

Optimizing Methane Oxidative Coupling over La2O3: Kinetic and Product Analysis


Abstract
1. Introduction
2. Results
2.1. Effect of Methane to Oxygen Ratio
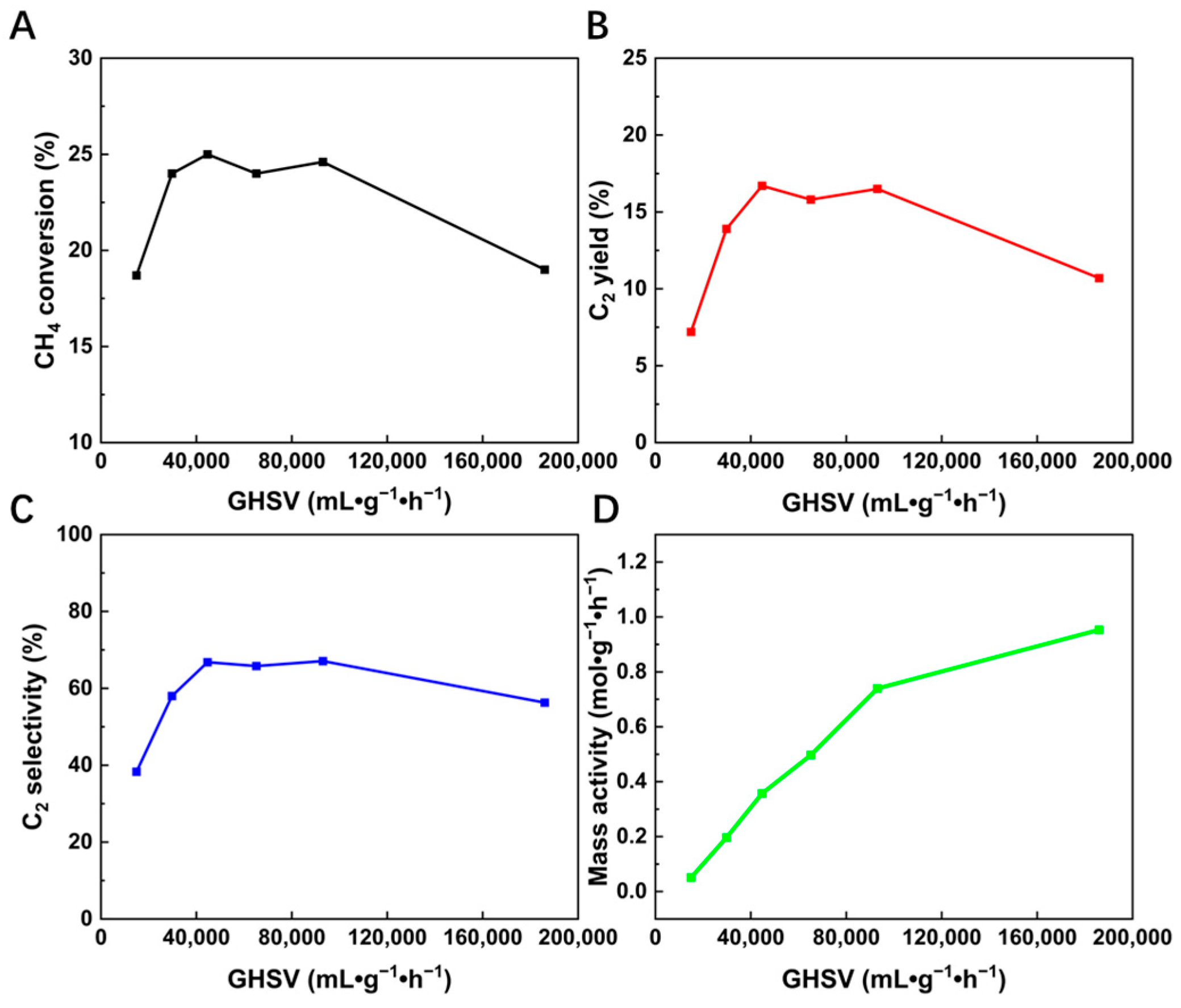
2.2. Effect of GHSV
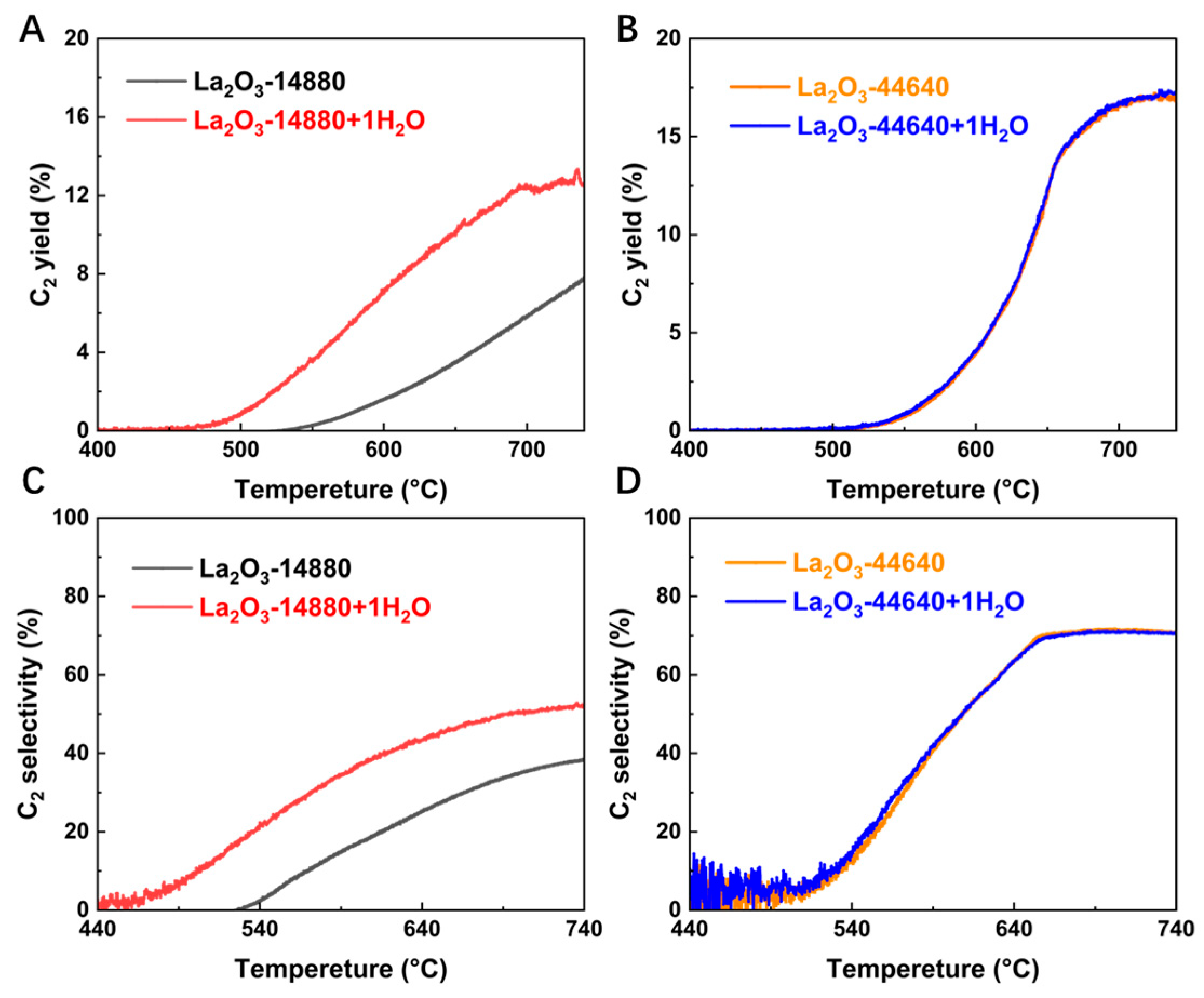
2.3. Effect of H2O in the Reaction Gas
2.4. Effect of CO in the Reaction Gas
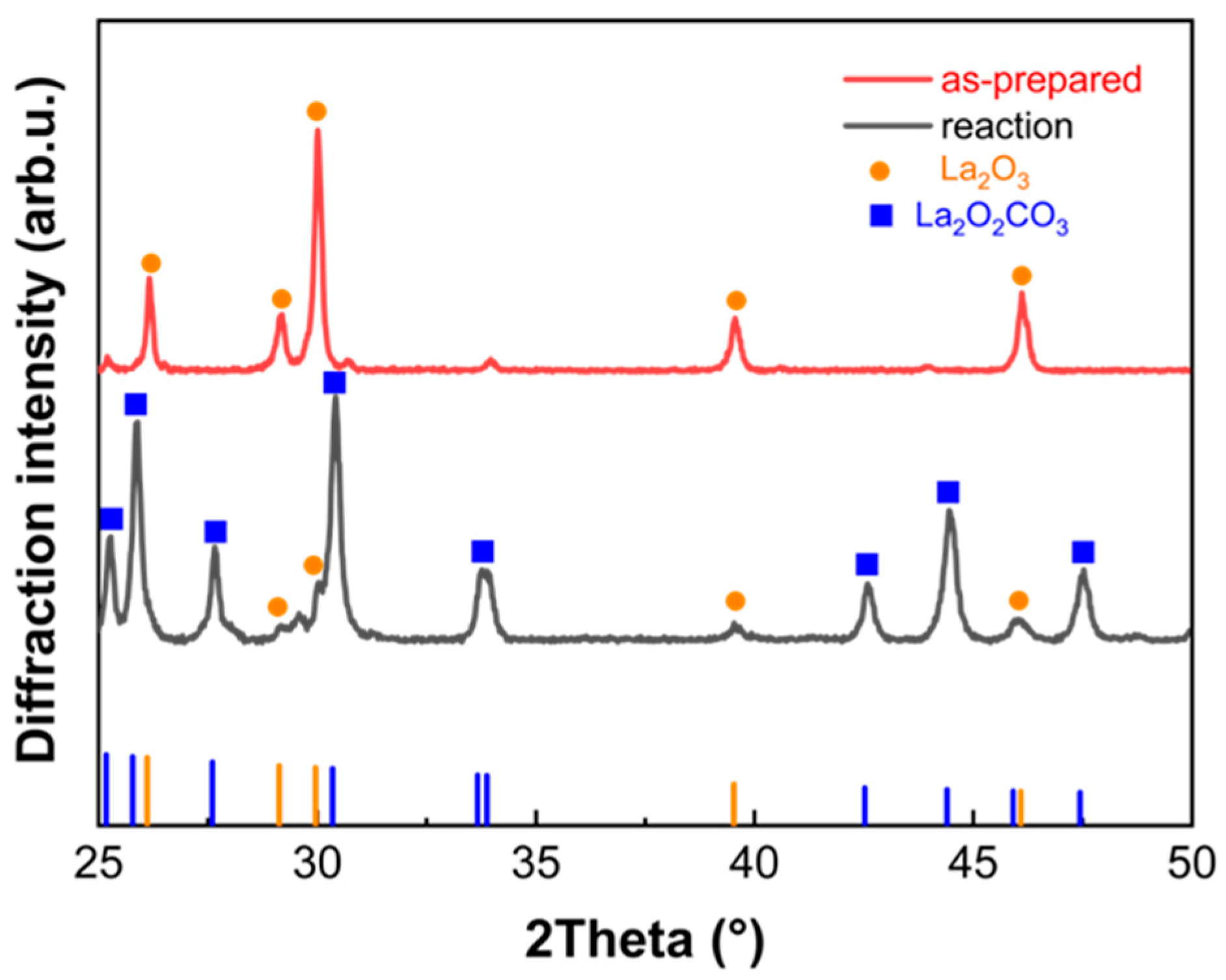
2.5. Structure Characterization
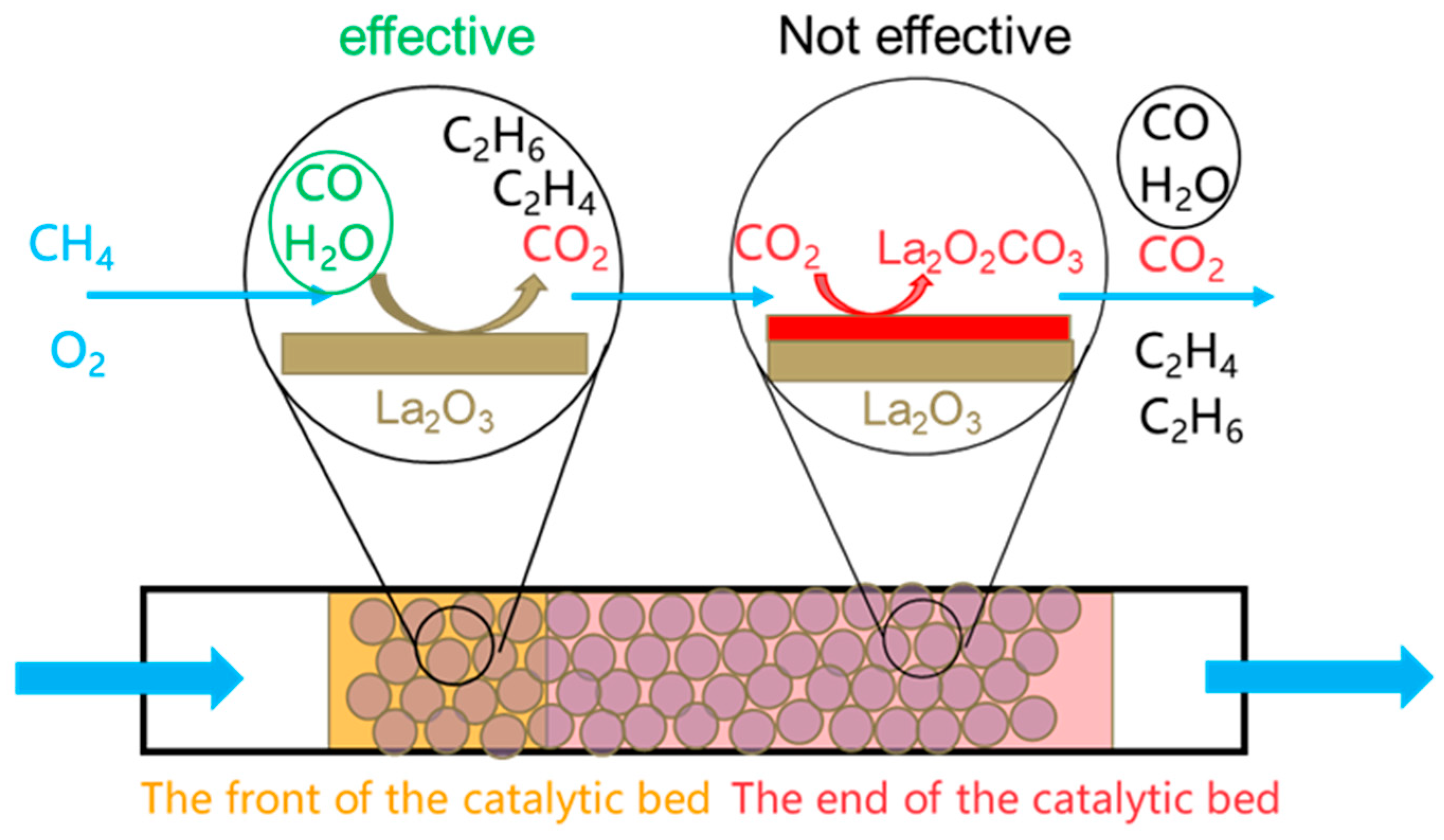
3. Discussion
4. Materials and Methods
4.1. Catalyst
4.2. Reactor
4.3. Online MS and Reaction Conditions
4.4. Characterization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lunsford, J.H. Catalytic conversion of methane to more useful chemicals and fuels: A challenge for the 21st century. Catal. Today 2000, 63, 165–174. [Google Scholar] [CrossRef]
- Schwach, P.; Pan, X.; Bao, X. Direct conversion of methane to value-added chemicals over heterogeneous catalysts: Challenges and prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef] [PubMed]
- Lunsford, J.H. The catalytic oxidative coupling of methane. Angew. Chem. Int. Ed. Engl. 1995, 34, 970–980. [Google Scholar] [CrossRef]
- Zavyalova, U.; Holena, M.; Schlögl, R.; Baerns, M. Statistical analysis of past catalytic data on oxidative methane coupling for new insights into the composition of high-performance catalysts. ChemCatChem 2011, 3, 1935–1947. [Google Scholar] [CrossRef]
