
Catalysis: Key Technology for the Conversion of CO2 into Fuels and Chemicals



Abstract

1. Introduction
2. Carbon Dioxide Capture, Utilization and Storage (CCUS)
3. Conversion of Carbon Dioxide into Chemicals and Fuels
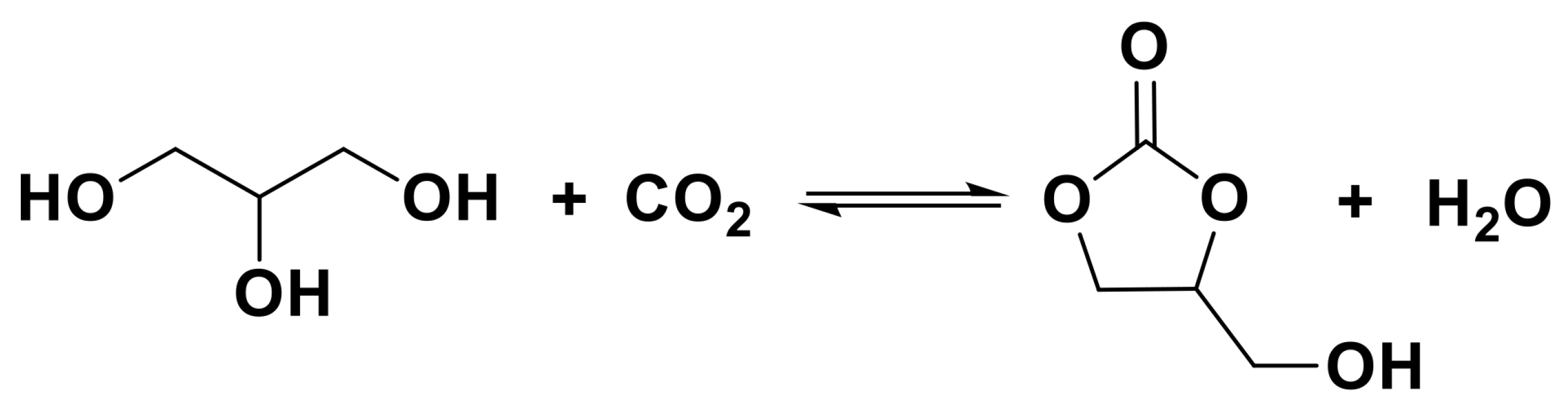
3.1. Synthesis of Carbonates from CO2
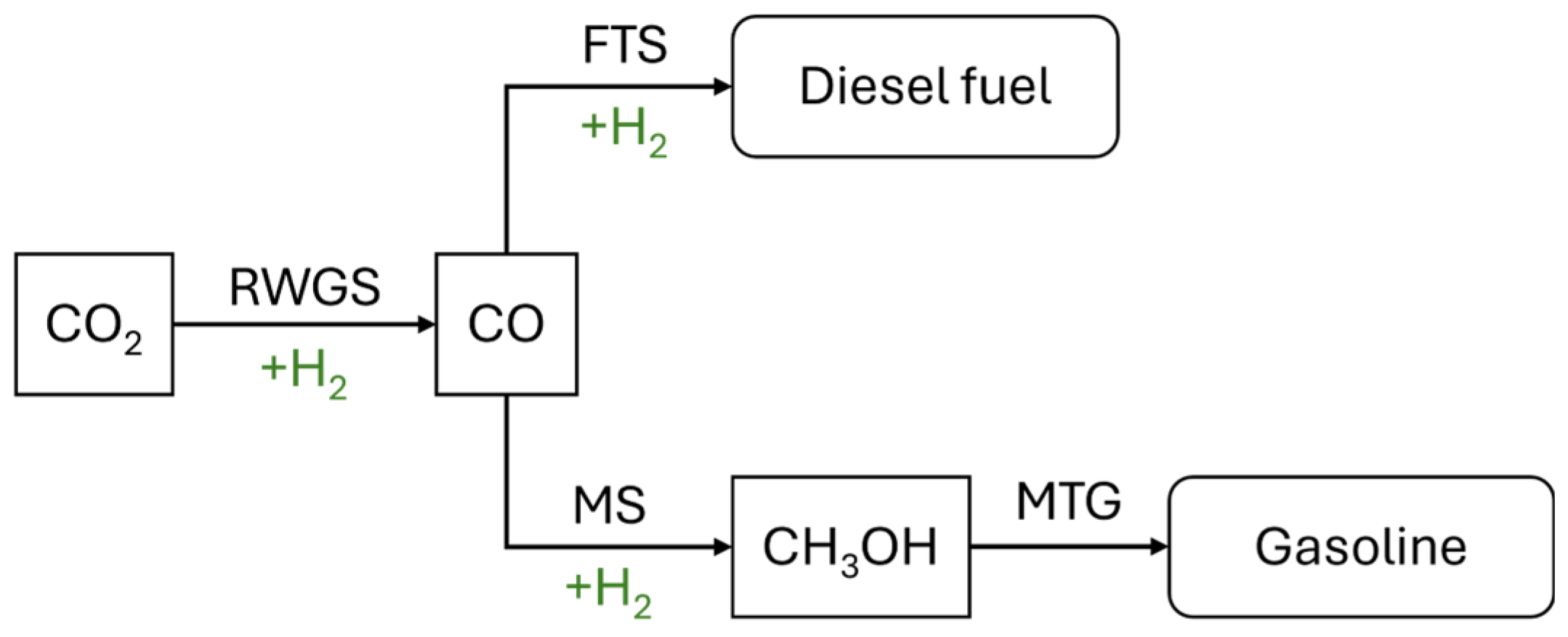
3.2. Conversion of CO2 into C1 and C2+ Chemicals and Fuels

{kind=link}
{kind=link}
{kind=link}
| Power-to-X | Products Obtained and Processes Involved |
|---|---|
| Power-to-gas | Methane: Sabatier reaction (CO2 methanation) |
| Power-to-liquids | Liquid hydrocarbons: “reverse water–gas shift” (RWGS), followed by Fischer–Tropsch synthesis |
| Power-to-chemicals | Synthesis of chemicals by catalytic hydrogenation or by electrocatalytic reduction |
3.2.1. Hydrogenation of Carbon Dioxide to Methanol
3.2.2. Multifunctional Catalysts for Tandem Conversion of CO2 to Hydrocarbons
3.2.3. Electrocatalysis
4. Summary and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wolfgang Eberhardt. Population, standard of living, pollution, and climate change—the drivers of the energy system. In Designing the Energy System of the Future; Elsevier: Amsterdam, The Netherlands, 2021; Chapter 1; pp. 1–34. Available online: https://shop.elsevier.com/books/designing-the-energy-system-of-the-future/eberhardt/978-0-08-102513-0 (accessed on 5 May 2022).
