–17. [Google Scholar] [PubMed]
- Foulkes, J.M.; Malone, K.J.; Coker, V.S.; Turner, N.J.; Lloyd, J.R. Engineering a biometallic whole cell catalyst for enantioselective deracemization reactions. ACS Catal. 2011, 1, 1589–1594. [Google Scholar] [CrossRef]
- Royer, G.P.; Chow, W.-S.; Hatton, K.S. Palladium/polyethylenimine catalysts. J. Mol. Catal. 1985, 31, 1–13. [Google Scholar] [CrossRef]
- Sirasani, G.; Tong, L.; Balskus, E.P. A biocompatible alkene hydrogenation merges organic synthesis with microbial metabolism. Angew. Chem. Int. Ed. 2014, 53, 7785–7788. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, J.; Chen, P.R. Transition metal-mediated bioorthogonal protein chemistry in living cells. Chem. Soc. Rev. 2014, 43, 6511–6526. [Google Scholar] [CrossRef] [PubMed]
- Volker, T.; Dempwolff, F.; Graumann, P.L.; Meggers, E. Progress towards bioorthogonal catalysis with organometallic compounds. Angew. Chem. Int. Ed. 2014, 53, 10536–10540. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Gamasa, M.; Martínez-Calvo, M.; Couceiro, J.R.; Mascareñas, J.L. Transition metal catalysis in the mitochondria of living cells. Nat. Commun. 2016, 7, 12538. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, J.; Zhao, J.; Wang, J.; Zheng, S.; Lin, S.; Chen, L.; Yang, M.; Jia, S.; Zhang, X.; et al. Palladium-triggered deprotection chemistry for protein activation in living cells. Nat. Chem. 2014, 6, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, D.; Grela, K. Aqueous olefin metathesis. Angew. Chem. Int. Ed. 2009, 48, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Binder, J.B.; Raines, R.T. Olefin metathesis for chemical biology. Curr. Opin. Chem. Biol. 2008, 12, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Gulajski, L.; Michrowska, A.; Naroznik, J.; Kaczmarska, Z.; Rupnicki, L.; Grela, K. A highly active aqueous olefin metathesis catalyst bearing a quaternary ammonium group. ChemSusChem 2008, 1, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Schaper, L.A.; Hock, S.J.; Herrmann, W.A.; Kuhn, F.E. Synthesis and application of water-soluble NHC transition-metal complexes. Angew. Chem. Int. Ed. 2013, 52, 270–289. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, D.; Barua, N.C. Recent developments on carbon-carbon bond forming reactions in water. Curr. Org. Synth. 2012, 9, 17–30. [Google Scholar] [CrossRef]
- Datta, S.; Christena, L.R.; Rajaram, Y.R.S. Enzyme immobilization: An overview on techniques and support materials. 3 Biotech 2013, 3, 1–9. [Google Scholar] [CrossRef]
- Engström, K.; Johnston, E.V.; Verho, O.; Gustafson, K.P.J.; Shakeri, M.; Tai, C.-W.; Bäckvall, J.-E. Co-immobilization of an enzyme and a metal into the compartments of mesoporous silica for cooperative tandem catalysis: An artificial metalloenzyme. Angew. Chem. Int. Ed. 2013, 52, 14006–14010. [Google Scholar] [CrossRef] [PubMed]
