1. Introduction
The development of biodiesel is highly desirable for meeting the needs of green chemistry and the energy industry. The production of biodiesel could generate glycerol (Gly) as a by-product. Although traditional petroleum-based Gly is an important raw material for various industrial uses, continuous biodiesel production has led to a serious surplus of bio-based Gly [
1]. A major drawback of bio-based Gly is its relatively low purity, commonly containing metal ion residues like Na
+ and K
+, and various fatty acids and their saponification products [
2,
3]. Such complex impurities make the energy consumption for the purification of bio-based glycerol considerably higher than that of petroleum-based Gly [
4]. In addition, the traditional Gly supply chain has already been well-established and the practical demand for Gly is still quite limited, which makes it impossible to solve the problem of Gly surplus. Therefore, there is an urgent need to develop novel technologies and methods for converting bio-based glycerol to highly evaluated products with wide application.
Recently, interest in developing green and efficient production processes for glycerol carbonate (GC) has been continuously growing, since it might be an ideal way to combine bio-based Gly valorization with greenhouse gas CO
2 utilization [
5]. As one of the most important platform compounds, GC has shown desirable features in a variety of application fields such as medicine, cosmetics, textiles, coatings, plastics, machinery, integrated circuits, new materials, and new energy [
6,
7,
8,
9]. In particular, the active groups (carbonyl, hydroxyl) in GC can react with compounds containing functional groups such as hydroxyl and thiol to synthesize important pharmaceutical and chemical intermediates. GC is also an ideal choice for replacing epoxides to modify polymer monomers and to produce functional polymer materials like polycarbonates and polyurethanes [
8]. In addition, GC has been proposed as a more suitable electrolyte carrier owing to its lower flammability and boiling point, as well as improved conductivity. However, the current high cost of GC, which is a result of high production costs, has limited its widespread application. Therefore, more recent effort has been devoted to developing a new efficient and low-cost GC production process that could meet the constantly growing demand in global resource utilization and environmental protection [
10,
11].
From the perspective of developing the green chemical industry, GC could be synthesized from Gly by several ecofriendly catalytic processes, such as transesterification with small organic carbonates, carbonation with carbon dioxide, and carbonylation with urea. At present, the transesterification of organic carbonates (e.g., dimethyl carbonate, ethylene carbonate) with Gly is the major method for the industrial production GC, which is conducted in the presence of alkaline catalysts such as calcium oxide and sodium hydroxide [
12,
13,
14,
15]. This reaction can occur spontaneously in thermodynamics with mild reaction conditions and simple operation procedures. The main disadvantage of this production process is the usage of expensive organic carbonates, leading to a higher production costs. In addition, the alkaline catalysts used in this process mainly exist in homogeneous form, making it difficult to realize product separation and scalable continuous production. Therefore, the transesterification production process of GC still faces multiple challenges such as catalyst innovation, cost control, and process improvement [
16,
17,
18,
19].
In principle, the direct coupling of glycerol with CO
2 (carbonation) would be the most straightforward and attractive method for producing GC. Numerous catalyst systems, including various metal oxides, ionic liquids, and supported solid base catalysts, have been investigated for catalyzing this process [
20,
21,
22]. However, such a direct route is not regarded as a feasible way for industrial implementation since the carbonation reaction is highly constrained by thermodynamics [
7,
23]. A relatively low GC yield is commonly obtained even when harsh operating conditions are adopted. For achieving successful industrial production, better heterogeneous catalysts and process control (e.g., shifting chemical balance) are still required, which are the major challenges faced by this research field.
Alternatively, the utilization of glycidol (GD), a derivative of Gly, to react with carbon dioxide seems to be a better choice for GC production [
24,
25]. A variety of metal-based and organocatalysts have been investigated for the coupling of CO
2 and GD, and some of them could act as effective homogeneous catalysts for converting GD to GC under mild conditions [
26]. In comparison with Gly, the carbonation of GD with CO
2 is much easier, although the search of more efficient and recyclable heterogeneous catalysts still remains a challenging subject for this process. In addition, the production of GD from bio-based Gly also deserves further study, and is a determinative factor for achieving large-scale production of GC through the carbonation of GD with CO
2. Traditionally, GD could be produced by using petroleum-based Gly as a raw material through a multiple-step route in the presence of Brønsted acid and Lewis acid [
27,
28,
29,
30,
31]. By establishing an intensified and scalable flow process, Monbaliu and co-workers have successfully achieved the effective conversion of bio-based Gly to GD with enhanced efficiency and selectivity in a two-step continuous flow setup [
32].
The carbonylation of Gly with urea is another choice for the effective production of GC. Urea itself is non-toxic and has a low price. The by-product of ammonia could be recycled to produce urea through the reaction with CO
2, thereby forming a glycerol–CO
2 comprehensive utilization system with ammonia as the medium. Essentially, the product of GC is produced by continuously consuming glycerol and CO
2. It was reported that a number of metal oxides, metal salts, hydrotalcites, and ionic liquids can catalyze the glycerol–urea carbonylation reaction [
33,
34,
35,
36,
37,
38]. Notably, some heterogenous Zn-based catalysts have received more attention owing to their relatively high catalytic activity and selectivity. However, most of them conducted the catalytic reaction in homogeneous rather than heterogenous conditions, since the dissolved Zn species has been identified as the main active site for the carbonylation reaction. Moreover, the reaction is also limited by thermodynamic equilibrium. It is necessary to shift the equilibrium towards the product by reducing the system pressure (i.e., removing the NH
3). Therefore, much work is still required in order to improve the stability of heterogeneous Zn-based catalysts without compromising catalytic activity and selectivity. In addition, more information revealing the nature of the catalytically active sites and reaction mechanisms needs to be explored, which can be helpful for designing novel and efficient catalysts for the carbonylation of Gly with urea.
In this review, we aim to summarize the very recent research advances involving the catalytic synthesis of GC through the pathways mentioned above. More attention has been drawn to the prominent progress made in the following aspects: (1) catalyst innovation, e.g., designing novel heterogeneous catalysts that can solve the common drawbacks like lower catalytic efficiency and poor stability/recyclability; (2) mechanism exploration, proposing a profound understanding clarifying the nature of catalytically active sites and reaction mechanisms by combining various characterization results and theory calculations; (3) catalytic process improvement, developing more efficient catalytic reaction units or equipment, which can achieve the sustainable and economical scale-up of GC production. In addition, the main challenges and opportunities in future work shall be proposed, which could hopefully provide appropriate guidance on rationally designing and developing more efficient catalysts and low-cost GC production processes.
2. Synthesis of GC from the Carbonation of Gly with CO2 and the Thermodynamic Analyses of GC Production Through Different Routes
The continuous increase of CO
2 concentration in the atmosphere is becoming a global crisis. Converting CO
2 into high-value-added chemicals provides an environmentally friendly approach to reduce carbon emissions [
39]. Significantly, the effective utilization of CO
2 and bio-based Gly to catalytically synthesize GC could not only reduce the level of atmospheric CO
2, but also significantly increase the appliable value of bio-based Gly. Many research works have demonstrated the great potential of this catalytic technology for achieving carbon capture and utilization [
40,
41].
As already mentioned above, the direct carbonation of Gly with CO
2 has been thought of as the most ideal method for producing GC. Various catalysts have been investigated for the direct carbonation reaction, including metal oxides, ionic liquids, and supported solid base catalysts. Examples include protic ionic liquids, Mg/ZIF-8 and CeZrO
2 solid solutions catalysts, etc. [
21,
42,
43]. In a few recent review articles, the research progress and the main challenges in the future involving the synthesis of GC through the direct carbonation of Gly with CO
2 have been well-summarized [
5]. As claimed by Manyar and coauthors, although a number of heterogeneous catalysts have shown relatively catalytic activity in the direct carbonation reaction, their catalytic efficiencies are still unsatisfactory. Relatively low GC yields are usually obtained, even when higher reaction temperatures and pressures have been adopted. The main reason should be attributed to the fact that direct carbonation is constrained by thermodynamics.
Previously, Wang et al. [
44] investigated the chemical equilibrium of GC synthesis from Gly. The results of thermodynamic analysis revealed that the direct carbonation of Gly with CO
2 is thermodynamically unfavorable under ambient conditions, showing that Δ
rG
θm = +23.92 kJ/mol at T = 298.15 K and 101,325 Pa. This phenomenon mainly stems from the high stability of CO
2 and the formation of water-induced reverse reactions. In general, the synthesis of GC via the direct carbonation route requires an increase in system pressure and temperature in order to overcome the energy barrier, which can shift the equilibrium toward GC production. In addition, the introduction of a dehydrating agent (e.g., acetonitrile) can significantly reduce the concentration of the product in the reaction system, thus contributing to a further shift in the equilibrium in the direction of GC generation.
For instance, Ke et al. [
45] reported that by using the hydrothermally synthesized CuO as catalyst, the Gly conversion and GC selectivity could reach 89.0% and 69.4%, respectively, under the optimized reaction conditions (Gly 0.92 g, 2-cyanopyridine 3.26 g, DMF 10 mL, 120 °C, 5 h, P(CO
2) = 3 Mpa), which are much higher than the previously reported catalyst systems. The enhanced performance of this CuO catalyst could be mainly related to the stabilized valence state of Cu
2+ and the modulation of its lattice oxygen to oxygen vacancy ratio. In another work reported recently, it was demonstrated that Mg-incorporated ZIF-8 (Mg/ZIF-8) could achieve 75.7% Gly conversion and 40% GC yield at 175 °C and 0.3 Mpa CO
2 for 6 h, upon using acetonitrile as dehydrating agent [
42]. The relatively high catalytic activity of Mg/ZIF-8 might be mainly attributed to the fact that the incorporated Mg
2+ could result in structural disruption, thus generating more exposed active Mg
2+ sites and coordinatively unsaturated nitrogen atoms, which could considerably boost the Lewis basicity of the catalyst. Despite this progress, more efforts are still required to conduct the efficient synthesis of GC from the direct carbonation route. The main challenge of this route is to develop more efficient heterogeneous catalysts which can work well (with high activity, selectivity, and stability) under the optimized operation conditions (suitable reaction temperature and pressure).
The coupling of CO
2 and glycerol-derived glycidol (GD) has been regarded as a more appropriate route for GC synthesis due to its relatively high atom economy and mild reaction pathway [
24]. Thermodynamic analysis reveals that, unlike the direct carbonation of Gly with CO
2, the coupling reaction between GD and CO
2 to form GC is thermodynamically spontaneous (Δ
rG
θm < 0). The activation energy barrier predominantly depends on the synergistic mechanism between CO
2 activation and epoxide ring opening [
44]. Crucially, the operated reaction temperature requires precise modulation: an insufficient thermodynamic driving force prevails at lower temperatures, while elevated temperatures promote the side reactions (e.g., hydrolysis, polymerization) or product decomposition. Consequently, optimizing temperature to reconcile thermodynamic and kinetic constraints could substantially enhance the coupling efficiency between GD and CO
2 for GC synthesis in the case where an effective catalyst is adopted.
