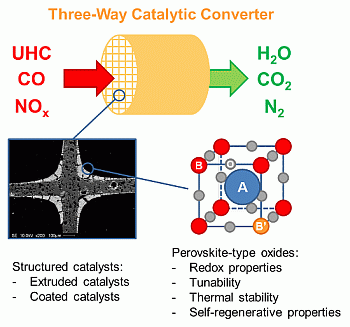
Structured Perovskite-Based Catalysts and Their Application as Three-Way Catalytic Converters—A Review


Abstract
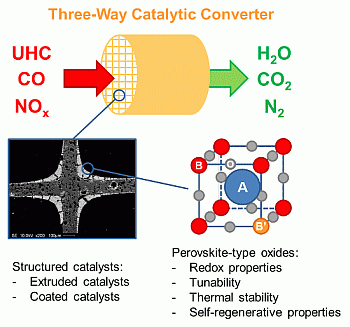
:
1. Introduction
2. Perovskite-Type Oxides
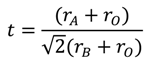
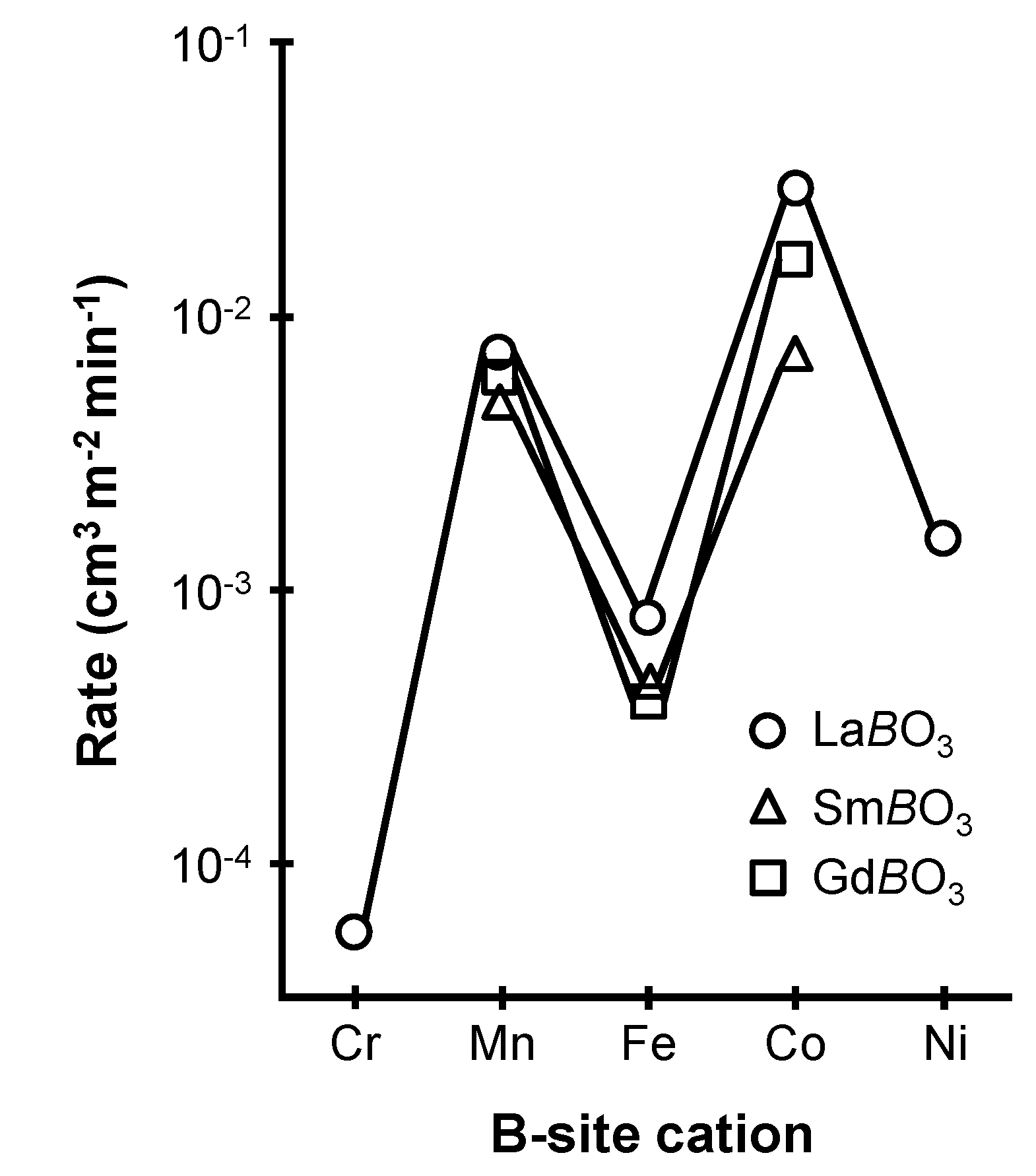
2.1. Parameters Influencing the Catalytic Activity
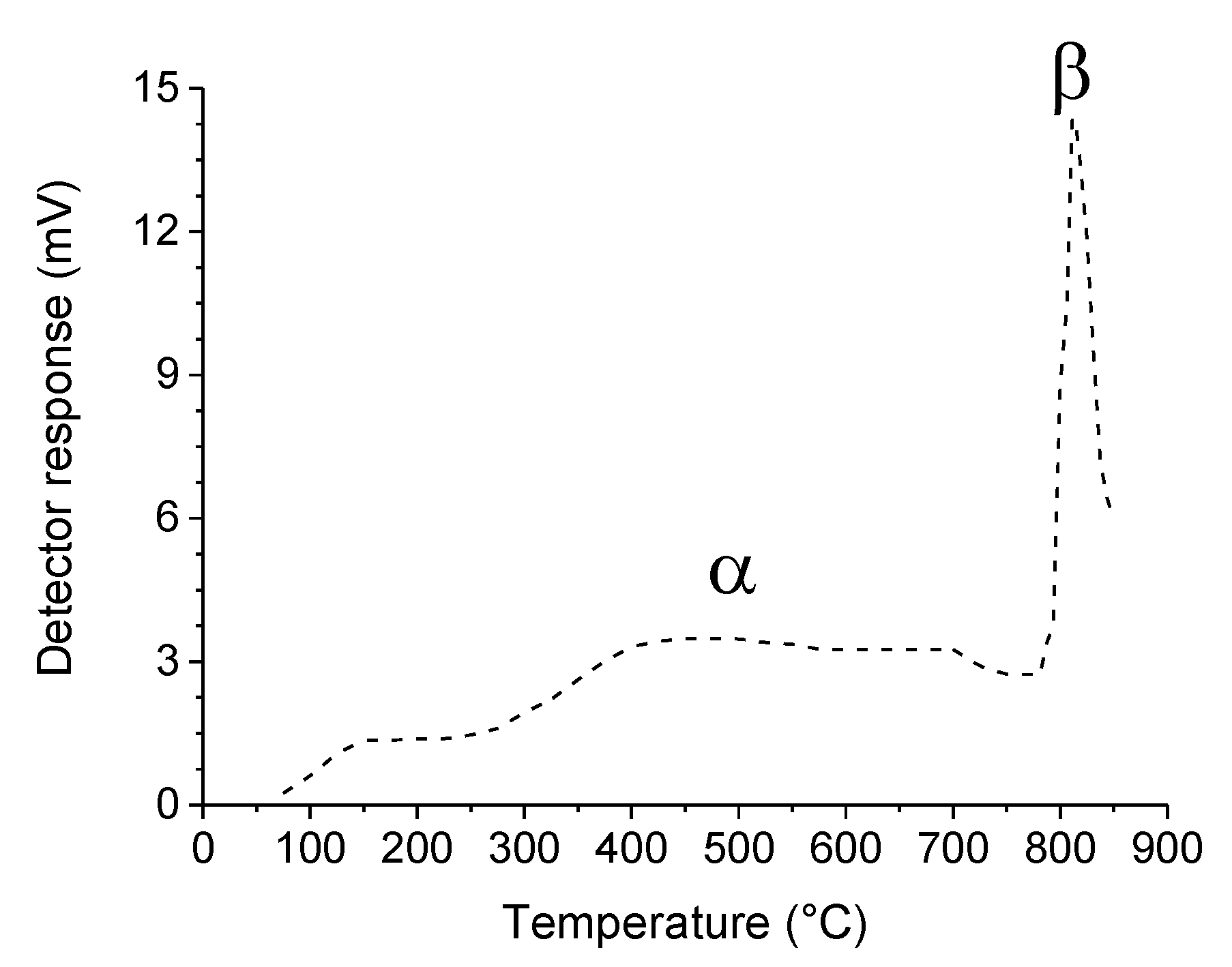
2.2. Reaction Mechanisms
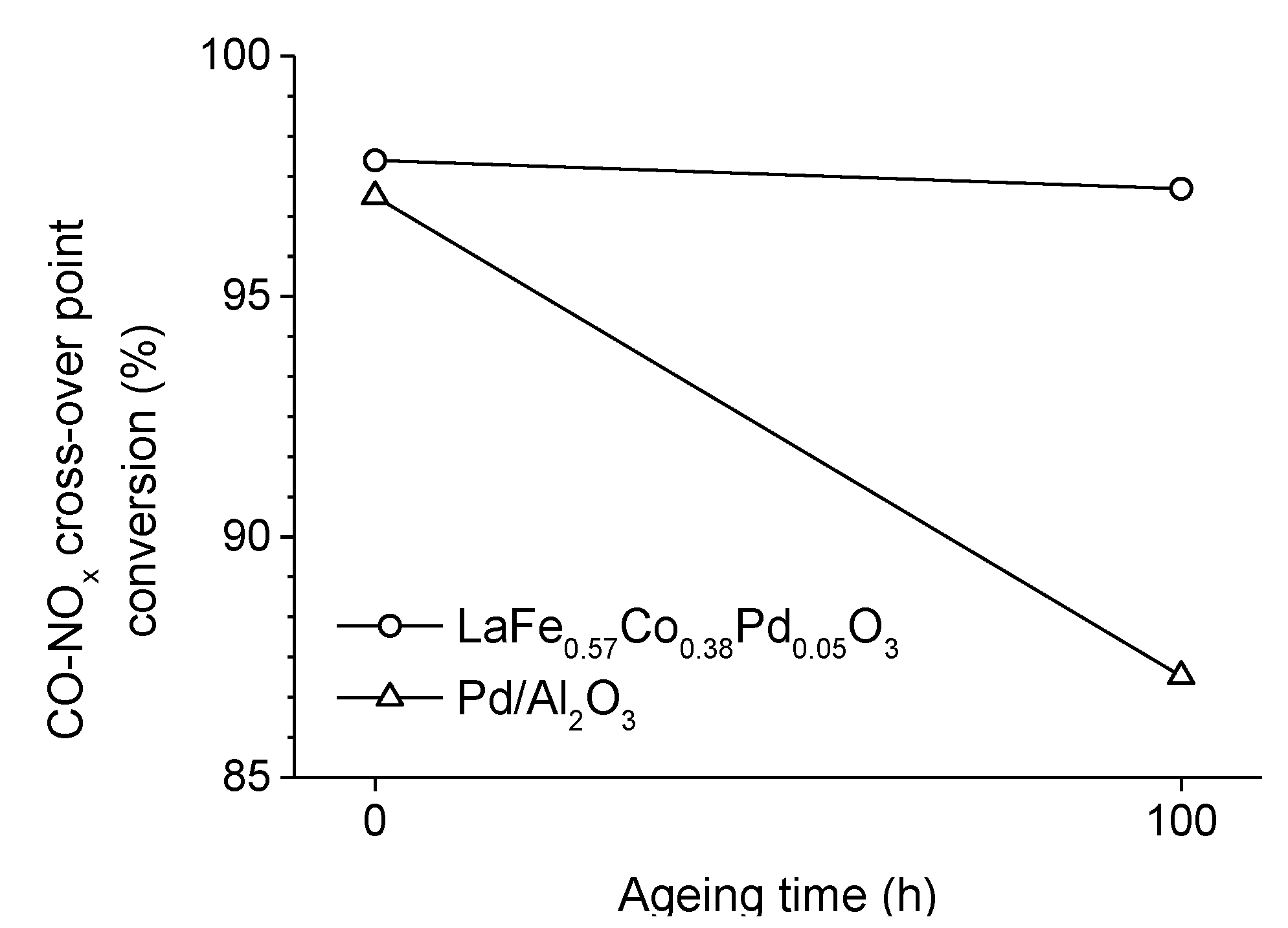
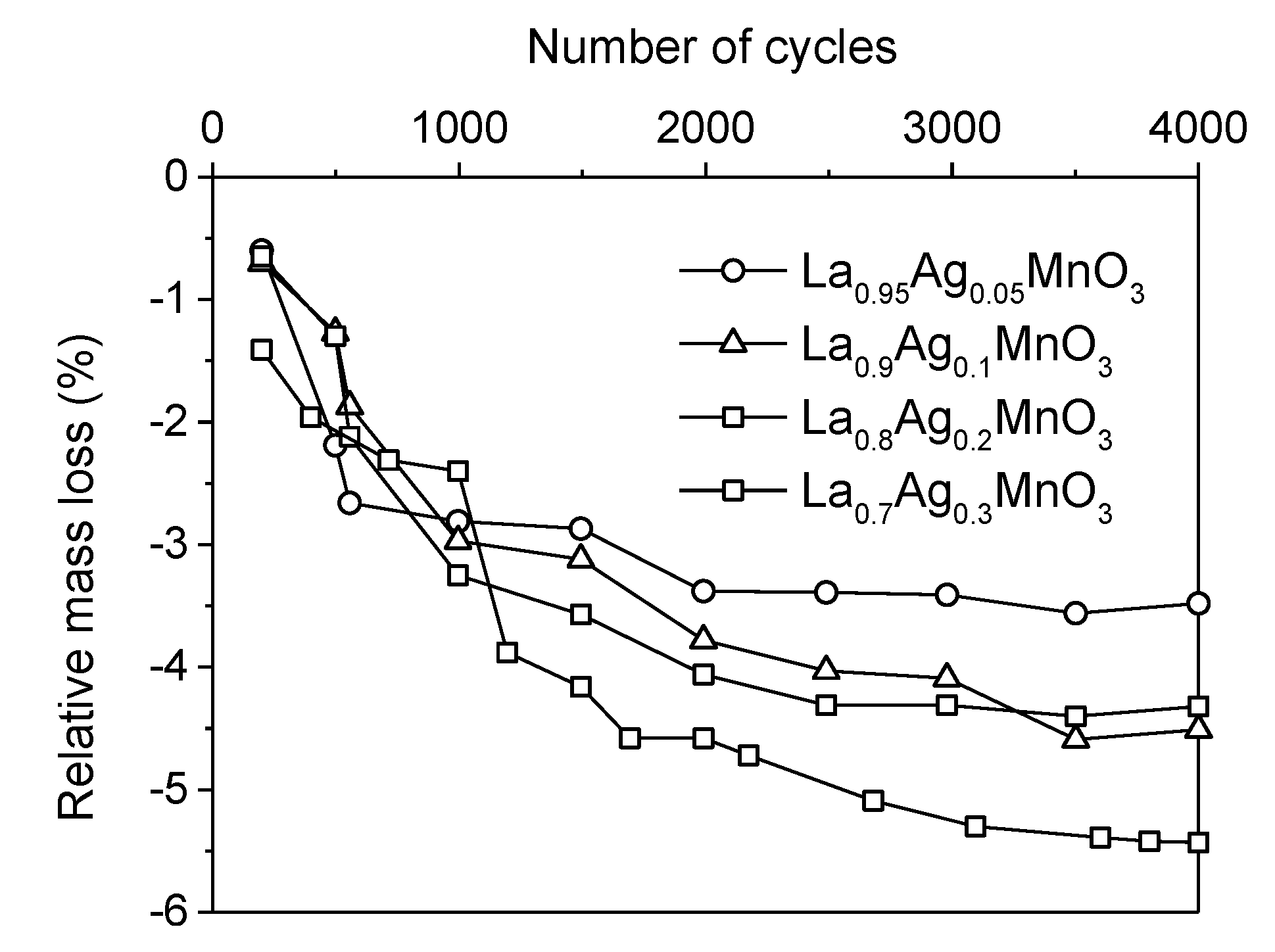
2.3. Incorporation of a Noble Metal
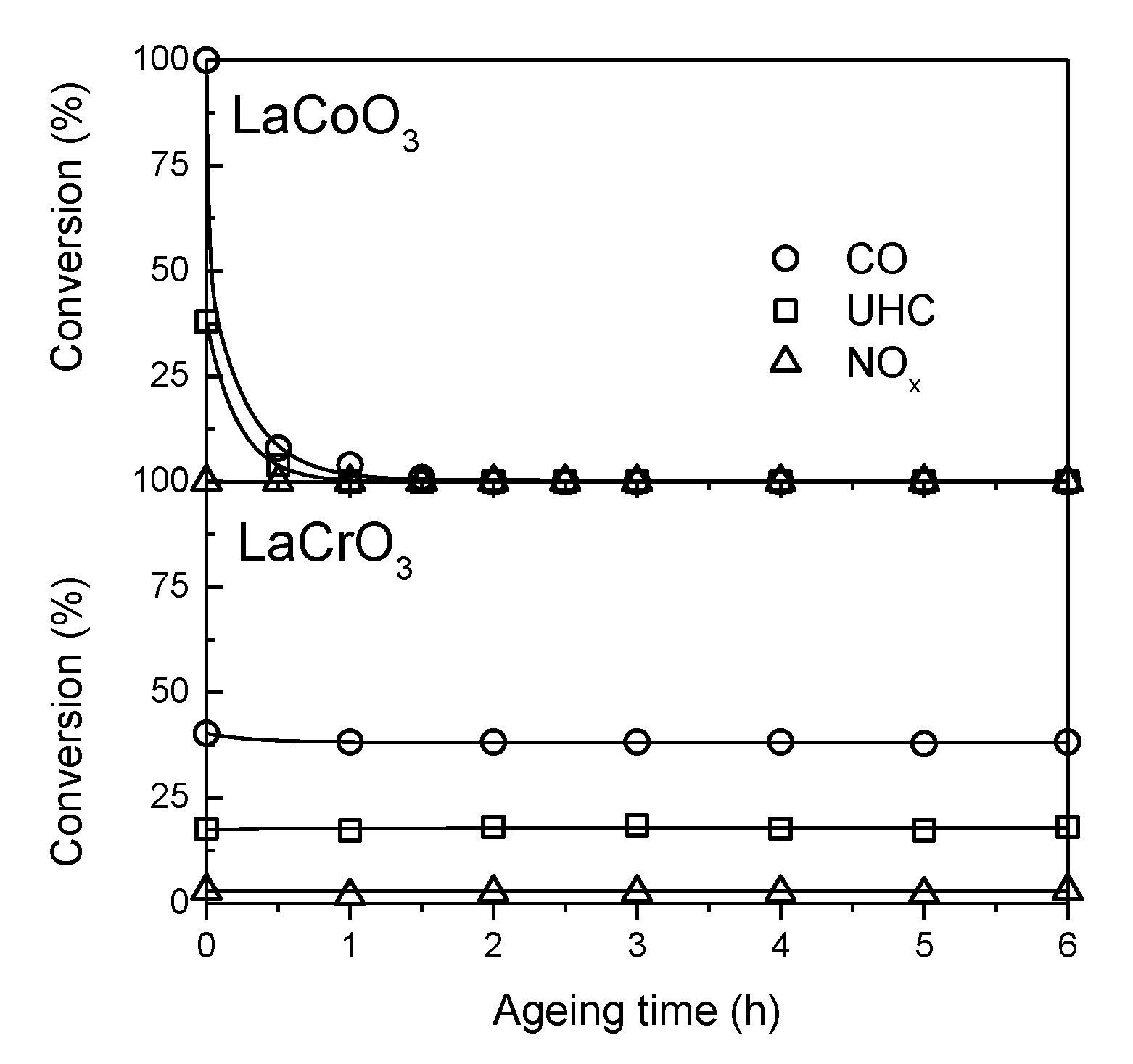
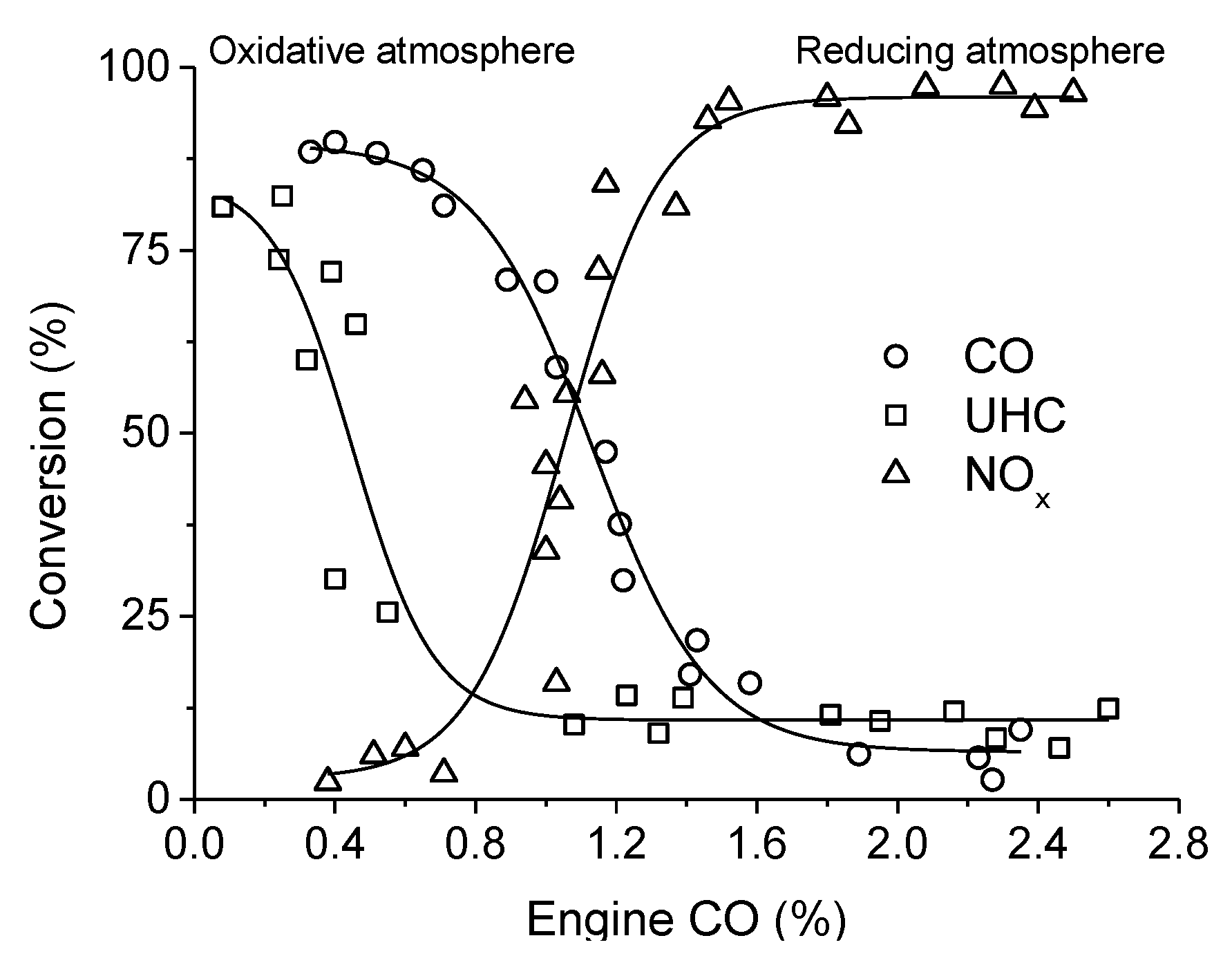
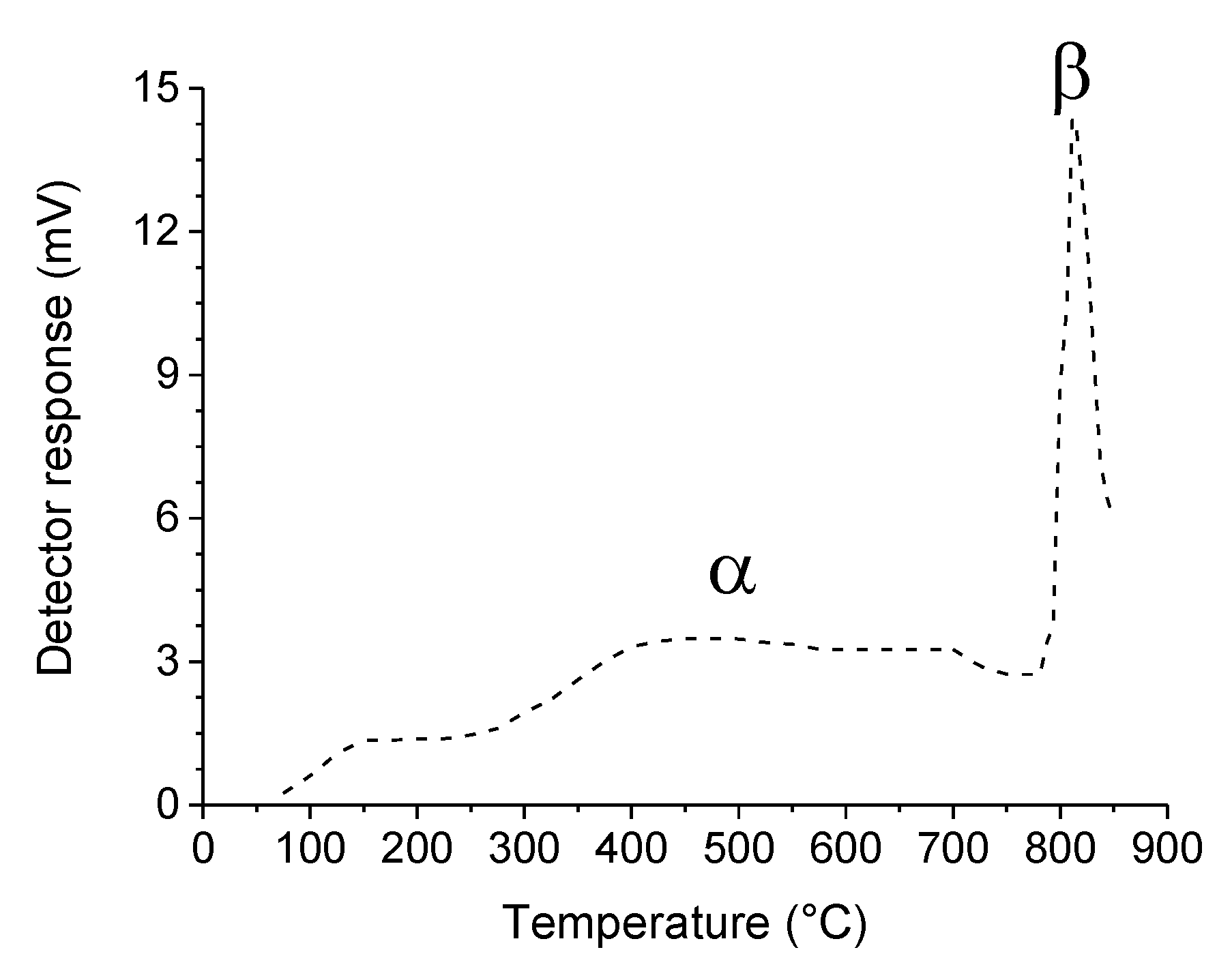
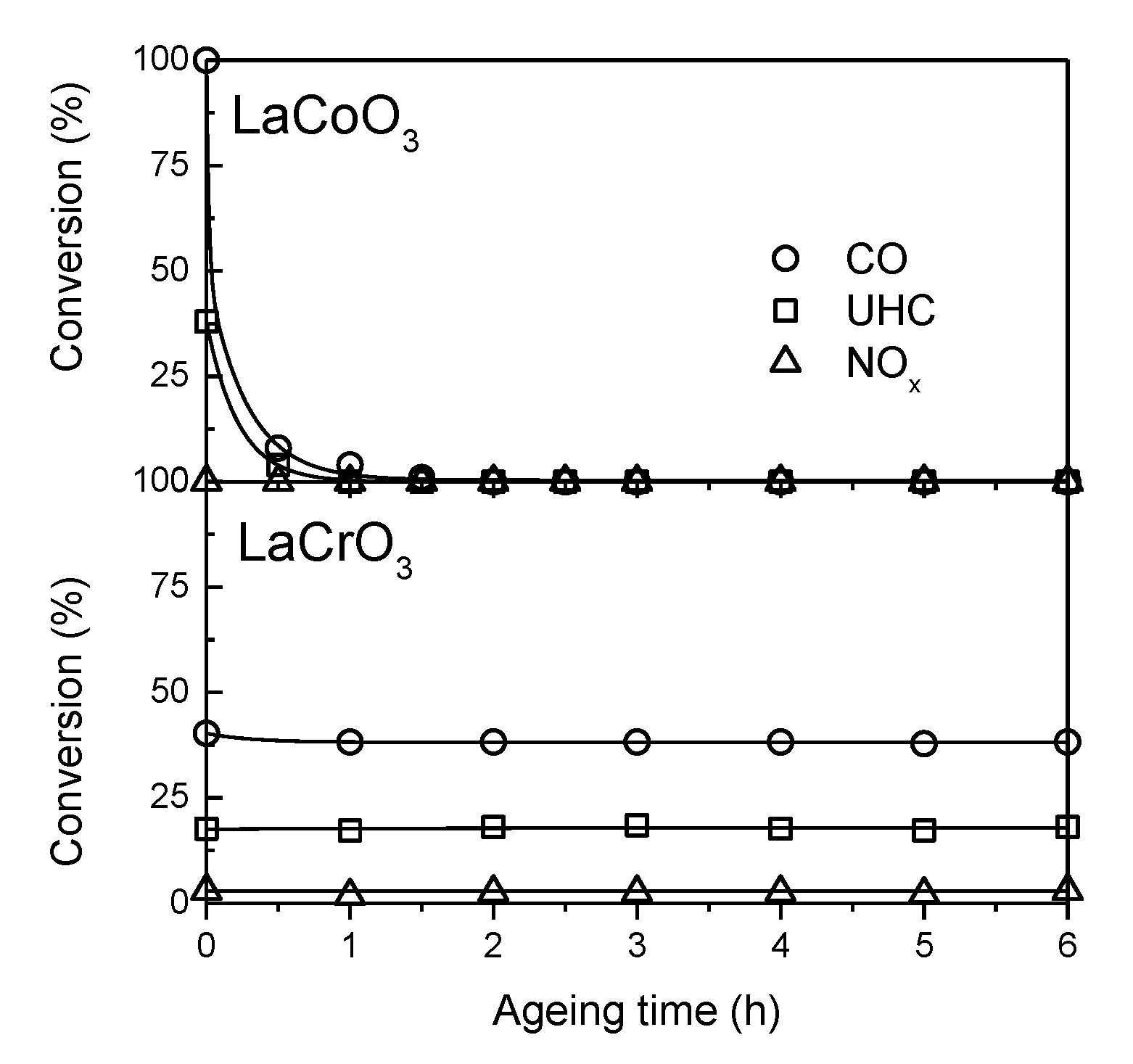
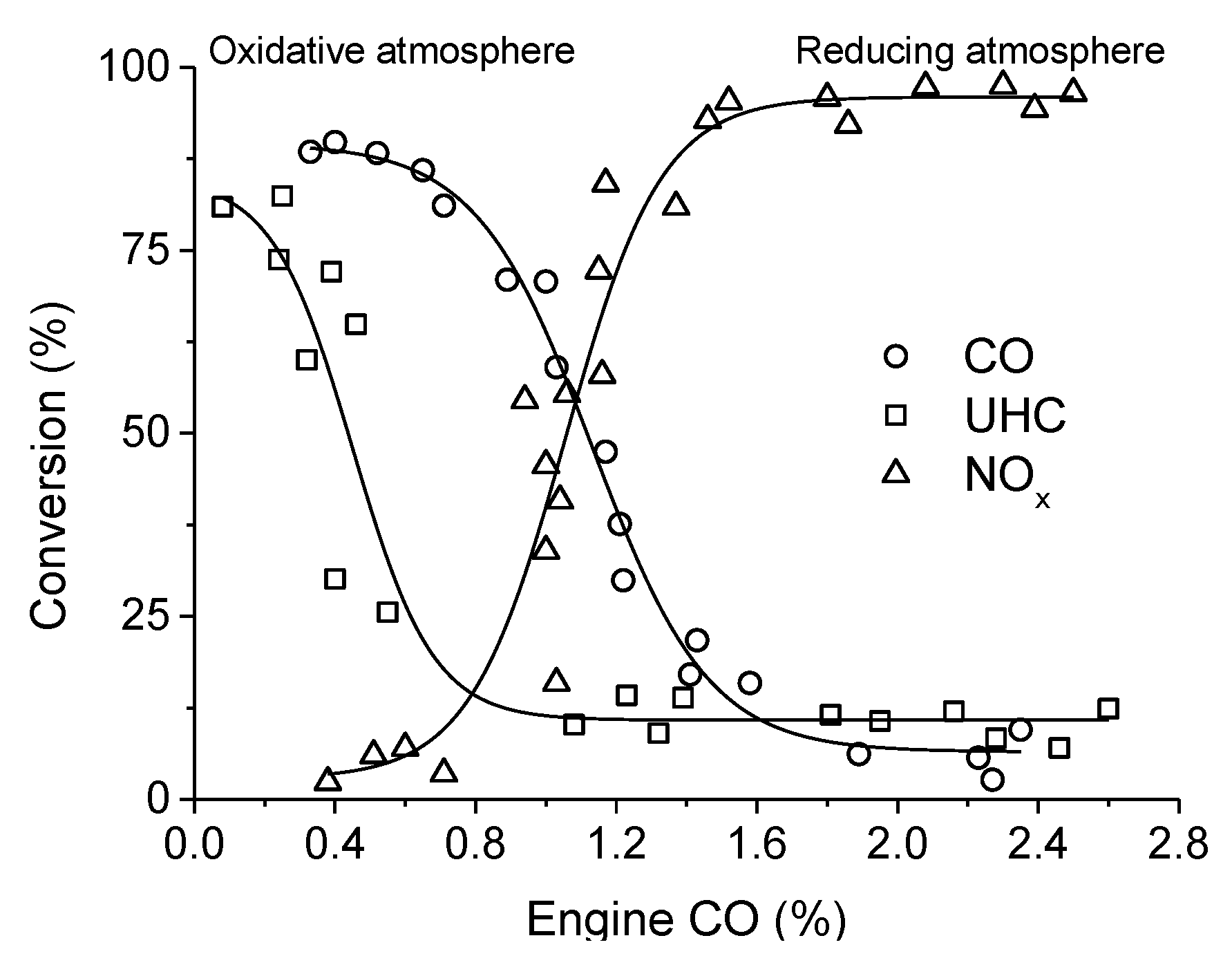
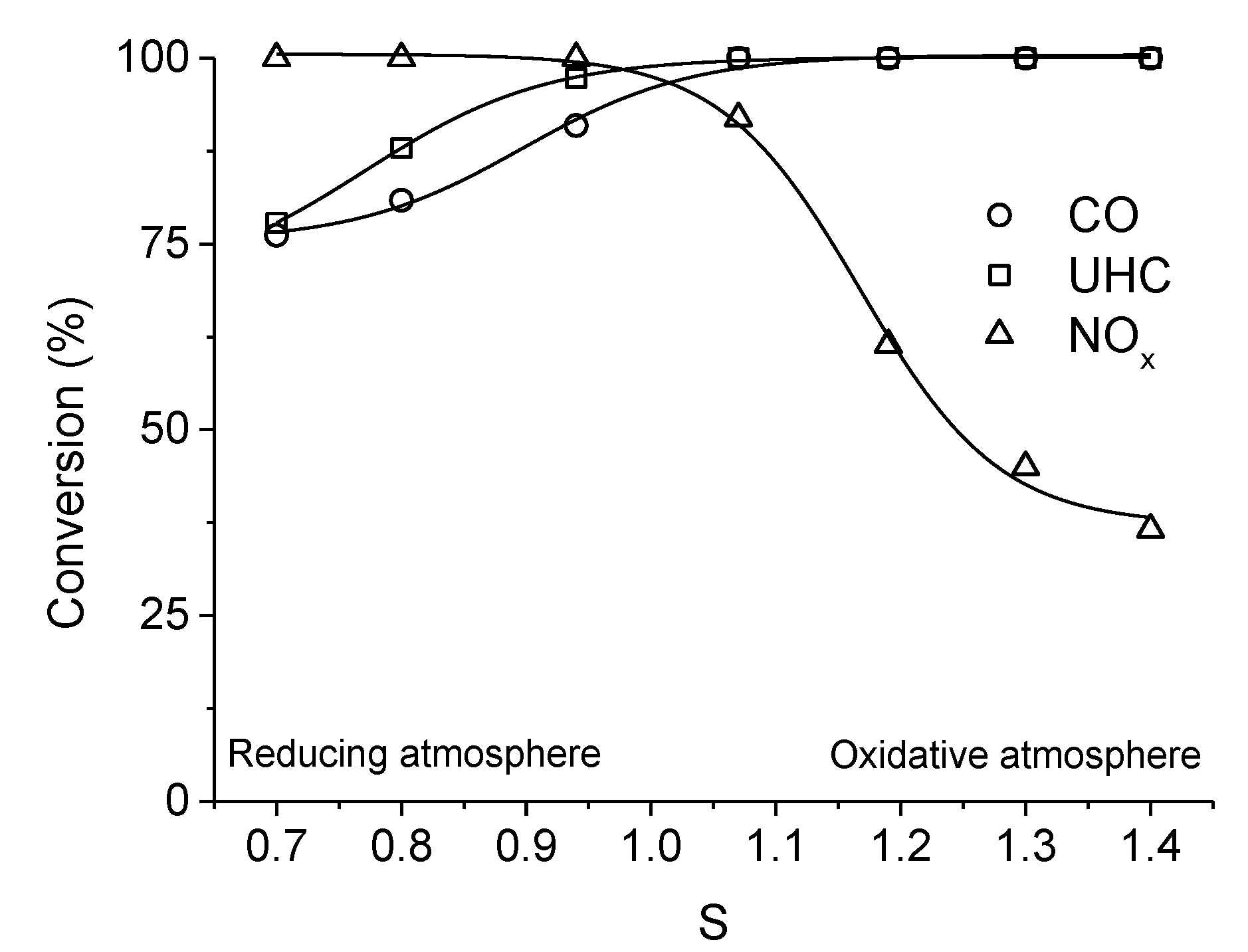
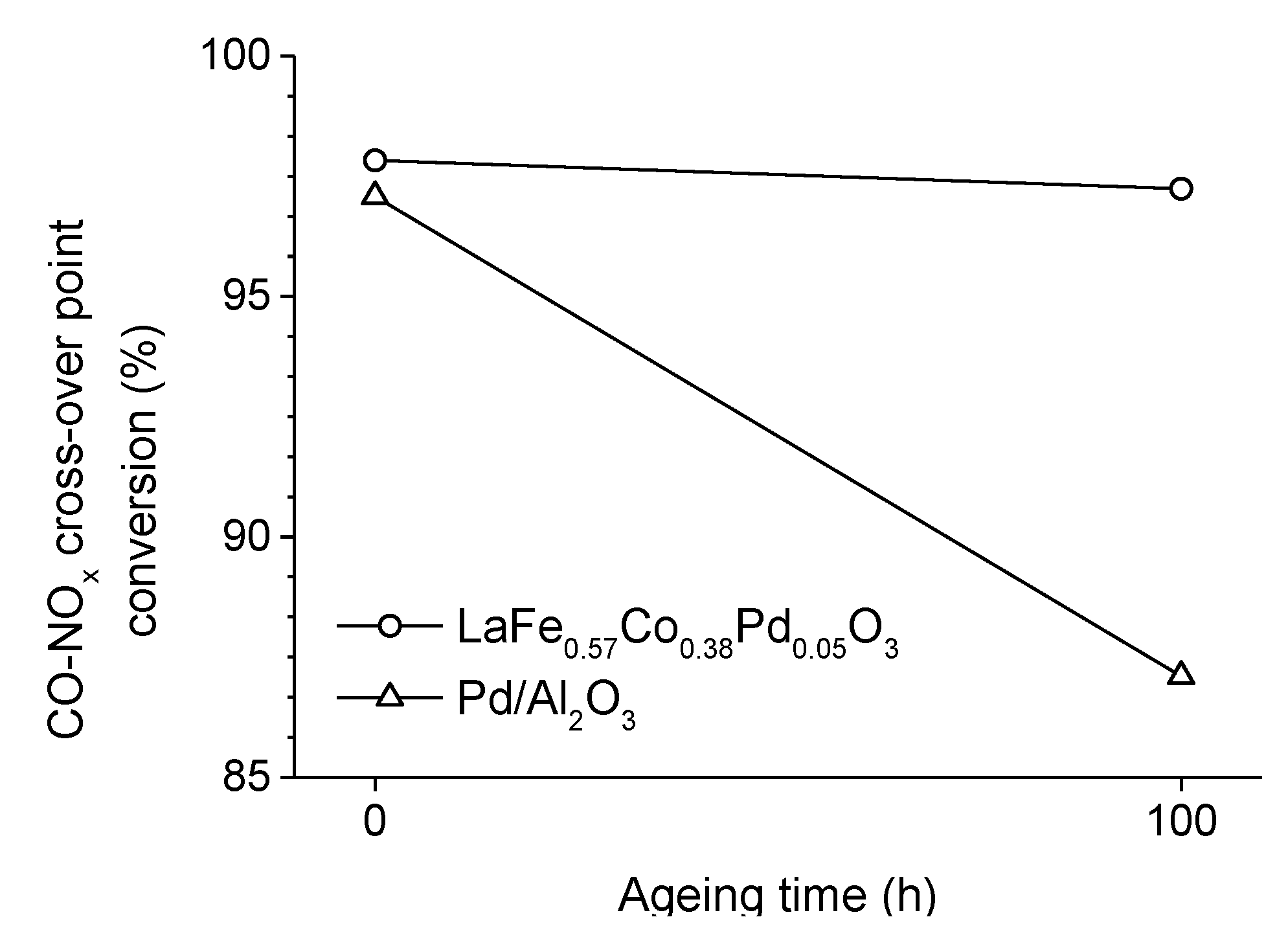
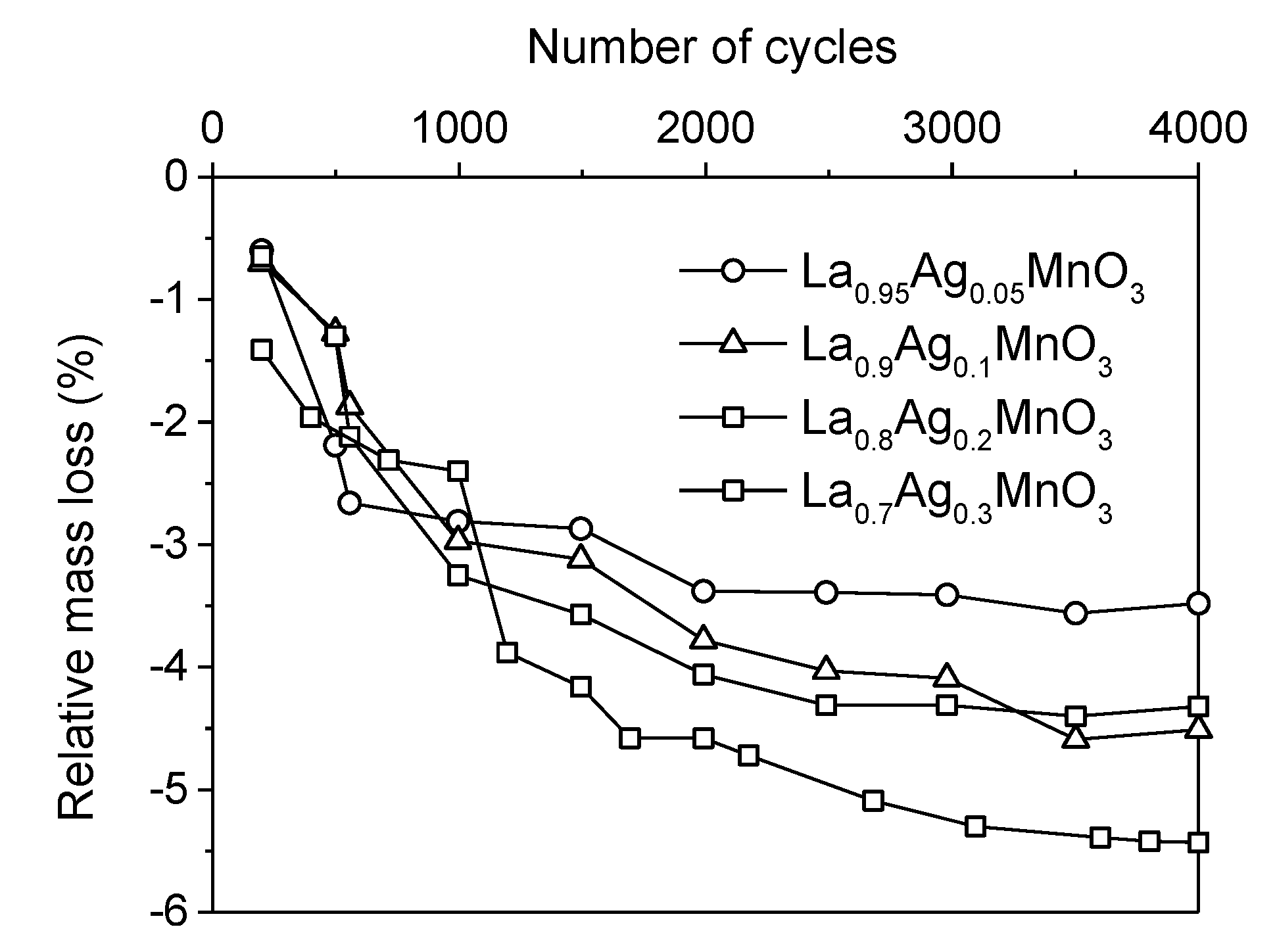
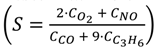 . Adapted with permission from Guilhaume et al. [13]. Copyright 1997, Elsevier.
. Adapted with permission from Guilhaume et al. [13]. Copyright 1997, Elsevier.
. Adapted with permission from Guilhaume et al. [13]. Copyright 1997, Elsevier.
. Adapted with permission from Guilhaume et al. [13]. Copyright 1997, Elsevier.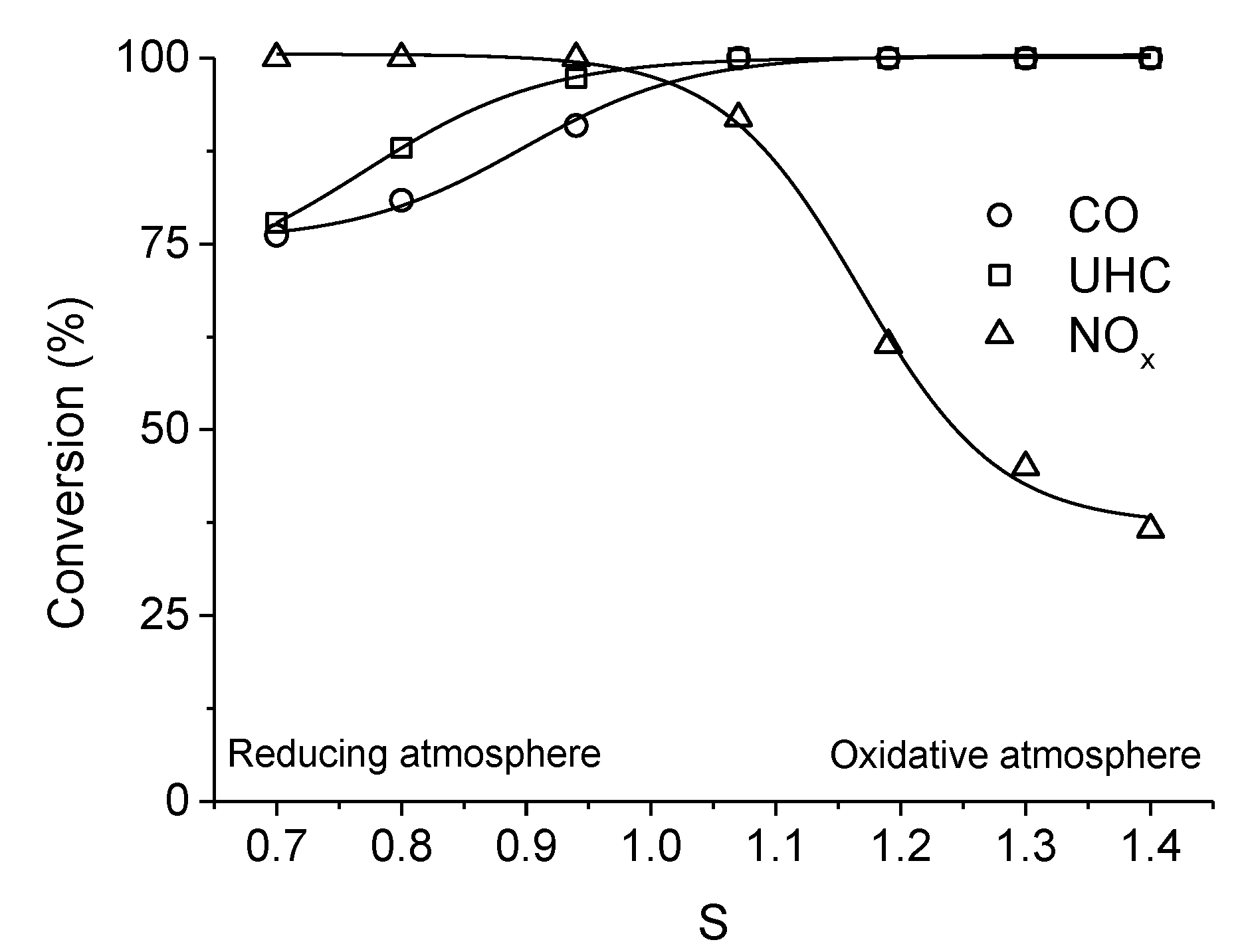

| Host structure | Noble metal | ||
|---|---|---|---|
| Pd | Rh | Pt | |
| LaFeO3 | 0%–100% | 63%–100% | 25%–83% |
| LaAlO3 | - | 69%–100% | - |
| CaTiO3 | x | 28%–100% | 20%–100% |
| CaZrO3 | - | 53%–100% | 27%–100% |
| SrTiO3 | - | 37%–100% | 50%–100% |
| SrZrO3 | - | x | x |
| BaTiO3 | - | 0%–100% | 25%–93%% |
| BaZrO3 | - | 47%–100% | x |

3. Preparation of Structured Catalysts
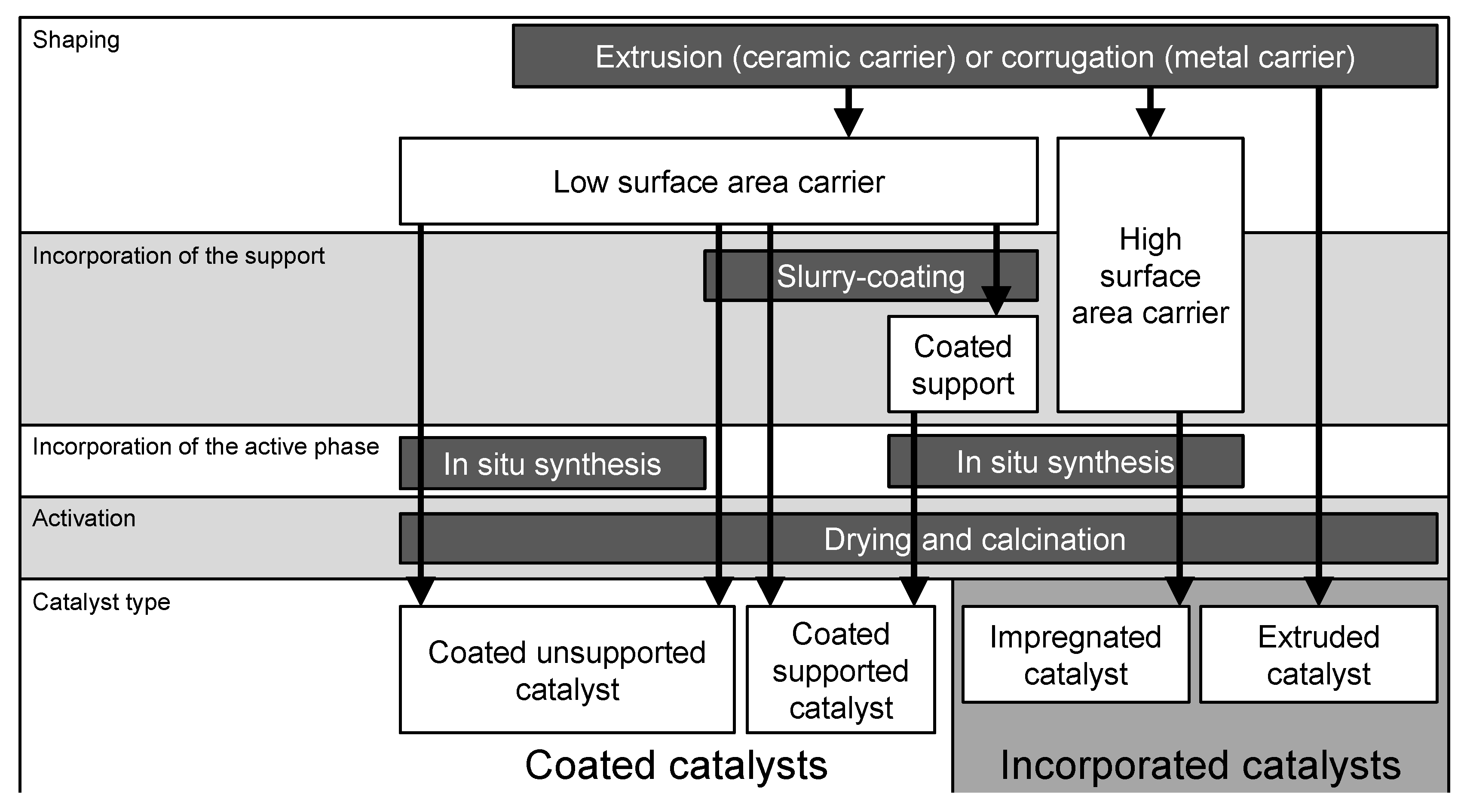
3.1. Incorporated Catalysts
3.2. Coated Catalysts
3.2.1. Precursor-Derived “On-Substrate-Wall” Synthesis of Catalysts
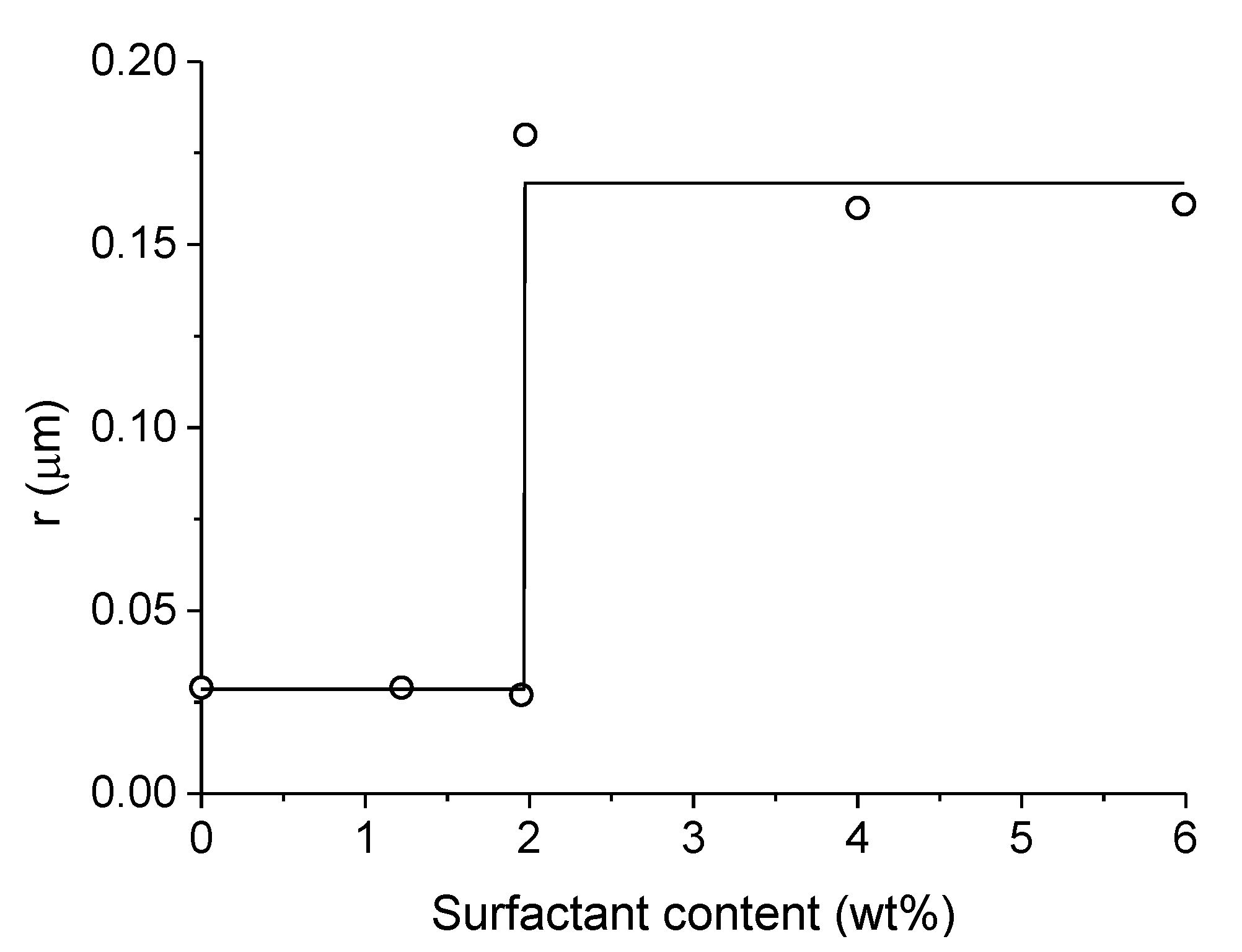
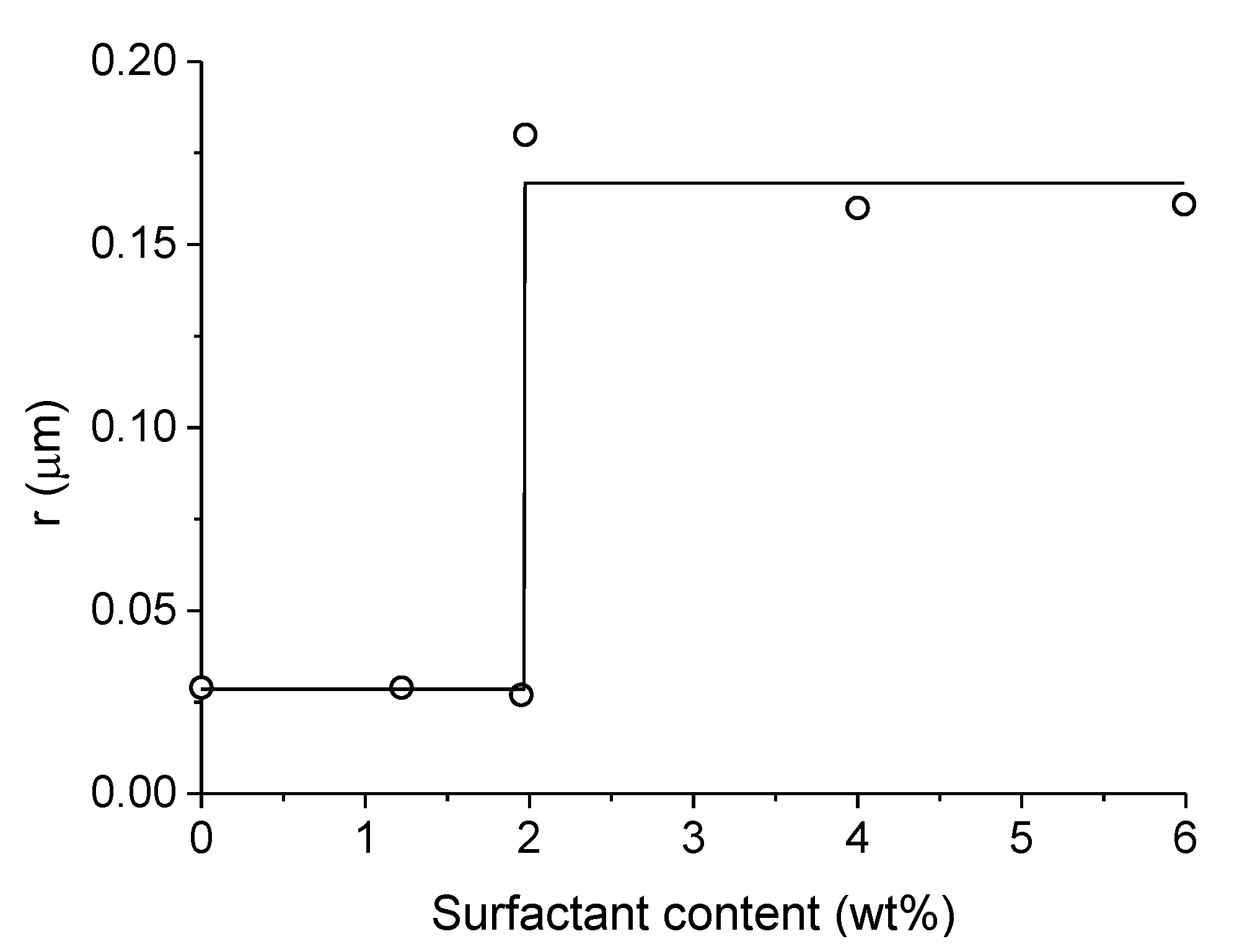
3.2.2. Washcoated Catalysts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
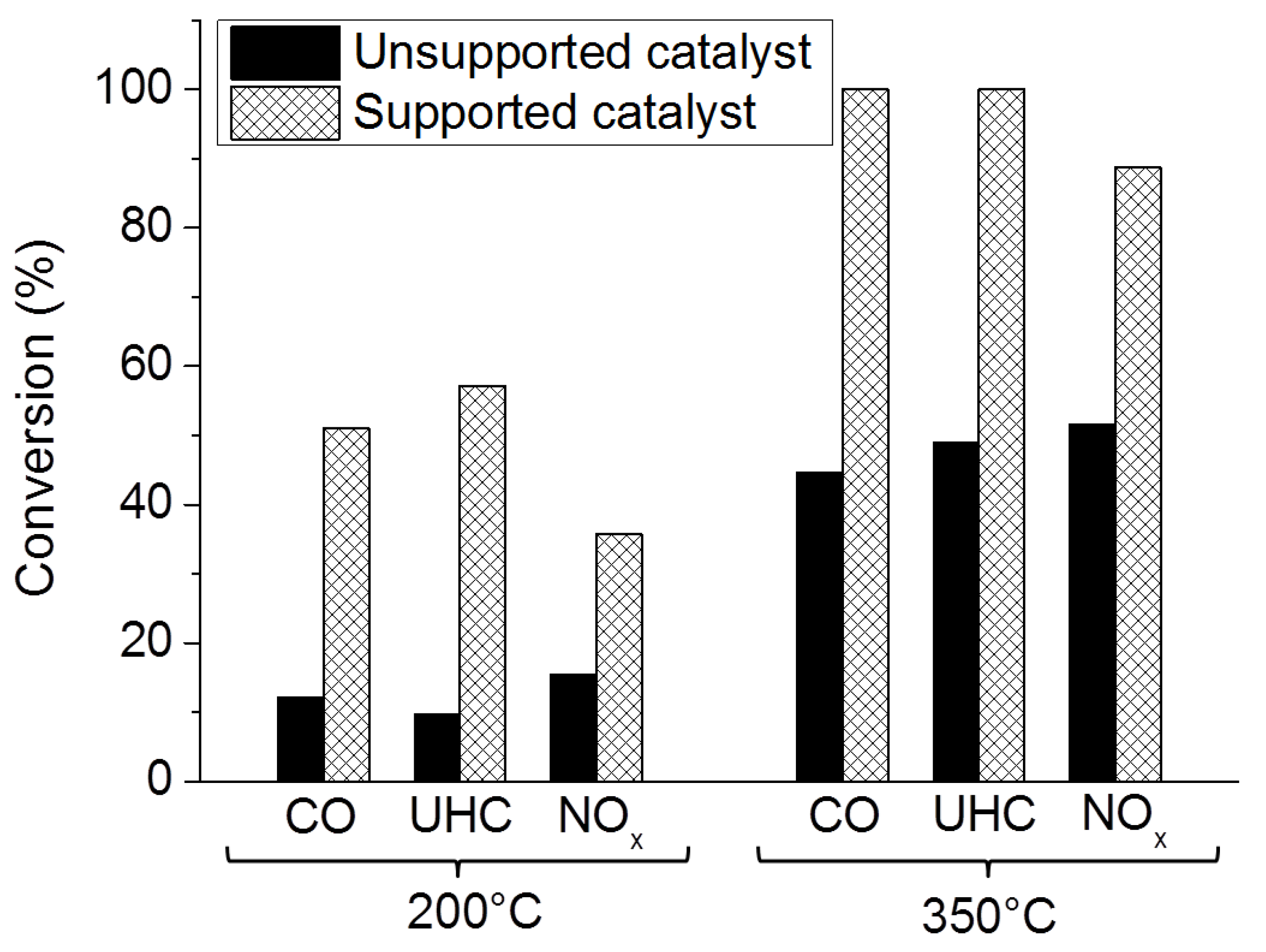
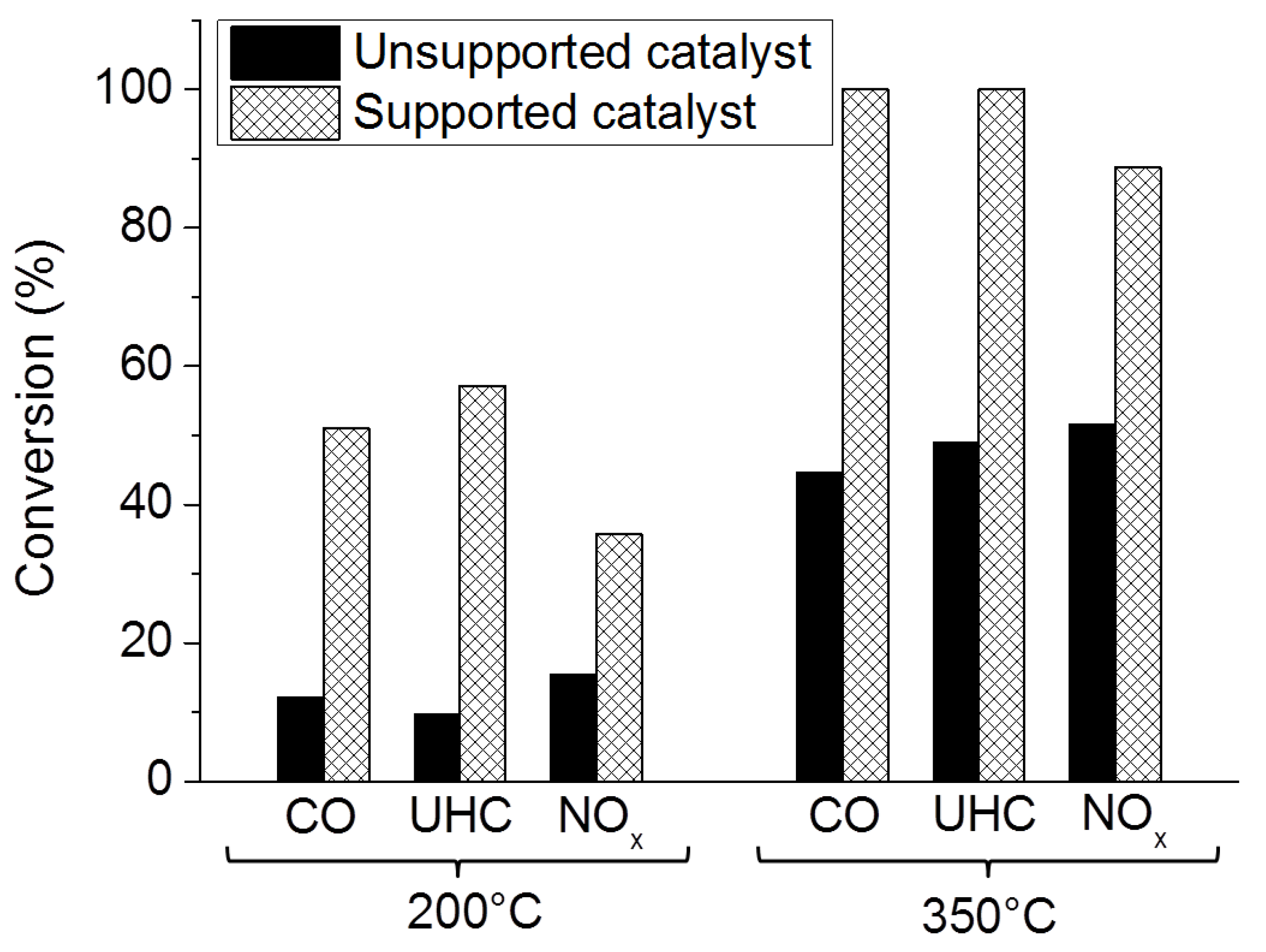
Washcoated Unsupported Catalysts

Washcoated Supported Catalysts
4. Implementation in TWC Converters
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Taylor, K.C. Automobile Catalytic Converters. Stud. Surf. Sci. Catal. 1987, 30, 97–116. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalysis by Ceria and Related Materials, 1st ed.; Imperial College Press: London, UK, 2002. [Google Scholar]
- Forster, P.; Ramaswamy, V.; Artaxo, P.; Berntsen, T.; Betts, R.; Fahey, D.W.; Haywood, J.; Lean, J.; Lowe, D.C.; Myhre, G.; et al. Changes in Atmospheric Constituents and in Radiative Forcing. In Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change; Solomon, S., Qin, D., Manning, M., Chen, Z., Marquis, M., Averyt, K.B., Tygnor, M., Miller, H.L., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2007; pp. 129–234. [Google Scholar]
- Fino, D.; Russo, N.; Saracco, G.; Specchia, V. Supported Pd-perovskite catalyst for CNG engines’ exhaust gas treatment. Prog. Solid State Chem. 2007, 35, 501–511. [Google Scholar] [CrossRef]
- Yao, H.C.; Japar, S.; Shelef, M. Surface Interactions in the System Rh/Al2O3. J. Catal. 1977, 50, 407–418. [Google Scholar] [CrossRef]
- Libby, W.F. Promising Catalyst for Auto Exhaust. Science 1971, 171, 499–500. [Google Scholar]
- Voorhoeve, R.J.H.; Remeika, J.P.; Freeland, P.E.; Matthias, B.T. Rare-Earth Oxides of Manganese and Cobalt Rival Platinum for the Treatment of Carbon Monoxide in Auto Exhaust. Science 1972, 177, 353–354. [Google Scholar]
- Voorhoeve, R.J.H.; Remeika, J.P.; Johnson, D.W., Jr. Rare-Earth Manganites: Catalysts with Low Ammonia Yield in the Reduction of Nitrogen Oxides. Science 1973, 180, 62–64. [Google Scholar]
- Voorhoeve, R.J.H.; Remeika, J.P.; Trimble, L.E. Perovskites containing ruthenium as catalysts for nitric oxide reduction. Mater. Res. Bull. 1974, 9, 1393–1404. [Google Scholar] [CrossRef]