- Engelmark Cassimjee, K.; Kadow, M.; Wikmark, Y.; Svedendahl Humble, M.; Rothstein, M.L.; Rothstein, D.M.; Backvall, J.E. A general protein purification and immobilization method on controlled porosity glass: Biocatalytic applications. Chem. Commun. 2014, 50, 9134–9137. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Cross-linked enzyme aggregates (CLEA®s): Stable and recyclable biocatalysts. Biochem. Soc. Trans. 2007, 35, 1583–1587. [Google Scholar] [CrossRef] [PubMed]
- Frushicheva, M.P.; Mills, M.J.; Schopf, P.; Singh, M.K.; Prasad, R.B.; Warshel, A. Computer aided enzyme design and catalytic concepts. Curr. Opin. Chem. Biol. 2014, 21, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Porebski, B.T.; Buckle, A.M. Consensus protein design. Protein Eng. Des. Sel. 2016, 29, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.S.; Liu, D.R. Methods for the directed evolution of proteins. Nat. Rev. Genet. 2015, 16, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Bistri, O.; Reinaud, O. Supramolecular control of transition metal complexes in water by a hydrophobic cavity: A bio-inspired strategy. Org. Biomol. Chem. 2015, 13, 2849–2865. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Ilie, A.; Sun, Z.; Lonsdale, R.; Xu, J.H.; Reetz, M.T. Whole-cell-catalyzed multiple regio- and stereoselective functionalizations in cascade reactions enabled by directed evolution. Angew. Chem. Int. Ed. 2016, 55, 12026–12029. [Google Scholar] [CrossRef] [PubMed]
