
Electrodeposited Co Crystalline Islands Shelled with Facile Spontaneously Deposited Pt for Improved Oxygen Reduction



Abstract
1. Introduction
2. Results and Discussion
2.1. The Preparation and Characterization of Co/GC and Co-Pt/GC Electrodes
2.1.1. The Preparation of Co/GC and Co-Pt/GC Electrodes
2.1.2. The Electrochemical Characterization of Cocryst/GC and Cocryst-Pt/GC Electrodes
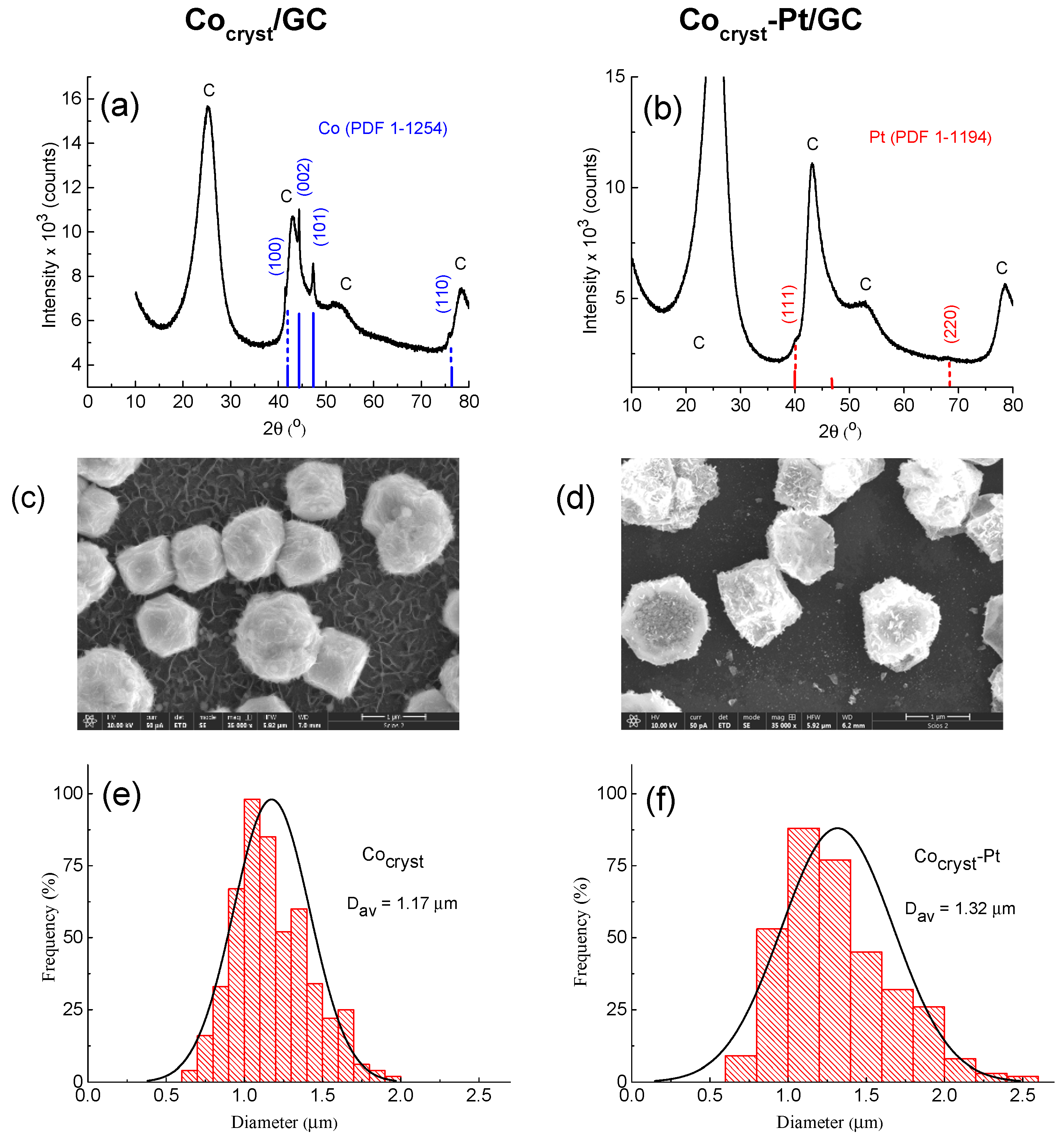
2.1.3. Structural, Morphological, and Chemical Characterization of the Cocryst/GC and Cocryst-Pt/GC
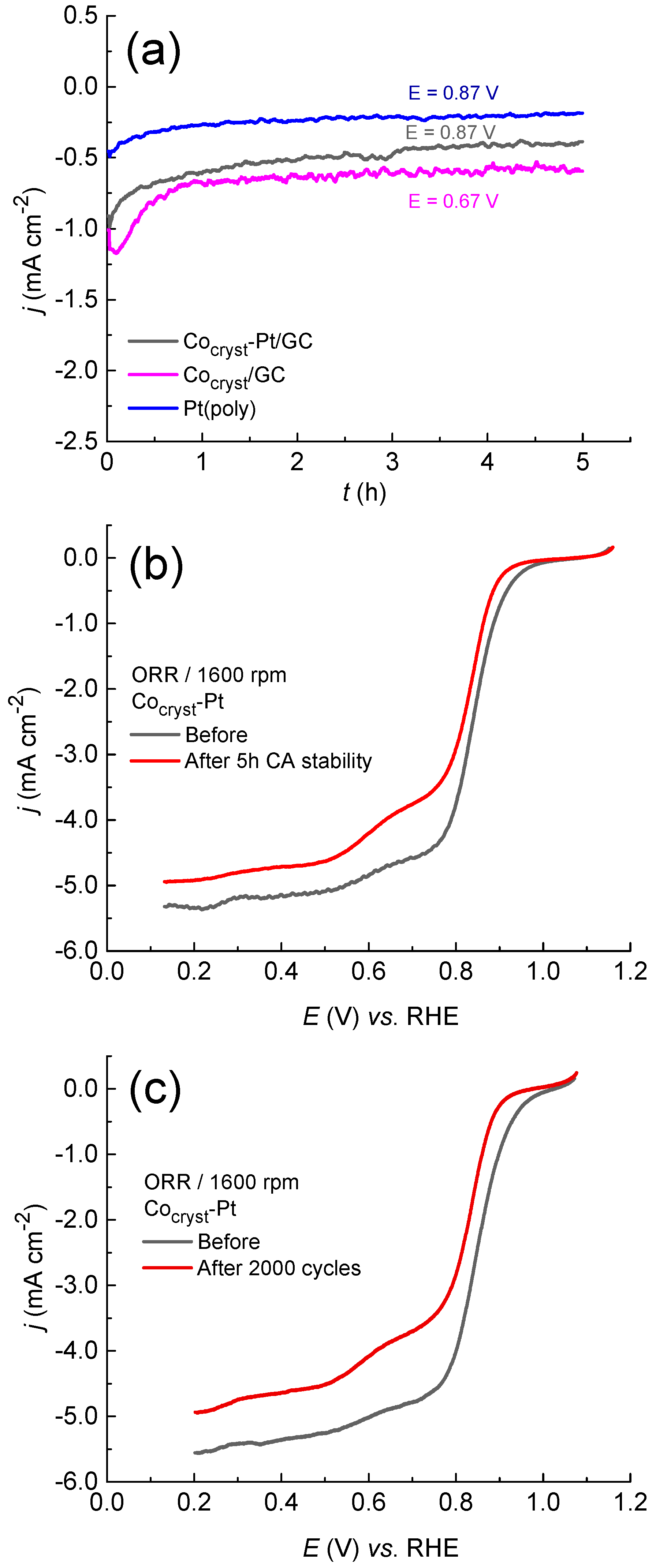
2.2. Oxygen Reduction Reaction on Cocryst/GC and Cocryst-Pt Catalysts
Comparison of the ORR Catalytic Activity and Stability of Cocryst-Pt/GC with That of Ptspont/GC, Pt(poly), and Cocryst/GC
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Material Characterization
3.3. Electrochemical Measurements
3.4. Chemicals
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zaman, S.; Huang, L.; Douka, A.I.; Yang, H.; You, B.; Xia, B.Y. Oxygen reduction electrocatalysts toward practical fuel cells: Progress and perspectives. Angew. Chem. Int. Ed. Engl. 2021, 9, 17832–17852. [Google Scholar] [CrossRef]
- Zhao, J.; Wei, D.; Zhang, C.; Shao, Q.; Murugadoss, V.; Guo, Z.; Jiang, Q.; Yang, X. An overview of oxygen reduction electrocatalysts for rechargeable zinc-air batteries enabled by carbon and carbon composites. Eng. Sci. 2021, 15, 1–19. [Google Scholar] [CrossRef]
- Wei, C.; Rao, R.R.; Peng, J.; Huang, B.; Stephens, I.E.L.; Risch, M.; Xu, Z.J.; Shao-Horn, Y. Recommended practices and benchmark activity for hydrogen and oxygen electrocatalysis in water splitting and fuel cells. Adv. Mater. 2019, 31, 1806296. [Google Scholar] [CrossRef] [PubMed]
- Norskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Wang, X.; Li, Z.; Qu, Y.; Yuan, T.; Wang, W.; Wu, Y.; Li, Y. Review of metal catalysts for oxygen reduction reaction: From nanoscale engineering to atomic design. Chem 2019, 5, 1486–1511. [Google Scholar] [CrossRef]
- Sui, S.; Wang, X.; Zhou, X.; Su, Y.; Riffat, S.; Liu, C.J. A comprehensive review of Pt electrocatalysts for the oxygen reduction reaction: Nanostructure, activity, mechanism and carbon support in PEM fuel cells. J. Mater. Chem. A 2017, 5, 1808. [Google Scholar] [CrossRef]
- Shao, M.; Chang, Q.; Dodelet, J.P.; Chenitz, R. Recent advances in electrocatalysts for oxygen reduction reaction. Chem. Rev. 2016, 116, 3594–3657. [Google Scholar] [CrossRef]
