
Oxygen Storage Capacity and Oxygen Mobility of Co-Mn-Mg-Al Mixed Oxides and Their Relation in the VOC Oxidation Reaction

Abstract
:1. Introduction
2. Results and Discussion
2.1. Precursors Characterization
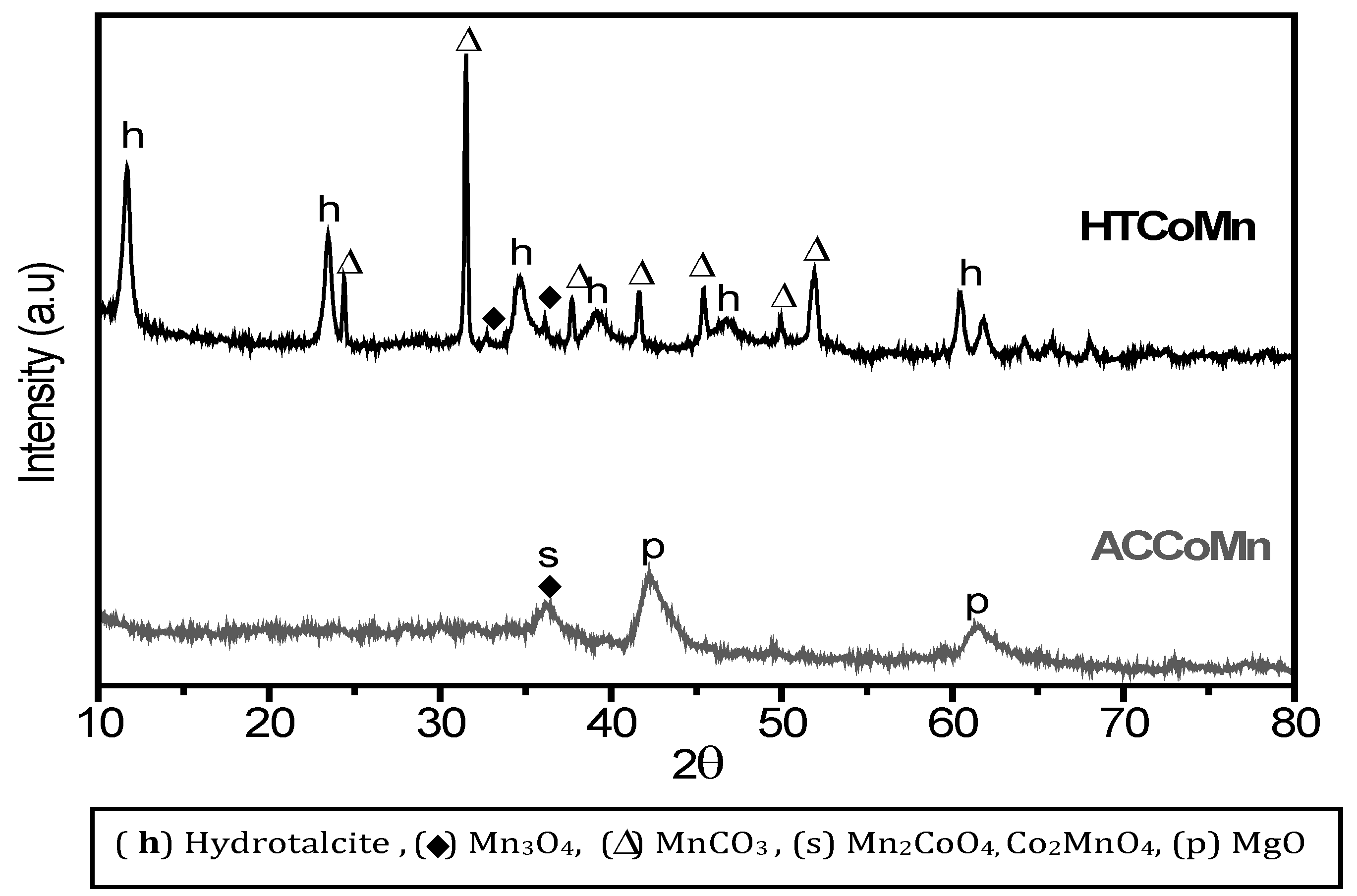
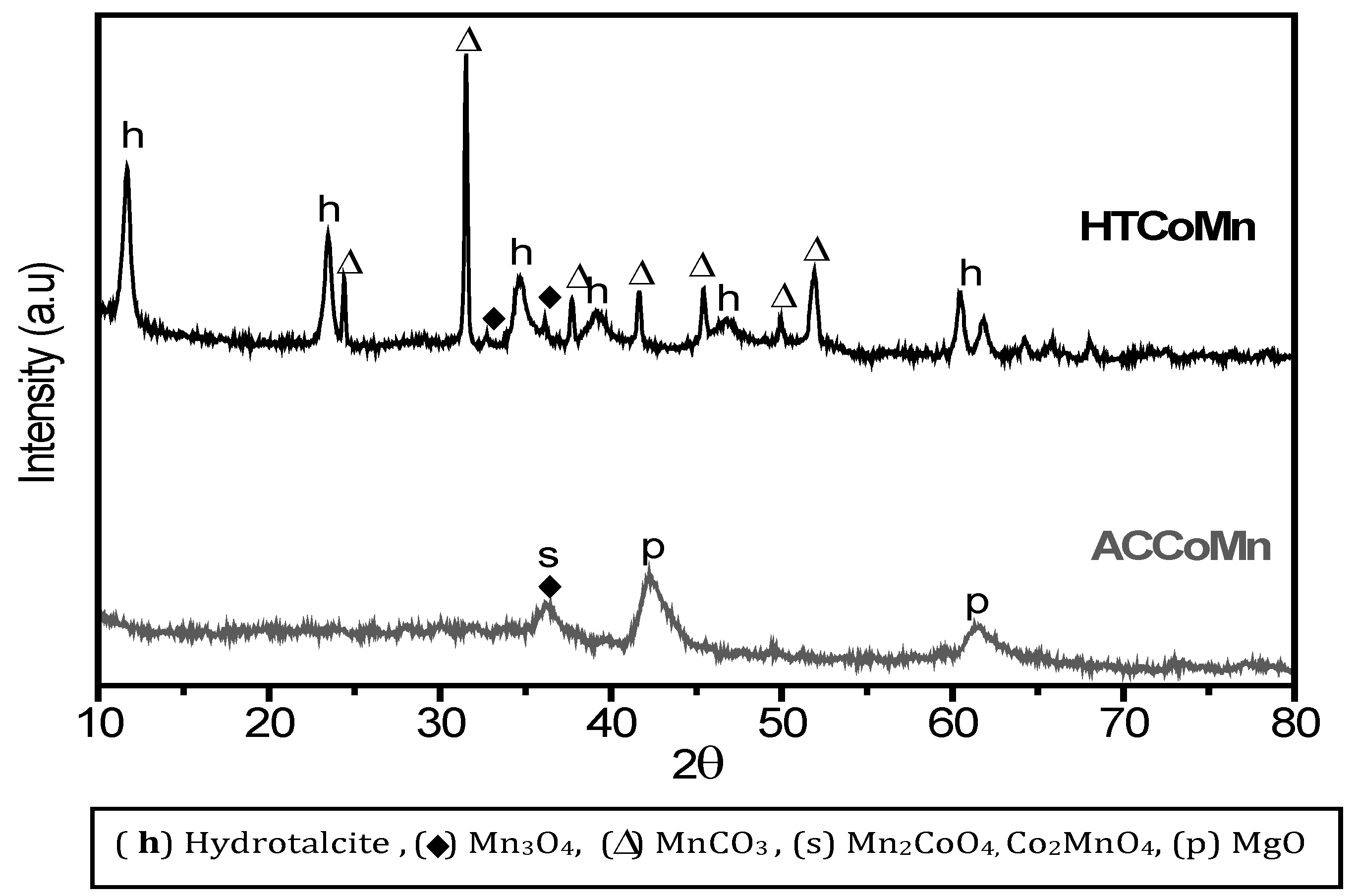
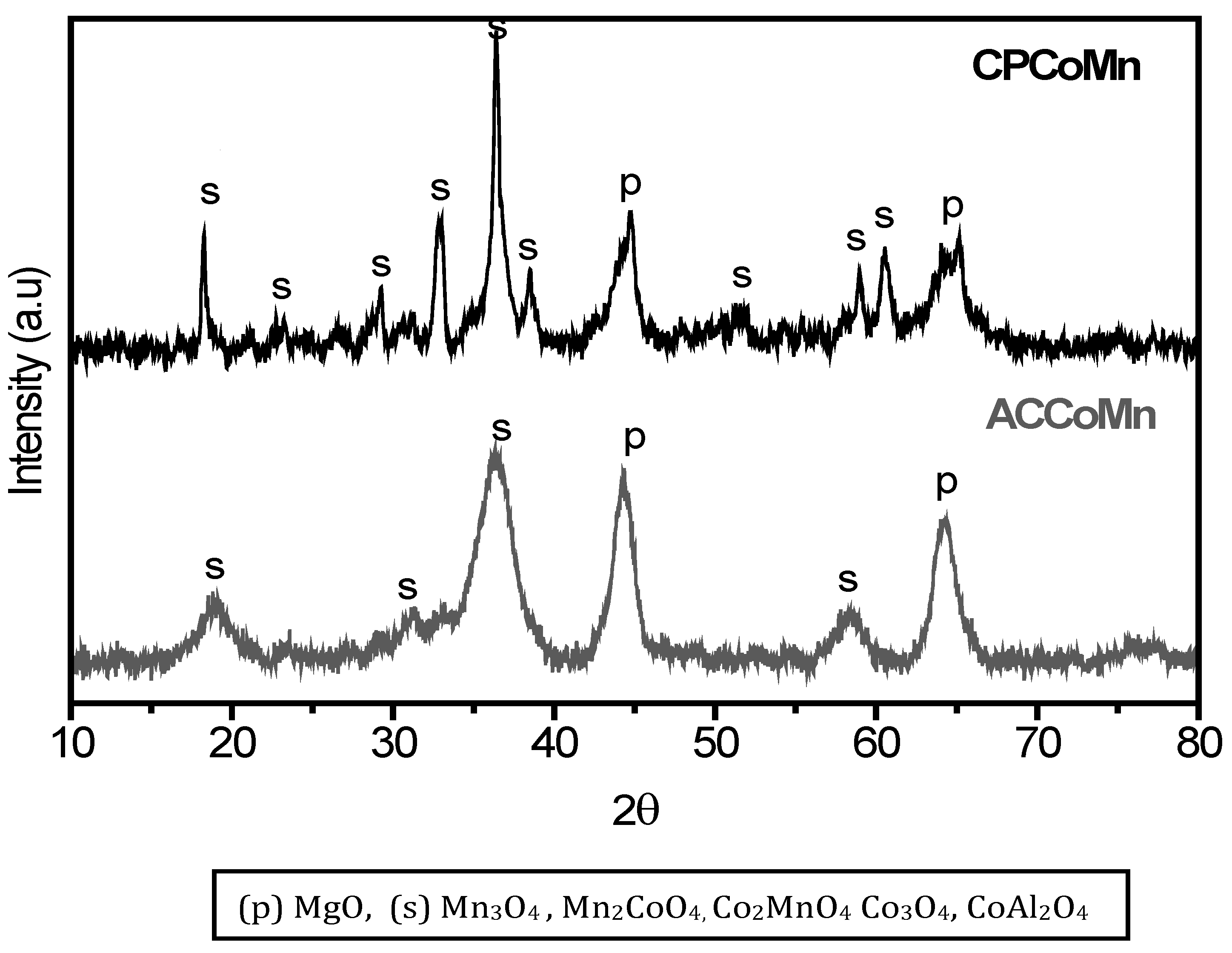
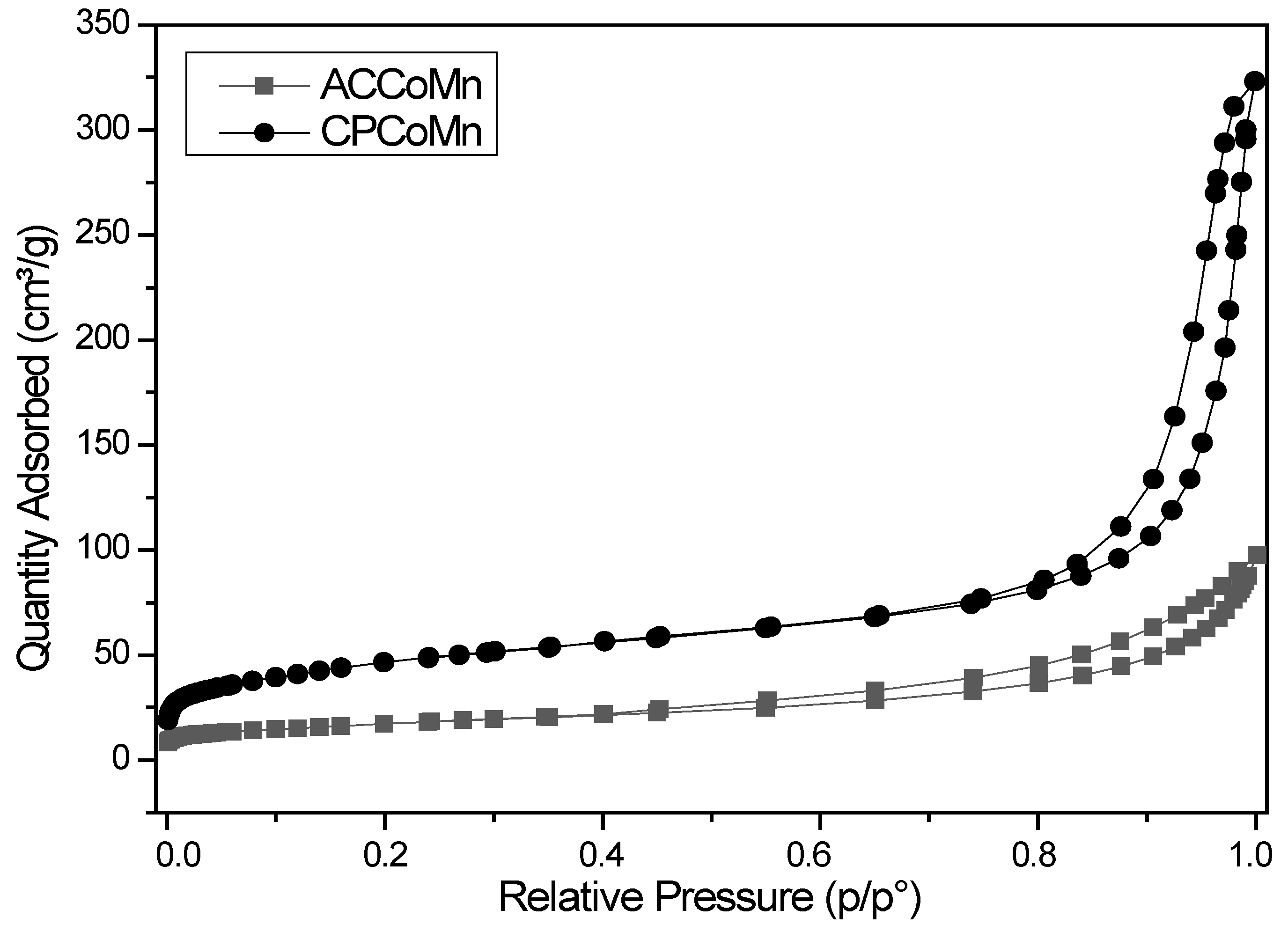
2.2. Characterization of the Mixed Oxides
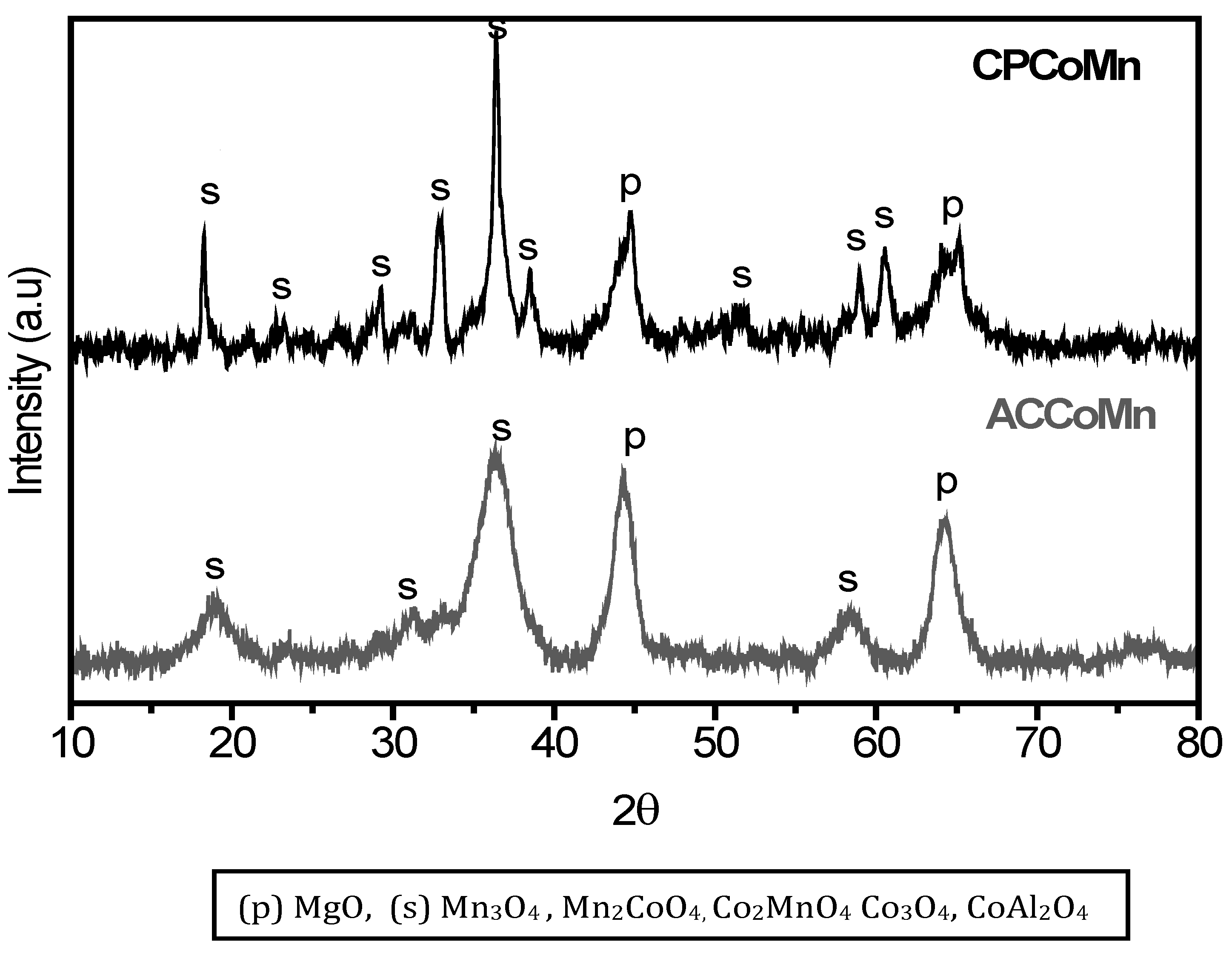
2.2.1. Chemical and Structural Composition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | (Mn2+ +Co2+ + Mg2+)/Al3+ | (Mn2+ + Co2+)/ Mg2+ | Co2+/Mn2+ | Dp (nm) ± 3nm | |
|---|---|---|---|---|---|
| ACCoMn | 3.3 | 1.6 | 0.5 | 7 | |
| CPCoMn | 2.9 | 1.4 | 0.6 | 20 | |
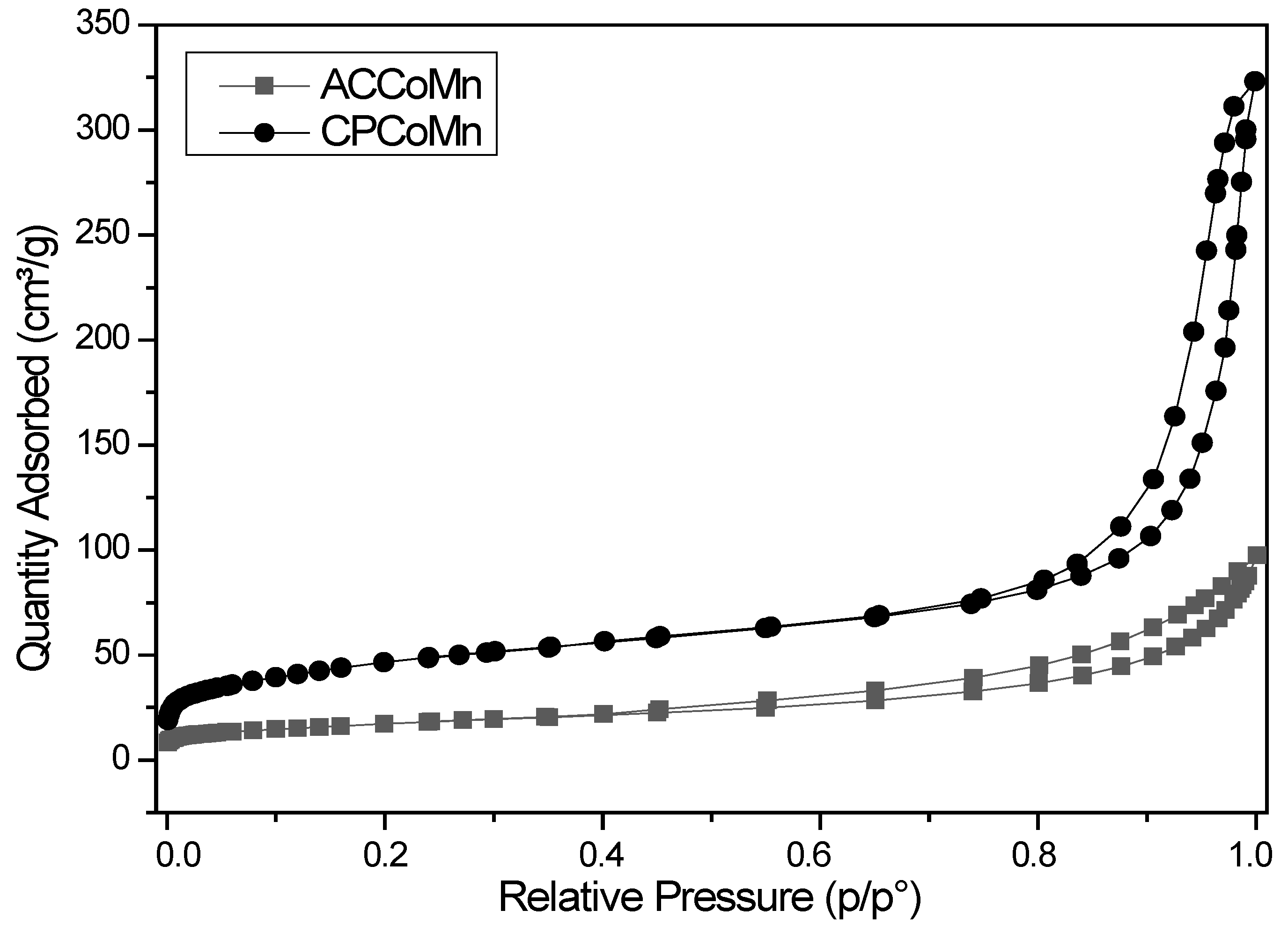
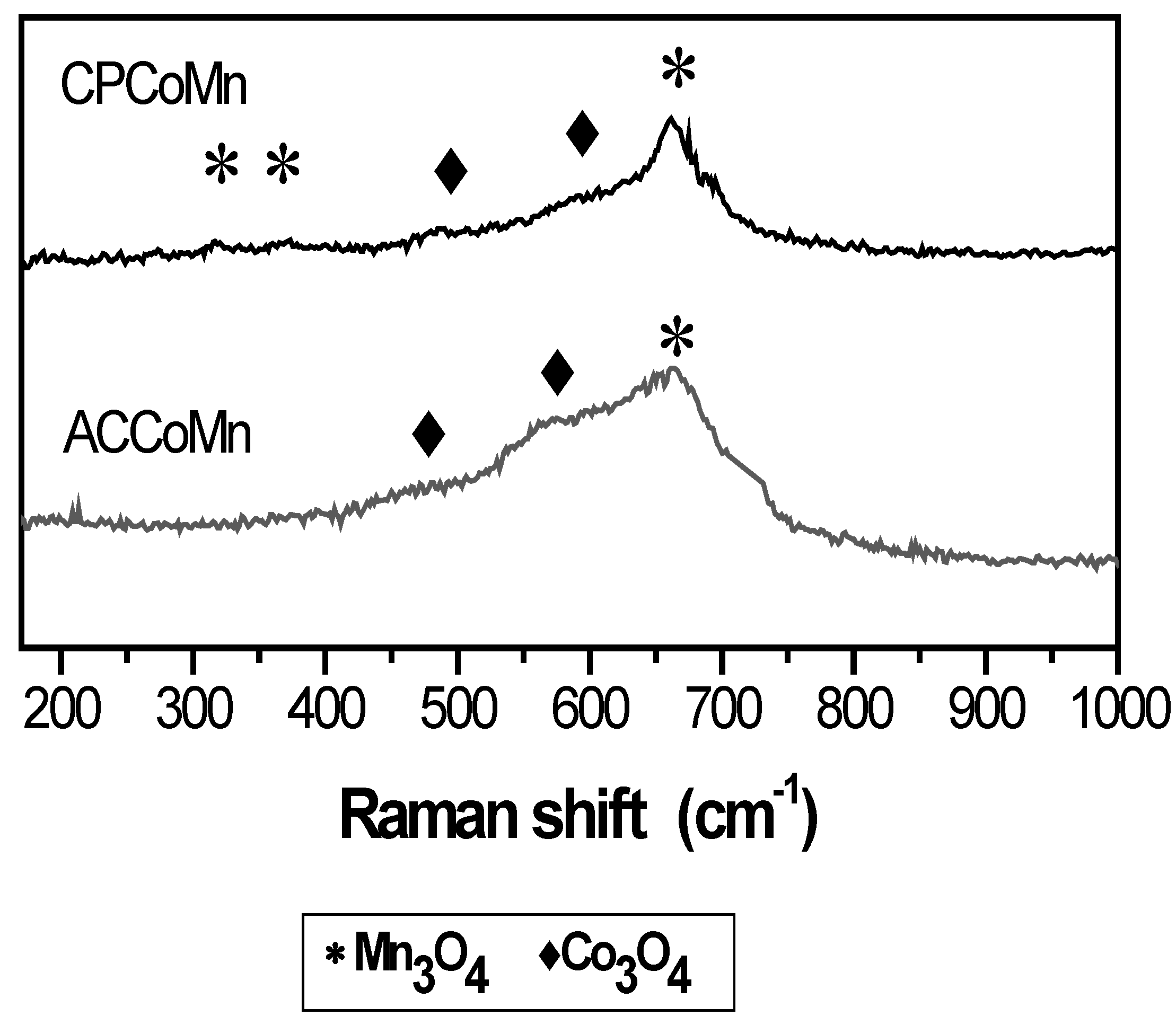

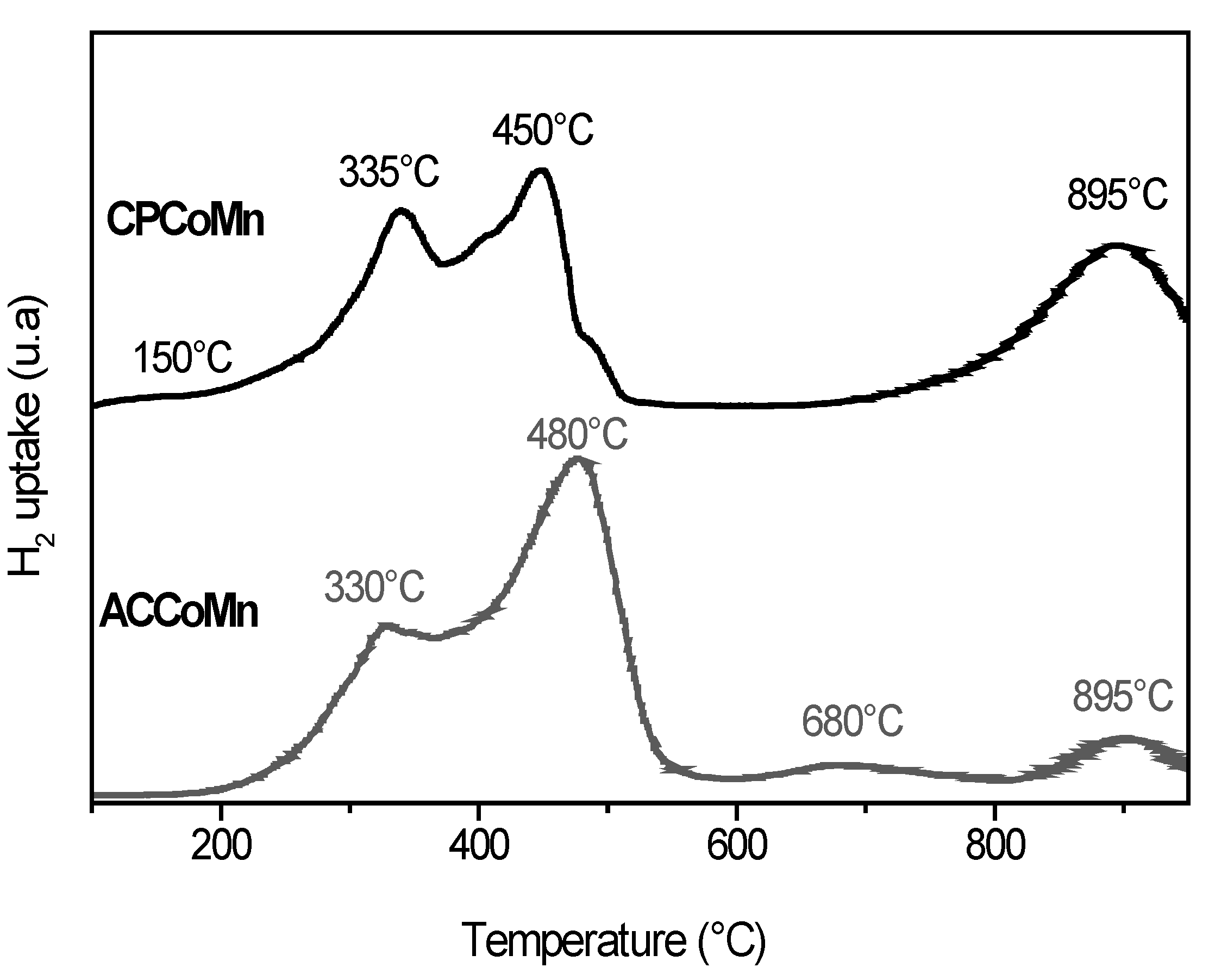
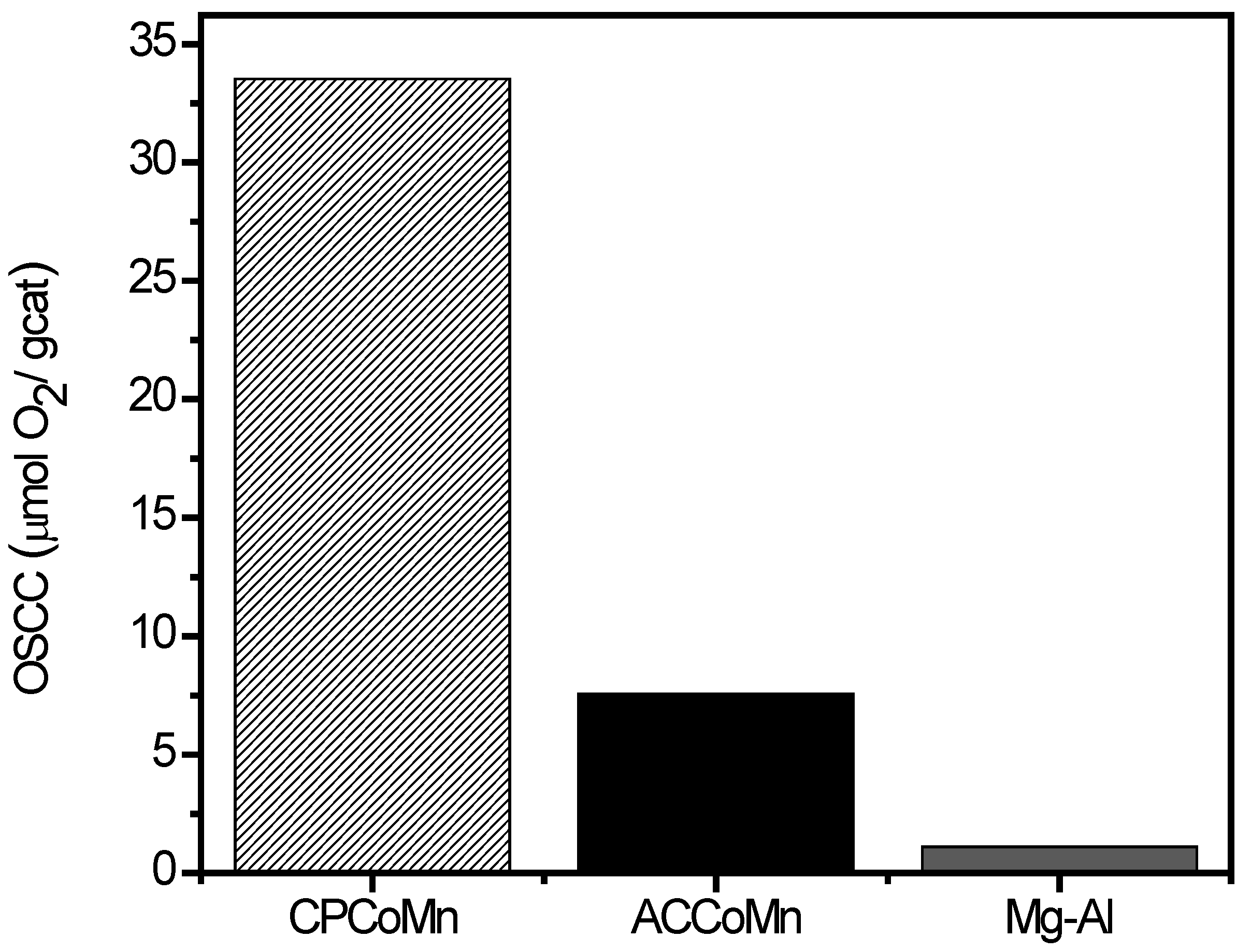
2.2.2. Redox Properties and Oxygen Mobility
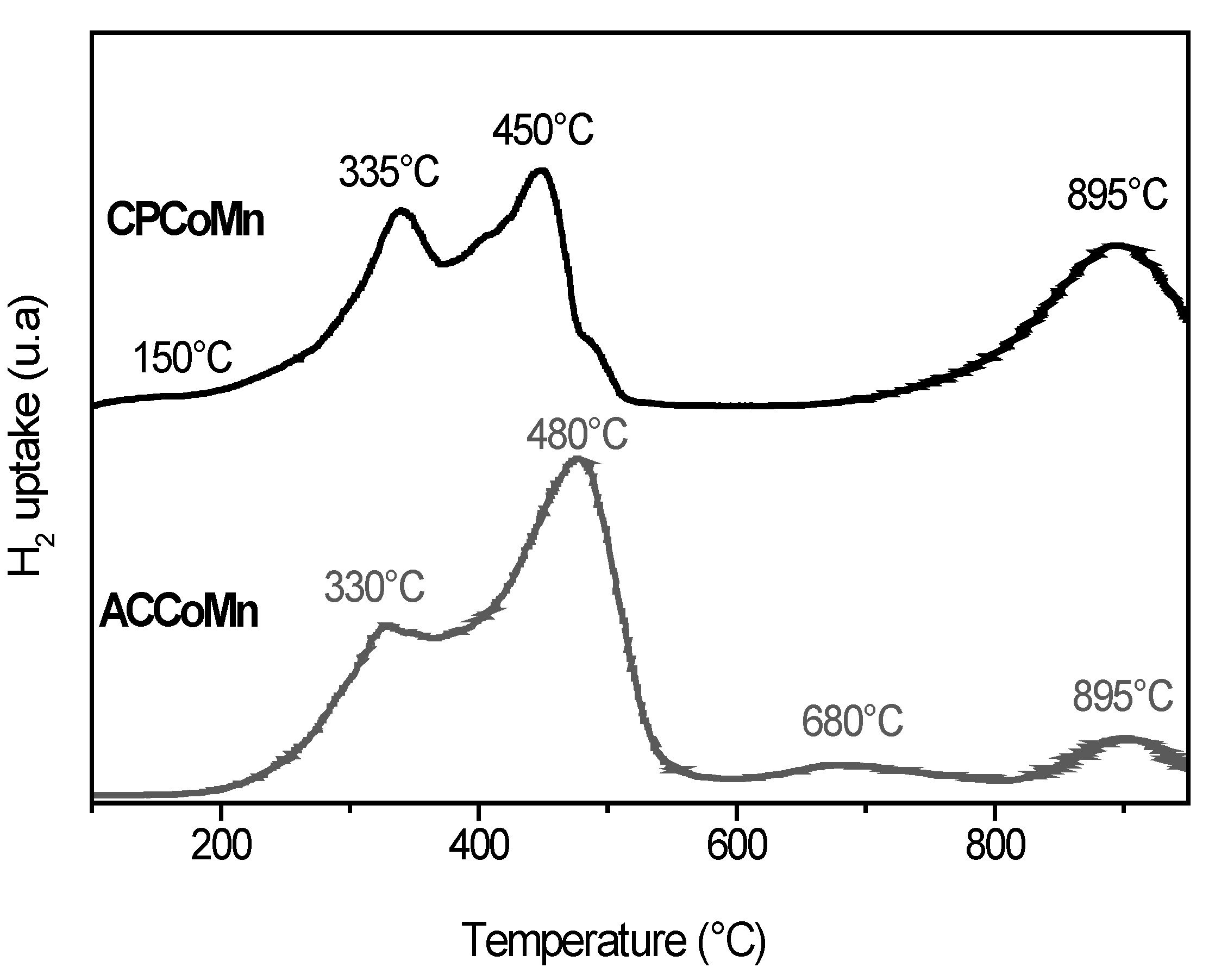
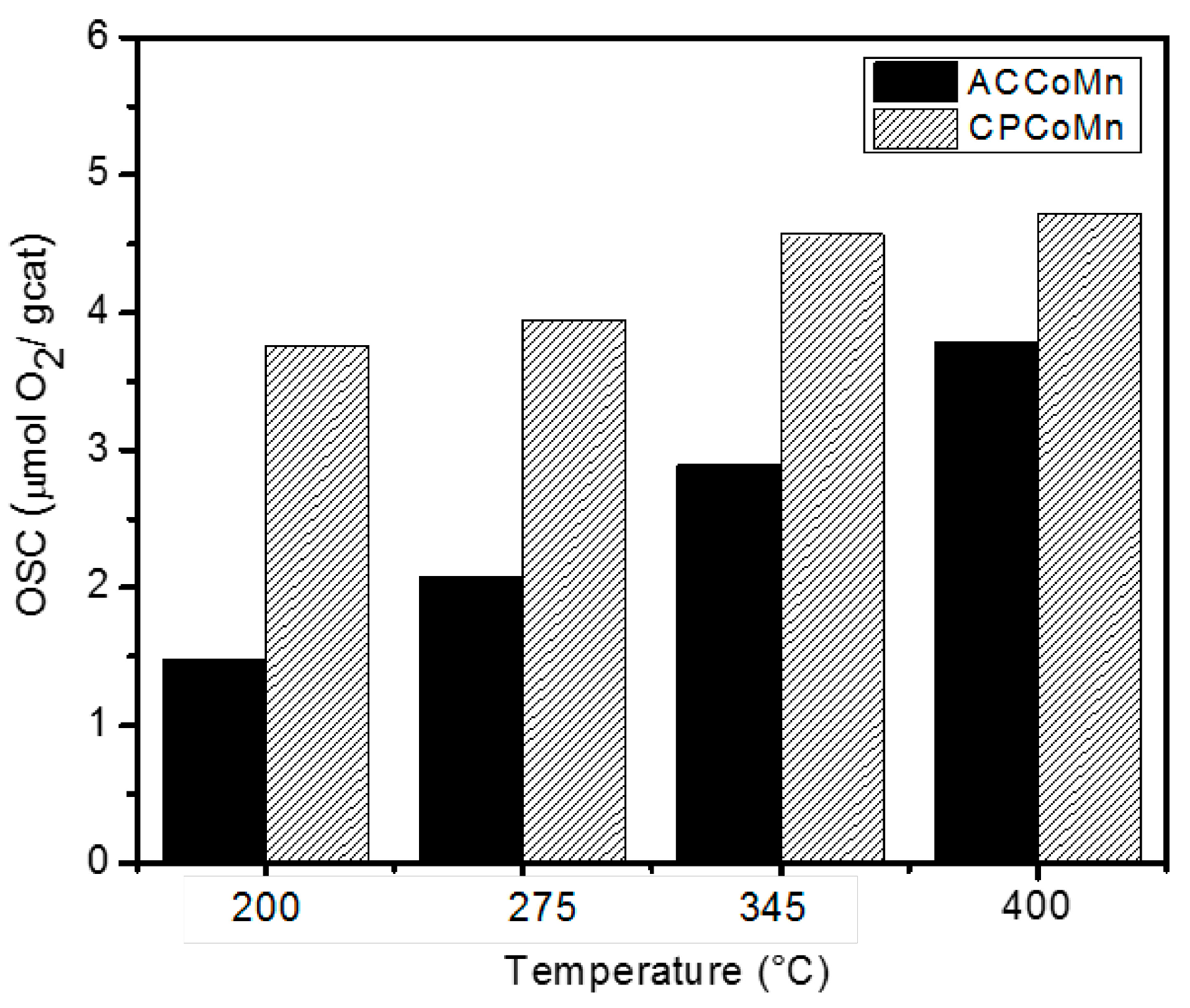
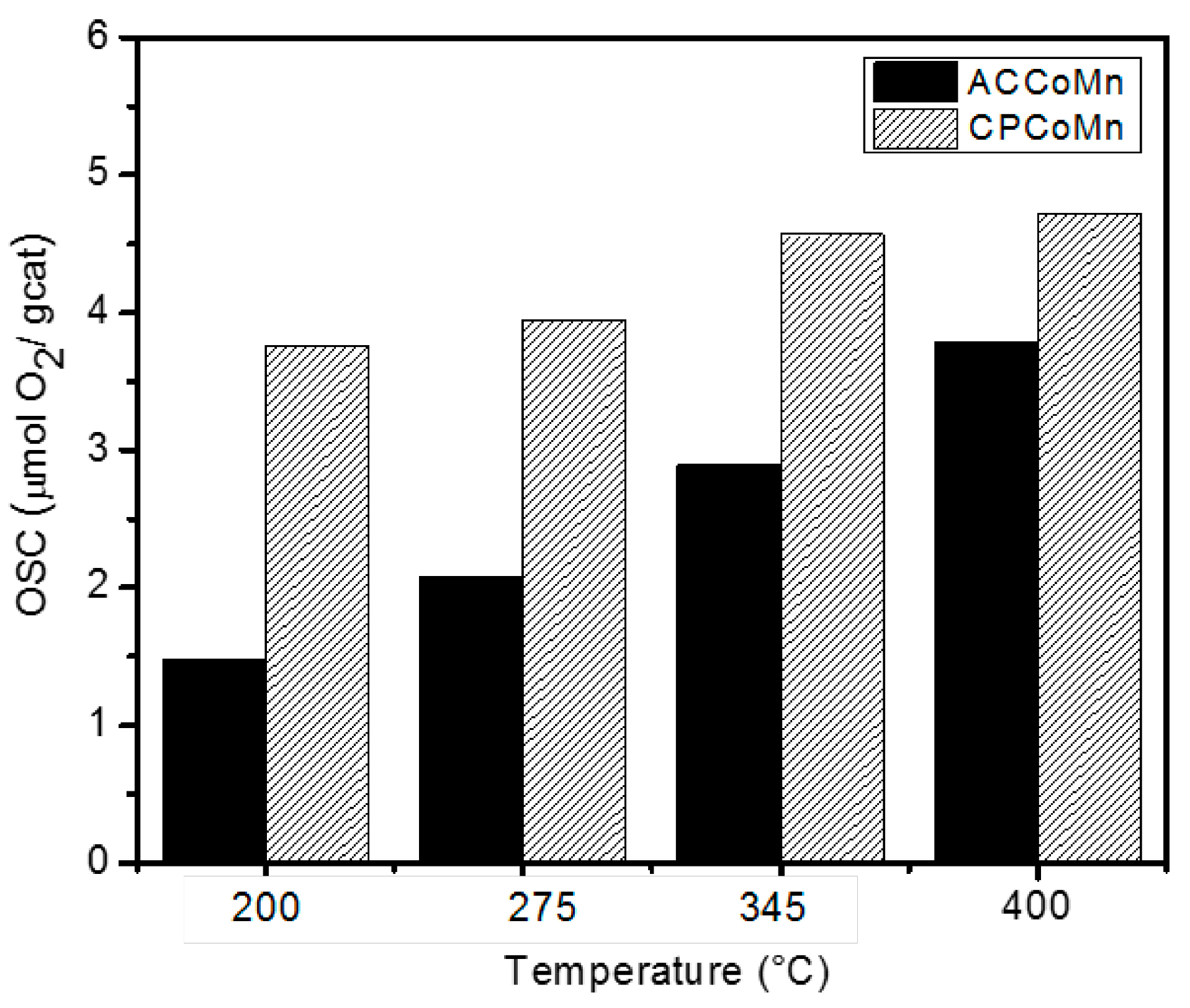
| Sample | OSC (µmolO2/g cat) two H2-O2 pulse cycles | |||||||
|---|---|---|---|---|---|---|---|---|
| 200 °C | 275 °C | 345 °C | 400 °C | |||||
| ACCoMn | 1.6 | 1.5 | 2.1 | 1.9 | 3.0 | 2.9 | 3.9 | 3.7 |
| CPCoMn | 3.9 | 3.7 | 4.0 | 4.2 | 4.7 | 4.5 | 4.7 | 4.8 |
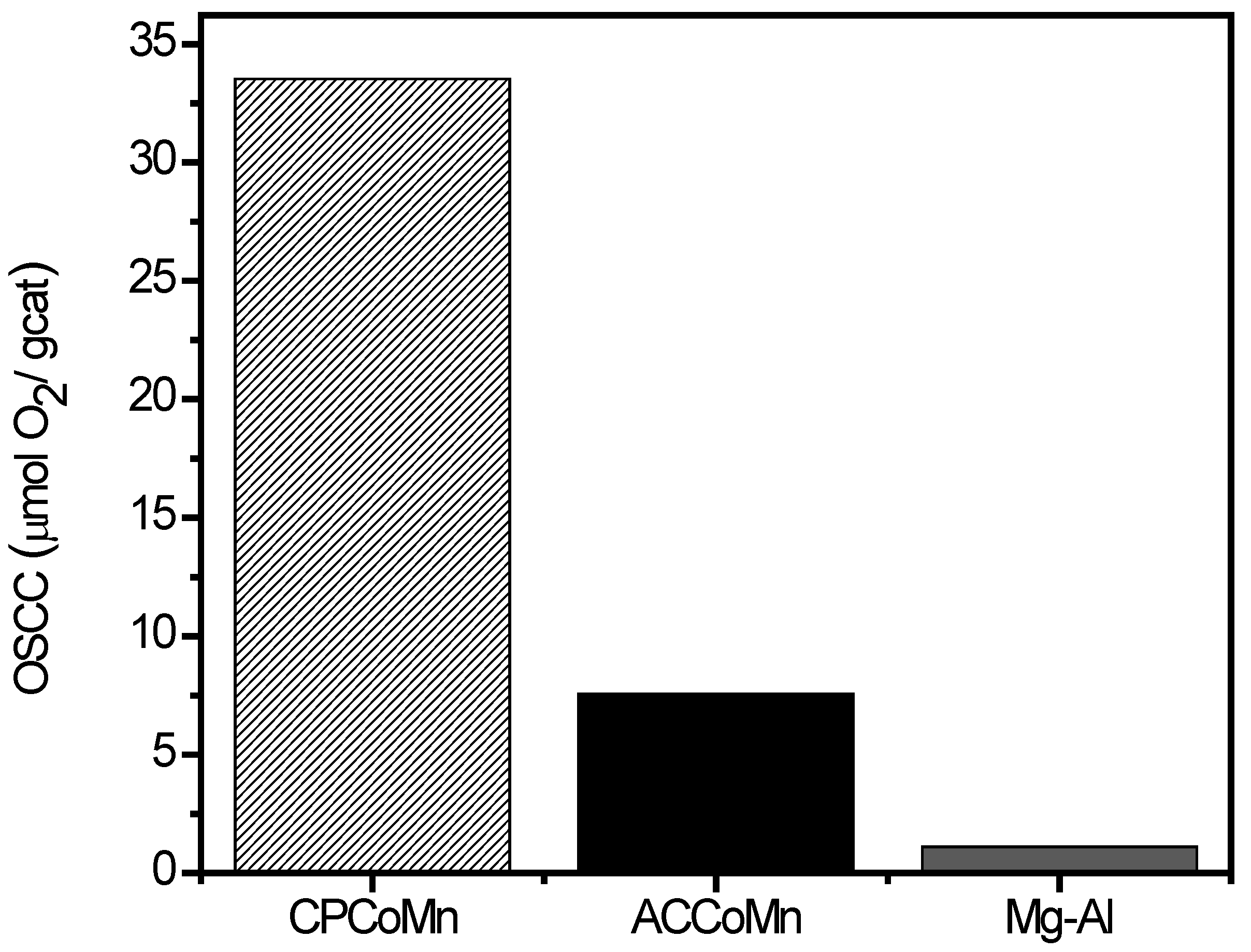
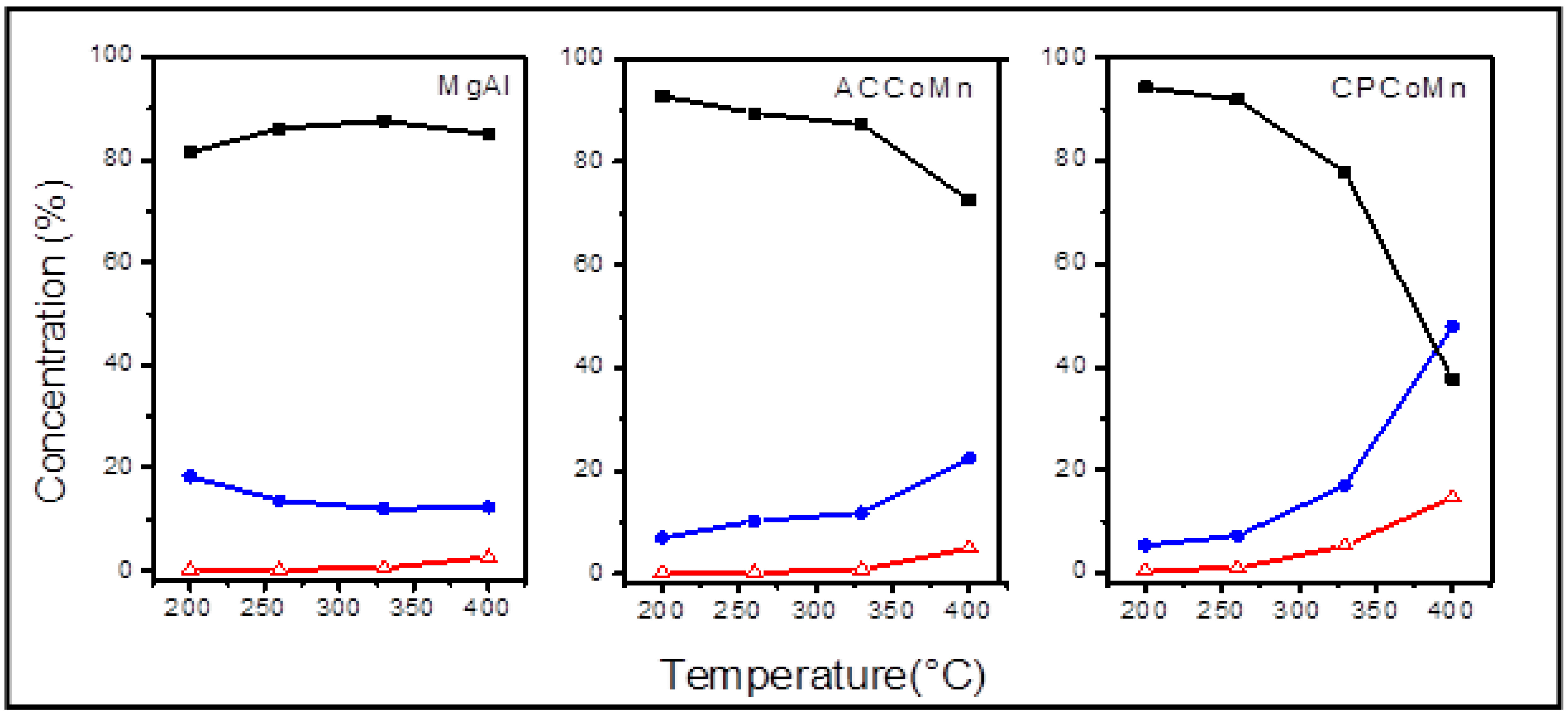
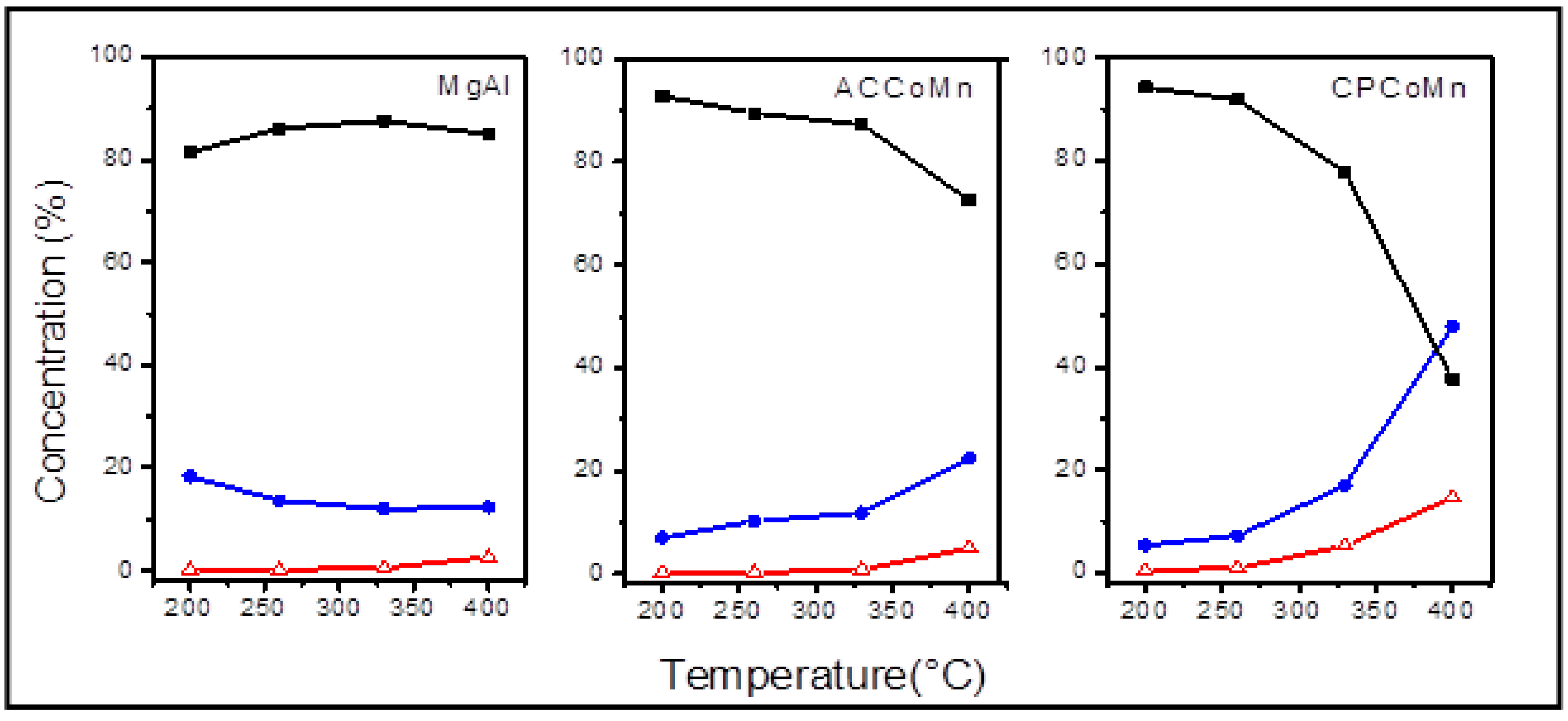
- Homogeneous exchange: this reaction does not require the participation of oxygen atoms in the solid, and the concentrations of 16O and 18O remain constant during the test:16O2(g) + 18O2(g)⇔218O16O(g)
- Heterogeneous single exchange: this reaction involves an oxygen atom from the oxide and an oxygen atom from the gas phase:18O18O(g) + 16O(s)⇔18O16O(g) + 18O(s)18O16O(g) + 16O(s)⇔16O16O(g) + 18O(s)