- Both, P.; Busch, H.; Kelly, P.P.; Mutti, F.G.; Turner, N.J.; Flitsch, S.L. Whole-cell biocatalysts for stereoselective C–H amination reactions. Angew. Chem. Int. Ed. 2016, 55, 1511–1513. [Google Scholar] [CrossRef] [PubMed]
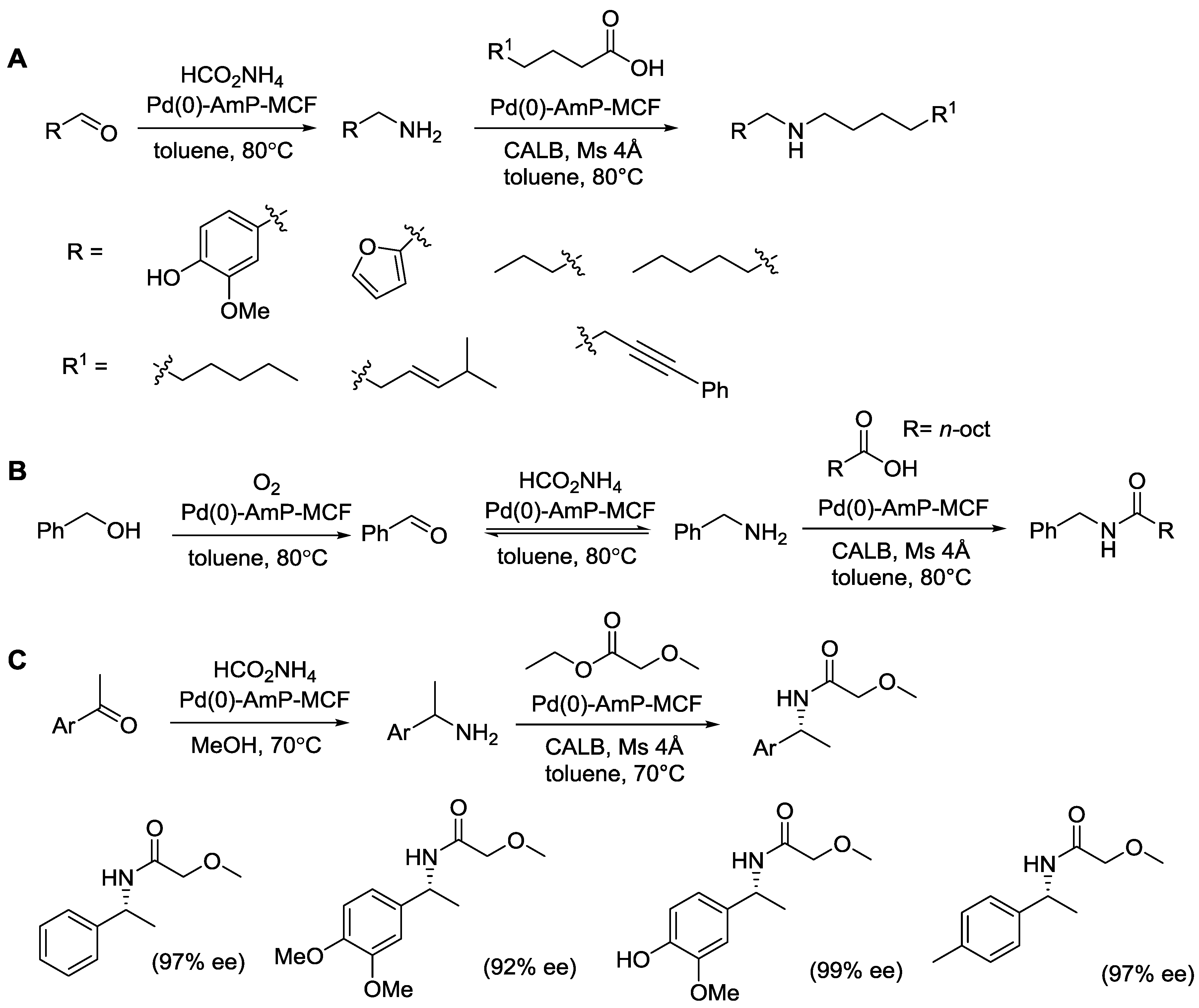
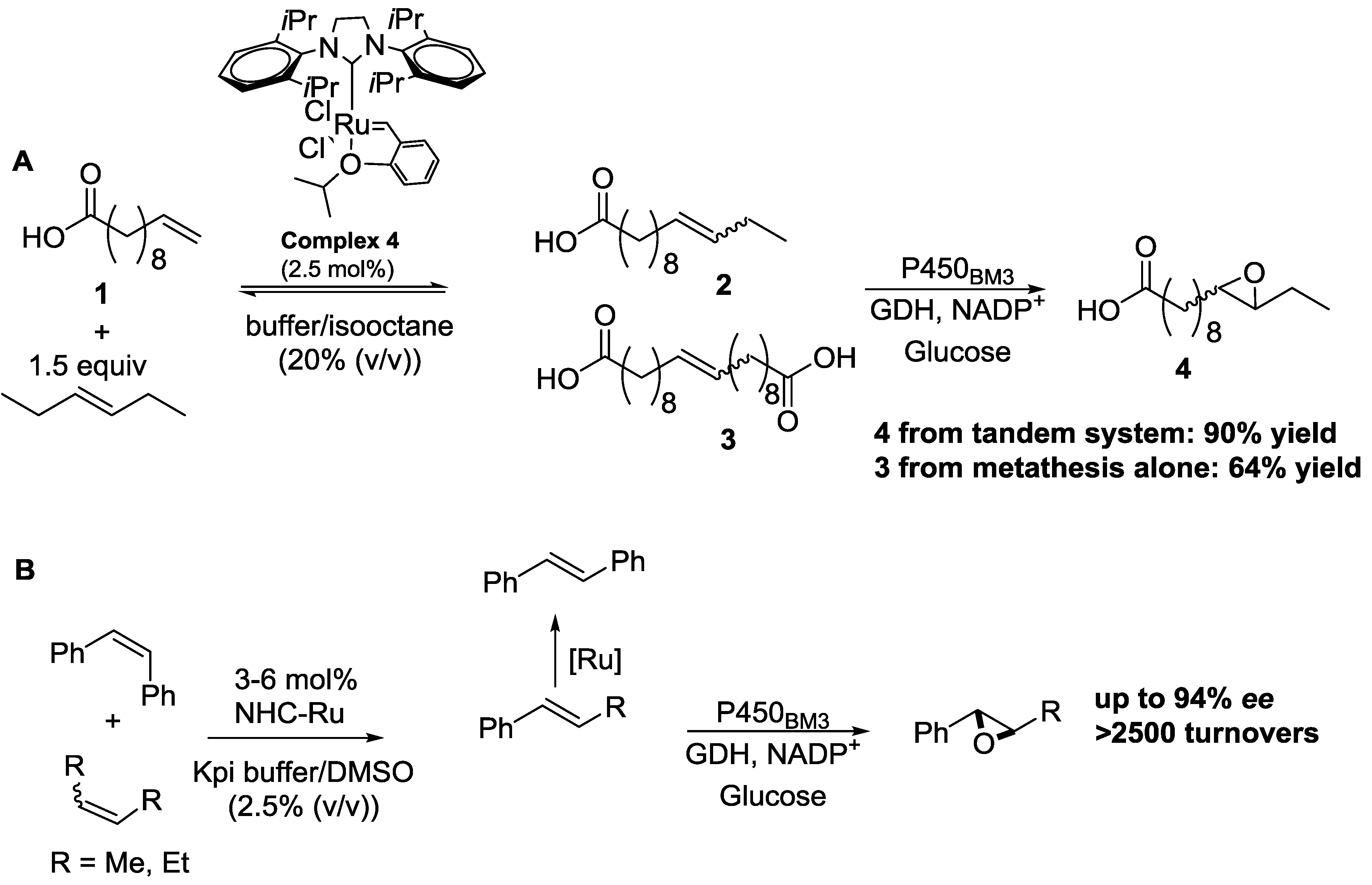
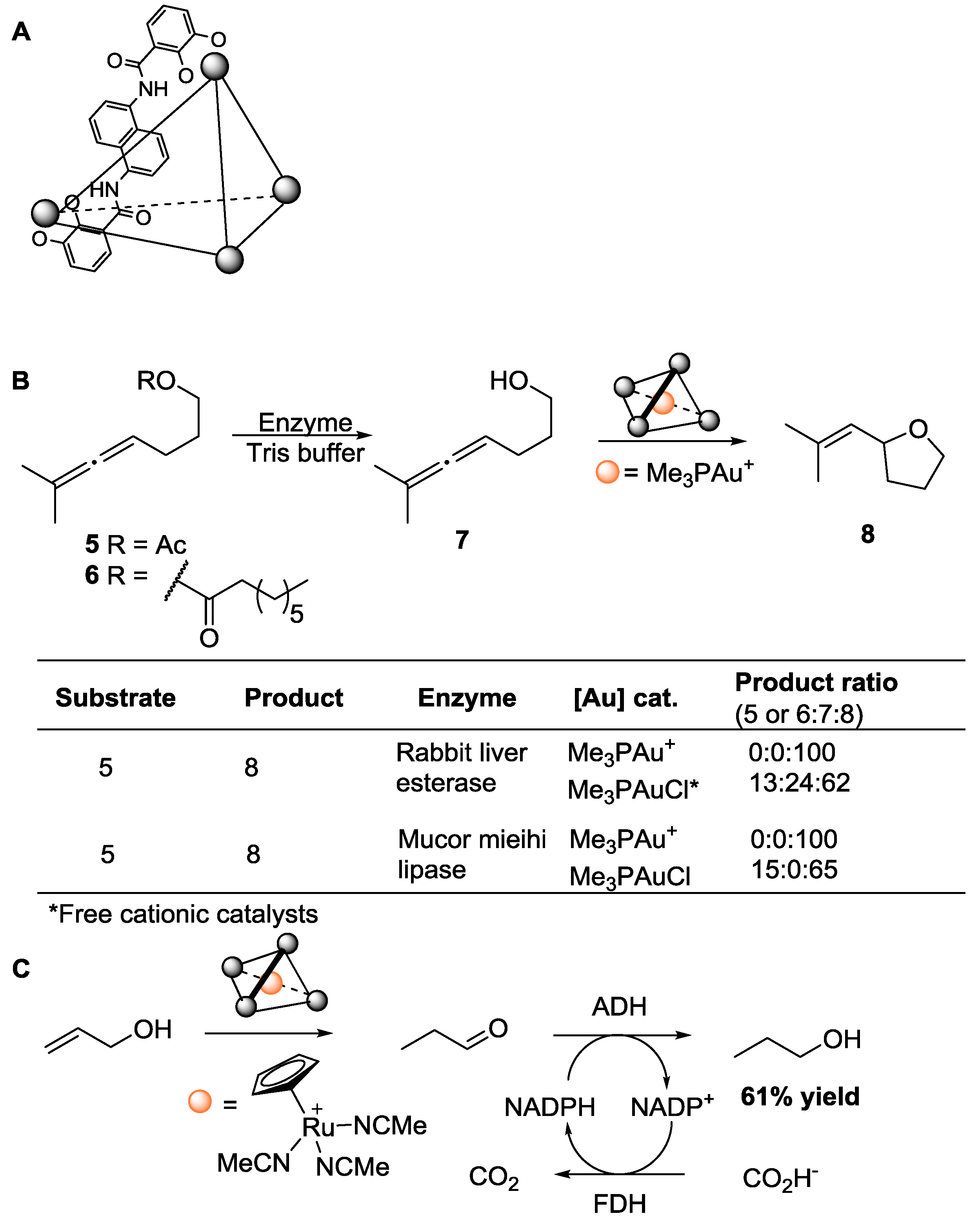
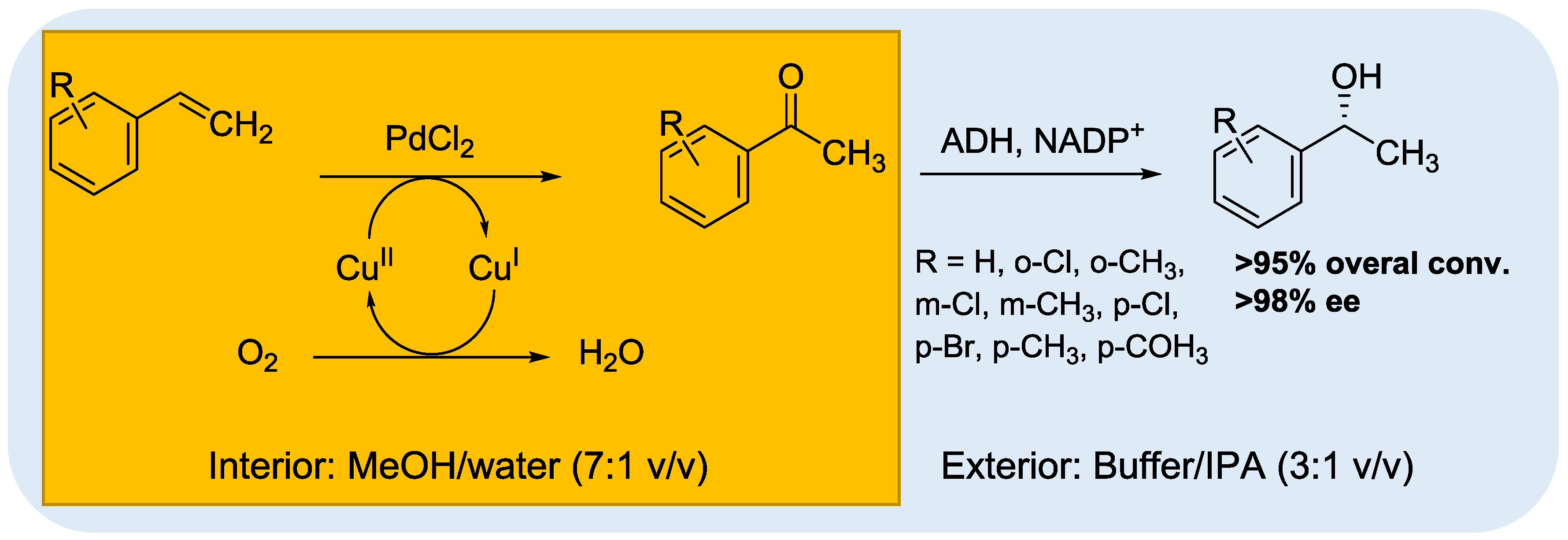
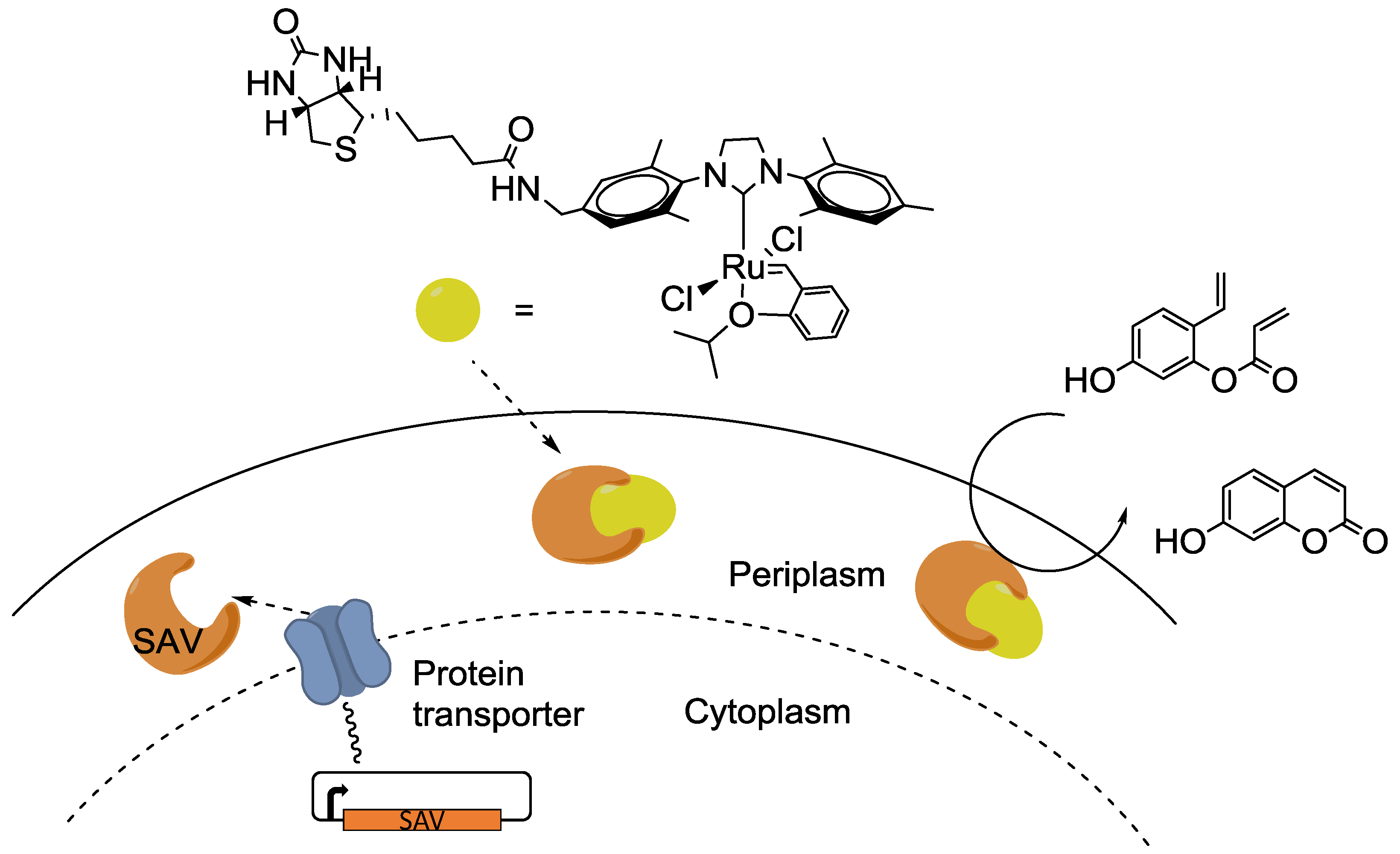
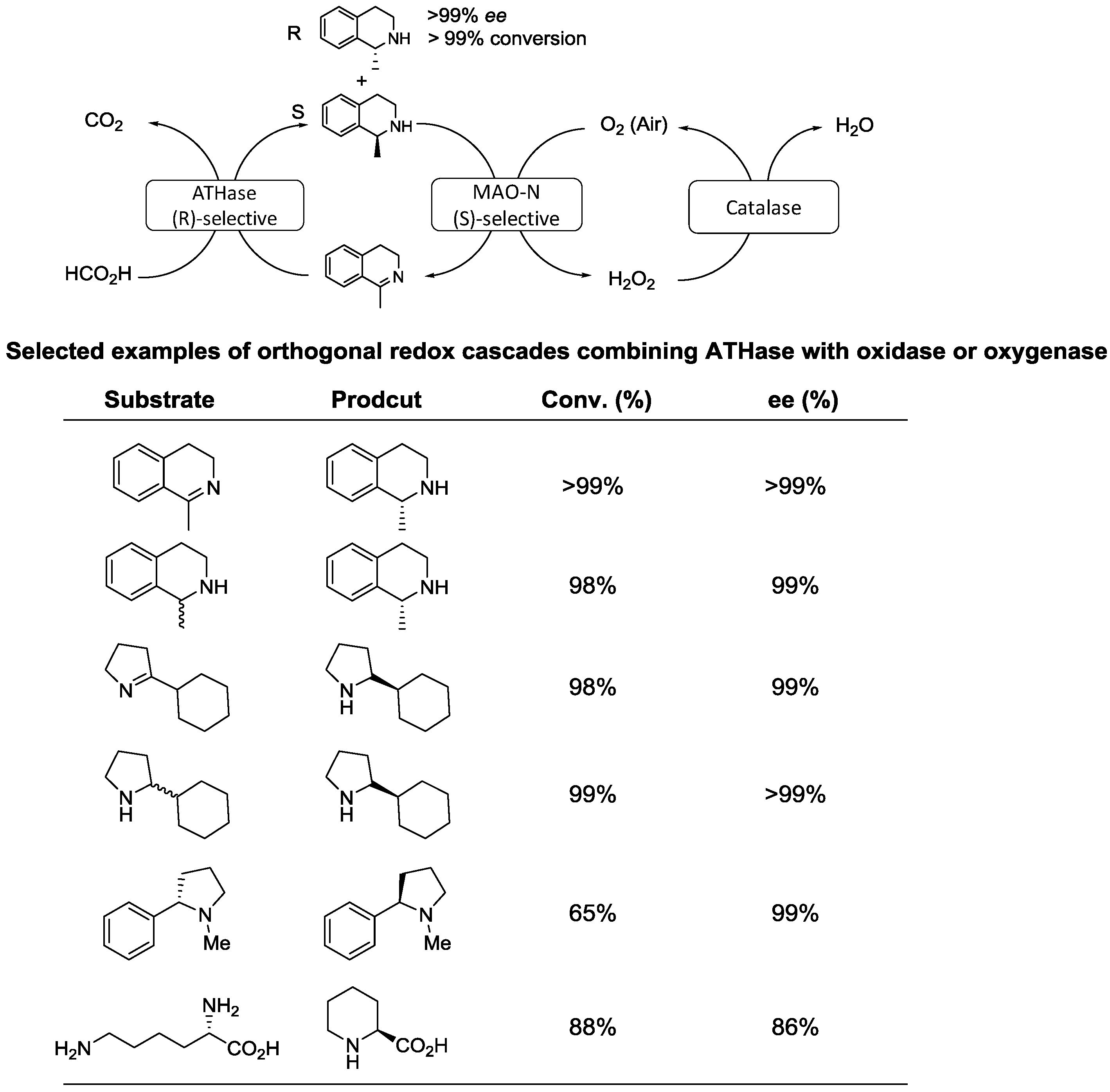
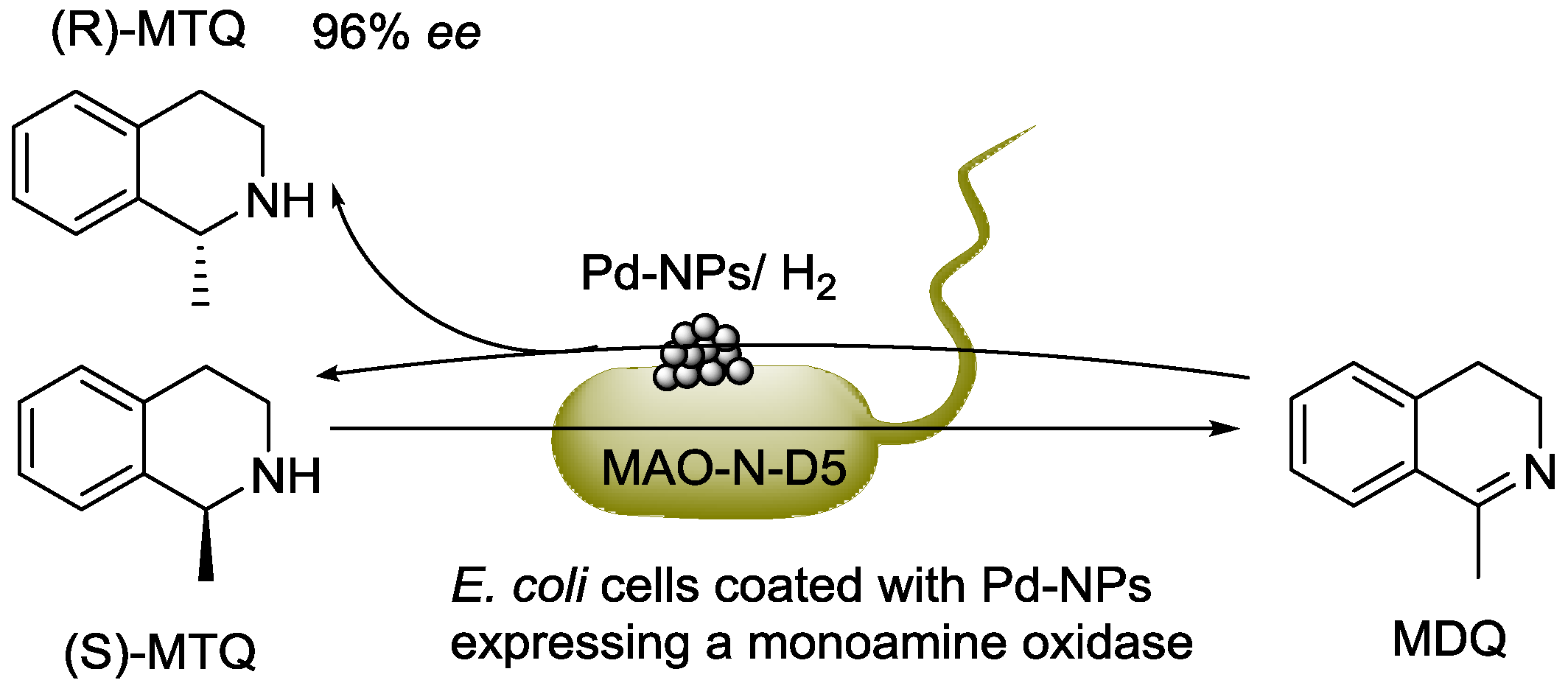
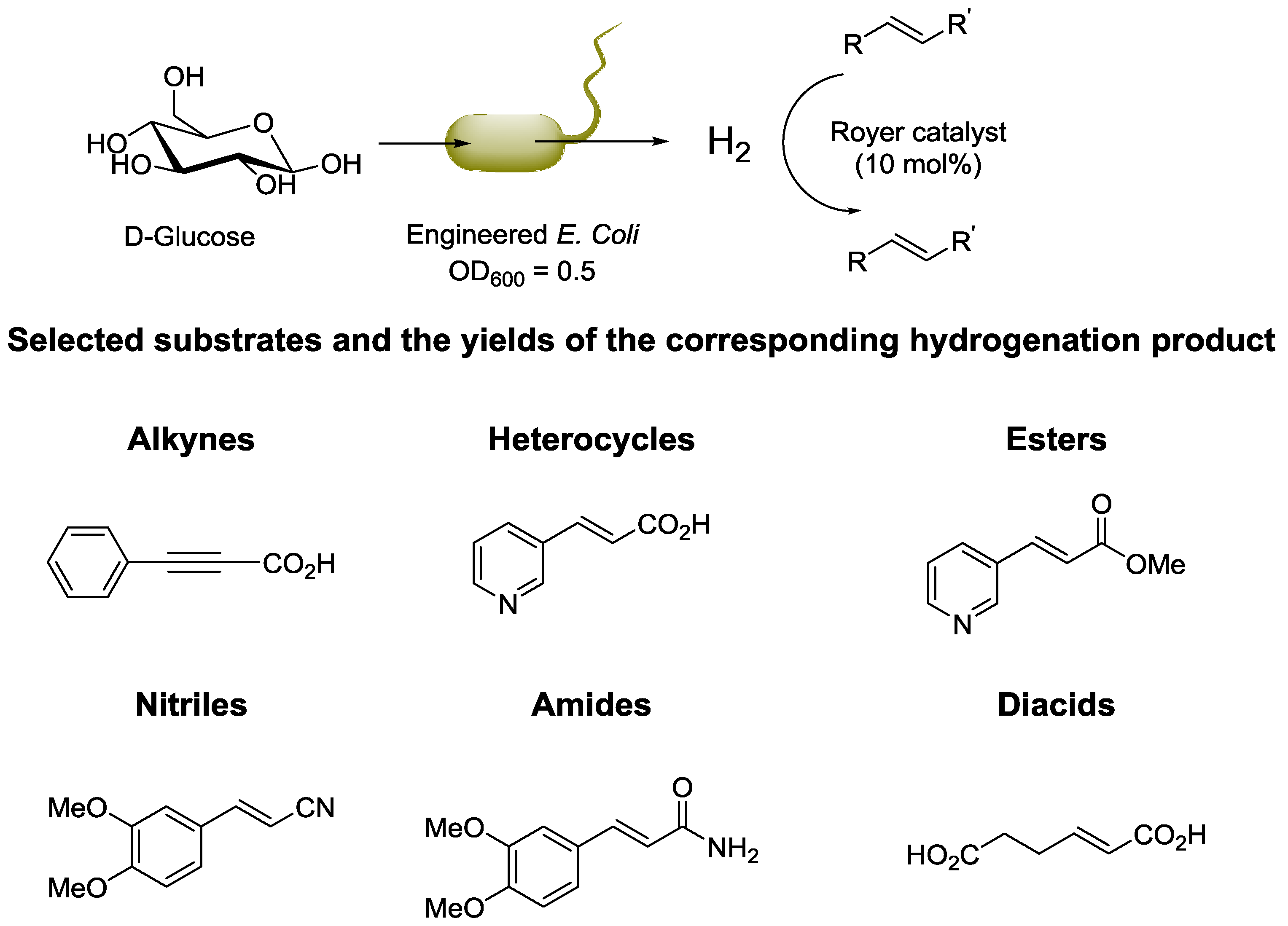
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhao, H. Tandem Reactions Combining Biocatalysts and Chemical Catalysts for Asymmetric Synthesis. Catalysts 2016, 6, 194. https://doi.org/10.3390/catal6120194
Wang Y, Zhao H. Tandem Reactions Combining Biocatalysts and Chemical Catalysts for Asymmetric Synthesis. Catalysts. 2016; 6(12):194. https://doi.org/10.3390/catal6120194
Chicago/Turabian StyleWang, Yajie, and Huimin Zhao. 2016. "Tandem Reactions Combining Biocatalysts and Chemical Catalysts for Asymmetric Synthesis" Catalysts 6, no. 12: 194. https://doi.org/10.3390/catal6120194
APA StyleWang, Y., & Zhao, H. (2016). Tandem Reactions Combining Biocatalysts and Chemical Catalysts for Asymmetric Synthesis. Catalysts, 6(12), 194. https://doi.org/10.3390/catal6120194