Alternatively, to improve the efficiency of GC generation, an indirect pathway can be adopted, for instance, converting CO
2 to urea or DMC firstly and then reacting it with Gly to synthesize GC. This cascade reaction pathway might greatly reduce the process’s energy barriers. As reported in the literature [
44], the transesterification route (glycerol + DMC) exhibits near-equilibrium thermodynamics (Δ
rG
θm = −1.83 kJ/mol at T = 298.15 K and 101,325 Pa), suggesting that this process is thermodynamically favorable. Notably, the chemical equilibrium constant increases with elevated temperature, indicating that adopting a heating strategy can effectively improve the thermodynamic driving force. Furthermore, removing the by-product of methanol may further overcome the equilibrium constraints, driving the reaction forward and thereby enhancing GC yield.
The thermodynamic analysis of Gly–urea carbonation reveals the positive Gibbs free energy (ΔrGθm = 32.43 kJ/mol) under ambient temperature and pressure conditions (T = 298.15 K, p = 101,325 Pa), confirming the non-spontaneous nature of this reaction pathway. Reduced system pressure facilitates the ammonia evolution step, thereby promoting GC formation. Further analysis of the temperature dependence indicates that the reaction equilibrium constant increases substantially with rising temperature. Consequently, the synergistic effect of elevated temperature and reduced pressure is thermodynamically imperative to shift the equilibrium toward products. Moreover, the prompt removal of gaseous ammonia by-products remains equally critical to circumventing equilibrium limitations and achieving viable process yields.
Overall, the reaction feasibility of GC synthesis depends on balancing thermodynamic driving forces (equilibrium conversion), by-product management (e.g., water/methanol removal), and operational conditions (temperature/pressure). Catalyst choice could play a crucial role in overcoming the kinetic barriers while maintaining high GC selectivity under optimized thermodynamic constraints.
3. Synthesis of GC from the Coupling of CO2 and Glycerol-Derived Glycidol
As mentioned above, the direct carbonation of Gly with CO
2 is the most attractive path for producing GC, since it is an ideal process targeting high-value-added chemicals and feeding upon bio-based Gly renewable sources and waste effluent CO
2 [
46,
47,
48,
49,
50]. However, this route is constrained by both thermodynamics and kinetics, and high reaction pressure and dehydrant are generally required in order to obtain a higher yield of GC. Currently, more efficient catalysts and operating processes are still desired for promoting industrial implementation, which remains a challenging research subject in the field of catalysis and the chemical industry.
Alternatively, the coupling of CO
2 and glycerol-derived glycidol (GD) has been proposed as an economically viable process [
24,
51,
52]. Compared to Gly, GD could react with CO
2 to form general GC under much milder conditions. Traditionally, GD could be produced by using petroleum-based Gly as a raw material through a multiple-step route in the presence of Brønsted acid and Lewis acid [
27,
28,
29,
30,
31]. In general, Gly is first converted into an active intermediate of chloropropanediol (or allyl alcohol) via methods such as chlorination (or oxidation) in the presence of catalysts like pimelic acid (or HZSM-5 zeolite), then GD can be obtained by further dichlorination (or epoxidation) of the corresponding intermediate. Significally, Monbaliu et al. developed a tandem continuous-flow process where Gly is first converted to chlorohydrins via concentrated aqueous hydrochloric acid and using pimelic acid as catalyst, then dehydrochlorinated with a concentrated aqueous solution of NaOH to yield a GD/epichlorohydrin mixture, with integrated extraction enhancing separation. This route seems quite attractive considering the fact that an intensified and scalable flow process for effectively converting Gly into GD has been established [
32]. In addition, recent progress has also revealed many types of catalysts, including metal catalysts, organic catalysts, and ionic liquid catalysts, that can catalyze the carbonation of GD with CO
2 to GC with relatively high yields [
53,
54,
55,
56,
57,
58,
59,
60,
61]. We list some of the catalysts for the coupling reaction of CO
2 and GD in
Table 1. Special attention has been paid to having intelligent control over the microenvironment of the catalysts to achieve a desirable performance, and to the integration and application of the technological processes [
62,
63,
64].
For instance, a simple and clean approach has been found to precisely control the defect density in Metal–Organic Frameworks (MOFs) to address the requirement of the carbonation of GD with CO
2 [
65]. Through functional group engineering on the linkers, the researchers systematically fabricated a series of Ce-UiO66-X materials with varying concentrations of ligand defects (
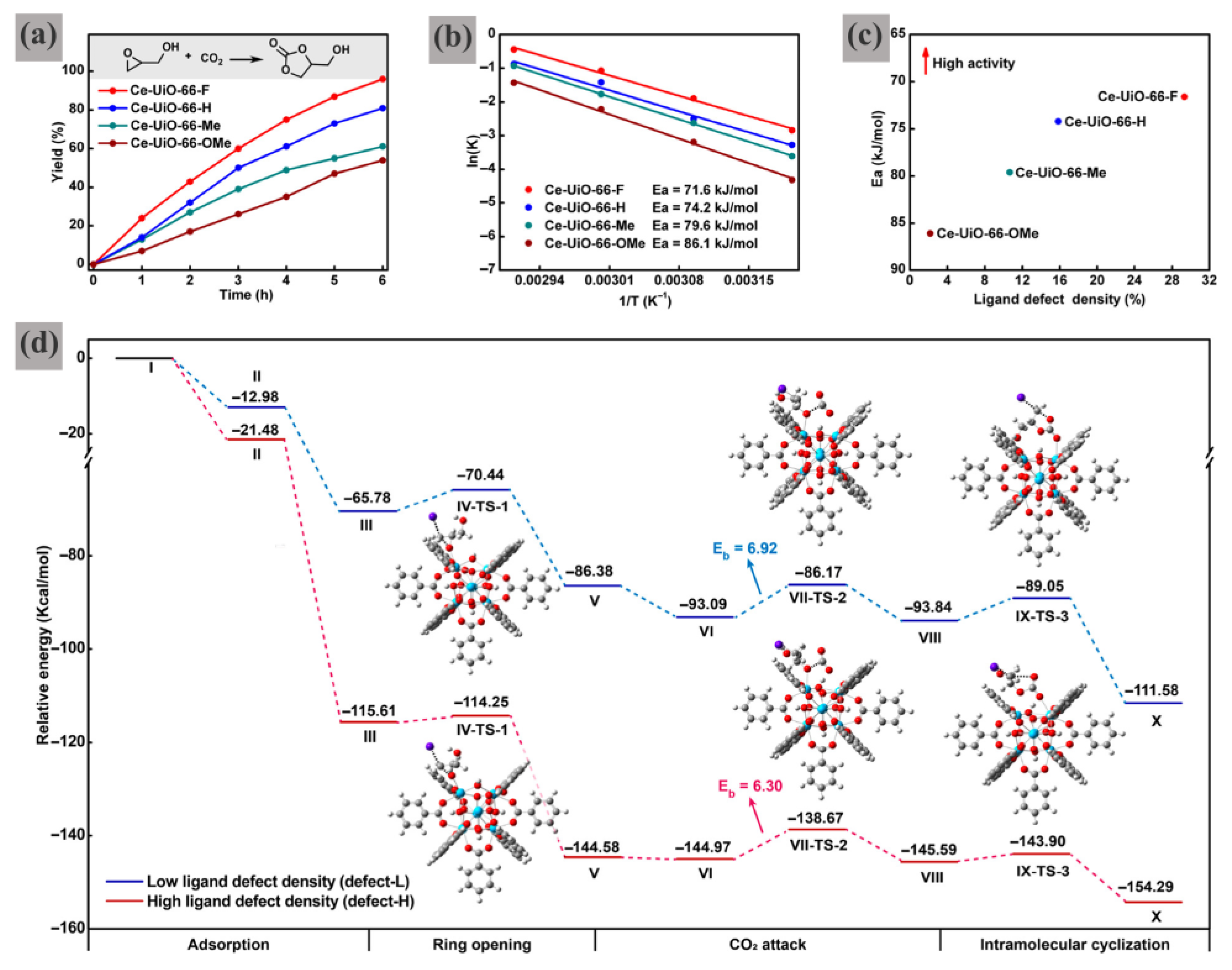
Figure 1a–c). The four isomorphic Ce-UiO-66-X samples (where X = OMe, Me, H, F) generated in this way have flexibly adjustable ligand defect densities (that is, unsaturated Ce sites). This characteristic is beneficial for establishing the structure−activity relationship. Among them, CeUiO-66-F, which contains the superior ligand defect density, presents the highest GC yield of 96% under mild reaction conditions, whereas Ce-UiO-66-OMe, with the lowest defect density, shows the lowest yield of 54% (
Figure 2a).
To comprehensively evaluate the catalytic performance, the activation energies (Ea) for the target reaction over Ce-UiO-66-X were calculated by the kinetic equations. The activation energy was calculated by analyzing the linear relationship between ln k and 1/T (
Figure 2b). An inverse correlation was observed between the MOF ligand defect concentration and Ea (
Figure 2c), indicating that coordinatively unsaturated Ce sites are responsible for catalyzing CO
2 cycloaddition.
Both experimental and theoretical studies have shown that the electron-withdrawing/donating properties of linker functional groups significantly influence the binding affinity between Ce-oxo clusters and BDC-X ligands, thereby enabling the tuning of ligand defect densities. When electron-donating groups (such as OMe and Me) are introduced to terephthalic acid (BDC), the coordination bonding strength between the Ce-oxo node and the BDC linker in Ce-UiO-66-OMe and Ce-UiO-66-Me is weakened. In contrast, electron-withdrawing substituents (e.g., fluorine) strengthen metal–ligand coordination bonds, leading to varied ligand defect concentrations (2.0–29.5%) in CeUiO-66-F (
Figure 1b). Notably, increased defect density substantially distorts MOF crystallinity while simultaneously decreasing both the coordination number and oxidation state of Ce centers (
Figure 1c). Furthermore, the inverse relationship between defect concentration and activation energy for CO
2 cycloaddition confirms that ligand-defect-generated unsaturated Ce sites enhance catalytic performance.