- Voorhoeve, R.J.H.; Trimble, L.E. Exploration of perovskite-like catalysts: Ba2CoWO6 and Ba2FeNbO6 in NO reduction and CO oxidation. Mater. Res. Bull. 1974, 9, 655–666. [Google Scholar] [CrossRef]
- Gallagher, P.K.; Johnson, D.W., Jr.; Schrey, F. Studies of some supported perovskite oxidation catalysts. Mater. Res. Bull. 1974, 9, 1345–1352. [Google Scholar] [CrossRef]
- Tabata, K.; Misono, M. Elimination of pollutant gases—Oxidation of CO, reduction and decomposition of NO. Catal. Today 1990, 8, 249–261. [Google Scholar] [CrossRef]
- Guilhaume, N.; Primet, M. Three-Way Catalytic Activity and Oxygen Storage Capacity of Perovskite LaMn0.976Rh0.024O3+δ. J. Catal. 1997, 165, 197–204. [Google Scholar] [CrossRef]
- Tanaka, H. An intelligent catalyst: The self-regenerative palladium-perovskite catalyst for automotive emissions control. Catal. Surv. Asia 2005, 9, 63–74. [Google Scholar] [CrossRef]
- Kim, C.H.; Qi, G.; Dahlberg, K.; Li, W. Strontium-Doped Perovskites Rival Platinum Catalysts for Treating NOx in Simulated Diesel Exhaust. Science 2010, 327, 1624–1627. [Google Scholar] [CrossRef]
- Goldschmidt, V.M. Die Gesetze der Krystallochemie. Die Naturwissenschaften 1926, 8, 477–485. [Google Scholar] [CrossRef]
- Ebbinghaus, S.G.; Abicht, H.P.; Dronskowski, R.; Müller, T.; Reller, A.; Weidenkaff, A. Perovskite-related oxynitrides—Recent developments in synthesis, characterisation and investigations of physical properties. Progr. Solid State Chem. 2009, 37, 173–205. [Google Scholar] [CrossRef]
- Yoon, S.; Maegli, A.E.; Karvonen, L.; Matam, S.K.; Shkabko, A.; Riegg, S.; Großmann, T.; Ebbinghaus, S.G.; Pokrant, S.; Weidenkaff, A. Bandgap tuning in SrTi(N,O,F)3 by anionic-lattice variation. J. Solid State Chem. 2013, 206, 226–232. [Google Scholar] [CrossRef]
- Weidenkaff, A.; Ebbinghaus, S.G.; Lippert, T.; Montenegro, M.J.; Soltmann, C.; Wessicken, R. Phase formation and phase transition of Ln1−xCaxCoO3−δ (Ln = La, Er) applied for bifunctional air electrodes. Cryst. Eng. 2002, 5, 449–457. [Google Scholar]
- Weidenkaff, A. Preparation and Application of Nanostructured Perovskite Phases. Adv. Eng. Mater. 2004, 6, 709–714. [Google Scholar] [CrossRef]
- Raveau, B. The perovskite history: More than 60 years of research from the discovery of ferroelectricity to colossal magnetoresistance via high TC superconductivity. Prog. Solid State Chem. 2007, 35, 171–173. [Google Scholar] [CrossRef]
- Weidenkaff, A.; Robert, R.; Aguirre, M.; Bochet, L.; Lippert, T.; Canulescu, S. Development of thermoelectric oxides for renewable energy conversion technologies. Renew. Energy 2008, 33, 342–347. [Google Scholar] [CrossRef]
- Voorhoeve, R.J.H.; Johnson, D.W., Jr.; Remeika, J.P.; Gallagher, P.K. Perovskite Oxides: Materials Science in Catalysis. Science 1977, 195, 827–833. [Google Scholar]
- Bhalla, A.S.; Guo, R.; Roy, R. The perovskite structure—A review of its role in ceramic science and technology. Mater. Res. Innov. 2000, 4, 3–26. [Google Scholar]
- Peña, M.A.; Fierro, J.L.G. Chemical Structures and Performance of Perovskite Oxides. Chem. Rev. 2001, 101, 1981–2017. [Google Scholar] [CrossRef]
- Tejuca, L.G.; Fierro, J.L.G.; Tascón, J.M.D. Structure and Reactivity of Perovskite-Type Oxides. Adv. Catal. 1989, 36, 237–328. [Google Scholar]
- Royer, S.; Duprez, D. Catalytic Oxidation of Carbon Monoxide over Transition Metal Oxides. ChemCatChem 2011, 3, 24–65. [Google Scholar] [CrossRef]
- Parravano, G. Ferroelectric Transitions and Heterogeneous Catalysis. J. Chem. Phys. 1952, 20, 342–343. [Google Scholar] [CrossRef]
- Dickens, P.G.; Whittingham, M.S. Recombination of oxygen atoms on oxide surfaces. Part 2.—Catalytic activities of the alkali metal tungsten bronzes. Trans. Faraday Soc. 1965, 61, 1226–1231. [Google Scholar] [CrossRef]
- Tascón, J.M.D.; Tejuca, L.G. Catalytic activity of perovskite-type oxides LaMeO3. React. Kinet. Catal. Lett. 1980, 15, 185–191. [Google Scholar]
