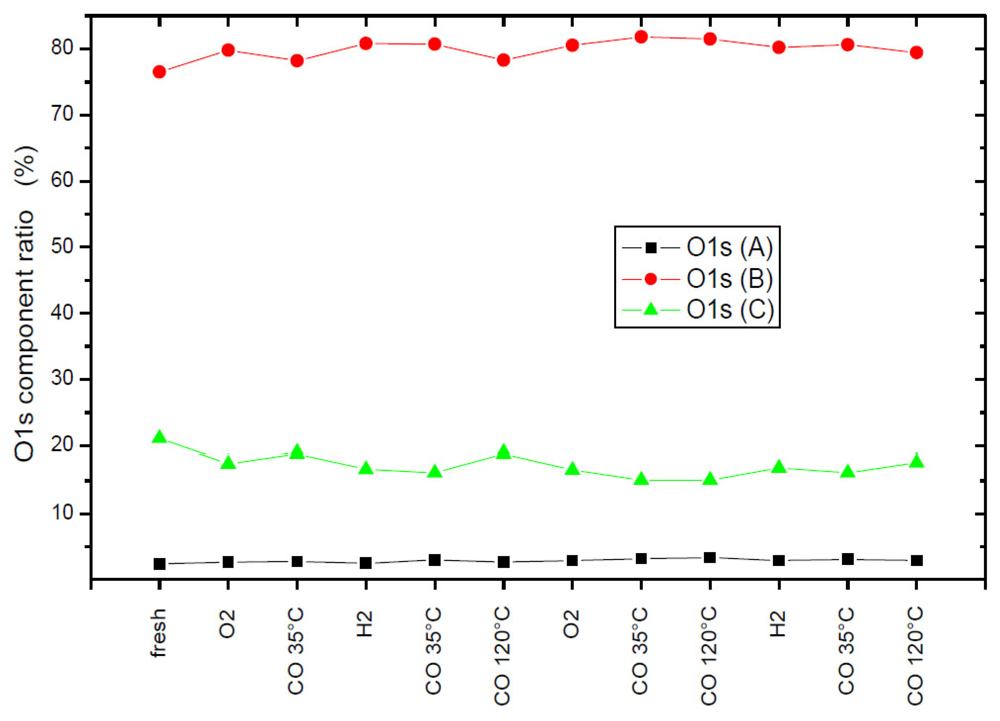
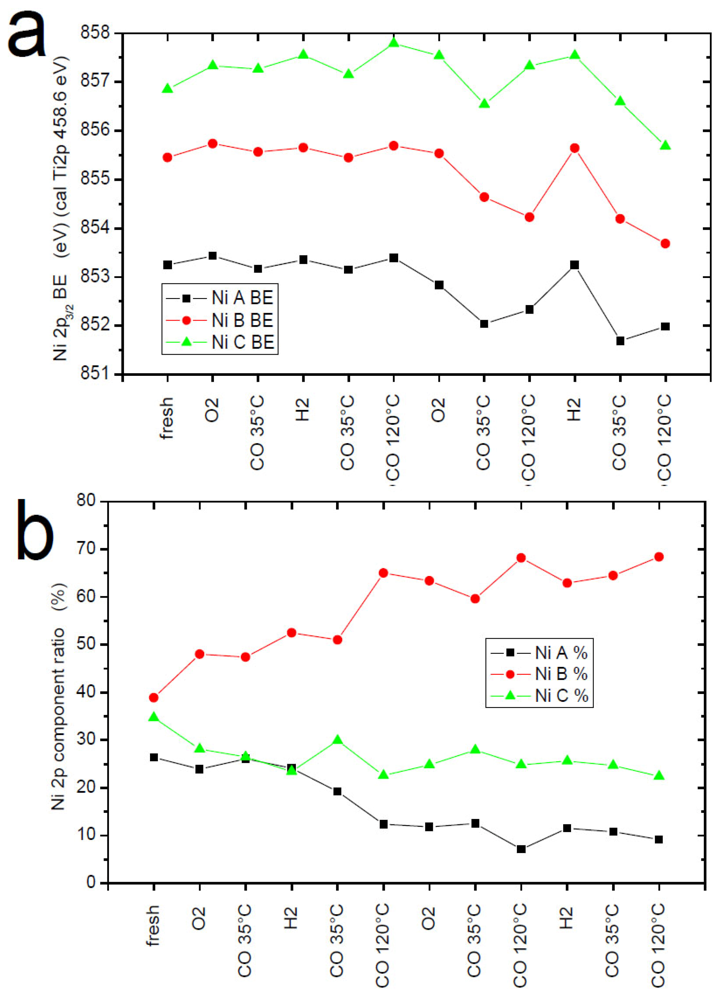
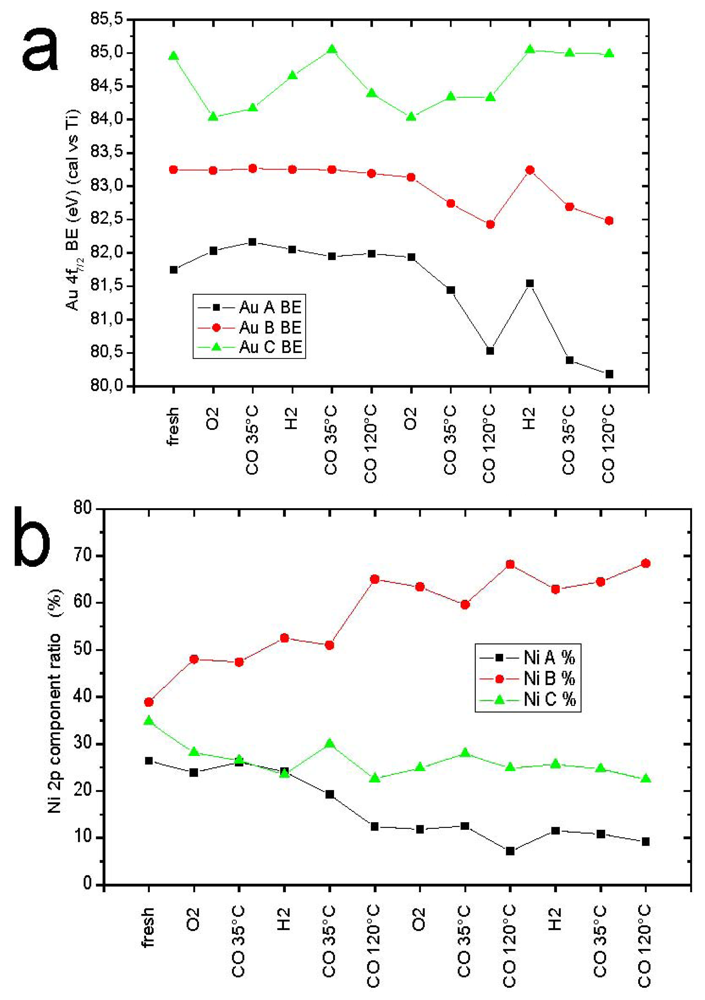
1. Introduction
It is hard to find a topic in the history of heterogeneous catalysis that has made such extreme progress over the last 20 years as is the case of gold catalysis. Regardless of the tiny harbingers published at the beginning of the 20th century [
1,
2] and the amazing discoveries presented in 1969 [
3] and 1973 [
4], until the mid of 1980s in the 20th century, scientists looked with disappointment at gold as an element which was useful in catalysis only as a diluent for other noble metals [
5]. However, since the revolutionary paper of Haruta [
6] in 1987, only within a few years, fresh air could be enjoyed in Japanese hospitals and restrooms thanks to the applied catalytic converters, burning VOC’s (Volatile Organic Compounds) on a Au/Fe
2O
3 catalyst bed [
7,
8] and some years later the first pilot plant for the gold catalyzed one-step direct production of methyl glycolate from ethylene glycol and methanol was started [
9]. Until today the activity of gold nanoparticles in several dozens of reactions has been proven and the scientific world knows more and more about the laws of nature ruling the catalytic system. Nevertheless, the industry can hardly wait to see reliable research showing active and stable catalysts for CO oxidation.
