In Situ XANES/XPS Investigation of Doped Manganese Perovskite Catalysts

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Doping Dependence of Virgin Samples
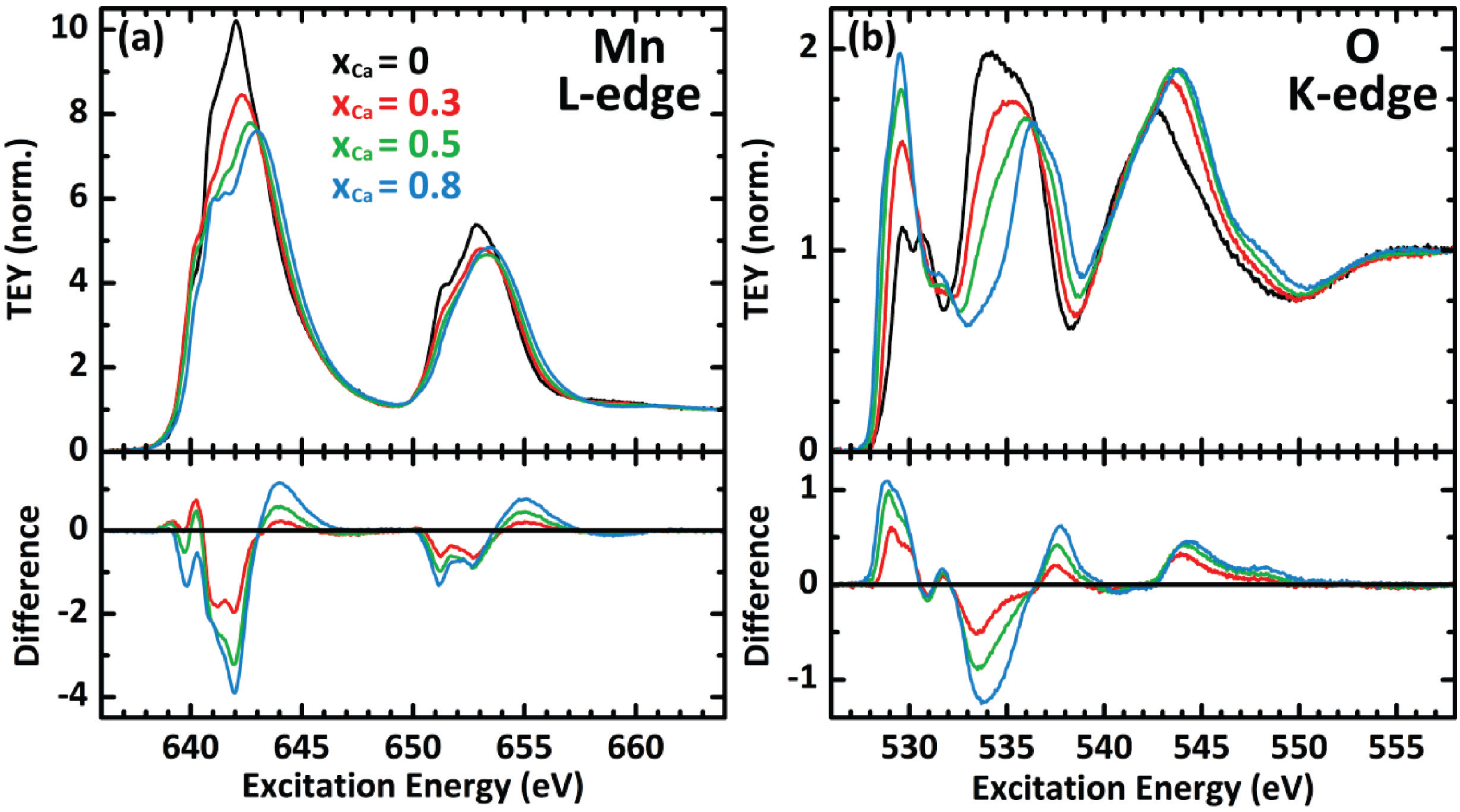

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| xCa | formal bulk valence | ΔEL (eV) | I3/I2 (BG1) | I3/I2 (BG2) | ΔEK (eV) |
|---|---|---|---|---|---|
| 0 | 3.0+ | 10.8 | 2.89 | 2.54 | 3.8 |
| 0.3 | 3.3+ | 10.8 | 2.88 | 2.53 | 5.8 |
| 0.5 | 3.5+ | 10.6 | 2.85 | 2.46 | 6.3 |
| 0.8 | 3.8+ | 10.4 | 2.75 | 2.32 | 6.8 |
2.2. In Situ Investigation
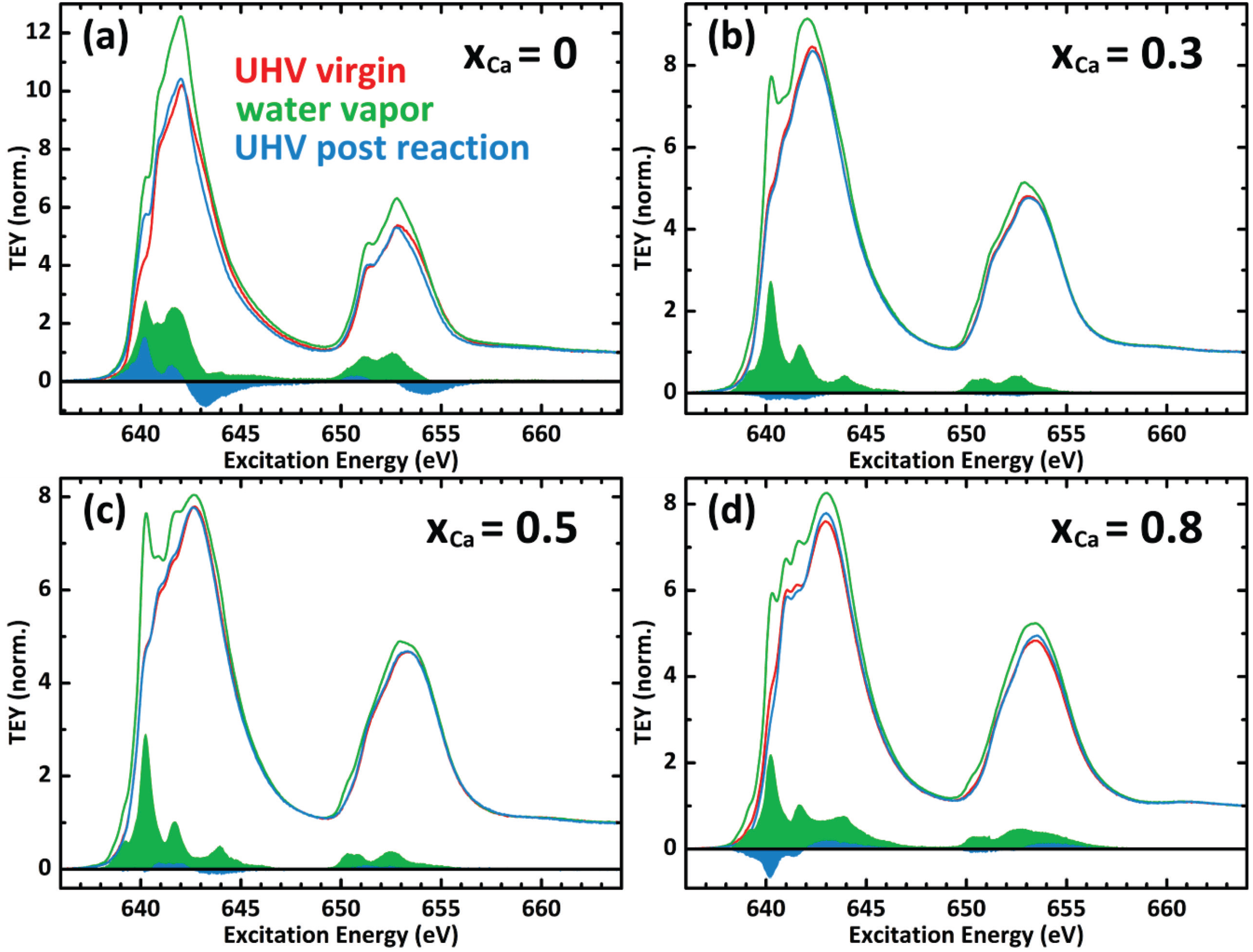
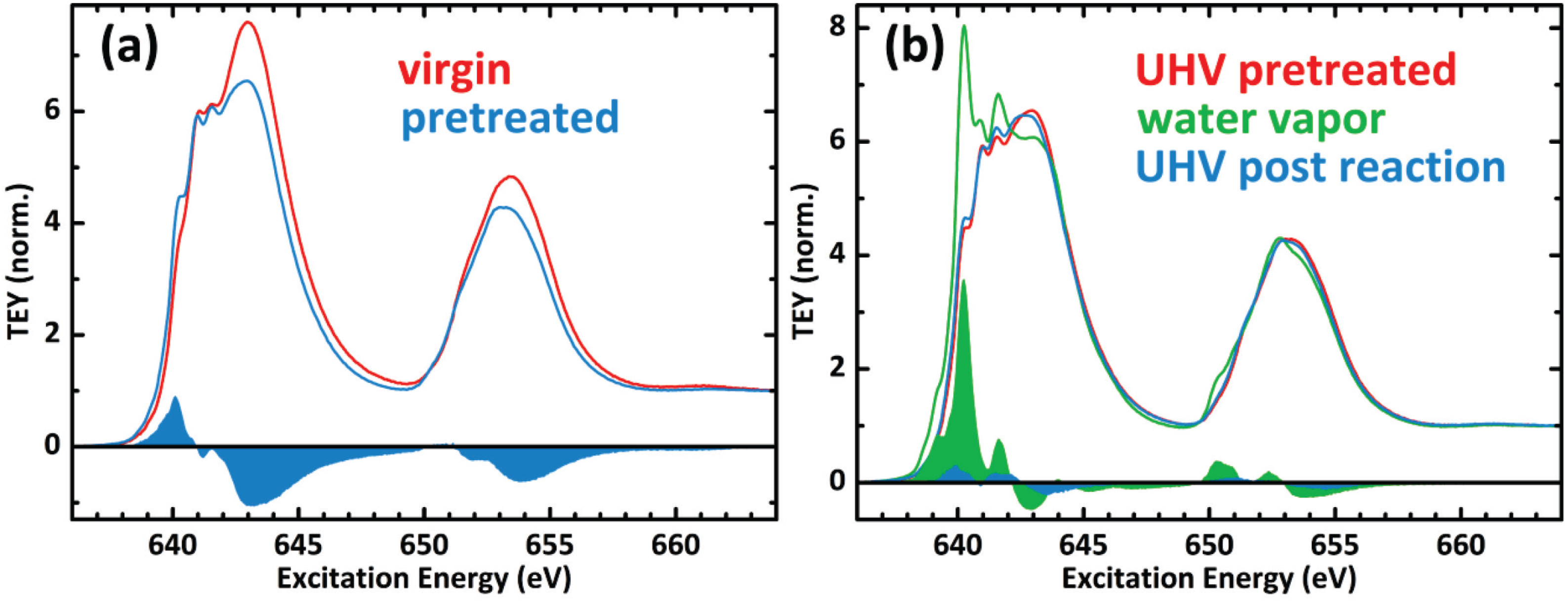
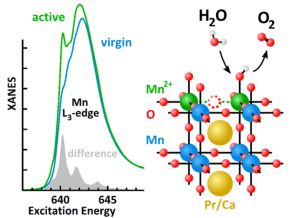
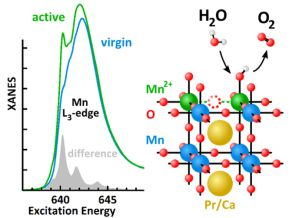
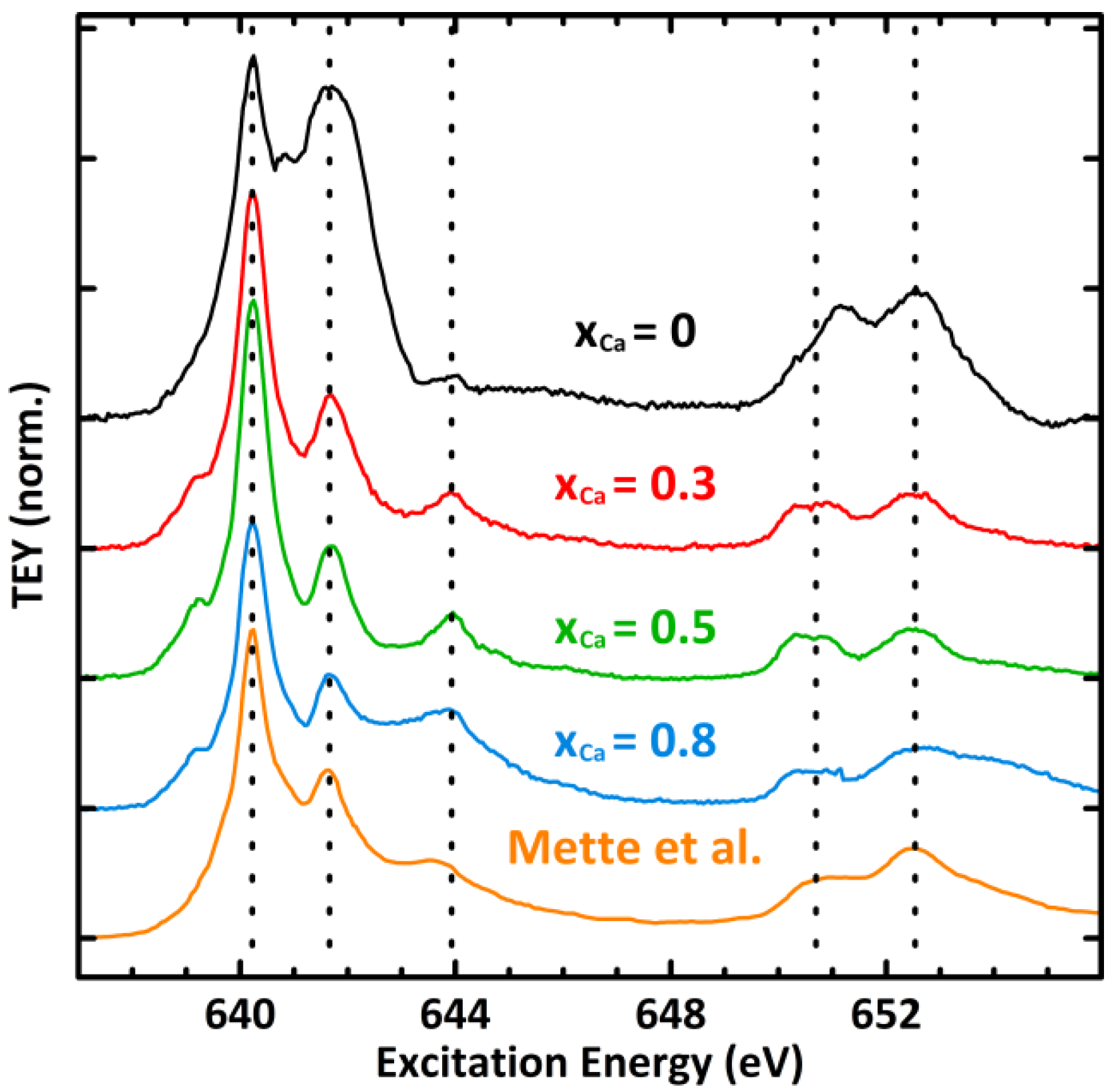
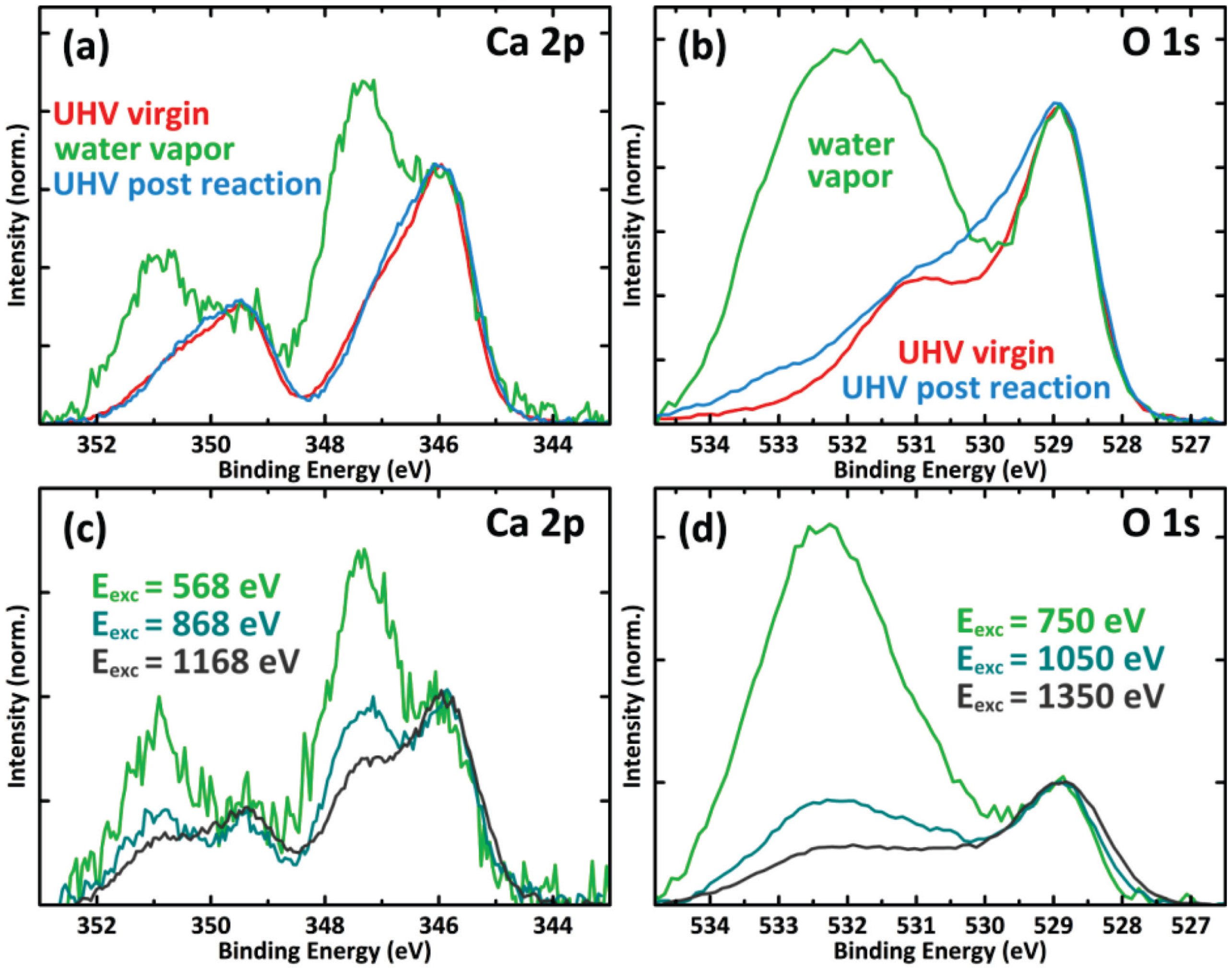
2.3. Mn Surface Species
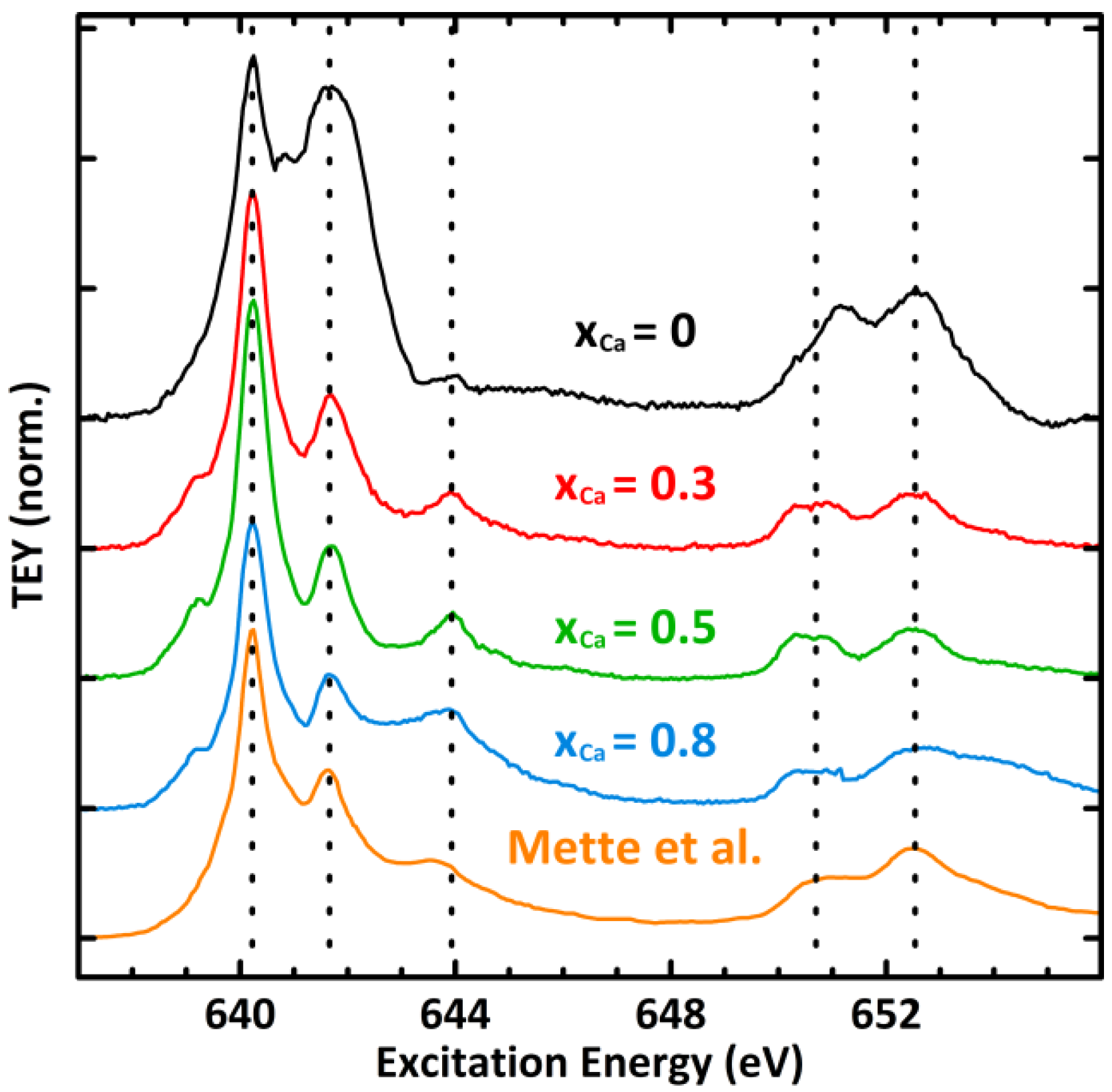
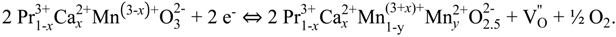
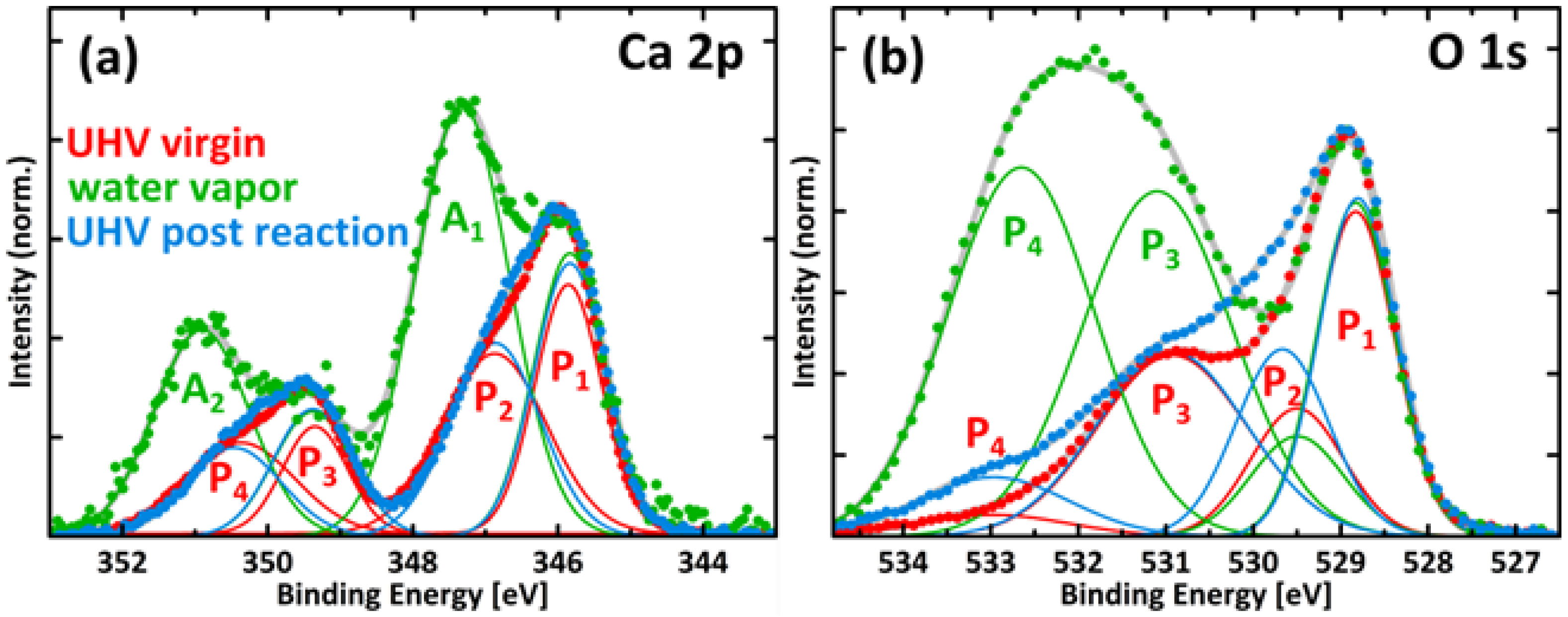
2.4. XPS

3. Experimental Section
3.1. Sample Preparation and Characterization
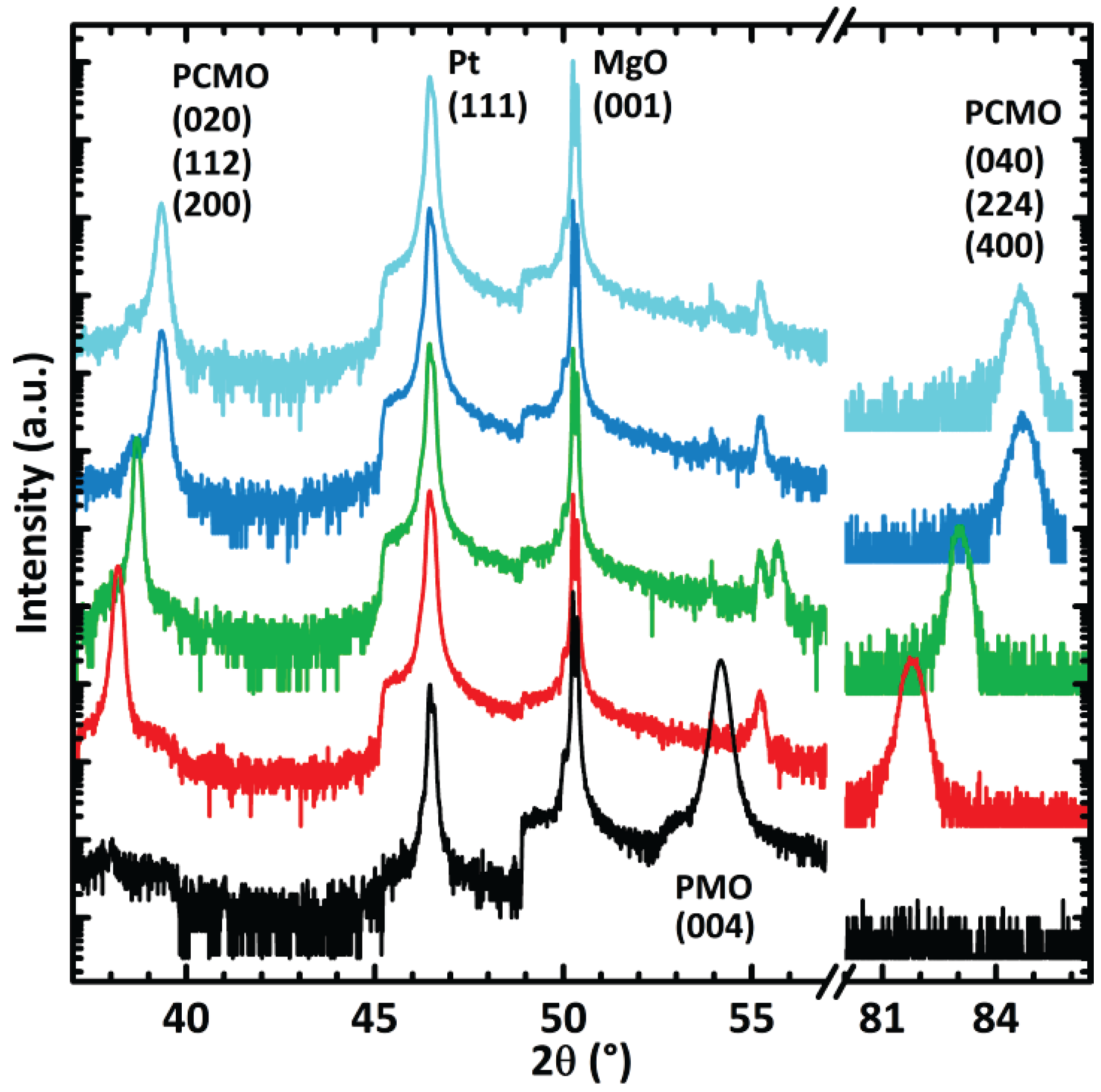

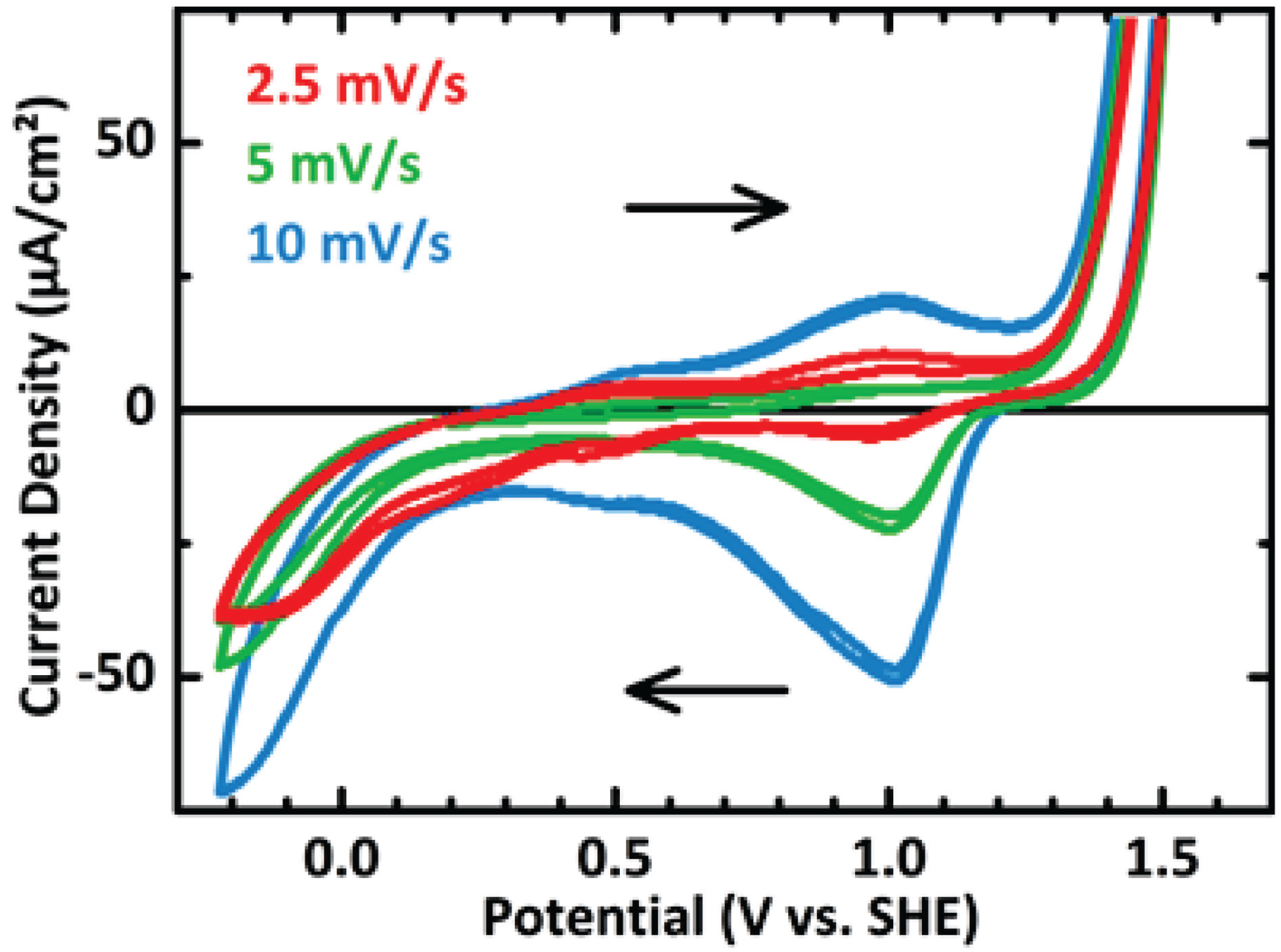
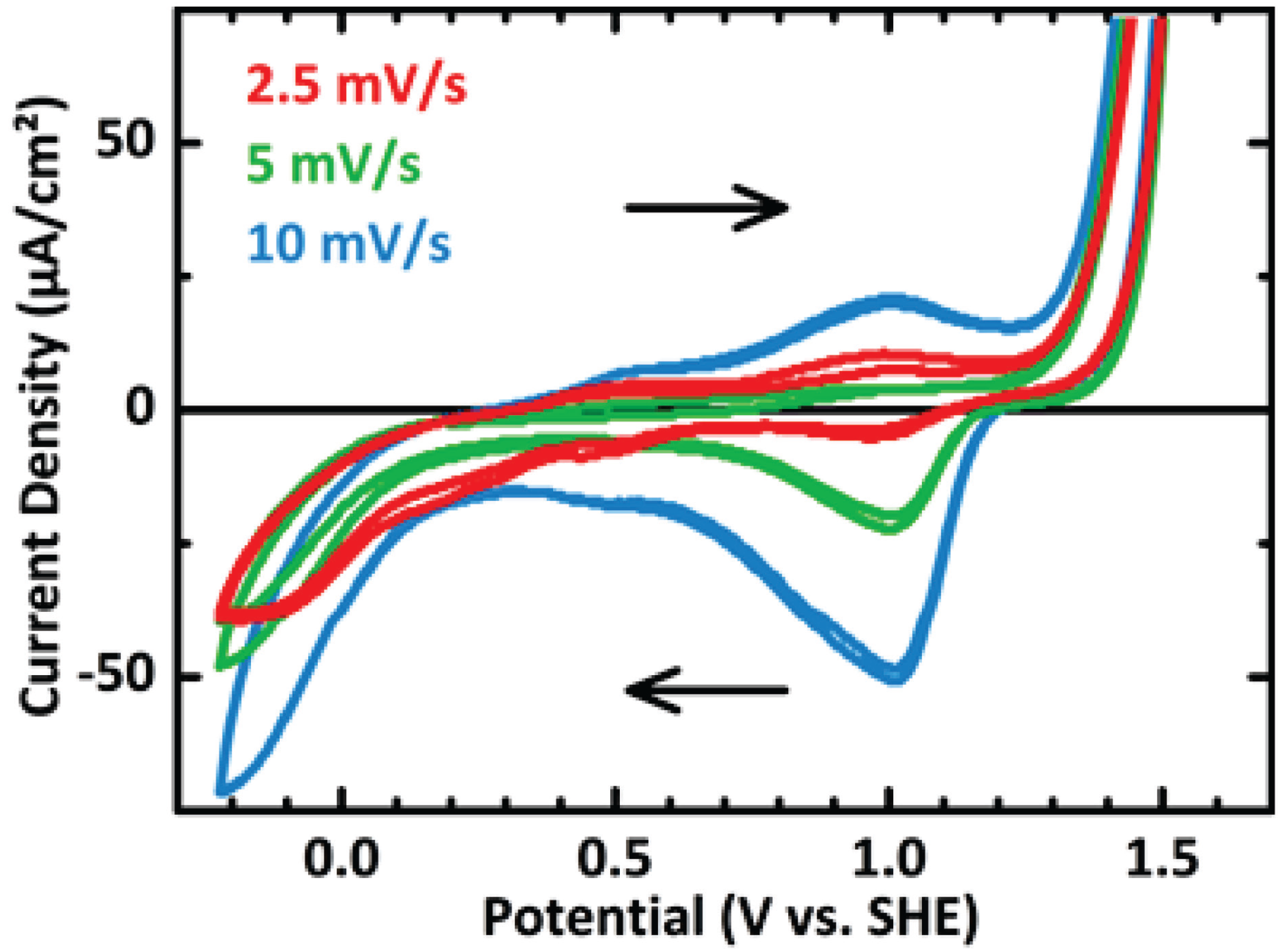
3.2. In Situ XANES/XPS Measurements
4. Conclusions
Acknowledgments
Conflicts of Interest
Appendix
A1. Linear Superposition of Experimental Mn L-Edges
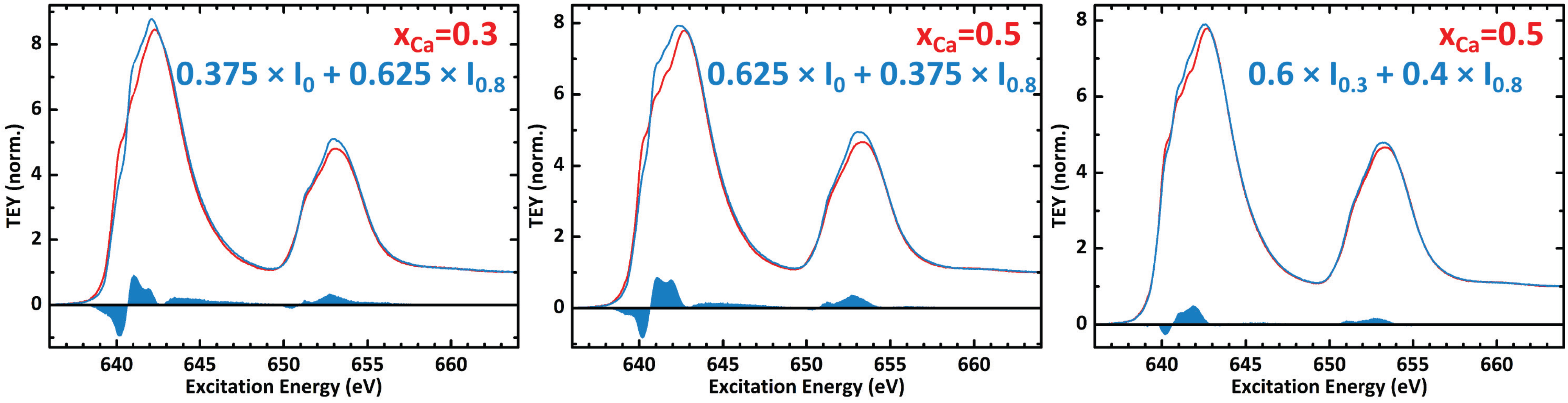
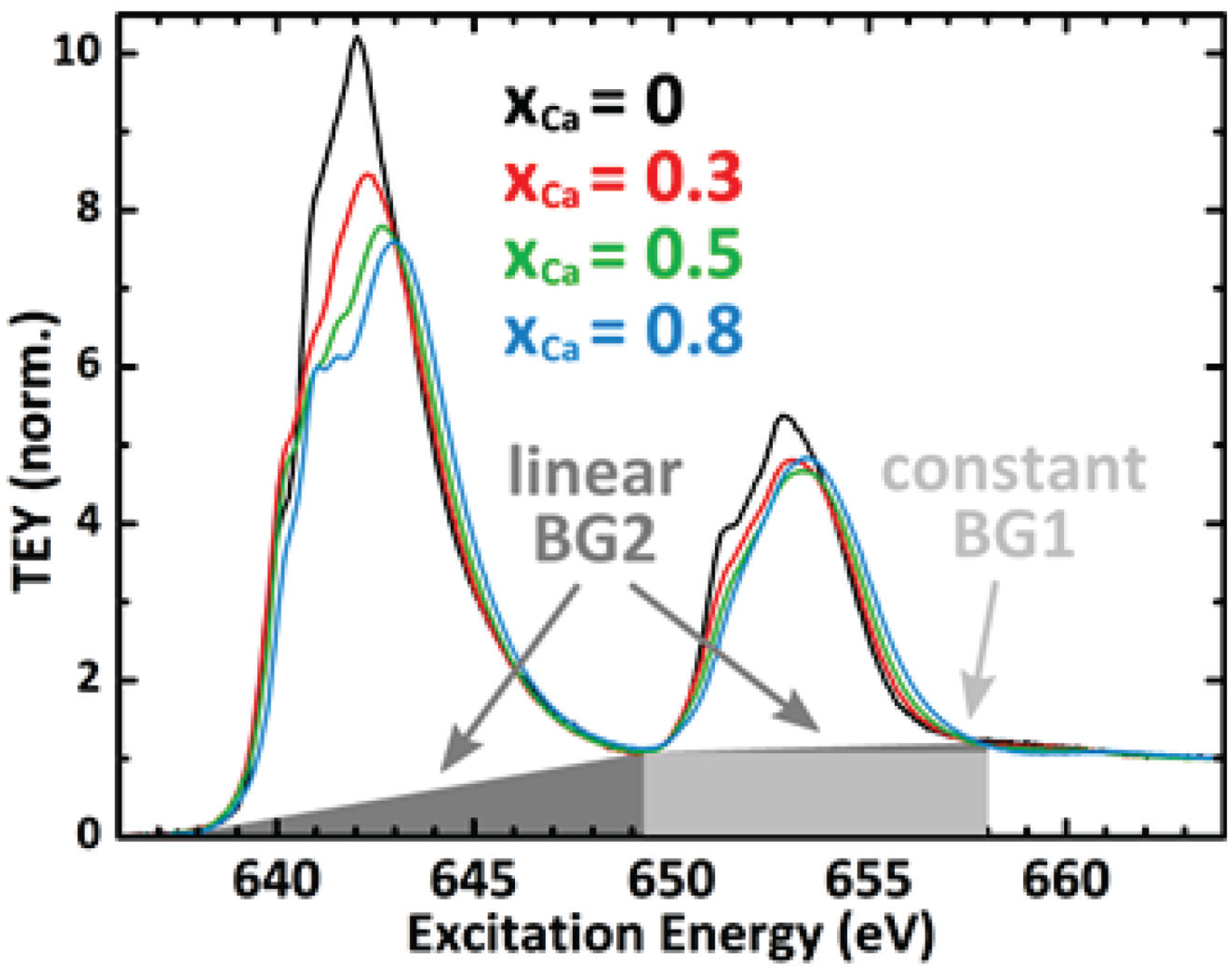
A2. Intensity Ratio of the Mn L3,2-Edges
