Abstract
Surface segregation in bimetallic systems plays a critical role in material functionality, as electrochemical activity and catalytic performance are governed by the surface composition. To explore the influence of atomic oxygen on the surface composition of Pd–Ti alloys, density functional theory (DFT) simulations were utilized to analyze Ti segregation within Pd matrices. The adsorption behavior of atomic oxygen on Pd–Ti low-index (111), (100), and (110) surfaces was systematically investigated through energetic and electronic analyses. Simulation results reveal that Ti atoms prefer to remain in the bulk of the alloy under vacuum conditions, whereas oxygen adsorption induces significant Ti segregation to the surface layer. This oxygen-driven segregation is mechanistically linked to oxygen-surface bonding strength, as evidenced by correlating adsorption energetics with electronic structure modifications. These results provide a theoretical basis for engineering Pd–Ti alloys as high-performance catalysts in the oxygen reduction reaction.
1. Introduction
Bimetallic alloys are known to improve stability and efficiency in catalytic processes, driving their growing relevance in sustainable energy applications. Therefore, they have become a key area of exploration in materials science and engineering [1,2]. The catalytic behavior of an alloy is intricately governed by its surface composition and structure, which often differ significantly from the bulk material due to elemental segregation [3,4]. However, under reactive conditions, the alloy surface undergoes dynamic restructuring as preferential interactions between specific components and gaseous adsorbates create localized chemical potential gradients, triggering adsorbate-induced segregation [5,6]. A multitude of studies have confirmed that adsorbate-induced segregation can markedly have a significant impact on catalytic performance. For example, McCue et al. [7] have prepared Pd-segregated Cu–Pd catalysts induced by CO adsorption, which have been shown to enhance catalytic activity for selective acetylene hydrogenation, with minimal impact on ethylene selectivity. Similarly, Hua et al. [8] investigated the effect of CO-induced Pt segregation on Pt–Ag catalysts and demonstrated that Pt-segregated surface could enhance the active site of the oxygen reduction reaction (ORR) and reduce the over-potential of the reaction. In contrast, the results obtained by Wang et al. [9] revealed that the segregation of Co induced by O2 reduced the catalytic efficiency of Pt–Co catalysts in the ORR. Therefore, a deep understanding of adsorbate-induced surface segregation is fundamental to optimizing the activity of bimetallic catalysts.
Pd serves as a high-performance catalyst for critical chemical processes, including methanol steam reforming [10], the partial oxidation of methanol [11], methanol synthesis [12], the selective catalytic reduction of NOx [13], the water-gas shift reaction [14], and the hydrogen evolution reaction [15]. Pd is particularly prevalent in fuel cell applications as the primary electrocatalyst for the ORR [16]. Nevertheless, the scarcity and high cost of Pd hinder its catalytic efficiency and scalability [17]. These limitations necessitate the development of alternative catalysts that balance affordability with enhanced activity. Research efforts have prioritized Pd-based bimetallic alloys, where the partial substitution of Pd with cheaper transition metals reduces costs while optimizing performance [18,19,20,21,22]. The catalytic performance of these alloys is governed by surface geometric and electronic properties, which can be tailored through compositional adjustments. By engineering optimal atomic configurations, significant improvements in catalytic efficacy are achievable. Notably, alloying Pd with transition metals, especially with Ti, enhances ORR performance [23,24,25], with surface segregation critically influencing catalytic behavior [19,26]. For instance, Shao et al. [19] indicated that the segregation of Pd enhances the catalytic efficiency of Pd-based bimetallic catalysts for the ORR. Similarly, Ou et al. [26] demonstrated that a Pd-segregated surface layer on a Pd–Ti alloy exhibits higher ORR activity in comparison to pure Pd. This exceptional catalytic behavior has motivated extensive experimental and theoretical investigations into the segregation phenomena in Pd-based alloys [19,26,27,28,29]. While most prior studies emphasize surface/subsurface structural and electronic properties in vacuum environments, practical catalytic systems, such as the ORR, operate under reactive conditions where O atoms may be present. The adsorption of O atoms on the alloy surface can alter the segregation tendency of the Pd element and even dynamically reconfigure the alloy surface compositions, which significantly reduces the catalytic activity of Pd alloys in the oxygen reduction reaction. Obviously, comprehensive theoretical calculations are essential for gaining a deeper understanding of the experimental observations.