- IEA. CCUS in Clean Energy Transitions; License: CC BY 4; IEA: Paris, France, 2020; Available online: https://www.iea.org/reports/ccus-in-clean-energy-transitions (accessed on 5 May 2022).
- Aresta, M. My journey in the CO2-chemistry wonderland. Coord. Chem. Rev. 2017, 334, 150–183. [Google Scholar] [CrossRef]
- Aresta, M.; Forti, G. Carbon Dioxide as a Source of Carbon: Biochemical and Chemical Uses; Nato Science Series C; D. Reidel Publishing Company: Dordrecht, The Netherlands, 1987; Available online: https://link.springer.com/book/10.1007/978-94-009-3923-3 (accessed on 6 May 2025).
- Bui, M.; Adjiman, C.S.; Bardow, A.; Anthony, E.J.; Boston, A.; Brown, S.; Fennell, P.S.; Fuss, S.; Galindo, A.; Hackett, L.A.; et al. Carbon capture and storage (CCS): The way forward. Energy Environ. Sci. 2018, 11, 1062–1176. [Google Scholar] [CrossRef]
- Ozkan, M. Direct air capture of CO2: A response to meet the global climate targets. MRS Energy Sustain. 2021, 8, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Willauer, H.D.; Hardy, D.R.; Williams, F.W. The feasibility and current estimated capital costs of producing jet fuel at sea using carbon dioxide and hydrogen (Memorandum Report NRL/MR/6180-10-9300, Naval Research Laboratory, Washington DC, 29 September, 2010). Available online: https://www.ourenergypolicy.org/wp-content/uploads/2012/07/GetTRDoc.pdf (accessed on 5 May 2022).
- Eisaman, M.D.; Parajuly, K.; Tuganov, A.; Eldershaw, C.; Chang, N.; Littau, K.A. CO2 extraction from seawater using bipolar membrane electrodialysis. Energy Environ. Sci. 2012, 5, 7346–7352. [Google Scholar] [CrossRef]
- Willauer, H.D.; DiMascio, F.; Hardy, D.R.; Williams, F.W. Feasibility of CO2 Extraction from Seawater and Simultaneous Hydrogen Gas Generation Using a Novel and Robust Electrolytic Cation Exchange Module Based on Continuous Electrodeionization Technology. Ind. Eng. Chem. Res. 2014, 53, 12192–12200. [Google Scholar] [CrossRef]
- Ozin, G. Offshore Oceanic CO2 Capture, Advanced Science News. Available online: https://www.advancedsciencenews.com/offshore-oceanic-co2-capture/ (accessed on 5 May 2022).
