

Exploring Perhydro-Benzyltoluene Dehydrogenation Using Sulfur-Doped PtMo/Al2O3 Catalysts




Abstract

1. Introduction
2. Results
2.1. Catalyst Metal Content and Metal Dispersion
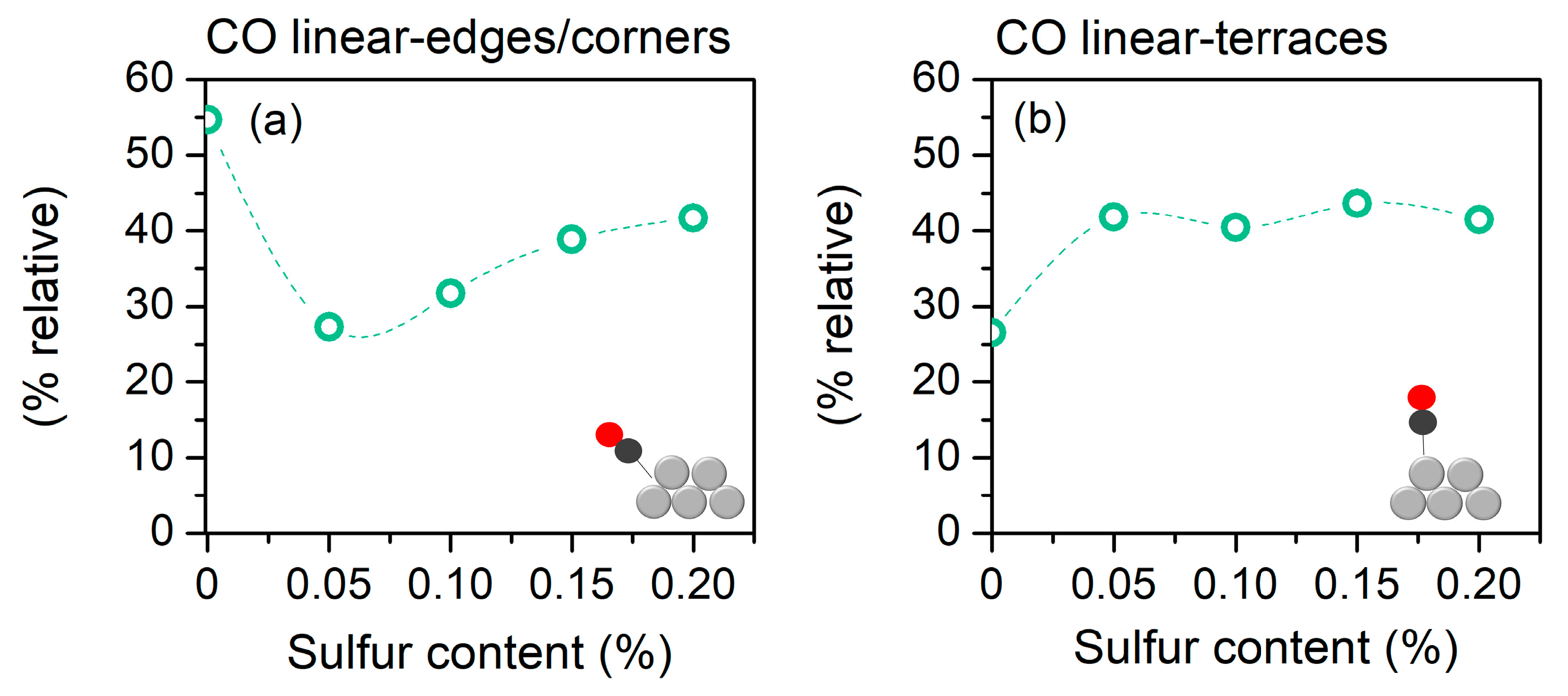
2.2. Metal Oxide Reducibility
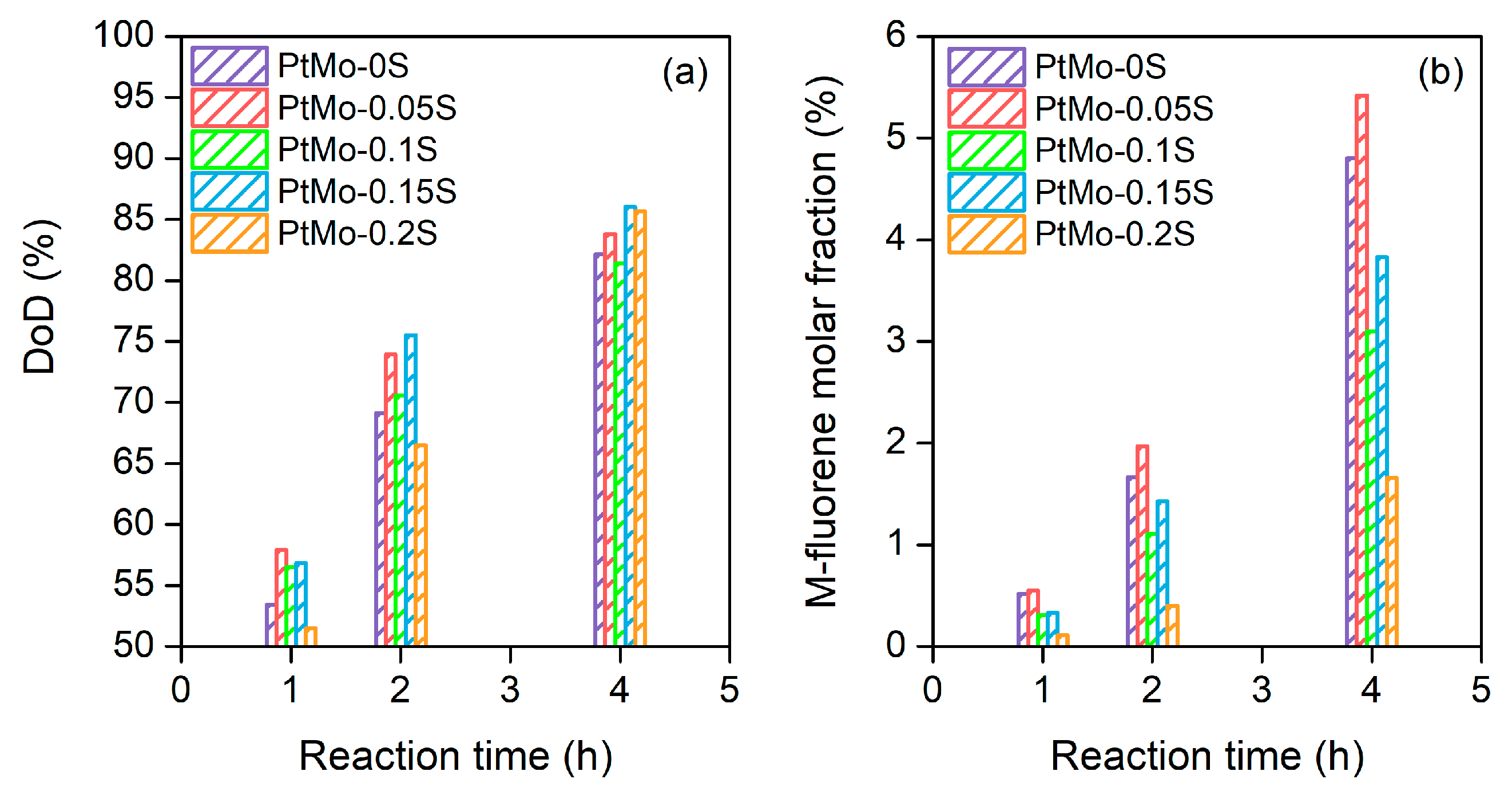
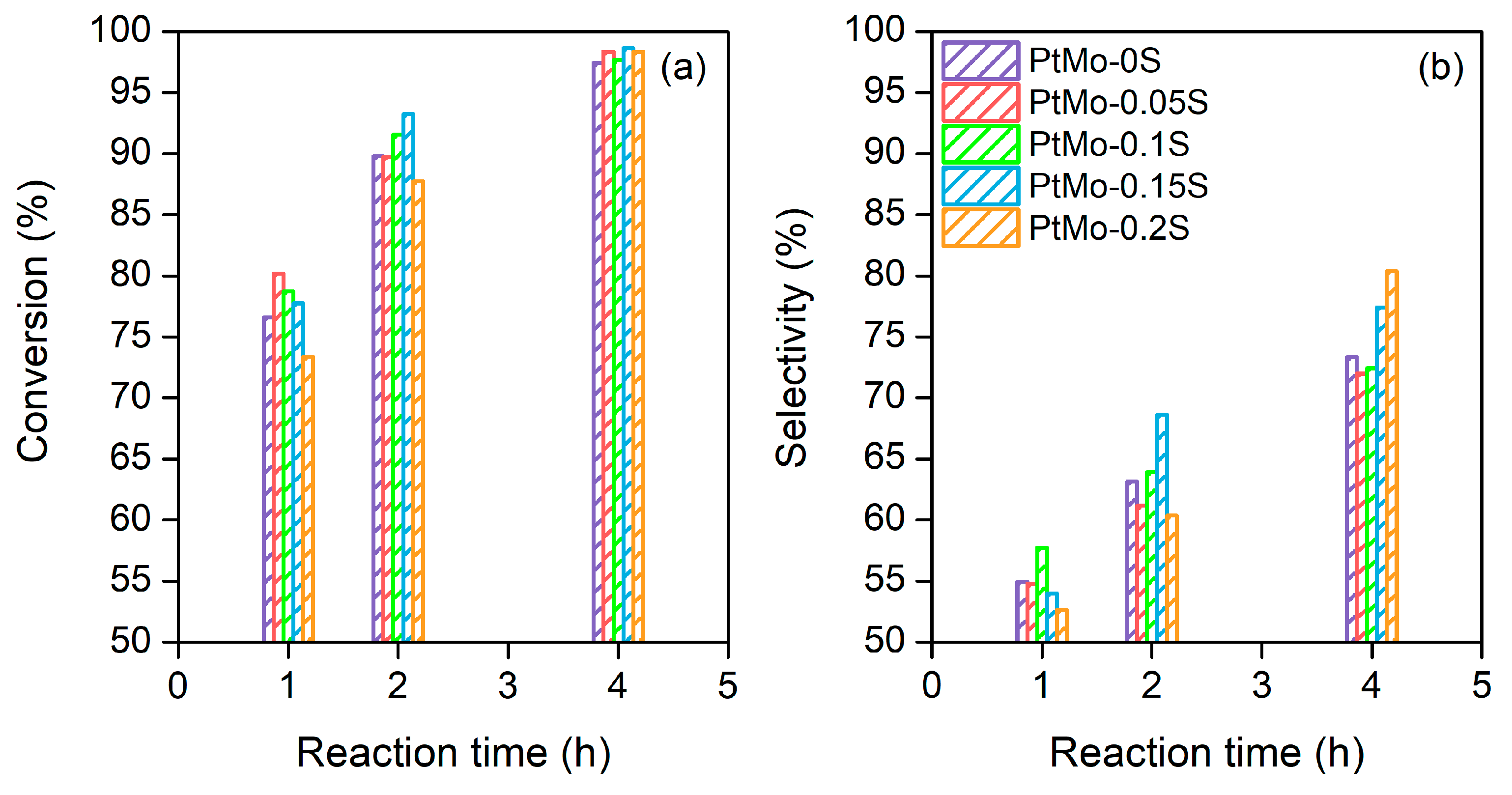
2.3. Dehydrogenation Activity
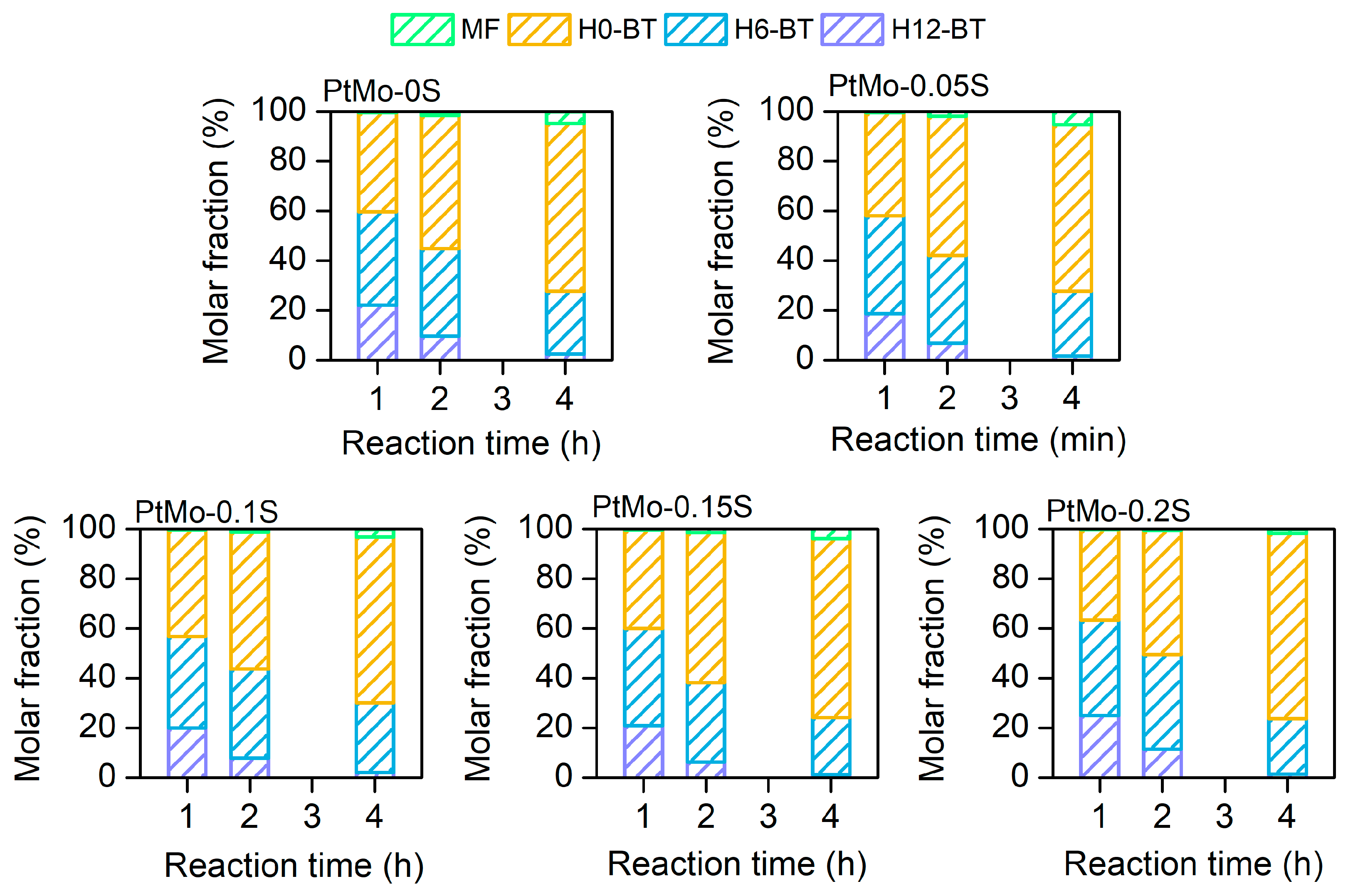
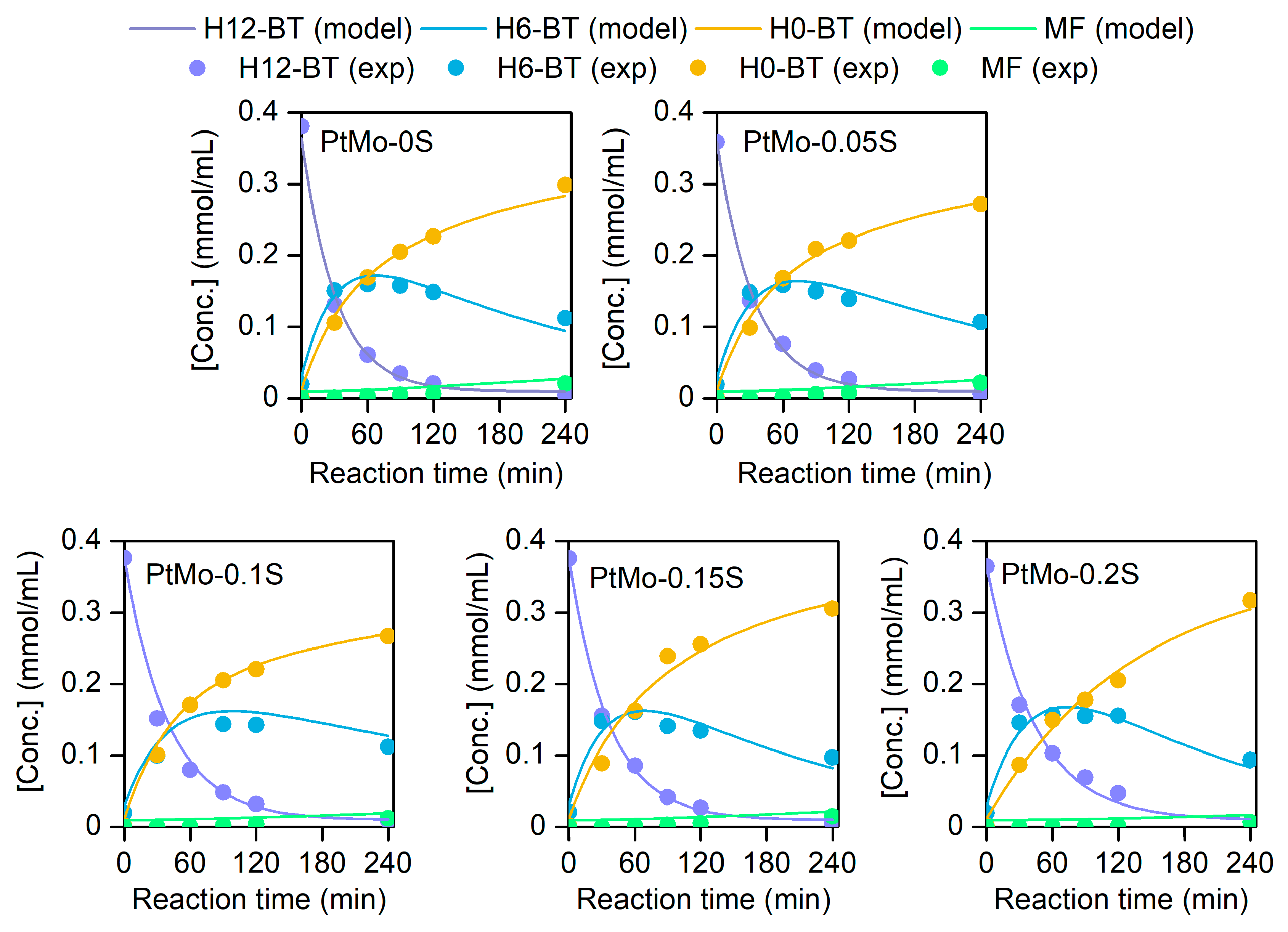
2.4. Dehydrogenation Pathway
H12-BT -> H6-BT
H12-BT -> H0-BT
H6-BT -> H0-BT
H0-BT -> MF
2.5. Effect of Sulfur Doping on Bimetallic PtMo/Al2O3 Catalyst Activity
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Dehydrogenation Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BET | Brunauer, Emmet, and Teller theory |
| BJH | Barrett–Joyner–Halenda |
| DRIFT | Diffuse Reflectance Infrared Fourier Transform Spectroscopy |
| GC-MS | Gas Chromatography–Mass Spectrometry |
| LOHC | Liquid Organic Hydrogen Carrier |
| H0-BT | Benzyltoluene |
| H6-BT | Cyclohexylmethyl-Methylbenzene |
| H12-BT | Perhydrobenzyltoluene |
| HAADF | High-Angle Annular Dark-Field PGMs (Platinum Group Metals) |
| STEM | Scanning Transmission Electron Microscopy |
| TPR | Temperature-Programmed Deduction |
| TPD | Temperature-Programmed Desorption |
| XRF | X-ray Fluorescence |
References
- Faye, O.; Szpunar, J.; Eduok, U. A critical review on the current technologies for the generation, storage, and transportation of hydrogen. Int. J. Hydrogen Energy 2022, 47, 13771–13802. [Google Scholar] [CrossRef]
- Durakovic, G.; del Granado, P.C.; Tomasgard, A. Are green and blue hydrogen competitive or complementary? Insights from a decarbonized European power system analysis. Energy 2023, 282, 128282. [Google Scholar] [CrossRef]
- Muhammed, N.S.; Gbadamosi, A.O.; Epelle, E.I.; Abdulrasheed, A.A.; Haq, B.; Patil, S.; Al-Shehri, D.; Kamal, M.S. Hydrogen production, transportation, utilization, and storage: Recent advances towards sustainable energy. J. Energy Storage 2023, 73, 109207. [Google Scholar] [CrossRef]
- Usman, M.R. Hydrogen storage methods: Review and current status. Renew. Sustain. Energy Rev. 2022, 167, 112743. [Google Scholar] [CrossRef]
- Hassan, I.A.; Ramadan, H.S.; Saleh, M.A.; Hissel, D. Hydrogen storage technologies for stationary and mobile applications: Review, analysis and perspectives. Renew. Sustain. Energy Rev. 2021, 149, 111311. [Google Scholar] [CrossRef]
- Zhang, L.; Jia, C.; Bai, F.; Wang, W.; An, S.; Zhao, K.; Li, Z.; Li, J.; Sun, H. A comprehensive review of the promising clean energy carrier: Hydrogen production, transportation, storage, and utilization (HPTSU) technologies. Fuel 2024, 355, 129455. [Google Scholar] [CrossRef]