- Wu, G.; Zelenay, P. Nanostructured nonprecious metal catalysts for oxygen reduction reaction. Acc. Chem. Res. 2013, 46, 1878–1889. [Google Scholar] [CrossRef]
- Goswami, C.; Hazarika, K.K.; Bharali, P. Transition metal oxide nanocatalysts for oxygen reduction reaction. Mater. Sci. Energy Technol. 2018, 1, 117–128. [Google Scholar] [CrossRef]
- Yang, C.C.; Zai, S.F.; Zhou, Y.T.; Du, L.; Jiang, Q. Fe3C-Co nanoparticles encapsulated in a hierarchical structure of N-doped carbon as a multifunctional electrocatalyst for ORR, OER, and HER. Adv. Funct. Mater. 2019, 29, 1901949. [Google Scholar] [CrossRef]
- Liang, Y.; Li, Y.; Wang, H.; Zhou, J.; Wang, J.; Regier, T.; Dai, H. Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction. Nat. Mater. 2011, 10, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Ge, K.; Wu, Y.; Zhang, B.; Duan, J. Synergistic catalysis of cobalt tetroxide and bamboo-shaped carbon nanotubes doped with nitrogen for oxygen reduction in Zn–air batteries. Inorg. Chem. 2023, 62, 13378–13386. [Google Scholar] [CrossRef] [PubMed]
- Khusnuriyalova, A.F.; Caporali, M.; Hey-Hawkins, E.; Sinyashin, O.G.; Yakhvarov, D.G. Preparation of cobalt nanoparticles. Eur. J. Inorg. Chem. 2021, 2021, 3023–3114. [Google Scholar]
- Gu, M.; Yao, S.B.; Zhou, S.M. Electrochemical study of cobalt electrocrystallization on glass carbon from sulfate solution. Electrochemistry 2006, 74, 309–314. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, B.; Zhang, N.; Ma, M. Electrosynthesis of Co3O4 and Co(OH)2 ultrathin nanosheet arrays for efficient electrocatalytic water splitting in alkaline and neutral media. Nano Res. 2018, 11, 323–333. [Google Scholar] [CrossRef]
- Lisnund, S.; Blay, V.; Muamkhunthod, P.; Thunyanon, K.; Pansalee, J.; Monkrathok, J.; Maneechote, P.; Chansaenpak, K.; Pinyou, P. Electrodeposition of cobalt oxides on carbon nanotubes for sensitive bromhexine sensing. Molecules 2022, 27, 4078. [Google Scholar] [CrossRef]
- Niveditha, C.V.; Aswini, R.; Jabeen Fatima, M.J.; Ramanarayanan, R.; Pullanjiyot, N.; Swaminathan, S. Feather like highly active Co3O4 electrode for supercapacitor application: A potentiodynamic approach. Mater. Res. Express 2018, 5, 065501. [Google Scholar] [CrossRef]
- Ling, T.; Yan, D.Y.; Jiao, Y.; Wang, H.; Zheng, Y.; Zheng, X.; Mao, J.; Du, X.-W.; Hu, Z.; Jaroniec, M.; et al. Engineering surface atomic structure of single-crystal cobalt (II) oxide nanorods for superior electrocatalysis. Nat. Commun. 2016, 7, 12876. [Google Scholar] [CrossRef]