- Nitadori, T.; Ichiki, T.; Misono, M. Catalytic Properties of Perovskite-Type Mixed Oxides (ABO3) Consisting of Rare Earth and 3d Transition Metals. The Roles of the A- and B-Site Ions. Bull. Chem. Soc. Jpn. 1988, 61, 621–626. [Google Scholar]
- Panich, N.M.; Pirogova, G.N.; Korosteleva, R.I.; Voronin, Y.V. Oxidation of CO and hydrocarbons over perovskite-type complex oxides. Russ. Chem. Bull. 1999, 48, 694–697. [Google Scholar]
- Voorhoeve, R.J.H.; Remeika, J.P.; Trimble, L.E.; Cooper, A.S.; Disalvo, F.J.; Gallagher, P.K. Perovskite-like La1−xKxMnO3 and related compounds: Solid state chemistry and the catalysis of the reduction of NO by CO and H2. J. Sol. State. Chem. 1975, 14, 395–406. [Google Scholar] [CrossRef]
- Hackenberger, M.; Stephan, K.; Kießling, D.; Schmitz, W.; Wendt, G. Influence of the preparation conditions on the properties of perovskite-type oxide catalysts. Solid State Ionics 1997, 101–103, 1195–1200. [Google Scholar] [CrossRef]
- Giannakas, A.E.; Ladavos, A.K.; Pomonis, P.J. Preparation, characterization and investigation of catalytic activity for NO + CO reaction of LaMnO3 and LaFeO3 perovskites prepared via microemulsion method. Appl. Catal. B 2004, 49, 147–158. [Google Scholar] [CrossRef]
- Lu, Y.; Eyssler, A.; Otal, E.H.; Matam, S.K.; Brunko, O.; Weidenkaff, A.; Ferri, D. Influence of the synthesis method on the structure of Pd-substituted perovskite catalysts for methane oxidation. Catal. Today 2013, 208, 42–47. [Google Scholar] [CrossRef]
- Campagnoli, E.; Tavares, A.; Fabbrini, L.; Rossetti, I.; Dubitsky, Y.A.; Zaopo, A.; Forni, A. Effect of preparation method on activity and stability of LaMnO3 and LaCoO3 catalysts for the flameless combustion of methane. Appl. Catal. B 2005, 55, 133–139. [Google Scholar]
- Oliva, C.; Bonoldi, L.; Cappelli, S.; Fabbrini, L.; Rossetti, I.; Forni, L. Effect of preparation parameters on SrTiO3±δ catalyst for the flameless combustion of methane. J. Mol. Catal. A 2005, 226, 33–40. [Google Scholar] [CrossRef]
- Stephan, K.; Hackenberger, M.; Kiessling, D.; Wendt, G. Supported perovskite-type oxide catalysts for the total oxidation of chlorinated hydrocarbons. Catal. Today 1999, 54, 23–30. [Google Scholar] [CrossRef]
- Ran, R.; Wu, X.; Weng, D. Effect of complexing species in a sol-gel synthesis on the physicochemical properties of La0.7Sr0.3Mn0.7Cu0.3O3+λ catalyst. J. Alloys Compd. 2006, 414, 169–174. [Google Scholar] [CrossRef]
- Kaliaguine, S.; van Neste, A. Process for Synthesizing Perovskites Using High Energy Milling. U.S. Patent 6,017,504, 25 January 2000. [Google Scholar]
- Kaliaguine, S.; van Neste, A.; Szabo, V.; Gallot, J.E.; Bassir, M.; Muzychuk, R. Perovskite-type oxides synthesized by reactive grinding—Part I. Preparation and characterization. Appl. Catal. A 2001, 209, 345–358. [Google Scholar] [CrossRef]
- Royer, S.; Bérubé, F.; Kaliaguine, S. Effect of the synthesis conditions on the redox and catalytic properties in oxidation reactions of LaCo1−xFexO3. Appl. Catal. A 2005, 282, 273–284. [Google Scholar] [CrossRef]
- Zhang, R.; Villanueva, A.; Alamdari, H.; Kaliaguine, S. Catalytic reduction of NO by propene over LaCo1−xCuxO3 perovskites synthesized by reactive grinding. Appl. Catal. B 2006, 64, 220–223. [Google Scholar] [CrossRef]
- Levasseur, B.; Kaliaguine, S. Methanol oxidation on LaBO3 (B = Co, Mn, Fe) perovskite-type catalysts prepared by reactive grinding. Appl. Catal. A 2008, 343, 29–38. [Google Scholar] [CrossRef]
- Shu, J.; Kaliaguine, S. Well-dispersed perovskite-type oxidation catalysts. Appl. Catal. B 1998, 16, L303–L308. [Google Scholar] [CrossRef]
- Ran, R.; Wu, X.; Weng, D.; Fan, J. Oxygen storage capacity and structural properties of Ni-doped LaMnO3 perovskites. J. Alloys Compd. 2013, 577, 288–294. [Google Scholar] [CrossRef]
- Chang, Y.F.; McCarty, J.G. Novel oxygen storage components for advanced catalysts for emission control in natural gas fueled vehicles. Catal. Today 1996, 30, 163–170. [Google Scholar] [CrossRef]
- Yamazoe, N.; Teraoka, Y.; Seiyama, T. TPD and XPS study on thermal behavior of adsorbed oxygen in La1−xSrxCoO3. Chem. Lett. 1981, 10, 1767–1770. [Google Scholar] [CrossRef]