The problem encumbering them is the relatively low time–on–stream stability caused by the reduction of the Au oxidation state and accumulation of carbonates and formates on the gold active sites [
10]. The most common trend to solve these problems was changing the support materials from common ones (titania, iron oxides) into more expensive ones like zirconia and rare earth metal oxides [
11]. Even if such an approach led to the discovery of general rules, describing the properties of a stable catalyst, these general rules would need to be further transferred into knowledge about tailoring stable catalysts with cheaper supports. Thus, it seems that tuning titania properties by doping it with other metal oxides appears to be a good shortcut on the way to discover an applicable gold catalyst. This approach seems to be reasonable not only because of the difference in prices of the mentioned supports, but also because of the possibility to use the ample knowledge that scientists already have about titania.
It was shown that the role of the support in gold catalysts is not limited to only being a material on which gold nanoparticles can be deposited. The intrinsic properties of the support, especially its reducibility, can influence the properties of gold nanoparticles such as the size and the electronic state [
12,
13]. Also these reducible supports generate more oxygen vacancies at the Au-support interface [
14,
15], which is believed to be the center activating oxygen molecules.
The idea of this research is to modify an active catalyst (Au/TiO2) with metal oxides (Fe, Ni, Cu), having different reducibility and the ability to generate oxygen vacancies in TiO2 to tune the interactions between gold nanoparticles and the support, leading to higher catalytic activity. The presented results approach some dependencies between the type of the applied doping metal, its oxidation state, and the changes in the catalytic performance of the promoted catalyst.
2. Results and Discussion
The synthesized catalysts and their basic properties are listed in
Table 1. The nominal content of gold was 1 wt% in all cases and the content of base metals was always equimolar with respect to gold. In the case of Au1/TiO
2 and its modifications the applied support—Titania—had a surface area of 22.4 m
2g
−1 and BET measurements showed that the modifications have no influence on the specific surface area. In the case of the rest of the samples the BET surface area was 32 m
2g
−1.
Table 1.
Basic properties of the synthesized catalysts.
Table 1.
Basic properties of the synthesized catalysts.
| Catalyst | Preparation method | Nominal composition (wt%) | BET | TPRmax |
|---|
| deposition | heating atmosphere (350 °C) | Au | M | | |
|---|
| Au1/TiO2 | DP * | air | 1 | | 22.4 | – |
| NiO/Au1/TiO2 | MaGSD * | air | 1 | 0.3 | 22.4 | – |
| Ni/Au1/TiO2 | MaGSD * | 5% H2/Ar | 1 | 0.3 | 22.4 | – |
| Au1/NiO/TiO2 | GaMSD * | air | 1 | 0.3 | 22.4 | – |
| Au1/Ni/TiO2 | GaMSD * | 5% H2/Ar | 1 | 0.3 | 22.4 | – |
| Au2/TiO2 | DP * | air | 1 | | 32 | reference sample |
| Au2/NiO/TiO2 | GaMSD * | air | 1 | 0.3 | 32 | 340 °C |
| Au2/FeOX/TiO2 | GaMSD * | air | 1 | 0.28 | 32 | 90 °C |
| Au2/CuO/TiO2 | GaMSD * | air | 1 | 0.32 | 32 | 70, 135 °C |
| Au3/TiO2 | DP * | air | 1 | | 32 | – |
| Au3 + NiO/TiO2 | CoD * | air | 1 | 0.3 | 32 | – |
| Au3 + FeOX/TiO2 | CoD * | air | 1 | 0.28 | 32 | – |
| Au3 + CuO/TiO2 | CoD * | air | 1 | 0.32 | 32 | – |
2.1. Catalyst Reducibility
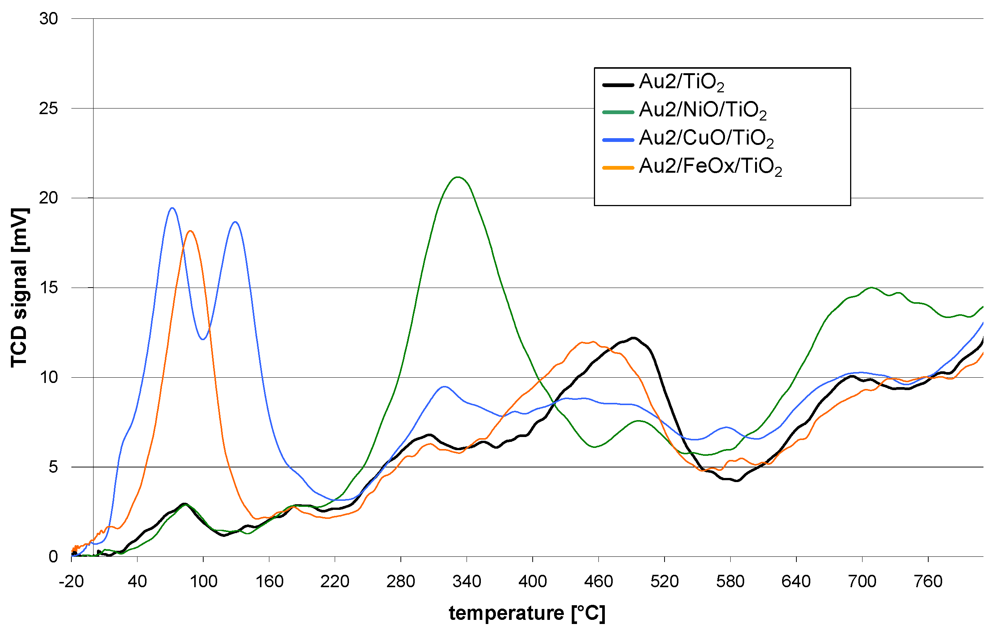
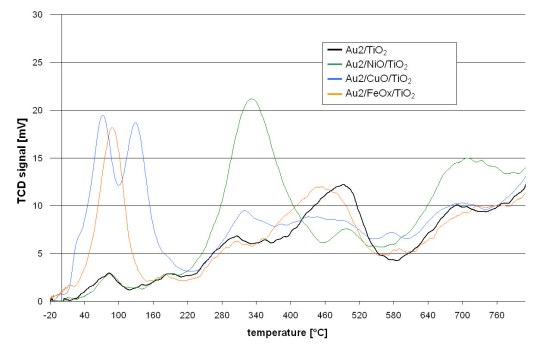
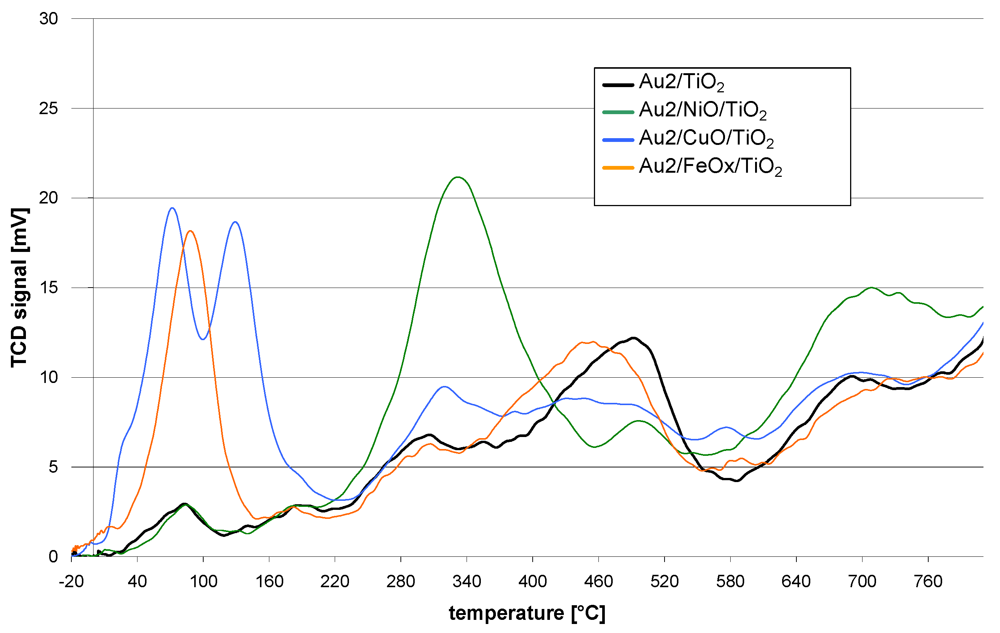
The redox properties of the catalysts are shown in
Figure 1. The course of the TPR curve of the reference catalyst (Au2/TiO
2) shows that the reduction starts just above room temperature. At further steps of the reduction process maxima can be observed at 90, 180, 300, 500 and 700 °C. The TPR curves of the promoted catalysts follow a similar course. However, they differ in the presence of additional reduction peaks, intensity or shift of the T
max with respect to the maxima observed for the reference catalyst.
