
Information-Driven Catalyst Design Based on High-Throughput Intrinsic Kinetics



Abstract
:
1. Introduction
2. Catalyst Design Methodologies
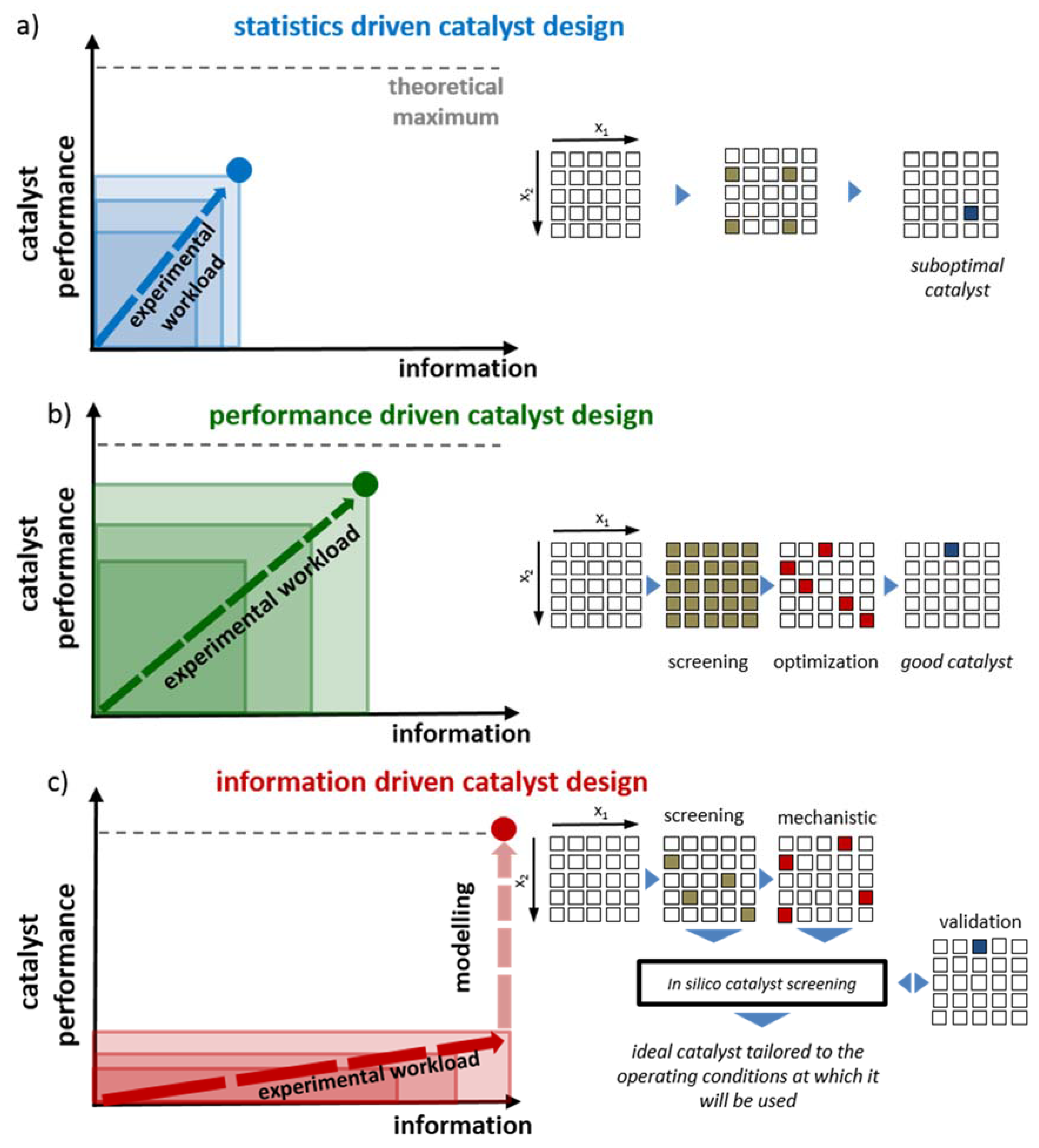
2.1. Statistics-Driven Catalyst Design
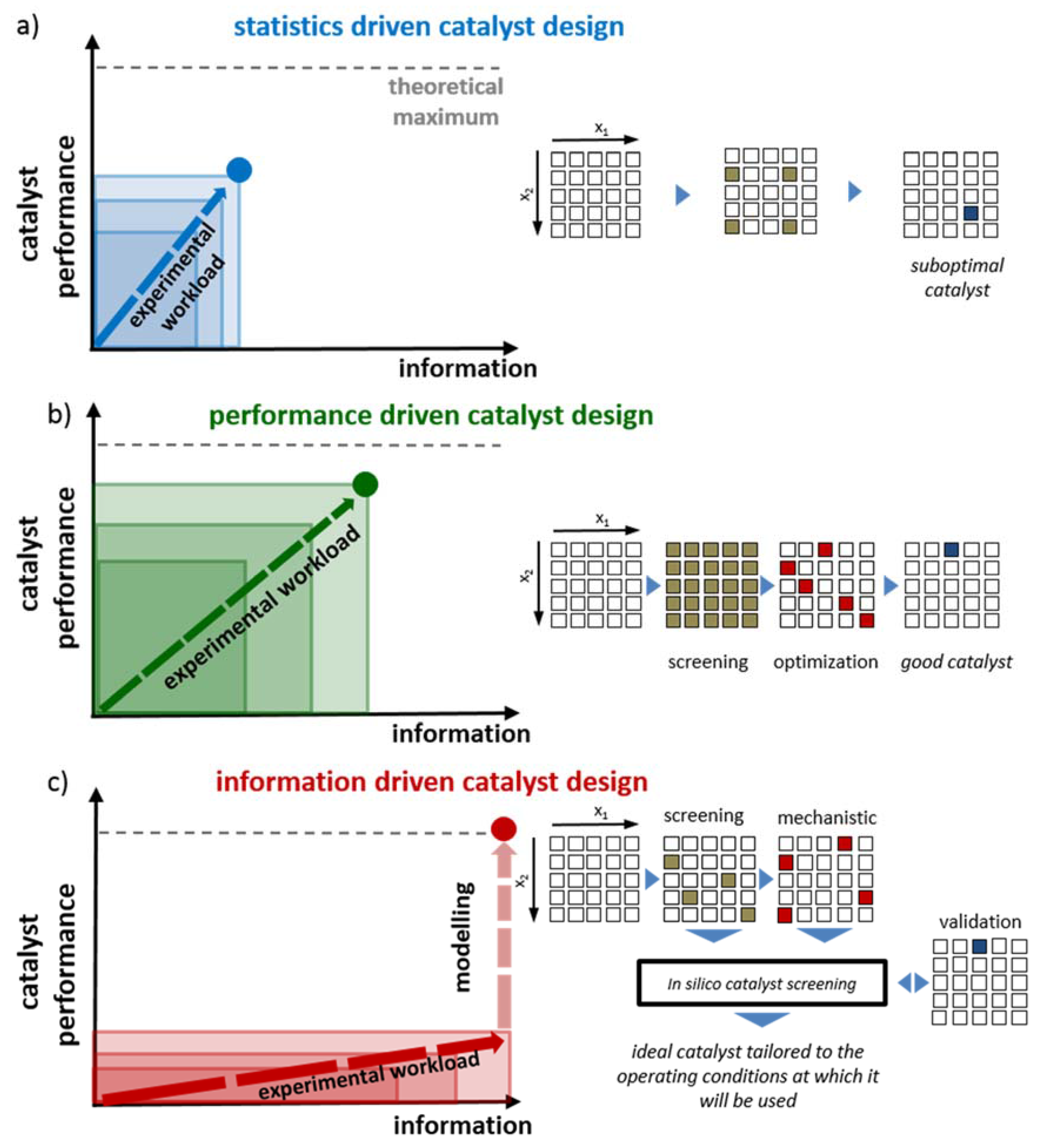
2.2. Performance-Driven Catalyst Design
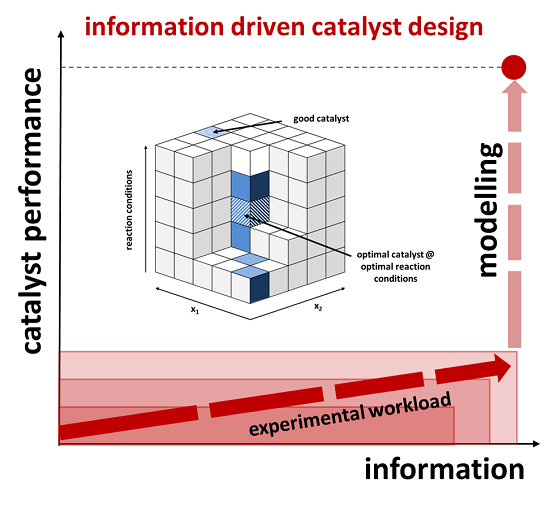
2.3. Information-Driven Catalyst Design
3. High-Throughput Kinetics Information Acquisition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature or Operating Condition | High-Throughput Kinetics Screening (HTK-S) | High-Throughput Kinetics Mechanistic Investigation (HTK-MI) |
|---|---|---|
| number of reactors | 16 | 8 |
| number of heating blocks | 4 | 4 |
| reactor type | Tubular | Tubular |
| reactor internal diameter did (10−3 m) | 2.1 | 11.0 |
| reactor length L (m) | 0.8 | 0.9 |
| feed flow rate control | per reactor block | per reactor |
| operating temperature range Tmin, Tmax (K) | 323–773 (SS) 323–1273 (Quartz) | 323–923 |
| operating pressure range pmin, pmax (105 Pa) | 1–100 (SS) 1–3 (Quartz) | 1–200 |
| catalyst mass W (10−3 kg) | 0.05–0.2 | 0.5–10 |
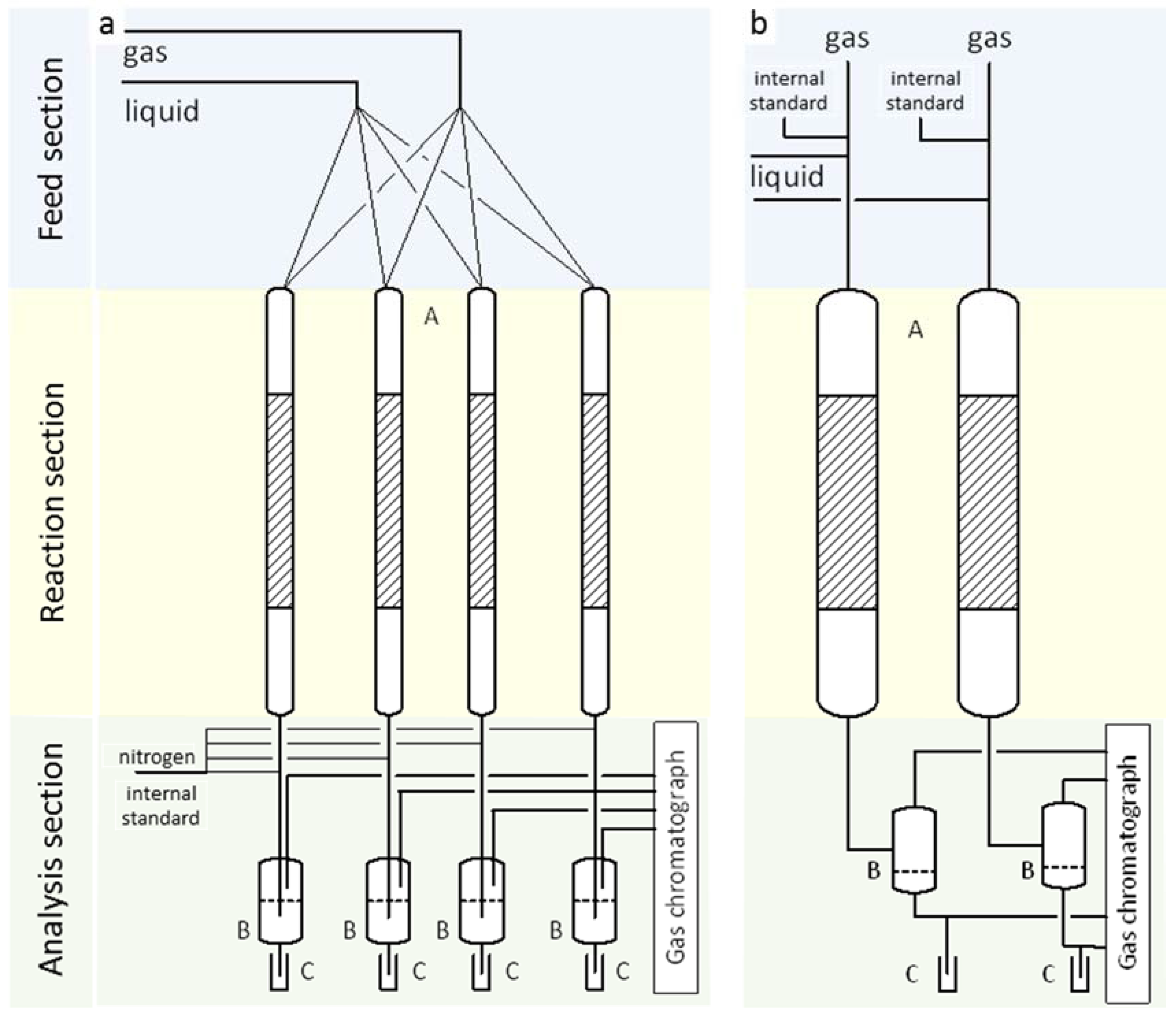
3.1. Screening (HTK-S)

3.1.1. Feed Section
3.1.2. Reaction Section
3.1.3. Analysis Section
3.2. Mechanistic Investigation (HTK-MI)
3.2.1. Feed Section

3.2.2. Reaction Section
3.2.3. Analysis Section
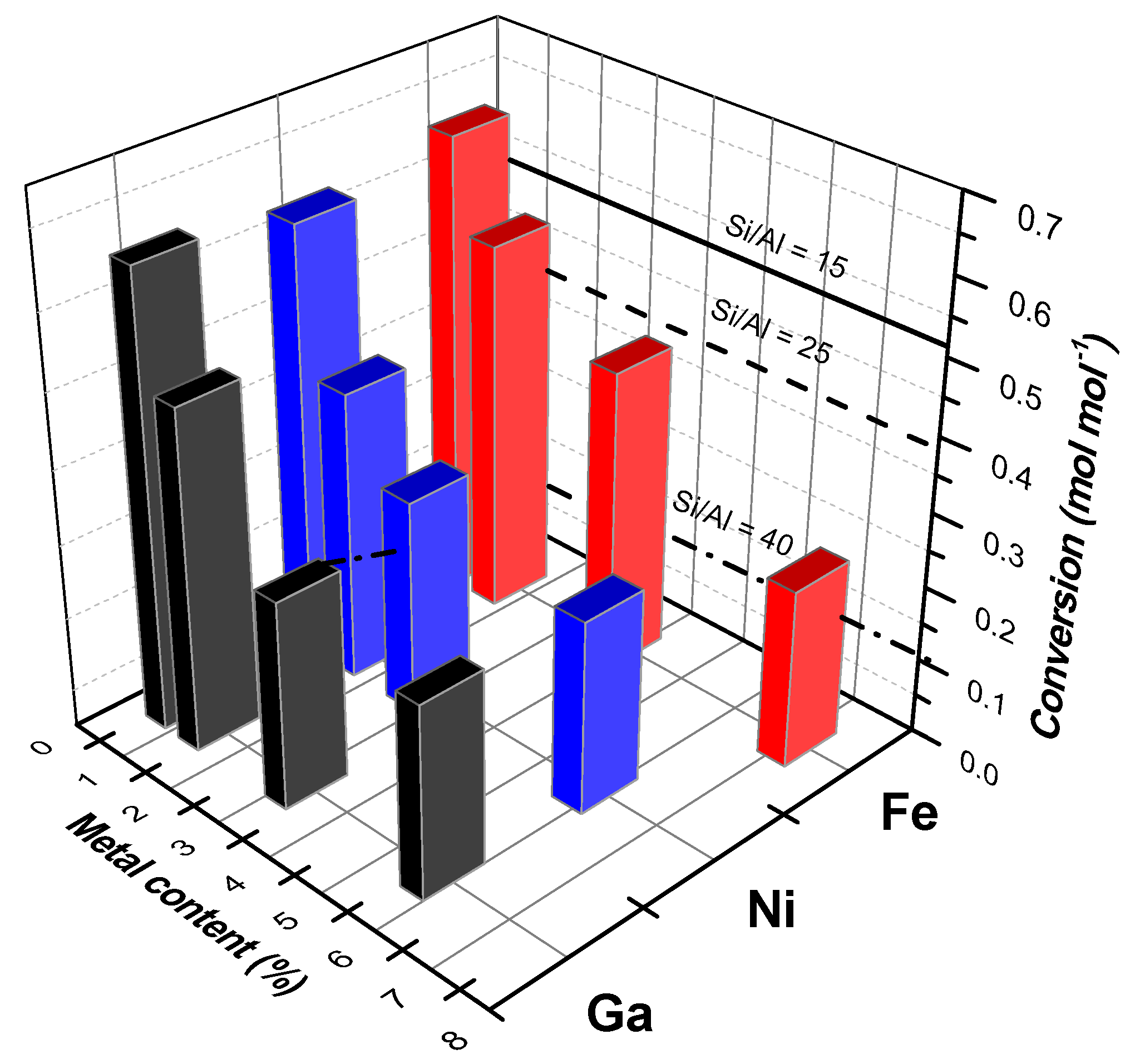
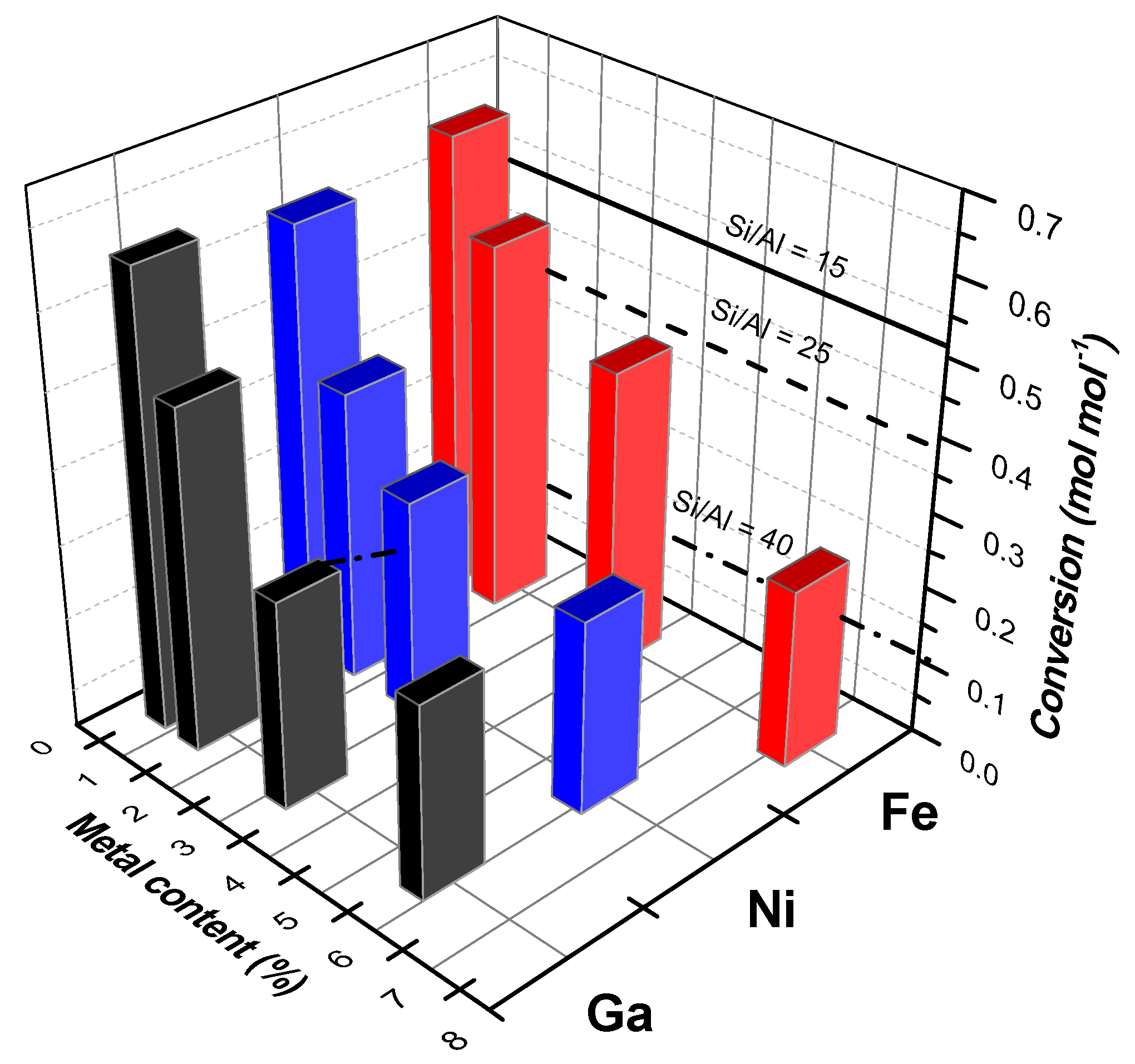
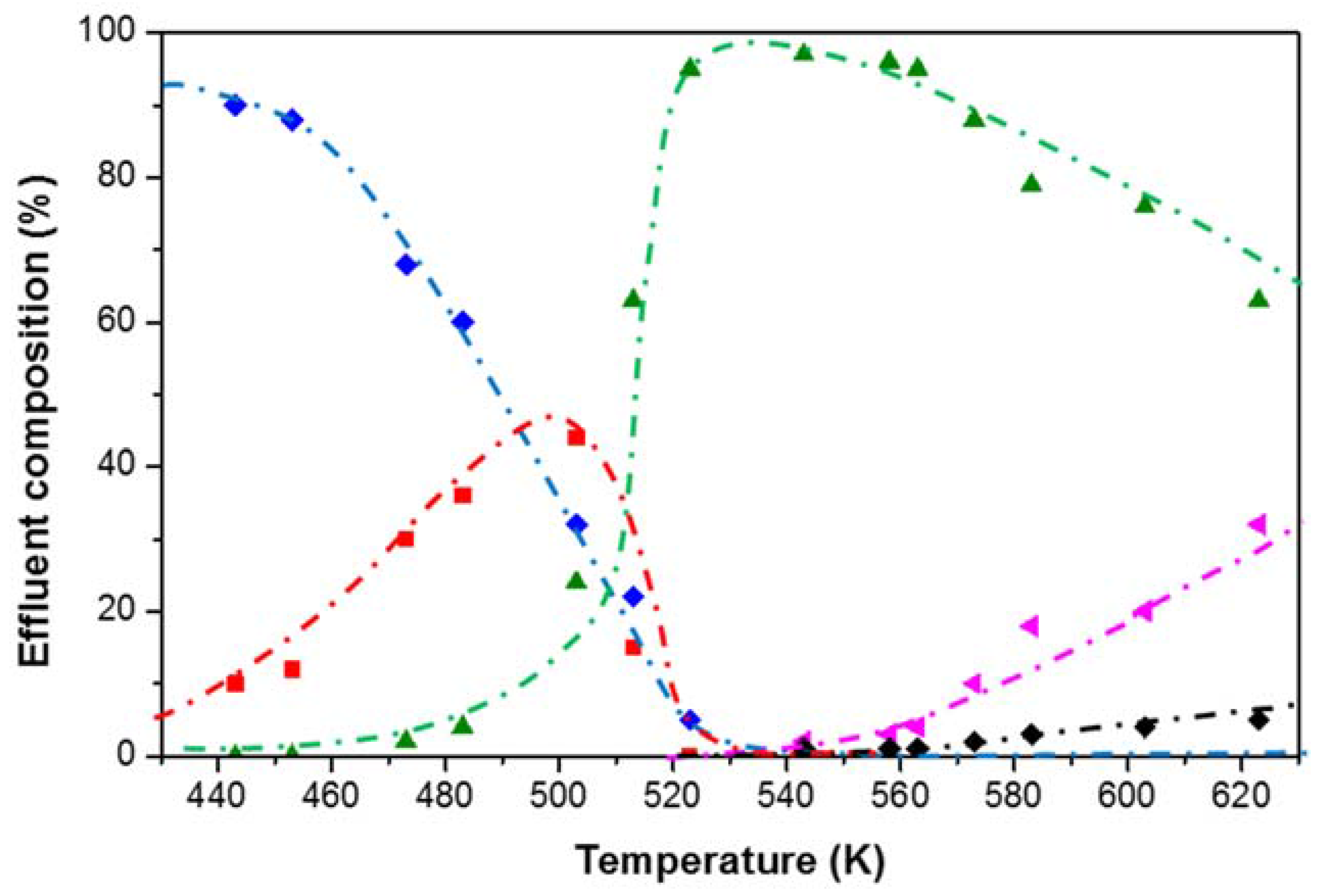
4. Case Study: Ethanol Conversion to Hydrocarbons