- Kremenić, G.; Nieto, J.M.L.; Tascón, J.M.D.; Tejuca, L.G. Chemisorption and catalysis on LaMO3 oxides. J. Chem. Soc. Faraday Trans. 1 1985, 81, 939–949. [Google Scholar] [CrossRef]
- Arai, H.; Yamada, T.; Eguchi, K.; Seiyama, T. Catalytic combustion of methane over various perovskite-type oxides. Appl. Catal. 1986, 26, 265–276. [Google Scholar] [CrossRef]
- Markova-Velichkova, M.; Lazarova, T.; Tumbalev, V.; Ivanov, G.; Kovacheva, D.; Stefanov, P.; Naydenov, A. Complete oxidation of hydrocarbons on YFeO3 and LaFeO3 catalysts. Chem. Eng. J. 2013, 231, 236–244. [Google Scholar] [CrossRef]
- Voorhoeve, R.J.H.; Remeika, J.P.; Trimble, L.E. Defect chemistry and catalysis in oxidation and reduction over perovskite-type oxides. Ann. N. Y. Acad. Sci. 1976, 272, 3–21. [Google Scholar] [CrossRef]
- Fierro, J.L.G. Structure and composition of perovskite surface in relation to adsorption and catalytic properties. Catal. Today 1990, 8, 153–174. [Google Scholar] [CrossRef]
- Tascón, J.M.D.; Fierro, J.L.G.; Tejuca, L.G. Kinetics and Mechanism of CO Oxidation on LaCoO3. Z. Phys. Chem. 1981, 124, 249–257. [Google Scholar] [CrossRef]
- Tascón, J.M.D.; Tejuca, L.G. Adsorption of CO2 on the perovskite-type oxide LaCoO3. J. Chem. Soc. Faraday Trans. 1 1981, 77, 591–602. [Google Scholar] [CrossRef]
- Forni, L.; Oliva, C.; Barzetti, T.; Selli, E.; Ezerets, A.M.; Vishniakov, A.V. FT-IR and EPR spectroscopic analysis of La1−xCexCoO3 perovskite-like catalysts for NO reduction by CO. Appl. Catal. B 1997, 13, 35–43. [Google Scholar] [CrossRef]
- Harada, T.; Teraoka, Y.; Kagawa, S. Perovskite-type oxides as catalysts for selective reduction of nitric oxide by ethylene. Appl. Surf. Sci. 1997, 121–122, 505–508. [Google Scholar] [CrossRef]
- Yu Yao, Y.-F. The oxidation of hydrocarbons and CO over metal oxides: IV. Perovskite-type oxides. J. Catal. 1975, 36, 266–275. [Google Scholar] [CrossRef]
- Jovanovic, D.; Dondur, V.; Terlecki-Baricevic, A.; Grbic, B. Three-way activity and sulfur tolerance of single phase perovskites. Stud. Surf. Sci. Catal. 1991, 71, 371–379. [Google Scholar] [CrossRef]
- Gallagher, P.K.; Johnson, D.W., Jr.; Vogel, E.M.; Schrey, F. Effect of the Pt content of La0.7Pb0.3MnO3 on its catalytic activity for the oxidation of CO in the presence of SO2. Mat. Res. Bull. 1975, 10, 623–628. [Google Scholar] [CrossRef]
- Tzimpilis, E.; Moschoudis, N.; Stoukides, M.; Bekiaroglou, P. Ageing and SO2 resistance of Pd containing perovskite-type oxides. Appl. Catal. B 2009, 87, 9–17. [Google Scholar] [CrossRef]
- Tzimpilis, E.; Moschoudis, N.; Stoukides, M.; Bekiaroglou, P. Preparation, active phase composition and Pd content of perovskite-type oxides. Appl. Catal. B 2008, 84, 607–615. [Google Scholar] [CrossRef]
- Mondragón Rodríguez, G.C.; Kelm, K.; Heikens, S.; Grünert, W.; Saruhan, B. Pd-integrated perovskites for TWC applications: Synthesis, microstructure and N2O-selectivity. Catal. Today 2012, 184, 184–191. [Google Scholar] [CrossRef]
- Sorenson, S.C.; Wronkiewicz, J.A.; Sis, L.B.; Wirtz, G.P. Properties of LaCoO3 as a Catalyst in Engine Exhaust Gases. Am. Ceram. Soc. Bull. 1974, 53, 446–449. [Google Scholar]
- Labhsetwar, N.K.; Watanabe, A.; Biniwale, R.B.; Kumar, R.; Mitsuhashi, T. Alumina supported, perovskite oxide based catalytic materials and their auto-exhaust application. Appl. Catal. B 2001, 33, 165–173. [Google Scholar] [CrossRef]
- Tanaka, H.; Mizuno, N.; Misono, M. Catalytic activity and structural stability of La0.9Ce0.1Co1−xFexO3 perovskite catalysts for automotive emission control. Appl. Catal. A 2003, 244, 371–382. [Google Scholar] [CrossRef]
- Bradow, R.; Jovanović, D.; Petrović, S.; Jovanović, Ž.; Terlecki-Baričević, A. Ruthenium Perovskite Catalysts for Lean NOx Automotive Emission Control. Ind. Eng. Chem. Res. 1995, 34, 1929–1932. [Google Scholar] [CrossRef]
- Shelef, M.; Gandhi, H.S. The Reduction of Nitric Oxide in Automobile Emissions—Stabilisation of catalysts containing ruthenium. Platin. Met. Rev. 1974, 18, 2–14. [Google Scholar]