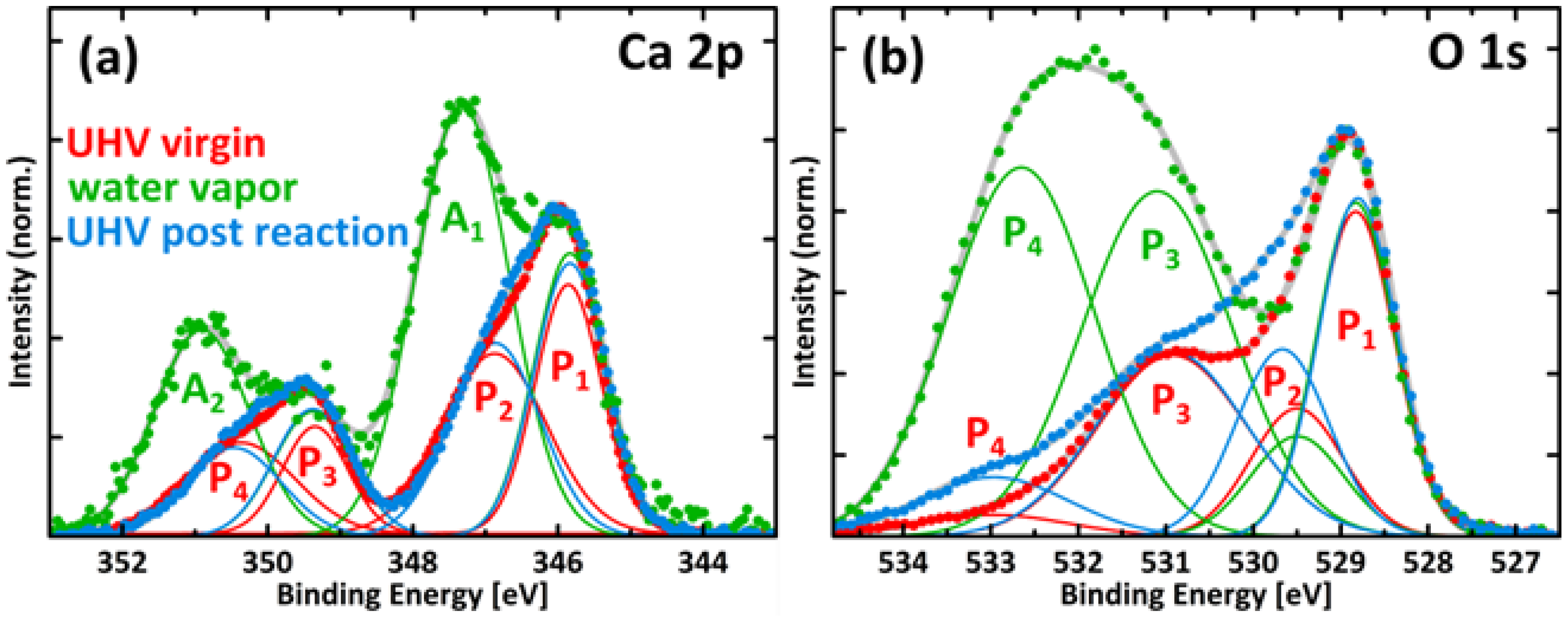
A3. XPS
References
- Armstrong, F.A. Why did nature choose manganese to make oxygen? Philos. T. Roy. Soc. B 2008, 363, 1263–1270. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Martínez, J.I.; García-Lastra, J.M.; Mogensen, M.; Rossmeisl, J. Trends in Stability of Perovskite Oxides. Angew. Chem. Int. Ed. 2010, 49, 7699–7701. [Google Scholar]
- Man, I.C.; Su, H.-Y.; Calle-Vallejo, F.; Hansen, H.A.; Martínez, J.I.; Inoglu, N.G.; Kitchin, J.; Jaramillo, T.F.; Nørskov, J.K.; Rossmeisl, J. Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces. ChemCatChem 2011, 3, 1159–1165. [Google Scholar] [CrossRef]
- Suntivich, J.; May, K.J.; Gasteiger, H.A.; Goodenough, J.B.; Shao-Horn, Y. A Perovskite Oxide Optimized for Oxygen Evolution Catalysis from Molecular Orbital Principles. Science 2011, 334, 1383–1385. [Google Scholar] [CrossRef]
- Raabe, S.; Mierwaldt, D.; Ciston, J.; Uijttewaal, M.; Stein, H.; Hoffmann, J.; Zhu, Y.; Blöchl, P.; Jooss, C. In Situ Electrochemical Electron Microscopy Study of Oxygen Evolution Activity of Doped Manganite Perovskites. Adv. Funct. Mater. 2012, 22, 3378–3388. [Google Scholar]
- Raveau, B. The crucial role of mixed valence in the magnetoresistance properties of manganites and cobaltites. Philos. T. Roy. Soc. A 2008, 366, 83–92. [Google Scholar] [CrossRef]