| Phenomenon | Criterion | HTK-S | HTK-MI | |
|---|---|---|---|---|
| Mass transfer | external | Ca | 2.9 × 10−4 < 0.05 | 5.3 × 10−3 < 0.05 |
| internal | Φ | 9.7 × 10−2 < 0.08 | 3.0 × 10−3 < 0.08 | |
| Heat transfer | external | ΔText | 1.3 × 10−2 < 2.0 K | 1.4 × 10−1 < 6.0 K |
| internal | ΔTint | 1.4 × 10−3 < 2.0 K | 5.1 × 10−3 < 6.0 K | |
| Flow pattern ideality Plug flow | radial dispersion | dt/dp | 24 > 8 | 16 > 8 |
| axial dispersion | LB/dp | 1074 > 50 | 519 > 50 | |
| pressure drop | ΔP/P | 7.5 × 10−2 < 0.2 | 1.1 × 10−3 < 0.2 | |
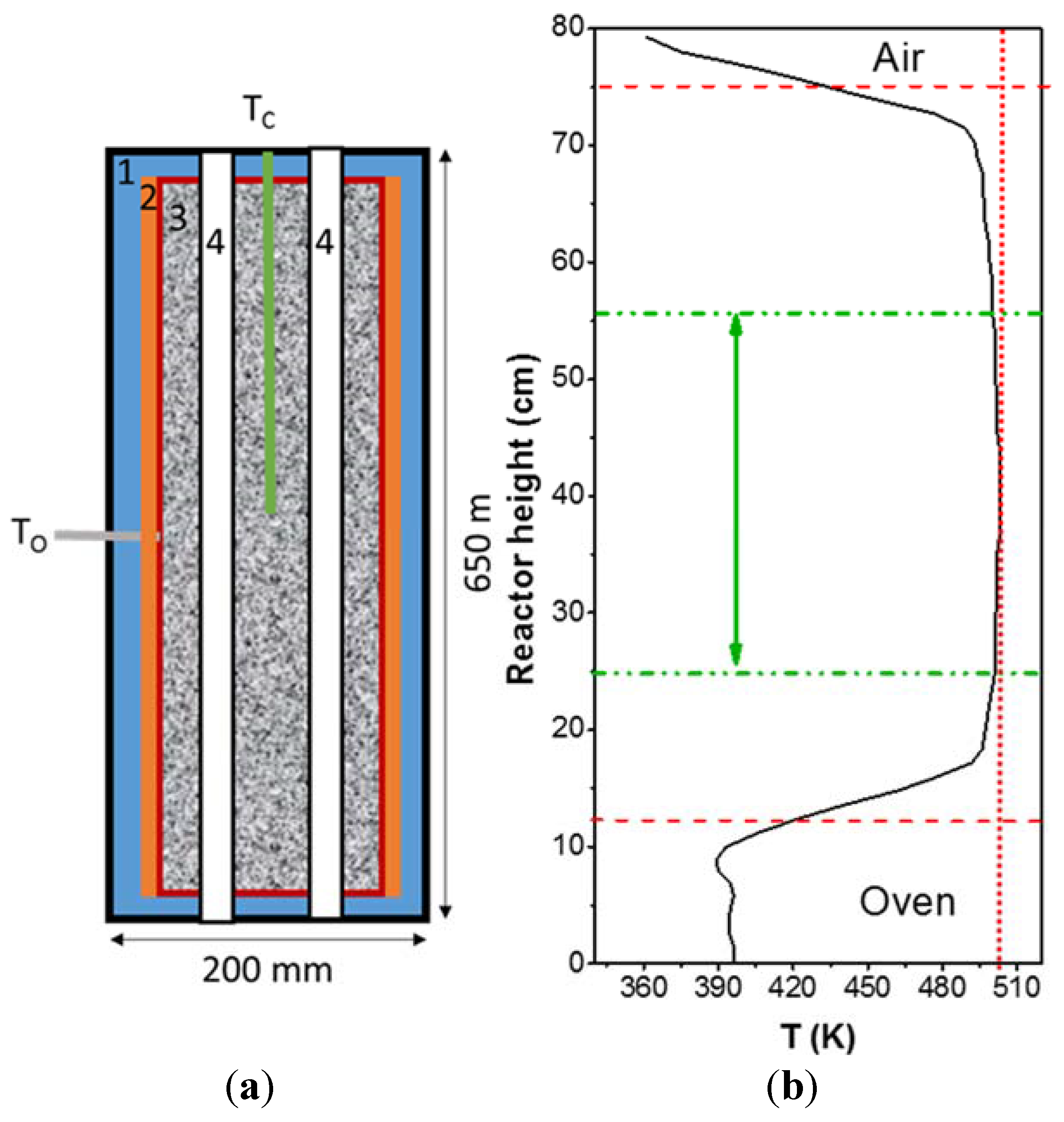
4.1. Catalyst Screening (HTK-S Set-Up)
4.2. Mechanistic Investigation (HTK-MI Set-Up)
5. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Nomenclature
| Roman | ||
| bi | parameter | - |
| Ca | Carberry number | - |
| Fi | molar flow rate of component i | mol·s−1 |
| L | length | m |
| d | diameter | m |
| p | pressure | Pa |
| pi | partial pressure of component i | Pa |
| T | temperature | K |
| t | time | h |
| W | catalyst mass | kg |
| x | factor | - |
| Y | output variable | - |
| Greek | ||
| Ф | Weisz modulus | - |
| Subscripts | ||
| B | bed | - |
| Cap | capillary | - |
| G | gas | - |
| EtOH | ethanol | - |
| Id | internal diameter | - |
| L | liquid | - |
| max | maximum | - |
| min | minimum | - |
| P | pellet | - |
| React | reaction | - |
| T | total | - |
| Superscripts | ||
| ° | inlet | |
References
- Norskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Senkan, S.M. High-throughput screening of solid-state catalyst libraries. Nature 1998, 394, 350–353. [Google Scholar] [CrossRef]