Figure 1.
TPR curves of catalysts obtained by sequential deposition of base metal oxides on TiO2 followed by deposition of gold nanoparticles.
Figure 1.
TPR curves of catalysts obtained by sequential deposition of base metal oxides on TiO2 followed by deposition of gold nanoparticles.
The Au2/NiO/TiO2 TPR curve is distinguished by the presence of a peak at 340 °C and also a lower intensity of that at 500 °C and higher intensity over 600 °C.
The Au2/CuO/TiO2 TPR profile revealed that the reduction process starts at −10 °C. Further temperature increase gives two low temperature peaks at 70 and 135 °C. Then at higher temperatures (300–400 °C) and also around 600 °C, the intensity of the peaks is higher while a lower intensity in the range 420–530 °C can be observed.
The TPR process of Au2/FeOX/TiO2 starts at −15 °C. There is a well marked peak at 90 °C but none other at higher temperatures. Only the peak, observed for the reference catalyst at 500 °C, is slightly shifted by 40 °C towards lower temperatures in the case of the iron promoted catalyst.
The TPR curves presented in
Figure 1 were integrated; the integral value of the reference catalyst (Au2/TiO
2) was subtracted. On the basis of calibration data the amount of consumed hydrogen and the reduction degree was calculated. The values are shown in
Table 2.
Table 2.
Reduction degrees of promoted catalyst calculated from TPR curves.
Table 2.
Reduction degrees of promoted catalyst calculated from TPR curves.
| Promoter | Ni | Fe | Cu |
| Presumed valence | 2 | 3 | 2 |
| Reduction degree (%) | <550 °C | 550–800 °C | sum | 38 | 106 |
| 55% | 35% | 90% |
The presented data show that only copper is reduced completely. Iron reduction degree equal to 38% (which is close to 1/3) suggests that the reduction of the Fe
3+ ion proceeds only up to Fe
2+. Reduction of pure iron oxides shows that metallic iron is formed at 600 °C at the latest [
16]. The reason why iron in the catalyst is not completely reduced may be due to an incorporation of the Fe
2+ ions in the TiO
2 structure and the creation of a species having much lower reduction equilibrium constant, close to that of ilmenite [
17]. It is possible that water vapor generated through the reduction of other species inhibits the reduction of the iron containing species.
The TPR curve of the nickel promoted catalyst and the calculated reduction degree show that below 550 °C half of the nickel is reduced, but above that temperature the reduction intensity is higher than for the reference catalyst. One explanation for that fact could be simply the continuation of the reduction process of nickel oxide. It can be due to an enhanced reduction of titania owing to an increase of the concentration of hydrogen atoms, activated not only on gold nanoparticles but also on the freshly emerged metallic nickel surface.
2.2. Catalytic Results of Catalysts Obtained by Deposition of Reduced or Oxidized Nickel Species on TiO2 Before and After Gold Nanoparticle Deposition
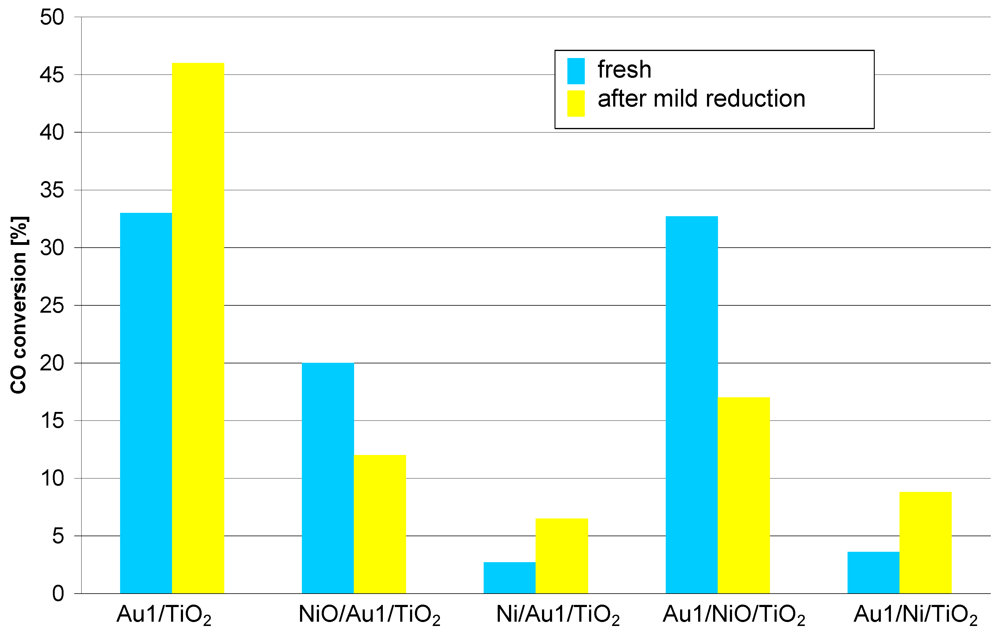
The catalytic results presented in
Figure 2 show the differences between catalysts modified with nickel. The catalysts differ in the order of gold and nickel compound deposition and also in the atmosphere (oxidizing or reducing) the catalysts were heated after nickel compound deposition.
Figure 2.
Catalytic activity at 35 °C measured for catalysts differing in the sequence of gold and nickel compound deposition and in the preparation atmosphere (oxidizing or reducing). Reaction conditions: contact time: 0.36 s, feed composition CO:O2:He ≈ 1:8:24.
Figure 2.
Catalytic activity at 35 °C measured for catalysts differing in the sequence of gold and nickel compound deposition and in the preparation atmosphere (oxidizing or reducing). Reaction conditions: contact time: 0.36 s, feed composition CO:O2:He ≈ 1:8:24.
The results show that the fresh Au1/NiO/TiO2 catalyst has a similar activity to the reference catalyst (Au1/TiO2), but the activity of the fresh NiO/Au1/TiO2 catalyst is much lower. At the same time the Ni/Au1/TiO2 catalyst shows a slightly lower activity than the Au1/Ni/TiO2 catalyst. It appears then that the deposition of gold after nickel compound results in better catalytic activity. On the other hand the catalysts prepared in reducing atmosphere (Au1/Ni/TiO2, Ni/Au1/TiO2), exhibit in both cases an activity lower than the reference catalyst. Their activity is also lower than the activity of the respective catalysts prepared in oxidizing atmosphere (Au1/NiO/TiO2, NiO/Au1/TiO2). This comparison shows that the preparation in reducing atmosphere at 350 °C generates, irrespective of the Au and Ni deposition sequence, worse catalysts, caused probably by nickel oxide and titania reduction.
