
Production of Sustainable Synthetic Natural Gas from Carbon Dioxide and Renewable Energy Catalyzed by Carbon-Nanotube-Supported Ni and ZrO2 Nanoparticles

 , , , , ,
, , , , , 
 and
and
Abstract

1. Introduction
2. Results and Discussion
2.1. Catalyst Characterization
2.1.1. Elemental Composition
2.1.2. The Textural Properties of the Samples
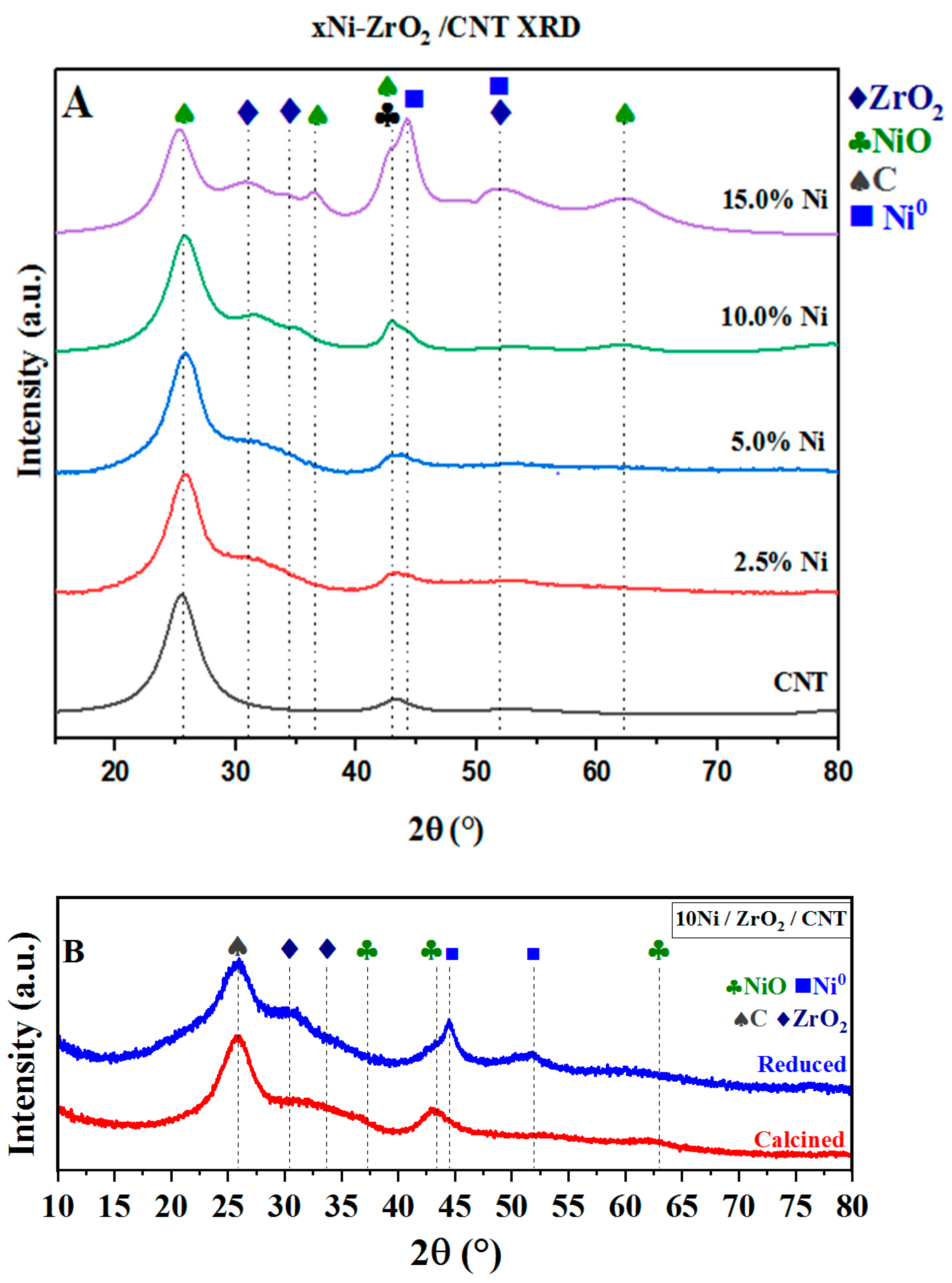
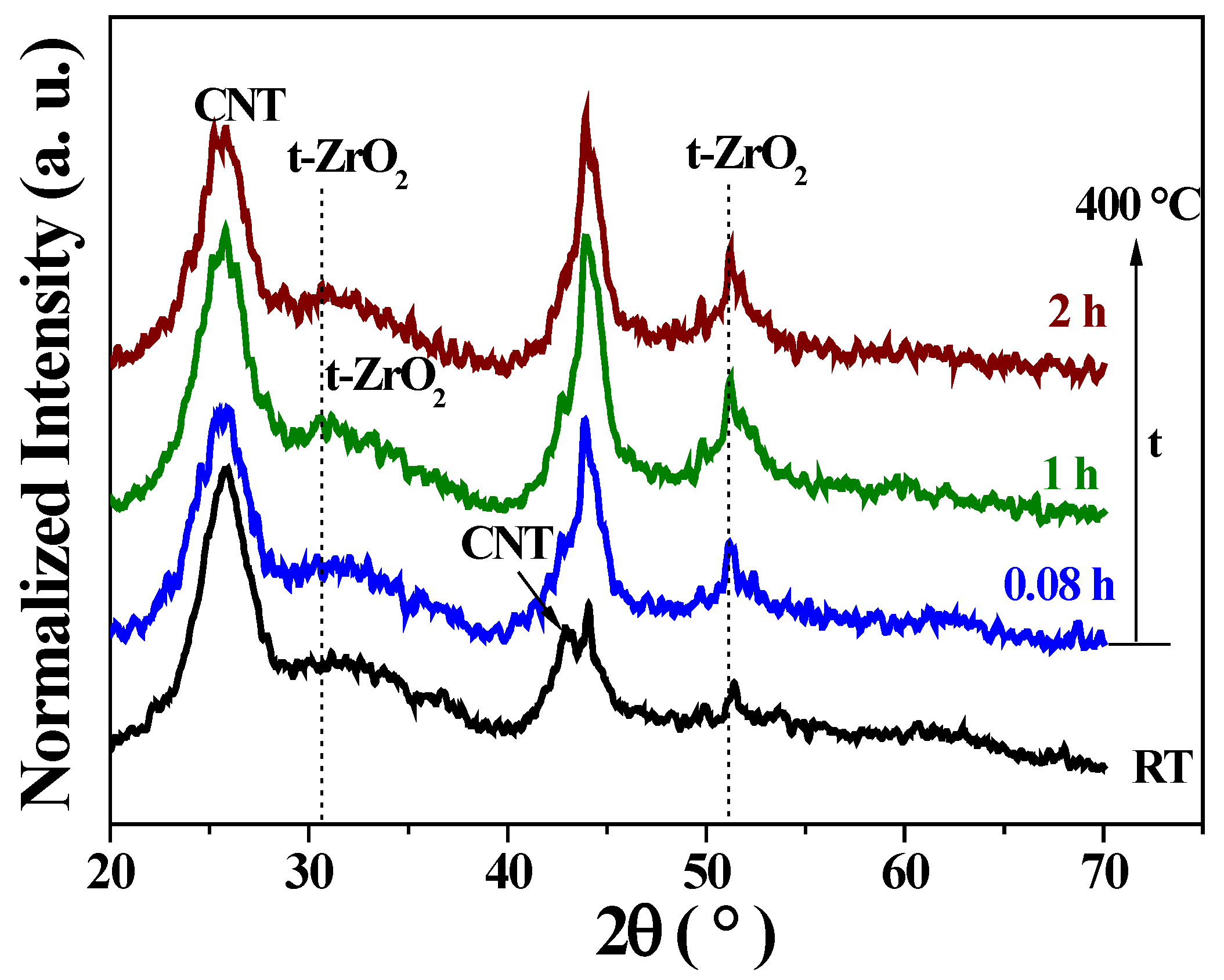
2.1.3. X-Ray Diffraction
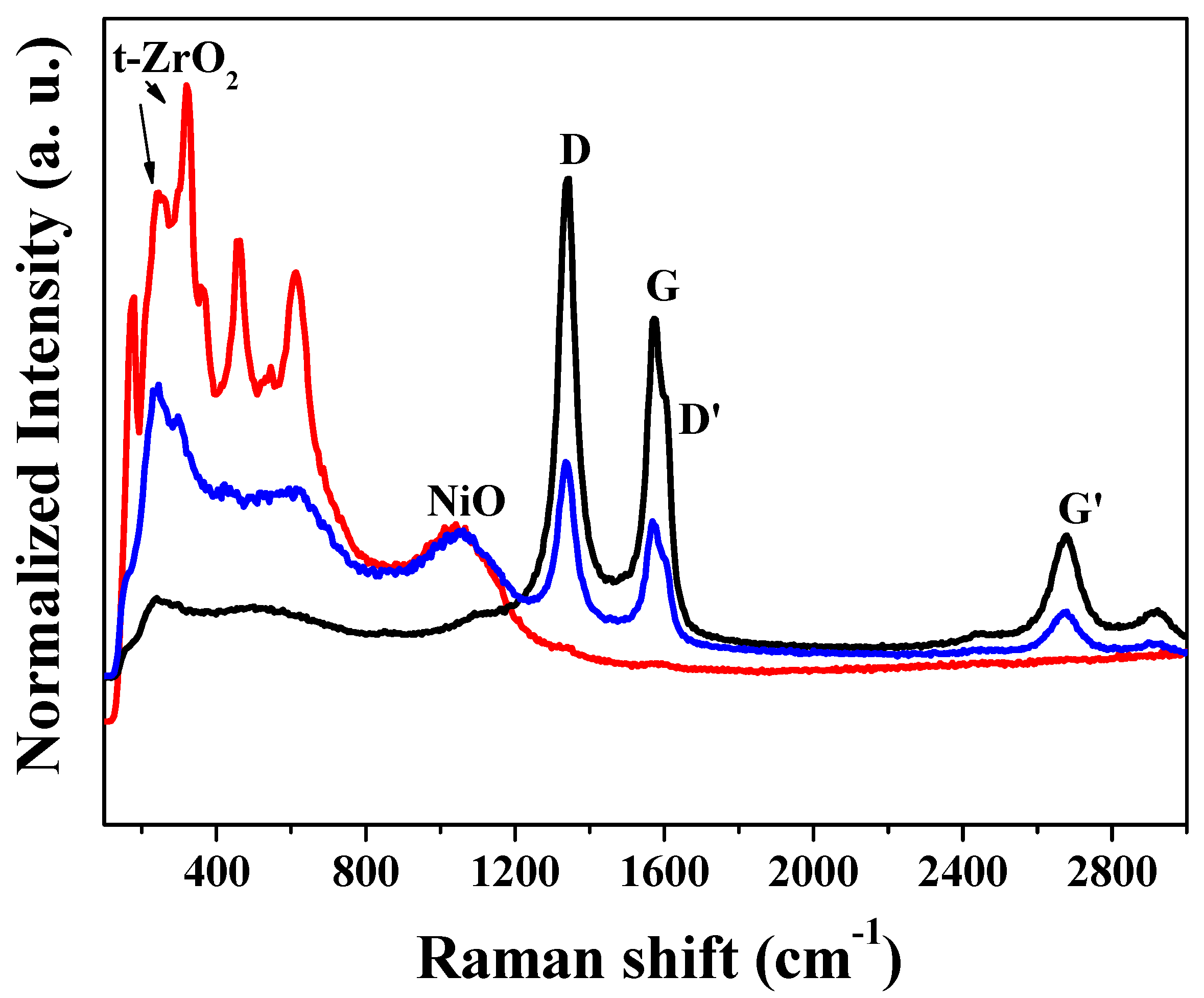
2.1.4. Raman Spectroscopy
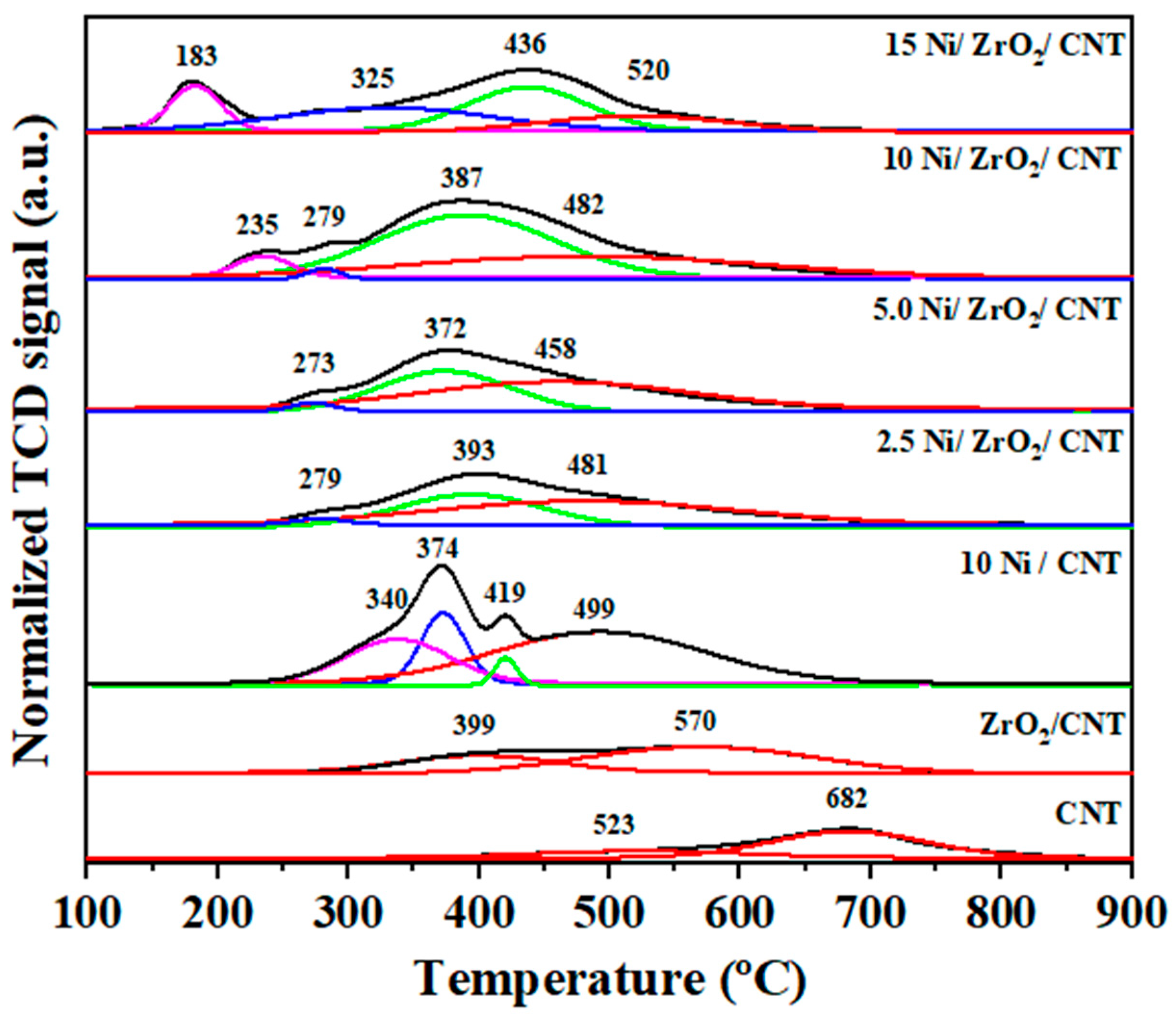
2.1.5. Temperature-Programmed Reduction (H2-TPR)
2.1.6. Transmission Electron Microscopy (TEM)
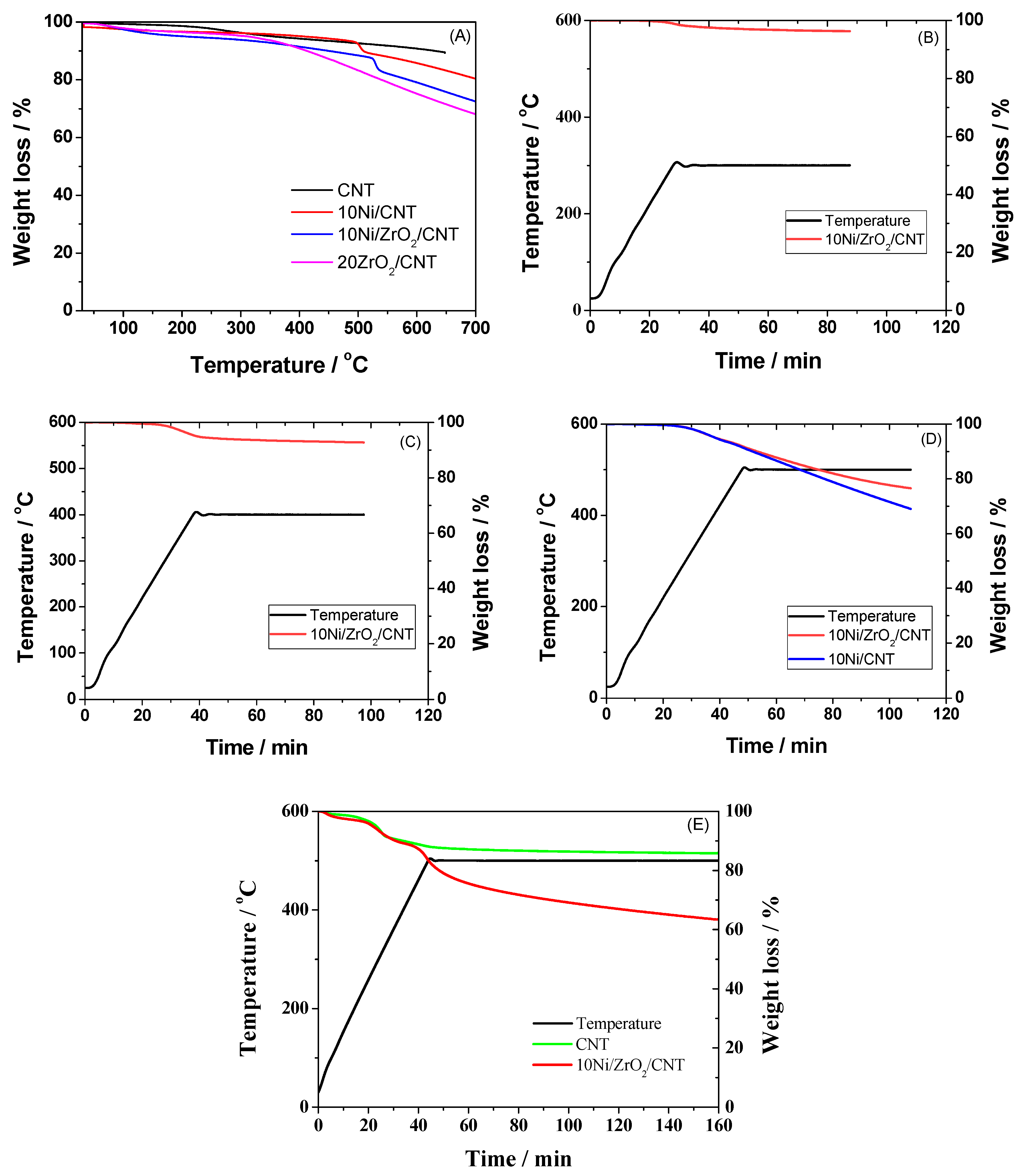
2.1.7. The Thermogravimetric Analysis
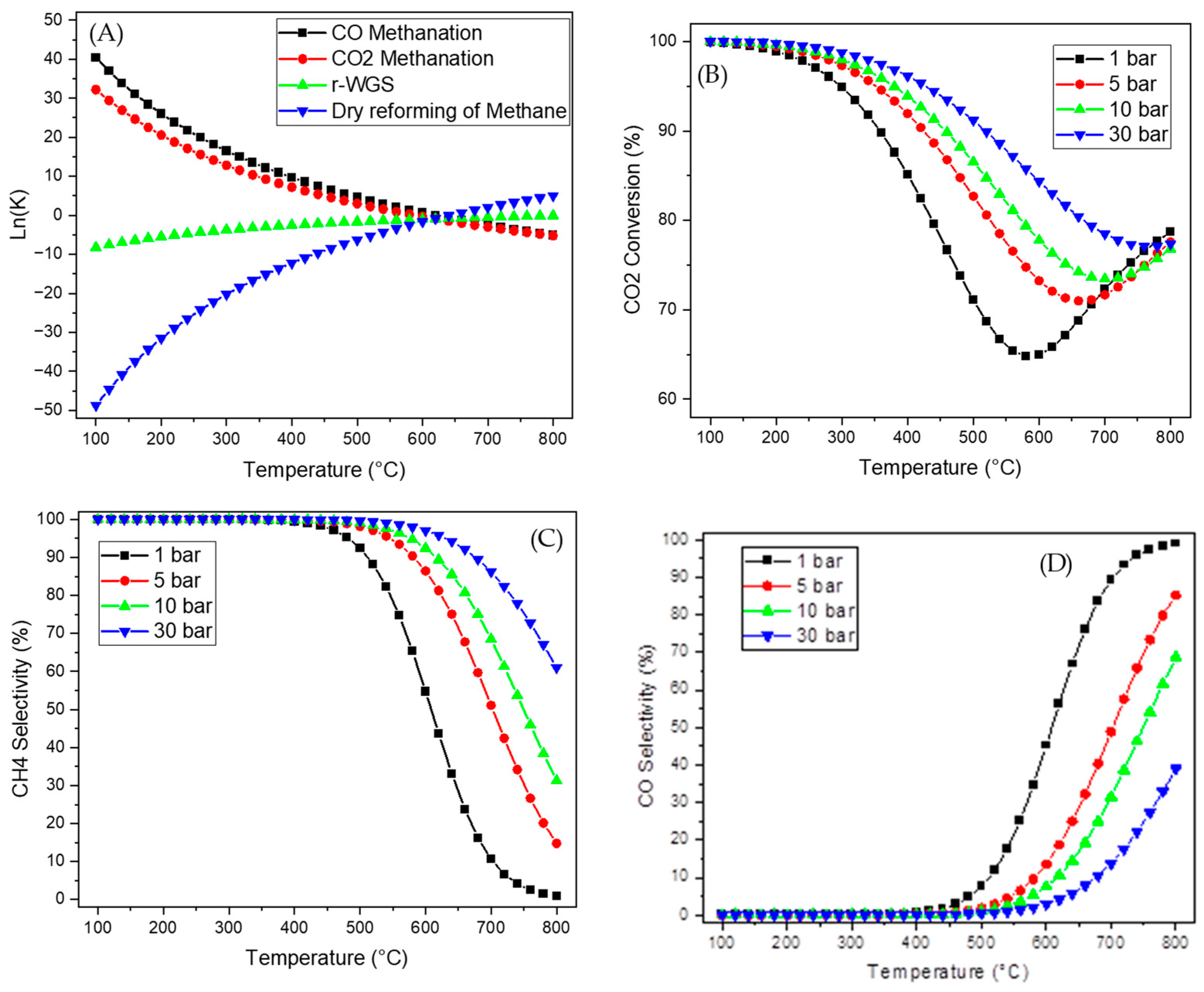
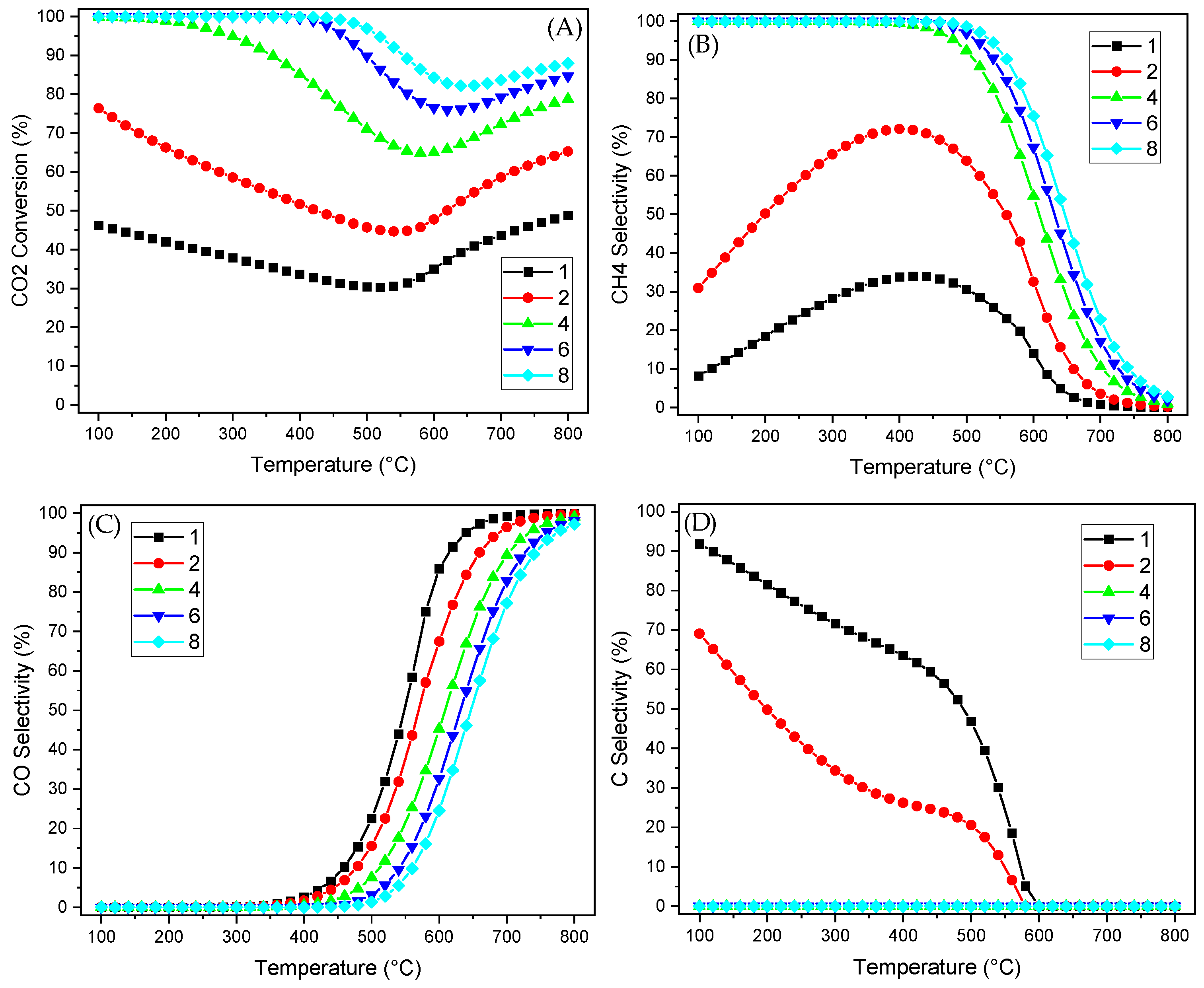
2.2. Thermodynamic Equilibrium
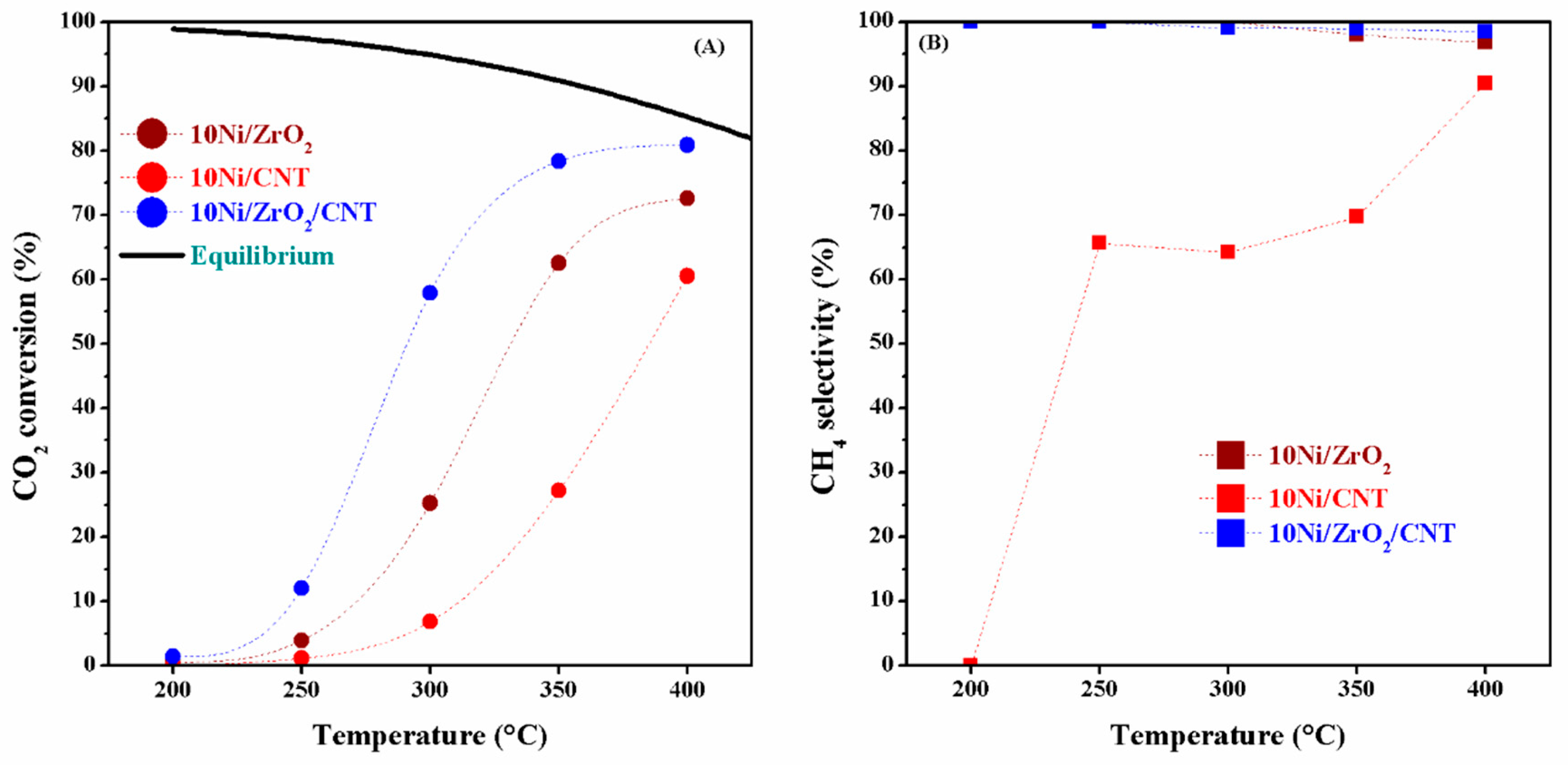
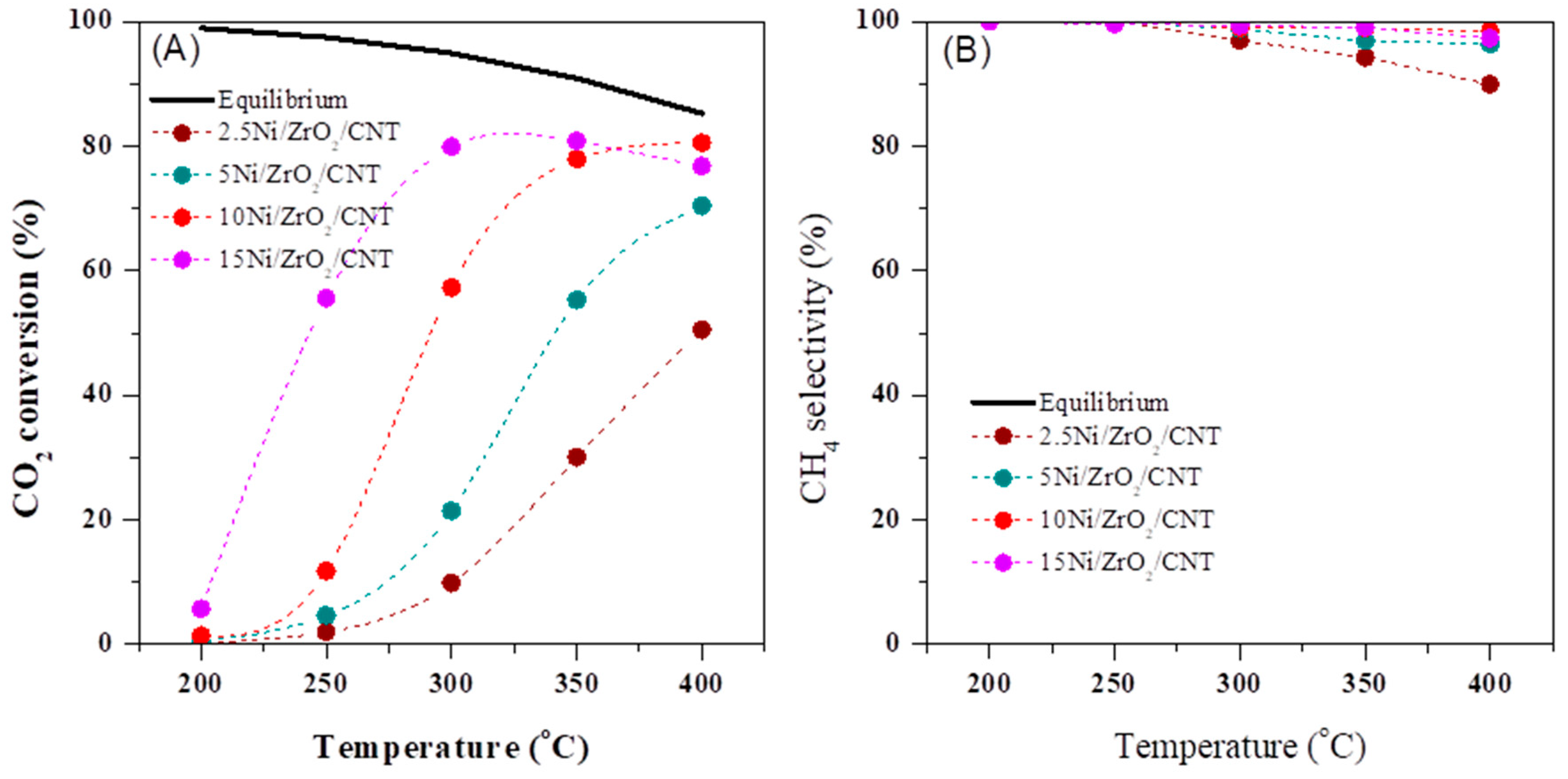
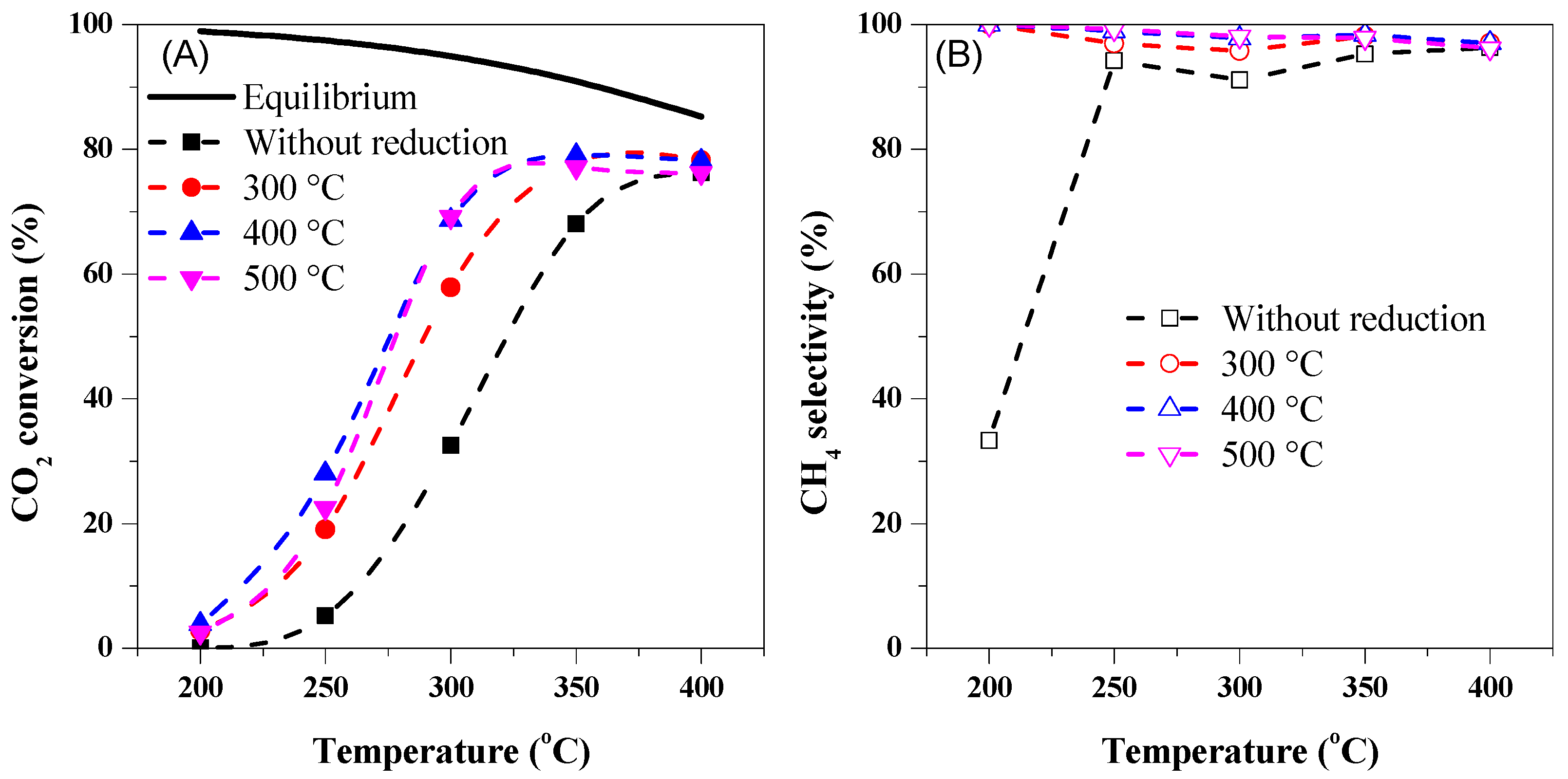
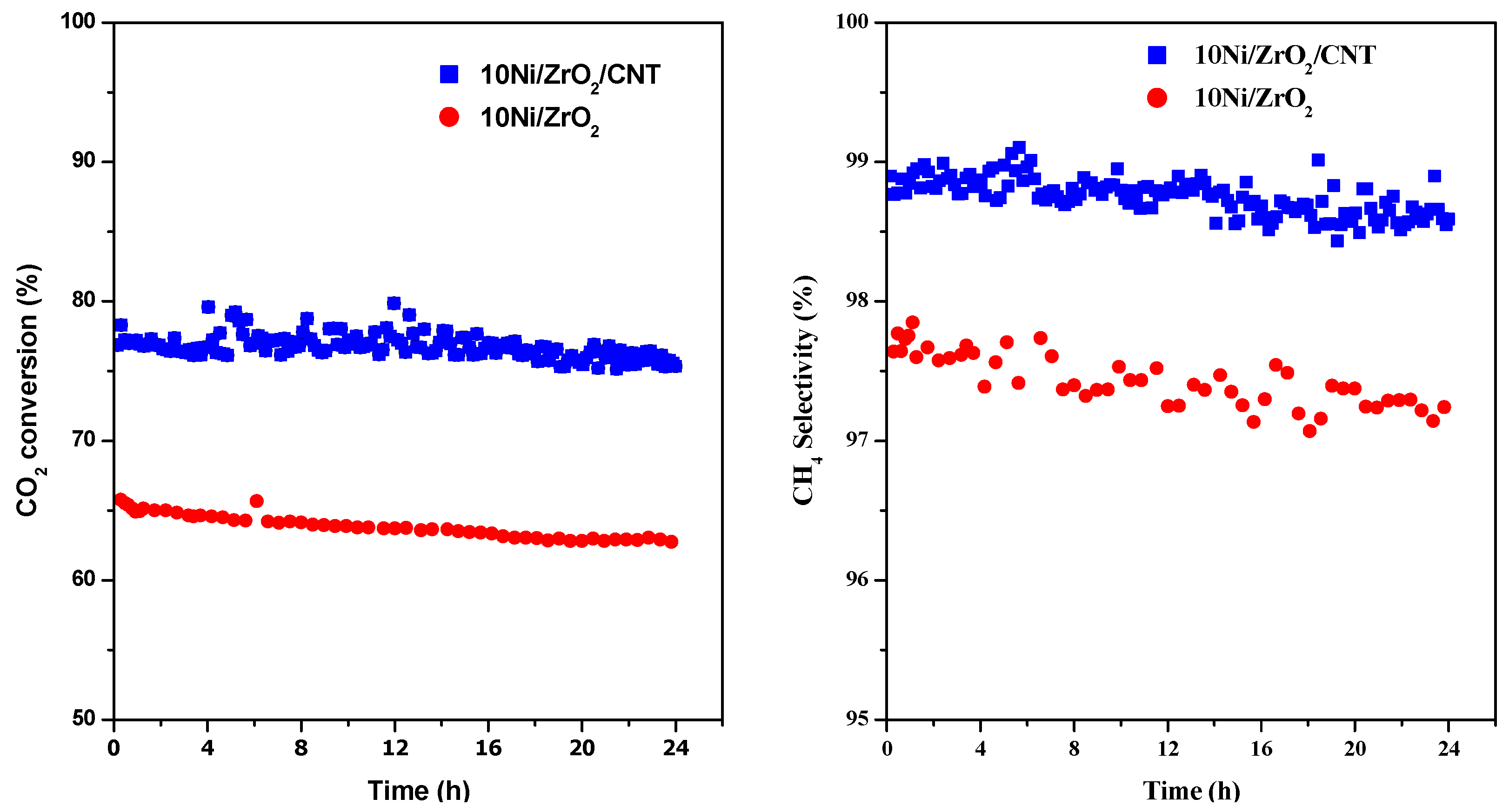
2.3. The Catalytic Activity
3. The Experimental Section
3.1. Material Synthesis
3.2. Characterization Methods
3.3. The Catalytic Performance Evaluation
3.4. Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ai, X.E.; Studer, A.S.; Sigman, D.M.; Martínez-García, D.A.; Fripiat, F.; Thöle, L.M.; Michel, E.; Gottschalk, J.; Arnold, L.; Moretti, S.; et al. Southern Ocean upwelling, Earth’s obliquity, and glacial-interglacial atmospheric CO2 change. Science 2020, 370, 1348–1352. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, H.; Fan, M. Stoichiometric theory in aquatic carbon sequestration under elevated carbon dioxide. Math. Biosci. 2024, 376, 109285. [Google Scholar] [CrossRef]
- Li, J.; Jia, K.; Wei, X.; Xia, M.; Chen, Z.; Yao, Y.; Zhang, X.; Jiang, H.; Yuan, B.; Tao, G.; et al. High-spatiotemporal resolution mapping of spatiotemporally continuous atmospheric CO2 concentrations over the global continent. Int. J. Appl. Earth Obs. Geoinf. 2022, 108, 102743. [Google Scholar] [CrossRef]
- Martens, J.A.; Bogaerts, A.; Kimpe, N.; Jacobs, P.A.; Marin, G.B.; Rabaey, K.; Saeys, M.; Verhelst, S. The Chemical Route to a Carbon Dioxide Neutral World. ChemSusChem 2017, 10, 1039–1055. [Google Scholar] [CrossRef]
- Antenucci, A.; Sansavini, G. Extensive CO2 recycling in power systems via Power-to-Gas and network storage. Renew. Sustain. Energy Rev. 2019, 100, 33–43. [Google Scholar] [CrossRef]
- Bailera, M.; Lisbona, P.; Romeo, L.M.; Espatolero, S. Power to Gas projects review: Lab, pilot and demo plants for storing renewable energy and CO2. Renew. Sustain. Energy Rev. 2017, 69, 292–312. [Google Scholar] [CrossRef]
- Ghaib, K.; Ben-Fares, F.Z. Power-to-Methane: A state-of-the-art review. Renew. Sustain. Energy Rev. 2018, 81, 433–446. [Google Scholar] [CrossRef]
- Götz, M.; Lefebvre, J.; Mörs, F.; Koch, A.M.; Graf, F.; Bajohr, S.; Reimert, R.; Kolb, T. Renewable Power-to-Gas: A technological and economic review. Renew. Energy 2016, 85, 1371–1390. [Google Scholar] [CrossRef]
- Leonzio, G. Design and feasibility analysis of a Power-to-Gas plant in Germany. J. Clean. Prod. 2017, 162, 609–623. [Google Scholar] [CrossRef]
- Martsinchyk, K.; Martsinchyk, A.; Łazor, M.; Shuhayeu, P.; Kupecki, J.; Niemczyk, A.; Błesznowski, M.; Milewski, J. Feasibility study and techno-economic assessment of power-to-gas (P2G) technology based on solid oxide electrolysis (SOE). J. Environ. Manag. 2024, 354, 120425. [Google Scholar] [CrossRef] [PubMed]
- Mazza, A.; Bompard, E.; Chicco, G. Applications of power to gas technologies in emerging electrical systems. Renew. Sustain. Energy Rev. 2018, 92, 794–806. [Google Scholar] [CrossRef]