- Keller, G.; Bhasin, M. Synthesis of ethylene via oxidative coupling of methane: I. Determination of active catalysts. J. Catal. 1982, 73, 9–19. [Google Scholar] [CrossRef]
- Ortiz-Bravo, C.A.; Chagas, C.A.; Toniolo, F.S. Oxidative coupling of methane (OCM): An overview of the challenges and opportunities for developing new technologies. J. Nat. Gas Sci. Eng. 2021, 96, 104254. [Google Scholar] [CrossRef]
- Huang, P.; Zhao, Y.; Zhang, J.; Zhu, Y.; Sun, Y. Exploiting shape effects of La2O3 nanocatalysts for oxidative coupling of methane reaction. Nanoscale 2013, 5, 10844–10848. [Google Scholar] [CrossRef]
- Kim, I.; Lee, G.; Na, H.B.; Ha, J.-M.; Jung, J.C. Selective oxygen species for the oxidative coupling of methane. Mol. Catal. 2017, 435, 13–23. [Google Scholar] [CrossRef]
- Xiong, Z.; Guo, J.; Deng, Y.; Liu, B.; Lou, H.; Zeng, M.; Wang, Z.; Zhou, Z.; Yuan, W.; Qi, F. Elucidating the mechanism for oxidative coupling of methane catalyzed by La2O3: Experimental and microkinetic modeling studies. ACS Catal. 2024, 14, 1267–1280. [Google Scholar] [CrossRef]
- Yue, J.; Xiao, Q.; Zhao, D.; Guan, Y.; Liu, J.; Xu, G. Surfactant-assisted preparation of nanorod La2O3 catalysts for oxidative coupling of methane. Energy Fuels 2023, 37, 15986–15994. [Google Scholar] [CrossRef]
- Kuo, J.; Kresge, C.; Palermo, R. Evaluation of direct methane conversion to higher hydrocarbons and oxygenates. Catal. Today 1989, 4, 463–470. [Google Scholar] [CrossRef]