- Grenier, S.; Hill, J.P.; Gibbs, D.; Thomas, K.J.; von Zimmermann, M.; Nelson, C.S.; Kiryukhin, V.; Tokura, Y.; Tomioka, Y.; Casa, D.; et al. Resonant X-ray diffraction of the magnetoresistant perovskite Pr0.6Ca0.4MnO3. Phys. Rev. B 2004, 69, 134419. [Google Scholar] [CrossRef]
- De Groot, F.M.F.; Fuggle, J.C.; Thole, B.T.; Sawatzky, G.A. 2p X-ray absorption of 3d transition-metal compounds: An atomic multiplet description including the crystal field. Phys. Rev. B 1990, 42, 5459–5468. [Google Scholar]
- Grush, M.M.; Chen, J.; Stemmler, T.L.; George, S.J.; Ralston, C.Y.; Stibrany, R.T.; Gelasco, A.; Christou, G.; Gorun, S.M.; Penner-Hahn, J.E.; et al. Manganese L-Edge X-ray Absorption Spectroscopy of Manganese Catalase from Lactobacillus plantarum and Mixed Valence Manganese Complexes. J. Am. Chem. Soc. 1996, 118, 65–69. [Google Scholar] [CrossRef]
- Imada, S.; Suga, S.; Muro, T.; Ueda, S.; Jung, R.-J.; Kotsugi, M.; Saitoh, Y.; Matsushita, T.; Kuwahara, H.; Moritomo, H.; et al. Local magnetic states in La1−xSrxMnO3 and Nd1−xSrxMnO3. Physica B 2000, 281–282, 498–499. [Google Scholar]
- Subías, G.; García, J.; Sánchez, M.C.; Blasco, J.; Proietti, M.G. Soft X-Ray Absorption Spectroscopy (Mn L2,3 and O K) in Mixed Valence Manganites. Surf. Rev. Let. 2002, 9, 1071–1078. [Google Scholar] [CrossRef]
- Kanamori, H.; Yoshioka, T.; Hirose, K.; Yamamoto, T. Determination of valence state of Mn ions in Pr1−xAxMnO3−δ (A = Ca, Sr) by Mn-L3 X-ray absorption near-edge structure analysis. J. Electron. Spectrosc. 2012, 185, 129–132. [Google Scholar] [CrossRef]