- Senkan, S. Combinatorial heterogeneous catalysis—A new path in an old field. Angew. Chem. Int. Ed. 2001, 40, 312–329. [Google Scholar] [CrossRef]
- Hoogenboom, R.; Meier, M.A.R.; Schubert, U.S. Combinatorial methods, automated synthesis and high-throughput screening in polymer research: Past and present. Macromol. Rapid. Commun. 2003, 24, 16–32. [Google Scholar] [CrossRef]
- Smil, V. Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production; MIT Press: Cambridge, MA, USA, 2001; p. 338. [Google Scholar]
- Maier, W.F.; Stowe, K.; Sieg, S. Combinatorial and high-throughput materials science. Angew. Chem. Int. Ed. 2007, 46, 6016–6067. [Google Scholar] [CrossRef] [PubMed]
- Cong, P.J.; Doolen, R.D.; Fan, Q.; Giaquinta, D.M.; Guan, S.H.; McFarland, E.W.; Poojary, D.M.; Self, K.; Turner, H.W.; Weinberg, W.H. High-throughput synthesis and screening of combinatorial heterogeneous catalyst libraries. Angew. Chem. Int. Ed. 1999, 38, 484–488. [Google Scholar] [CrossRef]
- Newsam, J.M.; Bein, T.; Klein, J.; Maier, W.F.; Stichert, W. High throughput experimentation for the synthesis of new crystalline microporous solids. Microporous Mesoporous Mater. 2001, 48, 355–365. [Google Scholar] [CrossRef]
- Boussie, T.R.; Diamond, G.M.; Goh, C.; Hall, K.A.; LaPointe, A.M.; Leclerc, M.; Lund, C.; Murphy, V.; Shoemaker, J.A.; Tracht, U.; et al. A fully integrated high-throughput screening methodology for the discovery of new polyolefin catalysts: Discovery of a new class of high temperature single-site group (IV) copolymerization catalysts. J. Am. Chem. Soc. 2003, 125, 4306–4317. [Google Scholar] [CrossRef] [PubMed]