- Aresta, M.; Dibenedetto, A. Carbon Recycling Through CO2-Conversion for Stepping Toward a Cyclic-C Economy. A Perspective. Front. Energy Res. 2020, 8. [Google Scholar] [CrossRef]
- Klankermayer, J.; Wesselbaum, S.; Beydoun, K.; Leitner, W. Selective Catalytic Synthesis Using the Combination of Carbon Dioxide and Hydrogen: Catalytic Chess at the Interface of Energy and Chemistry. Angew. Chem. Int. Ed. 2016, 55, 7296–7343. [Google Scholar] [CrossRef]
- Yan, T.; Liu, H.; Zeng, Z.X.; Pan, W.G. Recent progress of catalysts for synthesis of cyclic carbonates from CO2 and epoxides. J. CO2 Util. 2023, 68, 102355. [Google Scholar] [CrossRef]
- Pescarmona, P.P. Cyclic carbonates synthesised from CO2: Applications, challenges and recent research trends. Curr. Opin. Green Sustain. Chem. 2021, 29, 100457. [Google Scholar] [CrossRef]
- Ma, J.; Sun, N.; Zhang, X.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. A short review of catalysis for CO2 conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933. [Google Scholar] [CrossRef] [PubMed]
- Dabral, S.; Schaub, T. The Use of Carbon Dioxide (CO2) as a Building Block in Organic Synthesis from an Industrial Perspective. Adv. Synth. Catal. 2019, 361, 223–246. [Google Scholar] [CrossRef]
- Sonnati, M.O.; Amigoni, S.; Taffin de Givenchy, E.P.; Darmanin, T.; Choulet, O.; Guittard, F. Glycerol carbonate as a versatile building block for tomorrow: Synthesis, reactivity, properties and applications. Green Chem. 2013, 15, 283–306. [Google Scholar] [CrossRef]
- Inrirai, P.; Keogh, J.; Centeno-Pedrazo, A.; Artioli, N.; Manyar, H. Recent advances in processes and catalysts for glycerol carbonate production via direct and indirect use of CO2. J. CO2 Util. 2024, 80, 102693. [Google Scholar] [CrossRef]
- Li, J.; Wang, T. Chemical equilibrium of glycerol carbonate synthesis from glycerol. J. Chem. Thermodyn. 2011, 43, 731–736. [Google Scholar] [CrossRef]
- Su, X.; Lin, W.; Cheng, H.; Zhang, C.; Wang, Y.; Yu, X.; Wu, Z.; Zhao, F. Metal-free catalytic conversion of CO2 and glycerol to glycerol carbonate. Green Chem. 2017, 19, 1775–1781. [Google Scholar] [CrossRef]
- Lim, Y.N.; Lee, C.; Jang, H.-Y. Metal-Free Synthesis of Cyclic and Acyclic Carbonates from CO2 and Alcohols. Eur. J. Org. Chem. 2014, 2014, 1823–1826. [Google Scholar] [CrossRef]
- Ozorio, L.P.; Mota, C.J.A. Direct Carbonation of Glycerol with CO2 Catalyzed by Metal Oxides. ChemPhysChem 2017, 18, 3260–3265. [Google Scholar] [CrossRef]
- Ochoa-Gómez, J.R.; Gómez-Jiménez-Aberasturi, O.; Ramírez-López, C.; Belsué, M. A Brief Review on Industrial Alternatives for the Manufacturing of Glycerol Carbonate, a Green Chemical. Org. Process Res. Dev. 2012, 16, 389–399. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Zhang, J.; He, D. Glycerol carbonylation with CO2 to glycerol carbonate over CeO2 catalyst and the influence of CeO2 preparation methods and reaction parameters. Appl. Catal. A Gen. 2016, 513, 9–18. [Google Scholar] [CrossRef]
- Park, C.-Y.; Nguyen-Phu, H.; Shin, E.W. Glycerol carbonation with CO2 and La2O2CO3/ZnO catalysts prepared by two different methods: Preferred reaction route depending on crystalline structure. Mol. Catal. 2017, 435, 99–109. [Google Scholar] [CrossRef]
- Devi, P.; Das, U.; Dalai, A.K. Production of glycerol carbonate using a novel Ti-SBA-15 catalyst. Chem. Eng. J. 2018, 346, 477–488. [Google Scholar] [CrossRef]
- Ozorio, L.P.; Pianzolli, R.; da Cruz Machado, L.; Miranda, J.L.; Turci, C.C.; Guerra, A.C.O.; Souza-Aguiar, E.F.; Mota, C.J.A. Metal-impregnated zeolite Y as efficient catalyst for the direct carbonation of glycerol with CO2. Appl. Catal. A Gen. 2015, 504, 187–191. [Google Scholar] [CrossRef]
- Christy, S.; Noschese, A.; Lomelí-Rodriguez, M.; Greeves, N.; Lopez-Sanchez, J.A. Recent progress in the synthesis and applications of glycerol carbonate. Curr. Opin. Green Sustain. Chem. 2018, 14, 99–107. [Google Scholar] [CrossRef]
- Sahil; Gupta, N. Cyclic carbonates: Treasure of fine chemicals obtained from waste stream CO2 over carbon-based heterogeneous catalysts. Renew. Sustain. Energy Rev. 2024, 193, 114297. [Google Scholar] [CrossRef]