- Heterogeneous multiple exchange: this reaction assumes the participation of two atoms of the solid in each step:18O18O(g) + 216O(s)⇔16O16O(g) + 218O(s)18 O16 O(g) + 216 O(s)⇔16 O16O(g) + 18O(s) + 16O(s)18 O16 O(g) + 218 O(s)⇔18O18O(g) + 18O(s) + 16O(s)
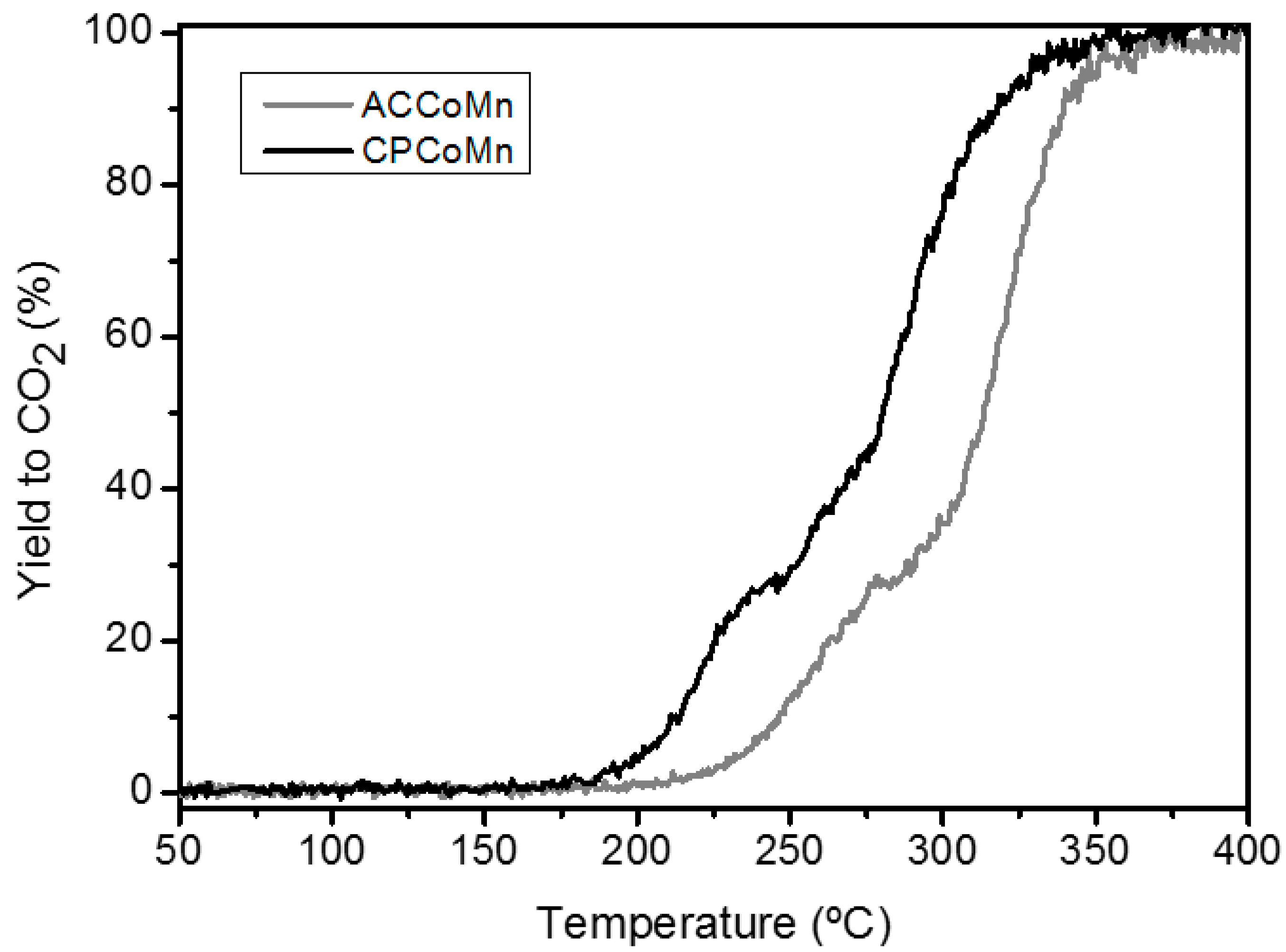
2.2.3. Oxidation Reaction
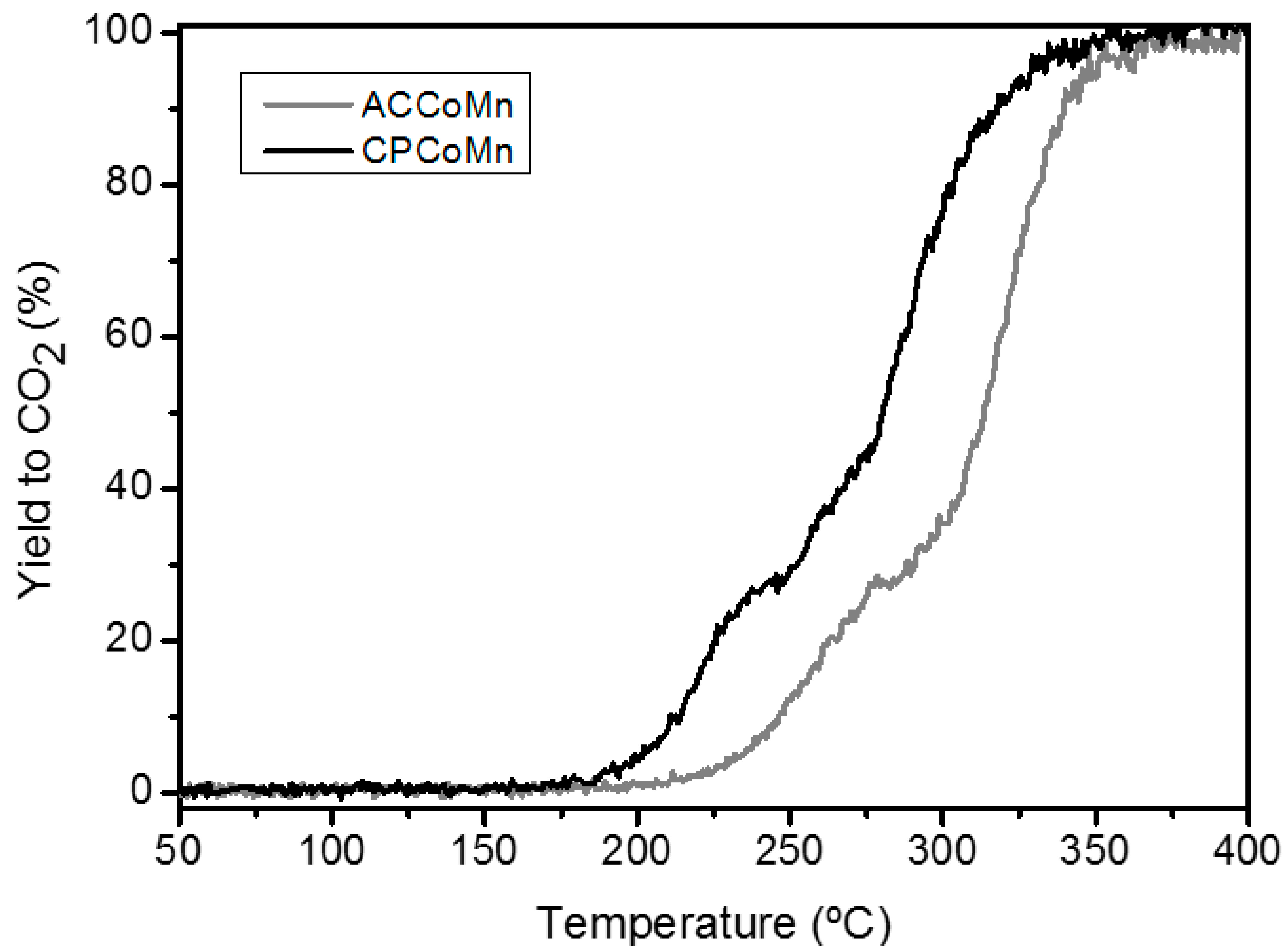
| Sample | Yield to CO2 temperatures (°C) | OSC (µmolO2/ g cat.)* | 16O2 concentration (%) * | |||
|---|---|---|---|---|---|---|
| T90 | T50 | T90 | T50 | T90 | T50 | |
| ACCoMn | 340 | 312 | 2.9 | 2.4 | 12 | 10 |
| CPCoMn 1%Pt/Al2O3 | 320 195 | 280 180 | 4.4 - | 4.1 - | 14 - | 8 - |
3. Experimental Section
3.1. Synthesis of Catalysts
3.1.1. Auto-Combustion
3.1.2. Co-Precipitation
3.2. Characterization
3.3. Catalytic Evaluation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bastos, S.S.T.; Carabineiro, S.A.C.; Órfão, J.J.M.; Pereira, M.F.R.; Delgado, J.J.; Figueiredo, J.L. Total oxidation of ethyl acetate, ethanol and toluene catalyzed by exotemplated manganese and cerium oxides loaded with gold. Catal. Today 2012, 180, 148–154. [Google Scholar] [CrossRef]
- Domínguez, M.I.; Sánchez, M.; Centeno, M.A.; Montes, M.; Odriozola, J.A. 2-Propanol oxidation over gold supported catalysts coated ceramic foams prepared from stainless steel wastes. J. Mol. Catal. A 2007, 277, 145–154. [Google Scholar] [CrossRef]
- Liu, S.Y.; Yang, S.M. Complete oxidation of 2-propanol over gold-based catalysts supported on metal oxides. Appl. Catal. A 2008, 334, 92–99. [Google Scholar] [CrossRef]
- Aguilera, D.A.; Perez, A.; Molina, R.; Moreno, S. Cu–Mn and Co–Mn catalysts synthesized from hydrotalcites and their use in the oxidation of VOCs. Appl. Catal. B 2011, 104, 144–150. [Google Scholar] [CrossRef]
- Tsou, J.; Magnoux, P.; Guisnet, M.; Órfão, J.J.M.; Figueiredo, J.L. Catalytic oxidation of volatile organic compounds: Oxidation of methyl-isobutyl-ketone over Pt/zeolite catalysts. Appl. Catal. B 2005, 57, 117–123. [Google Scholar] [CrossRef]