- Liang, Y.; Wang, H.; Diao, P.; Chang, W.; Hong, G.; Li, Y.; Gong, M.; Xie, L.; Zhou, J.; Wang, J.; et al. Oxygen reduction electrocatalyst based on strongly coupled cobalt oxide nanocrystals and carbon nanotubes. J. Am. Chem. Soc. 2012, 134, 15849–15857. [Google Scholar] [CrossRef]
- Mao, S.; Wen, Z.; Huang, T.; Hou, Y.; Chen, J. High-performance bifunctional electrocatalysts of 3d crumpled graphene cobalt oxide nanohybrids for oxygen reduction and evolution reactions. Energy Environ. Sci. 2014, 7, 609–616. [Google Scholar] [CrossRef]
- Zhang, S.; Shang, N.; Gao, S.; Meng, T.; Wang, Z.; Gao, Y.; Wang, C. Ultra dispersed Co supported on nitrogen-doped carbon: An efficient electrocatalyst for oxygen reduction reaction and Zn-air battery. Chem. Eng. Sci. 2021, 234, 116442. [Google Scholar] [CrossRef]
- Huang, D.; Luo, Y.; Li, S.; Zhang, B.; Shen, Y.; Wang, M. Active catalysts based on cobalt oxide@cobalt/N-C nanocomposites for oxygen reduction reaction in alkaline solutions. Nano Res. 2014, 7, 1054–1064. [Google Scholar] [CrossRef]
- Liu, L.; Liu, H.; Sun, X.; Li, C.; Bai, J. Efficient electrocatalyst of PteFe/CNFs for oxygen reduction reaction in alkaline media. Int. J. Hydrogen Energy 2020, 45, 15112–15120. [Google Scholar] [CrossRef]
- Alemany-Molina, G.; Lo Vecchio, C.; Baglio, V.; Aricò, A.S.; Morallón, E.; Cazorla-Amorós, D. Pt nanoparticles for improving the performance and durability of Fe-N-C based materials towards oxygen reduction reaction in alkaline direct methanol fuel cells. J. Colloid Sci. 2025, 691, 137426. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, K.; Wang, P.; He, Y.; Liu, Z. Pt-Fe-Co Ternary metal single atom catalyst for toward high efficiency alkaline oxygen reduction reaction. Energies 2023, 16, 3684. [Google Scholar] [CrossRef]
- Campos-Roldán, C.A.; Calvillo, L.; Granozzi, G.; Alonso-Vante, N. Alkaline hydrogen electrode and oxygen reduction reaction on PtxNi nanoalloys. J. Electroanal. Chem. 2020, 857, 113449. [Google Scholar] [CrossRef]
- Zysler, M.; Shokhen, V.; Hardisty, S.S.; Muzikansky, A.; Zitoun, D. Bifunctional Pt−Ni electrocatalyst synthesis with ultralow platinum seeds for oxygen evolution and reduction in alkaline medium. ACS Appl. Energy Mater. 2022, 5, 4212–4220. [Google Scholar] [CrossRef]
- Liao, Q.; Kuroki, H.; Tamaki, T.; Arao, M.; Matsumoto, M.; Imai, H.; Yamaguchi, T. Three-dimensionally connected platinum-cobalt nanoparticles as support-free electrocatalysts for oxygen reduction. ACS Appl. Nano Mater. 2025, 8, 3323–3332. [Google Scholar] [CrossRef]