- Lukato, S.; Kasozi, G.N.; Naziriwo, B.; Tebandeke, E. Glycerol carbonylation with CO2 to form glycerol carbonate: A review of recent developments and challenges. Curr. Res. Green Sustain. Chem. 2021, 4, 100199. [Google Scholar] [CrossRef]
- Samikannu, A.; Konwar, L.J.; Mäki-Arvela, P.; Mikkola, J.-P. Renewable N-doped active carbons as efficient catalysts for direct synthesis of cyclic carbonates from epoxides and CO2. Appl. Catal. B Environ. 2019, 241, 41–51. [Google Scholar] [CrossRef]
- Buckley, B.R.; Patel, A.P.; Wijayantha, K.G.U. Electrosynthesis of cyclic carbonates from epoxides and atmospheric pressure carbon dioxide. Chem. Commun. 2011, 47, 11888–11890. [Google Scholar] [CrossRef]
- Gallardo-Fuentes, S.; Contreras, R.; Isaacs, M.; Honores, J.; Quezada, D.; Landaeta, E.; Ormazábal-Toledo, R. On the mechanism of CO2 electro-cycloaddition to propylene oxides. J. CO2 Util. 2016, 16, 114–120. [Google Scholar] [CrossRef]
- Wu, L.-X. Electrosynthesis of Cyclic Carbonates from CO2 and Epoxides on Compacted Silver Nanoparticles Electrode. Int. J. Electrochem. Sci. 2017, 12, 8963–8972. [Google Scholar] [CrossRef]
- Al-Kurdhani, J.M.H.; Wang, H. The Synthesis of Glycerol Carbonate from Glycerol and Carbon Dioxide over Supported CuO-Based Nanoparticle Catalyst. Molecules 2023, 28, 4164. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A.; Nocito, F.; Pastore, C. A study on the carboxylation of glycerol to glycerol carbonate with carbon dioxide: The role of the catalyst, solvent and reaction conditions. J. Mol. Catal. A Chem. 2006, 257, 149–153. [Google Scholar] [CrossRef]
- George, J.; Patel, Y.; Pillai, S.M.; Munshi, P. Methanol assisted selective formation of 1,2-glycerol carbonate from glycerol and carbon dioxide using nBu2SnO as a catalyst. J. Mol. Catal. A Chem. 2009, 304, 1–7. [Google Scholar] [CrossRef]
- Zhang, Q.; Yuan, H.-Y.; Lin, X.-T.; Fukaya, N.; Fujitani, T.; Sato, K.; Choi, J.-C. Calcium carbide as a dehydrating agent for the synthesis of carbamates, glycerol carbonate, and cyclic carbonates from carbon dioxide. Green Chem. 2020, 22, 4231–4239. [Google Scholar] [CrossRef]
- Zhang, J.; He, D. Surface properties of Cu/La2O3 and its catalytic performance in the synthesis of glycerol carbonate and monoacetin from glycerol and carbon dioxide. J. Colloid Interface Sci. 2014, 419, 31–38. [Google Scholar] [CrossRef]
- Zhang, J.; He, D. Synthesis of glycerol carbonate and monoacetin from glycerol and carbon dioxide over Cu catalysts: The role of supports. J. Chem. Technol. Biotechnol. 2015, 90, 1077–1085. [Google Scholar] [CrossRef]
- Koranian, P.; Kumar Dalai, A.; Sammynaiken, R. Production of glycerol carbonate from glycerol and carbon dioxide using metal oxide catalysts. Chem. Eng. Sci. 2024, 286, 119687. [Google Scholar] [CrossRef]
- Ding, J.; Qin, J.; Zhou, S.; Wu, Y.; Fan, C.; Li, Y.; Wang, J. Lanthanum-based perovskite catalysts for the glycerol carbonylation to glycerol carbonate: Effects of oxygen defects and basicity. Mol. Catal. 2024, 558, 114001. [Google Scholar] [CrossRef]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef]
- Sabatier, P.; Senderens, J.-B. Comptes Rendus Des Seances De L’Academie Des Sciences Section VI—Chimie; Imprimerie Gauthier-Villars: Paris, France, 1902. [Google Scholar]
- Mills, G.A.; Steffgen, F.W. Catalytic Methanation. Catal. Rev. 1974, 8, 159–210. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Loiland, J.A.; Wulfers, M.J.; Marinkovic, N.S.; Lobo, R.F. Fe/γ-Al2O3 and Fe–K/γ-Al2O3 as reverse water-gas shift catalysts. Catal. Sci. Technol. 2016, 6, 5267–5279. [Google Scholar] [CrossRef]
- Zhu, M.; Ge, Q.; Zhu, X. Catalytic Reduction of CO2 to CO via Reverse Water Gas Shift Reaction: Recent Advances in the Design of Active and Selective Supported Metal Catalysts. Trans. Tianjin Univ. 2020, 26, 172–187. [Google Scholar] [CrossRef]
- Wang, J.; Wang, C.; Feng, Y.; Li, F.; Su, W.; Fang, Y.; Zhao, B. Cu/CeO2 catalysts for reverse water gas shift reactions: The effect of the preparation method. RSC Adv. 2024, 14, 16736–16746. [Google Scholar] [CrossRef]
- Martinelli, M.; Gnanamani, M.K.; LeViness, S.; Jacobs, G.; Shafer, W.D. An overview of Fischer-Tropsch Synthesis: XtL processes, catalysts and reactors. Appl. Catal. A Gen. 2020, 608, 117740. [Google Scholar] [CrossRef]