- Ozturk, M.; Dincer, I. A comprehensive review on power-to-gas with hydrogen options for cleaner applications. Int. J. Hydrogen Energy 2021, 46, 31511–31522. [Google Scholar] [CrossRef]
- Thema, M.; Bauer, F.; Sterner, M. Power-to-Gas: Electrolysis and methanation status review. Renew. Sustain. Energy Rev. 2019, 112, 775–787. [Google Scholar] [CrossRef]
- Zhang, U.; Zhang, X.; Feng, S. Power to gas: An option for 2060 high penetration rate of renewable energy scenario of China. Environ. Sci. Pollut. Res. 2022, 29, 6857–6870. [Google Scholar] [CrossRef] [PubMed]
- Estevez, R.; Aguado-Deblas, L.; Bautista, F.M.; López-Tenllado, F.J.; Romero, A.A.; Luna, D. A Review on Green Hydrogen Valorization by Heterogeneous Catalytic Hydrogenation of Captured CO2 into Value-Added Products. Catalysts 2022, 12, 1555. [Google Scholar] [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on methanation—From fundamentals to current projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Vogt, C.; Monai, M.; Kramer, G.K.; Weckhuysen, B.M. The renaissance of the Sabatier reaction and its applications on Earth and in space. Nat. Catal. 2019, 2, 188–197. [Google Scholar] [CrossRef]
- Gac, W.; Zawadzki, W.; Rotko, M.; Greluk, M.; Slowik, G.; Kolb, G. Effects of support composition on the performance of nickel catalysts in CO2 methanation reaction. Catal. Today 2020, 357, 468–482. [Google Scholar] [CrossRef]
- González-Rangulan, V.V.; Reyero, I.; Bimbela, F.; Romero-Sarria, F.; Daturi, M.; Gandía, L.M. CO2 Methanation over Nickel Catalysts: Support Effects Investigated through Specific Activity and Operando IR Spectroscopy Measurements. Catalysts 2023, 13, 448. [Google Scholar] [CrossRef]
- Martínez, J.; Hernández, E.; Alfaro, S.; López Medina, R.; Valverde Aguilar, G.; Albiter, E.; Valenzuela, M.A. High Selectivity and Stability of Nickel Catalysts for CO2 Methanation: Support Effects. Catalysts 2019, 9, 24. [Google Scholar] [CrossRef]
- Pandey, D.; Deo, G. Effect of support on the catalytic activity of supported Ni-Fe catalysts for the CO2 methanation reaction. J. Ind. Eng. Chem. 2016, 33, 99–107. [Google Scholar] [CrossRef]
- Wang, K.; Men, U.; Liu, S.; Wang, J.G.; Li, Y.U.; Tang, Y.H.; Li, Z.P.; An, W.; Pan, X.L.; Li, L. Decoupling the size and support/metal loadings effect of Ni/SiO2 catalysts for CO2 methanation. Fuel 2021, 304, 121388. [Google Scholar] [CrossRef]
- Chen, S.L.; Abdel-Mageed, A.M.; Li, M.R.; Cisneros, S.; Bansmann, J.; Rabeah, J.; Brückner, A.; Gross, A.; Behm, R.J. Electronic metal-support interactions and their promotional effect on CO2 methanation on Ru/ZrO2 catalysts. J. Catal. 2021, 400, 407–420. [Google Scholar] [CrossRef]
- El-Salamony, R.A.; Acharya, K.; Al-Fatesh, A.S.; Osman, A.I.; Alreshaidan, S.B.; Kumar, N.S.; Ahmed, H.; Kumar, R. Enhanced direct methanation of CO2 using Ni-based catalysts supported on ZrO2, CeO2-ZrO2, and La2O3-ZrO2: The effect of support material on the reducible NiO-interacted species and catalytic activity. Mol. Catal. 2023, 547, 113378. [Google Scholar] [CrossRef]
- Jia, X.; Zhang, X.; Rui, N.; Hu, X.; Liu, C.-J. Structural effect of Ni/ZrO2 catalyst on CO2 methanation with enhanced activity. Appl. Catal. B 2019, 244, 159–169. [Google Scholar] [CrossRef]
- Shen, L.; Xu, J.; Zhu, M.; Han, Y.-N. Essential Role of the Support for Nickel-Based CO2 Methanation Catalysts. ACS Catal. 2020, 10, 14581–14591. [Google Scholar] [CrossRef]
- Fan, W.K.; Tahir, M. Recent trends in developments of active metals and heterogeneous materials for catalytic CO2 hydrogenation to renewable methane: A review. J. Environ. Chem. Eng. 2021, 9, 105460. [Google Scholar] [CrossRef]