- Jorschick, H.; Preuster, P.; Bösmann, A.; Wasserscheid, P. Hydrogenation of aromatic and heteroaromatic compounds—A key process for future logistics of green hydrogen using liquid organic hydrogen carrier systems. Sustain. Energy Fuels 2021, 5, 1311–1346. [Google Scholar] [CrossRef]
- Bourane, A.; Elanany, M.; Pham, T.V.; Katikaneni, S.P. An overview of organic liquid phase hydrogen carriers. Int. J. Hydrogen Energy 2016, 41, 23075–23091. [Google Scholar] [CrossRef]
- Niermann, M.; Beckendorff, A.; Kaltschmitt, M.; Bonhoff, K. Liquid Organic Hydrogen Carrier (LOHC)—Assessment based on chemical and economic properties. Int. J. Hydrogen Energy 2019, 44, 6631–6654. [Google Scholar] [CrossRef]
- Aakko-Saksa, P.T.; Cook, C.; Kiviaho, J.; Repo, T. Liquid organic hydrogen carriers for transportation and storing of renewable energy—Review and discussion. J. Power Sources 2018, 396, 803–823. [Google Scholar] [CrossRef]
- Niermann, M.; Timmerberg, S.; Drünert, S.; Kaltschmitt, M. Liquid Organic Hydrogen Carriers and alternatives for international transport of renewable hydrogen. Renew. Sustain. Energy Rev. 2021, 135, 110171. [Google Scholar] [CrossRef]
- Alhumaidan, F.; Cresswell, D.; Garforth, A. Hydrogen Storage in Liquid Organic Hydride: Producing Hydrogen Catalytically from Methylcyclohexane. Energy Fuels 2011, 25, 4217–4234. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Y.; Yu, X.; Ma, X.; Boebinger, M.; Weber, J.; Wu, Z. Insights into size effects of Pt/Al2O3 catalysts on hydrogen production from methylcyclohexane dehydrogenation. Catal. Sci. Technol. 2024, 14, 1791–1801. [Google Scholar] [CrossRef]
- Lee, G.; Jeong, Y.; Kim, B.-G.; Han, J.S.; Jeong, H.; Na, H.B.; Jung, J.C. Hydrogen production by catalytic decalin dehydrogenation over carbon-supported platinum catalyst: Effect of catalyst preparation method. Catal. Commun. 2015, 67, 40–44. [Google Scholar] [CrossRef]
- Jander, J.H.; Kerscher, M.; Li, S.; Rausch, M.H.; Wasserscheid, P.; Fröba, A.P. Determination of hydrogen loading in the carrier system diphenylmethane/dicyclohexylmethane by depolarized Raman spectroscopy. Int. J. Hydrogen Energy 2022, 47, 9331–9345. [Google Scholar] [CrossRef]
- Jander, J.H.; Kerscher, M.; Cui, J.; Wicklein, J.; Rüde, T.; Preuster, P.; Rausch, M.H.; Wasserscheid, P.; Koller, T.M.; Fröba, A.P. Viscosity, surface tension, and density of the liquid organic hydrogen carrier system based on diphenylmethane, biphenyl, and benzophenone. Int. J. Hydrogen Energy 2022, 47, 22078–22092. [Google Scholar] [CrossRef]