- Abbate, M.; de Groot, F.M.F.; Fuggle, J.C.; Fujimori, A.; Strebel, O.; Lopez, F.; Domke, M.; Kaindl, G.; Sawatzky, G.A.; Takano, M.; et al. Controlled-valence properties of La1−xSrxFeO3 and La1−xSrxMnO3 studied by soft-X-ray absorption spectroscopy. Phys. Rev. B 1992, 46, 4511–4519. [Google Scholar] [CrossRef]
- Liu, R.S.; Wu, J.B.; Chang, C.Y.; Lin, J.G.; Huang, C.Y.; Chen, J.M.; Liu, R.G. Determination of Mn Valence from X-Ray Absorption Near Edge Structure and Study of Magnetic Behavior in Hole-Doped (Nd1−xCax)MnO3 System. J. Solid State Chem. 1996, 125, 112–115. [Google Scholar] [CrossRef]
- Schmid, H.K.; Mader, W. Oxidation states of Mn and Fe in various compound oxide systems. Micron 2006, 37, 426–432. [Google Scholar] [CrossRef]
- Riedl, T.; Gemming, T.; Gruner, W.; Acker, J.; Wetzig, K. Determination of manganese valency in La1−xSrxMnO3 using ELNES in the (S)TEM. Micron 2007, 38, 224–230. [Google Scholar] [CrossRef]
- De Groot, F.M.F.; Grioni, M.; Fuggle, J.C.; Ghijsen, J.; Sawatzky, G.A.; Petersen, H. Oxygen 1s X-ray-absorption edges of transition-metal oxides. Phys. Rev. B 1989, 40, 5715–5723. [Google Scholar] [CrossRef]
- Saucke, G.; Norpoth, J.; Jooss, C.; Su, D.; Zhu, Y. Polaron absorption for photovoltaic energy conversion in a manganite-titanate pn heterojunction. Phys. Rev. B 2012, 85, 165315. [Google Scholar] [CrossRef]