In this study, we employed DFT simulations to analyze Ti segregation on the Pd–Ti alloy surface in the presence of adsorbed atomic oxygen. The adsorption behavior of atomic oxygen on Pd–Ti low-index (111), (100), and (110) surfaces was systematically investigated through energetic and electronic analyses. Simulation results reveal that Ti atoms prefer to remain in the bulk of the alloy under vacuum conditions, whereas oxygen adsorption induces significant Ti segregation to the surface layer. This oxygen-driven segregation is mechanistically linked to oxygen-surface bonding strength, as evidenced by correlating adsorption energetics with electronic structure modifications. The remainder of this paper is structured as follows: Section 2 outlines the theoretical methods and computational details. Section 3 presents the results and discussion, and Section 4 provides a brief summary.
2. Computational Details
The spin-polarized DFT calculations in this study were performed using the Vienna Ab initio Simulation Package (VASP) code [30,31,32]. In all the calculations, we used the Perdew-Burke-Ernzerhof (PBE) functional [33] as the exchange-correlation functional to obtain the energy. The projector augmented-wave (PAW) potentials were employed to represent the interaction between ions and electrons [34,35], and a cutoff energy of 400 eV was selected to expand the electronic wave functions. The integrals in the reciprocal space were evaluated through the summations over 5 × 5 × 1 k-points in the Monkhorst–Pack grids [36]. We have tested the cutoff energy, the slab thickness, and the k-point mesh density to ensure the accuracy and speed of the calculations. The electric dipole correction was ignored in the slabs because of its negligible influence on the calculated results, and the converge criteria for electronic self-consistency was 10−5 eV.
In this study, three low-index crystallographic planes ((111), (100), and (110)) were selected for investigation due to their prevalence as dominant facets in bimetallic alloys [37,38], and the results showed that the surface energies for the (111), (100), and (110) surfaces of Pd were 1.33, 1.50, and 1.58 J/m2, respectively, in agreement with previous calculations [39]. Structural models comprised pure Pd and Pd–Ti alloy configurations that were constructed using a slab of six atomic layers representing 2 × 2 supercells containing 24 atoms for the (111), (100), and (110) surfaces. For the Pd–Ti alloy models, the configurations were generated by replacing one of the Pd atoms with a Ti atom in the first, second, third, and fourth layers of the pure metal Pd surface, respectively. Therefore, the concentrations of Pd and Ti are 23/24 and 1/24, respectively. Based on prior studies of bimetallic systems [38,40,41,42], a six-layer slab was shown to balance computational cost and accuracy in surface energy and adsorption property calculations. To minimize periodic boundary interactions, a 15 Å vacuum space was introduced between adjacent slabs, and a single oxygen atom was positioned on the alloy surface. The atomic positions for all layers except the bottom two were iteratively optimized until residual forces converged below 0.02 eV/Å. In contrast, the constrained bottom two layers retained bulk geometric parameters, including the equilibrium lattice constant of 3.94 Å for Pd, which is consistent with previously reported values [43]. By performing a convergence test on the slab thickness, we found that the number of layers fixed was sufficient for modeling the bulk characteristics.
To analyze the segregation behavior of Ti in Pd, the segregation energy () was introduced as the energy difference between a surface configuration and a bulk configuration [38,44]. Therefore, this parameter is computed by the following equation:
where the first term on the right-hand side denotes the total energy of the Pd–Ti system containing a Ti atom positioned within the upper x Pd layers (x = 1, 2, or 3). Conversely, the second one quantifies the total energy of the same alloy when the Ti atom resides in the fourth Pd atomic layer, which is analogous to the “bulk” Pd matrix.
The adsorption energy of an oxygen atom on metallic surfaces serves as a critical metric for evaluating thermodynamic behavior in surface science. This parameter is determined by the following equation:
In this equation, describes the total energy of the adsorbed system, corresponds to the bare surface energy, and characterizes the energy contribution from the reactive gas phase. Thermodynamically favorable adsorption is indicated by negative values of the .
3. Results and Discussion
3.1. Adsorption Behavior
Pd adopts a face-centered cubic structure, which gives rise to three low-index surfaces: (111), (110), and (100). To investigate atomic oxygen adsorption behavior on Pd and Pd–Ti surfaces, we systematically examined adsorption configurations across these surfaces, with the lowest adsorption energies summarized in Table 1. For the Pd(111) surface, prior experimental studies [45] have established that oxygen adsorption at the fcc site is more stable than at the hcp site, a trend corroborated by our computational results (−1.36 eV for fcc vs. −1.13 eV for hcp). Similarly, on the Pd(100) surface, oxygen preferentially occupies the fourfold hollow site, yielding an adsorption energy of −1.30 eV. In contrast, the fourfold hollow site is unstable on Pd(110) due to the significant lattice distortions required to achieve optimal O–Pd bonding distances [46]. Instead, oxygen stabilizes at a quasi-threefold coordinated hollow site with an adsorption energy of −1.20 eV.