- Nishihata, Y.; Mizuki, J.; Akao, T.; Tanaka, H.; Uenishi, M.; Kimura, M.; Okamoto, T.; Hamada, N. Self-regeneration of a Pd-perovskite catalyst for automotive emissions control. Nature 2002, 418, 164–167. [Google Scholar] [CrossRef]
- Tanaka, H.; Tan, I.; Uenishi, M.; Kimura, M.; Dohmae, K. Regeneration of palladium subsequent to solid solution and segregation in a perovskite catalyst: An intelligent catalyst. Top. Catal. 2001, 16/17, 63–70. [Google Scholar] [CrossRef]
- Uenishi, M.; Taniguchi, M.; Tanaka, H.; Kimura, M.; Nishihata, Y.; Mizuki, J.; Kobayashi, T. Redox behavior of palladium at start-up in the Perovskite-type LaFePdOx automotive catalysts showing a self-regenerative function. Appl. Catal. B 2005, 57, 267–273. [Google Scholar] [CrossRef]
- Uenishi, M.; Tanaka, H.; Taniguchi, M.; Tan, I.; Sakamoto, Y.; Matsunaga, S.; Yokota, K.; Kobayashi, T. The reducing capability of palladium segregated from perovskite-type LaFePdOx automotive catalysts. Appl. Catal. A 2005, 296, 114–119. [Google Scholar] [CrossRef]
- Tanaka, H.; Taniguchi, M.; Kajita, N.; Uenishi, M.; Tan, I.; Sato, N.; Narita, K.; Kimura, M. Design of the intelligent catalyst for Japan ULEV standard. Top. Catal. 2004, 30/31, 389–396. [Google Scholar] [CrossRef]
- Tanaka, H.; Taniguchi, M.; Uenishi, M.; Kajita, N.; Tan, I.; Nishihata, Y.; Mizuki, J.; Narita, K.; Kimura, M.; Kaneko, K. Self-Regenerating Rh- and Pt-Based Perovskite Catalysts for Automotive-Emissions Control. Angew. Chem. Int. Ed. 2006, 45, 5998–6002. [Google Scholar] [CrossRef]
- Dacquin, J.P.; Cabié, M.; Henry, C.R.; Lancelot, C.; Dujardin, C.; Raouf, S.R.; Granger, P. Structural changes of nano-Pt particles during thermal ageing: Support-induced effect and related impact on the catalytic performances. J. Catal. 2010, 270, 299–309. [Google Scholar] [CrossRef]
- Dacquin, J.P.; Lancelot, C.; Dujardin, C.; Cordier-Robert, C.; Granger, P. Support-Induced Effects of LaFeO3 Perovskite on the Catalytic Performances of Supported Pt Catalysts in DeNOx Applications. J. Phys. Chem. C 2011, 115, 1911–1921. [Google Scholar]
- Katz, M.B.; Graham, G.W.; Duan, Y.; Liu, H.; Adamo, C.; Schlom, D.G.; Pan, X. Self-Regeneration of Pd-LaFeO3 Catalysts: New Insight from Atomic-Resolution Electron Microscopy. J. Am. Chem. Soc. 2011, 133, 18090–18093. [Google Scholar]
- Katz, M.B.; Zhang, S.; Duan, Y.; Wang, H.; Fang, M.; Zhang, K.; Li, B.; Graham, G.W.; Pan, X. Reversible precipitation/dissolution of precious-metal clusters in perovskite-based catalyst materials: Bulk versus surface re-dispersion. J. Catal. 2012, 293, 145–148. [Google Scholar] [CrossRef]
- Zhou, K.; Chen, H.; Tian, Q.; Hao, Z.; Shen, D.; Xu, X. Pd-containing perovskite-type oxides used for three-way catalysts. J. Mol. Catal. A 2002, 189, 225–232. [Google Scholar] [CrossRef]
- Lu, Y.; Michalow, K.A.; Matam, S.K.; Winkler, A.; Maegli, A.E.; Yoon, S.; Heel, A.; Weidenkaff, A.; Ferri, D. Methane abatement under stoichiometric conditions on perovskite-supported palladium catalysts prepared by flame spray synthesis. Appl. Catal. B 2014, 144, 631–643. [Google Scholar] [CrossRef]
- Nijhuis, T.A.; Beers, A.E.W.; Vergunst, T.; Hoek, I.; Kapteijn, F.; Moulijn, J.A. Preparation of monolithic catalysts. Catal. Rev. Sci. Eng. 2001, 43, 345–380. [Google Scholar] [CrossRef]
- Tomašić, V.; Jović, F. State-of-the-art in the monolithic catalysts/reactors. Appl. Catal. A 2006, 311, 112–121. [Google Scholar] [CrossRef]
- Meille, V. Review on methods to deposit catalysts on structured surfaces. Appl. Catal. A 2006, 315, 1–17. [Google Scholar] [CrossRef]
- Avila, P.; Montes, M.; Miró, E.E. Monolithic reactors for environmental applications: A review on preparation technologies. Chem. Eng. J. 2005, 109, 11–36. [Google Scholar] [CrossRef]
- Lachman, I.M.; Williams, J.L. Extruded Monolithic Catalysts Supports. Catal. Today 1992, 14, 317–329. [Google Scholar] [CrossRef]