- Yaghobi, N. The role of gas hourly space velocity and feed composition for catalytic oxidative coupling of methane: Experimental study. J. King Saud Univ.-Eng. Sci. 2013, 25, 1–10. [Google Scholar] [CrossRef]
- Ahari, J.S.; Sadeghi, M.T.; Zarrinpashne, S. Effects of operating parameters on oxidative coupling of methane over Na-W-Mn/SiO2 catalyst at elevated pressures. J. Nat. Gas Chem. 2011, 20, 204–213. [Google Scholar] [CrossRef]
- Karimi, A.; Ahmadi, R.; Zadeh, H.B.; Jolodar, A.J.; Barkhordarion, A. Catalytic oxidative coupling of methane-experimental investigation and optimization of operational conditions. Pet. Coal 2007, 49, 36–40. [Google Scholar]
- Ni, J.; Lan, T.; Liu, P.; Lu, Y. Fluidized-bed OCM reaction: A promising Mn2O3-Na2WO4/TiO2 catalyst and a numerical study. Chem. Eng. J. 2024, 499, 155845. [Google Scholar] [CrossRef]
- Alturkistani, S.; Wang, H.; Gautam, R.; Sarathy, S.M. Importance of Process Variables and Their Optimization for Oxidative Coupling of Methane (OCM). ACS Omega 2023, 8, 21223–21236. [Google Scholar] [CrossRef]
- Aydin, Z.; Zanina, A.; Kondratenko, V.A.; Rabeah, J.; Li, J.; Chen, J.; Li, Y.; Jiang, G.; Lund, H.; Bartling, S. Effects of N2O and water on activity and selectivity in the oxidative coupling of methane over Mn–Na2WO4/SiO2: Role of oxygen species. ACS Catal. 2022, 12, 1298–1309. [Google Scholar] [CrossRef]