The catalysts react diversely to the mild reduction process (150 °C H2, 1 h). The activity of the NiO containing catalysts decreases but the Ni containing catalysts become more active. For the Au/TiO2 reference catalyst, the activity also increases after the mild reduction. As this phenomenon is observed not only for nickel promoted catalysts, this problem will be discussed later.
2.3. Catalytic Results of Catalysts Obtained by Deposition of Base Metal Oxides (Ni, Cu, Fe) on TiO2 Followed by Gold Nanoparticle Deposition
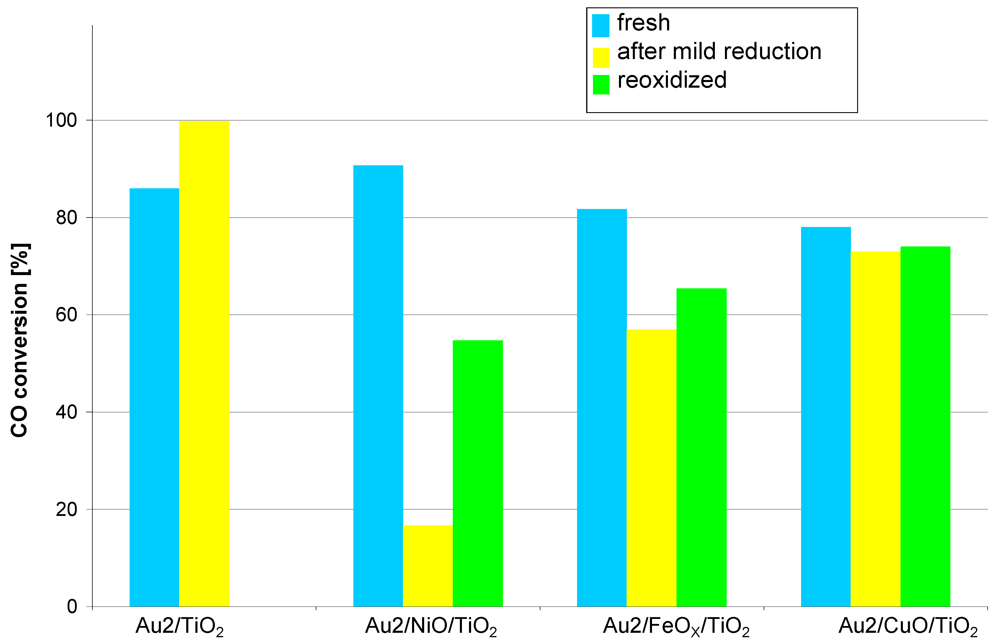
The bar graph in
Figure 3 compares the catalytic activity at 35 °C (conversion at higher temperatures was 100% in all cases) of catalysts promoted with different metals obtained by sequential deposition method. The activity data are given for fresh samples and after mild reduction at 150 °C, followed by reoxidation at 300 °C. As seen, the activity of all the fresh samples was comparable and it can be juxtaposed as follows:
Au2/NiO/TiO2 > Au2/TiO2 > Au2/FeOX/TiO2 > Au2/CuO/TiO2
Figure 3.
Catalytic activity at 35 °C measured for catalysts obtained by deposition of metal oxides (Ni, Fe, Cu) on TiO2 before gold nanoparticle deposition. Reaction conditions: contact time: 0.36 s, feed composition: CO:O2:He ≈ 1:8:24.
Figure 3.
Catalytic activity at 35 °C measured for catalysts obtained by deposition of metal oxides (Ni, Fe, Cu) on TiO2 before gold nanoparticle deposition. Reaction conditions: contact time: 0.36 s, feed composition: CO:O2:He ≈ 1:8:24.
There were several articles published in the literature concerning CO oxidation on catalysts obtained by doping of the Au/TiO
2 catalyst with base metals [
18,
19,
20,
21,
22]. In some cases, the effect is positive, sometimes it is negative or no effect is observed. The results of the group of Sheng Dai [
18] show that the doping with NiO improves the catalytic activity. In case of CuO and Fe
2O
3 doped samples, the activity was worse. However it is necessary to take into consideration the content of the dopant metal, which was approximately 10 times higher than in our research. The same content of iron oxide, presented in the paper of Moreau and Bond [
19], resulted in the same catalytic activity as the undoped catalyst. A wider spectrum of dopant content is presented in the paper of Parida
et al. [
21], which showed 5% iron content to be the best among 1, 3 and 7%. However, the NaBH
4 preparation method, which resulted in a generally low activity, also makes these results incomparable with ours. Solely the investigations published by Carrettin
et al. [
22] presented a catalyst where the surface concentration of iron was close to our catalyst. The synthesized 0.56% Fe-doped Au/TiO
2 catalysts showed several times better activity than the WGC (World Gold Council) reference catalyst (unfortunately the authors did not compare the doped sample with a self-synthesized reference catalyst). The authors ascribed the extraordinary activity to an increase of the number of oxygen defect sites reacting with molecular oxygen to form peroxide and superoxide species. Our TPR experiments suggest that the surface of the iron doped catalyst is more reducible than the surface of the reference catalyst so also in this catalyst more defect sites should be generated. However, the predicted phenomenon was not observed in our research.
The influence of the hydrogen treatment is different: the activity of the reference catalyst (Au2/TiO2) increased but that of the promoted catalysts decreased. The strongest effect was observed for the nickel promoted catalyst. In case of the iron promoted one, the impact was moderate and the activity of the copper promoted catalyst decreased only slightly. The following reoxidation process improved all promoted catalysts but the initial activity was not restored for any of them.