- Zhao, K.; Wang, W.; Li, Z. Highly efficient Ni/ZrO2 catalysts prepared via combustion method for CO2 methanation. J. CO2 Util. 2016, 16, 236–244. [Google Scholar] [CrossRef]
- Souza, E.F.; Appel, L.G. Oxygen vacancy formation and their role in the CO2 activation on Ca doped ZrO2 surface: An ab-initio DFT study. Appl. Surf. Sci. 2021, 553, 149589. [Google Scholar] [CrossRef]
- Niu, J.T.; Zhang, C.X.; Liu, H.Y.; Jin, Y.; Zhang, R.G. Enhanced performance of oxygen vacancies on CO2 adsorption and activation over different phases of ZrO2. Front. Energy 2023, 17, 545–554. [Google Scholar] [CrossRef]
- Yamaguchi, T. Application of ZrO2 as a catalyst and a catalyst support. Catal. Today 1994, 20, 199–217. [Google Scholar] [CrossRef]
- Hao, Z.; Shen, J.; Lin, S.; Han, X.; Chang, X.; Liu, J.; Li, M.; Ma, X. Decoupling the effect of Ni particle size and surface oxygen deficiencies in CO2 methanation over ceria supported Ni. Appl. Catal. B 2021, 286, 119922–119932. [Google Scholar] [CrossRef]
- Kuhaudomlap, S.; Srifa, A.; Koo-Amornpattana, W.; Fukuhara, W.; Ratchahat, S. Insight and comprehensive study of Ni-based catalysts supported on various metal oxides for CO2 methanation. Sci. Rep. 2024, 14, 23149. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szany, J. Heterogeneous Catalysis on Atomically Dispersed Supported Metals: CO2 Reduction on Multifunctional Pd Catalysts. ACS Catal. 2013, 3, 2094–2100. [Google Scholar] [CrossRef]
- Wu, H.C.; Chang, Y.C.; Wu, J.H.; Lin, J.H.; Lin, I.K.; Chen, C.S. Methanation of CO2 and reverse water gas shift reactions on Ni/SiO2 catalysts: The influence of particle size on selectivity and reaction pathway. Catal. Sci. Technol. 2015, 5, 4154–4163. [Google Scholar] [CrossRef]
- Wang, Z.; Shu, D.; Chen, H.; Ding, C.; Wang, J.; Lv, Y.; Gu, Q.; Wang, Q.; Yang, B. Dynamic Interplay between Particle Size and Strong Metal Support Interaction in Rh/TiO2 Tuning the Selectivity of CO2 Hydrogenation. ACS Catal. 2025, 15, 4101–4109. [Google Scholar] [CrossRef]
- Tauster, S.J.; Fung, S.C.; Garten, R.L. Strong metal-support interactions. Group 8 noble metals supported on titanium dioxide. J. Am. Chem. Soc. 1978, 100, 170–175. [Google Scholar] [CrossRef]
- Vayssilov, G.N.; Lykhach, Y.; Migani, A.; Staudt, T.; Petrova, G.P.; Tsud, N.; Skála, T.; Bruix, A.; Illa, F.; Prince, K.C.; et al. Support nanostructure boosts oxygen transfer to catalytically active platinum nanoparticles. Nat. Mat. 2011, 10, 310–315. [Google Scholar] [CrossRef]
- Franchini, C.A.; Llorca, J.; Kuznetsov, A.; Silva, A.M. Outstanding dispersion of CeO2 on reduced graphene oxide. Implications for highly dispersed Pd catalysts. Diam. Relat. Mater. 2020, 109, 108061–108071. [Google Scholar] [CrossRef]
- Puigdollers, A.R.; Pacchioni, G. Reducibility of ZrO2/Pt3Zr and ZrO2/Pt 2D films compared to bulk zirconia: A DFT+U study of oxygen removal and H2 adsorption. Nanoscale 2017, 9, 6866–6876. [Google Scholar] [CrossRef]
- Puigdollers, A.R.; Tosoni, S.; Pacchioni, G. Turning a Nonreducible into a Reducible Oxide via Nanostructuring: Opposite Behavior of Bulk ZrO2 and ZrO2 Nanoparticles Toward H2 Adsorption. J. Phys. Chem. C 2016, 120, 15329–15337. [Google Scholar] [CrossRef]
- Espino, O.E.; Zonetti, P.C.; Celin, R.R.; Costa, L.T.; Alves, O.C.; Spadotto, J.C.; Appel, L.G.; Avillez, R.R. The tendency of supports to generate oxygen vacancies and the catalytic performance of Ni/ZrO2 and Ni/Mg(Al)O in CO2 methanation. Catal. Sci. Technol. 2022, 12, 1324. [Google Scholar] [CrossRef]
- Gharegozloo, S.; Ataie, S.; Abdizadeh, H.; Mostafavi, E.; Parnian, M.J.; Khodadadi, A.A. High performance Ni–CNTs catalyst: Synthesis and characterization. RSC Adv. 2016, 6, 47072–47082. [Google Scholar] [CrossRef]