- He, C.; Li, P.; Cheng, J.; Hao, Z.-P.; Xu, Z.-P. A Comprehensive Study of Deep Catalytic Oxidation of Benzene, Toluene, Ethyl Acetate; their Mixtures over Pd/ZSM-5 Catalyst: Mutual Effects and Kinetics. Water Air Soil Pollut. 2010, 209, 365–376. [Google Scholar] [CrossRef]
- Burgos, N.; Paulis, M.a.; Mirari Antxustegi, M.; Montes, M. Deep oxidation of VOC mixtures with platinum supported on Al2O3/Al monoliths. Appl. Catal. B 2002, 38, 251–258. [Google Scholar] [CrossRef]
- Beauchet, R.; Mijoin, J.; Batonneau-Gener, I.; Magnoux, P. Catalytic oxidation of VOCs on NaX zeolite: Mixture effect with isopropanol and o-xylene. Appl. Catal. B 2010, 100, 91–96. [Google Scholar] [CrossRef]
- Sanz, O.; Delgado, J.J.; Navarro, P.; Arzamendi, G.; Gandía, L.M.; Montes, M. VOCs combustion catalysed by platinum supported on manganese octahedral molecular sieves. Appl. Catal. B 2011, 110, 231–237. [Google Scholar] [CrossRef]
- Fierro, J.L.G. Applications of Metal Oxides for Volatile Organic Compound Combustion. In Metal Oxides, Chemistry and Applications; CRC Press: New York, NY, USA, 2006; pp. 543–555. [Google Scholar]
- Liotta, L.F.; Wu, H.; Pantaleo, G.; Venezia, A.M. Co3O4 nanocrystals and Co3O4-MOx binary oxides for CO, CH4 and VOC oxidation at low temperatures: A review. Catal. Sci. Tech. 2013, 3, 3085–3102. [Google Scholar] [CrossRef]
- Santos, V.P.; Pereira, M.F.R.; Órfão, J.J.M.; Figueiredo, J.L. The role of lattice oxygen on the activity of manganese oxides towards the oxidation of volatile organic compounds. Appl. Catal. B 2010, 99, 353–363. [Google Scholar] [CrossRef]
