
Structural Evolution of Molybdenum Carbides in Hot Aqueous Environments and Impact on Low-Temperature Hydroprocessing of Acetic Acid

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of Mo Carbides

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | BET surface area, Sg/m2 g−1 | Pore volume /cm3 g−1 | Pore size /nm | CO uptake /µmol g−1 | Site density a /×1015 cm−2 | Particle size, Dp b /nm | Crystallite size, Dc c /nm |
|---|---|---|---|---|---|---|---|
| Mo2C-A fresh | 36 | 0.08 | 8 | 186 | 0.31 | 18 | 11/11 |
| Mo2C-A aged | 64 | 0.50 | 36 | 26 | 0.02 | 10 | 10/11 |
| Mo2C-A tested | 22 | 0.02 | 4 | 186 | 0.51 | 30 | 8/10 |
| Mo2C-B fresh | 6 | 0.03 | 23 | 5 | 0.05 | 110 | 13/19 |
| Mo2C-B aged | 15 | 0.02 | 4 | 12 | 0.05 | 44 | 12/16 |
| Ru/C fresh | 815 | NM d | NM d | NA e | 0.04 f | 1.1 g | NA e |
| Ru/C aged | 765 | NM d | NM d | NA e | 0.01 f | 2.0 g | NA e |
| Catalyst | Mocarb b /at.% | Mooxid c /at.% | Ccarb b /at.% | Ccont d /at.% | Ooxid c /at.% | Ocont d /at.% | C/Mo e | O/Mo f |
|---|---|---|---|---|---|---|---|---|
| Mo2C-A fresh | 31.5 | 6.4 | 9.9 | 16.2 | 25.7 | 10.5 | 0.31 | 4.03 |
| Mo2C-A aged | 8.3 | 24.1 | 1.4 | 4.4 | 34.8 | 27.0 | 0.17 | 1.44 |
| Mo2C-A tested | 13.2 | 14.6 | 4.4 | 31.8 | 22.3 | 13.8 | 0.33 | 1.53 |
| Mo2C-B fresh | 40.0 | 4.8 | 15.7 | 13.5 | 14.5 | 11.5 | 0.39 | 3.02 |
| Mo2C-B aged | 17.7 | 9.5 | 4.8 | 15.3 | 28.8 | 23.9 | 0.27 | 3.03 |


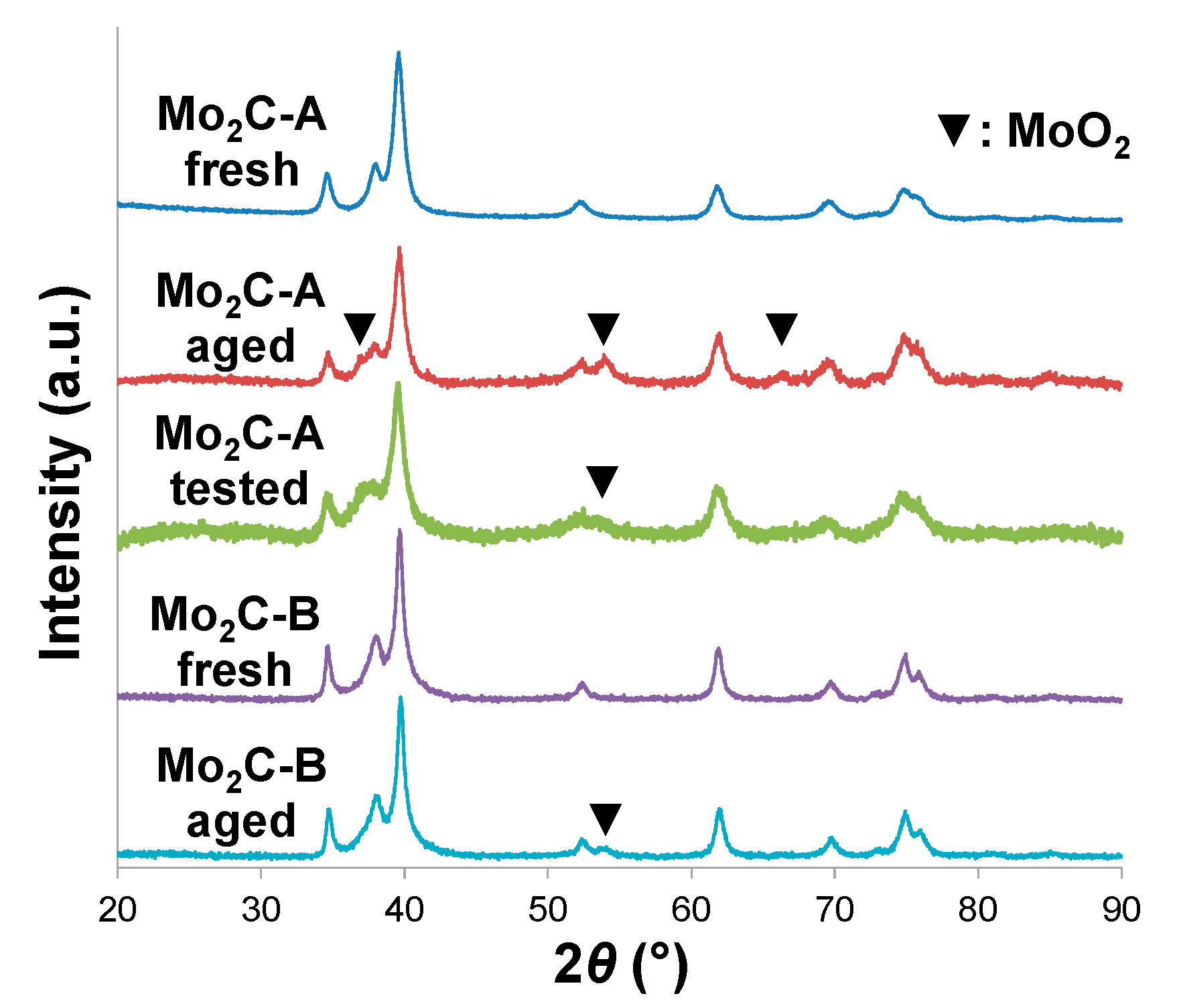
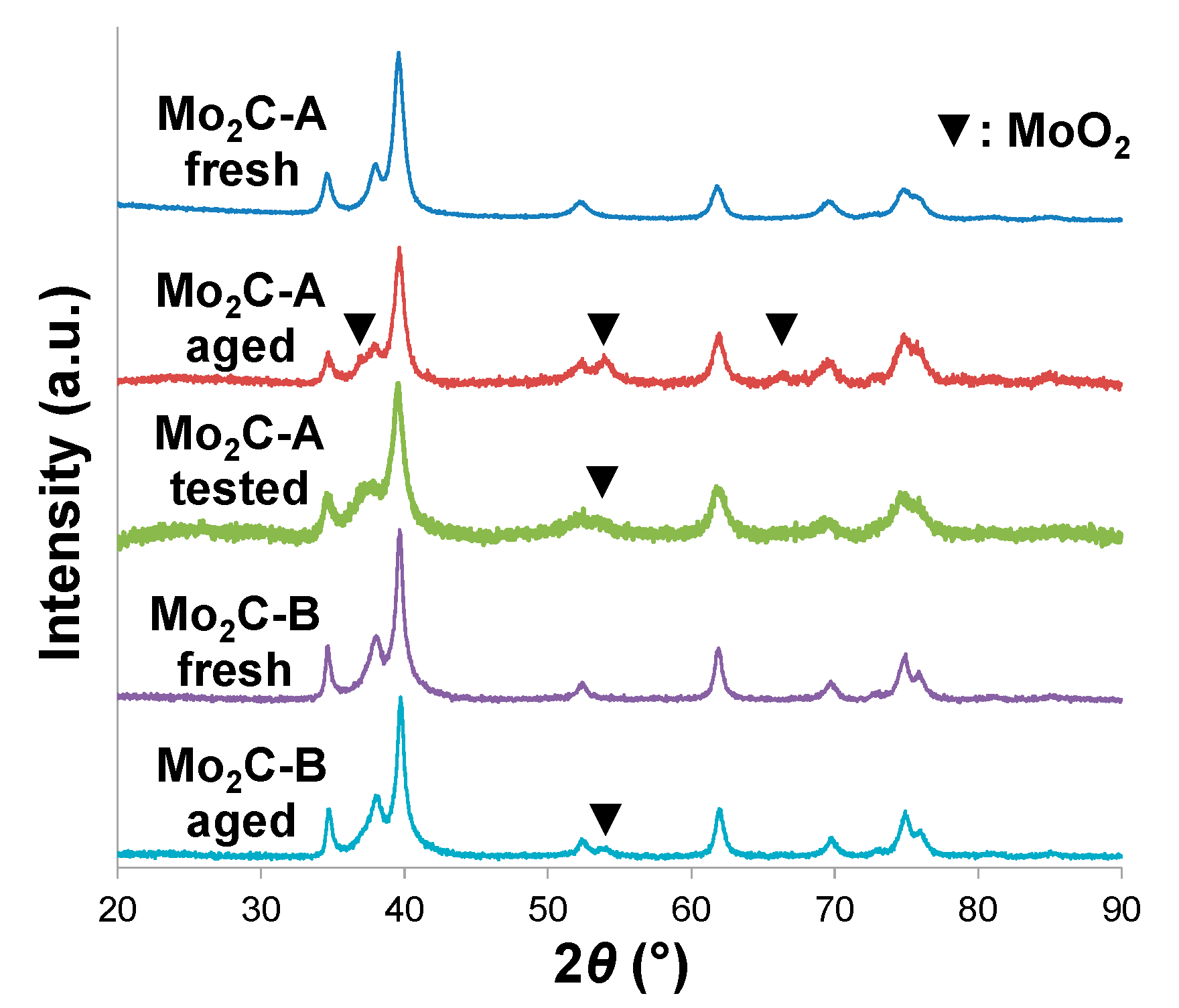
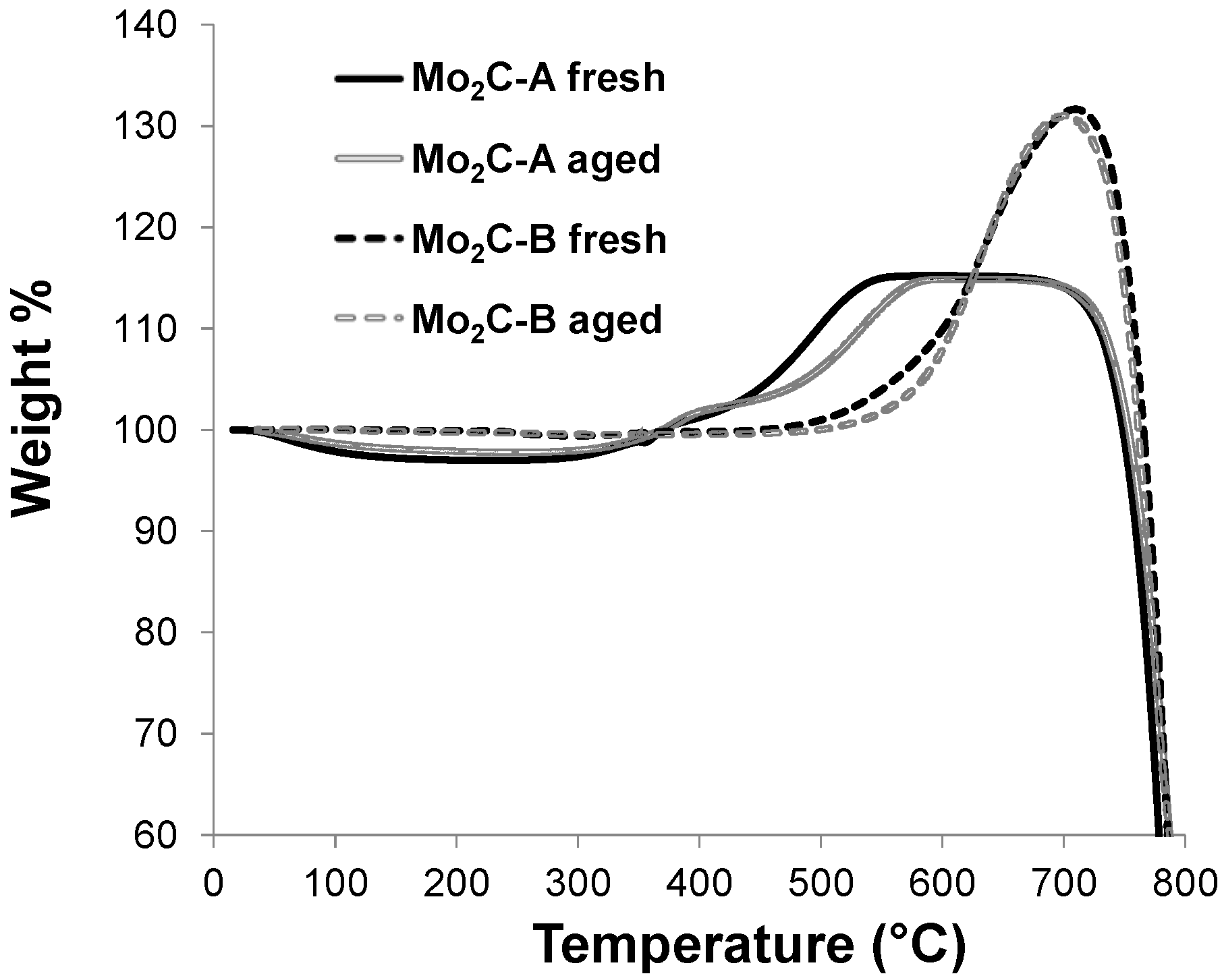
2.2. Structural Evaluation of Mo Carbides Subjected to Hot Aqueous Environments


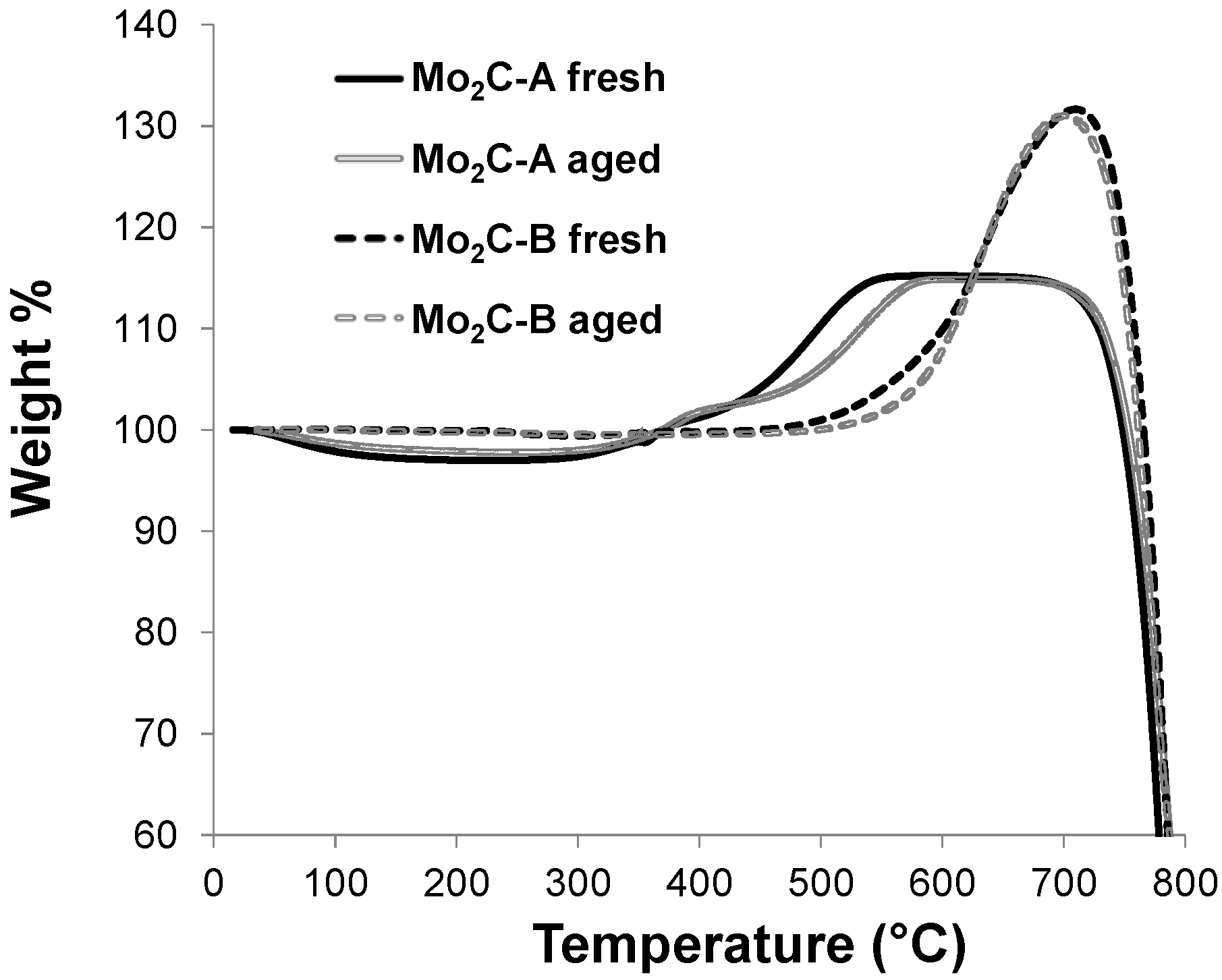
