1. Introduction
Titanium dioxide, a simple binary compound, possesses a long list of intriguing surface properties [
1,
2]. Some of the well-known examples include photo-splitting of water [
3], photo-degradation of organic molecules [
4,
5,
6], and enhanced catalytic reactivity of gold nanoparticles supported on TiO
2 [
7,
8,
9]. Because defects play a crucial role in photochemistry and in heterogeneous catalysis, these phenomena have been attributed to the unique surface or interfacial structure and properties imparted by defects.
Among intrinsic defects, oxygen vacancy has been the focus of intense investigations on reduced surfaces of TiO
2 [
10,
11,
12,
13]. It may be regarded as the simplest form of surface defects, created upon the loss of oxygen. However, a growing number of experiments have indicated that the presence of oxygen vacancy is not the only major characteristics of reduced TiO
2 surfaces. Besides oxygen vacancy, the experimental and theoretical studies all point to the significance of Ti interstitials and their role in forming surface defects during re-oxidation [
14,
15,
16,
17]. The formation of such defects driven by Ti interstitials and the dynamic restructuring of the surface can have profound influence on chemical reactivity of partially reduced and re-oxidized TiO
2 surface.
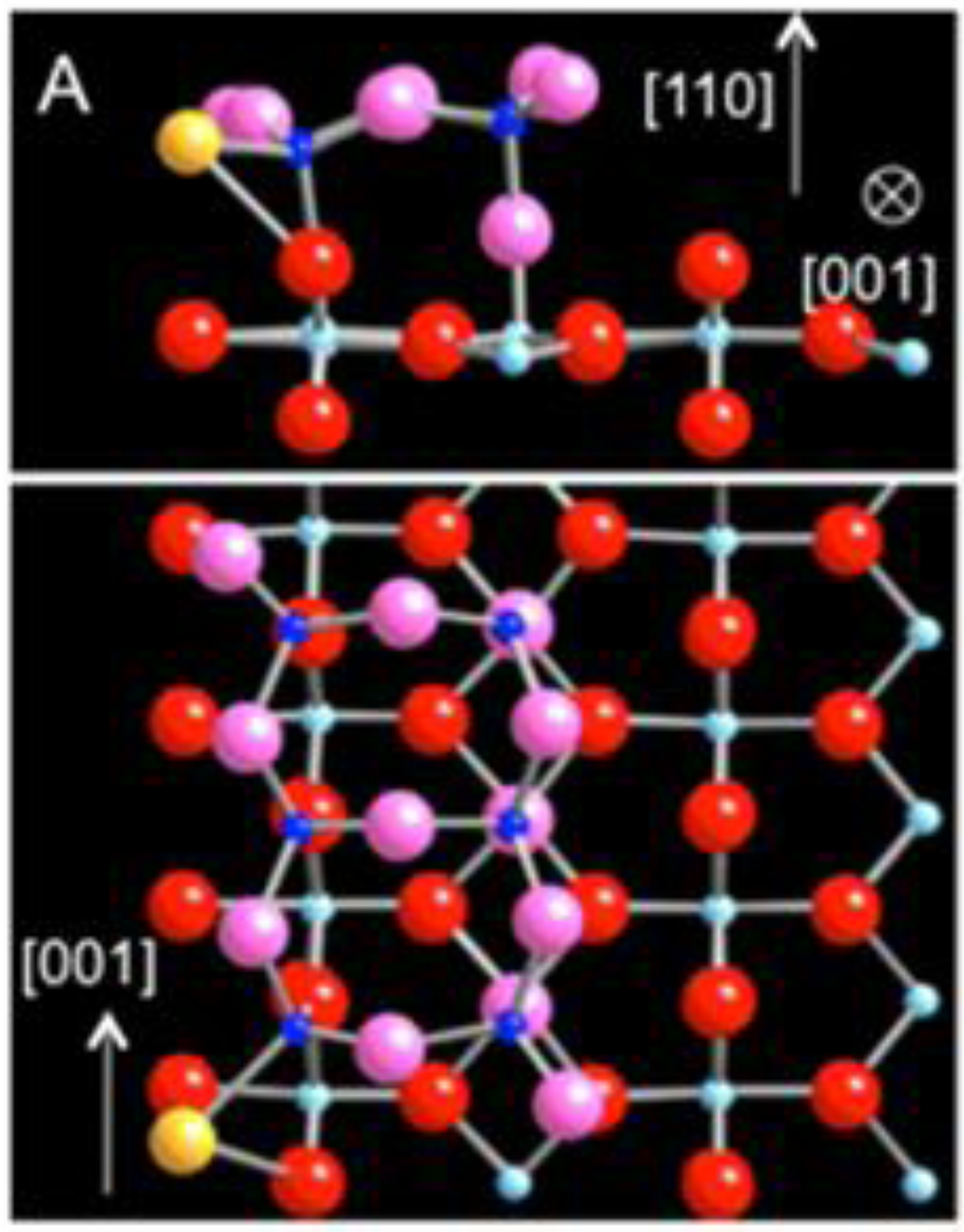
Bulk-like terminated TiO
2 (110) surfaces constitute an interesting case study. On these systems, it is now established that the linear defects observed on (1 × 1) terraces are sub-stoichiometric species (
Figure 1(a)). The linear defects can be characterized as either single or double strands depending on the local stoichiometry of TiO or Ti
2O, respectively [
18]. They can further assemble into (1 × 1) or (1 × 2) reconstructed surfaces. Either single or double strands can form depending on the local stoichiometry of TiO or Ti
2O, respectively. The proposed structural models (based on STM and DFT calculations) for the sub-stoichiometric defects have been independently confirmed by a high resolution TEM study [
19].
As these sub-oxide species possess structures that are markedly different from that of the mother compound, a partially reduced surface of TiO
2, even in a single crystal model system, can become increasingly complex and heterogeneous. However, these sub-stoichiometric defects possess distinctive structural features that are easily identifiable on bulk-like terminated surfaces. First, sub-stoichiometric defects on (110) surface are all aligned along the particular crystallographic direction [001] forming strands (
Figure 1(b)). In this geometry, a Ti row of a linear defect is sandwiched between the oxygen rows of the defect and the surface. As a result, the coordination number of Ti in the linear defect is kept as five like in stoichiometric surface despite a lower ratio of O to Ti [
18]. Second, whether they are single or double strands, and whether isolated or assembled to form a reconstructed surface, the apparent height of the linear defect is approximately 1.6 Å corresponding to half-step height on (110). This value is a direct consequence of forming face-sharing oxygen octahedra at the interface between the defects and the substrate. Similar face-sharing oxygen octahedra are well known in reduced TiO
2−x bulk phases, for example at the crystallographic shear planes (CSP) of the Ti
2O
2n−1 [
20] as well as the corundum Ti
2O
3 [
1].
In addition to the sub-stoichiometric defects, partially reduced surfaces of TiO
2 show another type of defects: a nanometer-sized, bright dot [
21]. It is imaged by STM as a 3 Å high protrusion on (110) surface (
Figure 1(a),(b)). Furthermore, it spans across both Ti (bright) and O (dark) rows of the terrace along [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10], resulting in circular or elliptical shape as seen in STM images. Being topographically distinct from the linear sub-stoichiometric defect, the dots are ascribed as stoichiometric TiO
2 nanoclusters with low coordination numbers. These topographical features of various types of defects make it possible to discriminate between them by scanning tunneling microscopy (STM). It follows that a partially reduced TiO
2 (110) provides an exciting opportunity to probe nanometer-sized defects with well-defined, local stoichiometry and structures and their chemical reactivity. In this paper, we present a comparative study of various defects formed on TiO
2 (110)—both sub-stoichiometric and stoichiometric—and their role as the nucleation sites for the initial adsorption of Au using STM and density functional theory (DFT).