- Blöchl, P.; Institute for Theoretical Physics, Clausthal, Germany; Norpoth, J.; Institut für Materialphysik, Göttingen, Germany; Jooss, C.; Institut für Materialphysik, Göttingen, Germany. First-Principles Study of Pr1−xCaxMnO3 with a Local Hybrid Functional. the authors' affiliation, unpublished work. 2014. [Google Scholar]
- Jung, J.H.; Kim, K.H.; Eom, D.J.; Noh, T.W.; Choi, E.J.; Yu, J.; Kwon, Y.S.; Chung, Y. Determination of electronic band structures of CaMnO3 and LaMnO3 using optical-conductivity analyses. Phys. Rev. B 1997, 55, 15489–15493. [Google Scholar]
- Mette, K.; Bergmann, A.; Tessonnier, J.-P.; Hävecker, M.; Yao, L.; Ressler, T.; Schlögl, R.; Strasser, P.; Behrens, M. Nanostructured Manganese Oxide Supported on Carbon Nanotubes for Electrocatalytic Water Splitting. ChemCatChem 2012, 4, 851–862. [Google Scholar] [CrossRef]
- Cressey, G.; Henderson, C.M.B.; Laan, G. Use of L-edge X-ray absorption spectroscopy to characterize multiple valence states of 3d transition metals; a new probe for mineralogical and geochemical research. Phys. Chem. Miner. 1993, 20, 111–119. [Google Scholar]
- Alonso, J.M.; Cortés-Gil, R.; Ruiz-González, L.; González-Calbet, J.M.; Hernando, A.; Vallet-Regí, M.; Dávila, M.E.; Asensio, M.C. Influence of the Synthetic Pathway on the Properties of Oxygen-Deficient Manganese-Related Perovskites. Eur. J. Inorg. Chem. 2007, 2007, 3350–3355. [Google Scholar]