- Sadjadi, S.; Jašo, S.; Godini, H.; Arndt, S.; Wollgarten, M.; Blume, R.; Görke, O.; Schomäcker, R.; Wozny, G.; Simon, U. Feasibility study of the Mn–Na2WO4/SiO2 catalytic system for the oxidative coupling of methane in a fluidized-bed reactor. Catal. Sci. Technol. 2015, 5, 942–952. [Google Scholar] [CrossRef]
- Takanabe, K.; Iglesia, E. Rate and selectivity enhancements mediated by OH radicals in the oxidative coupling of methane catalyzed by Mn/Na2WO4/SiO2. Angew. Chem. 2008, 120, 7803–7807. [Google Scholar] [CrossRef]
- Yoon, S.; Lim, S.; Choi, J.-W.; Suh, D.J.; Song, K.H.; Ha, J.-M. Study on the unsteady state oxidative coupling of methane: Effects of oxygen species from O2, surface lattice oxygen, and CO2 on the C2+ selectivity. RSC Adv. 2020, 10, 35889–35897. [Google Scholar] [CrossRef]
- Guan, C.; Yang, Y.; Pang, Y.; Liu, Z.; Li, S.; Vovk, E.I.; Zhou, X.; Li, J.P.H.; Zhang, J.; Yu, N. How CO2 poisons La2O3 in an OCM catalytic reaction: A study by in situ XRD-MS and DFT. J. Catal. 2021, 396, 202–214. [Google Scholar] [CrossRef]
- Stoychev, D.; Valov, I.; Stefanov, P.; Atanasova, G.; Stoycheva, M.; Marinova, T. Electrochemical growth of thin La2O3 films on oxide and metal surfaces. Mater. Sci. Eng. C 2003, 23, 123–128. [Google Scholar] [CrossRef]
- Sunding, M.; Hadidi, K.; Diplas, S.; Løvvik, O.; Norby, T.; Gunnæs, A. XPS characterisation of in situ treated lanthanum oxide and hydroxide using tailored charge referencing and peak fitting procedures. J. Electron Spectrosc. Relat. Phenom. 2011, 184, 399–409. [Google Scholar] [CrossRef]