By combining the DFT calculations, a deep mechanistic understanding of the aforementioned correlation has been obtained. As shown in
Figure 2d, the carbonation process may undergo three transition states to obtain the main product of GC. The high-defect model (defect-H, two missing linkers) demonstrates greater exothermicity compared to the low-defect model (defect-L, one missing linker), despite showing comparable activation energies (D
E = 6.30 vs. 6.92 kcal/mol). Notably, glycidol adsorption at the unsaturated Ce site in defect-H yields a more favorable binding energy (−21.48 kcal/mol), indicating enhanced reactivity at higher defect concentrations. Furthermore, the entire carbonation pathway remains more exothermic for defect-H, corroborating experimental observations that structural defects facilitate the CO
2–glycidol coupling reaction.
This work represents the first attempt at establishing a convenient and clean methodology to accurately control the defect density in MOFs. However, the strategy is primarily validated for UiO-66-type MOFs; its applicability to other MOF topologies or more complex reactions remains unexplored, which requires further validation for broader applicability and practical robustness.
In another work reported by Monbaliu et al., they developed a continuous flow process toward the large-scale production of GC, relying on readily available nitrogen-containing Barton’s base as a potent homogeneous organocatalyst, which is an extension and in-depth study of the work previously reported in bio-based Gly to produce GD [
26].
The effect of temperature, CO
2 flow rate, and counter-pressure on the catalytic conversion of GD (
2) to GC (
3) was first investigated in a microfluidic reactor with 5 mol% of TBD (1,5,7-Triazabicyclo[4.4.0]dec-5-ene) in MEK (methyl ethyl ketone). The reaction conditions were optimized, involving processing at 140 °C (10 bar) with a 2 min residence time and 10 mL
N/min of CO
2, which struck a balance between yield and selectivity under intensified conditions (
Figure 3a). Subsequently, a range of homogeneous organic bases were screened as organocatalysts, such as TMG (1,1,3,3-tetramethylguanidine), DBU (1,8-diazabicyclo[5.4.0]undec-7-ene), BB (2-
tert-butyl-1,1,3,3-tetra-methylguanidine), and so on. Among them, BB showed the best catalytic performance, with a 91% GC yield in the case of a 5 mol% loading of the catalyst (
Figure 3b).
The scalability of the carbonation process was then determined in commercial mesofluidic glass reactors (Corning Advanced-Flow Reactors) in the temperature range of 120–150 °C and 10 bar with a feed solution of GD (1.8 M in MEK) and 1 equiv. of CO
2 in the presence of 1 mol% of BB. Similar trends to the microfluidic experiments were obtained. After that, the carbonation process was further evaluated in a pilot mesofluidic reactor with BB, and the typical flow chart is present in
Figure 4. Further intensification could lead to a 78% GC yield within a much shorter residence time (i.e., 28 s) at 140 °C. This result outperforms all conditions reported in the previous literature. The significance of this process is emphasized by its high throughput and low footprint, leading to an E-factor of 4.7 and an STY (Space Time Yield) of 2.7 kg h
−1 L
−1 for the upstream carbonation process.
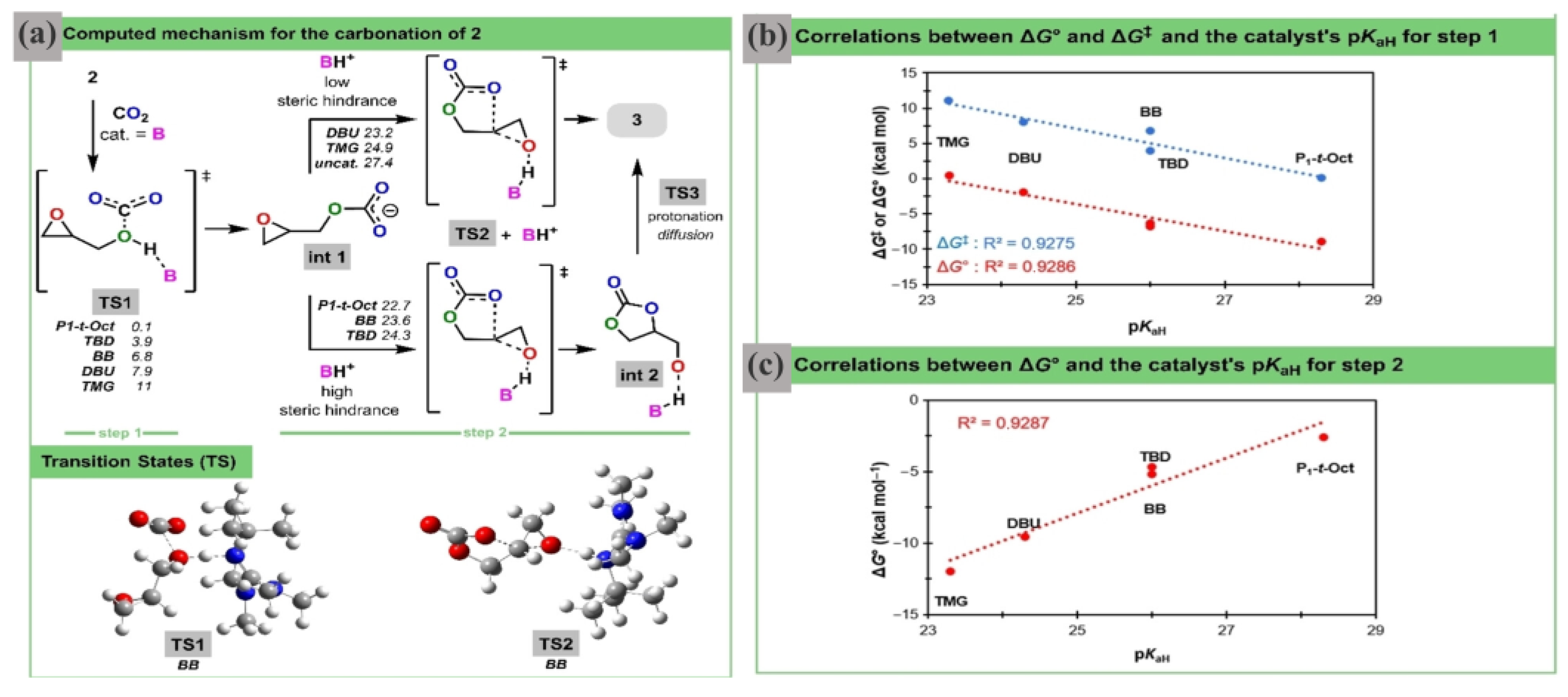
DFT calculations were employed to explain the necessity of both a β-hydroxyl group on the substrate and an organocatalyst with significant Brønsted basicity. Computation results highlighted the distinctive characteristics of a double H-shuffle mechanism for the coupling of CO
2 and GD following a two-step route (
Figure 5a). For step 1, the basicity of the catalyst is of the utmost significance (
Figure 5b). In fact, a high pK
aH guarantees a rapid and favorable CO
2 fixation, resulting in the formation of a highly stable conjugated acid along with a linear carbonate intermediate. In step 2, the presence of the conjugated acid significantly accelerates the reaction (
Figure 5c). This conjugated acid serves as a general Brønsted acid catalyst to activate the epoxide, although step 2 remains the overall rate-determining step. Opposing trends are seen in the activation energies of the two carbonation steps. This finding offers an understanding of why Barton’s base showed the most favorable outcomes for the entire transformation process. When a stronger base is used, it gives rise to a weaker conjugate acid, which then acts as a less efficient Brønsted acid for epoxide activation.
The above results verified that the carbonation of GD with CO2 was validated at the pilot scale in a commercial mesofluidic reactor upon using Barton’s base catalyst. Relatively high GC selectivity and yield could be achieved within a very short residence time with a low catalyst loading (1 mol%), demonstrating the unprecedented productivity of this intensified continuous flow process. This work represents a substantial enhancement of the existing conditions toward the intensified and scalable production of GC. Further efforts are still required in order to achieve large-scale industrial production, mainly involving the catalyst separation/recycling, solvent choices, and process optimization. In addition, the mechanistic insights should also be strengthened for promoting future catalyst development and the industrialization process.
In short, the coupling of CO2 and glycidol has shown significant potential for the industrial production of GC. Considerable efforts are still required in order to design and develop more efficient, stable, and recyclable catalysts for this process. To achieve this aim, it is necessary to deeply explore the catalytic mechanism and to analyze the key structural factors driving the generation of GC, which can provide guidance on further optimizing the catalytically active sites of the catalysts. Future research should also focus on driving technological development for producing glycidol from waste Gly, and on exploring low-cost, sustainable, and reusable catalysts that can adapt to low-purity glycidol and CO2 in industrial waste gas. Meanwhile, scalable process technologies need to be introduced in the early stage of technological research and development to promote the industrialization process in this field.
4. Synthesis of Glycerol Carbonate from Glycerol and Organic Carbonates
Currently, the transesterification of organic carbonates with glycerol (Gly) is a major route for industrial production of GC [
68,
69]. Several carbonates like ethylene carbonate (EC), dimethyl carbonate (DMC), propylene carbonate (PC), and diethyl carbonate (DEC) could be used for the transesterification process. Among them, the transesterification of Gly and DMC represents a highly attractive and sustainable route to GC, largely owing to DMC’s status as a “green reagent” [
70,
71]. In general, the transesterification of DMC with Gly could be conducted in the presence of homogeneous alkaline catalysts such as calcium oxide and sodium hydroxide [
12,
13,
14,
15]. In order to overcome the drawback of the homogeneous catalysis system, more recent effort has been devoted to the development of efficient heterogeneous catalysts with enhanced recyclability [
5,
72,
73]. To enlarge the scale of industrial implementation, it also needs to decrease the production cost of the reagents, since the organic carbonates, including DMC, are relatively expensive in comparison with other carbonylation agents (e.g., urea).
So far, a variety of metal oxides [
74,
75], mixed metal oxides [
76,
77], hydrotalcites [
78,
79], and zeolitic materials [
80,
81] have been investigated as solid catalysts for the transesterification reaction. We have listed in
Table 2 a selection of catalysts mentioned in the transesterification of glycerol and organic carbonates to facilitate a literature data comparison. However, most of them showed unsatisfied catalytic efficiency and/or poor recyclability. In addition, the nature of the catalytically active sites and the catalytic mechanisms remain inadequately explored. This knowledge gap can impede the rational design of cost-effective, high-efficiency catalysts for this transesterification reaction.
Recently, Fan et al. [
82] synthesized a series of La
2O
3-based catalysts doped with alkali and alkaline earth metals (M/La
2O
3), and systematically investigated their catalytic performance for the transesterification of DMC and Gly. In their work, a simple wet impregnation strategy was employed to modify La
2O
3 with alkali and alkaline earth metals, and synthetic mechanisms were investigated through Na/La
2O
3 catalysts (
Scheme 1). During impregnation, Na
+ anchors onto the La
2O
3 carrier via pore diffusion and surface adsorption, while partial ions can form surface ion pairs at the oxygen/hydroxyl sites. During the drying process, solvent evaporation induces dopant migration and promotes crystalline reconstruction, enabling the formation of uniformly distributed Na
x(NO
3)
y(OH)
z crystals on La
2O
3 [or La(OH)
3/La
2O
2CO
3] surfaces. During calcination, Na
x(NO
3)
y(OH)
z decomposes into a molten salt phase, facilitating the incorporation of Na
+ into the carrier lattice. The final products consist of sodium oxide (Na
2O) and reconstituted La
2O
3.