2. Results and Discussion
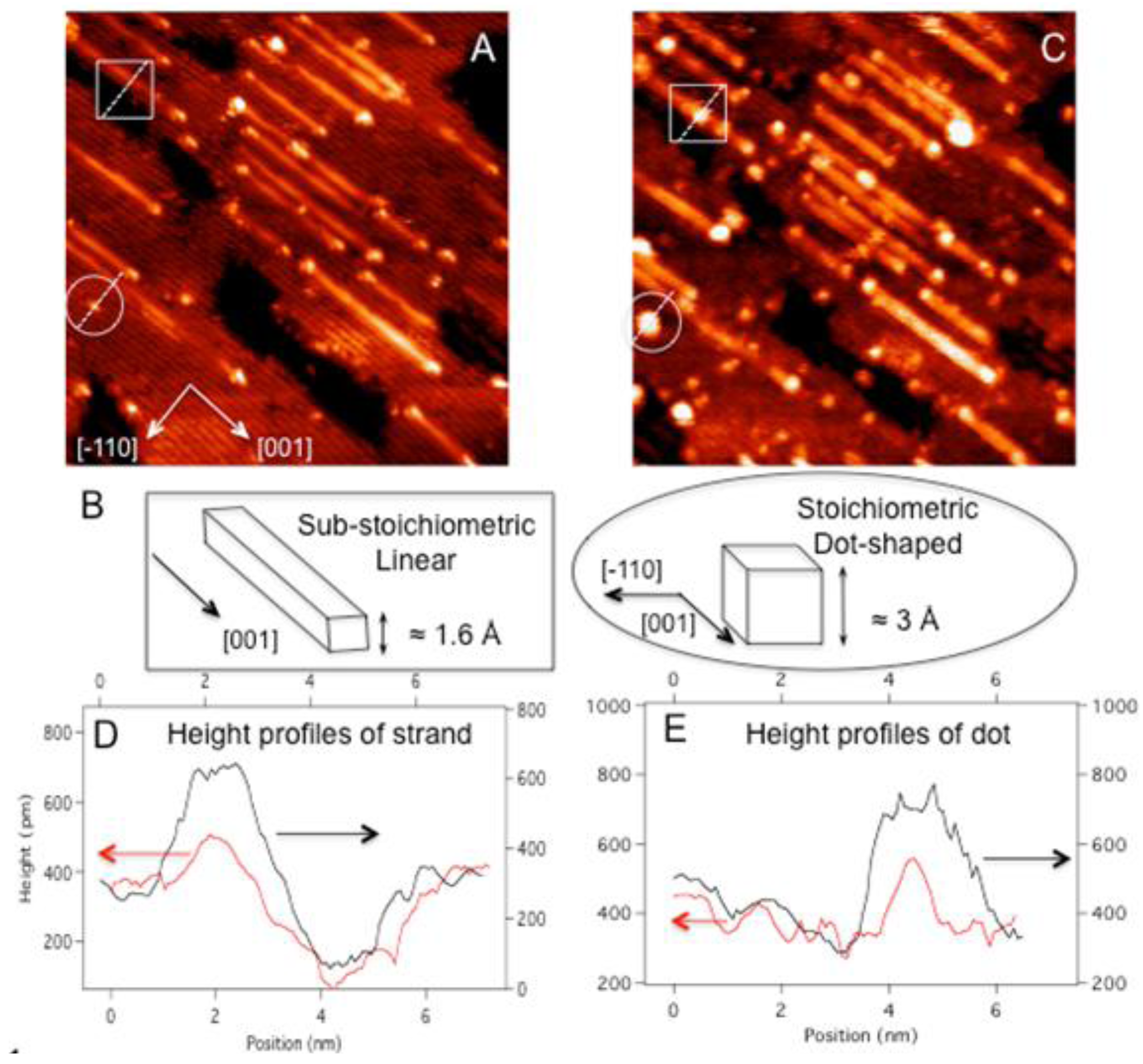
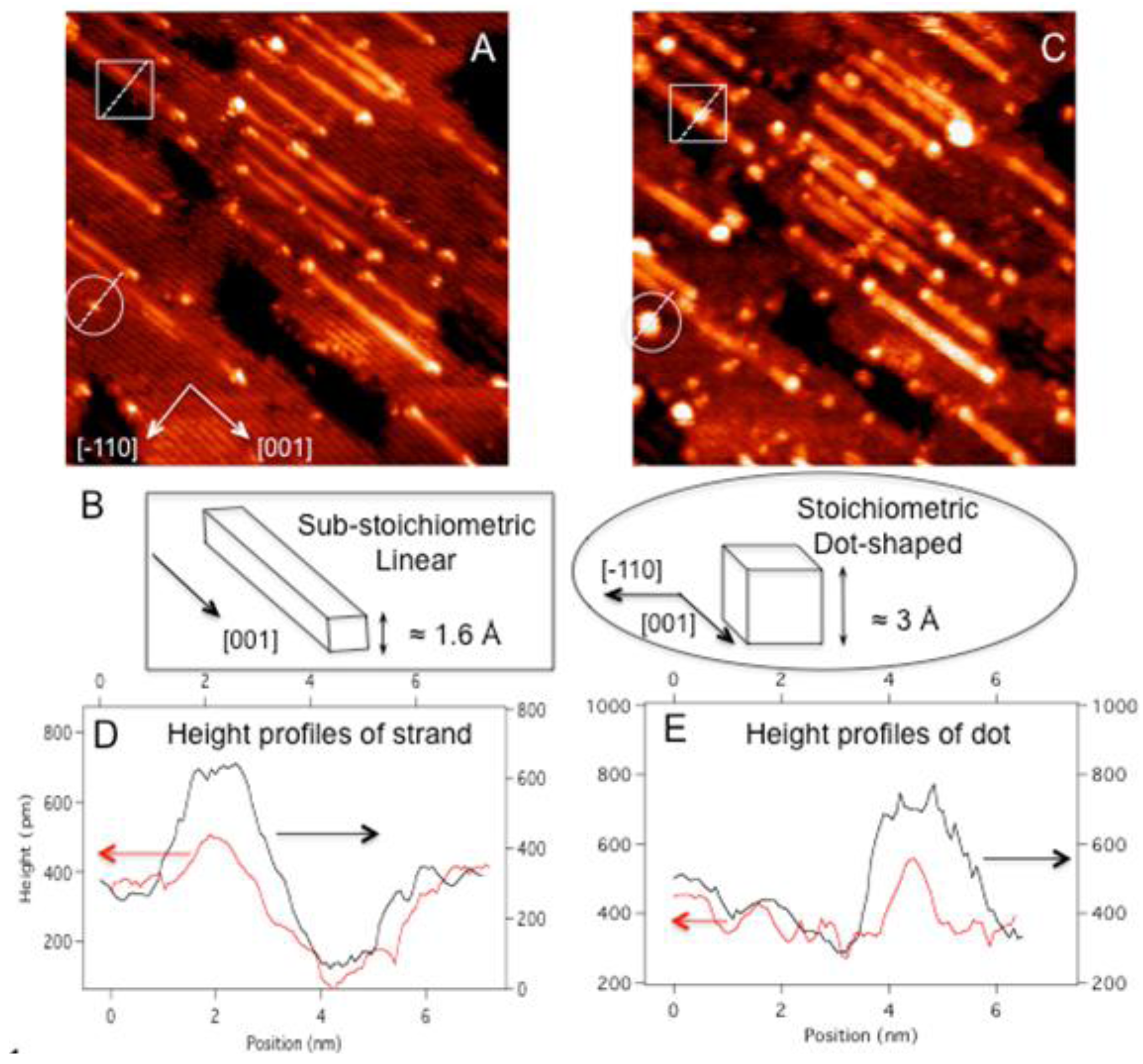
A typical STM image of reduced TiO
2 (110) showed that 5% of the surface area was covered by sub-stoichiometric strands (
Figure 1(a)). After one minute of Au deposition, numerous bright clusters are observed on partially reduced TiO
2 (110) (
Figure 1(c)). The large clusters have diameters of about 20 Å and heights of 5 Å on average. The comparison of the same area before and after Au deposition clearly shows that many of the large Au cluster formation started preferentially with pre-existing defects such as strands (highlighted by squares), their end structures, and dots on (1 × 1) terraces (highlighted in circles). Because these native defects already have significant heights (e.g., 1.5 Å to 3 Å tall) and widths, only the part of measured heights and widths can be assigned to adsorbed Au clusters. For example, the comparison in height profiles of the strand defect between before and after Au deposition reveals that only one layer thick Au cluster is adsorbed on top of the strand (
Figure 1(d)). Similarly for the dot defect, the height increased from 3 Å to 5 Å upon Au deposition, indicating that the adsorbed Au cluster is one monolayer thick (
Figure 1(e)). However, the full-width-at-half-maximum (FWHM) of the defect also increased deposition along [−110] from 8 Å to 20 Å, suggesting that the Au cluster grew laterally as well to form a quasi-2D raft with the height of 2 atomic layers.