Table 1.
The lowest adsorption energies (eV) for atomic oxygen on the (111), (100), and (110) surfaces of Pd and Pd–Ti.
In contrast to pure Pd surfaces, oxygen adsorption on Pd–Ti surfaces exhibits greater complexity. We performed an initial screening of oxygen adsorption sites on Pd–Ti surfaces with Ti atoms in the topmost layer. The most stable configurations (Figure 1) and corresponding adsorption energies (Table 1) reveal distinct trends compared to pure Pd surfaces. For Pd–Ti(111), oxygen preferentially occupies an fcc site adjacent to surface Ti atoms, with an adsorption energy of −2.61 eV. Similarly, on Pd–Ti(100), oxygen favors a fourfold hollow site near Ti, yielding −2.63 eV. However, on Pd–Ti(110), the fourfold hollow site is unstable, and adsorption stabilizes at a short bridge (‘s-bridge’) site near Ti (−2.75 eV). Further analysis reveals that Ti migration from the topmost layer to subsurface layers drastically reduces adsorption strength. For Pd–Ti(111), (100), and (110) surfaces with Ti in the second layer, adsorption energies decrease sharply to −1.17, −1.31, and −1.47 eV, respectively. When Ti resides in deeper bulk layers (third/fourth), the oxygen adsorption behavior converges with that of pure Pd surfaces, demonstrating that surface-exposed Ti atoms are critical for enhanced oxygen binding.
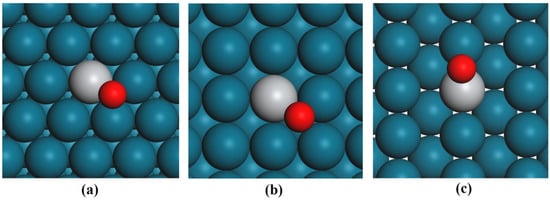
Figure 1.
Top views of the most stable adsorption configurations with Ti located at the topmost layer on the surfaces of (a) Pd–Ti(111), (b) Pd–Ti(100) and (c) Pd–Ti(110). The Pd, Ti and O atoms are represented by the blue-green, gray and red balls, respectively.
3.2. Segregation Behavior
To determine the location preference of Ti in Pd matrices, we calculated the segregation energies of a single Ti atom embedded in Pd using Equation (1). Figure 2 illustrates the segregation energy trends for Ti across Pd(111), (100), and (110) surfaces with and without adsorbed oxygen. In the absence of oxygen (Figure 2a), Ti exhibits positive segregation energies at the topmost surface layers, indicating thermodynamic instability at these positions. This behavior aligns with the higher surface energy of Ti (2.1 eV/atom) compared to Pd (2.05 eV/atom) [47], as elements with elevated surface energies typically resist surface segregation in metallic alloys [4,44]. Notably, Ti prefers subsurface or bulk regions: For Pd(111) and (100) surfaces, segregation energies at the subsurface layers are −0.23 eV and −0.24 eV, respectively, contrasting with positive values (0.61–0.69 eV) at the topmost layers. This suggests that the topmost Pd layer acts as a barrier against Ti segregation. However, on Pd(110), Ti’s most stable location shifts deeper into the bulk (fourth layer), with near-zero segregation energies at the third layer, reflecting bulk-like behavior. The distinct segregation trend on Pd(110) arises from its unique topology, where atoms in the second layer retain surface-like coordination [38].