The XRD patterns suggest that the alkali metals dispersed uniformly on the La
2O
3 surface without forming additional crystalline oxide phases. In contrast, the alkaline-earth-metal-doped La
2O
3 samples exhibited much weaker and broader diffraction signals, accompanied by the appearance of a CaO, SrCO
3, or BaCO
3 phase, indicating that heavy alkaline earth metal doping could inhibit the formation of the La
2O
3 phase. The XPS profiles shown in
Figure 6 reveal the changes in the binding energies of the La 3d and O 1s spectra induced by alkali metals or alkaline earth metals. The La 3d
5/2 peak shifted towards a lower binding energy for the alkali-metal-doped catalysts, whereas no obvious shift occurred for the alkaline-earth-metal-doped samples. Meanwhile, the O 1s binding energies shifted towards higher values for both types of doped catalysts, and the shift induced by alkaline earth metals was more pronounced, indicating that alkaline earth metals have stronger interactions with the O species existing in La
2O
3. In addition, the CO
2-TPD measurements revealed that doping alkali or alkaline earth metals could significantly enhance the surface basicity of La
2O
3. Compared to their alkaline-earth-metal-doped analogs, the La
2O
3 samples modified with alkali metals exhibited a higher density of basic sites.
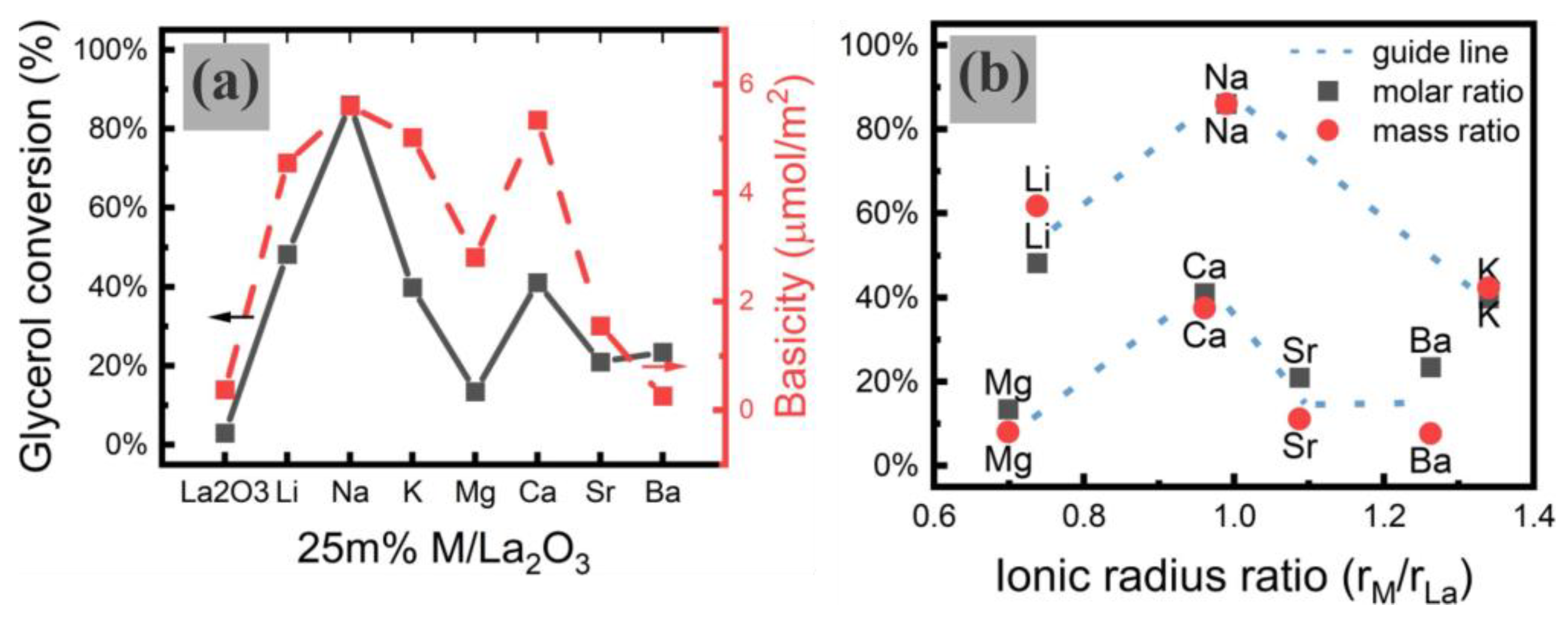
Among all the doped La
2O
3 catalysts, Na/La
2O
3 exhibit optimal catalytic performance (85% Gly conversion, 60% GC yield) for the transesterification of DMC with Gly. For La
2O
3 catalysts doped with different metals (
Figure 7a), the catalytic performance shows no proportionality to the surface basic site concentration. For instance, Na/ La
2O
3 and Ca/ La
2O
3 exhibit comparable basic site densities (5.60 vs. 5.34 μmol/m
2), yet Na/La
2O
3 achieves about 40% higher Gly conversion than Ca/La
2O
3. Similarly, the basic site densities of Li- and K-doped catalysts are also close to those of Na-doped La
2O
3 catalysts, but deliver significantly lower Gly conversions and GC yields.
Figure 7b shows the relationship between glycerol conversion and the ionic radius ratio of dopant and La, which could illustrate more clearly the reason why Na/La
2O
3 exhibits optimal catalytic performance. This enhancement may stem from the close match between the Na
+ ionic radius (1.02 Å) and the La
3+ ionic radius (1.03 Å), which promotes stable active site formation. In contrast, dopants with ionic radii significantly smaller or larger than that of La
3+ may cause structural deformation around the doped site, thus affecting the stability of the active site. Furthermore, XPS spectra (
Figure 6) demonstrate that alkali metals are more likely to donate their electrons than alkaline earth metals, while alkaline earth metals predominantly modulate electron distributions around O sites due to the extra phase of the alkaline earth metal oxides formed. This electronic behavior correlates with catalytic activity trends wherein alkali-metal-doped La
2O
3 catalysts exhibit superior performance. These results suggest that the ionic radius and valence of the dopant are the decisive factors, whereas the surface basicity, although crucial for the activation of Gly/DMC, does not determine the catalytic performance of the metal-doped La
2O
3 catalysts.
The recyclability of the spent Na/La2O3 catalyst was tested, showing that the catalyst could be reused with a slight decrease in catalytic activity after two cycles. To investigate the deactivation reason, the effects of the regeneration solvent and Na+ leaching were evaluated by additional experiments. The results indicated that the decrease in activity was due mainly to the surface wearing rather than the regeneration process or Na+ leaching. The surface wearing of the catalyst was mainly caused by the friction during the high-rate-stirring process, which was required for uniform dispersion in the immiscible Gly/DMC phase.
As shown in
Figure 8, the plausible reaction mechanism for Gly and DMC transesterification over Na/La
2O
3 catalysts was proposed. The carbonyl group of DMC and the hydroxyl group of Gly undergo activation at the La and O sites, respectively. Subsequently, the activated Gly anion attacks the carbonyl carbon of activated DMC, forming the intermediate of 1-(o-methoxy-carbonyl) glycerol complex (intermediate 1) and methanol. Intermediate 1 undergoes cyclization to form GC with another methanol molecule. The incorporation of alkali metals creates M-La centers that facilitate DMC activation and intermediate 1 cyclization, consequently leading to a higher GC yield than the pristine La
2O
3.
The authors believed that this research could help to understand the promotional role of alkali metals and alkaline earth metals and that the results may guide the future design of more efficient and stable metal oxide catalysts for the transesterification of DMC and Gly to produce GC. It is worth noting that the plausible mechanism (
Figure 8) is proposed based on the literature and has not yet been verified by in situ characterization of the intermediates (e.g., in situ FTIR). Verifying these intermediates would significantly advance the understanding of the reaction mechanism for alkali-/alkaline-earth-metal-doped oxides, offering new insights for rational catalyst design.
As part of the ongoing development of green GC synthesis methods, Wang et al. [
83] recently utilized an easily available and biocompatible sodium citrate (Na
3-citrate) catalyst for the efficient production of GC and ethylene glycol (EG) through the transesterification of Gly and ethylene carbonate (EC) (
Scheme 2).