2.3. Catalytic Performance in Aqueous-Phase Hydroprocessing of Acetic Acid
| Catalyst | Reaction temperature/°C | Acetic acid conversion/% | Selectivity to ethanol/% | Selectivity to ethyl acetate/% | Average rate of acetic acid conversion (areal) a /µmol m−2 |
|---|---|---|---|---|---|
| Mo2C-A fresh | 200 | 37 | NM b | NM b | 0.12 |
| Mo2C-A fresh | 250 | 44 | NM b | NM b | 0.14 |
| Mo2C-A aged | 250 | 40 | NM b | NM b | 0.07 |
| Mo2C-B fresh | 250 | 26 | 70 | 12 | 0.50 |
| Mo2C-B aged | 250 | 50 | 18 | 14 | 0.39 |
| Ru/C fresh | 200 | 61 | 28 | 4 | 0.39 |
| Ru/C fresh | 250 | 77 | 10 | 2 | 0.49 |
| Ru/C aged | 250 | 68 | 1 | 0 | 1.58 |
3. Experimental Section
3.1. Catalysts
3.2. Hydrothermal Aging
3.3. Characterization
3.4. Reactor Evaluation
4. Conclusions
- •
- Bulk structures of Mo2C were maintained after 48 h aging at 250 °C in liquid water;
- •
- Carbide surfaces underwent significant structural changes during hydrothermal aging, particularly oxidation of carbidic Mo sites to oxidic sites, transformation of non-carbidic carbon species deposited during the carbide synthesis, and increased surface roughness;
- •
- The removal of pore-blocking non-carbidic carbon and increased surface roughness during aging led to increased BET surface area;
- •
- The extent of the structural changes observed during aging was sensitive to the initial carbide structure and was less severe under actual hydroprocessing conditions;
- •
- Mo carbides showed excellent performance in the aqueous-phase hydroprocessing of acetic acid showing an intrinsic conversion activity comparable to that of Ru/C;
- •
- The intrinsic activity of Mo carbides was shown to be sensitive to the carbide structure;