In addition to large clusters, smaller clusters having diameters of about 7 Å and heights of 2 Å are also observed. They appear to be scattered at various sites, including strands, their ends, and step edges as well as on (1 × 1) terraces, presumably at oxygen vacancies or sites near sub-surface Ti interstitials. Because these small Au clusters have similar topographical features as the native stoichiometric dot defects, it would be a non-trivial task to distinguish them from native defects. However, the comparison of the same area used in this study allows one to unambiguously identify them as Au clusters. Counting all the Au clusters, the total surface coverage of Au on TiO2 (110) is estimated as 0.02 monolayers (ML), where 1 ML is equal to 1.39 × 1015 atoms/cm2. Using a larger data set, 69% of the new Au clusters are observed at the dot defects (including the end structures of the strands), 14% are at or on the side of line defects, and 17% are on seen on the (1 × 1) terraces, presumably on top of O vacancy sites.
For the investigation of the energetics of defect-structures of TiO
2 and subsequent Au adsorption, theoretical modeling was performed using VASP package based on DFT [
22,
23,
24]. The calculations were carried out using a slab model keeping the atoms of the bottom layers at bulk positions to simulate the semi-infinite crystal. The unit cells had the following lateral dimensions: 1.119 × 1.317 nm
2 for Au on a linear defect and 1.783 × 1.317 nm
2 for Au on a nanocluster defect. In the direction perpendicular to the slab the dimension was chosen to ensure a 1.5 nm vacuum between images. Core atomic states were represented by projector augmented wave pseudopotentials [
25,
26] within the Perdew-Wang (PW91) generalized gradient approximation (GGA) for the exchange-correlation functional [
27]. For selected systems, the calculations were also performed using the Perdew-Burke-Ernzerhof (PBE) GGA functional [
28] and spin polarization in order to verify the main results associated with the energetics of adsorption. Plane-wave basis with energy cut-off up to 400 eV, a fine k-point sampling of the surface Brillouin zone were chosen after careful convergence studies of the total energy. For each structure energy convergence down to 0.1 meV and forces smaller than 4 meV/Å were systematically reached.
Figure 1.
(A) STM scan of a 40 × 40 nm2 area of (1 × 1) TiO2 (110) with full of defects; (B) schematic diagrams of substoichiomeric strand and stoichiometric dot defects contrasting their distinct topographical features; (C) STM scan of the same area in (A) after 1 min Au deposition at room temperature (≈0.02 ML). The height profiles, across (D) the strand and (E) the dot defects ((highlighted in squares and circles, respectively), reveal a marked increase in the height as well as the FWHM along [−110] (along the dotted lines) due to Au adsorption. The height profiles before and after Au adsorption are shown in red and black lines, respectively. Both STM scans are taken with V = 1.80 V and I = 0.5 nA for (A) and V = 1.42 V and 0.5 nA for (C).
Figure 1.
(A) STM scan of a 40 × 40 nm2 area of (1 × 1) TiO2 (110) with full of defects; (B) schematic diagrams of substoichiomeric strand and stoichiometric dot defects contrasting their distinct topographical features; (C) STM scan of the same area in (A) after 1 min Au deposition at room temperature (≈0.02 ML). The height profiles, across (D) the strand and (E) the dot defects ((highlighted in squares and circles, respectively), reveal a marked increase in the height as well as the FWHM along [−110] (along the dotted lines) due to Au adsorption. The height profiles before and after Au adsorption are shown in red and black lines, respectively. Both STM scans are taken with V = 1.80 V and I = 0.5 nA for (A) and V = 1.42 V and 0.5 nA for (C).