- Chang, C.D.; Silvestri, A.J. The conversion of methanol and other O-compounds to hydrocarbons over zeolite catalysts. J. Catal. 1977, 47, 249–259. [Google Scholar] [CrossRef]
- Chang, C.D. Hydrocarbons from Methanol. Catal. Rev. 1983, 25, 1–118. [Google Scholar] [CrossRef]
- Stöcker, M. Methanol-to-hydrocarbons: Catalytic materials and their behavior1Dedicated to my wife Wencke Ophaug.1. Microporous Mesoporous Mater. 1999, 29, 3–48. [Google Scholar] [CrossRef]
- Keil, F.J. Methanol-to-hydrocarbons: Process technology. Microporous Mesoporous Mater. 1999, 29, 49–66. [Google Scholar] [CrossRef]
- Ilias, S.; Bhan, A. Mechanism of the Catalytic Conversion of Methanol to Hydrocarbons. ACS Catal. 2013, 3, 18–31. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the Valorization of Exhaust Carbon: From CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Sun, J.; Ge, Q.; Tsubaki, N. Recent advances in direct catalytic hydrogenation of carbon dioxide to valuable C2+ hydrocarbons. J. Mater. Chem. A 2018, 6, 23244–23262. [Google Scholar] [CrossRef]
- Saravanan, K.; Ham, H.; Tsubaki, N.; Bae, J.W. Recent progress for direct synthesis of dimethyl ether from syngas on the heterogeneous bifunctional hybrid catalysts. Appl. Catal. B Environ. 2017, 217, 494–522. [Google Scholar] [CrossRef]
- Li, Z.; Wang, J.; Qu, Y.; Liu, H.; Tang, C.; Miao, S.; Feng, Z.; An, H.; Li, C. Highly Selective Conversion of Carbon Dioxide to Lower Olefins. ACS Catal. 2017, 7, 8544–8548. [Google Scholar] [CrossRef]
- Wei, J.; Ge, Q.; Yao, R.; Wen, Z.; Fang, C.; Guo, L.; Xu, H.; Sun, J. Directly converting CO2 into a gasoline fuel. Nat. Commun. 2017, 8, 15174. [Google Scholar] [CrossRef]
- Yang, C.; Mu, R.; Wang, G.; Song, J.; Tian, H.; Zhao, Z.-J.; Gong, J. Hydroxyl-mediated ethanol selectivity of CO2 hydrogenation. Chem. Sci. 2019, 10, 3161–3167. [Google Scholar] [CrossRef]
- Ye, R.-P.; Ding, J.; Gong, W.; Argyle, M.D.; Zhong, Q.; Wang, Y.; Russell, C.K.; Xu, Z.; Russell, A.G.; Li, Q.; et al. CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Commun. 2019, 10, 5698. [Google Scholar] [CrossRef]
- Kattel, S.; Liu, P.; Chen, J.G. Tuning Selectivity of CO2 Hydrogenation Reactions at the Metal/Oxide Interface. J. Am. Chem. Soc. 2017, 139, 9739–9754. [Google Scholar] [CrossRef]
- Wang, J.; You, Z.; Zhang, Q.; Deng, W.; Wang, Y. Synthesis of lower olefins by hydrogenation of carbon dioxide over supported iron catalysts. Catal. Today 2013, 215, 186–193. [Google Scholar] [CrossRef]
- Gupta, S.; Jain, V.K.; Jagadeesan, D. Fine Tuning the Composition and Nanostructure of Fe-Based Core–Shell Nanocatalyst for Efficient CO2 Hydrogenation. ChemNanoMat 2016, 2, 989–996. [Google Scholar] [CrossRef]
- Mattia, D.; Jones, M.D.; O’Byrne, J.P.; Griffiths, O.G.; Owen, R.E.; Sackville, E.; McManus, M.; Plucinski, P. Towards Carbon-Neutral CO2 Conversion to Hydrocarbons. ChemSusChem 2015, 8, 4064–4072. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Lin, J.; Cheng, Y.; Tian, J.; Wang, S.; Xie, S.; Pei, Y.; Yan, S.; Qiao, M.; Xu, H.; et al. Porous Graphene-Confined Fe–K as Highly Efficient Catalyst for CO2 Direct Hydrogenation to Light Olefins. ACS Appl. Mater. Interfaces 2018, 10, 23439–23443. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, L.; Zhang, J.; Liu, X.; Wang, H.; Zhang, W.; Yang, Q.; Ma, J.; Dong, X.; Yoo, S.J.; et al. Selective Hydrogenation of CO2 to Ethanol over Cobalt Catalysts. Angew. Chem. Int. Ed. 2018, 57, 6104–6108. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Shao, Q.; Wang, P.; Dai, Q.; Wang, X.; Huang, X. Highly Active and Selective Hydrogenation of CO2 to Ethanol by Ordered Pd–Cu Nanoparticles. J. Am. Chem. Soc. 2017, 139, 6827–6830. [Google Scholar] [CrossRef]
- Gao, P.; Li, S.; Bu, X.; Dang, S.; Liu, Z.; Wang, H.; Zhong, L.; Qiu, M.; Yang, C.; Cai, J.; et al. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 2017, 9, 1019–1024. [Google Scholar] [CrossRef]
- Blay-Roger, R.; Nawaz, M.A.; Baena-Moreno, F.M.; Bobadilla, L.F.; Reina, T.R.; Odriozola, J.A. Tandem catalytic approaches for CO2 enriched Fischer-Tropsch synthesis. Prog. Energy Combust. Sci. 2024, 103, 101159. [Google Scholar] [CrossRef]