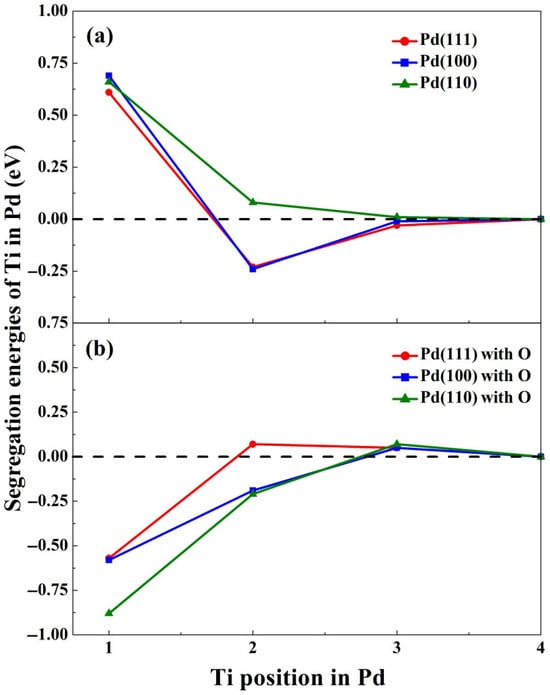
Figure 2.
Evolution of the segregation energies (eV) of Ti from the Pd ‘bulk’ (4th layer) to upper surface layers without (a) and with (b) oxygen adsorption.
The presence of adsorbed oxygen dramatically alters Ti segregation behavior. As shown in Figure 2b, oxygen chemisorption reduces Ti segregation energies at the topmost layers of Pd–Ti(111), (100), and (110) surfaces to −0.57, −0.58, and −0.88 eV, respectively. This destabilizes the Pd surface barrier, enabling Ti segregation. For Pd–Ti(110), oxygen adsorption shifts the segregation energy of Ti at the second layer from 0.08 eV to −0.21 eV, facilitating rapid Ti migration to the surface. These results agree with experimental observations of Ti enrichment on Pd-based catalysts under reactive conditions [48]. Adsorbate-induced segregation is governed by three competing factors: the alloying element’s surface energy, atomic radius, and adsorbate–surface binding strength [6,49]. While Ti’s higher surface energy normally inhibits segregation, the strong Pd–O binding and Pd’s smaller atomic radius (compared to Ti) dominate under oxygen-rich conditions, overriding this limitation and driving Ti to the surface.
3.3. Electronic Structure Analysis
The electronic density of states (DOS) serves as a critical tool for probing the electronic characteristics of transition metal alloys, particularly their catalytic behavior. To elucidate the role of local coordination and adsorbate interactions, we calculated the d-band DOS for the Ti atom in Pd–Ti systems without oxygen adsorption. Figure 3a illustrates the d-band DOS for the Ti atom located in the first layer of Pd–Ti(111), Ti(111), Pd–Ti(100), and Pd–Ti(110) surfaces under vacuum. Notably, embedding Ti within the Pd(111) surface induces significant narrowing of its d-band compared to the pure Ti(111) surface. This phenomenon is attributed to lattice strain effects. This narrowing arises from Ti’s larger atomic radius relative to Pd, generating localized tensile strain at the surface. Consequently, electron density occupancy decreases, compressing the d-band width and shifting the d-band center upward toward the Fermi level to preserve orbital filling integrity [40]. In addition, we observe that the alloyed Ti’s d-band center lies above the Fermi level. This further confirms that the high electronegativity of Pd leads to Ti charge loss. By comparing the positions of the d-band center for the Ti atom on the Pd–Ti(111), Pd–Ti(100), and Pd–Ti(110) surfaces under vacuum, we observe that peak positions correlate with the density of accessible d-states, where reduced atomic coordination elevates the d-band center toward higher energies. This electronic redistribution enhances orbital hybridization capacity at low-coordination sites, directly rationalizing the strengthened oxygen binding affinity quantified in Table 1. Figure 3b shows the d-band DOS variations in Ti atoms positioned on the first, second, and third layers of the Pd(110) surface in the absence of adsorbed oxygen. Significant alterations in the shape and energy alignment of the Ti d-band DOS occur as Ti migrates from bulk regions to surface sites. Specifically, the primary d-band peak associated with Ti in the topmost layer exhibits greater intensity and shifts markedly closer to the Fermi level, implying that surface energy considerations predominantly dictate Ti’s preferential occupation of bulk regions.
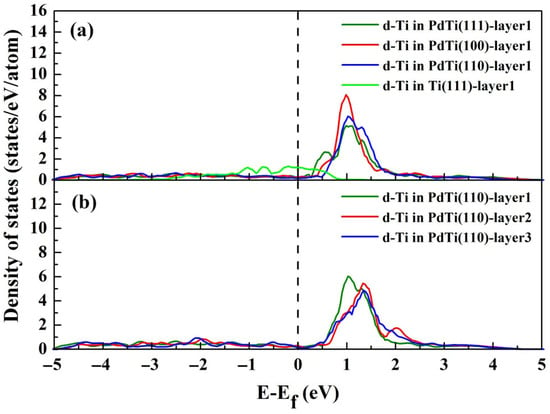
Figure 3.