2.4. Catalytic Results of Catalysts Obtained by Co-deposition of Base Metal Oxides with Oxidized Gold Compounds on TiO2
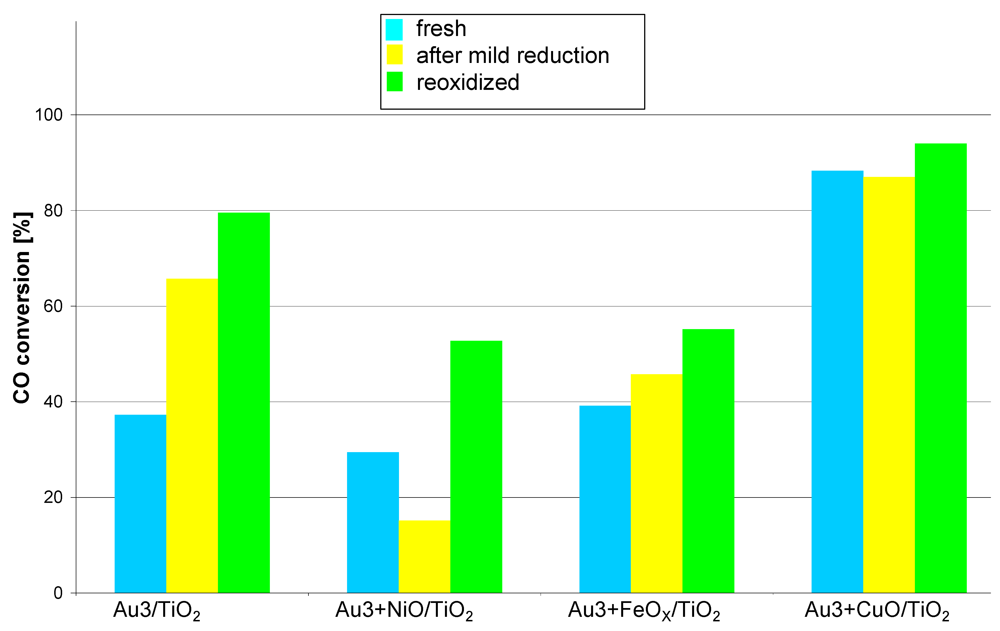
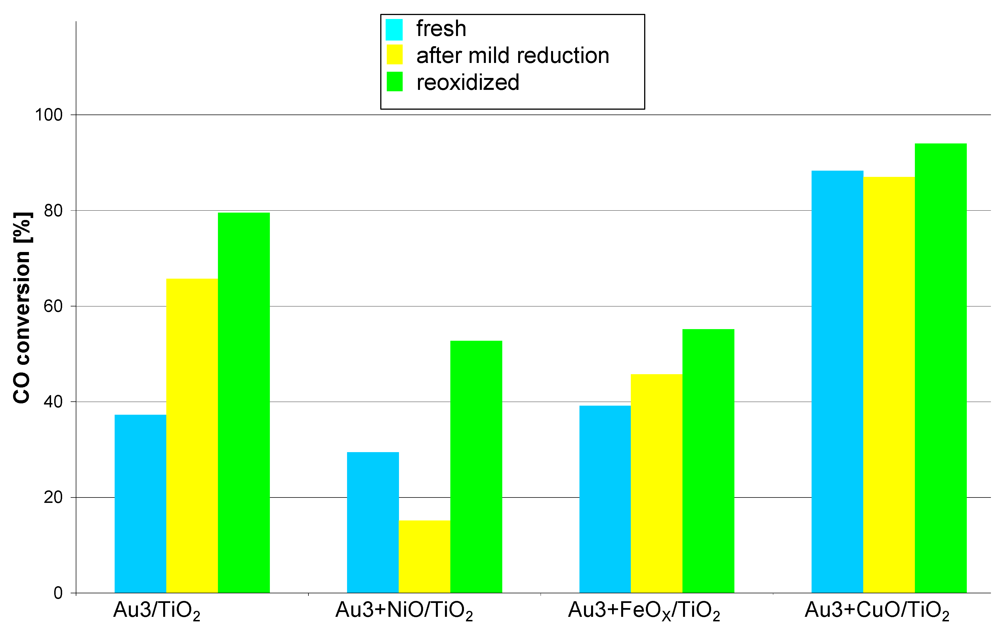
The activity of the co-deposited fresh catalysts, measured at 35 °C (
Figure 4) is generally much lower than that of the catalysts prepared by sequential deposition. The copper promoted catalyst surpasses the rest of that series, showing a higher conversion of 20%. The conversions measured at higher temperatures kept the same dependencies between the samples as these measured for 35 °C so the data were not plotted.
The mild reduction process (150 °C, H2/Ar, 1 h) significantly decreased the activity only in the case of Au3 + NiO/TiO2 while in the case of the copper promoted one (Au3 + CuO/TiO2) it remained practically stable. In contrast, the activity of the other catalysts clearly increased (Au3/TiO2, Au3 + FeOX/TiO2). The reoxidation process increased the activity in all cases especially that of Au3/TiO2, Au3 + NiO/TiO2, Au3 + FeOX/TiO2. The increase after the mild reduction, in the case of copper promoted catalysts, was hardly noticeable. This catalyst series has an exceptional feature i.e., the activity increases after treatments in various conditions. This behavior suggests that the active phase of the catalyst (Au + MOX) forms during individual steps of the experiment (i.e., mild reduction, reoxidation).
Figure 4.
Catalytic activity at 35 °C measured for catalysts obtained by co-deposition of gold and base metal compounds (Ni, Fe, Cu) on TiO2. Reaction conditions: contact time: 0.36 s, feed composition: CO:O2:He ≈ 1:8:24.
Figure 4.
Catalytic activity at 35 °C measured for catalysts obtained by co-deposition of gold and base metal compounds (Ni, Fe, Cu) on TiO2. Reaction conditions: contact time: 0.36 s, feed composition: CO:O2:He ≈ 1:8:24.
2.6. The Influence of the Mild Reduction Process (150 °C, H2/Ar, 1 h)
It has been shown in many articles that treatments in oxidizing or reducing atmospheres had a remarkable impact on gold catalysts. Some of these papers claim a positive effect of the hydrogen treatment; others report an activity decrease after this treatment [
23,
24,
25].
The presented catalytic results show that the mild reduction process has also a marked influence on all catalysts’ activities. Promoted catalysts, obtained by sequential deposition method and prepared in oxidizing atmosphere, lost their activities while the catalysts prepared in reducing atmosphere improved after this process (
Figure 2 and
Figure 3). However, the unpromoted catalysts of each series (Au1, Au2, Au3) were enhanced.
The co-deposited catalysts reacted more intricately to the mild reduction process: it was observed that it decreased significantly the activity only in the case of Au
3 + NiO/TiO
2, for the iron promoted catalyst (Au
3 + FeO
X/TiO
2) the activity increased, while in the case of the copper promoted one (Au
3 + CuO/TiO
2), it remained practically stable. It was stated in
Section 2.4 that the active phase of the co-deposited catalysts (Au + MO
X) develops along individual steps of the experiment (
i.e., mild reduction, reoxidation). From this it follows that the performed hydrogen treatment changes the catalyst surface (as is observed for the sequentially deposited catalysts) as well as generating the aforementioned active phase development. Hence, due to the active phase development, this catalyst series does not provide reliable data about the changes on the catalyst surface so the behavior of the co-deposited catalysts will not be further taken into consideration.
The first thing that needs to be dealt with is the different influence of hydrogen on the unpromoted catalysts and the promoted ones prepared in oxidizing atmosphere. A possible explanation of this difference is the formation of surface titanate phases [
26] in the promoted catalysts, and these titanate phases are less reducible than anatase, so the formation of oxygen vacancies is more difficult. Since the mild reduction has the strongest influence on the nickel promoted catalyst, weaker on iron and the weakest on the copper promoted one, it can be concluded that these metals are incorporated into the TiO
2 to a different extent. The higher the activity decrease, the more the dopant metal incorporated into the TiO
2 structure. It is also possible that the activity decrease is not a question of higher or lower incorporation, but rather a feature of the particular titanate phases and their reducibility.
Additional questions provide the catalytic results of catalysts obtained by the preparation in reducing atmosphere. As shown in
Figure 2, the influence of the mild reduction process is positive, which makes these catalysts similar to the unpromoted one.
A possible explanation for the diverse behaviors of all the investigated samples could be the twofold influence of the mild reduction on the catalytic system. Firstly, hydrogen reduces Au cations creating a more active species [
27,
28] or generates hydroxyl groups on the gold nanoparticle perimeter [
29,
30], resulting in an activity increase. Secondly, hydrogen reduces surface oxygen species making the support unable to donate oxygen species [
31,
32,
33], which results in an activity decrease. Based on these two assumptions, it can be suggested that, in the case of the promoted catalysts prepared in oxidizing atmosphere, both phenomena occur. In each case hydrogen generates a positive effect according to the first assumption. However depending on the aforementioned titanates formation and their different content or reducibility, hydrogen decreases the catalytic activity. In these catalysts containing the dopant metal in oxidized form, both phenomena compete, so the resultant activity depends on the extent of these two particular phenomena.