- Bardhan, P. Ceramic honeycomb filters and catalysts. Curr. Opin. Solid State Mater. Sci. 1997, 2, 577–583. [Google Scholar] [CrossRef]
- Isupova, L.A.; Sadykov, V.A.; Tikhov, S.F.; Kimkhai, O.N.; Kovalenko, O.N.; Kustova, G.N.; Ovsyannikova, I.A.; Dovbii, Z.A.; Kryukova, G.N.; Rozovskii, A.Y.; et al. Monolith perovskite catalysts for environmentally benign fuels combustion and toxic wastes incineration. Catal. Today 1996, 27, 249–256. [Google Scholar] [CrossRef]
- Isupova, L.A.; Sadykov, V.A.; Solovyova, L.P.; Andrianova, M.P.; Ivanov, V.P.; Kryukova, G.N.; Kolomiichuk, V.N.; Avvakumov, E.G.; Pauli, I.A.; Andryushkova, O.V.; et al. Monolith perovskite catalysts of honeycomb structure for fuel combustion. Stud. Surf. Sci. Catal. 1995, 91, 637–645. [Google Scholar] [CrossRef]
- Sadykov, V.A.; Isupova, L.A.; Tikhov, S.F.; Kimkhai, O.N. Perovskite catalysts: High surface area powders synthesis, monolith shaping and high-temperature applications. Mat. Res. Soc. Symp. Proc. 1995, 368, 293–298. [Google Scholar]
- Isupova, L.A.; Alikina, G.M.; Snegurenko, O.I.; Sadykov, V.A.; Tsybulya, S.V. Monolith honeycomb mixed oxide catalysts for methane oxidation. Appl. Catal. B 1999, 21, 171–181. [Google Scholar] [CrossRef]
- Isupova, L.A.; Sadykov, V.A.; Alikina, G.M.; Snegurenko, O.I.; Tsybulya, S.V.; Salanov, A.N.; Rogov, V.A. Monolith honeycomb mixed oxide catalysts for methane oxidation. Stud. Surf. Sci. Catal. 2000, 130, 3783–3788. [Google Scholar] [CrossRef]
- Ciambelli, P.; Palma, V.; Tikhov, S.F.; Sadykov, V.A.; Isupova, L.A.; Lisi, L. Catalytic activity of powder and monolith perovskites in methane combustion. Catal. Today 1999, 47, 199–207. [Google Scholar] [CrossRef]
- Schneider, R.; Kiessling, D.; Wendt, G.; Burckhardt, W.; Winterstein, G. Perovskite-type oxide monolithic catalysts for combustion of chlorinated hydrocarbons. Catal. Today 1999, 47, 429–435. [Google Scholar]
- Khan, H.R.; Frey, H. R.f. plasma spray deposition of LaMOx (M = Co, Mn, Ni) films and the investigations of structure, morphology and the catalytic oxidation of CO and C3H8. J. Alloys Compd. 1993, 190, 209–217. [Google Scholar] [CrossRef]
- Bahlawane, N.; Ngamou, P.H.T.; Kohse-Höinghaus, K. Structure, Electrical Properties, and Surface Reactivity of CVD-Made Functional Complex Oxides. J. Electrochem. Soc. 2010, 157, D16–D20. [Google Scholar] [CrossRef]
- Sansernnivet, M.; Laosiripojana, N.; Assabumrungrat, S.; Charojrochkul, S. Fabrication of La0.8Sr0.2CrO3-based Perovskite Film via Flame-Assisted Vapor Deposition for H2 Production by Reforming. Chem. Vap. Depos. 2010, 16, 311–321. [Google Scholar] [CrossRef]
- Jarligo, M.O.; Mauer, G.; Bram, M.; Baumann, S.; Vaßen, R. Plasma Spray Physical Vapor Deposition of La1−xSrxCoyFe1−yO3−δ Thin-Film Oxygen Transport Membrane on Porous Metallic Supports. J. Therm. Spray Technol. 2014, 23, 213–219. [Google Scholar] [CrossRef]
- Uusi-Esko, K.; Karppinen, M. Extensive Series of Hexagonal and Orthorhombic RMnO3 (R = Y, La, Sm, Tb, Yb, Lu) Thin Films by Atomic Layer Deposition. Chem. Mater. 2011, 23, 1835–1840. [Google Scholar] [CrossRef]
- Schneider, R.; Kiessling, D.; Wendt, G. Cordierite monolith supported perovskite-type oxides catalysts for the total oxidation of chlorinated hydrocarbons. Appl. Catal. B 2000, 28, 187–195. [Google Scholar] [CrossRef]
- Isupova, L.A.; Alikina, G.M.; Tsybulya, S.V.; Salanov, A.N.; Blodyreva, N.N.; Rusina, E.S.; Ovsyannikova, I.A.; Rogov, V.A.; Bunina, R.V.; Sadykov, V.A. Honeycomb-supported perovskite catalysts for high-temperature processes. Catal. Today 2002, 75, 305–315. [Google Scholar] [CrossRef]
- Li, L.; Shen, X.; Wang, P.; Meng, X.; Song, F. Soot capture and combustion for perovskite La-Mn-O based catalysts coated on honeycomb ceramic in practical diesel exhaust. Appl. Surf. Sci. 2011, 257, 9519–9524. [Google Scholar]
- Isupova, L.A.; Sutormina, E.F.; Kulilovskaya, N.A.; Plyasova, L.M.; Rudina, N.A.; Ovsyannikova, I.A.; Zootarskii, I.A.; Sadykov, V.A. Honeycomb supported perovskite catalysts for ammonia oxidation processes. Catal. Today 2005, 105, 429–435. [Google Scholar] [CrossRef]