- Jang, M.; Jo, Y.S.; Lee, W.J.; Shin, B.S.; Sohn, H.; Jeong, H.; Jang, S.C.; Kwak, S.K.; Kang, J.W.; Yoon, C.W. A High-Capacity, Reversible Liquid Organic Hydrogen Carrier: H2-Release Properties and an Application to a Fuel Cell. ACS Sustain. Chem. Eng. 2019, 7, 1185–1194. [Google Scholar] [CrossRef]
- Kwak, Y.; Moon, S.; Ahn, C.; Kim, A.-R.; Park, Y.; Kim, Y.; Sohn, H.; Jeong, H.; Nam, S.W.; Yoon, C.W.; et al. Effect of the support properties in dehydrogenation of biphenyl-based eutectic mixture as liquid organic hydrogen carrier (LOHC) over Pt/Al2O3 catalysts. Fuel 2021, 284, 119285. [Google Scholar] [CrossRef]
- Leinweber, A.; Müller, K. Hydrogenation of the Liquid Organic Hydrogen Carrier Compound Monobenzyl Toluene: Reaction Pathway and Kinetic Effects. Energy Technol. 2018, 6, 513–520. [Google Scholar] [CrossRef]
- Brückner, N.; Obesser, K.; Bösmann, A.; Teichmann, D.; Arlt, W.; Dungs, J.; Wasserscheid, P. Evaluation of Industrially Applied Heat-Transfer Fluids as Liquid Organic Hydrogen Carrier Systems. ChemSusChem 2014, 7, 229–235. [Google Scholar] [CrossRef]
- Oh, J.; Jo, Y.; Kim, T.W.; Bathula, H.B.; Yang, S.; Baik, J.H.; Suh, Y.-W. Highly efficient and robust Pt ensembles on mesoporous alumina for reversible H2 charge and release of commercial benzyltoluene molecules. Appl. Catal. B Environ. 2022, 305, 121061. [Google Scholar] [CrossRef]
- Jorschick, H.; Preuster, P.; Dürr, S.; Seidel, A.; Müller, K.; Bösmann, A.; Wasserscheid, P. Hydrogen storage using a hot pressure swing reactor. Energy Environ. Sci. 2017, 10, 1652–1659. [Google Scholar] [CrossRef]
- Modisha, P.; Bessarabov, D. Stress tolerance assessment of dibenzyltoluene-based liquid organic hydrogen carriers. Sustain. Energy Fuels 2020, 4, 4662–4670. [Google Scholar] [CrossRef]
- Garidzirai, R.; Modisha, P.; Bessarabov, D. Assessment of Reaction Kinetics for the Dehydrogenation of Perhydro-Dibenzyltoluene Using Mg- and Zn-Modified Pt/Al2O3 Catalysts. Catalysts 2024, 14, 32. [Google Scholar] [CrossRef]
- Ali, A.; Kumar, G.U.; Lee, H.J. Investigation of hydrogenation of Dibenzyltoluene as liquid organic hydrogen carrier. Mater. Today Proc. 2021, 45, 1123–1127. [Google Scholar] [CrossRef]
- García, A.; Marín, P.; Ordóñez, S. Hydrogenation of liquid organic hydrogen carriers: Process scale-up, economic analysis and optimization. Int. J. Hydrogen Energy 2024, 52, 1113–1123. [Google Scholar] [CrossRef]
- Sotoodeh, F.; Huber, B.J.M.; Smith, K.J. The effect of the N atom on the dehydrogenation of heterocycles used for hydrogen storage. Appl. Catal. A Gen. 2012, 419–420, 67–72. [Google Scholar] [CrossRef]