- Farrusseng, D. High-throughput heterogeneous catalysis. Surf. Sci. Rep. 2008, 63, 487–513. [Google Scholar] [CrossRef]
- Montgomery, D.C. Design and Analysis of Experiments, 2nd ed.; Wiley: New York, NY, USA, 1984; p. 538. [Google Scholar]
- Wolf, D.; Buyevskaya, O.V.; Baerns, M. An evolutionary approach in the combinatorial selection and optimization of catalytic materials. Appl. Catal. A 2000, 200, 63–77. [Google Scholar] [CrossRef]
- Caruthers, J.M.; Lauterbach, J.A.; Thomson, K.T.; Venkatasubramanian, V.; Snively, C.M.; Bhan, A.; Katare, S.; Oskarsdottir, G. Catalyst design: Knowledge extraction from high-throughput experimentation. J. Catal. 2003, 216, 98–109. [Google Scholar] [CrossRef]
- Baumes, L.A.; Serna, P.; Corma, A. Merging traditional and high-throughput approaches results in efficient design, synthesis and screening of catalysts for an industrial process. Appl. Catal. A 2010, 381, 197–208. [Google Scholar] [CrossRef]
- Huybrechts, W.; Mijoin, J.; Jacobs, P.A.; Martens, J.A. Development of a fixed-bed continuous-flow high-throughput reactor for long-chain N-alkane hydroconversion. Appl. Catal. A 2003, 243, 1–13. [Google Scholar] [CrossRef]
- Perez-Ramirez, J.; Berger, R.J.; Mul, G.; Kapteijn, F.; Moulijn, J.A. The six-flow reactor technology—A review on fast catalyst screening and kinetic studies. Catal. Today 2000, 60, 93–109. [Google Scholar] [CrossRef]