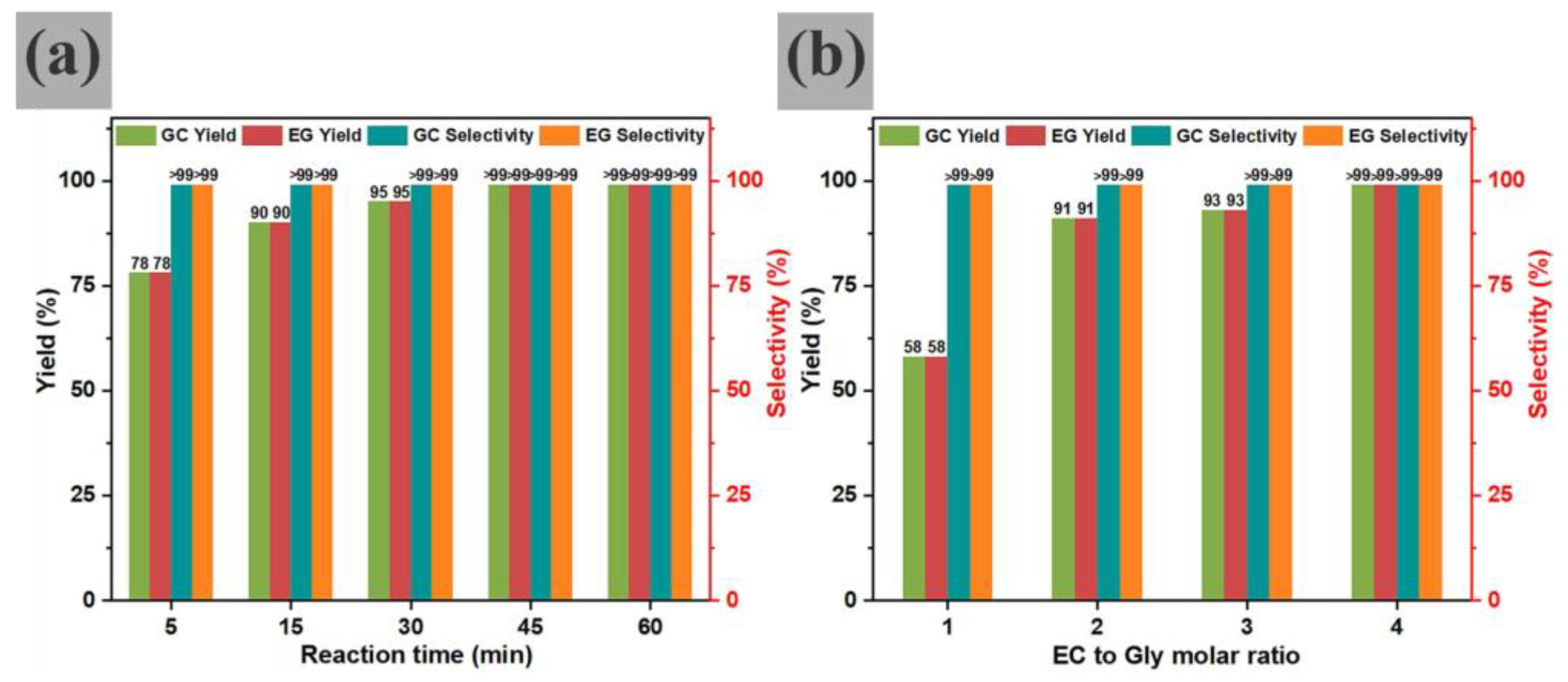
Under the test conditions, 0.01 wt% sodium citrate demonstrates exceptional catalytic activity, achieving >99% Gly conversion and >99% GC yield with a very high turnover frequency (TOF) of 16,925 g
GC·g
−1Cat·h
−1. The reaction time screening reveals that 45 min suffices for a >99% GC yield, while prolonging it to 60 min maintains >99% yields of both GC and EG. Notably, increasing the EC/Gly molar ratio from 1:1 to 4:1 elevates the GC yield from 58% to >99% (
Figure 9). Furthermore, a kinetic model for the transesterification reaction was established, revealing an activation energy of 51.3 kJ⋅mol
−1 for the Na
3-citrate catalyzation process. Combining the experimental results and the DFT calculations, it is demonstrated that Na
3-citrate promotes transesterification by forming strong hydrogen bonds with Gly.
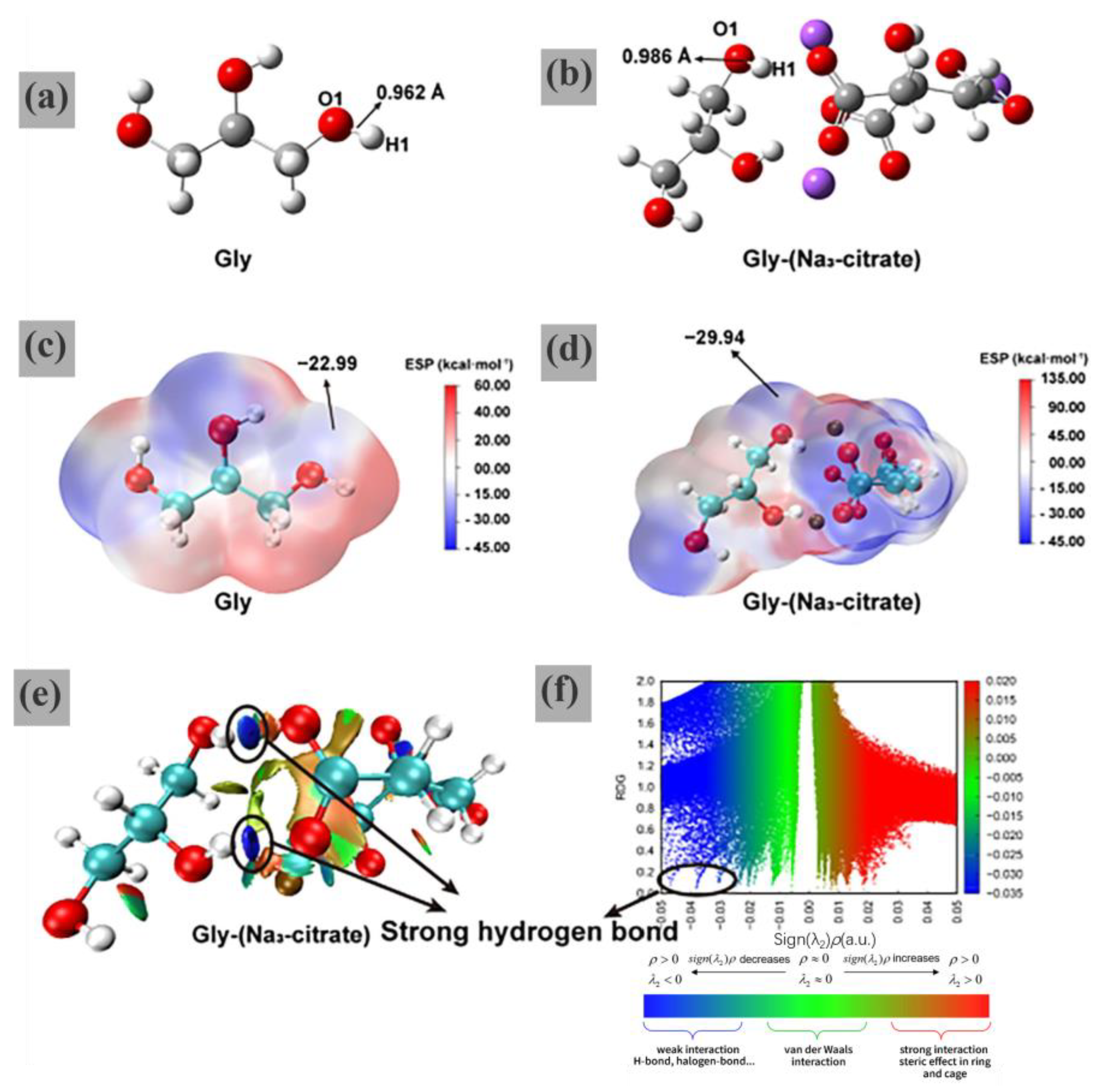
The FT-IR spectra show that the hydroxyl vibrational band of Gly is red-shifted in the Gly-(Na
3-citrate) complex, along with there being a decrease in peak intensity. It is tentatively suggested that Gly can be activated by sodium citrate through hydrogen bonding interactions. DFT calculations revealed that the O1-H1 bond length of Gly increased from 0.962 Å to 0.986 Å in the optimized structure of the Gly-(Na
3-citrate) complex, as shown in
Figure 10a,b. ESP analysis showed in
Figure 10c,d that the activated (Gly) exhibited a stronger negative potential near the O1 atom than that of the inactivated (Gly) (−29.94 vs. −22.99 kcal·mol
−1). The interaction between Gly and sodium citrate is mainly realized through a strong hydrogen bond (blue part in
Figure 10e) as a way to achieve the goal of activating the hydroxyl group of Gly. The characterization results reveal that Na
3-citrate activates the hydroxyl group of Gly, enhancing the nucleophilic attack on the electrophilic carbonyl carbon of EC to facilitate the transesterification process.
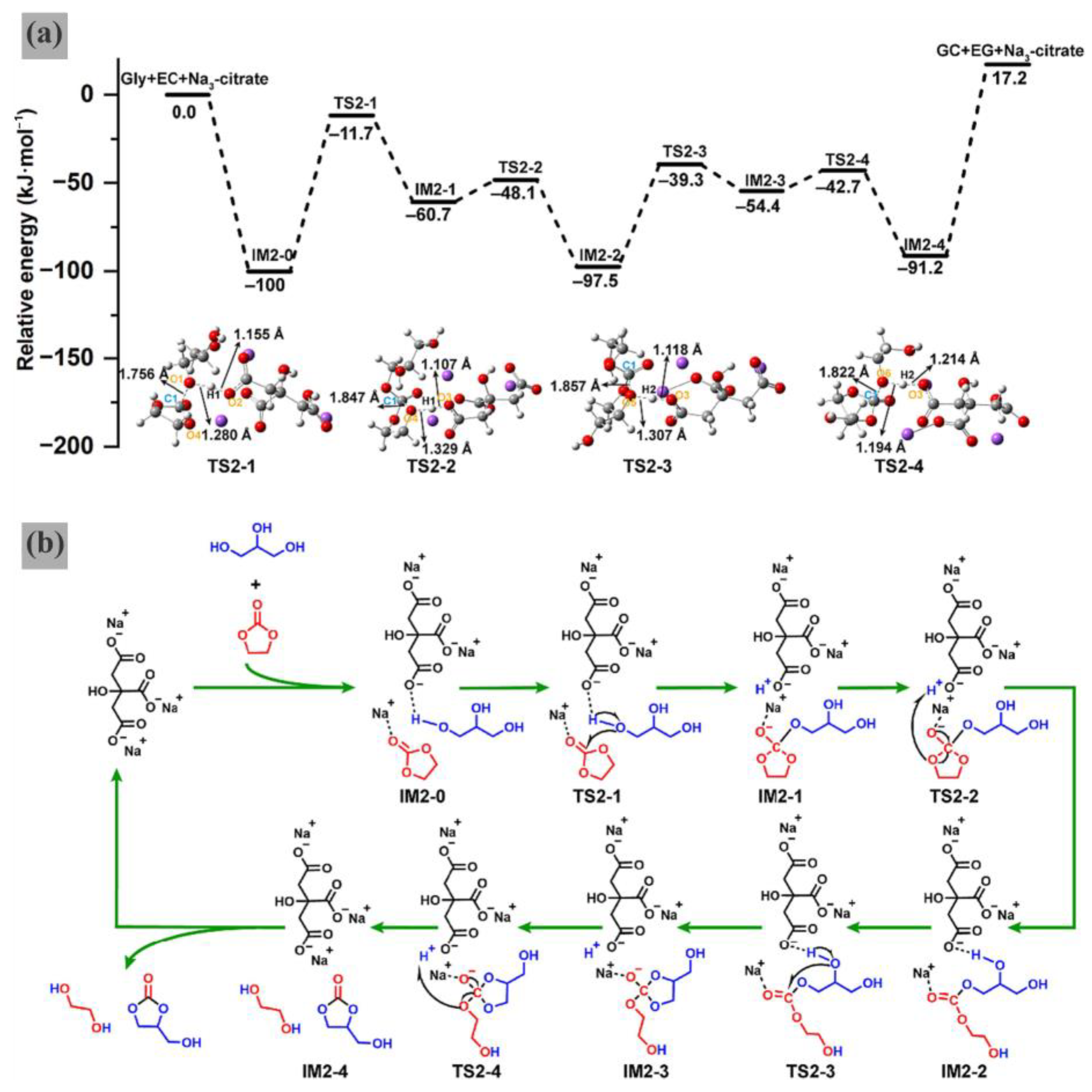
Notably, DFT calculations elucidated the potential energy profiles, optimized geometries, and possible catalytic mechanism for the Na
3-citrate-catalyzed transesterification of Gly and EC (
Figure 11a), which provide a clearer and more intuitive understanding of the reaction process. Reaction path analysis reveals a succession of steps separated by four transition states (TS2-1 to TS2-4) and five intermediates (IM2-0 to IM2-4). IRC analysis (
Figure 11b) describes in detail the process of bond breaking and formation at each step: First, the formation of a strong hydrogen bond between the H1 atom of Gly and the O1 of the -COO- group of Na
3-citrate leads to the breaking of the H1-O1 bond of Gly, and at the same time, the carbonyl oxygen (O1) of Gly becomes close to the carbon (C1) of EC. Subsequently, new O-H (O2-H1) and C-O (O1-C1) bonds are formed in IM2-1. The formation of IM2-2 involves the breakage of the C1-O4 and H1-O3 bonds, accompanied by the formation of the O4-H1 bond. After IM2-2 undergoes the breakage of the H2-O5 bond, both O5-C1 and O3-H2 bonds are formed to give the intermediate IM2-3. Finally, IM2-3 breaks the C1-O6 bond and the H2-O3 bond, forming the O6-H2 bond and giving rise to the intermediate IM2-4.