- Liotta, L.F.; Ousmane, M.; di Carlo, G.; Pantaleo, G.; Deganello, G.; Marcì, G.; Retailleau, L.; Giroir-Fendler, A. Total oxidation of propene at low temperature over Co3O4–CeO2 mixed oxides: Role of surface oxygen vacancies and bulk oxygen mobility in the catalytic activity. Appl. Catal. A 2008, 347, 81–88. [Google Scholar] [CrossRef]
- Zhu, L.; Lu, G.; Wang, Y.; Guo, Y.; Guo, Y. Effects of Preparation Methods on the Catalytic Performance of LaMn0.8Mg0.2O3 Perovskite for Methane Combustion. Chin. J. Catal. 2010, 31, 1006–1012. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T.; Matralis, H. Influence of the preparation method on the performance of CuO-CeO2 catalysts for the selective oxidation of CO. Appl. Catal. B 2005, 56, 87–93. [Google Scholar] [CrossRef]
- Kovanda, F.; Jirátová, K. Supported layered double hydroxide-related mixed oxides and their application in the total oxidation of volatile organic compounds. Appl. Clay. Sci. 2011, 53, 305–316. [Google Scholar] [CrossRef]
- Vaccari, A. Preparation and catalytic properties of cationic and anionic clays. Catal. Today 1998, 41, 53–71. [Google Scholar] [CrossRef]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Mukasyan, A.; Dinka, P. Novel approaches to solution-combustion synthesis of nanomaterials. Int. J. Self-Propag. High-Temp Synth. 2007, 16, 23–35. [Google Scholar] [CrossRef]
- Tahmasebi, K.; Paydar, M.H. The effect of starch addition on solution combustion synthesis of Al2O3-ZrO2 nanocomposite powder using urea as fuel. Mater. Chem. Phys 2008, 109, 156–163. [Google Scholar] [CrossRef]
- Castaño, M.H.; Molina, R.; Moreno, S. Catalytic oxidation of VOCs on MnMgAlOx mixed oxides obtained by auto-combustion. J. Mol. Catal. A 2015, 398, 358–367. [Google Scholar] [CrossRef]
- Tsyganok, A.; Sayari, A. Incorporation of transition metals into Mg–Al layered double hydroxides: Coprecipitation of cations vs. their pre-complexation with an anionic chelator. J. Solid State Chem. 2006, 179, 1830–1841. [Google Scholar] [CrossRef]