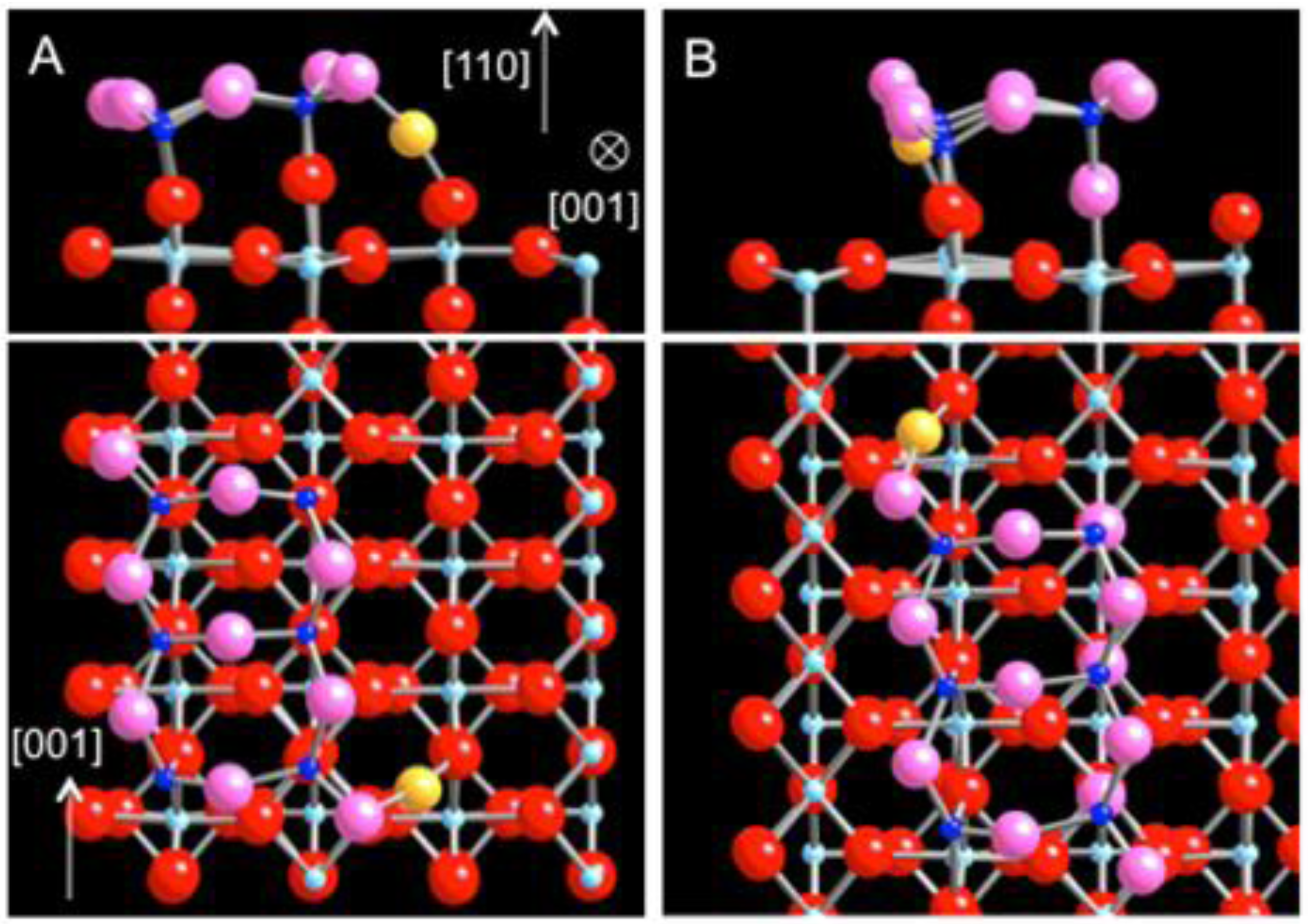
The initial adsorption of Au is examined on the strand and dot defects by comparing the energetics at various sites. The adsorption energy for Au is defined as the difference in the total energy before and after Au attachment per unit cell, EA = E(Au/TiO
2) – [E(TiO
2) + E(Au)]. As for a reference, the adsorption energy of an Au atom above a 2-coordinated oxygen (2c-O, also known as a bridging oxygen) on stoichiometric TiO
2 (110) is calculated using two differently functionals. The calculated values of −0.64 eV with PBE and −0.57 eV with PW91 are in good agreement with the reported value of −0.61 eV [
29].
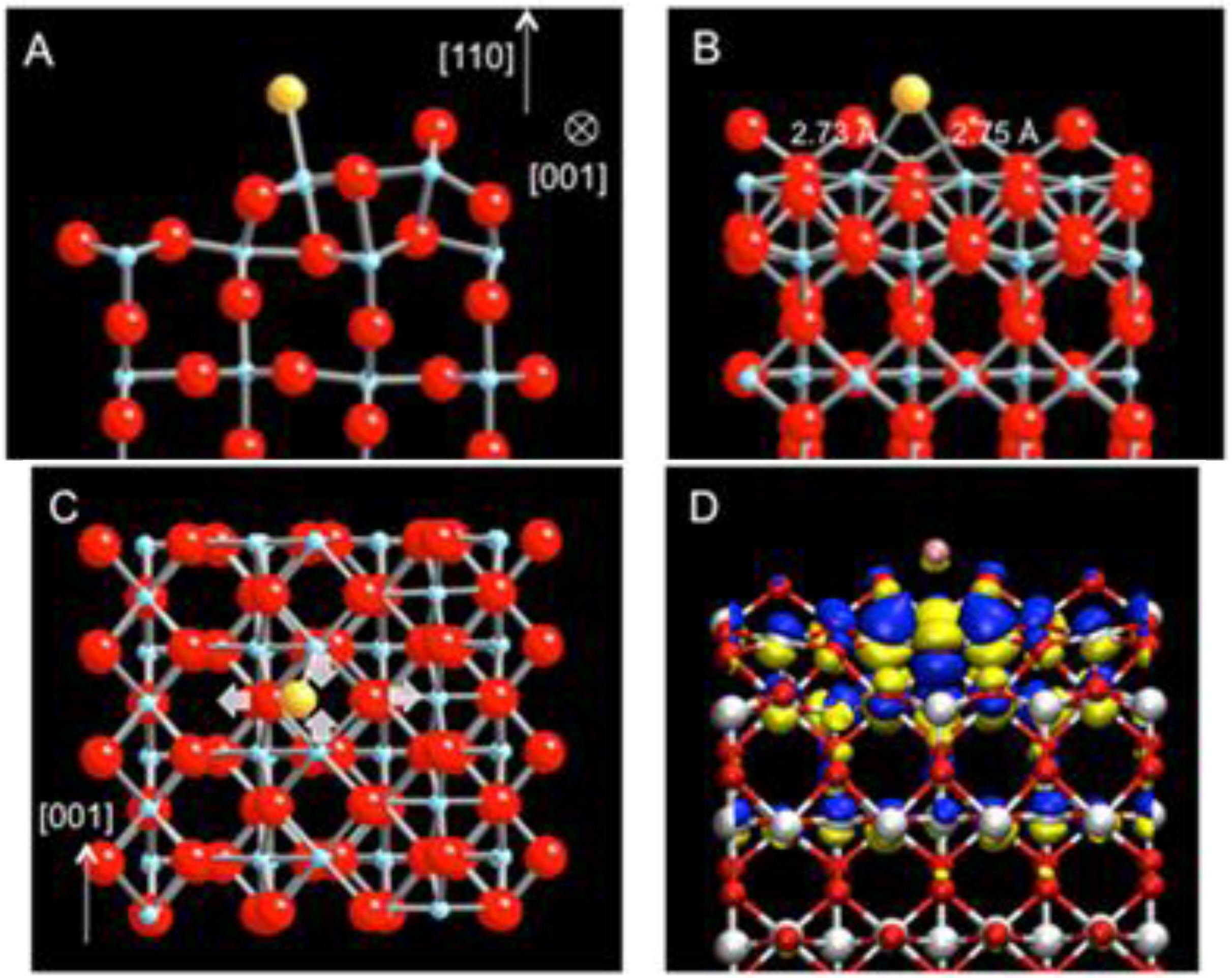
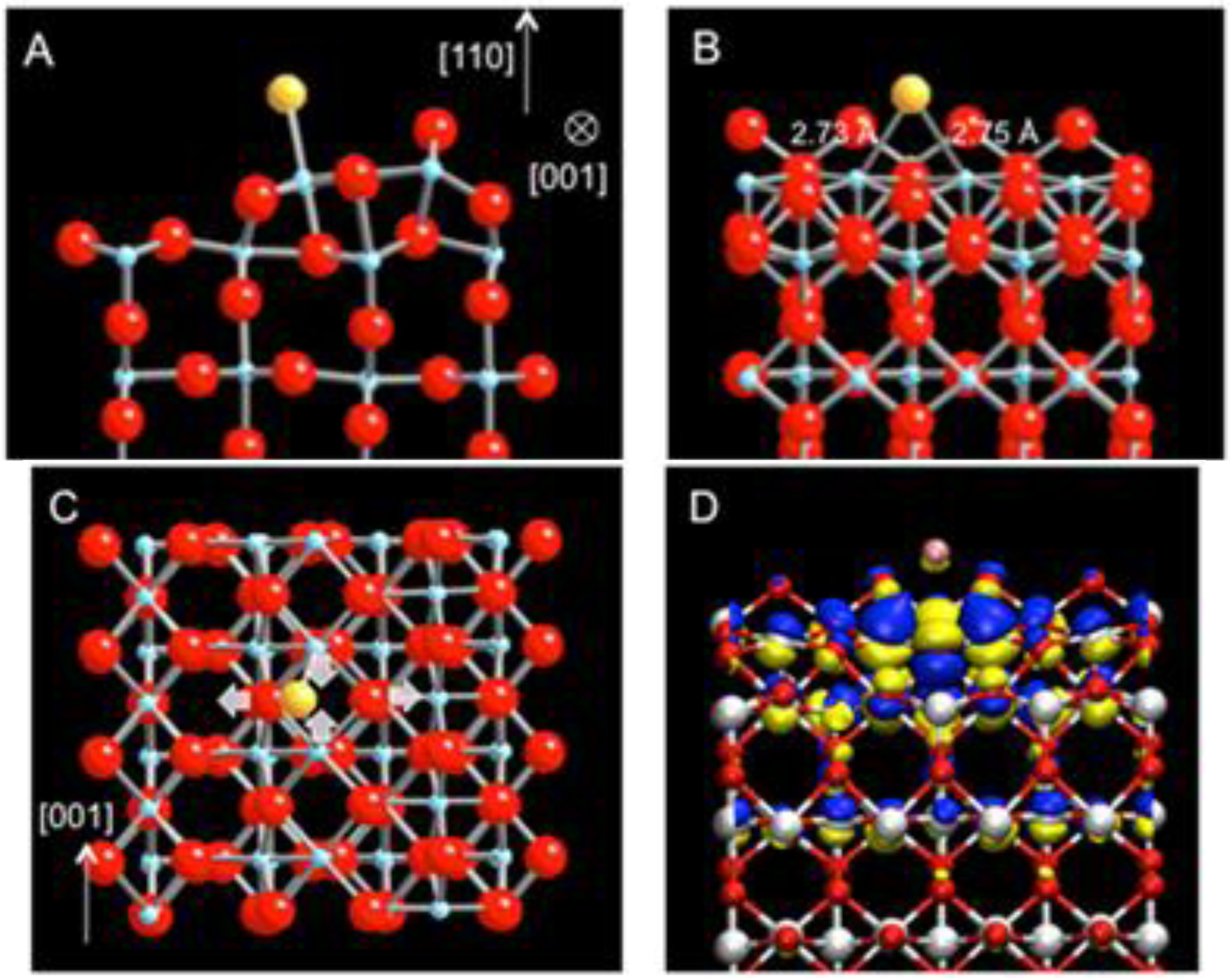
For the Au adsorption on the strands, the linear defect is modeled as a single strand (Ti
2O
2) [
18]. The energetically most favorable site for a single Au atom is found at the hollow site, 2.73 Å and 2.75 Å away from the two nearest 5c-Ti atoms and 3.00 Å and 3.03 Å from the two O atoms in-plane with the Ti atoms (
Figure 2(a),(b)). Upon the Au adsorption, the strand undergoes a substantial structural relaxation. The two 5c-Ti atoms are pulled toward each other while the two O atoms are slightly pushed apart (
Figure 2(c)). The adsorption energy for Au on the single strand is calculated as −3.11 eV, representing an exceptionally strong bonding between the Au atom and TiO
2 (110) via the strand defect. The large enhancement in bonding is also accompanied by a significant amount of charge transfer from the strand to the adsorbed Au atom. Using Bader’s charge analysis [
30,
31], it is calculated that Au acquires 0.54 e
− upon adsorption and becomes negatively charged.
The effect of the sub-stoichiometric strand on Au bonding is qualitatively in good agreement with recent DFT calculations of reporting similar effects of electron donor defects on Au adsorption on TiO
2 (110). Madsen and Hammer [