- Zeng, X.; Mitra, S.K.; Li, X. One-pot synthesis of supported PtCox bifunctional catalysts for oxygen reduction and hydrogen evolution reactions. Int. J. Hydrogen Energy 2024, 86, 577–585. [Google Scholar] [CrossRef]
- Yan, W.; Sun, P.; Luo, C.; Xia, X.; Liu, Z.; Zhao, Y.; Zhang, S.; Sun, L.; Du, F. PtCo-based nanocatalyst for oxygen reduction reaction: Recent highlights on synthesis strategy and catalytic mechanism. Chin. J. Chem. Eng. 2023, 53, 101–123. [Google Scholar] [CrossRef]
- Oezaslan, M.; Hasché, F.; Strasser, P. Oxygen electroreduction on PtCo3, PtCo and Pt3Co alloy nanoparticles for alkaline and acidic PEM fuel cells. J. Electrochem. Soc. 2012, 159, B394–B405. [Google Scholar] [CrossRef]
- Lima, F.H.B.; de Castro, J.F.R.; Santos, L.G.R.A.; Ticianelli, E.A. Electrocatalysis of oxygen reduction on carbon-supported Pt–Co nanoparticles with low Pt content. J. Power Sources 2009, 190, 293–300. [Google Scholar] [CrossRef]
- Miyatake, K.; Shimizu, Y. Pt/Co Alloy Nanoparticles Prepared by Nanocapsule Method Exhibit a High Oxygen Reduction Reaction Activity in the Alkaline Media. ACS Omega 2017, 2, 2085–2089. [Google Scholar] [CrossRef] [PubMed]
- Sravani, B.; Raghavendra, P.; Chandrasekhar, Y.; Veera Manohara Reddy, Y.; Sivasubramanian, R.; Venkateswarlu, K.; Madhavi, G.; Subramanyam Sarma, L. Immobilization of platinum-cobalt and platinum-nickel bimetallic nanoparticles on pomegranate peel extract-treated reduced graphene oxide as electrocatalysts for oxygen reduction reaction. Int. J. Hydrogen Energy 2020, 45, 7680–7690. [Google Scholar] [CrossRef]
- Yun, M.; Ahmed, M.S.; Han, H.S.; Jeon, S. Platinum-cobalt binary alloyed nanoparticles supported on thiolated graphene oxide for oxygen reduction reaction in alkaline media. J. Nanosci. Nanotechnol. 2016, 16, 9675–9682. [Google Scholar] [CrossRef]
- Rakočević, L.; Golubović, J.; Vasiljević Radović, D.; Rajić, V.; Štrbac, S. A comparative study of hydrogen evolution on Pt/GC and Pt/GNPs in acid solution. Int. J. Hydrogen Energy 2024, 51, 1240–1254. [Google Scholar] [CrossRef]
- Vanysek, P. Electrochemical series. In CRC Handbook of Chemistry and Physics, 93rd ed.; Haynes, W.M., Ed.; CRC Press Taylor & Francis Group: Boca Raton, FL, USA; London, UK, 2012; pp. 8–21. [Google Scholar]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions, 2nd ed.; National Association of Corrosion Engineers: Houston, TX, USA, 1974; p. 325. (In English) [Google Scholar]