- Xu, Z.P.; Zhang, J.; Adebajo, M.O.; Zhang, H.; Zhou, C. Catalytic applications of layered double hydroxides and derivatives. Appl. Clay. Sci. 2011, 53, 139–150. [Google Scholar] [CrossRef]
- Lamonier, J.-F.; Boutoundou, A.-B.; Gennequin, C.; Pérez-Zurita, M.; Siffert, S.; Aboukais, A. Catalytic Removal of Toluene in Air over Co–Mn–Al Nano-oxides Synthesized by Hydrotalcite Route. Catal. Lett. 2007, 118, 165–172. [Google Scholar] [CrossRef]
- Evans, D.G.; Duan, X. Structural Aspects of Layered Double Hydroxides, in Structure and Bonding; Springer-Verlag: Berlin & Heidelberg: Germany, 2006; p. 15. [Google Scholar]
- Tsai, Y.-T.; Mo, X.; Campos, A.; Goodwin, J.G., Jr.; Spivey, J.J. Hydrotalcite supported Co catalysts for CO hydrogenation. Appl. Catal. A 2011, 396, 91–100. [Google Scholar] [CrossRef]
- Hem John, D. Redox Coprecipitation Mechanisms of Manganese Oxides. In Particulates in Water; American Chemical Society: Menlo Park, CA, USA, 1980; pp. 45–72. [Google Scholar]
- Sinha, A.P.B.; Sanjana, N.R.; Biswas, A.B. On the structure of some manganites. Acta Crystallogr. 1957, 10, 439–440. [Google Scholar] [CrossRef]
- Leofanti, G.; Padovan, M.; Tozzola, G.; Venturelli, B. Surface area and pore texture of catalysts. Catal. Today 1998, 41, 207–219. [Google Scholar] [CrossRef]
- Castaño, M.H.; Molina, R.; Moreno, S. Cooperative effect of the Co–Mn mixed oxides for the catalytic oxidation of VOCs: Influence of the synthesis method. Appl. Catal. A 2015, 492, 48–59. [Google Scholar] [CrossRef]
- Julien, C.M.; Massot, M.; Poinsignon, C. Lattice vibrations of manganese oxides: Part I. Periodic structures. Spectrochim. Acta Part A 2004, 60, 689–700. [Google Scholar] [CrossRef]
- Jiang, J.; Li, L. Synthesis of sphere-like Co3O4 nanocrystals via a simple polyol route. Matter. Lett. 2007, 61, 4894–4896. [Google Scholar] [CrossRef]