In short, this work revealed that the readily available Na3-citrate could act as an efficient catalyst for the transesterification of Gly and EC to produce GC. The established kinetic model confirms that the reaction could be easily achieved under a lower activation energy of 51.3 kJ⋅mol−1. DFT calculations demonstrated that the catalyst of Na3-citrate facilitated transesterification via the activation of Gly through a strong hydrogen bond interaction. The authors believed that this study may have offered an effective strategy for the catalytic transformation of Gly to GC. However, as a homogeneous catalyst, sodium citrate still presents challenges in post-reaction separation. Future research should focus on developing sodium citrate immobilization techniques (e.g., on magnetic carriers), exploring continuous flow processes, optimizing by-product control, and conducting economic analysis.
Although significant progress has been made in the transesterification synthesis of GC, the development of novel catalysts with high activity, excellent cyclic stability, and low cost remains a core challenge in this field. To solve this challenge, it is necessary to deeply analyze the micro-mechanisms of multi-step reactions in heterogeneous catalytic systems, and to elucidate the reaction pathways, deceleration steps, and inactivation kinetics through in situ characterization combined with kinetic simulations. This would be helpful to integrate the catalyst design with the reaction mechanism, and to then guide the precise design of active sites and the synergistic optimization of reaction parameters. In addition, much effort should be devoted to further reducing feedstock costs and advancing industrial sustainability. For instance, the feedstock of DMC could be produced from the captured CO2, thus building a closed-loop industrial chain of “carbon capture–chemical conversion–high-value product synthesis”. In this case, the Gly-DMC route would become a truly green, intelligent, and low-cost industrial production process for GC.
5. Catalysts for Glycerol Carbonate Synthesis from Glycerol and Urea
The carbonylation of Gly with urea to produce GC has gained considerable attention for its mild conditions, low cost, and easy product separation [
6,
7,
84]. We list some of the catalysts for the carbonylation of Gly with urea in
Table 3. Up to now, various catalysts have been tested for this process, including zinc-based catalysts [
8,
38,
85,
86], magnesium-based catalysts [
87], tungsten-based catalysts [
88,
89], ionic liquid catalysts [
90,
91,
92], and so on. In particular, zinc-based catalysts dominate this field due to their tunable Lewis acidity, stability, and availability. Inorganic components such as ZnO, ZnCl
2, and ZnSO
4 have shown a tendency to achieve high Gly conversion and GC selectivity. This tendency stems from the easy reactivity of Zn species with urea, which causes them to dissolve in the reaction medium. In order to solve the separation problem of these homogeneous catalysts, various solid Zn-based catalysts have been prepared and investigated for the carbonylation reaction. However, most Zn-based catalysts claimed to be heterogeneous fundamentally operate homogeneously [
33,
36,
85,
93,
94,
95].
Consequently, the design of novel heterogeneous catalysts, characterized by an abundance of stable and efficient high-density active sites, remains a highly attractive endeavor in the field of glycerol carbonylation. Some new progress has been made in GC synthesis in terms of catalyst design, characterization, and reaction mechanism studies. For instance, Shin and his collaborators synthesized disordered ZnAl
2O
4 spinel catalysts using the polymeric citrate complex method with various polyol precursors, providing a conceptual representation of the partially inverted ZnAl
2O
4 catalysts [
96].
As shown in
Figure 12, long-chain polyols with multiple adjacent hydroxyl groups increased disordered ZnAl
2O
4 spinel formation. The disorder degree in the disordered ZnAl
2O
4 spinel structure was determined by the order parameters and lattice parameters shown in
Figure 12b.
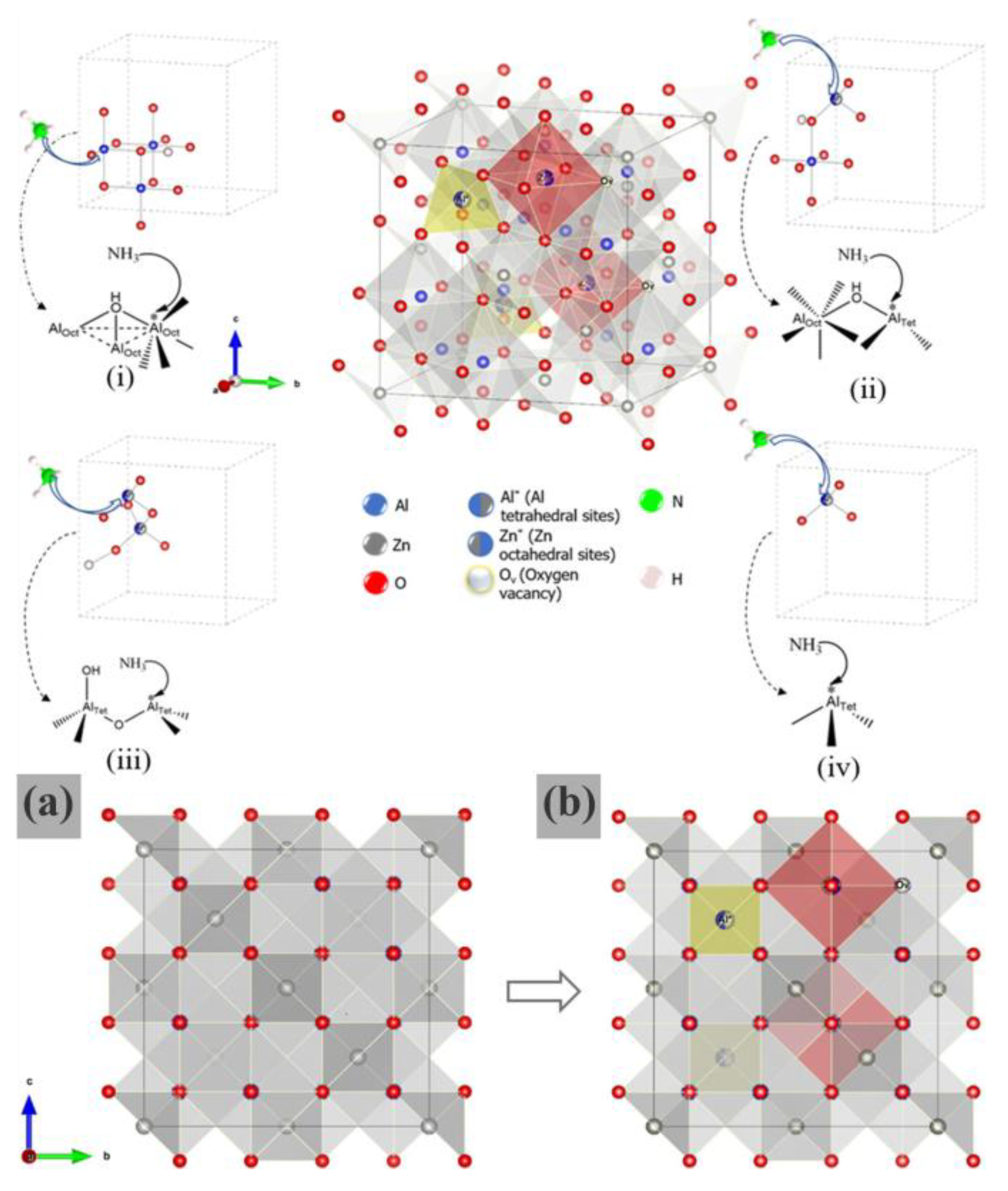
The differences between a normal ordered spinel lattice structure and a partially inverted one, which arise from the substitution of Zn
2+ and Al
3+ ions. The incorporation of ZnO
6 and AlO
4 sites via partial inversion enhances structural disorder and promotes surface acidity (
Scheme 3). The surface AlO
4 sites are associated with varying degrees of acidity, including medium, medium-strong, and strong acidic sites. In addition, the disordered s–ZnAl
2O
4 structure generated both acidic and basic sites. The superior performance of the s–ZnAl
2O
4 catalyst, as evidenced by its high GC yield and low (2) selectivity of 41% and 25%, respectively, further highlighted the relationship between the surface properties and the catalytic properties of the catalysts, as presented in
Figure 13.
The heightened degree of the disordered ZnAl
2O
4 structure makes it easier to form more acidic sites (
Figure 14). These acidic sites function as active sites for the urea molecule during the system. Furthermore, the disordered ZnAl
2O
4 generates more basic sites. These basic sites act as active sites for converting glycerol to accelerate the degradation of intermediate (2) and the formation of the main product GC. Consequently, the highly disordered s–ZnAl
2O
4 catalyst exhibits increased acidic and basic sites. These sites critically promote urea glycerolysis and subsequent GC generation, achieving the highest GC yield.
The above study demonstrates that polyols with longer chains and adjacent hydroxyl groups (e.g., sorbitol) may enhance the structural disorder of ZnAl2O4, thus leading to higher surface acidity and catalytic activity for GC production. However, the catalytic performance of the disordered ZnAl2O4 still needs to be further improved, since the selectivity of the ally by-product (i.e., 2,3-dihydroxypropyl carbamate) is relatively high (~25% selectivity) under the test conditions.