- Sannino, D.; Vaiano, V.; Ciambelli, P.; Isupova, L.A. Structured catalysts for photo-Fenton oxidation of acetic acid. Catal. Today 2011, 161, 255–259. [Google Scholar] [CrossRef]
- Pechini, M.P. Method of preparing lead and alkaline earth titanates and niobates and coating method using the same to form a capacitor. U.S. Patent 3,330,697, 11 July 1967. [Google Scholar]
- Qi, A.; Wang, S.; Fu, G.; Ni, C.; Wu, D. La-Ce-Ni-O monolithic perovskite catalysts potential for gasoline autothermal reforming system. Appl. Catal. A 2005, 281, 233–246. [Google Scholar] [CrossRef]
- Vergunst, T.; Kapteijn, F.; Moulijn, J.A. Monolithic catalysts—Non-uniform active phase distribution by impregnation. Appl. Catal. A 2001, 213, 179–187. [Google Scholar] [CrossRef]
- Cauda, E.; Fino, D.; Saracco, G.; Specchia, V. Nanosized Pt-perovskite catalyst for the regeneration of a wall-flow filter for soot removal from diesel exhaust gases. Top. Catal. 2004, 30/31, 299–303. [Google Scholar] [CrossRef]
- Russo, N.; Furfori, S.; Fino, D.; Saracco, G.; Specchia, V. Lanthanum cobaltite catalysts for diesel soot combustion. Appl. Catal. B 2008, 83, 85–95. [Google Scholar] [CrossRef]
- Russo, N.; Fino, D.; Saracco, G.; Specchia, V. Promotion effect of Au on perovskite catalysts for the regeneration of diesel particulate filters. Catal. Today 2008, 137, 306–311. [Google Scholar] [CrossRef]
- Mescia, D.; Caroca, J.C.; Russo, N.; Labhsetwar, N.; Fino, D.; Saracco, G.; Specchia, V. Towards a single brick solution for the abatement of NOx and soot from diesel engine exhausts. Catal. Today 2008, 137, 300–305. [Google Scholar] [CrossRef]
- Fino, D.; Russo, N.; Cauda, E.; Saracco, G.; Specchia, V. La-Li-Cr perovskite catalysts for diesel particulate combustion. Catal. Today 2006, 114, 31–39. [Google Scholar] [CrossRef]
- Furfori, S.; Russo, N.; Fino, D.; Saracco, G.; Specchia, V. NO SCR reduction by hydrogen generated in line on perovskite-type catalysts for automotive diesel exhaust gas treatment. Chem. Eng. Sci. 2010, 65, 120–127. [Google Scholar] [CrossRef]
- Cauda, E.; Fino, D.; Saracco, G.; Specchia, V. Preparation and regeneration of a catalytic diesel particulate filter. Chem. Eng. Sci. 2007, 62, 5182–5185. [Google Scholar]
- Mescia, D.; Cauda, E.; Russo, N.; Fino, D.; Saracco, G.; Specchia, V. Towards practical application of lanthanum chromite catalysts for diesel particulate combustion. Catal. Today 2006, 117, 369–375. [Google Scholar] [CrossRef]
- Fino, D.; Fino, P.; Saracco, G.; Specchia, V. Innovative means for the catalytic regeneration of particulate traps for diesel exhaust cleaning. Chem. Eng. Sci. 2003, 58, 951–958. [Google Scholar] [CrossRef]
- Arendt, E.; Maione, A.; Klisinska, A.; Sanz, O.; Montes, M.; Suarez, S.; Blanco, J.; Ruiz, P. Structuration of LaMnO3 perovskite catalysts on ceramic and metallic monoliths: Physico-chemical characterisation and catalytic activity in methane combustion. Appl. Catal. A 2008, 339, 1–14. [Google Scholar] [CrossRef]
- Fabbrini, L.; Rossetti, I.; Forni, L. Effect of primer on honeycomb-supported La0.9Ce0.1CoO3±δ perovskite for methane catalytic flameless combustion. Appl. Catal. B 2003, 44, 107–116. [Google Scholar] [CrossRef]
- Podyacheva, O.Y.; Ketov, A.A.; Ismagilov, Z.R.; Ushakov, V.A.; Bos, A.; Veringa, H.J. Metal foam supported perovskite catalysts. React. Kinet. Catal. Lett. 1997, 60, 243–250. [Google Scholar] [CrossRef]
- Forni, L.; Rossetti, I. Catalytic combustion of hydrocarbons over perovskites. Appl. Catal. B 2002, 38, 29–37. [Google Scholar] [CrossRef]
- Fabbrini, L.; Rossetti, I.; Forni, L. La2O3 as primer for supporting La0.9Ce0.1CoO3±δ on cordieritic honeycombs. Appl. Catal. B 2005, 56, 221–227. [Google Scholar] [CrossRef]
- Fabbrini, L.; Rossetti, I.; Forni, L. Effect of honeycomb supporting on activity of LaBO3±δ perovskite-like catalysts for methane flameless combustion. Appl. Catal. B 2006, 63, 131–136. [Google Scholar] [CrossRef]