- Li, J.; Zhou, Y.; Xiao, X.; Wang, W.; Wang, N.; Qian, W.; Chu, W. Regulation of Ni–CNT Interaction on Mn-Promoted Nickel Nanocatalysts Supported on Oxygenated CNTs for CO2 Selective Hydrogenation. ACS Appl. Mater. Interfaces 2018, 10, 41224–41236. [Google Scholar] [CrossRef]
- Wang, W.; Chu, W.; Wang, N.; Yang, W.; Jiang, C. Mesoporous nickel catalyst supported on multi-walled carbon nanotubes for carbon dioxide methanation. Int. J. Hydrogen Energy 2016, 41, 967–975. [Google Scholar] [CrossRef]
- Eschemann, T.O.; Lamme, W.S.; Manchester, R.L.; Parmentier, T.E.; Cognigni, A.; Rønning, M.; Jong, K.P. Effect of support surface treatment on the synthesis, structure, and performance of Co/CNT Fischer–Tropsch catalysts. J. Catal. 2015, 328, 130–138. [Google Scholar] [CrossRef]
- Huang, Y.; Cai, J.; Zheng, S.; Guo, Y. Fabrication of a high-performance Pb–PtCu/CNT catalyst for methanol electro-oxidation. J. Power Sources 2012, 210, 81–85. [Google Scholar] [CrossRef]
- Liu, L.; Lou, H.; Chen, M. Selective hydrogenation of furfural to tetrahydrofurfuryl alcohol over Ni/CNTs and bimetallic Cu Ni/CNTs catalysts. Int. J. Hydrogen Energy 2016, 41, 14721–14731. [Google Scholar] [CrossRef]
- Qu, Y.; Xu, G.; Yang, J.; Zhang, Z. Reduction of aromatic nitro compounds over Ni nanoparticles confined in CNTs. Appl. Catal. A 2020, 590, 117311. [Google Scholar] [CrossRef]
- Sun, Y.; Li, C.; Zhang, A. Preparation of Ni/CNTs catalyst with high reducibility and their superior catalytic performance in benzene hydrogenation. Appl. Catal. A 2016, 522, 180–187. [Google Scholar] [CrossRef]
- Tessonnier, J.P.; Pesant, L.; Ehret, G.; Ledoux, M.J.; Pham-Huu, C. Pd nanoparticles introduced inside multi-walled carbon nanotubes for selective hydrogenation of cinnamaldehyde into hydrocinnamaldehyde. Appl. Catal. A 2005, 288, 203–210. [Google Scholar] [CrossRef]
- Wei, Q.; Chen, X.; He, Y.; Fu, J.; Liang, J.; Wei, X.; Wang, L. Ni Nanoparticles supported on N-doped carbon nanotubes for efficient hydrogenation of C5 hydrocarbon resins under mild conditions. Microporous Mesoporous Mater. 2022, 222, 111727. [Google Scholar] [CrossRef]
- Zhang, A.M.; Dong, J.L.; Xu, K.H.; Rhee, H.K.; Li, X.L. Palladium cluster filled in inner of carbon nanotubes and their catalytic properties in liquid phase benzene hydrogenation. Catal. Today 2004, 93–95, 347–352. [Google Scholar] [CrossRef]
- Saravanan, A.; Prasad, K.; Gokulakrishnan, N.; Kalaivani, R.; Somanathan, T. Efficiency of Transition Metals in Combustion Catalyst for High Yield Helical Multi-Walled Carbon Nanotubes. Adv. Sci. Eng. Med. 2014, 6, 809–813. [Google Scholar] [CrossRef]
- Wepasnick, K.A.; Smith, B.A.; Bitter, J.L.; Fairbrother, H. Chemical and structural characterization of carbon nanotube surfaces. Anal. Bioanal. Chem. 2010, 396, 1003–1014. [Google Scholar] [CrossRef]
- Feng, Y.; Yang, W.; Chu, W. A Study of CO2 Methanation over Ni-Based Catalysts Supported by CNTs with Various Textural Characteristics. Int. J. Chem. Eng. 2015, 795386. [Google Scholar] [CrossRef]
- Hou, P.-X.; Xu, S.-T.; Ying, Z.; Yang, Q.-H.; Liu, C.; Cheng, H.-M. Hydrogen adsorption/desorption behavior of multi-walled carbon nanotubes with different diameters. Carbon 2003, 41, 2471–2476. [Google Scholar] [CrossRef]
- Zacharia, R.; Kim, K.Y.; Fazle Kibria, A.L.M.; Nahm, K.S. Enhancement of hydrogen storage capacity of carbon nanotubes via spill-over from vanadium and palladium nanoparticles. Chem. Phys. Lett. 2005, 412, 369–375. [Google Scholar] [CrossRef]
- Gonçalves, L.P.L.; Meledina, M.; Meledin, A.; Petrovykh, D.Y.; Sousa, J.P.S.; Soares, O.S.G.P.; Kolenko, Y.V.; Pereira, M.F.R. Understanding the importance of N−doping for CNT-supported Ni catalysts for CO2 methanation. Carbon 2022, 195, 35–43. [Google Scholar] [CrossRef]