- Xie, R.-C.; Batchelor-McAuley, C.; Rauwel, E.; Rauwel, P.; Compton, R.G. Electrochemical characterisation of Co@Co(OH)2 core-shell nanoparticles and their aggregation in solution. ChemElectroChem 2020, 7, 4259–4268. [Google Scholar] [CrossRef]
- Gomez Meier, H.; Vilche, J.R.; Arvía, A.J. The electrochemical behaviour of cobalt in alkaline solutions Part I. The potentiodynamic response in the potential region of the Co/CoO couple. J. Electroanal. Chem. Interf. Electrochem. 1982, 134, 251–272. [Google Scholar] [CrossRef]
- Gomez Meier, H.; Vilche, J.R.; Arvía, A.J. The electrochemical behaviour of cobalt in alkaline solutions part II. The potentiodynamic response of Co(OH)2 electrodes. J. Electroanal. Chem. Interf. Electrochem. 1982, 138, 367–379. [Google Scholar] [CrossRef]
- Burke, L.D.; Murphy, M.M. The electrocatalytic behavior of cobalt (and iron) electrodes at low potential in base. J. Electrochem. Soc. 1991, 138, 88–94. [Google Scholar] [CrossRef]
- Veggetti, E.; Kodintsev, I.M.; Trasatti, S. Hydrogen evolution on oxide electrodes: Co3O4 in alkaline solution. J. Electroanal. Chem. 1992, 339, 255–268. [Google Scholar] [CrossRef]
- Štrbac, S. The effect of pH on oxygen and hydrogen peroxide reduction on polycrystalline Pt electrode. Electrochim. Acta 2011, 56, 1597–1604. [Google Scholar] [CrossRef]
- Saeki, R.; Ohgai, T. Effect of growth rate on the crystal orientation and magnetization performance of cobalt nanocrystal arrays electrodeposited from aqueous solution. Nanomaterials 2018, 8, 566. [Google Scholar] [CrossRef]
- Manjunatha, M.; Reddy, G.S.; Mallikarjunaiah, K.J.; Ramakrishna, D.; Ramesh, K.P. Determination of phase composition of cobalt nanoparticles using 59Co internal field nuclear magnetic resonance. J. Supercond. Nov. Magn. 2019, 32, 3201–3209. [Google Scholar] [CrossRef]
- de la Pena O’Shea, V.A.; Piscina, P.R.; Homs, N.; Aromí, G.; Fierro, J.L.G. Development of hexagonal closed-packed cobalt nanoparticles stable at high temperature. Chem. Mater. 2009, 21, 5637–5643. [Google Scholar] [CrossRef]
- Meng, Q.; Guo, S.; Zhao, X.; Veintemillas-Verdaguer, S. Bulk metastable cobalt in fcc crystal structure. J. Alloys Comp. 2013, 580, 187–190. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Rajagopal, V.; Mehla, S.; Jones, L.A.; Bhargava, S.K. Nanoengineered cobalt electrocatalyst for alkaline oxygen evolution reaction. Nanomaterials 2024, 14, 946. [Google Scholar] [CrossRef]