- Li, J.P.H.; Zhou, X.; Pang, Y.; Zhu, L.; Vovk, E.I.; Cong, L.; van Bavel, A.P.; Li, S.; Yang, Y. Understanding of binding energy calibration in XPS of lanthanum oxide by in situ treatment. Phys. Chem. Chem. Phys. 2019, 21, 22351–22358. [Google Scholar] [CrossRef]
- Lee, H.; Northrop, W.F. Oxidative coupling of methane using oxidant mixtures of CO2 and O2 over Sr/La2O3. Appl. Catal. A Gen. 2024, 673, 119587. [Google Scholar] [CrossRef]
- Wang, H.; Yang, C.; Shao, C.; Alturkistani, S.; Magnotti, G.; Gascon, J.; Takanabe, K.; Sarathy, S.M. A Homogeneous-Heterogeneous Kinetic Study of Oxidative Coupling of Methane (OCM) on La2O3/CeO2 Catalyst. ChemCatChem 2022, 14, e202200927. [Google Scholar] [CrossRef]
- Zhang, Z.; Gong, Y.; Xu, J.; Zhang, Y.; Xiao, Q.; Xi, R.; Xu, X.; Fang, X.; Wang, X. Dissecting La2Ce2O7 catalyst to unravel the origin of the surface active sites devoting to its performance for oxidative coupling of methane (OCM). Catal. Today 2022, 400, 73–81. [Google Scholar] [CrossRef]
- Xie, Y.; Cheng, J.; Wang, W.; Han, Y.; Fan, Q.; Li, H.; Cheng, K.; Zhang, Q.; Wang, Y. Separation of MnWOx and NaWSiOx Phases Boosts Oxidative Coupling of Methane. Angew. Chem. 2025, e202503767. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, G.; Wang, Y.; Lu, Y. MnTiO3-driven low-temperature oxidative coupling of methane over TiO2-doped Mn2O3-Na2WO4/SiO2 catalyst. Sci. Adv. 2017, 3, e1603180. [Google Scholar] [CrossRef]