The unpromoted reference catalyst which does not contain any titanate phases is not negatively influenced due to the support reduction so the only effect which is observed is the positive effect. On the other hand, in the case of catalysts prepared in reducing atmosphere (350 °C), the support underwent a deep reduction generating a surface structure which is less abundant in free oxygen species. Then, during the mild reduction process (150 °C), the support cannot be reduced anymore, so the hydrogen treatment can only influence gold nanoparticles or generate hydroxyl groups (according to the first assumption).
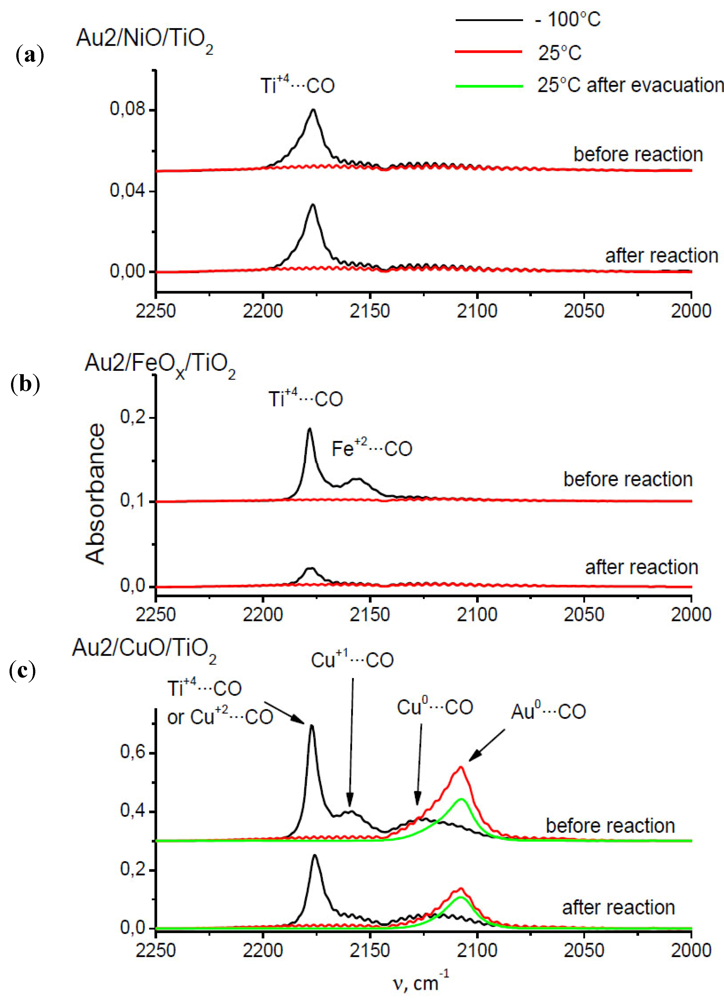
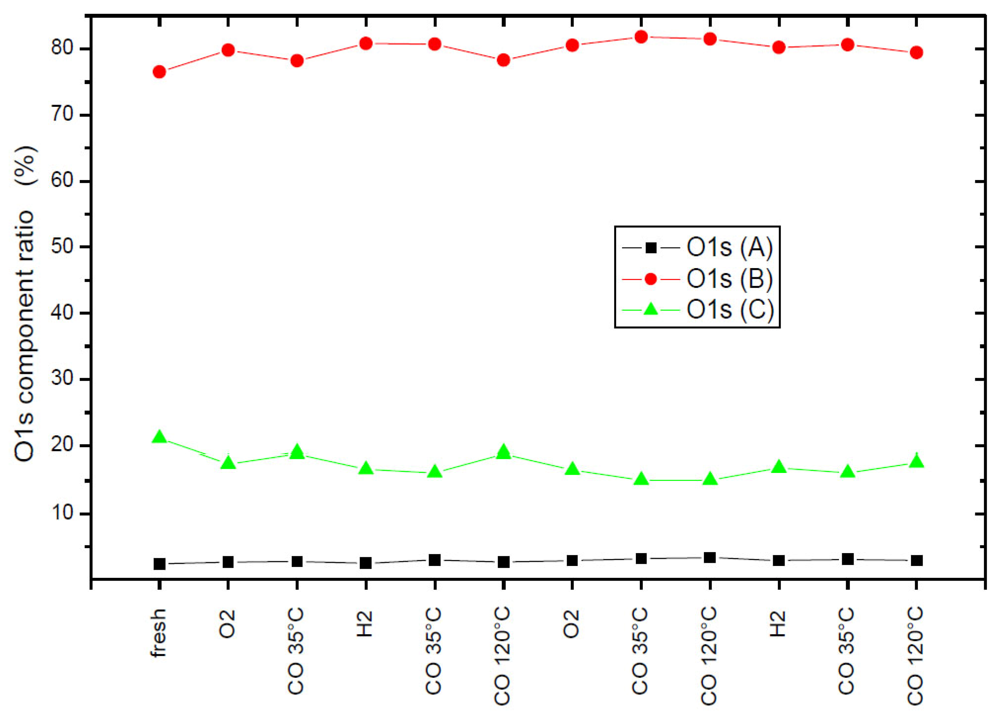
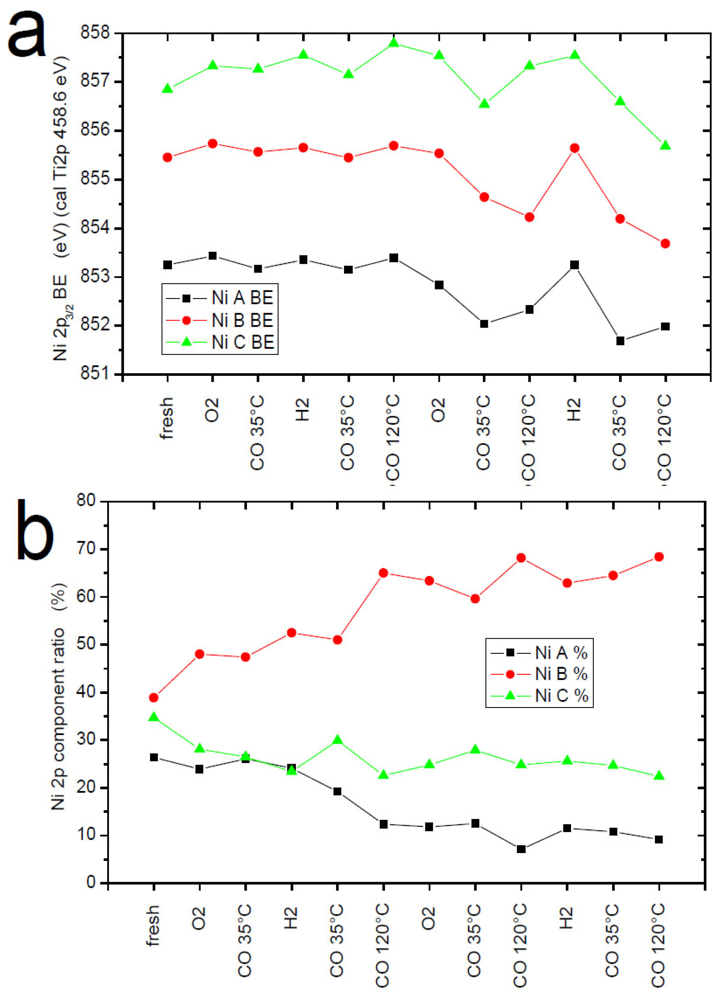
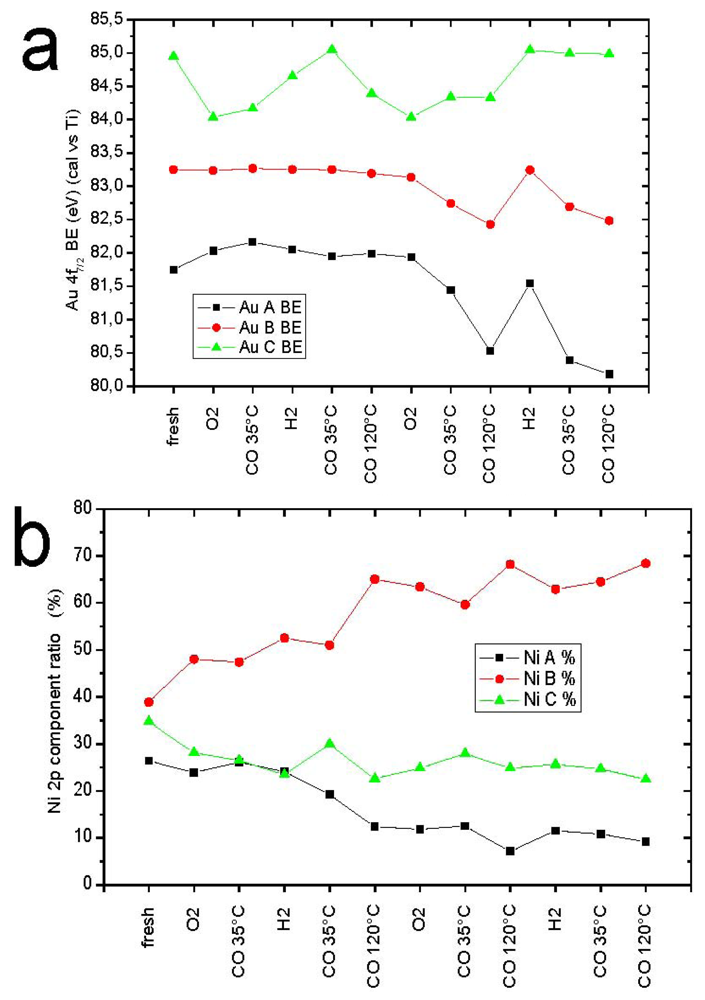
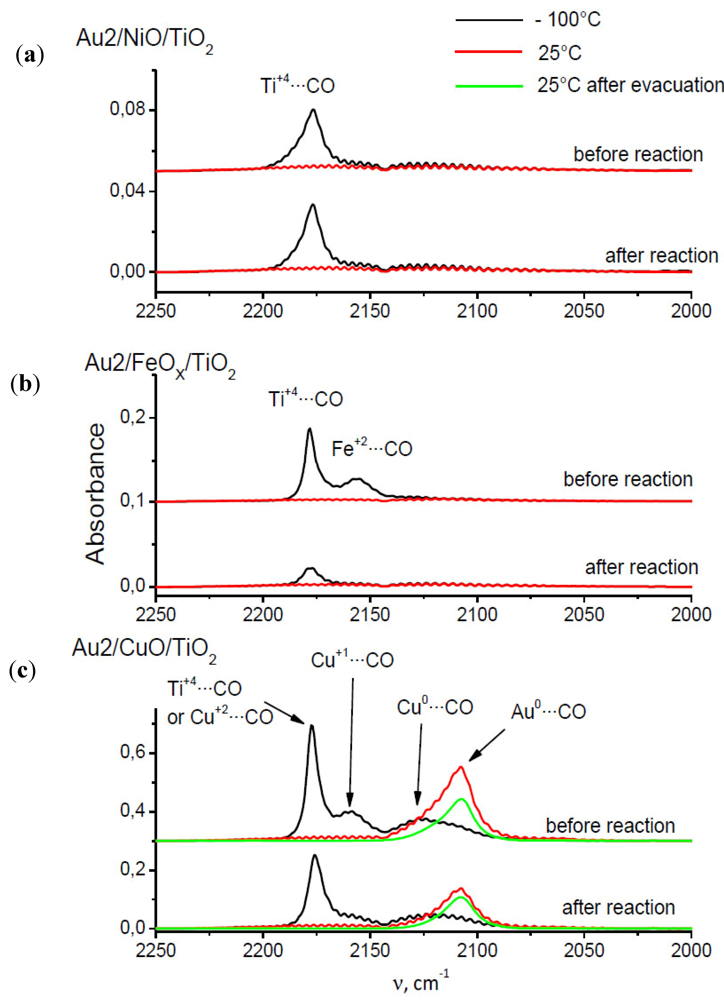
2.7. Catalysts Surface Changes Observed with FTIR
Further data indicating the changes inside the support were obtained by FTIR. The spectra collected for Au2/NiO/TiO
2, Au2/FeO/TiO
2, Au2/CuO/TiO
2 samples are plotted in
Figure 8. Each diagram consists of two sets of spectra where the upper one refers to the sample before catalytic reaction (heating at 120 °C in a CO–O
2–He mixture) and the lower one, after it. According to the literature [
34,
35], the observed frequencies were attributed to CO adsorption on the following centers: 2175 cm
−1—weak Lewis centers on Ti
+4 (β-center) [
35] and/or Cu
+2, 2158 cm
−1—Cu
+1, 2156 cm
−1—Fe
+2, 2127 cm
−1—Cu
0, 2108 cm
−1—Au
0.
Figure 8.
Comparison of CO adsorption FTIR spectra of Au/TiO2 catalysts promoted by (a) Ni; (b) Fe; (c) Cu.
Figure 8.
Comparison of CO adsorption FTIR spectra of Au/TiO2 catalysts promoted by (a) Ni; (b) Fe; (c) Cu.
The presented spectra show that CO adsorbs on the Ti+4 β-centers (2175 cm−1) at −100 °C in the case of each catalyst, but the peak intensity is different for each catalyst and also depends on whether CO was adsorbed on the catalyst before or after reaction. For the Au2/NiO/TiO2 catalyst, the peak intensity before and after the catalytic reaction, is comparable and amounts to about 0.036 