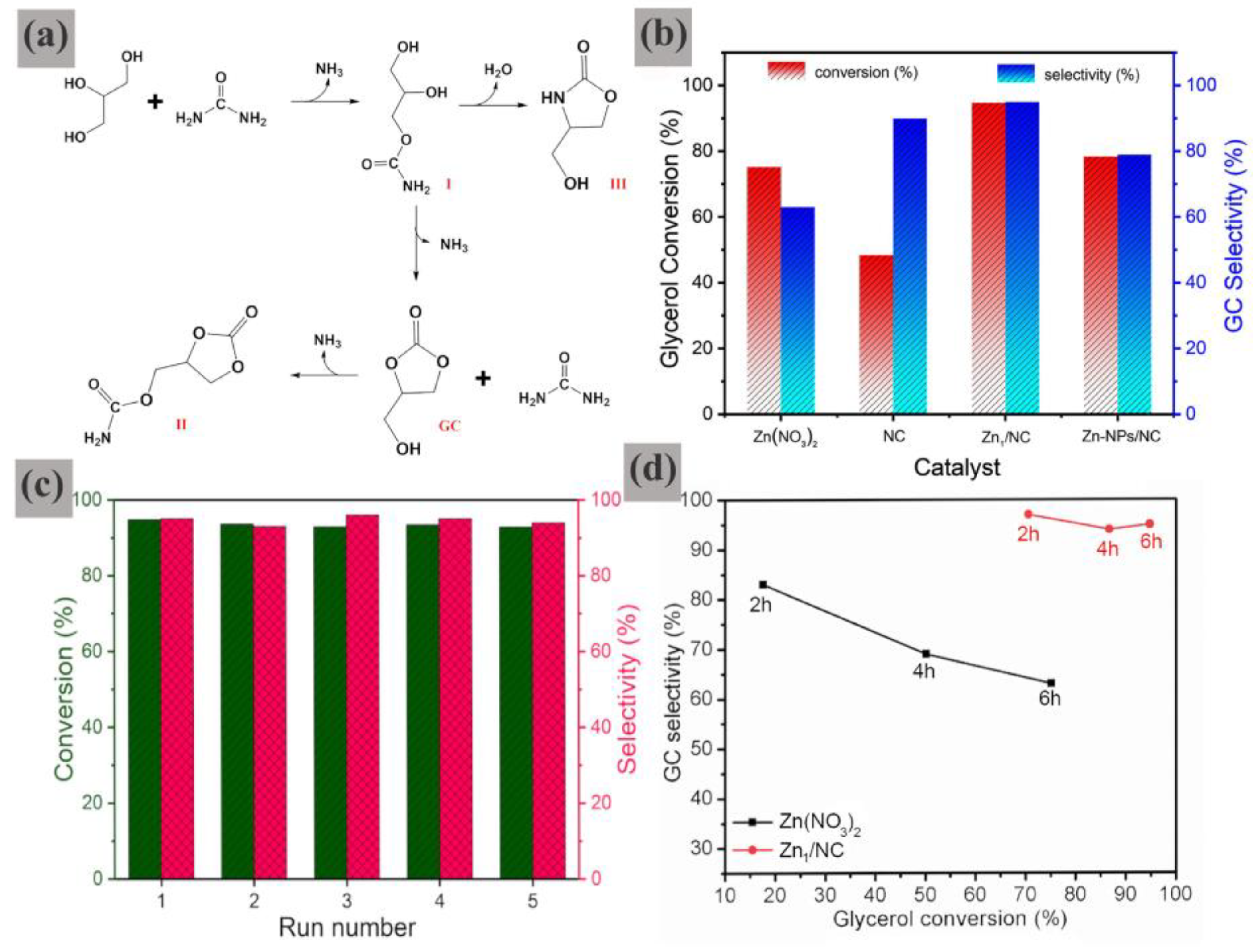
In another study reported by our research group, it was discovered that a single Zn atom catalyst supported on N-doped porous carbon (Zn
1/NC) demonstrated outstanding catalytic activity and stability in the carbonylation of Gly with urea to yield GC [
97]. As depicted in
Scheme 4, the Zn
1/NC catalyst was fabricated by immobilizing zinc salt on a type of N-doped porous carbon material (NC), followed by a thermal treatment procedure.
The single-atom structure of the Zn species could be validated through HR-TEM imaging, XANES, and EXAFS measurements (
Figure 15). The Zn
1/NC shows a single intensity peak at approximately 3.75 Å
−1, which can be attributed to the Zn-N/O contribution in the first nearest-neighbor coordination shell. The absence of detectable Zn-Zn signals indicates that the zinc atoms are isolated. These zinc atoms are anchored to the carbon support via Zn-O and/or Zn-N bonds. The fitting result of EXAFS suggests that the Zn atom is in a hexagonal coordination environment. Based on the structural characteristics of the NC support, Zn species in the Zn
1/NC catalyst likely adopt a coordination structure where single Zn
2+ ions are coplanarly coordinated to four N/O atoms from pyridine-like and carbonyl-like groups. This may involve additional weak coordination with two perpendicular O atoms from water molecules, forming a Zn
1-N
4-xO
x(H
2O)
2 configuration.
Interestingly, the Zn
1/NC catalyst demonstrates exceptionally high catalytic efficiency, achieving 94.8% glycerol conversion and 95.0% GC selectivity within 6 h, resulting in an impressive GC yield of 90.1%, and could be reused at least five times without any noticeable decline in either catalytic activity or GC selectivity (
Figure 16).
In principle, seven possible single-Zn-site models (Zn
1-N
4-xO
x, where x = 0–4) can be established using DFT, as illustrated in
Figure 17a. It is observed that only the adsorption energy (−2.42 kcal/mol) of urea on Zn
1-N
2O
2-1 is lower than that of glycerol (7.93 kcal/mol), indicating that urea molecules are more easily adsorbed on a single Zn site with this configuration (
Figure 17b). In the other cases, the single Zn sites preferentially adsorb glycerol molecules. Hence, it is reasonable to infer that the Zn
1-N
2O
2-1 model represents the preferred configuration of the primary active sites within the Zn
1/NC catalyst.
The FT-IR (3D-FTIR) profiles are also studied to obtain the clear evolution process of the carbonylation reaction in three-dimensional conditions, indicating that the Zn1/NC catalyst demonstrates a significantly greater capacity for effectively adsorbing and activating urea to produce the active intermediate and the primary product GC.
The study achieved highly efficient, selective, and stable catalytic performance in the glycerol carbonylation reaction with urea by designing a nitrogen-doped, porous, carbon-supported, and single-atom Zn catalyst. The mechanistic investigation was thorough, demonstrating significant academic value and application potential. However, the catalyst preparation involved multiple steps, including high-temperature carbonization, acid treatment, and wet impregnation, which may have increased the cost and difficulty of large-scale production.
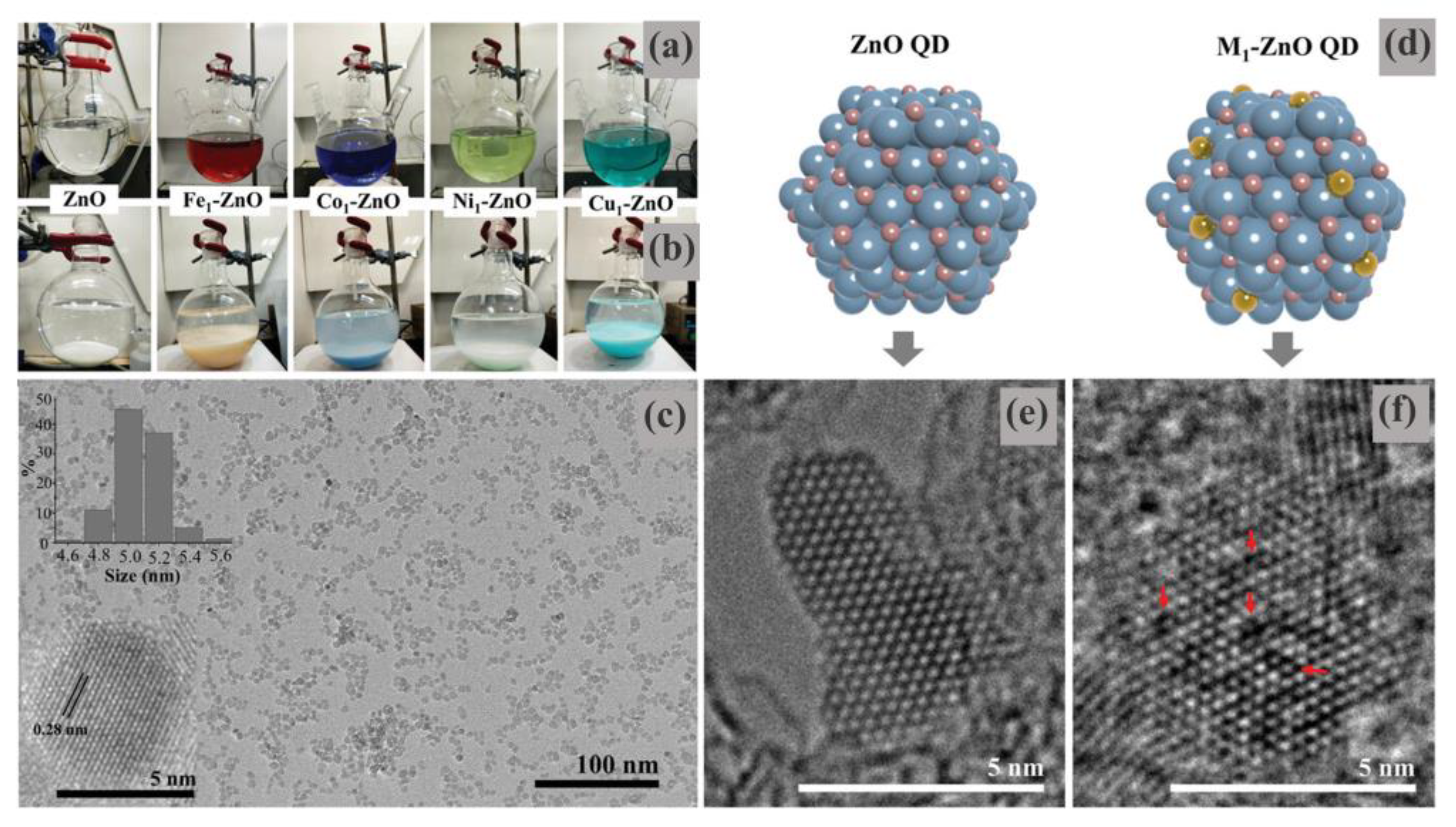
Similarly, Butburee et al. reported that a kind of transition metal single-atom-loaded zinc oxide quantum dots (M
1-ZnO QDs), which are synthesized by facile wet chemical synthesis (
Figure 18), may act as an efficient catalyst for the carbonylation reaction [
98]. Their structure has been downsized to a 0 D quantum dot to maximize the exposure of the active sites and promote solubility/homogeneity with the reactants. The trace amount of single metal atoms creates significant numbers of defects on ZnO QDs, which are beneficial for the carbonylation of Gly and urea molecules. As revealed by the TEM images (
Figure 18c,e), M
1-ZnO QDs (M = Fe, Co, Ni, and Cu) are uniform with an average size of 5.0 ± 0.6 nm, and the metal dopants are incorporated in the ZnO structure in an atomically dispersed form. In addition, M
1 could substitute some Zn atoms, as confirmed by the high-resolution TEM images (
Figure 18e, f). And the substitution of some Zn atoms by the metal dopants (M
1) could generate a significant number of defects in the structure, as evidently indicated by the dark-contrast spots, suggesting that some atoms are replaced or removed from the plan (red arrows in
Figure 18f).