- Nagy, A.J. Implementation of High Throughput Experimentation Techniques for Kinetic Reaction Testing. Comb. Chem. High Throughput Screen. 2012, 15, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Navidi, N.; Thybaut, J.W.; Marin, G.B. Experimental investigation of ethylene hydroformylation to propanal on Rh and Co based catalysts. Appl. Catal. A 2014, 469, 357–366. [Google Scholar] [CrossRef]
- Chheda, J.N.; Huber, G.W.; Dumesic, J.A. Liquid-phase catalytic processing of biomass-derived oxygenated hydrocarbons to fuels and chemicals. Angew. Chem. Int. Ed. 2007, 46, 7164–7183. [Google Scholar] [CrossRef] [PubMed]
- Hagemeyer, A.; Strasser, P.; Volpe, A. High-Throughput Screening in Chemical Catalysis: Technologies, Strategies and Applications; John Wiley & Sons: Weinheim; Germany, 2004. [Google Scholar]
- Shimizu, K.D.; Snapper, M.L.; Hoveyda, A.H. High-throughput strategies for the discovery of catalysts. Chem. Eur. J. 1998, 4, 1885–1889. [Google Scholar] [CrossRef]
- Cawse, J.N. Experimental Strategies for Combinatorial and High-Throughput Materials Development. Acc. Chem. Res. 2001, 34, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Massart, D.L. Handbook of Chemometrics and Qualimetrics; Elsevier: Amsterdam, The Nertherland; New York, NY, USA, 1997. [Google Scholar]
- Hendershot, R.J.; Snively, C.M.; Lauterbach, J. High-Throughput Heterogeneous Catalytic Science. Chem. Eur. J. 2005, 11, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Thybaut, J.W.; Marin, G.B. Single-Event MicroKinetics: Catalyst design for complex reaction networks. J. Catal. 2013, 308, 352–362. [Google Scholar] [CrossRef]