- Vovk, E.I.; Wang, D.; Qiu, Z.; Liu, Y.; He, M.; Zhou, X.; Guan, C.; Yu, N.; Lu, Y.; Lang, J. In situ structure study of a TiO2 doped MnOX-Na2WO4/SiO2 catalyst under Na2WO4 melting conditions. Top. Catal. 2024, 1–15. [Google Scholar] [CrossRef]
- Wang, D.; Lang, J.; Qiu, Z.; Ding, N.; Yang, Y. Investigating the Impact of Na2WO4 Doping in La2O3-Catalyzed OCM Reaction: A Structure–Activity Study via In Situ XRD-MS. Catalysts 2024, 14, 150. [Google Scholar] [CrossRef]
- Zhao, M.; Ke, S.; Wu, H.; Xia, W.; Wan, H. Flower-like Sr-La2O3 microspheres with hierarchically porous structures for oxidative coupling of methane. Ind. Eng. Chem. Res. 2019, 58, 22847–22856. [Google Scholar] [CrossRef]
- Au, C.; Zhou, X.; Liu, Y.; Ji, W.; Ng, C. The Characterization of BaF2/Y2O3 Catalysts for the OCM Reaction. J. Catal. 1998, 174, 153–163. [Google Scholar] [CrossRef]
- Zhou, X.; Pang, Y.; Liu, Z.; Vovk, E.I.; van Bavel, A.P.; Li, S.; Yang, Y. Active oxygen center in oxidative coupling of methane on La2O3 catalyst. J. Energy Chem. 2021, 60, 649–659. [Google Scholar] [CrossRef]
- Li, J.P.H.; Liu, Z.; Wu, H.; Yang, Y. Investigation of CO oxidation over Au/TiO2 catalyst through detailed temperature programmed desorption study under low temperature and Operando conditions. Catal. Today 2018, 307, 84–92. [Google Scholar] [CrossRef]