The defective structure of the doped ZnO QDs could be further confirmed by XRD, XAS, and XPS results (
Figure 19a–f). This feature is consistent with the previous reports, showing that a trace amount of Fe doping in ZnO can appear in a single hexagonal wurtzite phase, in which Fe replaces the Zn atom to be incorporated into the lattice oxygen atoms in ZnO in the form of Zn
1−xFe
xO [
99].
As shown in
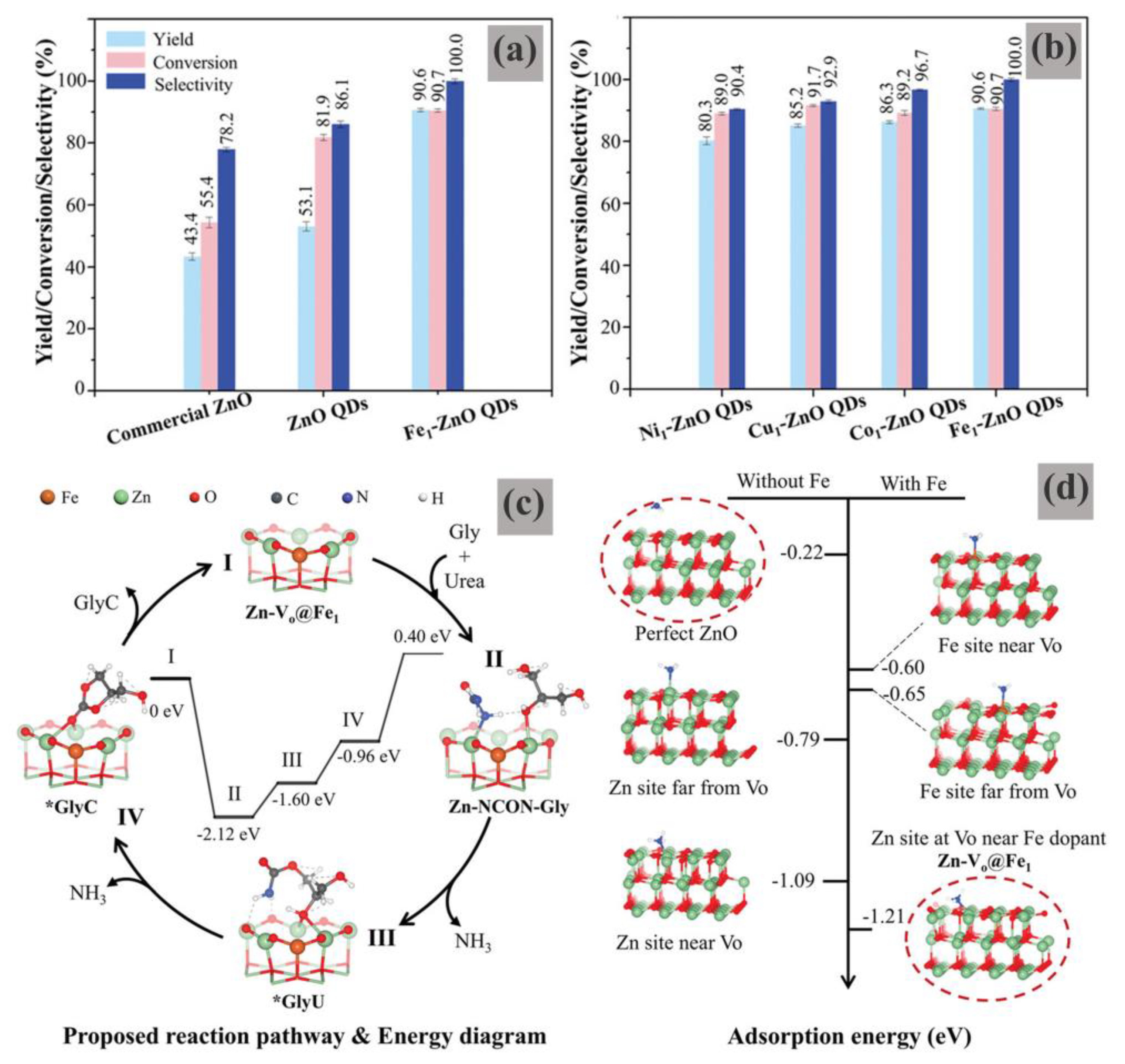
Figure 20, M
1-ZnO QDs exhibited both high activity and high selectivity for the carbonylation reaction, compared to the pristine ZnO QDs. Fe
1-ZnO QDs with enriched oxygen vacancies and isolated Fe atoms especially showed the highest performance among the investigated metals (Fe, Co, Ni, and Cu), with a 90.7% Gly conversion and a nearly 100% GC selectivity, respectively (
Figure 20a,b). The additional cycling experiments showed that the recovered catalyst was structurally stable, although the catalytic performance of the spent catalyst decreased somewhat during the recycling test, possibly related to the difficulty in separating the tiny particles from the reaction.
The catalytic behaviors were insightfully investigated by in situ Diffused Reflectance Infrared Fourier Transform Spectroscopy combined with the DFT calculations (
Figure 20c,d). The congruential results verified that the superior catalytic efficiency could be mainly attributed to the enriched Lewis acidity at Zn around the Fe1-Vo, which facilitated the formation of *GlyU, the key intermediate for the formation of GC. In addition, this active site was supported on the tiny ZnO QDs that could maximize the exposure to the reactants, leading to an impressive Gly-to-GC efficiency.
This study demonstrates that the highly efficient and selective conversion of Gly to GC could be conducted by designing Lewis acid defect sites through single-atom doping on ZnO quantum dots. However, the small size of the ZnO quantum dots (5 nm) may bring serious separation problems; this might be the main reason why approximately 20–30% of catalyst loss has been detected during the recycling tests, which is unfavorable for achieving practical applications.
In short, the above results suggest that the precise regulation of the structure and properties of the active centers may create highly active and stable heterogeneous Zn-based catalysts for the carbonylation of Gly with urea. In spite of this progress, more efforts are still required in order to further optimize the catalytic performance, especially the stability/recyclability of the heterogenous catalysts. In addition, some model catalysts with defined coordination structures and environments (e.g., Zn-containing complexes) should be synthesized to simulate the structure of active Zn species, which can be useful for deeply understanding the nature of catalytically active sites and reaction mechanisms. Moreover, integrated technologies, such as coupling the carbonylation processes with the recycling of the by-product ammonia, are highly attractive for developing more economic and effective GC production processes.
6. Conclusions
In this review, we summarize the recent research advances in the catalytic production of glycerol carbonate from bio-based glycerol involving different synthesis routes, including the coupling of CO2- and glycerol-derived glycidol, the transesterification of glycerol with organic carbonates, and the carbonylation of glycerol with urea. Significant progress has been made in catalyst design, reaction mechanism exploration, and catalytic process optimization. By choosing suitable catalyst preparation methods and combining this with catalytic process optimization, some novel homogeneous and heterogeneous catalytic systems with improved catalytic efficiency and stability/recyclability have been developed, which can achieve higher glycerol conversion and glycerol carbonate yields under optimized reaction conditions. This progress, though still quite limited, demonstrates clearly the great feasibility for the implementation of large-scale industrial applications in the near future. By combining a variety of characterization results and DFT calculations, the nature of the catalytic actives and the catalytic reaction mechanisms have also been explored; the obtained knowledge could guide the precise design and construction of more efficient catalysts for glycerol carbonate production.
To further promote the development of the related research and application fields, several challenges and opportunities involving catalyst preparation, reaction mechanism studies, and catalytic process optimization shall be addressed in future work. Firstly, maintaining the high catalytic efficiency of heterogeneous catalysts in long-term operations is challenging, especially in harsh reaction conditions (i.e., higher temperatures and pressures). This requires continuous innovation in catalyst design and preparation through systematically tuning the microstructure, composition, and surface acidity–basicity of the active centers.
Secondly, it is necessary to deeply explore the catalytic mechanisms involving a special reaction system for further guiding the design and preparation of new catalytic materials. For instance, there is still significant controversy regarding the understanding of the nature of active centers and reaction mechanisms for the heterogeneous Zn-based catalysts applied in the carbonylation of glycerol with urea. To deal with this issue, some model catalysts like Zn-containing complexes, which have a defined coordination structure and microenvironment, should be synthesized. This would be helpful for precisely simulating the structure of active Zn species and revealing the mechanism of active centers in multi-step reaction processes through the combination of various in situ characterization techniques, kinetic experiments, and DFT calculations.
Thirdly, much effort should be devoted to conducting the catalytic synthesis of glycerol carbonate using the true bio-based glycerol (crude biogenic glycerol) as a raw material. For a specific catalytic system, the utilization of the crude biogenic glycerol might be challenging, since it usually contains methanol, water, salts, fatty acids, and soaps, which may bring various negative effects, such as poisoning catalysts, promoting side reactions, and reducing product yields. In addition, the impurity profile varies depending on the biodiesel feedstock (e.g., vegetable oils vs. animal fats), leading to an unpredictable influence on the catalytic performance. Removing these contaminants (e.g., distillation, ion exchange) can certainly increase the energy consumption and process complexity, reducing economic viability. Hence, to enable large-scale deployment, it is necessary to develop cost-effective purification processes for a given bio-based glycerol, and to advance the catalytic technologies for the direct conversion of crude glycerol.
Moreover, it is highly desirable to integrate the related industrial production technologies, such as coupling the production process of glycerol carbonate (e.g., carbonylation or transesterification process) with the production process of the feedstock, for instance, to produce urea or DMC by reacting the by-product (ammonia or methanol) with the captured CO2. This would be highly attractive for developing a truly green, intelligent, and low-cost industrial production process for glycerol carbonate. Therefore, there is still a very large space and many opportunities in catalyst innovation, reaction mechanism research, and catalytic process optimization, which shall be essential to address the current challenges faced by the catalytic production of glycerol carbonate and to promote the development of the green chemical industry.



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}