- Cabrera, G.; Mora, M.; Gil-Burgos, J.P.; Visbal, R.; Machuca-Martínez, F.; Mosquera-Vargas, E. Liquid Organic Hydrogen Carrier Concepts and Catalysts for Hydrogenation and Dehydrogenation Reactions. Molecules 2024, 29, 4938. [Google Scholar] [CrossRef]
- Barthos, R.; Lónyi, F.; Shi, Y.; Szegedi, Á.; Vikár, A.; Solt, H.E.; Novodárszki, G. Catalytic Aspects of Liquid Organic Hydrogen Carrier Technology. Catalysts 2025, 15, 427. [Google Scholar] [CrossRef]
- Cho, J.-Y.; Kim, H.; Oh, J.-E.; Park, B.Y. Recent Advances in Homogeneous/Heterogeneous Catalytic Hydrogenation and Dehydrogenation for Potential Liquid Organic Hydrogen Carrier (LOHC) Systems. Catalysts 2021, 11, 1497. [Google Scholar] [CrossRef]
- Kim, C.; Lee, Y.; Kim, K.; Lee, U. Implementation of Formic Acid as a Liquid Organic Hydrogen Carrier (LOHC): Techno-Economic Analysis and Life Cycle Assessment of Formic Acid Produced via CO2 Utilization. Catalysts 2022, 12, 1113. [Google Scholar] [CrossRef]
- Geißelbrecht, M.; Mrusek, S.; Müller, K.; Preuster, P.; Bösmann, A.; Wasserscheid, P. Highly efficient, low-temperature hydrogen release from perhydro-benzyltoluene using reactive distillation. Energy Environ. Sci. 2020, 13, 3119–3128. [Google Scholar] [CrossRef]
- Müller, K.; Skeledzic, T.; Wasserscheid, P. Strategies for Low-Temperature Liquid Organic Hydrogen Carrier Dehydrogenation. Energy Fuels 2021, 35, 10929–10936. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, M.; You, X.; Li, Y.; Lin, H.; Chen, J.P. A novel bimetallic Fe-Cu-CNT catalyst for effective catalytic wet peroxide oxidation: Reaction optimization and mechanism investigation. Chem. Eng. J. 2024, 479, 147320. [Google Scholar] [CrossRef]
- Mitländer, K.; Henseler, J.; Rullo, F.; Nathrath, P.; Geißelbrecht, M.; Wasserscheid, P.; Schühle, P. Continuous (hydro-)dechlorination of aromatic chloride compounds in benzyltoluene. Int. J. Hydrogen Energy 2025, 128, 674–683. [Google Scholar] [CrossRef]
- Jorschick, H.; Geißelbrecht, M.; Eßl, M.; Preuster, P.; Bösmann, A.; Wasserscheid, P. Benzyltoluene/dibenzyltoluene-based mixtures as suitable liquid organic hydrogen carrier systems for low temperature applications. Int. J. Hydrogen Energy 2020, 45, 14897–14906. [Google Scholar] [CrossRef]
- Jo, Y.; Oh, J.; Kim, D.; Park, J.H.; Baik, J.H.; Suh, Y.-W. Recent progress in dehydrogenation catalysts for heterocyclic and homocyclic liquid organic hydrogen carriers. Korean J. Chem. Eng. 2022, 39, 20–37. [Google Scholar] [CrossRef]
- Qi, S.-C.; Wei, X.-Y.; Zong, Z.-M.; Wang, Y.-K. Application of supported metallic catalysts in catalytic hydrogenation of arenes. RSC Adv. 2013, 3, 14219–14232. [Google Scholar] [CrossRef]