(a) The d-band DOS for the Ti atom positioned on the first layer of Pd–Ti(111), Pd–Ti(100), and Pd–Ti(110) surfaces without oxygen adsorption. (b) The d-band DOS for the Ti atom positioned on the first, second, and third Pd–Ti(110) layers without oxygen adsorption.
Figure 4 demonstrates significant modifications to the d-band DOS of Ti in the Pd–Ti(110) system upon oxygen interaction compared to the pristine surface (i.e., without adsorbed oxygen). Calculations show that oxygen adsorption leads to a significant upward shift in the center of the d-band of Ti atoms on the surface of Pd–Ti(110). This upward shift in the d-band center will induce an increase in the number of unoccupied anti-bonding states and enhance the strength of adsorption between Ti and O, which is conducive to driving the segregation of sub-surface Ti towards the surface and the formation of the Ti-rich surface layer. Notably, oxygen adsorption also induces a marked broadening of the Ti d-band DOS. The narrowed DOS distributions observed for surface atoms are straightforward to interpret, as reduced coordination numbers relative to bulk counterparts limit orbital overlap. In adsorption scenarios, the increased local coordination of surface atoms caused by adsorbates counteracts this narrowing effect, leading to DOS broadening through enhanced orbital hybridization [50]. Additionally, pronounced overlap between O p-states and Ti d-states is observed, with unoccupied O–Ti anti-bonding states positioned above the Fermi level. This electronic configuration facilitates robust covalent hybridization between Ti and oxygen, a critical feature for catalytic interaction strength.
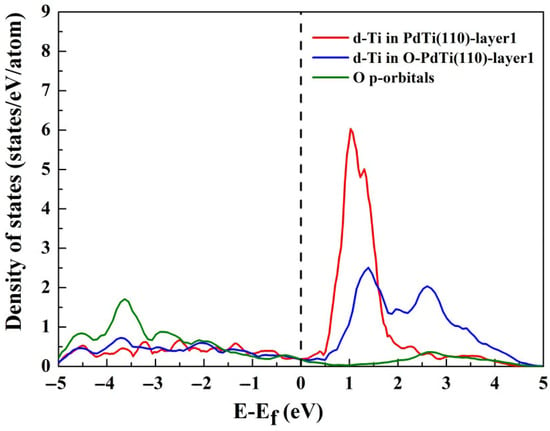
Figure 4.
The d-band DOS for the Ti atom positioned on the first layer of Pd–Ti(110) surfaces with and without oxygen adsorption and the oxygen p-band DOS.
In order to investigate the effect of oxygen adsorption on the electronic structure of the Pd–Ti(110) surface, we carried out a Bader charge analysis of Ti atoms positioned on the first layer of the alloy surface in the presence and absence of oxygen adsorption. In the absence of oxygen adsorption, the Pd atoms gained 1.2 electrons from their neighboring Ti atom, suggesting that the charge transfer is significant and that the Ti atom is less likely to segregate to the alloy surface because of strong covalent Pd–Ti bonds. However, oxygen adsorption significantly alters this charge balance. When atomic oxygen adsorbs to the surface, the O and Pd atoms gain 0.84 and 0.62 electrons, respectively, from the Ti atoms. This electron depletion weakens the covalent bonds of Pd–Ti and strengthens the adsorption strength between O and Ti atoms, creating a thermodynamic driving force for Ti to segregate towards the surface.