- Bogdan, V.I.; Pokusaeva, Y.A.; Koklin, A.E.; Savilov, S.V.; Chernyak, S.A.; Lunin, V.V.; Kustov, L.M. Carbon Dioxide Reduction with Hydrogen on Carbon-Nanotube-Supported Catalysts under Supercritical Conditions. Energy Technol. 2019, 7, 3–8. [Google Scholar] [CrossRef]
- Romero-Sáez, M.; Dongil, A.B.; Benito, N.; Espinoza-González, R.; Escalona, N.; Gracia, F. CO2 methanation over nickel-ZrO2 catalyst supported on carbon nanotubes: A comparison between two impregnation strategies. App. Catal. B 2018, 237, 817–825. [Google Scholar] [CrossRef]
- Hu, Y.H.; Ruckenstein, E. Pore Size Distribution of Single-Walled Carbon Nanotubes. Ind. Eng. Chem. Res. 2004, 43, 708–711. [Google Scholar] [CrossRef]
- Duong, H.M.; Einarsson, E.; Okawa, J.; Xiang, R.; Maruyama, S. Thermal Degradation of Single-Walled Carbon Nanotubes. Jpn. J. Appl. Phys. 2008, 47, 1994. [Google Scholar] [CrossRef]
- Bom, D.; Andrews, R.; Jacques, D.; Anthony, J.; Chen, B.; Meier, B.S.; Selegue, J.P. Thermogravimetric Analysis of the Oxidation of Multiwalled Carbon Nanotubes: Evidence for the Role of Defect Sites in Carbon Nanotube Chemistry. Nano Lett. 2002, 2, 615–619. [Google Scholar] [CrossRef]
- Mahajan, A.; Kingon, A.; Kukovecz, Á.; Konya, Z.; Vilarinho, P.M. Studies on the thermal decomposition of multiwall carbon nanotubes under different atmospheres. Mater. Lett. 2013, 90, 165–168. [Google Scholar] [CrossRef]
- Łamacz, A.; Matus, K.; Liszka, B.; Silvestre-Albero, J.; Lafjah, M.; Dintzer, T.; Janowska, I. The impact of synthesis method of CNT supported CeZrO2 and Ni-CeZrO2 on catalytic activity in WGS reaction. Catal. Today 2018, 301, 172–182. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, D. Influence of geometries of multi-walled carbon nanotubes on the pore structures of Buckypaper. Compos. Part A Appl. Sci. Manuf. 2012, 43, 469–474. [Google Scholar] [CrossRef]
- Sun, C.H.; Li, F.; Cheng, H.M.; Lu, C.Q. Packing-dependent pore structures in single-walled carbon nanotube arrays. Appl. Phys. Lett. 2005, 87, 243109. [Google Scholar] [CrossRef]
- Yang, Q.-H.; Hou, P.-X.; Bai, S.; Wang, M.-Z.; Cheng, H.-M. Adsorption and capillarity of nitrogen in aggregated multi-walled carbon nanotubes. Chem. Phys. Lett. 2001, 345, 18–24. [Google Scholar] [CrossRef]
- Leofanti, G.; Padovan, M.; Tozzola, G.; Venturelli, B. Surface area and pore texture of catalysts. Catal. Today 1998, 41, 207–219. [Google Scholar] [CrossRef]
- Fujiwara, A.; Ishii, K.; Suematsu, H.; Kataura, H.; Maniwa, Y.; Suzuki, S.; Achiba, Y. Gas adsorption in the inside and outside of single-walled carbon nanotubes. Chem. Phys. Lett. 2001, 336, 205–211. [Google Scholar] [CrossRef]
- Kocabas, S.; Kopac, T.; Dogu, G.; Dogu, T. Effect of thermal treatments and palladium loading on hydrogen sorption characteristics of single-walled carbon nanotubes. Int. J. Hydrogen Energy 2008, 33, 1693–1699. [Google Scholar] [CrossRef]
- Srinivasan, R.; Davis, B.H.; Cavin, O.B.; Hubbard, C.R. Crystallization and Phase Transformation Process in Zirconia: An in situ High-Temperature X-ray Diffraction Study. J. Am. Ceram. Soc. 1992, 75, 1217–1222. [Google Scholar] [CrossRef]
- Ramamoorthy, R.; Sundararaman, D.; Ramasamy, S. X-ray diffraction study of phase transformation in hydrolyzed zirconia nanoparticles. J. Eur. Ceram. Soc. 1999, 19, 1827–1833. [Google Scholar] [CrossRef]
- Srinivasan, R.; De Angelis, R.; Davis, B.H. Factors influencing the stability of the tetragonal form of zirconia. J. Mater. Res. 1986, 1, 583–588. [Google Scholar] [CrossRef]