- Milikić, J.; Knežević, S.; Ognjanović, M.; Stanković, D.; Rakočević, L.; Šljukić, B. Template-based synthesis of Co3O4 and Co3O4/SnO2 bifunctional catalysts with enhanced electrocatalytic properties for reversible oxygen evolution and reduction reaction. Int. J. Hydrogen Energy 2023, 48, 27568–27581. [Google Scholar] [CrossRef]
- Paul, A.; Gusmão, F.; Mahmoud, A.G.; Hazra, S.; Rakočević, L.; Šljukić, B.; Khan, R.A.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Catalyzing towards clean energy: Tuning the oxygen evolution reaction by amide-functionalized Co(II) and Ni(II) pristine coordination polymers. CrystEngComm 2024, 26, 2755–2764. [Google Scholar] [CrossRef]
- Rakočević, L.; Simatović Stojković, I.; Maksić, A.; Rajić, V.; Štrbac, S.; Srejić, I. PtAu nanoparticles supported by reduced graphene oxide as a highly active catalyst for hydrogen evolution. Catalysts 2022, 12, 43. [Google Scholar] [CrossRef]
- Štrbac, S.; Adzić, R.R. The influence of OH− chemisorption on the catalytic properties of gold single crystal surfaces for oxygen reduction in alkaline solutions. J. Electroanal. Chem. 1996, 403, 169–181. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef]
- Markovic, N.; Gasteiger, H.; Ross, P.N. Kinetics of oxygen reduction on Pt(hkl) electrodes: Implications for the crystallite size effect with supported Pt electrocatalysts. J. Electrochem. Soc. 1997, 144, 1591. [Google Scholar] [CrossRef]
- Siddika, M.; Hosen, N.; Althomali, R.H.; Al-Humaidi, J.Y.; Rahman, M.M.; Hasnat, M.A. Kinetics of electrocatalytic oxygen reduction reaction over an activated glassy carbon electrode in an alkaline medium. Catalysts 2024, 14, 164. [Google Scholar] [CrossRef]
- Xiao, J.; Kuang, Q.; Yang, S.; Xiao, F.; Wang, S.; Guo, L. Surface structure dependent electrocatalytic activity of Co3O4 anchored on graphene sheets toward oxygen reduction reaction. Sci. Rep. 2013, 3, 2300. [Google Scholar] [CrossRef]
- Zhan, Y.; Lu, M.; Yang, S.; Xu, C.; Liu, Z.; Lee, J.Y. Activity of transition-metal (manganese, iron, cobalt, and nickel) phosphates for oxygen electrocatalysis in alkaline solution. ChemCatChem 2016, 8, 372–379. [Google Scholar] [CrossRef]
- Huang, K.; Xu, P.; He, X.; Wang, R.; Wang, Y.; Yang, H.; Zhang, R.; Lei, M.; Tang, H. Annealing-free platinum cobalt alloy nanoparticles on nitrogen-doped mesoporous carbon with boosted oxygen electroreduction performance. ChemElectroChem 2020, 7, 3341–3346. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, J.; Zhao, Y.; Wang, F. Composition-controlled synthesis of carbon-supported Pt–Co alloy nanoparticles and the origin of their ORR activity enhancement. Phys. F Chem. Chem. Phys. 2014, 16, 19298–19306. [Google Scholar] [CrossRef]