- Sermon, P.A.; Bond, G.C. Hydrogen Spillover. Catal. Rev. 1974, 8, 211–239. [Google Scholar] [CrossRef]
- Rozanov, V.V.; Krylov, O.V. Hydrogen spillover in heterogeneous catalysis. Russ. Chem. Rev. 1997, 66, 107–119. [Google Scholar] [CrossRef]
- Kim, C.H.; Lee, M.-W.; Jang, J.S.; Lee, S.H.; Lee, K.-Y. Enhanced activity of a WOx-incorporated Pt/Al2O3 catalyst for the dehydrogenation of homocyclic LOHCs: Effects of impregnation sequence on Pt–WOx interactions. Fuel 2022, 313, 122654. [Google Scholar] [CrossRef]
- Shi, L.; Qi, S.; Qu, J.; Che, T.; Yi, C.; Yang, B. Integration of hydrogenation and dehydrogenation based on dibenzyltoluene as liquid organic hydrogen energy carrier. Int. J. Hydrogen Energy 2019, 44, 5345–5354. [Google Scholar] [CrossRef]
- Bulgarin, A.; Jorschick, H.; Preuster, P.; Bösmann, A.; Wasserscheid, P. Purity of hydrogen released from the Liquid Organic Hydrogen Carrier compound perhydro dibenzyltoluene by catalytic dehydrogenation. Int. J. Hydrogen Energy 2020, 45, 712–720. [Google Scholar] [CrossRef]
- Auer, F.; Hupfer, A.; Bösmann, A.; Szesni, N.; Wasserscheidpeter, P. Influence of the nanoparticle size on hydrogen release and side product formation in liquid organic hydrogen carrier systems with supported platinum catalysts. Catal. Sci. Technol. 2020, 10, 6669–6678. [Google Scholar] [CrossRef]
- Musavuli, K.C.; Modisha, P.; Everson, R.C.; Malakhov, A.; Bessarabov, D. Metal Foam as Surface-Extended Catalyst Support Structure for Process Intensification in the Dehydrogenation of Perhydro-Dibenzyltoluene on a Pt/Al2O3 Catalyst. Catalysts 2025, 15, 44. [Google Scholar] [CrossRef]
- Li, H.; Zhang, X.; Zhang, C.; Ding, Z.; Jin, X. Application and Analysis of Liquid Organic Hydrogen Carrier (LOHC) Technology in Practical Projects. Energies 2024, 17, 1940. [Google Scholar] [CrossRef]
- Clematis, D.; Bellotti, D.; Rivarolo, M.; Magistri, L.; Barbucci, A. Hydrogen Carriers: Scientific Limits and Challenges for the Supply Chain, and Key Factors for Techno-Economic Analysis. Energies 2023, 16, 6035. [Google Scholar] [CrossRef]
- Auer, F.; Blaumeiser, D.; Bauer, T.; Bösmann, A.; Szesni, N.; Libuda, J.; Wasserscheid, P. Boosting the activity of hydrogen release from liquid organic hydrogen carrier systems by sulfur-additives to Pt on alumina catalysts. Catal. Sci. Technol. 2019, 9, 3537–3547. [Google Scholar] [CrossRef]
- Alconada, K.; Barrio, V.L. Enhancing perhydrobenzyltoluene dehydrogenation performance with Co, Mo and Mn metal oxides: A comparative study with Pt/Al2O3 catalyst. Appl. Catal. B Environ. Energy 2024, 357, 124349. [Google Scholar] [CrossRef]