- Ulla, M.A.; Spretz, R.; Lombardo, E.; Daniell, W.; Knözinger, H. Catalytic combustion of methane on Co/MgO: Characterisation of active cobalt sites. Appl. Catal. B 2001, 29, 217–229. [Google Scholar] [CrossRef]
- Rezaei, E.; Soltan, J. Low temperature oxidation of toluene by ozone over MnOx/γ-alumina and MnOx/MCM-41 catalysts. Chem. Eng. J. 2012, 198–199, 482–490. [Google Scholar] [CrossRef]
- Mars, P.; van Krevelen, D.W. Oxidations carried out by means of vanadium oxide catalysts. Chem. Eng. Sci. 1954, 3, 41–59. [Google Scholar] [CrossRef]
- Fierro, J.L.G. Metal Oxides, Chemistry and Applications; CRC Press: New York, NY, USA, 2006. [Google Scholar]
- Ran, R.; Weng, D.; Wu, X.; Fan, J.; Wang, L.; Wu, X. Structure and oxygen storage capacity of Pr-doped Ce0.26Zr0.74O2 mixed oxides. J. Rare Earth 2011, 29, 1053–1059. [Google Scholar] [CrossRef]
- Muñoz, M.; Moreno, S.; Molina, R. Promoting effect of Ce and Pr in Co catalysts for hydrogen production via oxidative steam reforming of ethanol. Catal. Today 2013, 213, 33–41. [Google Scholar] [CrossRef]
- Coq, B.; Tichit, D.; Ribet, S. Co/Ni/Mg/Al Layered Double Hydroxides as Precursors of Catalysts for the Hydrogenation of Nitriles: Hydrogenation of Acetonitrile. J. Catal. 2000, 189, 117–128. [Google Scholar] [CrossRef]
- Pérez, A.; Lamonier, J.-F.; Giraudon, J.-M.; Molina, R.; Moreno, S. Catalytic activity of Co-Mg mixed oxides in the VOC oxidation: Effects of ultrasonic assisted in the synthesis. Catal. Today 2011, 176, 286–291. [Google Scholar] [CrossRef]
- Döbber, D.; Kießling, D.; Schmitz, W.; Wendt, G. MnOx/ZrO2 catalysts for the total oxidation of methane and chloromethane. Appl. Catal. B 2004, 52, 135–143. [Google Scholar] [CrossRef]
- Stobbe, E.R.; de Boer, B.A.; Geus, J.W. The reduction and oxidation behaviour of manganese oxides. Catal. Today 1999, 47, 161–167. [Google Scholar] [CrossRef]