- Liu, Z.; Ho Li, J.P.; Vovk, E.; Zhu, Y.; Li, S.; Wang, S.; van Bavel, A.P.; Yang, Y. Online kinetics study of oxidative coupling of methane over La2O3 for methane activation: What is behind the distinguished light-off temperatures? ACS Catal. 2018, 8, 11761–11772. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GHSV (mL·g−1·h−1) | CH4 Conversion at 740 °C (%) | C2 Selectivity at 740 °C (%) |
|---|---|---|
| 14,880 | 18.7 | 38.3 |
| 29,760 | 24 | 58.1 |
| 44,640 | 25 | 71 |
| 65,100 | 24 | 70.3 |
| 93,000 | 24.6 | 70.4 |
| 186,000 | 19 | 56.3 |
| Sample | C2 Yield at 550 °C (%) | C2 Yield at 740 °C (%) |
|---|---|---|
| La2O3-44,640 | 0.77 | 17.0 |
| La2O3-44,640 + 1%CO | 2.5 | 15.6 |
| La2O3-44,640 + 5%CO | 0.46 | 14.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, Z.; Cai, Y. Optimizing Methane Oxidative Coupling over La2O3: Kinetic and Product Analysis. Catalysts 2025, 15, 499. https://doi.org/10.3390/catal15050499
Qiu Z, Cai Y. Optimizing Methane Oxidative Coupling over La2O3: Kinetic and Product Analysis. Catalysts. 2025; 15(5):499. https://doi.org/10.3390/catal15050499
Chicago/Turabian StyleQiu, Zhehao, and Yulu Cai. 2025. "Optimizing Methane Oxidative Coupling over La2O3: Kinetic and Product Analysis" Catalysts 15, no. 5: 499. https://doi.org/10.3390/catal15050499
APA StyleQiu, Z., & Cai, Y. (2025). Optimizing Methane Oxidative Coupling over La2O3: Kinetic and Product Analysis. Catalysts, 15(5), 499. https://doi.org/10.3390/catal15050499