- Dao, Q.N.; On, E.; Ramadhani, S.; Lee, K.; Sohn, H.; Choi, S.H.; Lee, S.Y.; Jeong, H.; Kim, Y. Catalytic insights into perhydro-benzyltoluene dehydrogenation: Probing surface characteristics revealed by DRIFTS study. Int. J. Hydrogen Energy 2024, 56, 1284–1293. [Google Scholar] [CrossRef]
- Jo, Y.; Kim, D.; Kim, T.W.; Yoon, D.; Suh, Y.-W. Highly dispersed Pt–MnOx nanoclusters for the promoted activity of mesoporous Pt–MnOx–Al2O3 in dehydrogenation of perhydro-benzyltoluene. Appl. Catal. B Environ. 2023, 334, 122848. [Google Scholar] [CrossRef]
- Gillan, C.; Fowles, M.; French, S.; Jackson, S.D. Ethane Steam Reforming over a Platinum/Alumina Catalyst: Effect of Sulfur Poisoning. Ind. Eng. Chem. Res. 2013, 52, 13350–13356. [Google Scholar] [CrossRef]
- Lonergan, W.W.; Vlachos, D.G.; Chen, J.G. Correlating extent of Pt–Ni bond formation with low-temperature hydrogenation of benzene and 1,3-butadiene over supported Pt/Ni bimetallic catalysts. J. Catal. 2010, 271, 239–250. [Google Scholar] [CrossRef]
- Bergeret, G.; Gallezot, P. Particle Size and Dispersion Measurements. In Handbook of Heterogeneous Catalysis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 738–765. ISBN 978-3-527-61004-4. [Google Scholar]
- Thompson, A.C.; Attwood, D.T.; Gullikson, E.M.; Howells, M.R.; Kortright, J.B.; Robinson, A.L.; Underwood, J.H. X-RAY DATA BOOKLET, 2nd ed.; Lawrence Berkeley National Laboratory University of California: Berkeley, CA, USA, 2001. [Google Scholar]
- Nam, S.W.; Gavalas, G.R. Reduction of sulfur dioxide and surface sulfate on γ-alumina. Appl. Catal. 1991, 74, 53–64. [Google Scholar] [CrossRef]
- Li-Dun, A.; Quan, D.Y. Mechanism of sulfur poisoning of supported Pd(Pt)/Al2O3 catalysts for H2-O2 reaction. Appl. Catal. 1990, 66, 219–234. [Google Scholar] [CrossRef]
- Xu, B.-Q.; Sachtler, W.M.H. Reduction of SO4=Ions in Sulfated Zirconia Catalysts. J. Catal. 1997, 167, 224–233. [Google Scholar] [CrossRef]
- Lieske, H.; Lietz, G.; Spindler, H.; Völter, J. Reactions of platinum in oxygen- and hydrogen-treated Ptγ-Al2O3 catalysts: I. Temperature-programmed reduction, adsorption, and redispersion of platinum. J. Catal. 1983, 81, 8–16. [Google Scholar] [CrossRef]
- Wang, Q.; Le, K.; Lin, Y.; Yin, W.; Lin, Y.; Alekseeva, M.V.; Yakovlev, V.A.; Koskin, A.P.; Yang, C.; Qiu, T. Investigation on catalytic distillation dehydrogenation of perhydro-benzyltoluene: Reaction kinetics, modeling and process analysis. Chem. Eng. J. 2024, 482, 148591. [Google Scholar] [CrossRef]