a.u. The height of this peak in the case of the Au2/FeO/TiO2 catalyst before the reaction is about three times higher than for Au2/NiO/TiO2, but after the reaction it is comparable to the peak height of Au2/NiO/TiO2. For the Au2/CuO/TiO2 catalyst, the intensity of this peak cannot clearly be determined due to the fact that the vibration of CO adsorbed on Cu+2 has a similar frequency. However, it can be concluded that the intensity of the band before the reaction is nearly 1.6 times higher than after the reaction.
The spectra of CO adsorption on the catalyst Au2/NiO/TiO
2 do not show any characteristic vibrations, with the exception of the aforementioned CO–β (Ti
+4) vibration centers. The adsorption of CO on Au2/FeO/TiO
2 leads to the appearence of the 2162 cm
−1 band which can be attributed to the interaction of CO with Fe
+2. In the Au2/CuO/TiO
2 spectra, besides the CO–β-centers vibration, two additional bands 2168 cm
−1 and 2127 cm
−1 were recorded at −100 °C, which correspond to the CO–Cu
+1 and CO–Cu
0 vibrations respectively. At room temperature a broadened peak appears, consisting probably of two vibrations 2127 cm
−1–Cu
0 and 2108 cm
−1, attributed to CO–Au
0 vibration [
35]. For this catalyst, such dependencies can be stated:
(a) CO–Cu0 vibration is observed at both temperatures (−100 °C and room temperature).
(b) the assignment of 2108 cm−1 frequency to CO–Au0 vibration, although it has its justification in the literature, seems to be questionable because at the same time it should be observed for other catalysts.
(c) the ratio of peaks at 2175 cm−1 recorded in the spectra before and after the reaction is approximately equal to the ratio of peaks at 2108 cm−1 in the spectra collected before and after the reaction, which may indicate that the catalytic reaction leads to a decrease of the amount of both adsorption centers in a uniform extent.
A general feature, which is common for the iron and the copper promoted catalyst, is the lower absorbance before reaction for all frequencies. The nickel promoted catalyst exhibits low peaks before and after reaction.