- Gilbert, B.; Frazer, B.H.; Belz, A.; Conrad, P.G.; Nealson, K.H.; Haskel, D.; Lang, J.C.; Srajer, G.; de Stasio, G. Multiple Scattering Calculations of Bonding and X-ray Absorption Spectroscopy of Manganese Oxides. J. Phys. Chem. A 2003, 107, 2839–2847. [Google Scholar] [CrossRef]
- Cramer, S.P.; de Groot, F.M.F.; Ma, Y.; Chen, C.T.; Sette, F.; Kipke, C.A.; Eichhorn, D.M.; Chan, M.K.; Armstrong, W.H. Ligand field strengths and oxidation states from manganese L-edge spectroscopy. J. Am. Chem. Soc. 1991, 113, 7937–7940. [Google Scholar] [CrossRef]
- Frazer, B.H.; Gilbert, B.; Sonderegger, B.R.; de Stasio, G. The probing depth of total electron yield in the sub-keV range: TEY-XAS and X-PEEM. Surf. Sci. 2003, 537, 161–167. [Google Scholar] [CrossRef]
- Montenegro, M.J.; Lippert, T.; Müller, S.; Weidenkaff, A.; Willmott, P.R.; Wokaun, A. Pulsed laser deposition of electrochemically active perovskite films. Appl. Surf. Sci. 2002, 197–198, 505–511. [Google Scholar]
- Montenegro, M.J.; Döbeli, M.; Lippert, T.; Müller, S.; Schnyder, B.; Weidenkaff, A.; Willmott, P.R.; Wokaun, A. Pulsed laser deposition of La0.6Ca0.4CoO3 (LCCO) films. A promising metal-oxide catalyst for air based batteries. Phys. Chem. Chem. Phys. 2002, 4, 2799–2805. [Google Scholar] [CrossRef]
- Niu, J.; Deng, J.; Liu, W.; Zhang, L.; Wang, G.; Dai, H.; He, H.; Zi, X. Nanosized perovskite-type oxides La1−xSrxMO3−δ (M = Co, Mn; x = 0, 0.4) for the catalytic removal of ethylacetate. Catal. Today 2007, 126, 420–429. [Google Scholar] [CrossRef]