- Bazin, P.; Saur, O.; Lavalley, J.C.; Daturi, M.; Blanchard, G. FT-IR study of CO adsorption on Pt/CeO2: Characterisation and structural rearrangement of small Pt particles. Phys. Chem. Chem. Phys. 2005, 7, 187–194. [Google Scholar] [CrossRef]
- Ivanova, E.; Mihaylov, M.; Thibault-Starzyk, F.; Daturi, M.; Hadjiivanov, K. FTIR spectroscopy study of CO and NO adsorption and co-adsorption on Pt/TiO2. J. Mol. Catal. A Chem. 2007, 274, 179–184. [Google Scholar] [CrossRef]
- Haaland, D.M. Infrared studies of CO adsorbed on Pt/Al2O3: Evidence for CO bonded in 3-fold coordination. Surf. Sci. 1987, 185, 1–14. [Google Scholar] [CrossRef]
- Dablemont, C.; Lang, P.; Mangeney, C.; Piquemal, J.-Y.; Petkov, V.; Herbst, F.; Viau, G. FTIR and XPS Study of Pt Nanoparticle Functionalization and Interaction with Alumina. Langmuir 2008, 24, 5832–5841. [Google Scholar] [CrossRef]
- Bourane, A.; Dulaurent, O.; Bianchi, D. Comparison of the Coverage of the Linear CO Species on Pt/Al2O3 Measured under Adsorption Equilibrium Conditions by Using FTIR and Mass Spectroscopy. J. Catal. 2000, 195, 406–411. [Google Scholar] [CrossRef]
- Arteaga, G.; Anderson, J.; Rochester, C. FTIR study of CO adsorption on coked Pt–Sn/Al2O3 catalysts. Catal. Lett. 1999, 58, 189–194. [Google Scholar] [CrossRef]
- Allian, A.D.; Takanabe, K.; Fujdala, K.L.; Hao, X.; Truex, T.J.; Cai, J.; Buda, C.; Neurock, M.; Iglesia, E. Chemisorption of CO and Mechanism of CO Oxidation on Supported Platinum Nanoclusters. J. Am. Chem. Soc. 2011, 133, 4498–4517. [Google Scholar] [CrossRef] [PubMed]
- Frenking, G.; Fernández, I.; Holzmann, N.; Pan, S.; Krossing, I.; Zhou, M. Metal–CO Bonding in Mononuclear Transition Metal Carbonyl Complexes. JACS Au 2021, 1, 623–645. [Google Scholar] [CrossRef]
- Chiou, J.-F.; Huang, Y.-L.; Lin, T.-B.; Chang, J.-R. Aromatics Reduction over Supported Platinum Catalysts. 1. Effect of Sulfur on the Catalyst Deactivation of Tetralin Hydrogenation. Ind. Eng. Chem. Res. 1995, 34, 4277–4283. [Google Scholar] [CrossRef]
- Nasri, N.S.; Jones, J.M.; Dupont, V.A.; Williams, A. A Comparative Study of Sulfur Poisoning and Regeneration of Precious-Metal Catalysts. Energy Fuels 1998, 12, 1130–1134. [Google Scholar] [CrossRef]
- Zhang, N.; Li, L.; Wu, R.; Song, L.; Zheng, L.; Zhang, G.; He, H. Activity enhancement of Pt/MnOx catalyst by novel β-MnO2 for low-temperature CO oxidation: Study of the CO–O2 competitive adsorption and active oxygen species. Catal. Sci. Technol. 2019, 9, 347–354. [Google Scholar] [CrossRef]
- Mahayni, Y.; Maurer, L.; Auer, F.; Hutzler, A.; Wasserscheid, P.; Wolf, M. Structure sensitivity of the low-temperature dehydrogenation of perhydro dibenzyltoluene on supported platinum nanoparticles. Catal. Sci. Technol. 2024, 14, 5464–5473. [Google Scholar] [CrossRef]