- Zhong, J.; Yang, X.; Wu, Z.; Liang, B.; Huang, Y.; Zhang, T. State of the art and perspectives in heterogeneous catalysis of CO2 hydrogenation to methanol. Chem. Soc. Rev. 2020, 49, 1385–1413. [Google Scholar] [CrossRef]
- Bowker, M. Methanol Synthesis from CO2 Hydrogenation. ChemCatChem 2019, 11, 4238–4246. [Google Scholar] [CrossRef]
- Ye, J.; Dimitratos, N.; Rossi, L.M.; Thonemann, N.; Beale, A.M.; Wojcieszak, R. Hydrogenation of CO2 for sustainable fuel and chemical production. Science 2025, 387, eadn9388. [Google Scholar] [CrossRef]
- Arena, F.; Barbera, K.; Italiano, G.; Bonura, G.; Spadaro, L.; Frusteri, F. Synthesis, characterization and activity pattern of Cu–ZnO/ZrO2 catalysts in the hydrogenation of carbon dioxide to methanol. J. Catal. 2007, 249, 185–194. [Google Scholar] [CrossRef]
- Martin, O.; Martín, A.J.; Mondelli, C.; Mitchell, S.; Segawa, T.F.; Hauert, R.; Drouilly, C.; Curulla-Ferré, D.; Pérez-Ramírez, J. Indium Oxide as a Superior Catalyst for Methanol Synthesis by CO2 Hydrogenation. Angew. Chem. Int. Ed. 2016, 55, 6261–6265. [Google Scholar] [CrossRef] [PubMed]
- Frei, M.S.; Mondelli, C.; García-Muelas, R.; Kley, K.S.; Puértolas, B.; López, N.; Safonova, O.V.; Stewart, J.A.; Curulla Ferré, D.; Pérez-Ramírez, J. Atomic-scale engineering of indium oxide promotion by palladium for methanol production via CO2 hydrogenation. Nat. Commun. 2019, 10, 3377. [Google Scholar] [CrossRef] [PubMed]
- Potter, M.E.; Mediavilla Madrigal, S.; Campbell, E.; Allen, L.J.; Vyas, U.; Parry, S.; García-Zaragova, A.; Martínez-Prieto, L.M.; Oña-Burgos, P.; Lützen, M.; et al. A High Pressure Operando Spectroscopy Examination of Bimetal Interactions in ‘Metal Efficient’ Palladium/In2O3/Al2O3 Catalysts for CO2 Hydrogenation. Angew. Chem. Int. Ed. 2023, 62, e202312645. [Google Scholar] [CrossRef] [PubMed]
- Kubas, D.; Gierse, M.; Salem, O.; Krossing, I. Is Direct DME Synthesis Superior to Methanol Production in Carbon Dioxide Valorization? From Thermodynamic Predictions to Experimental Confirmation. ACS Catal. 2023, 13, 3960–3970. [Google Scholar] [CrossRef]
- Yang, G.; Tsubaki, N.; Shamoto, J.; Yoneyama, Y.; Zhang, Y. Confinement Effect and Synergistic Function of H-ZSM-5/Cu-ZnO-Al2O3 Capsule Catalyst for One-Step Controlled Synthesis. J. Am. Chem. Soc. 2010, 132, 8129–8136. [Google Scholar] [CrossRef]
- Das, S.; Pérez-Ramírez, J.; Gong, J.; Dewangan, N.; Hidajat, K.; Gates, B.C.; Kawi, S. Core–shell structured catalysts for thermocatalytic, photocatalytic, and electrocatalytic conversion of CO2. Chem. Soc. Rev. 2020, 49, 2937–3004. [Google Scholar] [CrossRef]
- Li, T.; Shoinkhorova, T.; Gascon, J.; Ruiz-Martínez, J. Aromatics Production via Methanol-Mediated Transformation Routes. ACS Catal. 2021, 11, 7780–7819. [Google Scholar] [CrossRef]
- Olah, G.A. Beyond Oil and Gas: The Methanol Economy. Angew. Chem. Int. Ed. 2005, 44, 2636–2639. [Google Scholar] [CrossRef]
- Riedel, T.; Schulz, H.; Schaub, G.; Jun, K.-W.; Hwang, J.-S.; Lee, K.-W. Fischer–Tropsch on Iron with H2/CO and H2/CO2 as Synthesis Gases: The Episodes of Formation of the Fischer–Tropsch Regime and Construction of the Catalyst. Top. Catal. 2003, 26, 41–54. [Google Scholar] [CrossRef]
- Chang, Q.; Zhang, C.; Liu, C.; Wei, Y.; Cheruvathur, A.V.; Dugulan, A.I.; Niemantsverdriet, J.W.; Liu, X.; He, Y.; Qing, M.; et al. Relationship between Iron Carbide Phases (ε-Fe2C, Fe7C3, and χ-Fe5C2) and Catalytic Performances of Fe/SiO2 Fischer–Tropsch Catalysts. ACS Catal. 2018, 8, 3304–3316. [Google Scholar] [CrossRef]
- Wei, J.; Yao, R.; Ge, Q.; Wen, Z.; Ji, X.; Fang, C.; Zhang, J.; Xu, H.; Sun, J. Catalytic Hydrogenation of CO2 to Isoparaffins over Fe-Based Multifunctional Catalysts. ACS Catal. 2018, 8, 9958–9967. [Google Scholar] [CrossRef]
- Wei, J.; Yao, R.; Han, Y.; Ge, Q.; Sun, J. Towards the development of the emerging process of CO2 heterogenous hydrogenation into high-value unsaturated heavy hydrocarbons. Chem. Soc. Rev. 2021, 50, 10764–10805. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qu, Y.; Wang, J.; Liu, H.; Li, M.; Miao, S.; Li, C. Highly Selective Conversion of Carbon Dioxide to Aromatics over Tandem Catalysts. Joule 2019, 3, 570–583. [Google Scholar] [CrossRef]