32] reported that the presence of a sub-surface Ti interstitial significantly enhances the bonding of Au on otherwise, defect-free TiO
2 (110). The energy of Au on TiO
2 (110) was lowered by about 1 eV with the Ti interstitial defect, and Au gained about 0.3 e
− regardless of the types of functionals. They further concluded that other types of sub-surface defects, such as oxygen vacancy and CSP, had the same effect on Au bonding on surface. Similarly, Chrétien and Metiu [
33] reported that co-adsorption of electron donating molecules as well as artificial addition of an extra electron increase the binding energy of an Au atom on 5c-Ti increased from 0.45 eV for defect-free TiO
2 (110) to 1.2 < 1.3 eV. However, the very large enhancement in bonding and charge accumulation at Au observed in this study is unique to the strand defect by its sub-stoichiometric and spatially extended nature. Although the Ti atoms underneath the Au atom exhibit the largest change in charge by 0.021 e
−, all the Ti atoms around the adsorbed Au as well as sub-surface Ti atoms contribute to the charge transfer. The spatial map of difference in valence charge density clearly shows the extended nature of charge transfer not only along the strand but also into sub-surface, providing exceptional stability of Au atom (
Figure 2(d)).
Figure 2.
DFT-relaxed structure of Au adsorbed on the sub-stoichiometric strand defect on TiO
2 (110) viewed along (
a) [001], (
b) [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10], and (
c) [−1-10] (Au: yellow, Ti: blue; O: red). (
d) The charge density difference isosurface plot of (b) revealing electron surplus (blue) and depletion (yellow).
Figure 2.
DFT-relaxed structure of Au adsorbed on the sub-stoichiometric strand defect on TiO
2 (110) viewed along (
a) [001], (
b) [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10], and (
c) [−1-10] (Au: yellow, Ti: blue; O: red). (
d) The charge density difference isosurface plot of (b) revealing electron surplus (blue) and depletion (yellow).