2.8. Comparison of Preparation Methods
Comparison of the preparation methods showed that the gold after metal sequential deposition method is better than the metal after gold sequential deposition method and, in most cases, is also better than the co-deposition method. However the co-deposited copper promoted catalyst exhibited the highest activity of all the prepared samples.
3. Experimental Section
Titanium dioxide (100% anatase [
36]) from TIOXIDE with specific surface area of 32 m
2g
−1 or 22.4 m
2g
−1 was used as support. Au/MO
X/TiO
2 and MO
X/Au/TiO
2 systems were synthesized by sequential deposition of each component but in different order. Gold nanoparticles were deposited through deposition-precipitation method (DP), using ammonium hydroxide at pH = 9.0 as precipitating agent and chloroauric acid as Au source. Metal oxides were deposited by impregnation of the support with nitrates (Cu, Fe) or ammonates (Ni) followed by heating at 350 °C in oxidizing or reducing atmosphere. The nominal concentration of gold is 1 wt% and the amount of the additive metal is equimolar.
Au-MOX/TiO2 systems were obtained by co-deposition of gold and base metal compounds on the support. The applied method was similar to the DP method, but a mixture of chloroauric acid and a base metal chloride was used instead of chloroauric acid only.
The specific surface areas of the samples were determined with the use of Quantachrome–Autosorb-1 instrument. Samples were degassed at 150 °C under vacuum (10−6 mmHg). N2 was used as adsorbate at 77.4 K. BET isotherm was used to determine the specific surface area.
The surface composition and texture study was performed using a high resolution scanning electron microscope JEOL JSM-7500F with extra-INCA EDS PentaFETx3. Secondary electron imaging detector-SEI was used to determine the catalyst morphology, for gold particles imaging on the catalyst surface Back-Scattered Electron detector-BSE was used.
Catalyst reducibility measurements were carried out with the use of Quantachrome Chembet-3000 apparatus, in a flow of 30 mL/min of 5% H2/Ar mixture. The amount of the sample (0.68 g) was selected in such a way that limitations by heat and mass transfer were avoided and good resolution was obtained. The heating rate was 10 °C/min. Water vapor was removed by freezing. Reduction peak maximum Tmax was the reducibility describing factor.
FT-IR spectra were recorded in absorption mode on a Bruker Tensor 27 spectrometer equipped with a MCT detector, with a spectral resolution of 2 cm−1. The cuvette was connected online to a gas feed system and equipped with a temperature control system to perform measurements from −100 to 150 °C in controlled atmosphere at pressures of 10−4 to 1 bar. The measurement was carried out according to the following procedure:
Degassing under vacuum at 150°C;
CO adsorption at −100°C and collecting FTIR spectra;
Warming up up to room temperature and collecting FTIR spectra;
Degassing and collecting FTIR spectra;
Catalytic reaction in 2%CO, 2%O2/He atmosphere at 100°C for 4h;
Collecting FTIR spectra like in points 2–4.
The catalytic experiments were performed in a stainless steel reactor (i.d. = 8 mm, h = 19 mm) connected online with an SRI 8610C Gas Chromatograph equipped with two columns for reactant analysis. Helium Ionization Detector (HID) was used for CO an O2 analysis. The elution of them was performed on a packed column with 13X molecular sieves. CO2 analysis was performed on a Porapak Q column connected to a TCD detector. The gas feed was controlled by β-ERG Mass Flow Controllers. The feed gas mixture total flow was 58 mL/min and its composition was CO—3%, O2—24%, He—73%. The amount of the catalyst was approximately 0.33 g. The catalytic experiments were performed according to the following steps:
Standardizing (oxidation at 150°C in O2/He for 1h);
Catalytic CO oxidation at 35, 50 and 60°C;
Mild reduction at 150°C in H2/He for 1h;
Catalytic CO oxidation at 35, 50 and 60°C;
Reoxidation at 300°C in O2/He for 1h;
Catalytic CO oxidation at 35, 50 and 60°C.
In case of Au1/TiO
2 catalyst and its modification (
Figure 2), the steps 1–4 were only performed and the catalytic activity was measured only at 35 °C. All of the catalyst prepared by gold after metal sequential deposition method (
Figure 3) exhibited 100% conversion at temperatures 50 and 60 °C so the diagram shows only the results obtained at 35 °C.
XPS studies were carried out in an ultrahigh vacuum (UHV) multichamber system equipped with a high-pressure reactor (Prevac). A pellet of powdered sample was placed on a holder containing resistive heater covered with ceramic coating. The experiments were carried out according to the following procedure:
Standardizing (oxidation at 150°C in O2/He for 1h);
Catalytic CO oxidation at 35°C;
Mild reduction at 150°C in H2/He for 1h;
Catalytic CO oxidation at (a) 35 °C (b) 120°C;
Reoxidation at 300°C in O2/He for 1h;
Catalytic CO oxidation at (a) 35°C (b) 120°C;
Mild reduction at 150°C in H2/He for 1h;
Catalytic CO oxidation at (a) 35°C (b) 120°C.
The sample was not active until the catalytic reaction at 120 °C was performed, so the steps 7 and 8 were carried out as a repetition. For the standardizing and reoxidation processes a mixture of 5% O
2/He 30 mL/min was used. The mild reduction process was carried out with 33% H
2/He. The feed mixture used in the CO oxidation process contained 2% CO, 2% O
2 in He. At every step the gas flow was 30 mL/min and the reaction temperature was monitored by a thermocouple attached to the sample. The reaction progress was followed qualitatively by mass spectrometer (SRS 200), linked to the reactor by registering masses 2, 12, 16, 18, 32, 40, 44 and 48. After each treatment, the sample was cooled down, the reactor was evacuated to 2·× 10
−7 mbar and then the sample was transferred to the XPS spectrometer chamber (5 × 10
−10 mbar). The XPS measurements were performed with Al Kα (1486.6 eV) X-ray source and hemispherical analyzer R4000 (Gammadata Scienta). The spectrometer was calibrated according to ISO 15472:2001. The energy resolution of the system, measured as a full width at half maximum (FWHM) for Ag 3d
5/2 excitation line, was 0.9 eV. Titanium 2p
3/2 core excitation at binding energy of 458.6 eV, was taken as an internal standard. The assignment of the obtained BE values was done basing on references [
37,
38,
39].
The spectra were processed in CasaXPS 2.3.12 program. In the spectra, the background was approximated by a Shirley profile. The spectra deconvolution into a minimum number of the components was done by application of the Voigt-type line shapes (70:30 Gaussian/Lorentzian product).
4. Conclusions
The presented results show that doping of the gold/titania catalyst with copper, iron and nickel results in obtaining samples with quite diverse properties. Firstly, they reveal different redox properties. The reducibility of the copper promoted gold catalyst is the highest, the iron promoted one has a slightly lower reducibility and that of the nickel promoted catalyst is the lowest. The redox behavior was attributed to the extent of incorporation of the dopant metals into the titania structure.
The activity of the fresh catalysts was comparable (with the exception of the catalysts prepared by Metal after Gold Sequential Deposition) but the mild reduction process demonstrated distinct differences: nickel promoted catalysts lost their activities significantly, while the iron promoted ones lost only slightly; and the influence on the copper promoted one is hardly noticeable. It was concluded that catalytic activity depends considerably on the reducibility of the supports. The formation of surface titanates decreases the amount of free oxygen species.
The FTIR investigation showed a higher concentration of active sites adsorbing CO on iron and copper promoted catalysts. However, the catalytic reaction reduces the amount of these sites.
From the three presented preparation methods, the gold after metal sequential deposition method seems to be the most effective.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}