- •
- Further investigation of carbide structures and performance in complex model or real bio-oil feedstock is needed to design robust, active, and inexpensive Mo carbide catalysts for bio-oil hydroprocessing.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Czernik, S.; Bridgwater, A.V. Overview of applications of biomass fast pyrolysis oil. Energy Fuels 2004, 18, 590–598. [Google Scholar] [CrossRef]
- Connatser, R.M.; Lewis, S.A.L., Sr.; Keiser, J.R.; Choi, J.-S. Measuring bio-oil upgrade intermediates and corrosive species with polarity-matched analytical approaches. Biomass Bioenergy 2014, 70, 557–563. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Farrauto, R.J. Fundamentals of Industrial Catalytic Processes, 2nd ed.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2006. [Google Scholar]
- Bridgwater, A.V. Catalysis in thermal biomass conversion. Appl. Catal. A 1994, 116, 5–47. [Google Scholar] [CrossRef]
- Bridgwater, A.V. Production of high grade fuels and chemicals from catalytic pyrolysis of biomass. Catal. Today 1996, 29, 285–295. [Google Scholar] [CrossRef]
- Elliott, D.C. Historical developments in hydroprocessing bio-oils. Energy Fuels 2007, 21, 1792–1815. [Google Scholar] [CrossRef]
- Elliott, D.C.; Sealock, L.J., Jr.; Baker, E.G. Chemical processing in high-pressure aqueous environments. 2. Development of catalysts for gasification. Ind. Eng. Chem. Res. 1993, 32, 1542–1548. [Google Scholar] [CrossRef]
- Baker, E.G.; Elliott, D.C. Catalytic upgrading of biomass pyrolysis oils. In Research in Thermochemical Biomass Conversion; Bridgwater, A.V., Kuester, J.L., Eds.; Elsevier Science Publishers Ltd.: Barking, UK, 1988; pp. 883–895. [Google Scholar]