- Jo, Y.; Wan Kim, T.; Oh, J.; Kim, D.; Suh, Y.-W. Mesoporous sulfur-decorated Pt–Al2O3 for dehydrogenation of perhydro benzyltoluenes: Activity-favorable adsorption of reaction species onto electron-deficient Pt atoms. J. Catal. 2022, 413, 127–137. [Google Scholar] [CrossRef]
- Ellert, A.; Herold, F.; Rønning, M.; Hutzler, A.; Piccirilli, L.; Janssens, T.V.W.; Vennestrøm, P.N.R.; Wasserscheid, P.; Schühle, P. Phosphorus-modification of Pt/Al2O3 catalysts improves dispersion and cycloalkane dehydrogenation activity. J. Catal. 2024, 436, 115607. [Google Scholar] [CrossRef]
- Alconada, K.; Barrio, V.L. Evaluation of bimetallic Pt–Co and Pt–Ni catalysts in LOHC dehydrogenation. Int. J. Hydrogen Energy 2024, 51, 243–255. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Pt wt.% | Mo wt.% | S wt.% | Pt:Mo | Pt:S |
|---|---|---|---|---|---|
| PtMo-0S | 0.20 | 0.45 | 0 | 0.22 | 0 |
| PtMo-0.05S | 0.23 | 0.52 | 0.06 | 0.22 | 0.61 |
| PtMo-0.10S | 0.23 | 0.52 | 0.11 | 0.22 | 0.34 |
| PtMo-0.15S | 0.17 | 0.43 | 0.15 | 0.19 | 0.19 |
| PtMo-0.20S | 0.21 | 0.50 | 0.20 | 0.22 | 0.17 |
| Sample | BET, m2/g | Pore Volume, cm3/g | Pore Radius, nm |
|---|---|---|---|
| PtMo-0S | 249.37 | 0.86 | 3.91 |
| PtMo-0.05S | 219.11 | 0.81 | 3.92 |
| PtMo-0.10S | 208.47 | 0.75 | 3.93 |
| PtMo-0.15S | 233.23 | 0.83 | 4.83 |
| PtMo-0.20S | 231.81 | 0.82 | 3.91 |
| Sample | CO, µmol/g | Pt, µmol/g | D, % |
|---|---|---|---|
| PtMo-0S | 8.93 | 10.3 | 87.1 |
| PtMo-0.05S | 8.93 | 11.8 | 75.8 |
| PtMo-0.10S | 8.49 | 11.8 | 72.0 |
| PtMo-0.15S | 8.49 | 8.7 | 97.4 |
| PtMo-0.20S | 9.38 | 10.8 | 87.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alconada, K.; Mariño, F.; Agirre, I.; Barrio, V.L. Exploring Perhydro-Benzyltoluene Dehydrogenation Using Sulfur-Doped PtMo/Al2O3 Catalysts. Catalysts 2025, 15, 485. https://doi.org/10.3390/catal15050485
Alconada K, Mariño F, Agirre I, Barrio VL. Exploring Perhydro-Benzyltoluene Dehydrogenation Using Sulfur-Doped PtMo/Al2O3 Catalysts. Catalysts. 2025; 15(5):485. https://doi.org/10.3390/catal15050485
Chicago/Turabian StyleAlconada, Kevin, Fatima Mariño, Ion Agirre, and Victoria Laura Barrio. 2025. "Exploring Perhydro-Benzyltoluene Dehydrogenation Using Sulfur-Doped PtMo/Al2O3 Catalysts" Catalysts 15, no. 5: 485. https://doi.org/10.3390/catal15050485
APA StyleAlconada, K., Mariño, F., Agirre, I., & Barrio, V. L. (2025). Exploring Perhydro-Benzyltoluene Dehydrogenation Using Sulfur-Doped PtMo/Al2O3 Catalysts. Catalysts, 15(5), 485. https://doi.org/10.3390/catal15050485