- Li, Q.; Meng, M.; Xian, H.; Tsubaki, N.; Li, X.; Xie, Y.; Hu, T.; Zhang, J. Hydrotalcite-Derived MnxMg3−xAlO Catalysts Used for Soot Combustion, NOx Storage and Simultaneous Soot-NOx Removal. Environ. Sci. Technol. 2010, 44, 4747–4752. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.-C.; Wu, T.-Y.; Wan, J.; Tsai, J.-S. Development of a novel combustion synthesis method for synthesizing of ceramic oxide powders. Mater. Sci. Eng. B 2004, 111, 49–56. [Google Scholar] [CrossRef]
- Gonzalez Castaño, M.; Reina, T.R.; Ivanova, S.; Centeno, M.A.; Odriozola, J.A. Pt vs. Au in water–gas shift reaction. J. Catal. 2014, 314, 1–9. [Google Scholar] [CrossRef]
- Zhao, M.; Shen, M.; Wang, J. Effect of surface area and bulk structure on oxygen storage capacity of Ce0.67Zr0.33O2. J.Catal. 2007, 248, 258–267. [Google Scholar] [CrossRef]
- Wang, J.; Shen, M.; Wang, J.; Gao, J.; Ma, J.; Liu, S. CeO2–CoOx mixed oxides: Structural characteristics and dynamic storage/release capacity. Catal. Today 2011, 175, 65–71. [Google Scholar] [CrossRef]
- de Rivas, B.; López-Fonseca, R.; Jiménez-González, C.; Gutiérrez-Ortiz, J.I. Synthesis, characterisation and catalytic performance of nanocrystalline Co3O4 for gas-phase chlorinated VOC abatement. J. Catal. 2011, 281, 88–97. [Google Scholar] [CrossRef]
- Cui, M.; Li, Y.; Wang, X.; Wang, J.; Shen, M. Effect of preparation method on MnOx-CeO2 catalysts for NO oxidation. J. Rare Earth 2013, 31, 572–576. [Google Scholar] [CrossRef]
- Kamiuchi, N.; Haneda, M.; Ozawa, M. Enhancement of OSC property of Zr rich ceria–zirconia by loading a small amount of platinum. Catal. Today 2014, 232, 179–184. [Google Scholar] [CrossRef]
- Mamontov, E.; Egami, T.; Brezny, R.; Koranne, M.; Tyagi, S. Lattice Defects and Oxygen Storage Capacity of Nanocrystalline Ceria and Ceria-Zirconia. J. Phys. Chem. B 2000, 104, 11110–11116. [Google Scholar] [CrossRef]