4. Conclusions
First-principles computational methods were employed to analyze Ti segregation on the Pd–Ti alloy surface in the presence of adsorbed atomic oxygen. The adsorption behavior of atomic oxygen on Pd–Ti low-index (111), (100), and (110) surfaces was systematically investigated through energetic and electronic analyses. Our results reveal that Ti atoms prefer to remain in the bulk of the alloy under vacuum conditions, whereas oxygen adsorption induces significant Ti segregation to the surface layer. Examining the adsorption behavior of atomic oxygen on Pd–Ti low-index surfaces demonstrates a binding strength order of (110) > (100) > (111), with all three low-index surfaces exhibiting stronger affinity for Ti than Pd. Electronic structure analysis for Pd–Ti systems shows that embedding Ti within the Pd surface layer induces significant narrowing of its d-band compared to pure Ti surfaces. This phenomenon is attributed to lattice strain effects. The alloyed Ti’s d-band center lies above the Fermi level, further confirming charge depletion caused by Pd’s higher electronegativity. Notably, significant alterations in the shape and energy alignment of the Ti d-band DOS occur as Ti migrates from bulk regions to surface sites. Specifically, the primary d-band peak associated with Ti in the topmost layer exhibits greater intensity and shifts markedly closer to the Fermi level, implying that surface energy considerations predominantly dictate Ti’s preferential occupation of bulk regions. Interestingly, oxygen adsorption induces a marked broadening of the Ti d-band DOS. The narrowed DOS distributions observed for surface atoms are straightforward to interpret, as reduced coordination numbers relative to bulk counterparts limit orbital overlap. These results provide a theoretical basis for engineering Pd–Ti alloys as high-performance catalysts in the oxygen reduction reaction.
Author Contributions
Conceptualization, Y.Y.; methodology, Y.W.; software, G.Z.; validation, Y.S.; formal analysis, H.G.; investigation, Y.S.; resources, H.G.; data curation, Y.L.; writing—original draft preparation, Y.W.; writing—review and editing, Y.Y.; visualization, Y.L.; supervision, Y.Y.; project administration, Q.H.; funding acquisition, Y.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Specialized Fund for the Doctoral of Kaili University, grant number: BS202502013, and Guizhou Provincial Basic Research Program (Natural Science) of Youth Guidance (Qian Ke He Jichu-[2024]Qingnian 024).
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article.
Acknowledgments
The authors would like to acknowledge the support of the Specialized Fund for the Doctoral of Kaili University (BS202502013), and Guizhou Provincial Basic Research Program (Natural Science) of Youth Guidance (Qian Ke He Jichu-[2024]Qingnian 024), and the National Natural Science Foundation of China (12064019).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chen, S.; Jelic, J.; Rein, D.; Najafishirtari, S.; Schmidt, F.-P.; Girgsdies, F.; Kang, L.; Wandzilak, A.; Rabe, A.; Doronkin, D.E.; et al. Highly loaded bimetallic iron-cobalt catalysts for hydrogen release from ammonia. Nat. Commun. 2024, 15, 871. [Google Scholar] [CrossRef]
- Gholinejad, M.; Bashirimousavi, S.; Sansano, J.M. Novel magnetic bimetallic AuCu catalyst for reduction of nitroarenes and degradation of organic dyes. Sci. Rep. 2024, 14, 5852. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Hu, Q.; Xiao, W.; Wang, J.; Wang, L. Design of highly efficient Ni-based water-electrolysis catalysts by a third transition metal addition into Ni3Mo. Intermetallics 2018, 94, 99–105. [Google Scholar] [CrossRef]
- Yu, Y.; Xiao, W.; Wang, J.; Wang, L. Understanding the surface segregation behavior of transition metals on Ni (111): A first-principles study. Phys. Chem. Chem. Phys. 2016, 18, 26616–26622. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, Z.; Huang, W.; Zhou, S.; Hu, Z.; Wang, L. Density functional theory study of Ni segregation in CuNi (111) alloy with chemisorbed CO, O, or H. J. Phys. Chem. Solids 2022, 171, 111021. [Google Scholar] [CrossRef]