- Todoroki, N.; Wadayama, T. Oxygen reduction and oxygen evolution reaction activity on Co/Pt(111) surfaces in alkaline solution. ECS Trans. 2018, 86, 569. [Google Scholar] [CrossRef]
- Hu, S.; Goenaga, G.; Melton, C.; Zawodzinski, T.A.; Mukherjee, D. PtCo/CoOx nanocomposites: Bifunctional electrocatalysts for oxygen reduction and evolution reactions synthesized via tandem laser ablation synthesis in solution-galvanic replacement reactions. Appl. Catal. B Environ. 2016, 182, 286–296. [Google Scholar] [CrossRef]
- Ribeiro, E.L.; Davis, E.M.; Mokhtarnejad, M.; Hu, S.; Mukherjee, D.; Khomami, B. MOF-derived PtCo/Co3O4 nanocomposites in carbonaceous matrices as high-performance ORR electrocatalysts synthesized via laser ablation techniques. Catal. Sci. Technol. 2021, 11, 3002–3013. [Google Scholar] [CrossRef]
- Ni, C.; Chen, X.; Chen, Y.; Li, S.; Zhou, T.; Yang, J.; Liu, M.; Su, H. Ultrafine intermetallic platinum-cobalt with a contracted Pt–Pt pair for efficient acidic oxygen reduction reactions. Nanoscale 2025, 17, 10380–10388. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Liu, Q.; Nichols, F.; Mercado, R.; Morris, D.; Li, N.; Zhang, P.; Gao, P.; Ping, Y.; Chen, S. Oxygen reduction reaction catalyzed by carbon-supported platinum few-atom clusters: Significant enhancement by doping of atomic cobalt. Research 2020, 2020, 9167829. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wei, M.; Qi, R.; Dong, C.-L.; Dang, D.; Yang, C.-C.; Xia, C.; Chen, C.; Zaman, S.; Li, F.-M.; et al. An integrated platinum-nanocarbon electrocatalyst for efficient oxygen reduction. Nat. Commun. 2022, 13, 6703. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, J.H.; Lee, D.; Oh, S.; Park, J.; Im, K.; Yoo, S.J.; Kim, J. Low-loading platinum–cobalt electrocatalyst supported on hollow carbon for enhanced oxygen reduction reaction. Chem. Eng. J. 2024, 500, 157072. [Google Scholar] [CrossRef]
- Li, F.; Liu, Y.; Dong, Y.; Wang, H.; Wang, T.; Lu, J.; Huang, W.; Chen, X.; Guo, Y.; Zheng, X.; et al. Electronic modulation of PtCo catalysts by topological carbon defects boosting durable oxygen reduction reaction in acidic media. Fundam. Res. 2025; in press. [Google Scholar] [CrossRef]