- Zacher, A.H.; Olarte, M.V.; Santosa, D.M.; Elliott, D.C.; Jones, S.B. A review and perspective of recent bio-oil hydrotreating research. Green Chem. 2014, 16, 491–515. [Google Scholar] [CrossRef]
- Elliott, D.C.; Wang, H.; French, R.; Deutch, S.; Iisa, K. Hydrocarbon liquid production from biomass via hot-vapor-filtered fast pyrolysis and catalytic hydroprocessing of the bio-oil. Energy Fuels 2014, 28, 5909–5917. [Google Scholar] [CrossRef]
- Elliott, D.C.; Hart, T.R. Catalytic hydroprocessing of chemical models for bio-oil. Energy Fuels 2009, 23, 631–637. [Google Scholar] [CrossRef]
- Sinfelt, J.H.; Yates, D.J.C. Effect of carbiding on hydrogenolysis activity of molybdenum. Nature Phys. Sci. 1971, 229, 27–28. [Google Scholar] [CrossRef]
- Levy, R.B.; Boudart, M. Platinum-like behavior of tungsten carbide in surface catalysis. Science 1973, 181, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Oyama, S.T. Preparation and catalytic properties of transition metal carbides and nitrides. Catal. Today 1992, 15, 179–200. [Google Scholar] [CrossRef]
- Lee, J.S.; Locatelli, S.; Oyama, S.T.; Boudart, M. Molybdenum carbide catalysts. 3. Turnover rates for the hydrogenolysis of n-butane. J. Catal. 1990, 125, 157–170. [Google Scholar] [CrossRef]
- Choi, J.-S.; Bugli, G.; Djéga-Mariadassou, G. Influence of the degree of carburization on the density of sites and hydrogenating activity of molybdenum carbides. J. Catal. 2000, 193, 238–247. [Google Scholar] [CrossRef]
- Volpe, L.; Boudart, M. Compounds of molybdenum and tungsten with high specific surface area. II. Carbides. J. Solid State Chem. 1985, 59, 348–356. [Google Scholar] [CrossRef]