- Srinivasan, R.; Harris, M.B.; Simpson, S.F.; Deangelis, R.J.; Davis, B.H. Zirconium-oxide crystal phase—The role fo the pH and the time to attain the final pH for precipitation of hydrous oxide. J. Mater. Res. 1988, 3, 787–797. [Google Scholar] [CrossRef]
- Denkewicz, R.P.; Tenhuisen, K.S.; Adair, J.H. Hydrothermal crystallization kinetics of meta-ZrO2 and t-ZrO2. J. Mater. Res. 1990, 5, 2698–2705. [Google Scholar] [CrossRef]
- Collins, D.E.; Bowman, K.J. Influence of atmosphere on crystallization of zirconia from a zirconium alkoxide. J. Mater. Res. 1998, 13, 1230–1237. [Google Scholar] [CrossRef]
- Basahel1, S.N.; Ali, T.T.; Mokhtar, M.; Narasimharao, K. Influence of crystal structure of nanosized ZrO2 on the photocatalytic degradation of methyl orange. Res. Lett. 2015, 10, 73. [Google Scholar] [CrossRef]
- Román-Martínez, M.C.; Cazorla-Amorós, D.; Linares-Solano, A.; Lecea, C.S.M. Tpd and TPR characterization of carbonaceous supports and Pt/C catalysts. Carbon 1993, 31, 895–902. [Google Scholar] [CrossRef]
- Pan, X.; Bao, X. The Effects of Confinement inside Carbon Nanotubes on Catalysis. Acc. Chem. Res. 2011, 44, 553–562. [Google Scholar] [CrossRef]
- Chen, W.; Fan, Z.; Pan, X.; Bao, X. Effect of Confinement in Carbon Nanotubes on the Activity of Fischer−Tropsch Iron Catalyst. J. Am. Chem. Soc. 2008, 130, 9414–9419. [Google Scholar] [CrossRef]
- Bian, J.; Xiao, M.; Wang, S.-J.; Lu, Y.-X.; Meng, Y.-Z. Carbon nanotubes supported Cu–Ni bimetallic catalysts and their properties for the direct synthesis of dimethyl carbonate from methanol and carbon dioxide. Appl. Surf. Sci. 2009, 255, 7188–7196. [Google Scholar] [CrossRef]
- Kundu, S.; Wang, Y.; Xia, W.; Muhler, M. Thermal Stability and Reducibility of Oxygen-Containing Functional Groups on Multiwalled Carbon Nanotube Surfaces: A Quantitative High-Resolution XPS and TPD/TPR Study. J. Phys. Chem. C. 2008, 112, 16869–16878. [Google Scholar] [CrossRef]
- Holstein, W.L.; Boudart, M. Hydrogenolysis of carbon and its catalysis by platinum. J. Catal. 1981, 72, 328–337. [Google Scholar] [CrossRef]
- Cao, J.R.; Back, M.H. Kinetics of the reaction of hydrogen with thin films of carbon. Carbon 1982, 20, 505–512. [Google Scholar] [CrossRef]
- Treptau, M.H.; Miller, D.J. Hydrogen gasification of HNO3-oxidized carbons. Carbon 1991, 29, 531–539. [Google Scholar] [CrossRef]
- Zhou, Z.; Orcutt, E.K.; Anderson, H.C.; Stowers, K.J. Hydrogen surface modification of a carbon nanotube catalyst for the improvement of ethane oxidative dehydrogenation. Carbon 2019, 152, 924–931. [Google Scholar] [CrossRef]
- Herold, F.; Gläsel, J.; Etzold, B.J.M.; Rønning, M. Can Temperature-Programmed Techniques Provide the Gold Standard for Carbon Surface Characterization? Chem. Mater. 2022, 34, 8490–8516. [Google Scholar] [CrossRef]
- Lobo, L.S.; Carabineiro, S.A.C. Kinetics and mechanism of catalytic carbon gasification. Fuel 2016, 183, 457–469. [Google Scholar] [CrossRef]
- Gardner, D.C.; Bartholomew, C.H. Kinetics of carbon deposition during methanation of carbon monoxide. IEC Prod. Res. Dev. 1981, 20, 80–87. [Google Scholar] [CrossRef]
- Schmider, D.; Maier, L.; Deutschmann, O. Reaction Kinetics of CO and CO2 Methanation over Nickel. Ind. Eng. Chem. Res. 2021, 60, 5792–5805. [Google Scholar] [CrossRef]
- Lalinde, J.A.H.; Roongruangsree, P.; Ilsemann, J.; Bäumer, M.; Kopyscinski, J. CO2 methanation and reverse water gas shift reaction. Kinetic study based on in situ spatially-resolved measurements. Chem. Eng. J. 2020, 390, 124629. [Google Scholar] [CrossRef]
- Ghaib, K.; Nitz, K.; Ben-Fares, F.-Z. Chemical Methanation of CO2: A Review. ChemBioEng Rev. 2016, 3, 266–275. [Google Scholar] [CrossRef]