- Gao, X.; Atchimarungsri, T.; Ma, Q.; Zhao, T.-S.; Tsubaki, N. Realizing efficient carbon dioxide hydrogenation to liquid hydrocarbons by tandem catalysis design. EnergyChem 2020, 2, 100038. [Google Scholar] [CrossRef]
- Overa, S.; Ko, B.H.; Zhao, Y.; Jiao, F. Electrochemical Approaches for CO2 Conversion to Chemicals: A Journey toward Practical Applications. Acc. Chem. Res. 2022, 55, 638–648. [Google Scholar] [CrossRef]
- Lu, Q.; Jiao, F. Electrochemical CO2 reduction: Electrocatalyst, reaction mechanism, and process engineering. Nano Energy 2016, 29, 439–456. [Google Scholar] [CrossRef]
- Senocrate, A.; Battaglia, C. Electrochemical CO2 reduction at room temperature: Status and perspectives. J. Energy Storage 2021, 36, 102373. [Google Scholar] [CrossRef]
- Sánchez, O.G.; Birdja, Y.Y.; Bulut, M.; Vaes, J.; Breugelmans, T.; Pant, D. Recent advances in industrial CO2 electroreduction. Curr. Opin. Green Sustain. Chem. 2019, 16, 47–56. [Google Scholar] [CrossRef]
- Chung, W.; Jeong, W.; Lee, J.; Kim, J.; Roh, K.; Lee, J.H. Electrification of CO2 conversion into chemicals and fuels: Gaps and opportunities in process systems engineering. Comput. Chem. Eng. 2023, 170, 108106. [Google Scholar] [CrossRef]
- Jhong, H.-R.M.; Ma, S.; Kenis, P.J.A. Electrochemical conversion of CO2 to useful chemicals: Current status, remaining challenges, and future opportunities. Curr. Opin. Chem. Eng. 2013, 2, 191–199. [Google Scholar] [CrossRef]
- Romero Cuellar, N.S.; Scherer, C.; Kaçkar, B.; Eisenreich, W.; Huber, C.; Wiesner-Fleischer, K.; Fleischer, M.; Hinrichsen, O. Two-step electrochemical reduction of CO2 towards multi-carbon products at high current densities. J. CO2 Util. 2020, 36, 263–275. [Google Scholar] [CrossRef]
- Ye, W.; Guo, X.; Ma, T. A review on electrochemical synthesized copper-based catalysts for electrochemical reduction of CO2 to C2+ products. Chem. Eng. J. 2021, 414, 128825. [Google Scholar] [CrossRef]
- Hao, J.; Shi, W. Transition metal (Mo, Fe, Co, and Ni)-based catalysts for electrochemical CO2 reduction. Chin. J. Catal. 2018, 39, 1157–1166. [Google Scholar] [CrossRef]
- Yue, Y.; Sun, Y.; Tang, C.; Liu, B.; Ji, Z.; Hu, A.; Shen, B.; Zhang, Z.; Sun, Z. Ranking the relative CO2 electrochemical reduction activity in carbon materials. Carbon 2019, 154, 108–114. [Google Scholar] [CrossRef]
- Xing, Y.; Cui, M.; Fan, P.; Ren, J.; Zhang, C.; Li, N.; Wen, X.; Ji, X. Efficient and selective electrochemical reduction of CO2 to formate on 3D porous structured multi-walled carbon nanotubes supported Pb nanoparticles. Mater. Chem. Phys. 2019, 237, 121826. [Google Scholar] [CrossRef]
- Takeda, H.; Cometto, C.; Ishitani, O.; Robert, M. Electrons, Photons, Protons and Earth-Abundant Metal Complexes for Molecular Catalysis of CO2 Reduction. ACS Catal. 2017, 7, 70–88. [Google Scholar] [CrossRef]
- Pérez-Sequera, A.C.; Díaz-Pérez, M.A.; Serrano-Ruiz, J.C. Recent Advances in the Electroreduction of CO2 over Heteroatom-Doped Carbon Materials. Catal 2020, 10, 1179. [Google Scholar] [CrossRef]
- Hao, X.; An, X.; Patil, A.M.; Wang, P.; Ma, X.; Du, X.; Hao, X.; Abudula, A.; Guan, G. Biomass-Derived N-Doped Carbon for Efficient Electrocatalytic CO2 Reduction to CO and Zn–CO2 Batteries. ACS Appl. Mater. Interfaces 2021, 13, 3738–3747. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Xiao, N.; Li, H.; Ji, Y.; Guo, Z.; Liu, C.; Qiu, J. Nitrogen-doped porous carbon from coal for high efficiency CO2 electrocatalytic reduction. Carbon 2019, 151, 46–52. [Google Scholar] [CrossRef]
- Huang, P.; Cheng, M.; Zhang, H.; Zuo, M.; Xiao, C.; Xie, Y. Single Mo atom realized enhanced CO2 electro-reduction into formate on N-doped graphene. Nano Energy 2019, 61, 428–434. [Google Scholar] [CrossRef]
- Song, Y.; Chen, W.; Zhao, C.; Li, S.; Wei, W.; Sun, Y. Metal-Free Nitrogen-Doped Mesoporous Carbon for Electroreduction of CO(2) to Ethanol. Angew. Chem. Int. Ed. Engl. 2017, 56, 10840–10844. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, S.; Takayama, T.; Yamaguchi, Y.; Iwase, A.; Kudo, A. CO2 Reduction Using Water as an Electron Donor over Heterogeneous Photocatalysts Aiming at Artificial Photosynthesis. Acc. Chem. Res. 2022, 55, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pei, X.; Wang, Z.-j.; Shi, L.; Song, H.; Ye, J. Photothermal Catalytic CO2 Conversion to Value-Added Chemicals: Progress and Prospects. ACS Sustain. Chem. Eng. 2024, 12, 17069–17097. [Google Scholar] [CrossRef]