- Lee, J.S.; Oyama, S.T.; Boudart, M. Molybdenum carbide catalysts. 1. Synthesis of unsupported powders. J. Catal. 1987, 106, 125–133. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Y.; Zhao, L.; Wei, W. Catalytic activities of NiMo carbide supported on SiO2 for the hydrodeoxygenation of ethyl benzoate, acetone, and acetaldehyde. Energy Fuels 2010, 24, 2052–2059. [Google Scholar] [CrossRef]
- Jongerius, A.L.; Gosselink, R.W.; Dijkstra, J.; Bitter, J.H.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Carbon nanofiber supported transition-metal carbide catalysts for the hydrodeoxygenation of guaiacol. ChemCatChem 2013, 5, 2964–2972. [Google Scholar]
- Yu, W.; Salciccioli, M.; Xiong, K.; Barteau, M.A.; Vlachos, D.G.; Chen, J.G. Theoretical and experimental studies of C−C versus C−O bond scission of ethylene glycol reaction pathways via metal-modified molybdenum carbides. ACS Catal. 2014, 4, 1409–1418. [Google Scholar] [CrossRef]
- Xiong, K.; Lee, W.-S.; Bhan, A.; Chen, J.G. Molybdenum carbide as a highly selective deoxygenation catalyst for converting furfural to 2-methylfuran. ChemSusChem 2014, 7, 2146–2151. [Google Scholar] [CrossRef] [PubMed]
- Rerez-Romo, P.; Potvin, C.; Manoli, J.-M.; Chehimi, M.M.; Djéga-Mariadassou, G. Phosphorus-doped molybdenum oxynitrides and oxygen-modified molybdenum carbides: synthesis, characterization, and determination of turnover rates for propene hydrogenation. J. Catal. 2002, 208, 187–196. [Google Scholar] [CrossRef]
- Ravenelle, R.M.; Copeland, J.R.; Kim, W.-G.; Crittenden, J.C.; Sievers, C. Structural changes of γ-Al2O3-supported catalysts in hot liquid water. ACS Catal. 2011, 1, 552–561. [Google Scholar] [CrossRef]