- Lim, J.U.; McGregor, J.; Sederman, A.J.; Dennis, J.S. The role of the Boudouard and water–gas shift reactions in the methanation of CO or CO2 over Ni/γ-Al2O3 catalyst. Chem. Eng. Sci. 2016, 152, 754–766. [Google Scholar] [CrossRef]
- Ashok, J.; Pati, S.; Hongmanorom, P.; Tianxi, Z.; Junmei, C.; Kawi, S. A review of recent catalyst advances in CO2 methanation processes. Catal. Today 2020, 356, 471–489. [Google Scholar] [CrossRef]
- Vogt, C.; Monai, M.; Sterk, E.B.; Palle, J.; Melcherts, A.E.M.; Zijlstra, B.; Groeneveld, E.; Berben, P.H.; Boereboom, J.M.; Hensen, E.J.M.; et al. Understanding carbon dioxide activation and carbon–carbon coupling over nickel. Nat. Commun. 2019, 10, 5330. [Google Scholar] [CrossRef]
- Wang, W.; Duong-Viet, C.; Ba, H.; Baaziz, W.; Tuci, G.; Caporali, S.; Nguyen-Dinh, L.; Ersen, O.; Giambastiani, G.; Pham-Huu, C. Nickel Nanoparticles Decorated Nitrogen-Doped Carbon Nanotubes (Ni/N-CNT); a Robust Catalyst for the Efficient and Selective CO2 Methanation. ACS Appl. Energy Mater. 2019, 2, 1111–1120. [Google Scholar] [CrossRef]
- Le, T.; Striolo, S.; Turner, C.H.; Cole, D.R. Confinement Effects on Carbon Dioxide Methanation: A Novel Mechanism for Abiotic Methane Formation. Sci. Rep. 2017, 7, 9021. [Google Scholar] [CrossRef]
- Kattel, S.; Liu, P.; Chen, J.G. Tuning Selectivity of CO2 Hydrogenation Reactions at the Metal/Oxide Interface. J. Am. Chem. Soc. 2017, 139, 9739–9754. [Google Scholar] [CrossRef]
- Ye, R.-P.; Li, Q.; Gong, W.; Wang, T.; Razink, J.J.; Lin, L.; Qin, Y.-Y.; Zhou, Z.; Adidharma, H.; Tang, J.; et al. High-performance of nanostructured Ni/CeO2 catalyst on CO2 methanation. Appl. Catal. B 2020, 268, 118474. [Google Scholar] [CrossRef]
- Mutz, B.; Belimov, M.; Wang, W.; Sprenger, P.; Serrer, M.-A.; Wang, D.; Pfeifer, P.; Kleist, W.; Grunwaldt, J.-D. Potential of an Alumina-Supported Ni3Fe Catalyst in the Methanation of CO2: Impact of Alloy Formation on Activity and Stability. ACS Catal. 2017, 7, 6802–6814. [Google Scholar] [CrossRef]
- Simons, J.F.M.; de Heer, T.J.; van de Poll, R.C.J.; Muravev, V.; Kosinov, N.; Hensen, E.J.M. Structure Sensitivity of CO2 Hydrogenation on Ni Revisited. J. Am. Chem. Soc. 2023, 145, 20289–20301. [Google Scholar] [CrossRef] [PubMed]
- Visser, N.L.; Daoura, O.; Plessow, P.N.; Smulders, L.C.J.; Rijk, J.W.; Stewart, J.A.; Vandegehuchte, B.D.; Studt, F.; van der Hoeven, J.E.S.; Jongh, P.E. Particle Size Effects of Carbon Supported Nickel Nanoparticles for High Pressure CO2 Methanation. ChemCatChem 2022, 14, e202200665. [Google Scholar] [CrossRef]
- Riani, P.; Spennati, E.; Garcia, E.M.V.; Escribano, V.S.; Busca, G.; Garbarino, G. Ni/Al2O3 catalysts for CO2 methanation: Effect of silica and nickel loading. Int. J. Hydrogen Energy 2023, 48, 24976–24995. [Google Scholar] [CrossRef]
- De Coster, V.; Srinath, N.V.; Yazdani, P.; Poelman, H.; Galvita, V.V. Does CO2 Oxidize Ni Catalysts? A Quick X-ray Absorption Spectroscopy Answer. J. Phys. Chem. Lett. 2022, 13, 7947–7952. [Google Scholar] [CrossRef]
- Foppa, L.; Margossian, T.; Kim, S.M.; Müller, C.; Copéret, C.; Larmier, K.; Comas-Vives, A. Contrasting the Role of Ni/Al2O3 Interfaces in Water–Gas Shift and Dry Reforming of Methane. J. Am. Chem. Soc. 2017, 139, 17128–17139. [Google Scholar] [CrossRef]
- Silaghi, M.-C.; Comas-Vives, A.; Copéret, C. CO2 Activation on Ni/γ–Al2O3 Catalysts by First-Principles Calculations: From Ideal Surfaces to Supported Nanoparticles. ACS Catal. 2016, 6, 4501–4505. [Google Scholar] [CrossRef]
- Shirsath, A.B.; Schulte, M.L.; Kreitz, B.; Tischer, S.; Grunwaldt, J.-D.; Deutschmann, O. Spatially-resolved investigation of CO2 methanation over Ni/γ-Al2O3 and Ni3.2Fe/γ-Al2O3 catalysts in a packed-bed reactor. J. Chem. Eng. 2023, 469, 143847. [Google Scholar] [CrossRef]