- Thybaut, J.W.; Choudhury, I.R.; Denayer, J.F.; Baron, G.V.; Jacobs, P.A.; Martens, J.A.; Marin, G.B. Design of Optimum Zeolite Pore System for Central Hydrocracking of Long-Chain N-Alkanes based on a Single-Event Microkinetic Model. Top. Catal. 2009, 52, 1251–1260. [Google Scholar] [CrossRef]
- Alexiadis, V.I.; Thybaut, J.W.; Kechagiopoulos, P.N.; Chaar, M.; Van Veen, A.C.; Muhler, M.; Marin, G.B. Oxidative coupling of methane: Catalytic behaviour assessment via comprehensive microkinetic modelling. Appl. Catal. B 2014, 150, 496–505. [Google Scholar] [CrossRef]
- Vandegehuchte, B.D.; Thybaut, J.W.; Marin, G.B. Unraveling Diffusion and Other Shape Selectivity Effects in ZSM5 Using N-Hexane Hydroconversion Single-Event Microkinetics. Ind. Eng. Chem. Res. 2014, 53, 15333–15347. [Google Scholar] [CrossRef]
- Vandegehuchte, B.D.; Choudhury, I.R.; Thybaut, J.W.; Martens, J.A.; Marin, G.B. Integrated Stefan-Maxwell, Mean Field, and Single-Event Microkinetic Methodology for Simultaneous Diffusion and Reaction inside Microporous Materials. J. Phys. Chem. C 2014, 118, 22053–22068. [Google Scholar] [CrossRef]
- Dewachtere, N.V.; Santaella, F.; Froment, G.F. Application of a single-event kinetic model in the simulation of an industrial riser reactor for the catalytic cracking of vacuum gas oil. Chem. Eng. Sci. 1999, 54, 3653–3660. [Google Scholar] [CrossRef]