- Zhang-Steenwinkel, Y.; Beckers, J.; Bliek, A. Surface properties and catalytic performance in CO oxidation of cerium substituted lanthanum-manganese oxides. Appl. Catal. A 2002, 235, 79–92. [Google Scholar] [CrossRef]
- Choi, J.; Zhang, J.; Liou, S.-H.; Dowben, P.A.; Plummer, E.W. Surfaces of the perovskite manganites La1−xCaxMnO3. Phys. Rev. B 1999, 59, 13453–13459. [Google Scholar] [CrossRef]
- Dupin, J.-C.; Gonbeau, D.; Vinatier, P.; Levasseur, A. Systematic XPS studies of metal oxides, hydroxides and peroxides. Phys. Chem. Chem. Phys. 2000, 2, 1319–1324. [Google Scholar] [CrossRef]
- Fierro, J.L.G.; Tejuca, L.G. Non-stoichiometric surface behaviour of LaMO3 oxides as evidenced by XPS. Appl. Surf. Sci. 1987, 27, 453–457. [Google Scholar] [CrossRef]
- Somorjai, G.A.; Li, Y. Introduction to Surface Chemistry and Catalysis; Wiley: New York, NY, USA, 1994; p. 383. [Google Scholar]
- Jirák, Z.; Krupička, S.; Šimša, Z.; Dlouhá, M.; Vratislav, S. Neutron diffraction study of Pr1−xCaxMnO3 perovskites. J. Magn. Magn. Mater. 1985, 53, 153–166. [Google Scholar] [CrossRef]
- Knop-Gericke, A.; Kleimenov, E.; Hävecker, M.; Blume, R.; Teschner, D.; Zafeiratos, S.; Schlögl, R.; Bukhtiyarov, V.I.; Kaichev, V.V.; Prosvirin, I.P.; et al. Chapter 4 X-ray Photoelectron Spectroscopy for Investigation of Heterogeneous Catalytic Processes. Adv. Catal. 2009, 52, 213–272. [Google Scholar] [CrossRef]