- Navarro-Ruiz, J.; Audevard, J.; Vidal, M.; Campos, C.H.; Rosal, I.D.; Serp, P.; Gerber, I.C. Mechanism of Hydrogen Spillover on Metal-Doped Carbon Materials: Surface Carboxylic Groups Are Key. ACS Catal. 2024, 14, 7111–7126. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, F.H.; Yang, R.T. Enhanced Hydrogen Spillover on Carbon Surfaces Modified by Oxygen Plasma. J. Phys. Chem. C 2010, 114, 1601–1609. [Google Scholar] [CrossRef]
- Munnik, P.; Jongh, P.E.; Jong, K.P. Recent Developments in the Synthesis of Supported Catalysts. Chem. Rev. 2015, 115, 6687–6718. [Google Scholar] [CrossRef] [PubMed]
- Mehrabadi, B.A.T.; Eskandari, S.; Khan, U.; White, R.D.; Regalbuto, J.R. Chapter One—A Review of Preparation Methods for Supported Metal Catalysts. In Advances in Catalysis; Song, C., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 1–35. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. J. Am. Chem. Soc. 1952, 73, 373–380. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Residual Weight a (%) | XRF b (%) | Actual Amount c (%) | |||
|---|---|---|---|---|---|---|
| NiO | ZrO2 | NiO | Ni | ZrO2 | ||
| CNT | 0 | - | - | - | - | - |
| 10Ni/CNT | 13.1 | 100 | 0.00 | 13.1 | 9.9 | 0.0 |
| 10Ni/ZrO2/CNT | 31.3 | 43.8 | 56.2 | 13.7 | 10.7 | 17.6 |
| 20ZrO2/CNT | 20.7 | 0.00 | 100 | - | 0.0 | 20.7 |
| 2.5Ni/ZrO2/CNT | 24.3 | 16.1 | 83.9 | 3.9 | 3.0 | 20.4 |
| 5Ni/ZrO2/CNT | 24.3 | 31.4 | 68.6 | 7.6 | 6.0 | 16.6 |
| 15Ni/ZrO2/CNT | 41.1 | 49.2 | 50.8 | 20.2 | 15.9 | 20.7 |
| Sample | Calcined Samples | Reduced Samples | ||||
|---|---|---|---|---|---|---|
| SBET (m2/g) | VP (cm3/g) | DP (nm) | SBET (m2/g) | VP (cm3/g) | DP (nm) | |
| CNT | 323 | 0.8 | 11 | 413 | 1.1 | 11 |
| 10Ni/CNT | 269 | 0.7 | 12 | 475 | 1.5 | 14 |
| 10Ni/ZrO2/CNT | 218 | 0.5 | 11 | 317 | 0.8 | 11 |
| 20ZrO2/CNT | 233 | 0.8 | 15 | 374 | 1.0 | 11 |
| 10Ni/ZrO2 | 114 | 0.1 | 4.5 | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira, J.P.B.; Iwasaki, M.T.; Milanezi, H.C.; Barros, J.L.M.; Simionato, A.A.; Marques, B.d.S.; Franchini, C.A.; Urquieta-González, E.A.; Chimentão, R.J.; Bueno, J.M.C.; et al. Production of Sustainable Synthetic Natural Gas from Carbon Dioxide and Renewable Energy Catalyzed by Carbon-Nanotube-Supported Ni and ZrO2 Nanoparticles. Catalysts 2025, 15, 675. https://doi.org/10.3390/catal15070675
de Oliveira JPB, Iwasaki MT, Milanezi HC, Barros JLM, Simionato AA, Marques BdS, Franchini CA, Urquieta-González EA, Chimentão RJ, Bueno JMC, et al. Production of Sustainable Synthetic Natural Gas from Carbon Dioxide and Renewable Energy Catalyzed by Carbon-Nanotube-Supported Ni and ZrO2 Nanoparticles. Catalysts. 2025; 15(7):675. https://doi.org/10.3390/catal15070675
Chicago/Turabian Stylede Oliveira, João Pedro Bueno, Mariana Tiemi Iwasaki, Henrique Carvalhais Milanezi, João Lucas Marques Barros, Arnaldo Agostinho Simionato, Bruno da Silva Marques, Carlos Alberto Franchini, Ernesto Antonio Urquieta-González, Ricardo José Chimentão, José Maria Corrêa Bueno, and et al. 2025. "Production of Sustainable Synthetic Natural Gas from Carbon Dioxide and Renewable Energy Catalyzed by Carbon-Nanotube-Supported Ni and ZrO2 Nanoparticles" Catalysts 15, no. 7: 675. https://doi.org/10.3390/catal15070675
APA Stylede Oliveira, J. P. B., Iwasaki, M. T., Milanezi, H. C., Barros, J. L. M., Simionato, A. A., Marques, B. d. S., Franchini, C. A., Urquieta-González, E. A., Chimentão, R. J., Bueno, J. M. C., da Silva, A. M., & dos Santos, J. B. O. (2025). Production of Sustainable Synthetic Natural Gas from Carbon Dioxide and Renewable Energy Catalyzed by Carbon-Nanotube-Supported Ni and ZrO2 Nanoparticles. Catalysts, 15(7), 675. https://doi.org/10.3390/catal15070675