- Arai, H.; Machida, M. Thermal stabilization of catalyst supports and their application to high-temperature catalytic combustion. Appl. Catal. A 1996, 138, 161–176. [Google Scholar] [CrossRef]
- Zhang-Steenwinkel, Y.; van der Zande, L.M.; Castricum, H.L.; Bliek, A.; van den Brink, R.W.; Elzinga, G.D. Microwave-assisted in-situ regeneration of a perovskite coated diesel soot filter. Chem. Eng. Sci. 2005, 60, 797–804. [Google Scholar] [CrossRef]
- Kucharczyk, B.; Tylus, W. Effect of Pd or Ag additive on the activity and stability of monolithic LaCoO3 perovskites for catalytic combustion of methane. Catal. Today 2004, 90, 121–126. [Google Scholar] [CrossRef]
- Kucharczyk, B.; Tylus, W. Partial substitution of lanthanum with silver in the LaMnO3 perovskite: Effect of the modification on the activity of monolithic catalysts in the reactions of methane and carbon oxide oxidation. Appl. Catal. A 2008, 335, 28–36. [Google Scholar] [CrossRef]
- Cimino, S.; Pirone, R.; Russo, G. Thermal Stability of Perovskite-Based Monolithic Reactors in the Catalytic Combustion of Methane. Ind. Eng. Chem. Res. 2001, 40, 80–85. [Google Scholar] [CrossRef]
- Cimino, S.; Pirone, R.; Lisi, L. Zirconia supported LaMnO3 monoliths for the catalytic combustion of methane. Appl. Catal. B 2002, 35, 243–254. [Google Scholar] [CrossRef]
- Cimino, S.; Lisi, L.; Pirone, R.; Russo, G.; Turco, M. Methane combustion on perovskites-based structured catalysts. Catal. Today 2000, 59, 19–31. [Google Scholar] [CrossRef]
- Jiratova, K.; Moravkova, L.; Malecha, J.; Koutsky, B. Ceramic foam-supported perovskites as catalysts for combustion of methane. Collect. Czechoslov. Chem. Commun. 1997, 62, 875–883. [Google Scholar] [CrossRef]
- Zwinkels, M.F.M.; Haussner, O.; Govind Menon, P.; Järås, S.G. Preparation and characterization of LaCrO3 and Cr2O3 methane combustion catalysts supported on LaAl11O18- and Al2O3-coated monoliths. Catal. Today 1999, 47, 73–82. [Google Scholar] [CrossRef]
- Cimino, S.; Di Benedetto, A.; Pirone, R.; Russo, G. Transient behaviour of perovskite-based monolithic reactors in the catalytic combustion of methane. Catal. Today 2001, 69, 95–103. [Google Scholar] [CrossRef]
- Muroi, T.; Nojiri, N.; Deguchi, T. Recent progress in catalytic technology in Japan—II (1994–2009). Appl. Catal. A 2010, 389, 27–45. [Google Scholar] [CrossRef]
- Dimopoulos Eggenschwiler, P.; Tsinoglou, D.N.; Seyfert, J.; Bach, C.; Vogt, U.; Gorbar, M. Ceramic foam substrates for automotive catalyst applications: Fluid mechanic analysis. Exp. Fluids 2009, 47, 209–222. [Google Scholar] [CrossRef]
- Patcas, F.C.; Garrido, G.I.; Kraushaar-Czanetzki, B. CO oxidation over structured carriers: A comparison of ceramic foams, honeycombs and beads. Chem. Eng. Sci. 2007, 62, 3984–3990. [Google Scholar] [CrossRef]
- Populoh, S.; Trottmann, M.; Brunko, O.C.; Thiel, P.; Weidenkaff, A. Construction of a high temperature TEG measurement system for the evaluation of thermoelectric oxide modules. Funct. Mater. Lett. 2013, 6. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Keav, S.; Matam, S.K.; Ferri, D.; Weidenkaff, A. Structured Perovskite-Based Catalysts and Their Application as Three-Way Catalytic Converters—A Review. Catalysts 2014, 4, 226-255. https://doi.org/10.3390/catal4030226
Keav S, Matam SK, Ferri D, Weidenkaff A. Structured Perovskite-Based Catalysts and Their Application as Three-Way Catalytic Converters—A Review. Catalysts. 2014; 4(3):226-255. https://doi.org/10.3390/catal4030226
Chicago/Turabian StyleKeav, Sylvain, Santhosh Kumar Matam, Davide Ferri, and Anke Weidenkaff. 2014. "Structured Perovskite-Based Catalysts and Their Application as Three-Way Catalytic Converters—A Review" Catalysts 4, no. 3: 226-255. https://doi.org/10.3390/catal4030226
APA StyleKeav, S., Matam, S. K., Ferri, D., & Weidenkaff, A. (2014). Structured Perovskite-Based Catalysts and Their Application as Three-Way Catalytic Converters—A Review. Catalysts, 4(3), 226-255. https://doi.org/10.3390/catal4030226