- Lozano-Blanco, G.; Thybaut, J.W.; Surla, K.; Galtier, P.; Marin, G.B. Simulation of a Slurry-Bubble Column Reactor for Fischer-Tropsch Synthesis Using Single-Event Microkinetics. AIChE J. 2009, 55, 2159–2170. [Google Scholar] [CrossRef]
- Froment, G.F. Modeling of catalyst deactivation. Appl. Catal. A 2001, 212, 117–128. [Google Scholar] [CrossRef]
- Vandegehuchte, B.D.; Thybaut, J.W.; Martens, J.A.; Marin, G.B. Maximizing N-alkane hydroisomerization: The interplay of phase, feed complexity and zeolite catalyst mixing. Catal. Sci. Technol. 2015, 5, 2053–2058. [Google Scholar] [CrossRef]
- Van Herk, D.; Kreutzer, M.T.; Makkee, M.; Moulijn, J.A. Scaling down trickle bed reactors. Catal. Today 2005, 106, 227–232. [Google Scholar] [CrossRef]
- Nagy, A. Integrated Lab Solutions. Available online: http://www.integratedlabsolutions.com/ (accessed on 23 September 2015).
- Berger, R.J.; Stitt, E.H.; Marin, G.B.; Kapteijn, F.; Moulijn, J.A. Eurokin—Chemical reaction kinetics in practice. Cattech 2001, 5, 30–60. [Google Scholar] [CrossRef]
- Al-Dahhan, M.H.; Wu, Y.; Dudukovic, M.P. Reproducible Technique for Packing Laboratory-Scale Trickle-Bed Reactors with a Mixture of Catalyst and Fines. Ind. Eng. Chem. Res. 1995, 34, 741–747. [Google Scholar] [CrossRef]
- Van Herk, D.; Castaño, P.; Quaglia, M.; Kreutzer, M.T.; Makkee, M.; Moulijn, J.A. Avoiding segregation during the loading of a catalyst—Inert powder mixture in a packed micro-bed. Appl. Catal. A 2009, 365, 110–121. [Google Scholar] [CrossRef]
- Zeton. Available online: http://www.zeton.com/site/home.html (accessed on 23 September 2015).