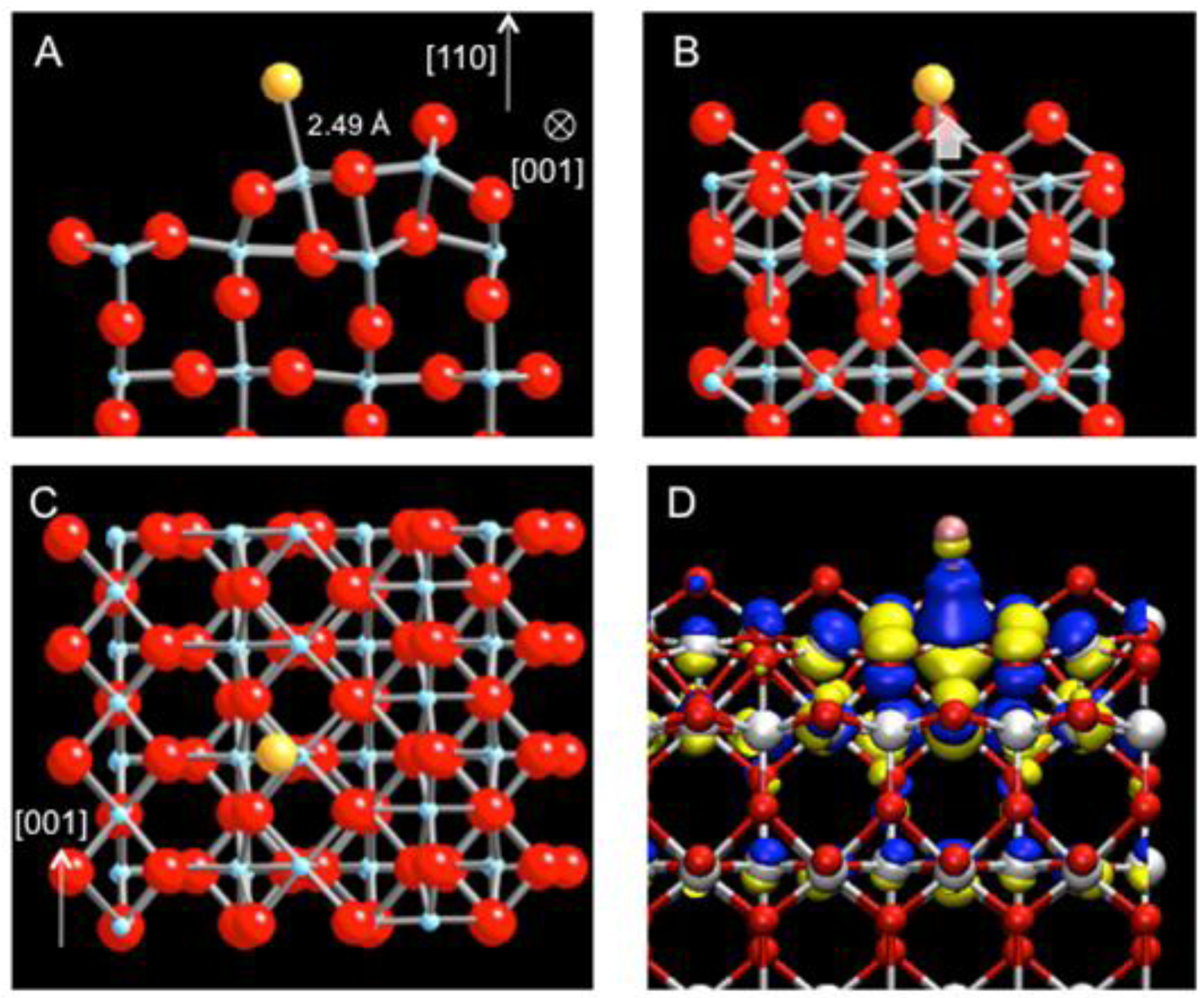
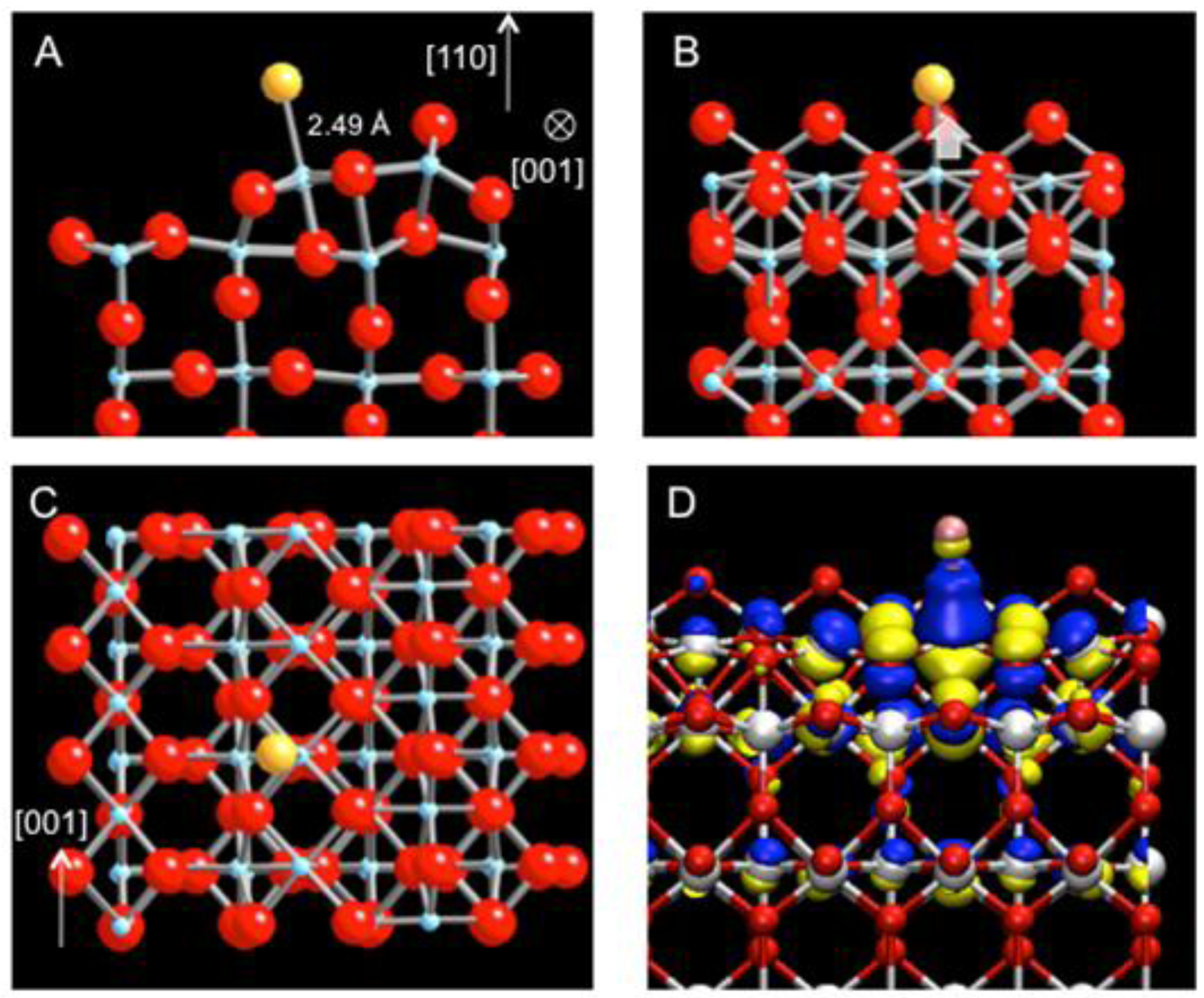
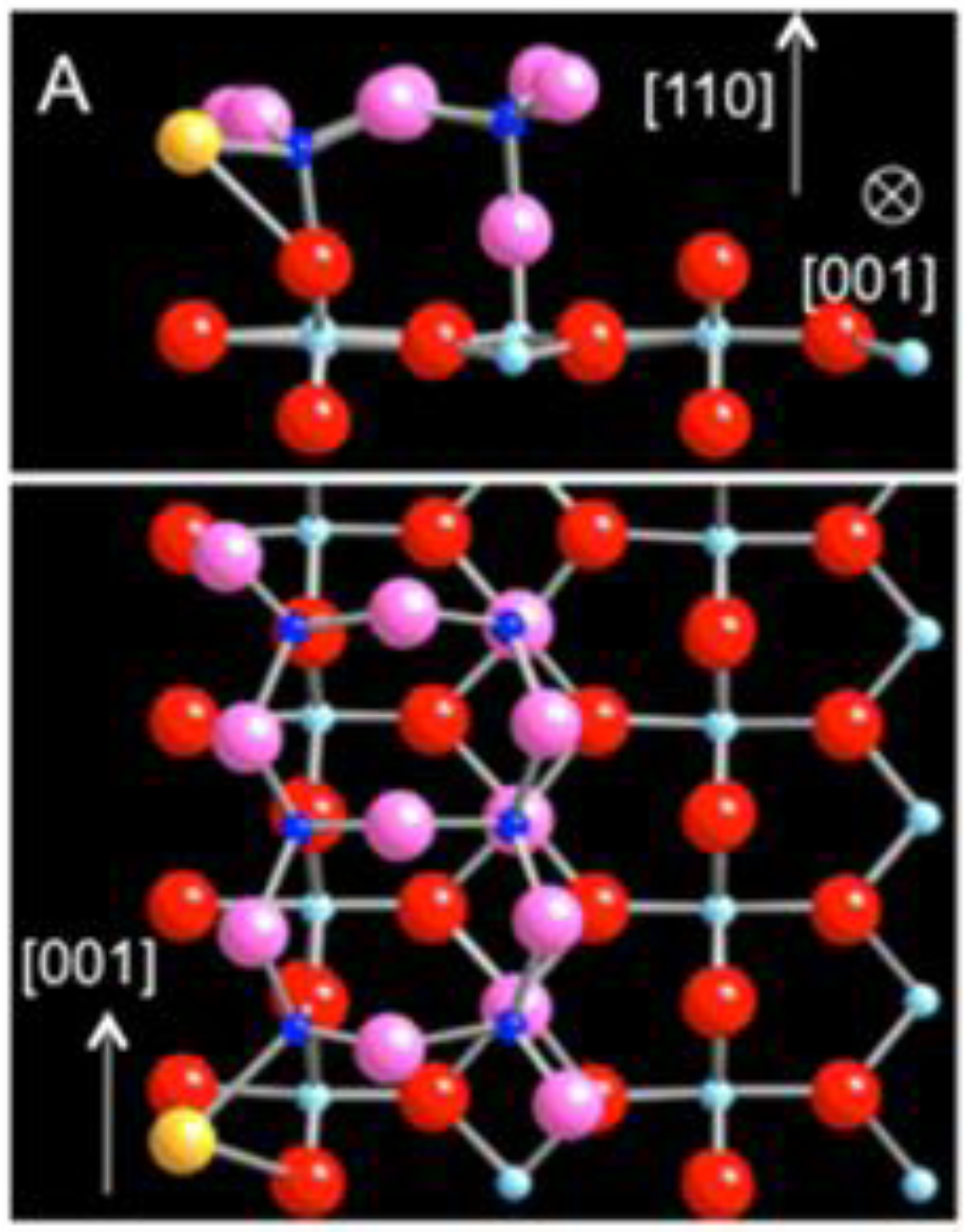
The participation of neighboring Ti atoms in Au bonding further suggests that the variation in energetics of Au adsorption along the strand may be relatively small. Indeed, there is another energetically favorable site for the Au atom, displaced approximately by half the lattice constant along [001]. The Au atom is directly located at 2.49 Å above the 5-coordinated Ti (5c-Ti) of the linear defect (
Figure 3(a)–(c)). The adsorption energy at the top site is −2.91 eV, the difference of only 200 meV compared to the previous hollow site. Upon the Au adsorption, the strand also undergoes a structural relaxation. In particular, the 5c-Ti is pulled up toward the Au atom, increasing its bond distance to the oxygen underneath from 1.95 Å to 2.12 Å (
Figure 3(b)). The isosurface map of charge density difference shows the electron accumulation along the bond axis between Ti and Au as the major effect but also clearly reflects the delocalized nature of charge density variation along the strand for this adsorption site as well (
Figure 3(d)). As the energetics of the Au atom is predominantly influenced by the Ti 4d states with large dispersion along [001], the adsorbed Au is expected to diffuse easily along the strand, analogous to a facile diffusion of Au on TiO
2 (110) along the Ti trough [
29].
Figure 3.
DFT-relaxed structure of Au adsorbed at an alternative site on the sub-stoichiometric strand defect viewed along (
a) [001], (
b) [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10], and (
c) [−1-10] (Au: yellow, Ti: blue; O: red). (
d) The charge density difference isosurface plot of (
b) revealing electron surplus (blue) and depletion (yellow).
Figure 3.
DFT-relaxed structure of Au adsorbed at an alternative site on the sub-stoichiometric strand defect viewed along (
a) [001], (
b) [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10], and (
c) [−1-10] (Au: yellow, Ti: blue; O: red). (
d) The charge density difference isosurface plot of (
b) revealing electron surplus (blue) and depletion (yellow).
Another type of surface defects at which Au atoms adsorb and nucleate, is expressed as a bright dot. The strand defect is usually terminated with the dots as the end structure. However, the dots are also observed as isolated alone on a terrace. Because in both cases, the dots exhibit the same topographical features and reactivity toward Au, the initial adsorption of Au on a dot is investigated with a fully stoichiometric nanocluster of (TiO
2)
6 isolated on (1 × 1) TiO
2 (110) [
21].
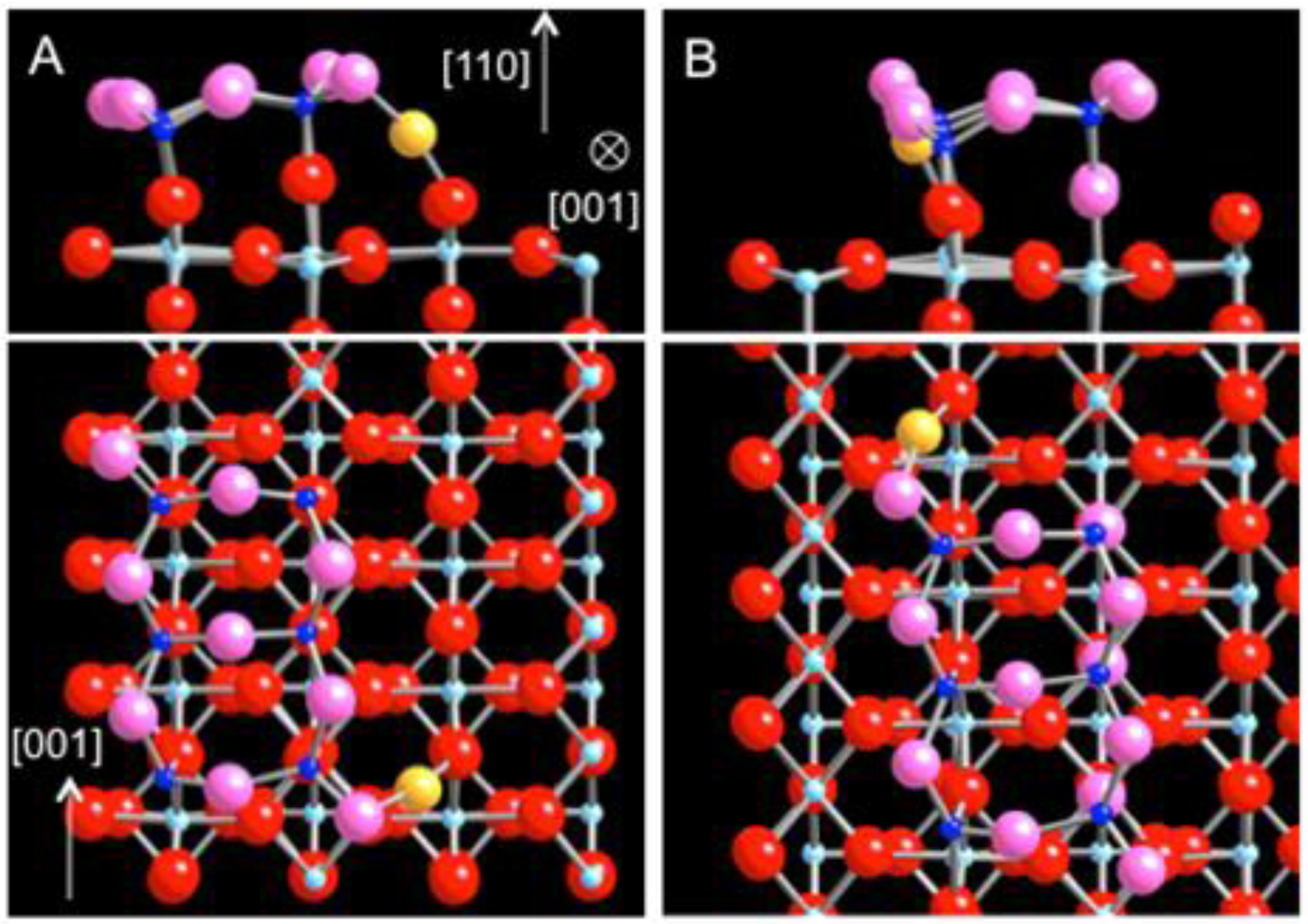
The energetically most favorable position for the gold adsorption site is found between the 1c-O atom of the nanocluster defect and the surface 2c-O atom (
Figure 4(a)). The adsorption energy for the Au atom at the cluster defect is calculated as −2.74 eV using either PW91 or PBE. The large adsorption energy can be understood from the unique bond geometry and electronic structure the nanocluster provides for Au. The adsorbed Au atom is nearly at equidistance from the 1c-O atom of the nanocluster defect and the surface 2c-O atom, 2.03 Å and at 2.05 Å, respectively. Moreover, at the adsorption site substantial density of states derived from O 2p are available just above the Fermi level. Ordinarily for rutile TiO