- Holade, Y.; Sahin, N.E.; Servat, K.; Napporn, T.W.; Kokoh, K.B. Recent advances in carbon supported metal nanoparticles preparation for oxygen reduction reaction in low temperature fuel cells. Catalysts 2015, 5, 310–348. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Line | Cocryst/GC | Cocryst-Pt/GC | ||
|---|---|---|---|---|
| at% | wt% | at% | wt% | |
| C 1s | 11.6 | 6.5 | 50.5 | 24.7 |
| O 1s | 74.4 | 55.2 | 36.2 | 23.6 |
| Co 2p | 14.0 | 38.3 | - | - |
| Pt 4f7/2 | - | - | 5.0 | 39.7 |
| Cl 2p | - | - | 8.3 | 12.0 |
| Catalyst | Solution | Eonset (V) | E1/2 (V) | Tafel Slope (mV dec−1) | Reference |
|---|---|---|---|---|---|
| Pt3Co/r-GO | 0.1 M KOH | 1.13 | 0.76 | - | [34] |
| ER/PtCo-tG | 0.1 M KOH | 0.94 | 0.86 | - | [35] |
| PtCo@NMC | 0.1 M KOH | 1.04 | 0.96 | 67.4 | [60] |
| Pt76Co24 | 1 M NaOH | 0.98 | - | 59.98 | [61] |
| Co/Pt(111) | 0.1 M KOH | - | - | 51 | [62] |
| PtCo-3 | 1 M KOH | - | 0.86 | 39.2 | [63] |
| 125-PtCo@C–Co3O4 | 1 M KOH | 0.95 | - | 38 | [64] |
| Pt3Co/NC | 1 M KOH | - | 0.91 | - | [65] |
| PtCo-NC-4 | 0.1 M HClO4 | 1.03 | 0.93 | 53.7 | [66] |
| PtCo@CoNC/NTG | 0.1 M HClO4 | - | 0.94 | 71 | [67] |
| 5-wt% Pt/h-Co-NC | 0.1 M HClO4 | 1.00 | 0.87 | 91 | [68] |
| PtCo-DPC | 0.1 M HClO4 | 1.02 | 0.85 | 87.5 | [69] |
| Cocryst-Pt | 0.1 M NaOH | 1.07 | 0.87 | 63 | This work |
| Pt/C | 0.1 M KOH | 1.01 | 0.89 | 102.6 | [60] |
| 20 wt%Pt/C | 0.1 M NaOH | - | 0.85 | 69 | [70] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golubović, J.; Rakočević, L.; Rajić, V.; Milović, M.; Štrbac, S. Electrodeposited Co Crystalline Islands Shelled with Facile Spontaneously Deposited Pt for Improved Oxygen Reduction. Catalysts 2025, 15, 490. https://doi.org/10.3390/catal15050490
Golubović J, Rakočević L, Rajić V, Milović M, Štrbac S. Electrodeposited Co Crystalline Islands Shelled with Facile Spontaneously Deposited Pt for Improved Oxygen Reduction. Catalysts. 2025; 15(5):490. https://doi.org/10.3390/catal15050490
Chicago/Turabian StyleGolubović, Jelena, Lazar Rakočević, Vladimir Rajić, Miloš Milović, and Svetlana Štrbac. 2025. "Electrodeposited Co Crystalline Islands Shelled with Facile Spontaneously Deposited Pt for Improved Oxygen Reduction" Catalysts 15, no. 5: 490. https://doi.org/10.3390/catal15050490
APA StyleGolubović, J., Rakočević, L., Rajić, V., Milović, M., & Štrbac, S. (2025). Electrodeposited Co Crystalline Islands Shelled with Facile Spontaneously Deposited Pt for Improved Oxygen Reduction. Catalysts, 15(5), 490. https://doi.org/10.3390/catal15050490