- Van der Borght, K.; Galvita, V.V.; Marin, G.B. Ethanol to higher hydrocarbons over Ni, Ga, Fe-modified ZSM-5: Effect of metal content. Appl. Catal. A 2015, 492, 117–126. [Google Scholar] [CrossRef]
- Madeira, F.F.; Gnep, N.S.; Magnoux, P.; Maury, S.; Cadran, N. Ethanol transformation over HFAU, HBEA and HMFI zeolites presenting similar Brønsted acidity. Appl. Catal. A 2009, 367, 39–46. [Google Scholar] [CrossRef]
- Machado, N.R.C.F.; Calsavara, V.; Astrath, N.G.C.; Medina, A.; Baesso, M.L. Hydrocarbons from ethanol using [Fe,Al]ZSM-5 zeolites obtained by direct synthesis. Appl. Catal. A 2006, 311, 193–198. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Alonso, A.; Valle, B.; Aguayo, A.T.; Olazar, M.; Bilbao, J. Kinetic modelling for the transformation of bioethanol into olefins on a hydrothermally stable Ni-HZSM-5 catalyst considering the deactivation by coke. Chem. Eng. J. 2011, 167, 262–277. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Alonso, A.; Valle, B.; Aguayo, A.T.; Bilbao, J. Selective production of olefins from bioethanol on HZSM-5 zeolite catalysts treated with NaOH. Appl. Catal. B 2010, 97, 299–306. [Google Scholar] [CrossRef]
- Xin, H.; Li, X.; Fang, Y.; Yi, X.; Hu, W.; Chu, Y.; Zhang, F.; Zheng, A.; Zhang, H.; Li, X. Catalytic dehydration of ethanol over post-treated ZSM-5 zeolites. J. Catal. 2014, 312, 204–215. [Google Scholar] [CrossRef]
- Song, Z.; Takahashi, A.; Nakamura, I.; Fujitani, T. Phosphorus-modified ZSM-5 for conversion of ethanol to propylene. Appl. Catal. A 2010, 384, 201–205. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Tarrío, A.M.; Aguayo, A.T.; Olazar, M.; Bilbao, J. Kinetic Modelling of the Transformation of Aqueous Ethanol into Hydrocarbons on a HZSM-5 Zeolite. Ind. Eng. Chem. Res. 2001, 40, 3467–3474. [Google Scholar] [CrossRef]
- Johansson, R.; Hruby, S.L.; Rass-Hansen, J.; Christensen, C.H. The Hydrocarbon Pool in Ethanol-to-Gasoline over HZSM-5 Catalysts. Catal. Lett. 2009, 127, 1–6. [Google Scholar] [CrossRef]
- Madeira, F.F.; Gnep, N.S.; Magnoux, P.; Vezin, H.; Maury, S.; Cadran, N. Mechanistic insights on the ethanol transformation into hydrocarbons over HZSM-5 zeolite. Chem. Eng. J. 2010, 161, 403–408. [Google Scholar] [CrossRef]
- Mears, D.E. Tests for Transport Limitations in Experimental Catalytic Reactors. Ind. Eng. Chem. Process. Dis. Dev. 1971, 10, 541–547. [Google Scholar] [CrossRef]
- Weisz, P.B.; Prater, C.D. Interpretation of Measurements in Experimental Catalysis. In Advances in Catalysis; Frankenburg, V.I.K., Rideal, E.K., Eds.; Academic Press: New York, NY, USA, 1954; Volume 6, pp. 143–196. [Google Scholar]
- Mears, D.E. Diagnostic Criteria for Heat Transport Limitations in Fixed Bed Reactors. J. Catal. 1971, 20, 127–131. [Google Scholar] [CrossRef]
- Mears, D.E. The role of axial dispersion in trickle-flow laboratory reactors. Chem. Eng. Sci. 1971, 26, 1361–1366. [Google Scholar] [CrossRef]
- Chu, C.F.; Ng, K.M. Flow in packed tubes with a small tube to particle diameter ratio. AIChE J. 1989, 35, 148–158. [Google Scholar] [CrossRef]
- Reyniers, M.F.; Marin, G.B. Experimental and Theoretical Methods in Kinetic Studies of Heterogeneously Catalyzed Reactions. Annu. Rev. Chem. Biomol. Eng. 2014, 5, 563–594. [Google Scholar] [CrossRef] [PubMed]
- Rakopoulos, D.C.; Rakopoulos, C.D.; Giakoumis, E.G.; Dimaratos, A.M. Characteristics of performance and emissions in high-speed direct injection diesel engine fueled with diethyl ether/diesel fuel blends. Energy 2012, 43, 214–224. [Google Scholar] [CrossRef]
- Thybaut, J.W.; Marin, G.B.; Baron, G.V.; Jacobs, P.A.; Martens, J.A. Alkene protonation enthalpy determination from fundamental kinetic modeling of alkane hydroconversion on Pt/H-(US)Y-zeolite. J. Catal. 2001, 202, 324–339. [Google Scholar] [CrossRef]
- Toch, K.; Thybaut, J.W.; Marin, G.B. Ethene oligomerization on Ni-SiO2-Al2O3: Experimental investigation and Single-Event MicroKinetic modeling. Appl. Catal. A 2015, 489, 292–304. [Google Scholar] [CrossRef]
- Lozano-Blanco, G.; Thybaut, J.W.; Surla, K.; Galtier, P.; Marin, G.B. Single-event microkinetic model for Fischer-Tropsch synthesis on iron-based catalysts. Ind. Eng. Chem. Res. 2008, 47, 5879–5891. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borght, K.V.d.; Toch, K.; Galvita, V.V.; Thybaut, J.W.; Marin, G.B. Information-Driven Catalyst Design Based on High-Throughput Intrinsic Kinetics. Catalysts 2015, 5, 1948-1968. https://doi.org/10.3390/catal5041948
Borght KVd, Toch K, Galvita VV, Thybaut JW, Marin GB. Information-Driven Catalyst Design Based on High-Throughput Intrinsic Kinetics. Catalysts. 2015; 5(4):1948-1968. https://doi.org/10.3390/catal5041948
Chicago/Turabian StyleBorght, Kristof Van der, Kenneth Toch, Vladimir V. Galvita, Joris W. Thybaut, and Guy B. Marin. 2015. "Information-Driven Catalyst Design Based on High-Throughput Intrinsic Kinetics" Catalysts 5, no. 4: 1948-1968. https://doi.org/10.3390/catal5041948
APA StyleBorght, K. V. d., Toch, K., Galvita, V. V., Thybaut, J. W., & Marin, G. B. (2015). Information-Driven Catalyst Design Based on High-Throughput Intrinsic Kinetics. Catalysts, 5(4), 1948-1968. https://doi.org/10.3390/catal5041948