- Follath, R.; Senf, F.; Gudat, W. Plane-grating monochromator at BESSY II using collimated light. J. Synchrotron Rad. 1998, 5, 769–771. [Google Scholar] [CrossRef]
- Shirley, D.A. High-Resolution X-ray Photoemission Spectrum of the Valence Bands of Gold. Phys. Rev. B 1972, 5, 4709–4714. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mierwaldt, D.; Mildner, S.; Arrigo, R.; Knop-Gericke, A.; Franke, E.; Blumenstein, A.; Hoffmann, J.; Jooss, C. In Situ XANES/XPS Investigation of Doped Manganese Perovskite Catalysts. Catalysts 2014, 4, 129-145. https://doi.org/10.3390/catal4020129
Mierwaldt D, Mildner S, Arrigo R, Knop-Gericke A, Franke E, Blumenstein A, Hoffmann J, Jooss C. In Situ XANES/XPS Investigation of Doped Manganese Perovskite Catalysts. Catalysts. 2014; 4(2):129-145. https://doi.org/10.3390/catal4020129
Chicago/Turabian StyleMierwaldt, Daniel, Stephanie Mildner, Rosa Arrigo, Axel Knop-Gericke, Emanuel Franke, Andreas Blumenstein, Jörg Hoffmann, and Christian Jooss. 2014. "In Situ XANES/XPS Investigation of Doped Manganese Perovskite Catalysts" Catalysts 4, no. 2: 129-145. https://doi.org/10.3390/catal4020129
APA StyleMierwaldt, D., Mildner, S., Arrigo, R., Knop-Gericke, A., Franke, E., Blumenstein, A., Hoffmann, J., & Jooss, C. (2014). In Situ XANES/XPS Investigation of Doped Manganese Perovskite Catalysts. Catalysts, 4(2), 129-145. https://doi.org/10.3390/catal4020129