- Wu, W.; Wu, Z.; Liang, C.; Ying, P.; Feng, Z.; Li, C. An IR study on the surface passivation of Mo2C/Al2O3 catalyst with O2, H2O and CO2. Phys. Chem. Chem. Phys. 2004, 6, 5603–5608. [Google Scholar] [CrossRef]
- Choi, J.-S.; Krafft, J.-M.; Krzton, A.; Djéga-Mariadassou, G. Study of residual oxygen species over molybdenum carbide prepared during in situ DRIFTS experiments. Catal. Lett. 2002, 81, 175–180. [Google Scholar] [CrossRef]
- Ravenelle, R.M.; Copeland, J.R.; van Pelt, A.H.; Crittenden, J.C.; Sievers, C. Stability of Pt/γ-Al2O3 catalysts in model biomass solutions. Top. Catal. 2012, 55, 162–174. [Google Scholar] [CrossRef]
- Elliott, D.C.; Hart, T.R.; Neuenschwander, G.G. Chemical processing in high-pressure aqueous environments. 8. Improved catalysts for hydrothermal gasification. Ind. Eng. Chem. Res. 2006, 45, 3376–3781. [Google Scholar] [CrossRef]
- Olcay, H.; Xu, L.; Xu, Y.; Huber, G.W. Aqueous-phase hydrogenation of acetic acid over transition metal catalysts. ChemCatChem 2010, 2, 1420–1424. [Google Scholar] [CrossRef]
- Wan, H.; Chaudhari, R.V.; Subramaniam, B. Aqueous phase hydrogenation of acetic acid and its promotional effect on p-cresol hydrodeoxygenation. Energy Fuels 2013, 27, 487–493. [Google Scholar] [CrossRef]
- Lee, J.S.; Yeom, M.H.; Park, K.Y.; Nam, I.-S.; Chung, J.S.; Kim, Y.G.; Moon, S.H. Preparation and benzene hydrogenation activity of supported molybdenum carbide catalysts. J. Catal. 1991, 128, 126–136. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, Y.; Zheng, H.; Zhang, C.; Zhang, B.; Li, Y. Aqueous-phase hydrodeoxygenation of carboxylic acids to alcohols or alkanes over supported Ru catalysts. J. Mol. Catal. A 2011, 351, 217–227. [Google Scholar] [CrossRef]