- Yu, Y.; Huang, W.; Liu, Z.; Hu, Z.; Wang, L. First-principles study of surface segregation in bimetallic Cu3M (1 1 1)(M= Au, Ag, and Zn) alloys in presence of adsorbed CO. Comput. Mater. Sci. 2022, 212, 111550. [Google Scholar] [CrossRef]
- Mccue, A.J.; Anderson, J.A. CO induced surface segregation as a means of improving surface composition and enhancing performance of CuPd bimetallic catalysts. J. Catal. 2015, 329, 538–546. [Google Scholar] [CrossRef]
- Hua, M.; Tian, X.; Li, S.; Shao, A.; Lin, X. Theoretical design of platinum–sliver single atom alloy catalysts with CO adsorbate-induced surface structures. Phys. Chem. Chem. Phys. 2022, 24, 19488–19501. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Gu, X.-K.; Li, W.-X. First-principles study on the origin of the different selectivities for methanol steam reforming on Cu (111) and Pd (111). J. Phys. Chem. C 2010, 114, 21539–21547. [Google Scholar] [CrossRef]
- Wojcieszak, R.; Gaigneaux, E.M.; Ruiz, P. Low Temperature-High Selectivity Process over Supported Pd Nanoparticles in Partial Oxidation of Methanol. ChemCatChem 2012, 4, 72–75. [Google Scholar] [CrossRef]
- Koizumi, N.; Jiang, X.; Kugai, J.; Song, C. Effects of mesoporous silica supports and alkaline promoters on activity of Pd catalysts in CO2 hydrogenation for methanol synthesis. Catal. Today 2012, 194, 16–24. [Google Scholar] [CrossRef]
- Zhang, Y.; Zeng, H.; Jia, B.; Wang, Z.; Liu, Z. Selective catalytic reduction of NOx by H2 over Pd/TiO2 catalyst. Chin. J. Catal. 2019, 40, 849–855. [Google Scholar] [CrossRef]
- Chutia, A.; Thetford, A.; Stamatakis, M.; Catlow, C.R.A. A DFT and KMC based study on the mechanism of the water gas shift reaction on the Pd(100) surface. Phys. Chem. Chem. Phys. 2020, 22, 3620–3632. [Google Scholar] [CrossRef]
- Liao, H.; Wei, C.; Wang, J.; Fisher, A.; Sritharan, T.; Feng, Z.; Xu, Z.J. A multisite strategy for enhancing the hydrogen evolution reaction on a nano-Pd surface in alkaline media. Adv. Energy Mater. 2017, 7, 1701129. [Google Scholar] [CrossRef]
- Antolini, E. Palladium in fuel cell catalysis. Energy Environ. Sci. 2009, 2, 915–931. [Google Scholar] [CrossRef]
- Xiao, L.; Zhuang, L.; Liu, Y.; Lu, J. Activating Pd by morphology tailoring for oxygen reduction. J. Am. Chem. Soc. 2009, 131, 602–608. [Google Scholar] [CrossRef]
- Savadogo, O.; Lee, K.; Oishi, K.; Mitsushima, S.; Kamiya, N.; Ota, K.-I. New palladium alloys catalyst for the oxygen reduction reaction in an acid medium. Electrochem. Commun. 2004, 6, 105–109. [Google Scholar] [CrossRef]
- Shao, M.; Huang, T.; Liu, P.; Zhang, J.; Sasaki, K.; Vukmirovic, M.; Adzic, R. Palladium monolayer and palladium alloy electrocatalysts for oxygen reduction. Langmuir 2006, 22, 10409–10415. [Google Scholar] [CrossRef]
- Lee, K.; Savadogo, O.; Ishihara, A.; Mitsushima, S.; Kamiya, N.; Ota, K.-I. Methanol-tolerant oxygen reduction electrocatalysts based on Pd-3D transition metal alloys for direct methanol fuel cells. J. Electrochem. Soc. 2005, 153, A20. [Google Scholar] [CrossRef]
- Zhang, L.; Lee, K.; Zhang, J. The effect of heat treatment on nanoparticle size and ORR activity for carbon-supported Pd–Co alloy electrocatalysts. Electrochim. Acta 2007, 52, 3088–3094. [Google Scholar] [CrossRef]
- Shao, M.-H.; Sasaki, K.; Adzic, R.R. Pd−Fe nanoparticles as electrocatalysts for oxygen reduction. J. Am. Chem. Soc. 2006, 128, 3526–3527. [Google Scholar] [CrossRef]
- Fernández, J.L.; Raghuveer, V.; Manthiram, A.; Bard, A.J. Pd−Ti and Pd−Co−Au electrocatalysts as a replacement for platinum for oxygen reduction in proton exchange membrane fuel cells. J. Am. Chem. Soc. 2005, 127, 13100–13101. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, C. Nanoporous PdTi alloys as non-platinum oxygen-reduction reaction electrocatalysts with enhanced activity and durability. ChemSusChem 2013, 6, 78–84. [Google Scholar] [CrossRef]