- Zhang, F.; Li, Y.-H.; Qi, M.-Y.; Yamada, Y.M.A.; Anpo, M.; Tang, Z.-R.; Xu, Y.-J. Photothermal catalytic CO2 reduction over nanomaterials. Chem Catal. 2021, 1, 272–297. [Google Scholar] [CrossRef]
- Ding, X.; Liu, W.; Zhao, J.; Wang, L.; Zou, Z. Photothermal CO2 Catalysis toward the Synthesis of Solar Fuel: From Material and Reactor Engineering to Techno-Economic Analysis. Adv. Mater. 2025, 37, 2312093. [Google Scholar] [CrossRef]
- Xiao, Z.; Zhang, L.; Tan, X.; Sun, K.; Li, J.; Pan, L.; Zou, J.-J.; Li, G.; Wang, D. Advances in Oxygen Defect-Mediated Photothermal Catalytic CO2 Hydrogenation Reduction. Adv. Funct. Mater. 2025, 2500339. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, H.; Yu, Q.; Roy, S.; Yu, X. Photo-/electrocatalytic approaches to CO2 conversion on Cu2O-based catalysts. Appl. Catal. A Gen. 2023, 667, 119445. [Google Scholar] [CrossRef]
- Kumaravel, V.; Bartlett, J.; Pillai, S.C. Photoelectrochemical Conversion of Carbon Dioxide (CO2) into Fuels and Value-Added Products. ACS Energy Lett. 2020, 5, 486–519. [Google Scholar] [CrossRef]
- Guo, F.; Zhao, Y.-F.; Li, R.-X.; Xu, H.; Sun, W.-Y. Photo/electrocatalytic Reduction of CO2 to C2+ Products on MOF-Based Catalysts. ChemNanoMat 2023, 9, e202300313. [Google Scholar] [CrossRef]
- Xiao, Z.; Zhang, H.; Tan, X.; Ye, F.; Zhang, Y.; Gu, J.; Li, J.; Sun, K.; Zhang, S.; Zou, J.-J.; et al. Comprehensive Insight into External Field-Driven CO2 Reduction to CO: Recent Progress and Future Prospects. Adv. Energy Mater. 2025, 15, 2500988. [Google Scholar] [CrossRef]
| CO2 Capture Technology | Conditions |
|---|---|
| Absorption | Aqueous solutions of alkanolamines, NH3 or K2CO3 |
| Adsorption | Solid porous adsorbents |
| Calcium looping | CaO + CO2 ⇆ CaCO3 in two fluidized bed reactors |
| Chemical looping combustion | Metal oxide used to supply oxygen for the combustion |
| Oxyfuel | Oxygen used for the combustion (instead of air) |
| Emerging technologies | Membrane separation, ionic liquids, clathrate hydrates |
| Type of Transformation | Carbon Oxidation State | Chemicals and Fuels |
|---|---|---|
| Incorporation | +4 | Cyclic carbonates, polycarbonates |
| Synthesis of functionalized compounds | intermediate | C1 (formic acid, formaldehyde, methanol) C2+ (alcohols, acids, aldehydes) |
| Complete reduction | −4 | Methane, >C1 hydrocarbons |
| Catalyst | Temperature (°C)/ Pressure (Bar) | Solvent/Dehydrating Agent | GC Yield (%) | Ref. |
|---|---|---|---|---|
| n-Bu2Sn(OMe)2 | 180/50 | -/Molecular sieves | 5.72 | [37] |
| nBu2SnO | 120/138 | MeOH/13X (soda) zeolite | 35 | [38] |
| Zn(OTf)2/phen | 180/80 | N-methyl-2-pyrrolidone/CaC2 | 92 | [39] |
| ZnY | 180/100 | -/- | 5.8 | [28] |
| ZnO | 180/150 | -/- | 8 | [23] |
| La2O2CO3/ZnO | 170/40 | CH3CN | 14.4 | [26] |
| Cu/La2O3 | 150/70 | CH3CN | 45.4 | [40] |
| Cu/MgO Cu/La2O3 | 150/40 | CH3CN | 26.1 29.3 | [41] |
| CeO2 nanorods | 150/40 | DMF/2-cyanopyridine | 78.9 | [25] |
| CeO2 nanopolyhedra | 180/150 | 2-cyanopyridine | 14.2 | [21] |
| MgO | 150/80 | DMF/2-cyanopyridine | 10.6 | [42] |
| LaCoO3 | 150/30 | DMF/2-cyanopyridine | 72.4 | [43] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha, R.P.; Figueiredo, J.L. Catalysis: Key Technology for the Conversion of CO2 into Fuels and Chemicals. Catalysts 2025, 15, 614. https://doi.org/10.3390/catal15070614
Rocha RP, Figueiredo JL. Catalysis: Key Technology for the Conversion of CO2 into Fuels and Chemicals. Catalysts. 2025; 15(7):614. https://doi.org/10.3390/catal15070614
Chicago/Turabian StyleRocha, Raquel Pinto, and José Luís Figueiredo. 2025. "Catalysis: Key Technology for the Conversion of CO2 into Fuels and Chemicals" Catalysts 15, no. 7: 614. https://doi.org/10.3390/catal15070614
APA StyleRocha, R. P., & Figueiredo, J. L. (2025). Catalysis: Key Technology for the Conversion of CO2 into Fuels and Chemicals. Catalysts, 15(7), 614. https://doi.org/10.3390/catal15070614