2, the O 2p states are mainly occupied forming the valence band, and the empty conduction band consists largely of the Ti 4d states. However, for the nanocluster defect, a part of the O 2p states is pushed up above the Fermi level due to Coulomb repulsion. The partially unoccupied states from the 1c-O of the cluster defect and the surface 2c-O become available for the hybridization with the Au 6s state. In this case, the Au atom loses about 0.2 e−, which are redistributed mostly to the two oxygen atoms. It follows that the enhancement of Au bonding originates from the lowering of the energy due to the hybridization between the two types of O atoms along with a lowering of the Coulomb repulsion by having the Au adsorption site between the defect and surface O atoms.
Figure 4.
DFT-relaxed structures of Au adsorbed on the dot defect, modeled as (TiO2)6 on TiO2 (110). (a) The energetically most favorable site and (b) the alternative 1-coordinated O site. At both locations, Au is found between the 1c-O of the defect and the 2c-O of the surface. Au becomes anionic, losing electrons to the oxygen atoms of low coordination (defect Ti: dark blue; defect O: pink).
Figure 4.
DFT-relaxed structures of Au adsorbed on the dot defect, modeled as (TiO2)6 on TiO2 (110). (a) The energetically most favorable site and (b) the alternative 1-coordinated O site. At both locations, Au is found between the 1c-O of the defect and the 2c-O of the surface. Au becomes anionic, losing electrons to the oxygen atoms of low coordination (defect Ti: dark blue; defect O: pink).
There is another 1-c O atom at the opposite end of the cluster defect (
Figure 4(b)). This site would be equivalent to the previous site if one considered the cluster geometry alone. However this site is located next to the 5-c Ti rows of the surface, thus breaking the symmetry. The Au atom bonds to the 1-c O atom of the (TiO
2)
6 nanocluster and the 2-c O atom of the surface with nearly the same bond lengths of 2.02 Å. The adsorption energy (using the PW91 functional) is calculated as −2.10 eV for this site. Although this site is energetically less stable than the previous site by 0.64 eV, the enhancement of Au bonding is based on the same mechanism: the hybridization between the partially unoccupied O 2p states and the Au 6s state to facilitate charge transfer. Then, the wedge sites around the nanocluster defect, i.e. between the 1c-O of the nanocluster and the 2c-O of the surface, may be viewed as an electron acceptor center, whereas the strand defect as a strong electron donor site with its ability to reduce adsorbates.
In addition to the O atoms of low coordination, the nanocluster of (TiO
2)
6 also possesses a 3-c Ti atom. The stable adsorption site for Au is found at 2.57 Å from the 3-c Ti atom (
Figure 5). The adsorbed Au atom is also coordinated to the 2-c O atom of the surface at 2.98 Å away in a similar manner observed for the Au in