- Pham-Huu, C.; Ledoux, M.J.; Guille, J. Reactions of 2- and 3-methylpentane, methylcyclopentane, cyclopentane, and cyclohexane on activated Mo2C. J. Catal. 1993, 143, 249–261. [Google Scholar] [CrossRef]
- Blekkan, E.A.; Pham-Huu, C.; Ledoux, M.J.; Guille, J. Isomerization of n-heptane on an oxygen-modified molybdenum carbide catalyst. Ind. Eng. Chem. Res. 1994, 33, 1657–1664. [Google Scholar] [CrossRef]
- Ledoux, M.J.; del Gallo, P.; Pham-Huu, C.; York, A.P.E. Molybdenum oxycarbide isomerization catalysts for cleaner fuel production. Catal. Today 1996, 27, 145–150. [Google Scholar] [CrossRef]
- Iglesia, E.; Ribeiro, F.H.; Boudart, M.; Baumgartner, J.E. Synthesis, characterization, and catalytic properties of clean and oxygen-modified tungsten carbides. Catal. Today 1992, 15, 307–337. [Google Scholar] [CrossRef]
- Ribeiro, F.H.; Betta, R.A.D.; Guskey, G.J.; Boudart, M. Preparation and surface composition of tungsten carbide powders with high specific surface area. Chem. Mater. 1991, 3, 805–812. [Google Scholar] [CrossRef]
- Yang, P.; Zhao, D.; Margolese, D.I.; Chmelka, B.F.; Stucky, G.D. Block copolymer templating syntheses of mesoporous metal oxides with large ordering lengths and semicrystalline framework. Chem. Mater. 1999, 11, 2813–2826. [Google Scholar] [CrossRef]
- Boudart, M.; Djéga-Mariadassou, G. Kinetics of Heterogeneous Catalytic Reactions; Princeton University Press: Princeton, NJ, USA, 1984; pp. 25–26. [Google Scholar]
- Ramanathan, S.; Oyama, S.T. New catalysts for hydroprocessing: transition metal carbides and nitrides. J. Phys. Chem. 1995, 99, 16365–16372. [Google Scholar] [CrossRef]