- Sun, Y.; Alpuche-Aviles, M.; Bard, A.; Zhou, J.; White, J. Preparation and characterization of Pd-Ti electrocatalyst on carbon supports for oxygen reduction. J. Nanosci. Nanotechnol. 2009, 9, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Ou, L. Design of Pd-Based Bimetallic Catalysts for ORR: A DFT Calculation Study. J. Chem. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Ruban, A.V.; Skriver, H.L.; Norskov, J.K. Surface segregation energies in transition-metal alloys. Phys. Rev. B 1999, 59, 15990–16000. [Google Scholar] [CrossRef]
- Ruban, A.V.; Skriver, H.L. Calculated surface segregation in transition metal alloys. Comput. Mater. Sci. 1999, 15, 119–143. [Google Scholar] [CrossRef]
- Løvvik, O. Surface segregation in palladium based alloys from density-functional calculations. Surf. Sci. 2005, 583, 100–106. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 1993, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter 1994, 50, 2665–2668. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Hendrik, J.; James, D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Dai, Z.; Borghetti, P.; Chenot, S.; David, P.; Koltsov, A.; Jupille, J.; Cabailh, G.; Goniakowski, J.; Lazzari, R. Aluminium segregation profiles in the (110), (100) and (111) surface regions of the Fe0.85Al0.15 random body-centered cubic alloy. Appl. Surf. Sci. 2019, 492, 886–895. [Google Scholar] [CrossRef]
- Dhifallah, M.; Dhouib, A.; Aldulaijan, S.; Di Renzo, F.; Guesmi, H. First-principles study of Au–Cu alloy surface changes induced by gas adsorption of CO, NO, or O2. J. Chem. Phys. 2016, 145, 024701. [Google Scholar] [CrossRef]
- Singh-Miller, N.E.; Marzari, N. Surface energies, work functions, and surface relaxations of low-index metallic surfaces from first principles. Phys. Rev. B Condens. Matte 2009, 80, 235407. [Google Scholar] [CrossRef]
- Guesmi, H.; Louis, C.; Delannoy, L. Chemisorbed atomic oxygen inducing Pd segregation in PdAu(111) alloy: Energetic and electronic DFT analysis. Chem. Phys. Lett. 2011, 503, 97–100. [Google Scholar] [CrossRef]
- Dhouib, A.; Guesmi, H. DFT study of the M segregation on MAu alloys (M = Ni, Pd, Pt) in presence of adsorbed oxygen O and O2. Chem. Phys. Lett. 2012, 521, 98–103. [Google Scholar] [CrossRef]
- Sansa, M.; Dhouib, A.; Guesmi, H. Density functional theory study of CO-induced segregation in gold-based alloys. J. Chem. Phys. 2014, 141, 064709. [Google Scholar] [CrossRef]
- Sankarasubramanian, S.; Singh, N.; Mizuno, F.; Prakash, J. Ab initio investigation of the oxygen reduction reaction activity on noble metal (Pt, Au, Pd), Pt3M (M= Fe, Co, Ni, Cu) and Pd3M (M= Fe, Co, Ni, Cu) alloy surfaces, for LiO2 cells. J. Power Sources 2016, 319, 202–209. [Google Scholar] [CrossRef]
- Yu, Y.; Xiao, W.; Wang, J.; Wang, L. First-Principles Study of Mo Segregation in MoNi(111): Effects of Chemisorbed Atomic Oxygen. Materials 2016, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Borg, A.; Dunphy, J.; Mitsui, T.; Ogletree, D.; Salmeron, M. Chemisorption of atomic oxygen on Pd (1 1 1) studied by STM. Surf. Sci. 2004, 561, 69–78. [Google Scholar] [CrossRef]
- Bukas, V.J.; Reuter, K. A comparative study of atomic oxygen adsorption at Pd surfaces from Density Functional Theory. Surf. Sci. 2017, 658, 38–45. [Google Scholar] [CrossRef]
- DeBoer, F.R.; Boom, R.; Miedema, A.R. Cohesion in Metals, 2nd ed.; North-Holland Physics Publishing: Amsterdam, The Netherlands, 1989; pp. 657–660. [Google Scholar]
- Chen, W.; Dalach, P.; Schneider, W.F.; Wolverton, C. Interplay between subsurface ordering, surface segregation, and adsorption on Pt–Ti(111) near-surface alloys. Langmuir 2012, 28, 4683–4693. [Google Scholar] [CrossRef]
- Ma, Y.; Balbuena, P.B. Surface segregation in bimetallic Pt3M (M = Fe, Co, Ni) alloys with adsorbed oxygen. Surf. Sci. 2009, 603, 349–353. [Google Scholar] [CrossRef]
- Wang, L.G.; Tsymbal, E.Y.; Jaswal, S.S. Structural and magnetic properties of clean and methylthiolate-adsorbed Co(0001) surfaces: A first-principles study. J. Magn. Magn. Mater. 2005, 286, 119–123. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).