Figure 4(b). The bond distances are much larger than those observed at the previous sites: 2.49 Å for Au-Ti and 2.02 ≈ 2.05 Å for Au-O, reflecting a much smaller interaction at the adsorption site. Indeed, the adsorption energy for this site is calculated as −1.37 eV, significantly smaller than those for the previous two sites, where the Au atom is coordinated to both O atoms of the defect and the surface.
Figure 5.
DFT calculation of Au adsorbed at the 3-coordinated Ti site of the dot defect, modeled as (TiO2)6 on TiO2 (110).
Figure 5.
DFT calculation of Au adsorbed at the 3-coordinated Ti site of the dot defect, modeled as (TiO2)6 on TiO2 (110).
The comparable bond enhancement of Au observed at both stoichiometric and sub-stoichiometric defects suggests that they may compete for the formations of cationic and anionic Au
−δ species for a given condition. Thus for low temperature CO oxidation, both types of defects and bond mechanisms must be carefully examined. However, considering a typical catalyst preparation via wet chemistry followed by heating in air, the stabilization of Au particles by stoichometric nanoclusters may be favored over by the sub-stoichoimetric, extended defects. In fact, the strong adsorption of Au at the perimeter of the stoichiometric nanocluster defect is consistent with key characteristics, recently suggested for active gold species supported metal oxides. A number of experimental and theoretical studies have indicated that cationic Au species are the catalytically active form [
34,
35,
36,
37,
38,
39,
40,
41]. In particular, Camellone and Fabris [
40] reported DFT calculations showing the selective adsorption of CO on positively charged Au and further proposed the formation of Au-COO* with the closest lattice O atom of CeO
2. We have separately shown that the positively charged Au
3 cluster formed at the nanocluster strongly binds CO with the adsorption energy of −1.11 eV [
21].
Another common characteristics emerging from recent investigations is the presence of small Au particles (≤1 nm) identified in active catalyst samples only. Unlike the earlier belief of 2- to 5-nm Au particles as the most active form, a number of studies have now indicated the continued increase in the CO oxidation activity as the Au particle size decreases. Using aberration-corrected scanning transmission electron microscopy, Herzing
et al. [
41] highlighted the strong correlation between high catalytic activity of Au/FeO
x for CO oxidation and the presence of bilayer clusters of <5 Å in diameter that contain about 10 Au atoms only. Likewise, Rashkeev
et al. [
42] reported evidence that most Au particles are only one to two layers thick in the most active form of Au/TiO
2 catalysts. The topographical features of quasi-2D Au rafts grown (
Figure 1) in this model study are strikingly similar to the reported shapes and size for the active Au species supported on oxides. The interface between Au cluster and the stoichiometric nanocluster defect could well be the site, where the crucial step of the CO oxidation reaction takes place as envisioned by Haruta and co-workers [
43].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}