- Nováková, J. Isotopic exchange of oxygen 18O between the gaseous phase and oxide catalysts. Catal. Rev. 1971, 4, 77–113. [Google Scholar]
- Royer, S.; Duprez, D.; Kaliaguine, S. Role of bulk and grain boundary oxygen mobility in the catalytic oxidation activity of LaCo1–xFexO3. J. Catal. 2005, 234, 364–375. [Google Scholar] [CrossRef]
- Dı́ez, V.K.; Apesteguı́a, C.R.; Di Cosimo, J.I. Acid–base properties and active site requirements for elimination reactions on alkali-promoted MgO catalysts. Catal. Today 2000, 63, 53–62. [Google Scholar] [CrossRef]
- Cimino, S.; Nigro, R.; Weidmann, U.; Holzner, R. Catalytic combustion of methanol over La, Mn-hexaaluminate catalysts. Fuel Process. Technol. 2015, 133, 1–7. [Google Scholar] [CrossRef]
- Golodets, G.I. Heterogeneous Catalytic Reactions Involving Molecular Oxygen; Elsevier: Amsterdam, The Netherlands, 1983. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castaño, M.H.; Molina, R.; Moreno, S. Oxygen Storage Capacity and Oxygen Mobility of Co-Mn-Mg-Al Mixed Oxides and Their Relation in the VOC Oxidation Reaction. Catalysts 2015, 5, 905-925. https://doi.org/10.3390/catal5020905
Castaño MH, Molina R, Moreno S. Oxygen Storage Capacity and Oxygen Mobility of Co-Mn-Mg-Al Mixed Oxides and Their Relation in the VOC Oxidation Reaction. Catalysts. 2015; 5(2):905-925. https://doi.org/10.3390/catal5020905
Chicago/Turabian StyleCastaño, María Haidy, Rafael Molina, and Sonia Moreno. 2015. "Oxygen Storage Capacity and Oxygen Mobility of Co-Mn-Mg-Al Mixed Oxides and Their Relation in the VOC Oxidation Reaction" Catalysts 5, no. 2: 905-925. https://doi.org/10.3390/catal5020905
APA StyleCastaño, M. H., Molina, R., & Moreno, S. (2015). Oxygen Storage Capacity and Oxygen Mobility of Co-Mn-Mg-Al Mixed Oxides and Their Relation in the VOC Oxidation Reaction. Catalysts, 5(2), 905-925. https://doi.org/10.3390/catal5020905