- St. Clair, T.P.; Dhandapani, B.; Oyama, S.T. Cumene hydrogenation turnover rates on Mo2C: CO and O2 as probes of the active site. Catal. Lett. 1999, 58, 169–171. [Google Scholar] [CrossRef]
© 2015 by U.S. Government; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.-S.; Schwartz, V.; Santillan-Jimenez, E.; Crocker, M.; Lewis, S.A., Sr.; Lance, M.J.; Meyer, H.M., III; More, K.L. Structural Evolution of Molybdenum Carbides in Hot Aqueous Environments and Impact on Low-Temperature Hydroprocessing of Acetic Acid. Catalysts 2015, 5, 406-423. https://doi.org/10.3390/catal5010406
Choi J-S, Schwartz V, Santillan-Jimenez E, Crocker M, Lewis SA Sr., Lance MJ, Meyer HM III, More KL. Structural Evolution of Molybdenum Carbides in Hot Aqueous Environments and Impact on Low-Temperature Hydroprocessing of Acetic Acid. Catalysts. 2015; 5(1):406-423. https://doi.org/10.3390/catal5010406
Chicago/Turabian StyleChoi, Jae-Soon, Viviane Schwartz, Eduardo Santillan-Jimenez, Mark Crocker, Samuel A. Lewis, Sr., Michael J. Lance, Harry M. Meyer, III, and Karren L. More. 2015. "Structural Evolution of Molybdenum Carbides in Hot Aqueous Environments and Impact on Low-Temperature Hydroprocessing of Acetic Acid" Catalysts 5, no. 1: 406-423. https://doi.org/10.3390/catal5010406
APA StyleChoi, J.-S., Schwartz, V., Santillan-Jimenez, E., Crocker, M., Lewis, S. A., Sr., Lance, M. J., Meyer, H. M., III, & More, K. L. (2015). Structural Evolution of Molybdenum Carbides in Hot Aqueous Environments and Impact on Low-Temperature Hydroprocessing of Acetic Acid. Catalysts, 5(1), 406-423. https://doi.org/10.3390/catal5010406